

Key Points
Cholesterol crystals induce coagulation through monocyte TF.
Monocyte TF induction is blocked by complement inhibitors to C3, C5, or C5aR1.
Monocyte TF induction is dependent on complement C5aR1.
Visual Abstract
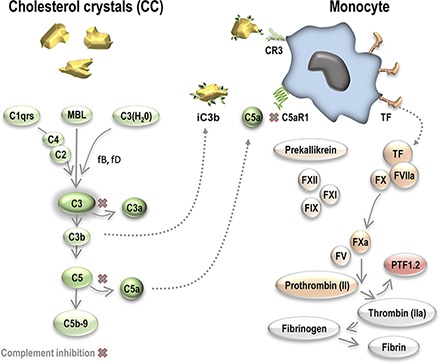
Abstract
Cholesterol crystals (CC) are strong activators of complement and could potentially be involved in thromboinflammation through complement–coagulation cross-talk. To explore the coagulation-inducing potential of CC, we performed studies in lepirudin-based human whole blood and plasma models. In addition, immunohistological examinations of brain thrombi and vulnerable plaque material from patients with advanced carotid atherosclerosis were performed using polarization filter reflected light microscopy to identify CC. In whole blood, CC exposure induced a time- and concentration-dependent generation of prothrombin fragment 1+2 (PTF1.2), tissue factor (TF) mRNA synthesis, and monocyte TF expression. Blocking Abs against TF abolished CC-mediated coagulation, thus indicating involvement of the TF-dependent pathway. Blockade of FXII by corn trypsin inhibitor had a significant inhibitory effect on CC-induced PTF1.2 in platelet-free plasma, although the overall activation potential was low. CC exposure did not induce platelet aggregation, TF microparticle induction, or TF on granulocytes or eosinophils. Inhibition of complement C3 by CP40 (compstatin), C5 by eculizumab, or C5aR1 by PMX53 blocked CC-induced PTF1.2 by 90% and reduced TF+ monocytes from 18–20 to 1–2%. The physiologic relevance was supported by birefringent CC structures adjacent to monocytes (CD14), TF, and activated complement iC3b and C5b–9 in a human brain thrombus. Furthermore, monocyte influx and TF induction in close proximity to CC-rich regions with activated complement were found in a vulnerable plaque. In conclusion, CC could be active, releasable contributors to thrombosis by inducing monocyte TF secondary to complement C5aR1 signaling.
Introduction
The World Health Organization estimates that cardiovascular disease causes more than 17 million deaths every year. Of these, 80% are due to myocardial infarction or ischemic stroke as a result of thrombosis obstructing blood flow to the heart or brain, respectively (1). Thrombosis occurs following atherosclerotic plaque rupture and involves lesion-derived tissue factor (TF) from endothelial cells (2), plaque macrophages (3, 4), and blood monocytes following exposure to oxidized LDL and C-reactive protein (5, 6). Cholesterol crystals (CC) formed upon oxidized LDL accumulation at the lesion sites are hallmarks of atherosclerotic plaques (7). Their sharp, needle-like structure promotes erosion of the fibrous cap and thereby plaque rupture with release into blood (8). Except for their role in promoting plaque rupture, CC are considered passive participators in the thrombotic events.
CC activate the complement system (9, 10), whereas their role as direct promoters of coagulation has not yet been investigated. The recent finding of CC being strong inducers of complement-dependent inflammatory processes (11) raises the question of whether CC could activate coagulation through thromboinflammation. This question is further justified in light of evidence of cross-talk between the complement and coagulation cascades (12–16).
The complement and coagulation cascades are first-line defense systems mediating inflammation and clot formation, respectively. Both consist of circulating zymogens converting to reactive proteases upon proteolytic activation (12). Complement can be activated through the classical, lectin, or alternative pathways, converging in the formation of C3 convertases. Downstream formation of C5 convertases cleaves C5 and leads to formation of C5a, a highly potent proinflammatory mediator (17), and the terminal complement complex (TCC) C5b–9. Of note, CC induce activation of all three complement pathways (18).
The coagulation system is activated either through the intrinsic pathway, activating FXII to FXIIa on a surface, or through the extrinsic pathway by binding of FVIIa to cellular expressed TF. Both pathways converge in the activation of FX to FXa, with subsequent cleavage of prothrombin to thrombin. This leads to the conversion of circulating fibrinogen to insoluble fibrin. Together with activated platelets, this process builds up a stable thrombus. TF is the major initiator of blood coagulation in vivo and is, under normal conditions, only expressed on extravascular cells. However, circulating monocytes are capable of expressing TF on their surface when exposed to proinflammatory stimuli, including LPS, TNF, C-reactive protein, and C5a (19). The latter demonstrates one of the most important points of cross-talk between the complement and coagulation systems (12).
Activated factors of complement and coagulation are evident within aspirates of human atherosclerotic plaques and thrombi. More specifically, TCC as well as mRNA of complement C1r, C1s, and C4 are found within atherosclerotic lesions (20). C3 has been detected in thrombi (21), and C3a and C5a have been shown to be increased in surrounding plasma at the site of thrombus formation (22). This suggests local complement activation within the plaque and thrombi. Leukocyte-associated TF has also been found in thrombi and is thought to contribute to clot propagation and growth (23).
Because CC activate complement, and there is an increased recognition of complement–coagulation cross-talk, the aim of the current study was to investigate the role of CC in thrombosis by studying their activation of coagulation and the dependency of complement activation in this process.
Materials and Methods
Reagents
This work includes the following reagents: anticoagulant lepirudin (Refludan) from Celgene Europe (Boudry, Switzerland) and sterile PBS (PBS with or without Ca2+ and Mg2+) and EDTA from Sigma-Aldrich (St. Louis, MO). Human serum albumin (HSA) was from Octapharma (Jessheim, Norway). Heat-inactivated Escherichia coli strain LE392 (ATCC 33572) was from the American Type Culture Collection (Manassas, VA). CryoTubes (polypropylene) were from Nunc (Roskilde, Denmark). Complement inhibitors were as follows: compstatin analog CP40 (sequence: 14-aa cyclic peptide [D]Tyr-Ile-[Cys-Val-Trp(Me)-Gln-Asp-Trp-Sar-Ala-His-Arg-Cys]-mIle-NH2) and a control peptide (Sar-Sar-Trp(Me)-Ala-Ala-Asp-Ile-His-Val-Gln-Arg-mlle-Trp-Ala-NH2), synthesized as described (24); C5 inhibitor eculizumab (Soliris) from Alexion Pharmaceuticals (New Haven, CT) and control rituximab (Roche, Basel, Switzerland); and cyclic peptide C5aR1 antagonist PMX53 (AcPhe-[Orn-ProdCha-Trp-Arg]), synthesized as described (25). TF inhibitors used included the functional grade purified inhibitory Ab against human TF (HTF-1) from eBioscience (San Diego, CA) and Ultra-LEAF Purified Mouse IgG1κ (MG1-45, 400165) from BioLegend (San Diego, CA). Corn trypsin inhibitor (CTI; CTI-01) from Haematologic Technologies (Essex Junction, VT) was used as a factor XII inhibitor. Flow cytometry Abs included anti-CD14 FITC (MΦP9) from BD Biosciences (345784), anti–HLA-DR allophycocyanin–Cy7 (L243) from BioLegend (307602), anti–Siglec-8 allophycocyanin (7C9) from BioLegend (347106), anti-TF PE (CD142, NY2) from BioLegend (365204), and anti-mouse IgG1, κ PE (MOPC-21) BioLegend (981804). FACS Lysing Solution was from BD Biosciences. Abs for histology included murine monoclonal neoepitope anti-human C9 (DIA011-01, clone E11, isotype IgG2a) from BioPorto Diagnostics (DK-2820; Gentofte, Denmark), neoepitope anti-human iC3b (A209, isotype IgG1) from Quidel (San Diego, CA), mAb against human TF (CD142, ADG4509, clone IIID8, isotype IgG1) from Sekisui (American Diagnostica, Pfungstdt, Germany), a monoclonal anti-human CD14 (18D11, isotype IgG1) synthesized in-house, monoclonal mouse anti-human CD68 (clone KP1, M0814, isotype IgG1), and anti-human CD61 (clone Y2/51, M0753, isotype IgG1) from Dako (Glostrup, Denmark). Isotype controls were purified mouse Igs IgG1 (clone MOPC-21), IgG2a (clone MOPC-172), and IgG2b (clone MPC-11) from BioLegend.
Preparation of CC and endotoxin measurement
CC were prepared as described by Samstad et al. (10). Briefly, ultrapure cholesterol (100 mg) was dissolved in 1-propanol (50 ml and aggregated by the addition of distilled water. CC were air-dried and suspended in 0.05% (w/v) HSA/PBS. After preparation, the endotoxin contamination was measured in a Chromogenic LAL Assay (Lonza). The LPS was below the detection limit (0.1 endotoxin units/ml) in a sample size of 50 mg/ml CC, and thus, the endotoxin content would be below 0.18 pg/mg CC.
Experiments in human whole blood
Human whole blood from healthy volunteers, anticoagulated with lepirudin (50 μg/ml), was used as first described by Mollnes et al. (26). Final concentrations of CC or controls were as follows: CC (500, 1000, and 2000 μg/ml), E. coli (1 × 107 particles/ml), or glass control (200 μl of PBS with Ca2+/Mg2+ in BD Vacutainer glass tubes). Final concentrations of inhibitors were as follows: CP40 or its peptide control (20 μM), eculizumab and its Ab control (100 μg/ml), anti-TF (10 μg/ml) or control Ig (10 μg/ml), and PMX53 (10 μM). The incubation time was between 60 and 240 min, as stated for the individual experiments. Complement and coagulation activity was terminated by addition of EDTA (10 mM final concentration). Whole blood was retracted for flow cytometric analysis. Plasma was harvested after centrifugation at 2000 × g for 15 min and stored in aliquots at −20°C for analyses of prothrombin fragment 1+2 (PTF1.2). After plasma harvesting, the pellets were added to 350 μl of PBS (equal to plasma harvesting) and further processed as described in TF mRNA below.
Experiments in platelet-free plasma
Platelet-free plasma (PFP) anticoagulated with lepirudin (50 μg/ml) was made by centrifugation at 2500 × g for 15 min twice at room temperature (RT). Each time the upper plasma fraction was retained while discarding the lowest 0.5 ml of rich fraction with platelets. The fresh plasma was incubated with or without CTI at a final concentration of 40 μg/ml in the presence of CC (2000 μg/ml) or in glass vials with incubation lengths of 60 and 240 min.
PTF1.2 quantification
PTF1.2 levels were quantified in duplicate using the Enzygnost Prothrombin F1+2 ELISA kit (Monoclonal OPBD03; Siemens Healthcare, Marburg, Germany) following the manufacturer’s instructions. OD was measured using a PolarStar OMEGA plate reader (BMG Labtech, Ortenberg, Germany).
TF mRNA
Cell pellets (550 μl) were added to 1.53 ml of PaxGene Blood RNA tubes solution (PreAnalytiX, Hombrechtikon, Switzerland) and stored at −80°C until RNA extraction. Total RNA was extracted using the MagNA Pure 96 Cellular RNA large volume kit (Roche Diagnostics, Mannheim, Germany) according to the manufacturer’s instructions, and the concentration was measured using the NanoDrop 2000c spectrophotometer (Thermo Fisher Scientific, Wilmington, DE). cDNA was synthesized using the High-Capacity cDNA Reverse Transcription Kit and 2720 Thermal Cycler (Applied Biosystems, Foster City, CA) and stored at −80°C. TF mRNA levels were measured using the StepOne Plus Real-Time PCR System (Applied Biosystems). Gene expression was detected by TaqMan Fast Universal PCR Master Mix reagents and predeveloped TaqMan gene expression assays using the target gene TF (TF, Hs 0017225) with reference gene human β-2-microglobulin (TaqMan B2M Probe Dye FAM, 4333766–0804015) analyzed using quantitative PCR with cycle conditions according to the manual.
TF-bearing microparticle quantification
TF-bearing microparticles (TF-MP) with procoagulant activity were quantified using Aniara ZYMUPHEN MP-TF (521196; Hyphen BioMed, Neuville Sur Oise, France) according to the producer’s protocol. Beforehand, whole blood samples were added to sodium citrate 3.2% (from Vacuette blood collection tubes; Greiner Bio-One) in 1:10 dilution and thereafter centrifuged at 1500 × g 15 min at RT before the plasma fractions were further centrifuged at 13,000 × g for 2 min at RT. PFP was then harvested by retracting 400 μl from the uppermost plasma fraction. Samples were stored at −80°C until analysis.
Flow cytometry
EDTA-inactivated blood samples (50 μl) were added to a mixture of anti-CD14 FITC (2.5 μl), anti–Siglec-8 allophycocyanin (1.25 μl), anti–HLA-DR allophycocyanin–Cy7 (1.25 μl), and anti-TF PE (IgG1; 1.25 μl) or isotype control (0.625 μl, concentration-matched) for anti-TF (anti-mouse IgG1κ PE), and RBCs were lysed by 1 ml of FACS Lysing Solution (diluted 1:10 in H2O). Samples were analyzed using a BD FACSCanto II flow cytometer with data processing in FlowJo software (version 10; Tree Star). To separate the monocytes from granulocytes and eosinophils emitting autofluorescence following CC exposure, a flow cytometric gating strategy based on the following settings was used: monocytes defined by side scatter (SSC)+(medium)/CD14+(high)/Siglec-8−/MHC class II (MHCII)+, granulocytes by SSC+(high)/CD14+(low)/Siglec-8−/ MHCII−, and eosinophils by SSC+(high)/Siglec-8+/MHCII−. The percentages of TF+ leukocytes were determined in a histogram plot set by the control Ab. The gating strategy is given in Fig. 1.
FIGURE 1.

Flow cytometric gating strategy for separating monocytes from granulocytes and eosinophils. (A) Nonstimulated blood. Monocytes were easily distinguished as the SSC+(medium)/CD14+(high), Siglec-8−, and MHCII+ population (red color in fourth picture). (B) CC-stimulated blood. Monocyte and eosinophil populations merged in the CD14+/SSC+ plot; thus, a large gate was set to include the most granular monocytes. Thereafter, monocytes were selected by a negative selection of the Siglec-8− population (based on settings from nonstimulated blood) and a positive selection of the MHCII+ cells (red color, second and third pictures), resulting in a distinguished monocyte population (red color in fourth picture). (C) E. coli–stimulated blood. Monocytes and granulocytes were separated, and the monocytes were distinguished further by Siglec-8− and MHCII+ staining. The percentage of TF+ cells was determined by a histogram plot of fluorescence intensity, determining baseline setting from nonstimulated blood, and the isotype control was subtracted from the positive staining obtained from the specific TF Ab. The granulocytes and eosinophils were further distinguished by negative and positive selection, respectively, of Siglec-8–stained cells (data not shown) and further analyzed for TF expression.
Platelet aggregation assay
Platelet aggregation was analyzed on a Multiplate electrical impedance aggregometry analyzer (Roche). Blood was collected in 4.5-ml plastic blood collection tubes containing the anticoagulant hirudin (25 μg/ml; Roche). After a 30-min rest at 20°C, the samples were diluted with 5% (v/v) PBS without Ca2+ and Mg2+ with 0.05% (w/v) HSA (Octapharma) or CC (500–2000 μg/ml). The samples were analyzed as recommended by the manufacturer, adding 0.3 ml of preheated (37°C) isotonic saline (0.9%) to 0.3 ml of whole blood in the Multiplate test cell, followed by incubation for 3 min at 37°C. The aggregation response over 6 min was expressed by the area under the curve as arbitrary units × min, either by the CC or its background control alone or following additional stimulation with 20 μl of ADP or thrombin receptor activating peptide-6 (TRAP) test as described by the producer (Roche).
Human thrombus and plaque material
Intracranial thrombus material was obtained by arterial recanalization or thrombectomy (27) from a single-center cohort study (28). Carotid plaques were obtained by carotid endarterectomy. Study inclusion criteria were acute stroke with clinical ischemic symptoms corresponding to an angiographically proven large intracranial vessel occlusion, absence of intracranial hemorrhage on computed tomography scan, and a clearly defined time of symptom onset within 6 h of inclusion. Mechanical thrombectomy involved arterial catheterization from the groin artery to the level of the intracranial vessel occlusion and thrombus retraction with the use of a retrievable stent delivered through a microcatheter. After retraction, the thrombi were stored at 4°C in Allprotect tissue reagent (Qiagen) or snap-frozen.
Histology
Histology processing was performed at the Cellular and Molecular Imaging Core Facility Histology laboratory at Norwegian University of Science and Technology. We chose frozen tissue material without preservations because of the possibility of preserving neoepitopes of activated complement and preserving the CC structures. Briefly, frozen thrombi tissue sections (4 μm) were stained for hematoxylin erythrosine saffron (HES) or with specific Abs. The HES procedure, in brief, was as follows: rehydration through descending grades of ethanol to water (1 min), staining in aqueous hematoxylin (5 min) followed by bluing in water (8 min), erythrosine treatment (5 min) and rinsing in water (1 min), dehydrating in ethanol (80, 96, and 100%), and staining in saffron (5 min) before rinsing in absolute ethanol and clearing in Tissue Clear. Cover-slipping used Sakura Tissue-Tek Glas. Specific Ab staining, in brief, was as follows: frozen sections on SuperFrost Plus slides were dried (10 min) and washed (soaking in Dako S3006 buffer), quenched for endogenous peroxidase activity in Peroxidase Block (Dako K4011), and treated with Protein Block Serum-Free (Dako X0909) for 20 min. The primary Abs were incubated overnight at 4°C by the following dilutions: anti-C9 (IgG2a, 1 mg/ml) neoepitope in 1:10,000 (plaque) and 1:5000 (thrombi) dilutions, anti-iC3b neoepitope (IgG2b, 1.04 mg/ml) in 1:1500 dilution, anti-TF (IgG1, 0.5 mg/ml) in 1:1000 dilution, anti-CD14 (IgG1, 165 μg/ml) in 1:200 dilution, anti-CD68 (IgG1, 156 mg/l) in 1:5000 dilution, and anti-CD61 (IgG1, 647 mg/l) in 1:5000 dilution. Negative controls were isotype- and concentration-matched. Finally, washing buffer was added followed by 30 min in labeled polymer HRP anti-mouse (Dako K4011), rinsing, and incubation in DAB (Dako K4011). Counterstaining was by a nonalcoholic aqueous hematoxylin (2 min) and mounting medium. Images were taken by an inverted fluorescence microscope (Olympus IX71) using a polarization filter and an Olympus XC30 CCD color camera and 4×–40× objectives, processed with Cell^P image visualization software, and optimized in Adobe Illustrator version 10.0.1 using linear stretch adjustments.
Statistical analysis
GraphPad Prism software (version 7.0a) was used for statistical analyses. Two-way ANOVA with multiple comparisons was used to evaluate the effects between stimuli, controls, and inhibitors. The Dunnett multiple comparisons test was used for the time course studies, the Tukey multiple comparisons test was used when comparing each condition of CC with its inhibitor and inhibitor control, and the Sidak multiple comparisons test was for additional data comparing each CC condition with inhibitor. The data generated by continuous scales were log-transformed beforehand to ensure normal distribution. A Wilcoxon matched-pairs signed-rank test was used for comparisons between two conditions.
Ethical approval
The use of human whole blood was approved and followed the guidelines as recommended by the Regional Committees for Medical and Health Research Ethics under Research Ethics Committee Central (REK 2009/2245) and in accordance with the Helsinki Declaration. The use of thrombi and plaque material was approved by the South East Norway Research Ethics Committee (REK 2014/2078), and written informed consent was obtained from all patients.
Results
CC induce PTF1.2 in human lepirudin whole blood
Lepirudin specifically inhibits thrombin and thus avoids fibrinogen activation and downstream coagulation while still allowing the coagulation system to be activated upstream. Despite the inhibition of the thrombin feedback loop, the upstream coagulation proteins together with all complement proteins are able to be activated, thereby allowing interaction with leukocytes. The conversion of prothrombin to thrombin results in a cleavage product, PTF1.2, that serves as an indirect estimate of the initial coagulation activation by either of the two coagulation pathways. CC 2000 μg/ml induced a significant increase (p < 0.001) of PTF1.2 compared with the PBS/HSA control following 240 min of incubation (Fig. 2A). Significant elevation was achieved for CC 1000 μg/ml (p < 0.05), whereas a nonsignificant (p > 0.05) elevation was obtained under exposure to CC 500 μg/ml (data not shown). Glass significantly elevated PTF1.2 levels by 60 min (p < 0.0001) and E. coli by 120 min (p < 0.001) (Fig. 2A). Glass is a highly potent activator of coagulation FXII and can be inhibited by CTI (29), whereas E. coli activates TF that can be explored by TF-inhibiting Abs (14). Inhibiting TF prior to addition of CC using a TF-blocking Ab efficiently and significantly (p < 0.0001) blocked the CC- and E. coli–induced PTF1.2 (Fig. 2B). These data pointed to the TF-driven pathway as most important for the CC-induced PTF1.2. To test for the FXII dependency, CC or glass were incubated in lepirudin PFP with or without blockage of FXII using CTI. In PFP, the overall PTF1.2 induction by CC or glass was low (just above baseline) but could still be significantly (p < 0.05) inhibited by CTI (Fig. 2C). The ability of CC to induce platelet aggregation was investigated by electrical impedance aggregometry (Fig. 2D–F). CC did not induce platelet aggregation as compared with its background control (HSA/PBS) and positive control, TRAP (Fig. 2D). The CC did not induce additional activation or inhibition in activated platelets, as investigated by the ADP test (Fig. 2E) or the TRAP test (Fig. 2F). Altogether, these data point to the TF pathway as the main pathway triggered by CC, with a small contribution from FXII.
FIGURE 2.

CC induce PTF1.2 dependent on the TF pathway. (A) PTF1.2 induction over time (60, 120, and 240 min) upon exposure to CC 2000 μg/ml, PBS/HSA, E. coli (1 × 107 particles/ml), or glass (n = 7 donors) in human whole blood. (B) Effect of TF inhibition following exposure of CC (500, 1000, and 2000 μg/ml) or E.coli after addition of the functional-grade inhibitory Ab against TF (anti-TF) or the corresponding Ultra-LEAF purified IgG1κ control Ab (Ctr IgG) for 240 min in human whole blood (n = 6 donors). (C) Effect of factor XII inhibition by CTI (40 μg/ml) in PFP following incubation with CC (2000 μg/ml) or glass for 60 min (n = 5). Effect of CC on platelet aggregation in human whole blood measured by electrical impedance aggregometry in (D) by CC (2000 μg/ml) or its controls, HSA/PBS, or TRAP (n = 6), (E) by additional stimulation with ADP (n = 3), and in (F) with TRAP following 6 min of incubation (n = 3), given as arbitrary units × min. In all experiments, thrombin was inhibited with lepirudin. All data are given as means ± SEM. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001.
CC induce coagulation through monocytic TF
The procoagulant activity of TF detected in the inhibition studies (Fig. 2B) could be a consequence of cellular expressed TF or TF-MP. Measuring TF at the transcriptional level (TF mRNA) after CC 2000 μg/ml exposure in the whole blood resulted in a highly significant (p < 0.0001) increase as compared with the PBS/HSA control, peaking after 60 min of incubation (Fig. 3A). Similar patterns, although less prominent, were found following exposure of CC 1000 μg/ml and CC 500 μg/ml (Supplemental Table I). In comparison, E. coli induced a significant (p < 0.0001) and stronger response but also peaking after 60 min (Fig. 3A). Measuring the TF-MP activity in human whole blood (Fig. 3B) resulted in the lack of activity following CC exposure, although the E. coli induction was consistent with previous data (14).
FIGURE 3.

CC induce TF mRNA and TF expression on the monocyte surface. Human whole blood was incubated with CC (500, 1000, and 2000 μg/ml) or controls, PBS/has, or E. coli (1 × 107 particles/ml) at different time points (60, 120, and 240 min). (A) TF mRNA in whole blood measured by quantitative RT-PCR (n = 6 donors); additionally, CC 1000 and 500 μg/ml are in Supplemental Table II. (B) The procoagulant activity of TF-MP (n = 3 donors). (C) Monocyte TF expression exemplified with flow cytometry histogram after 120 min and quantified as percentage of monocytes expressing surface TF at all time points (n = 6 donors). Additionally, CC 1000 and 500 μg/ml are shown in Supplemental Table II. (D) The lack of granulocyte TF expression exemplified with flow cytometry histogram after 120 min and quantified as percentage of granulocytes expressing surface TF at all time points (n = 6 donors). Histogram baselines (T0) are stippled lines, isotype controls are filled gray, and the TF expression are black lines. Graphs are given as means ± SEM. Significant differences between the stimuli and inhibitors at the given time points are marked with asterisks. **p < 0.01, ***p < 0.001, ****p < 0.0001.
We further investigated the TF expression on the surface of the monocyte and granulocyte populations using flow cytometry. The monocytic population (CD14+/Siglec-8−/MHCII+) histogram showed a clear shift to the right for TF following CC exposure (Fig. 3C) and stronger for E. coli. Quantification from six donors indicated an average of 23% of the monocytic population expressing TF at the surface upon CC 2000 μg/ml exposure and between 18 and 20% at the lower concentrations (Supplemental Table II). Of note, a significant increase was detected already after 60 min for CC 2000 μg/ml (p < 0.001) and CC 1000 μg/ml (p < 0.05) and after 120 and 240 min for all concentrations of CC (Supplemental Table II). E. coli induced the fastest and strongest TF expression, involving between 70 and 80% of the monocyte population, in agreement with previous findings. In contrast, no shift in TF was found on the surface of the granulocytes (SSChigh/CD14−/Siglec-8−) for either CC or E. coli stimulation (Fig. 3D) (14). Equally, no TF induction could be detected on the eosinophilic population (SSChigh/MHCII−/Siglec-8+; data not shown). In conclusion, CC induces synthesis of TF mRNA, whereas only monocytes express TF at the surface.
CC induce coagulation through an initial complement activation
CC are strong activators of complement, leading to TCC formation (10). Complement-induced TF has previously been demonstrated (13–15), suggesting a possibility also for CC-induced coagulation. Inhibiting complement by blocking C3 (CP40) or C5 (eculizumab) significantly (p < 0.0001) reduced the CC-induced PTF1.2 and monocyte TF expression close to baseline (Fig. 4A–D). Of note, the percentage of monocytes expressing TF was <1% upon inhibition of C3 and <2% upon inhibition of C5. Upon E. coli exposure, the inhibition was less efficient, in accordance with previous data demonstrating the need for dual blockage of CD14 and complement C5 to avoid E. coli–induced coagulation (14). Because the inhibition of CC was efficient also at the level of C5, we further investigated the involvement of the anaphylatoxin C5a receptor, C5aR1, using the inhibitor PMX53. The effective blocking of the CC-induced PTF1.2 and monocyte TF by PMX53 points to an important role of the C5a–C5aR1 axis in the CC-induced coagulation (Fig. 4E, 4F). In comparison, blocking of E. coli–induced PTF1.2 and monocytic TF by PMX53 was less effective (Fig. 4E, 4F). In conclusion, the CC-induced coagulation activation measured by PTF1.2 and monocytic TF is dependent on initial complement activation, in which the anaphylatoxin receptor C5aR1 is central.
FIGURE 4.

CC induce coagulation activation dependent on an initial complement activation. Whole blood was incubated with CC (500, 1000, or 2000 μg/ml), PBS/HSA, or E. coli (1 × 107 particles/ml) for 240 min with the C3 inhibitor CP40, the C5 inhibitor eculizumab, or their respective controls (scrambled peptide for CP40 or rituximab as control for eculizumab). (A) PTF1.2 production following C3 inhibition (CP40; n = 7 donors). (B) Monocyte TF expression at 120 min of incubation following C3 inhibition (n = 6 donors). (C) PTF1.2 production following complement C5 inhibition (eculizumab; n = 7 donors). (D) Monocyte TF expression at 120 min of incubation following C5 inhibition (n = 6 donors). (E) PTF1.2 following inhibition of complement receptor C5aR1 with PMX53 after 240 min (n = 5 donors). (F) Monocyte TF expression after 120 min of incubation following inhibition of C5aR1 with PMX53 (n = 5 donors). Data are given as means ± SEM. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001.
CC are found within human intracranial vessel thrombus material and are associated with activated complement, monocytes, and TF+ leukocytes
Our studies in whole blood pointed to complement and monocytes as essential participators in CC-induced coagulation. To further explore the physiological relevance of these findings, we next explored thrombus material retracted from the intracranial vessel of patients with advanced carotid atherosclerosis. First, we investigated the possibility of detecting CC structures within the thrombus material and next in the association with complement and monocytes. Because CC are one of few materials showing birefringence in human tissue (30), it was possible to detect CC structures by polarization filter reflected light microscopy. Birefringent structures similar to CC were detected within hematoxylin-stained samples (Fig. 5A, 5B). In the corresponding HES sections, the birefringent structures were absent (Fig. 5C, 5D). Crystalized cholesterol dissolves in ethanol (31), which is one of the constituents of HES. The disappearance of birefringent structures upon HES staining suggested that the structures could be of cholesterol origin.
FIGURE 5.

Histology from frozen sections of human intracranial thrombus retracted from a patient with advanced carotid atherosclerosis and explored by polarization filter reflected light microscopy. (A and B) Hematoxylin-stained sections with easily identifiable birefringent CC marked with orange arrows. (C and D) HES-stained sections of corresponding areas. Corresponding areas are (A) and (D), and (B) and (C). The thicknesses of the sections are 4 μm. Images were taken with original magnification ×40 objective by a polarization filter to visualize birefringent structures. Scale bar, 30 μm.
Of note, the CC areas were richly populated with cells having bean-shaped nuclei typical for monocytes (Fig. 5). Immunohistochemistry staining confirmed the presence of monocytes within the CC-rich areas, as marked by yellow arrows (Fig. 6A, 6B). The corresponding adjacent section stained prominently for TF (Fig. 6C, middle picture). Furthermore, complement neoepitopes C5b–9 (Fig. 5D) were densely present and strongly associated with the birefringent CC structures (yellow arrows). The neoepitope iC3b was also found within the CC-rich areas (Fig. 5E), in addition to being present in the outermost regions of the thrombus (Fig. 5E, left picture, blue arrows). The presence of monocytes/macrophages in the thrombus was further confirmed by positive staining for CD68 in areas rich in CC (Supplemental Fig. 1A). Platelets (CD61), in contrast, were present throughout the thrombus (Supplemental Fig. 1B). The Ab specificities were manifested by the areas free of staining and further confirmed by the lack of staining by their respective isotype control, IgG1 (Supplemental Fig. 1C), IgG2a, and IgG2b (data not shown). In conclusion, the histological examinations pointed to a physiological relevance of CC and complement in thrombosis through the activation of monocytic TF.
FIGURE 6.

Immunohistochemistry of frozen sections of human intracranial thrombus retracted from a patient with advanced carotid atherosclerosis and explored with polarization filter reflected light microscopy. (A and B) Staining against the monocyte marker CD14. (C) Staining against TF. (D) Staining against the complement neoepitopes C5b–9. (E) Staining against the complement neoepitope iC3b. The thicknesses of the sections are 4 μm. Pictures in the left column are taken by original magnification ×4 objective, and the CC-rich area (blue frame) is further augmented with an original magnification ×40 objective (middle and right columns). Examples of birefringent structures are shown by yellow arrows, whereas blue arrows point to the outer thrombi sections stained heavily for neoepitope iC3b. Scale bars, 200 μm for pictures in left column and 30 μm for pictures in middle and right columns.
Monocyte TF/CD14 expression patterns are connected to CC and activated complement in vulnerable plaque and in whole blood
Vulnerable plaques were examined for ruptures, the presence of CC, complement, and monocytes. One sample showed signs of an open lumen and had large CC present near the adventitia (Fig. 7A, Supplemental Fig. 2). Briefly, within the open lumen bright staining with the monocyte marker CD14 (CD14bright) suggested monocyte influx and blood exposure (Fig. 7A, yellow arrows). In the innermost region of the lumen, a layer of CD14low-staining monocytes was detected (Fig. 7A, first picture, blue arrows). The corresponding layer stained positive for TF (second picture, blue arrows). Of note, bright CC structures were observed in the adjacent region (marked CC). The CC-rich areas stained heavily for complement iC3b and C5b–9 (Fig. 7A, third and fourth pictures). The monocytic origin of the TF+ cells was further confirmed by positive staining for the monocyte/macrophage marker CD68 and negative staining for CD34 and CD61 in corresponding areas (Supplemental Fig. 2A). Larger magnifications confirmed the monocytes’ origin by the presence of bean-shaped nuclei (Supplemental Fig. 2B) both for the CD14bright and the CD14low/TF+ areas (Supplemental Fig. 2C, 2D). The presence of CC in the area adjacent to the CD14low/TF+ cells is clearly shown (Supplemental Fig. 2C–E). Endothelial staining was only found in the area of CD14bright monocytes, with irregular staining patterns typical for endothelial renewing (Supplemental Fig. 2E). The Ab specificities were confirmed by negative controls (data not shown).
FIGURE 7.

Human vulnerable plaque and whole blood show corresponding patterns in monocyte TF/CD14 expression and complement activation to CC. (A) Immunohistochemistry of the vulnerable plaque with the monocyte marker CD14, TF, and activated complement neoepitope iC3b and neoepitopes C5b–9. Staining areas for CD14bright monocytes are marked with yellow arrows, and CD14low/TF+-expressing monocytes are marked by blue arrows; specified also are the CC-rich areas, lumen, and adventitia. Sections are 4 μm and are taken by original magnification ×4 objective and given for two adjacent areas in each staining. Scale bar, 200 μm. Whole blood following CC exposure showing effects on (B) monocyte CD14 expression, (C) correlation between CD14 and TF in monocytes, (D) the increase in monocyte granularity, (E) monocyte CD14 following inhibition of complement C3 (120 min), (F) monocyte CD14 following inhibition of complement C5 (120 min), (G) monocyte granularity following inhibition of C3 (120 min), and (H) monocyte granularity following inhibition of C5 (120 min). Data are given as mean fluorescence intensity (MFI; mean ± SEM, n = 6 donors). *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001.
The findings of CD14low/TF+-expressing leukocytes within the plaque could be explained by further observations in whole blood upon CC exposure (Fig. 7B–H). Briefly explained, in the monocytes, a reduction of CD14 from the initial expression level over time was observed, with a significant lowering for CC 2000 μg/ml after 120 and 240 min (p < 0.001–0.0001) and for CC 1000 μg/ml after 240 min (p < 0.01) (Fig. 7B). Moreover, a negative correlation (r = −0.697) between CD14 expression and the TF+ monocytes (percentage) was identified (Fig. 7C). The CC exposure led to a substantial and significant (p < 0.0001) increase in the granularity, indicating phagocytosis (10) (Fig. 7D). The reduction in CD14 (Fig. 7E, 7F) and increase in granularity (Fig. 7G, 7H) could be inhibited by blocking complement at the level of C3 and C5, thus linking both events to complement. From these findings, we would suggest that CC in a vulnerable plaque situation activates complement, with a subsequent influx of monocytes, phagocytosis of CC, and induction of TF in monocytes. During this process, CD14 is internalized, in part through complement-mediated activity.
Discussion
CC have emerged as important contributors to atherosclerotic inflammation (11), but their potential role in thrombosis is unexplored. In this study, we show that CC induce coagulation through a complement-dependent expression of TF in monocytes by using an experimental whole blood model. The physiological relevance of these findings is supported by human intracranial vessel thrombus and vulnerable plaque histology demonstrating CC in association with monocyte TF (CD14/TF+ leukocytes) and activated complement (iC3b and C5b–9). The present data thus point to an active role of CC and complement in thrombosis.
Atherosclerotic plaques can contain dense amounts of CC (32), suggesting relatively high concentrations of CC at the releasing site. In this study, we show that CC in concentrations of 500–2000 μg/ml induce coagulation by cleavage product PTF1.2, mediated mainly through the TF pathway and TF surface expression by monocytes. Our data indicate a time span of CC-induced TF expression of 1–2 h, with TF mRNA peaking after 1 h and full surface expression after 2 h. Although endothelial damage could induce a faster response in concert with platelets, these findings point to releasable CC as active contributors in thrombus formation and growth. The demonstration of monocyte influx and TF-expressing monocytes in a vulnerable human plaque further support monocytes as important effectors in thrombosis. The time frame from plaque debris release to clinical symptoms is not well defined and depends on thrombus size and localization. According to clinical guidelines (33), patients with advanced carotid atherosclerosis should undergo carotid endarterectomy preferably within 48 h after a transient ischemic attack because of the high risk of recurrent infarction. The clinical picture with small ischemic occlusions such as transient ischemic attack promotes a high-risk period for up to 2 mo, in which patients are particularly vulnerable to new and often larger attacks. The release of CC during this period could thus promote thrombosis by inducing TF expression on monocytes.
Cholesterol embolization syndrome is a phenomenon characterized by a multitude of small emboli occurring over time in association with CC, with an incidence of 22–27% during surgical treatment of atherosclerosis (34). Of note, this phenomenon was connected to complement consumption, which fits well with the complement-dependent CC-induced coagulation activation demonstrated in this study. Our data might contribute to a mechanistic explanation by demonstrating that the coagulation activation is mediated predominantly by complement, with more than 90% inhibition at either the level of C3 or C5. The efficient prevention of coagulation by inhibiting the anaphylatoxin receptor C5aR1 points to an essential role of the C5a–C5aR1 signaling axis in CC-induced coagulation. This clear effect was in contrast to the partial inhibition of coagulation by C5aR1 under exposure to E. coli and clearly points to a specific complement-mediated activation by CC. Bacteria-induced coagulation is, in addition to complement, also dependent on CD14 (26) and, furthermore, connected to the TLR pathway (35). The lack of direct activation of CD14/TLRs by CC might explain the lower potency of CC to induce TF mRNA, the lower number of TF+ monocytes, and the lack of TF-MP activity presently demonstrated. We observed, however, a rapid loss of CD14 in correlation with TF induction following CC exposure. This phenomenon was concentration-dependent and linked to complement and could possibly be the consequence of phagocytosis of CC. Whether CD14 is involved in the signal induction leading to TF expression remains to be elucidated. Previous exploration by our group has shown that the CC-induced complement activation mediates upregulation of CR3 (CD11b/CD18) through C5a/C5aR1 (10). The additional opsonization with complement fragments at the level of C3 leads to efficient phagocytosis of CC. This was also demonstrated in the current study by the concentration-dependent granulation in monocytes after CC exposure, which could be blocked by the complement inhibitors both at the level of C3 and C5. Although we did not explore the link between monocyte TF and the phagocytic events, the rapid phagocytosis prior to TF mRNA synthesis, as well as the complement dependency for both, could indicate that these events are connected.
The inflammatory cytokines TNF and IL-1β, among others, are triggered by CC exposure (10). The induction of TF has also been connected to TNF (36), which could suggest its involvement in CC-mediated coagulation. However, the time span between CC exposure and TF mRNA induction of 1 h points to a shorter induction time than is needed for activation of TNF. It is, thus, more likely that these processes are initiated in parallel in connection to complement than that TNF plays a central role in the initial TF induction by CC. The NLRP3 inflammasome has been linked to CC-induced IL-1β secretion mediated by C5a (10). Further on, NLRP3 has been linked to induction of TF-MP. However, our investigation in whole blood using the specific NLRP3 inhibitor MCC950, caspase-1 inhibitor VX-765, and pan-caspase inhibitor Z-VAD-FMK did not provide evidence for its involvement in CC-induced TF expression or downstream coagulation activation (data not shown). Additionally, no induction of TF-MP by CC was observed in our system. As regulation of TF has been shown to occur mainly on the transcriptional level (37), our findings imply that CC-induced generation of C5a activates the C5a–C5aR1 axis to drive synthesis of TF mRNA, which has been shown to occur both in monocytes (38) and in endothelial cells (39).
From the present and previous data, CC can be regarded as participators in thromboinflammation with a strong dependency on complement. Our investigation points to the monocytes as the only leukocytes able to induce TF upon exposure to CC, as surface expression on granulocytes and eosinophils was absent. CR3 expression is, in contrast, induced both on the monocytes and granulocytes (10), and our explorations also revealed increased granulocyte granularity upon CC exposure (data not shown). This further points to the ability of CC to activate the granulocytes but not induce surface expression of TF. Although there is agreement regarding the capability of monocytes to express TF, the expression of TF on other cell types is debated (40–42). These discrepancies in cellular sources could be related to the health status of the donors as well as the type of stimuli (36, 42, 43). Platelets contribute in the initial clot-formation process by binding to damaged vascular endothelium (44), and connections between platelets and monocyte TF are commonly accepted (45). We found no direct effect of CC on platelet aggregation or in the induction of PTF1.2 following CC exposure in platelet-containing plasma (data not shown). The tiny induction of PTF1.2 following CC exposure in PFP, which was still significantly inhibited by CTI, suggests a small contribution of FXII in CC-induced coagulation. This could also explain the ∼10% of PTF1.2 activity remaining following complement inhibition. A limitation of the lepirudin anticoagulant is the inhibition of thrombin, the major platelet activator (46). However, because the platelets also contain complement receptors (47), a complement-dependent coagulation activation by the platelets could be possible. The presence of iC3b in the outermost parts of the thrombus also opens a role for complement–platelet interactions in thrombus growth, as previously described (22, 48).
CC are normally observed as imprints (CC clefts) in plaques because of their dissolution when specimens are treated with ethanol during regular immunohistochemical staining (31). CC accumulation over time gives rise to cleft structures within atherosclerotic plaques that are easily observed under the microscope. However, upon plaque rupture, CC will be subjected to blood flow and, therefore, more challenging to detect during thrombus formation. CC can be observed using polarization filter reflected light microscopy because of their high birefringence (30). The detection of complement products is also more efficient in frozen samples, as used in this study. A recent paper suggests that freezing biopsy specimens from atherosclerotic plaques may produce birefringent structures in frozen section samples that do not represent CC (49). An exclusion of such artifacts from the present images is not possible, but still, several findings support the birefringent structures being of CC origin: 1) the birefringent structures were associated with activated complement, monocytes, and TF; 2) monocytes or TF+ cells were adjacent to CC structures; 3) the shape of the birefringent structures resembled the square CC made in vitro for the smaller structures, whereas the larger needle-shaped or larger assemblies were similar to those seen in atherosclerotic plaques; and 4) upon HES staining of stepwise ethanol treatment, the birefringent structures disappeared, thereby supporting that the structures contained cholesterol. Altogether, these findings point to a CC nature of the birefringent structures.
Based on the efficiency of complement inhibition in reducing CC-induced coagulation, targeting complement warrants further exploration as a future treatment option for prevention of thrombosis. Currently, the complement C3 inhibitor CP40 is under clinical development (50), whereas the C5 inhibitor eculizumab is in clinical use for treatment of paroxysmal nocturnal hemoglobinuria (51). These patients often suffer from thromboembolism, and targeted treatment of C5 has proven to reduce the thrombotic events by 81% (51). In a preclinical porcine model of myocardial infarction, C5 treatment also reduced the infarction size (52). Targeting IL-1β has recently been proven to reduce the incidence of thrombosis in patients with underlying atherosclerosis (53). Of note, complement activation is an upstream event to the inflammatory cytokines, including IL-1β (10), and thus, targeting complement could represent an alternative strategy. This might also avoid a compromised immune activation upon infections, as is the concern with anti–IL-1β therapy (53).
In conclusion, the current study reveals a role for CC in thromboinflammation. CC possess a procoagulant feature dependent on complement and monocytic TF, and following plaque rupture, releasable CC might contribute to thrombosis. The use of complement inhibitors targeting either C3, C5, or C5aR1 could be an alternative clinical strategy for prophylactic treatment of atherosclerotic plaques or after thrombosis and warrants further exploration.
Supplementary Material
Acknowledgments
We acknowledge the Cellular and Molecular Imaging Core Facility at Norwegian University of Science and Technology, Ingunn Nervik for the immunohistochemistry, and Bjørnar Sporsheim for image optimization. Liv Ryan and Hilde Fure are acknowledged for technical help and valuable discussions in the initial experimental phase. Technical assistance on the electrical impedance aggregometry by Judith Krey Ludviksen is also greatly acknowledged as well as the production of CC by Nathalie Niyonzima. We are grateful to the department head of pathology, Harald Aarset, for evaluating the prepared samples from vulnerable plaque histology.
This work was supported by The Liaison Committee for Education, Research and Innovation in Central Norway (Regional Health Authority, Samarbeidsorganet) under Grant 46056819, the Joint Research Committee between St. Olav's Hospital and the Faculty of Medicine and Health Science, Norwegian University of Science and Technology, under Grant 46082800, The Research Council of Norway through its Centers of Excellence funding scheme (Project 223255), the European Community’s Seventh Framework Program under Grant Agreement 602699 (DIREKT), National Institutes of Health Grants AI068730 and AI030040, the Odd Fellow Foundation (Grant OFF-2014), and the Simon Fougner Hartmann Family Fund (SFHF-12/14).
The online version of this article contains supplemental material.
- CC
- cholesterol crystal
- CTI
- corn trypsin inhibitor
- HES
- hematoxylin erythrosine saffron
- HSA
- human serum albumin
- MHCII
- MHC class II
- PFP
- platelet-free plasma
- PTF1.2
- prothrombin fragment 1+2
- RT
- room temperature
- SSC
- side scatter
- TCC
- terminal complement complex
- TF
- tissue factor
- TF-MP
- TF-bearing microparticle
- TRAP
- thrombin receptor activating peptide-6.
Disclosures
J.D.L. is the inventor of patents and/or patent applications that describe the use of complement inhibitors for therapeutic purposes, the founder of Amyndas Pharmaceuticals, which is developing complement inhibitors (i.e., third-generation compstatins) for clinical applications, and the inventor of the compstatin technology licensed to Apellis Pharmaceuticals (i.e., 4[1MeW]7W/POT-4/APL-1 and PEGylated derivatives). The other authors have no financial conflicts of interest.
References
- 1.World Health Organization 2016. Cardiovascular diseases (CVDs).
- 2.Breitenstein A., Camici G. G., Tanner F. C. 2009. Tissue factor: beyond coagulation in the cardiovascular system. 118: 159–172. [DOI] [PubMed] [Google Scholar]
- 3.Nishi K., Itabe H., Uno M., Kitazato K. T., Horiguchi H., Shinno K., Nagahiro S. 2002. Oxidized LDL in carotid plaques and plasma associates with plaque instability. Arterioscler. Thromb. Vasc. Biol. 22: 1649–1654. [DOI] [PubMed] [Google Scholar]
- 4.Yasojima K., Schwab C., McGeer E. G., McGeer P. L. 2001. Generation of C-reactive protein and complement components in atherosclerotic plaques. Am. J. Pathol. 158: 1039–1051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Owens A. P., III, Byrnes J. R., Mackman N. 2014. Hyperlipidemia, tissue factor, coagulation, and simvastatin. Trends Cardiovasc. Med. 24: 95–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cai H., Song C., Lim I. G., Krilis S. A., Geczy C. L., McNeil H. P. 2005. Importance of C-reactive protein in regulating monocyte tissue factor expression in patients with inflammatory rheumatic diseases. J. Rheumatol. 32: 1224–1231. [PubMed] [Google Scholar]
- 7.Grebe A., Latz E. 2013. Cholesterol crystals and inflammation. Curr. Rheumatol. Rep. 15: 313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Abela G. S., Aziz K., Vedre A., Pathak D. R., Talbott J. D., Dejong J. 2009. Effect of cholesterol crystals on plaques and intima in arteries of patients with acute coronary and cerebrovascular syndromes. Am. J. Cardiol. 103: 959–968. [DOI] [PubMed] [Google Scholar]
- 9.Vogt W., von Zabern I., Damerau B., Hesse D., Lühmann B., Nolte R. 1985. Mechanisms of complement activation by crystalline cholesterol. Mol. Immunol. 22: 101–106. [DOI] [PubMed] [Google Scholar]
- 10.Samstad E. O., Niyonzima N., Nymo S., Aune M. H., Ryan L., Bakke S. S., Lappegård K. T., Brekke O. L., Lambris J. D., Damås J. K., et al. 2014. Cholesterol crystals induce complement-dependent inflammasome activation and cytokine release. J. Immunol. 192: 2837–2845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Niyonzima N., Halvorsen B., Sporsheim B., Garred P., Aukrust P., Mollnes T. E., Espevik T. 2017. Complement activation by cholesterol crystals triggers a subsequent cytokine response. Mol. Immunol. 84: 43–50. [DOI] [PubMed] [Google Scholar]
- 12.Markiewski M. M., Nilsson B., Ekdahl K. N., Mollnes T. E., Lambris J. D. 2007. Complement and coagulation: strangers or partners in crime? Trends Immunol. 28: 184–192. [DOI] [PubMed] [Google Scholar]
- 13.Gravastrand C., Hamad S., Fure H., Steinkjer B., Ryan L., Oberholzer J., Lambris J. D., Lacík I., Mollnes T. E., Espevik T., et al. 2017. Alginate microbeads are coagulation compatible, while alginate microcapsules activate coagulation secondary to complement or directly through FXII. Acta Biomater. 58: 158–167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Landsem A., Fure H., Christiansen D., Nielsen E. W., Østerud B., Mollnes T. E., Brekke O. L. 2015. The key roles of complement and tissue factor in Escherichia coli-induced coagulation in human whole blood. Clin. Exp. Immunol. 182: 81–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Langer F., Spath B., Fischer C., Stolz M., Ayuk F. A., Kröger N., Bokemeyer C., Ruf W. 2013. Rapid activation of monocyte tissue factor by antithymocyte globulin is dependent on complement and protein disulfide isomerase. Blood 121: 2324–2335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Øvstebø R., Hellum M., Aass H. C. D., Trøseid A. M., Brandtzaeg P., Mollnes T. E., Henriksson C. E. 2014. Microparticle-associated tissue factor activity is reduced by inhibition of the complement protein 5 in Neisseria meningitidis-exposed whole blood. Innate Immun. 20: 552–560. [DOI] [PubMed] [Google Scholar]
- 17.Ricklin D., Hajishengallis G., Yang K., Lambris J. D. 2010. Complement: a key system for immune surveillance and homeostasis. Nat. Immunol. 11: 785–797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pilely K., Rosbjerg A., Genster N., Gal P., Pál G., Halvorsen B., Holm S., Aukrust P., Bakke S. S., Sporsheim B., et al. 2016. Cholesterol crystals activate the lectin complement pathway via ficolin-2 and mannose-binding lectin: implications for the progression of atherosclerosis. J. Immunol. 196: 5064–5074. [DOI] [PubMed] [Google Scholar]
- 19.Chu A. J. 2011. Tissue factor, blood coagulation, and beyond: an overview. Int. J. Inflamm. 2011: 367284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yasojima K., Schwab C., McGeer E. G., McGeer P. L. 2001. Complement components, but not complement inhibitors, are upregulated in atherosclerotic plaques. Arterioscler. Thromb. Vasc. Biol. 21: 1214–1219. [DOI] [PubMed] [Google Scholar]
- 21.Howes J. M., Richardson V. R., Smith K. A., Schroeder V., Somani R., Shore A., Hess K., Ajjan R., Pease R. J., Keen J. N., et al. 2012. Complement C3 is a novel plasma clot component with anti-fibrinolytic properties. Diab. Vasc. Dis. Res. 9: 216–225. [DOI] [PubMed] [Google Scholar]
- 22.Distelmaier K., Adlbrecht C., Jakowitsch J., Winkler S., Dunkler D., Gerner C., Wagner O., Lang I. M., Kubicek M. 2009. Local complement activation triggers neutrophil recruitment to the site of thrombus formation in acute myocardial infarction. Thromb. Haemost. 102: 564–572. [DOI] [PubMed] [Google Scholar]
- 23.Palmerini T., Tomasi L., Barozzi C., Della Riva D., Mariani A., Taglieri N., Leone O., Ceccarelli C., De Servi S., Branzi A., et al. 2013. Detection of tissue factor antigen and coagulation activity in coronary artery thrombi isolated from patients with ST-segment elevation acute myocardial infarction. PLoS One 8: e81501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Qu H., Magotti P., Ricklin D., Wu E. L., Kourtzelis I., Wu Y. Q., Kaznessis Y. N., Lambris J. D. 2011. Novel analogues of the therapeutic complement inhibitor compstatin with significantly improved affinity and potency. Mol. Immunol. 48: 481–489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kumar V., Lee J. D., Clark R. J., Woodruff T. M. 2018. Development and validation of a LC-MS/MS assay for pharmacokinetic studies of complement C5a receptor antagonists PMX53 and PMX205 in mice. Sci. Rep. 8: 8101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mollnes T. E., Brekke O. L., Fung M., Fure H., Christiansen D., Bergseth G., Videm V., Lappegård K. T., Köhl J., Lambris J. D. 2002. Essential role of the C5a receptor in E coli-induced oxidative burst and phagocytosis revealed by a novel lepirudin-based human whole blood model of inflammation. [Published erratum appears in 2002 Blood 100: 2691.] Blood 100: 1869–1877. [PubMed] [Google Scholar]
- 27.Powers E. R. 2016. Aspiration thrombectomy: the possible importance of effective thrombus removal and minimal residual thrombus burden. JACC Cardiovasc. Interv. 9: 2012–2013. [DOI] [PubMed] [Google Scholar]
- 28.Skagen K., Skjelland M., Russell D., Jacobsen E. A. 2015. Large-vessel occlusion stroke: effect of recanalization on outcome depends on the National Institutes of Health stroke scale score. J. Stroke Cerebrovasc. Dis. 24: 1532–1539. [DOI] [PubMed] [Google Scholar]
- 29.Hansson K. M., Nielsen S., Elg M., Deinum J. 2014. The effect of corn trypsin inhibitor and inhibiting antibodies for FXIa and FXIIa on coagulation of plasma and whole blood. J. Thromb. Haemost. 12: 1678–1686. [DOI] [PubMed] [Google Scholar]
- 30.James J., Tanke H. J. 2012. Biomedical Light Microscopy. Springer, the Netherlands. [Google Scholar]
- 31.Nasiri M., Janoudi A., Vanderberg A., Frame M., Flegler C., Flegler S., Abela G. S. 2015. Role of cholesterol crystals in atherosclerosis is unmasked by altering tissue preparation methods. Microsc. Res. Tech. 78: 969–974. [DOI] [PubMed] [Google Scholar]
- 32.Janoudi A., Shamoun F. E., Kalavakunta J. K., Abela G. S. 2016. Cholesterol crystal induced arterial inflammation and destabilization of atherosclerotic plaque. Eur. Heart J. 37: 1959–1967. [DOI] [PubMed] [Google Scholar]
- 33.Kernan W. N., Ovbiagele B., Black H. R., Bravata D. M., Chimowitz M. I., Ezekowitz M. D., Fang M. C., Fisher M., Furie K. L., Heck D. V., et al. American Heart Association Stroke Council, Council on Cardiovascular and Stroke Nursing, Council on Clinical Cardiology, and Council on Peripheral Vascular Disease 2014. Guidelines for the prevention of stroke in patients with stroke and transient ischemic attack: a guideline for healthcare professionals from the American Heart Association/American Stroke Association. [Published erratum appears in 2015 Stroke 46: e54.] Stroke 45: 2160–2236. [DOI] [PubMed] [Google Scholar]
- 34.Kronzon I., Saric M. 2010. Cholesterol embolization syndrome. Circulation 122: 631–641. [DOI] [PubMed] [Google Scholar]
- 35.Barratt-Due A., Pischke S. E., Brekke O. L., Thorgersen E. B., Nielsen E. W., Espevik T., Huber-Lang M., Mollnes T. E. 2012. Bride and groom in systemic inflammation--the bells ring for complement and Toll in cooperation. Immunobiology 217: 1047–1056. [DOI] [PubMed] [Google Scholar]
- 36.Kambas K., Markiewski M. M., Pneumatikos I. A., Rafail S. S., Theodorou V., Konstantonis D., Kourtzelis I., Doumas M. N., Magotti P., Deangelis R. A., et al. 2008. C5a and TNF-alpha up-regulate the expression of tissue factor in intra-alveolar neutrophils of patients with the acute respiratory distress syndrome. J. Immunol. 180: 7368–7375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gregory S. A., Morrissey J. H., Edgington T. S. 1989. Regulation of tissue factor gene expression in the monocyte procoagulant response to endotoxin. Mol. Cell. Biol. 9: 2752–2755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Skjeflo E. W., Christiansen D., Fure H., Ludviksen J. K., Woodruff T. M., Espevik T., Nielsen E. W., Brekke O. L., Mollnes T. E. 2018. Staphylococcus aureus-induced complement activation promotes tissue factor-mediated coagulation. J. Thromb. Haemost. 16: 905–918. [DOI] [PubMed] [Google Scholar]
- 39.Ikeda K., Nagasawa K., Horiuchi T., Tsuru T., Nishizaka H., Niho Y. 1997. C5a induces tissue factor activity on endothelial cells. Thromb. Haemost. 77: 394–398. [PubMed] [Google Scholar]
- 40.Nakamura S., Imamura T., Okamoto K. 2004. Tissue factor in neutrophils: yes. J. Thromb. Haemost. 2: 214–217. [DOI] [PubMed] [Google Scholar]
- 41.Østerud B. 2004. Tissue factor in neutrophils: no. J. Thromb. Haemost. 2: 218–220. [DOI] [PubMed] [Google Scholar]
- 42.Maugeri N., Manfredi A. A. 2015. Tissue factor expressed by neutrophils: another piece in the vascular inflammation puzzle. Semin. Thromb. Hemost. 41: 728–736. [DOI] [PubMed] [Google Scholar]
- 43.Ritis K., Doumas M., Mastellos D., Micheli A., Giaglis S., Magotti P., Rafail S., Kartalis G., Sideras P., Lambris J. D. 2006. A novel C5a receptor-tissue factor cross-talk in neutrophils links innate immunity to coagulation pathways. J. Immunol. 177: 4794–4802. [DOI] [PubMed] [Google Scholar]
- 44.Palta S., Saroa R., Palta A. 2014. Overview of the coagulation system. Indian J. Anaesth. 58: 515–523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lösche W. 2005. Platelets and tissue factor. Platelets 16: 313–319. [DOI] [PubMed] [Google Scholar]
- 46.Kahn M. L., Zheng Y. W., Huang W., Bigornia V., Zeng D., Moff S., Farese R. V., Jr., Tam C., Coughlin S. R. 1998. A dual thrombin receptor system for platelet activation. Nature 394: 690–694. [DOI] [PubMed] [Google Scholar]
- 47.Patzelt J., Mueller K. A., Breuning S., Karathanos A., Schleicher R., Seizer P., Gawaz M., Langer H. F., Geisler T. 2015. Expression of anaphylatoxin receptors on platelets in patients with coronary heart disease. Atherosclerosis 238: 289–295. [DOI] [PubMed] [Google Scholar]
- 48.Del Conde I., Crúz M. A., Zhang H., López J. A., Afshar-Kharghan V. 2005. Platelet activation leads to activation and propagation of the complement system. J. Exp. Med. 201: 871–879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ho-Tin-Noé B., Vo S., Bayles R., Ferrière S., Ladjal H., Toumi S., Deschildre C., Ollivier V., Michel J. B. 2017. Cholesterol crystallization in human atherosclerosis is triggered in smooth muscle cells during the transition from fatty streak to fibroatheroma. J. Pathol. 241: 671–682. [DOI] [PubMed] [Google Scholar]
- 50.Ricklin D., Reis E. S., Mastellos D. C., Gros P., Lambris J. D. 2016. Complement component C3 - the “Swiss Army Knife” of innate immunity and host defense. Immunol. Rev. 274: 33–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hillmen P., Muus P., Röth A., Elebute M. O., Risitano A. M., Schrezenmeier H., Szer J., Browne P., Maciejewski J. P., Schubert J., et al. 2013. Long-term safety and efficacy of sustained eculizumab treatment in patients with paroxysmal nocturnal haemoglobinuria. Br. J. Haematol. 162: 62–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Pischke S. E., Gustavsen A., Orrem H. L., Egge K. H., Courivaud F., Fontenelle H., Despont A., Bongoni A. K., Rieben R., Tønnessen T. I., et al. 2017. Complement factor 5 blockade reduces porcine myocardial infarction size and improves immediate cardiac function. Basic Res. Cardiol. 112: 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ridker P. M., Everett B. M., Thuren T., MacFadyen J. G., Chang W. H., Ballantyne C., Fonseca F., Nicolau J., Koenig W., Anker S. D., et al. CANTOS Trial Group 2017. Antiinflammatory therapy with canakinumab for atherosclerotic disease. N. Engl. J. Med. 377: 1119–1131. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.