

ABSTRACT
Chromatin‐remodeling factors are required for a wide range of cellular and biological processes including development and cognition, mainly by regulating gene expression. As these functions would predict, deregulation of chromatin‐remodeling factors causes various disease syndromes, including neurodevelopmental disorders. Recent reports have linked mutations in several genes coding for chromatin‐remodeling factors to intellectual disability (ID). Here, we used exome sequencing and identified a nonsynonymous de novo mutation in BAZ1A (NM_182648.2:c.4043T > G, p.Phe1348Cys), encoding the ATP‐utilizing chromatin assembly and remodeling factor 1 (ACF1), in a patient with unexplained ID. ACF1 has been previously reported to bind to the promoter of the vitamin D receptor (VDR)‐regulated genes and suppress their expression. Our results show that the patient displays decreased binding of ACF1 to the promoter of the VDR‐regulated gene CYP24A1. Using RNA sequencing, we find that the mutation affects the expression of genes involved in several pathways including vitamin D metabolism, Wnt signaling and synaptic formation. RNA sequencing of BAZ1A knockdown cells and Baz1a knockout mice revealed that BAZ1A carry out distinctive functions in different tissues. We also demonstrate that BAZ1A depletion influence the expression of genes important for nervous system development and function. Our data point to an important role for BAZ1A in neurodevelopment, and highlight a possible link for BAZ1A to ID.
Keywords: ACF1, BAZ1A, CYP24A1, epilepsy, intellectual disability, neurodevelopment, vitamin D metabolism, Wnt signaling
Introduction
Chromatin organization is a dynamic and regulated process that allows differential accessibility of DNA to different regulatory proteins to activate or repress transcription. The establishment of different chromatin states is enabled by a group of ATP‐dependent chromatin‐remodeling complexes that utilize ATP hydrolysis to alter the state, placement, or position of the nucleosomes, thereby regulating transcription [Hargreaves and Crabtree, 2011]. Together with other chromatin organizers, they represent the first layer of regulatory control, which ascertains a stable and correct temporal and spatial gene expression [Zentner and Scacheri, 2012]. Most ATP‐dependent chromatin remodelers are multisubunit complexes and share a conserved catalytic ATPase subunit, which contains an ATPase domain and other specialized domains related to their mode of action. Additionally, they harbor several noncatalytic proteins, also known as accessory subunits, required to regulate the activity of the catalytic subunit and to ensure accurate recruitment of chromatin remodelers to their target genomic regions [Flaus et al., 2006]. Chromatin‐remodeling complexes can be divided into four families based on the composition of their ATPase subunit. These families are referred to as the SWI/SNF, ISWI, CHD, and INO80 families of remodelers [Becker and Horz, 2002]. Several studies have illustrated a fundamental role of chromatin‐remodeling factors during development, differentiation, and homeostasis [Day and Sweatt, 2011; Ronan et al., 2013]. In line with these observations, dysfunction of chromatin remodelers has been predicted to cause multisystem disorders.
ISWI, also known as the imitation switch, is among the most conserved chromatin remodeling families [Li and Reinberg, 2011]. In humans, complexes belonging to ISWI possess one of two possible ATPase catalytic subunits, Snf2H and Snf2L, and various noncatalytic subunits [Mohrmann and Verrijzer, 2005]. The differential incorporation of the several accessory units with the two ATPases in ISWI leads to the formation of complexes with distinct functions and tissue specificity [Lessard et al., 2007; He et al., 2008; Ho et al., 2009]. ISWI is involved in various biological functions, including transcriptional activation and repression, chromatin assembly and nucleosome spacing, and DNA replication [Langst and Becker, 2001; Narlikar et al., 2002; Eberharter and Becker, 2004; Banting et al., 2005]. Recently, several studies highlighted essential roles for members of ISWI during embryogenesis and neurodevelopment and for cognition [Strohner et al., 2001; Stopka and Skoultchi, 2003; Dirscherl et al., 2005; Yoshimura et al., 2009; Alvarez‐Saavedra et al., 2014]. Knockout mice for components of this complex displays growth and cell proliferation abnormalities, cognitive defects, neurodevelopmental deficits, and in some cases compromise mice viability [Stopka and Skoultchi, 2003; Landry et al., 2008; Alvarez‐Saavedra et al., 2014]. In line with this, dysfunction of members of ISWI has been predicted to cause cancer and developmental disorders such as intellectual disability (ID) [Perez Jurado, 2003; Ye et al., 2009; Buganim et al., 2010; Langst and Manelyte, 2015].
ID is a common neurodevelopmental condition that affects around 1%–3 % of the population [Leonard and Wen, 2002; Shevell et al., 2003]. It is generally characterized by reduced intellectual and behavioral abilities, which imposes a significant burden on patients and their families. ID is a complex condition and considered to be clinically and genetically heterogeneous. Several human exome sequencing studies have linked mutations in genes encoding chromatin‐remodeling complexes to general ID, to distinct ID disorders such as Coffin‐Siris syndrome (CSS) and to autism spectrum disorder [Lopez and Wood, 2015]. Although the majority of these mutations have been found in the ARID and SMARC gene families, it is likely that more mutations will be identified in other genes coding for chromatin‐remodeling factors [Hargreaves and Crabtree, 2011; Wilson and Roberts, 2011; Santen et al., 2012; Santen et al., 2013].
Using exome sequencing of a trio family with two healthy parents and a child suffering from ID, epilepsy, ataxia, and hyper mobile joints, we identified a single nonsynonymous de novo variant in the gene BAZ1A (see Supp. Methods for detailed phenotypic description of the patient). BAZ1A (NM_182648, OMIM 605680) codes for the chromatin‐remodeling factor ACF1, a member of the ISWI chromatin‐remodeling complexes ACF and CHRAC. ACF1 has been implicated in different functions including chromatin assembly and remodeling, and in double‐strand DNA damage repair [Ito et al., 1999 Lan et al., 2010]. It has also been shown to have a function in transcriptional repression of vitamin D3 receptor (VDR)‐regulated genes by blocking the accessibility of the transcription factors to VDR in the absence of vitamin D3 (VD3) [Ewing et al., 2007]. Moreover, ACF1 was recently shown to contribute to the susceptibility to depression, and in regulating stress related behaviors [Sun et al., 2015]. Here, we found that the identified variant in BAZ1A affects the function of ACF1 as a transcription regulator, causing differential expression in genes important in vitamin D metabolism, the Wnt signaling pathway and in the development of proper synaptic function. We further investigate the general function of Baz1a by RNA‐seq in knockout mice.
Materials and Methods
DNA Extraction
Genomic DNA was extracted from peripheral blood lymphocytes according to standard procedures. Patient samples were studied with the approval of the local Research Ethics Committee, Faculty of Medicine, Uppsala University, Sweden.
Copy Number Variation Analysis
Chromosomal microarray analysis (CMA) was performed using Affymetrix 250K Nsp 1 array according to the standard Affymetrix GeneChip protocol (Affymetrix Inc., Santa Clara, CA). Data analysis was carried out using Genotyping Console 4.0.
Exome Capture
Exome enrichments for the trio family were prepared from 3.0 μg genomic DNA from the trio. The DNA samples were sheared with Covaris instrument (Covaris, Inc., Wobum, MA). Libraries were created from the sheared DNA using AB Library Builder System and captured using the Agilent SureSelect 50 Mb exome enrichment kit, according to the manufacturer's protocols. Exome capture was conducted by hybridizing the DNA libraries with biotinylated RNA baits for 24 hr followed by extraction using streptavidin‐coated magnetic beads. Captured fragments were amplified and emulsion PCR was conducted using Bead System and sequenced using SOLiD4 producing 68–80 million reads per sample.
Exome Capture Data Analysis
Exome sequence data was aligned using BioScope (Applied Biosystems, Foster City, CA). SNP calls were made using DiBAYES with default settings and resulted in 16,723 nonsynonymous single‐nucleotide variants (SNVs) with a sequence read coverage >3 in the patient. Filtering was performed using ANNOVAR [Wang et al., 2010] and dbSNP 132 [Sherry et al., 2001]. Further filtering was done using the parental samples and our in‐house database of previously analyzed exomes. Manual inspection of each SNP remaining was then performed resulting in three putative de novo coding variant. Sanger sequencing only validated a single de novo mutation in the BAZ1A gene (NM_182648.2:c.4043T>G, g.35231163A>C). The other two variants were found to be false positives. The DNA mutation numbering system is based on genomic DNA. The de novo variant in BAZ1A identified in the patient is submitted to LOVD 3 (www.lovd.nl).
PCR and Sanger Sequencing
The de novo mutation in BAZ1A was validated using standard PCR and Sanger sequencing. Briefly, PCR was carried out with initial denaturation at 95°C for 3 min and 35 cycles of 95°C for 15 sec, 60°C for 30 sec, and 72°C for 1 min. The reaction contained 10x DreamTaq buffer (Fermentas, Waltham, MA), 200 μM of each dNTP, 1.25 U of DreamTaq DNA polymerase (Fermentas), primers harboring the mutation site (Supp. Table S1), and 12.5 ng DNA. PCR products were visualized on 1.5% agarose and purified using PCR purification kit (QiaGen, Hilden, Germany). The mutation was then validated in the patient by Sanger sequencing, whereas the parents confirmed not to carry the mutation.
In Silico Prediction Methods
The combined annotation‐dependent depletion
This score was assigned using the v1.0. Precalculated combined annotation‐dependent depletion (CADD) scores were found at http://cadd.gs.washington.edu/.
MutationTaster (build; NCBI 37/Ensembl 69)
This test was used to predict the disease‐causing potential of the variant (http://www.mutationtaster.org). BAZ1A transcript (ID ENST00000382422) was used in the analysis.
MUpro (MUpro 1.0)
This test was used to evaluate if the de novo variant compromises protein stability (http://mupro.proteomics.ics.uci.edu).
RNA Extraction and cDNA Synthesis
Total RNA was extracted from transfected cells or from trio member's whole blood using Ribopure kit (Ambion, Waltham, MA). RNA samples were then treated with TURBO DNase (Ambion). Single‐stranded cDNA was synthesized from 1 μg RNA using Maxima© first‐strand cDNA synthesis kit for RT‐qPCR (Thermo Scientific, Waltham, MA).
Quantitative Real‐Time PCR
Quantitative real‐time PCR (qrtPCR) was used to measure the relative expression of differentially expressed genes (DEGs) between the patient and the parents and between the wt‐ACF1 or mut‐ACF1‐transfected cells. The qrtPCR was carried out using Stratagene Mx3000P in 96 well plates. The reactions were conducted with initial denaturation at 95°C for 10 min, followed by 40 cycles of 95°C for 15 sec, 60°C for 30 sec, and 72°C for 30 sec. The cycles were followed by a melting curve step. The reaction contained 10 ng single‐stranded cDNA, 0.4 μM of each primer (Supp. Table S1), and 12.5 μl Maxima SYBR green/ROX master mix (Fermentas) in a total reaction volume of 25 μl. The relative expression levels in the samples were determined using ΔΔCT method normalized to the levels of β‐actin. All samples were run in triplicates and the average values were used to calculate the expression levels for each target. Raw data were analyzed using MxPro software (Stratagene, Santa Clara, CA).
Vitamin D Test
A test for vitamin‐D (25‐(OH)D) levels in serum revealed an insufficiency (70 nmol/l, normal range is 125–200 nmol/l). Calcium level was normal (2.34 nmol/l).
Cloning and Mutagenesis
Wild‐type (wt) and mutant (mut) hACF1 transcripts were cloned in pcDNA3.1 expression vector (Invitrogen, Carlsbad, CA). The original acf1 clone in pEGFP‐N1 expression vector (CLONTECH) was a generous gift from Prof. PD Varga‐Weisz. Briefly, the hACF1 transcript was amplified from the original transcript using full‐length primers (Supp. Table S1). The hACF1 transcript was then cloned into pcDNA3.1/V5‐his‐TOPO vector by TOPO Cloning reaction, using pcDNA3.1 Directional TOPO expression kit (Invitrogen). The mutant hACF1 construct was produced by PCR‐based site‐directed mutagenesis using PFU Turbo (Stratagene). Positive hACF1 constructs were further validated by Sanger sequencing.
Transient Transfection
Saos‐2 cells were maintained in DMEM medium (Sigma, Saint Louis, MO) supplemented with 10% fetal bovine serum (Sigma), 2 mM of l‐glutamin and 1× penicillin/streptomycin (Sigma). All transfection experiments were performed using Lipofectamine 2000 (Invitrogen) according to the manufacturer recommendations. Briefly, 1×106 cells were transfected in 6‐wells plates using 4 μg of wt or mut hACF1pcDNA3.1 plasmid. All transfections were performed in duplicate. Cells were collected for analysis 48 hr post‐transfection.
RNA Sequencing and Mapping for the Trio Family
RNA sequencing of the trio family was performed using a SOLiD4 instrument. Each parent was sequenced once and two replicates were sequenced for the child. Sequenced reads were mapped to the HG19 version of the human reference genome using the BioScope software (Applied Biosystems). The number of reads mapping to each RefSeq gene (based on the refFlat table from the UCSC genome browser) was then counted using htseq‐count (http://wwwhuber.embl.de/users/anders/HTSeq/doc/count.html). Differential expression was calculated using the DESeq2 [Love et al., 2014] and EdgeR [Robinson et al., 2010], comparing the replicated patient samples to the parents, generating a lists of DEGs. Candidate genes were then chosen using an adjusted P value of 0.0001 as cutoff and required that each gene had a P value below the cut‐off in both analysis programs. Gene ontology (GO) analysis was performed using the R‐package GOseq, and the REVIGO Webserver was used to identify nonredundant GO categories [Supek et al., 2011]. The list of Wnt target genes used for enrichment analysis was compiled from the Nousse laboratory (http://web.stanford.edu/group/nusselab/cgi‐bin/wnt/target_genes), for genes found in both humans and other organisms (if a human orthologue could be identified)
BAZ1A siRNA Knockdown
siRNA‐mediated knockdown of BAZ1A expression was performed in Saos‐2 cells using ON‐TARGETplus BAZ1A siRNA SMARTpool (Cat#: L‐006941; GE Dharmacon, Lafayette, CO) according to the manufacturer recommendations. ON‐TARGETplus nontargeting pool (Cat#: D‐001810‐10‐05; GE Dharmacon) was used as negative control for the siRNA experiment. Briefly, 5×104 cells were seeded in 24‐wells plate 24 hr before transfection with complete growth media (DMEM; Cat#: D 5546; Sigma–Aldrich). Cells were then transfected with either BAZ1A siRNA SMARTpool or nontargeting pool to a final concentration of 25 nm and 0.65 μl DharmaFECT transfection reagent 1 were added to the cells to a final concentration of 25 nM. The media containing the siRNA and the transfection reagent was replaced with complete growth media 24 hr after transfection. The cells were harvested at 48 hr post‐transfection. All transfections were performed in triplicates.
SiRNA Sequencing
Poly(A) selected RNA extracted from the siRNA‐treated Saos‐2 cells and controls was sequenced on an Illumina HiSEQ instrument. Sequenced reads were aligned using the program STAR and reads for each gene was counted using the Ensembl human gene list (version 70). Differential expression was detected using the DESeq2 R package, using an adjusted P value of 0.05. GO enrichment analysis was performed as described for the trio family. All further analysis was made using in‐house R scripts.
Mouse Transcriptome Sequencing
Baz1a knockout mice (C57BL/6N, Baz1atm1b(EUCOMM)Hmgu) were generated by the International Knockout Mouse Consortium [Skarnes et al., 2011; Bradley et al., 2012]. Mouse tissue was obtained from The Canadian Mouse Mutant Repository at The Hospital for Sick Children (Toronto, Canada). Four knockout mice (two males, two females) and four littermate controls (two males, two females) were euthanized at 6 weeks of age. Heart and brain tissue was removed and each brain was sliced into four pieces (corresponding to frontal cortex, striatum/hippocampus, cerebellum, and brain stem). Tissue pieces were immediately snap frozen in liquid nitrogen. Total RNA was extracted from tissues using Ribopure kit (Ambion). Baz1a knockout was validated in RNA sequencing data and using qrtPCR (Supp. Fig. S1). RNA was sequenced using Illumina HiSeq2500, generating 100‐bp paired‐end strand‐specific sequence from polyA+ RNA. Standard protocols were used for library preparation and sequencing and an average of 97 million reads per sample was produced. Reads were aligned using the same method as described for the siRNA sequencing but using the GRCm38 version of the mouse reference genome. Reads was counted using the Gencode GRCm38 mouse gene list. Differential expression analysis was performed using the R package DESeq2, and DEGs were identified using an adjusted P value of 0.05 as cutoff. GO analysis was performed using the R‐package GOseq, all further analysis of the data was made using in‐house R scripts. In order to compare overlaps between DEGs in knockout mice and previously published ChIP‐seq data, DEGs (P value < 0.05) in the knockout mice brain (n = 2,160) and knockout mice heart (n = 1,586) were compared with the list of regions indicated in Sun et al. (2015). All sequencing data are deposited in the EMBL‐EBI Sequence Read Archive (European Nucleotide Archive) under accession number PRJNA319519.
Results
Exome Sequencing
Aiming to identify the disease‐causing mutation in the patient, we first performed CMA using Affymetrix 250K Nsp 1 array and found no evidence of any chromosomal aberrations. We then performed target enrichment of exome sequences on DNA from the patient and his parents using the Agilent SureSelect 50 Mb kit. Captured targets were then sequenced using SOLiD4. Analysis of variants was performed to identify potential recessive‐causative variants as well as de novo mutations in the patient. We found no potentially causative homozygous or compound heterozygous recessive variants in ID candidate genes. After filtering the exome sequencing data followed by Sanger sequencing of candidate de novo mutations, one single de novo variant could be validated in the child, a mutation in the chromatin‐remodeling gene BAZ1A (NM_182648). The de novo variant is a T > G substitution in the gene BAZ1A (NM_182648.2:c.4043T > G, NC_000014.8:g.35231163A>C), and it leads to a phenylalanine (Phe) to cysteine (Cys) change at amino acid 1,348 (p.Phe1348Cys) in ACF1, the protein product of BAZ1A. The substitution thus changes a nonpolar hydrophobic residue (Phe) to a polar uncharged residue (Cys). The variant is located in an evolutionarily conserved region in the oligopeptide linker motif that connects the PHD domain and the bromodomain (Brd) (Fig. 1). We evaluated the potentially damaging effect of the variant using CADD [Kircher et al., 2014], which yielded a CADD‐score of 16.71, indicating that it is among the 10% most damaging substitutions in the genome. We also used MutationTaster [Schwarz et al., 2010] to predict the disease‐causing potential of the variant and it was scored as likely pathogenic. To test whether the variant compromises protein stability, we used the prediction of protein stability changes for single‐site mutation from sequences software (MUpro) [Cheng et al., 2006]. The analysis predicted a decreased stability with a confidence score −0.799. The confidence score of MUpro ranges between −1 and 1. A score less than zero is indication of decreased protein stability, and a score of more than zero is an indication that the mutation increases protein stability. This observation further suggests that the variant has a negative effect on the function of the protein.
Figure 1.
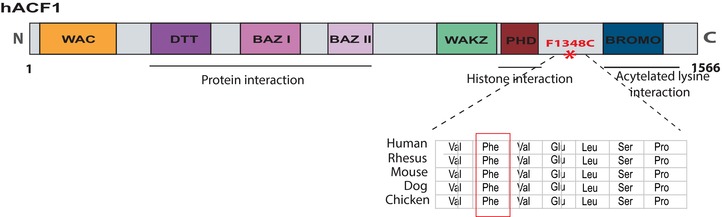
A schematic representation of the human ACF1 protein indicating known interaction domains. ACF1 domains include a WAC motif necessary for the binding of ACF1 to the DNA and involved in ACF‐mediated chromatin assembly) [Fyodorov and Kadonaga, 2002], and a DTT motif (DNA binding homeobox and transcription factors). It further contains three conserved motifs, two BAZ and a WAKZ (responsible for the binding of ACF1 to ISWI, the other subunit of the ACF complex) [Jones et al., 2000; Eberharter and Becker, 2004], a PHD finger (plant homeodomain) and bromodomain (Brds) motifs. The mutation (p.Phe1348Cys) is located in the linker region between the PHD and Brd domains. The evolutionary conservation is shown for the mutation and the surrounding amino acids.
Effect of BAZ1A De Novo Variant on Vitamin D‐Regulated Genes
ACF1 has been previously reported to interact with the nuclear receptor corepressor (N‐CoR) and act as a suppressor of the VDR‐regulated genes in the absence of VD3 [Ewing et al., 2007]. The PHD and brd domains in ACF1 are critical for the repression process through binding to histone structures of VDR‐regulated genes. To test the impact of the variant on the expression of VDR‐regulated genes, we performed targeted qrtPCR on RNA extracted from blood samples from the trio and measured the expression of RANKL, VDR, and CYP24A1. Interestingly, CYP24A1 [OMIM 126065], which has been implicated in the metabolism of VD3 levels [Jones et al., 2012], was significantly upregulated (sevenfold, P value = 0.005) in the patient compared with both parents (Fig. 2A). Meanwhile, BAZ1A itself showed no difference in expression between the patient and the parents. The patient showed equal biallelic expression of the reference allele and the mutation, as determined by Sanger sequencing of the qrtPCR product, indicating no effect of the mutation on the expression level of BAZ1A itself. No significant difference was detected in VDR and RANKL expression.
Figure 2.
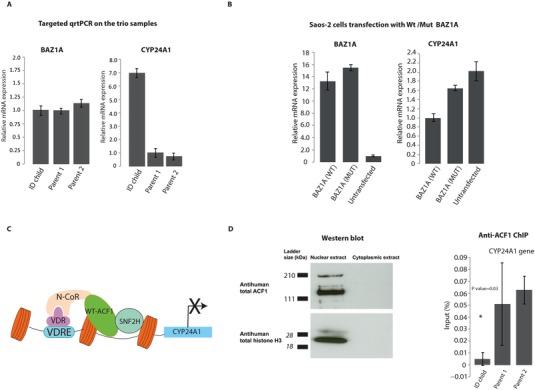
Differential expression of CYP24A1. A: The left graph shows levels of BAZ1A expression in the trio. To the right are the results of targeted qrtPCR validation of the CYP24A1 expression in the affected child and the healthy parents. B: The left graph shows overexpression of the BAZ1A in the transfected Saos‐2 cells compared with untransfected cells. The right graph shows the qrtPCR validation of the CYP24A1 expression in Saos‐2 cells transfected with either wt‐BAZ1A or mut‐BAZ1A. Saos‐2 cells transfected with the wt‐BAZ1A is considered as control. All expression values were normalized to the level of B‐actin in each respective sample. Expression levels represent mean values of the biological and technical replicates and error bars are ±SD. C: Simplified schematic model showing the role of ACF1 in the regulation of expression of CYP24A1. In the absence of vitamin D3, ACF1 and SNF2H (the other member of the ACF complex) interact with N‐CoR and suppress the expression of CYP24A1 through binding to the VDR. D: ACF‐1 protein is localized to the nucleus of Saos‐2 cells. To the left, protein expression analysis was performed by western blot of nuclear versus cytoplasmic protein lysates using specific antibodies against ACF‐1 and total histone H3. Histone H3 was used as a positive control for the nuclear fraction purification. To the right, ChIP analysis of anti‐ACF1 precipitated DNA measured by qrtPCR from fresh blood monocytes of the trio family. Chromatin precipitated from each sample is reported as a percentage of input. Error bars are ±SD of the biological and technical replicates. Asterisks indicate significant statistical difference (P ≤ 0.05) of CYP24A1 enrichment in anti‐ACf1‐precipitated DNA between the ID patient and the parents. Student's t‐test was used to calculate P values.
To test whether the elevated level of CYP24A1 in the patient also affected the levels of VD3, we performed a clinical test to measure the levels of VD3 in patient blood. The test showed that the patient suffers from moderate VD3 insufficiency (70 nmol/l). To determine whether the de novo variant is the cause of CYP24A1 upregulation, and to exclude the possibility that the upregulation is caused by other variants in the patient, we performed transient transfection of wild‐type and mutant ACF1 transcripts (wt‐ACF1, mut‐ACF1) in Saos‐2 cells. The mut‐ACF1 harbors the same mutation identified in the patient. Saos‐2 cells were chosen due to their well‐characterized VD3 pathway. qrtPCR results showed that CYP24A1 is indeed upregulated in the cells transfected with mut‐ACF1 (Fig. 2B), highlighting the role of the de novo variant on the expression levels of CYP24A1.
ACF1 is known to directly interact with a sequence motif upstream of the CYP24A1 gene through VDR, causing repression of CYP24A1 transcription (Fig. 2C) [Ewing et al., 2007; Luo et al., 2010]. As further validation on the effect of the variant, we sought to evaluate whether the de novo mutation decreases the association of ACF1 to the CYP24A1 promoter. To test this, we conducted chromatin‐immunoprecipitation (ChIP) using human ACF1 antibodies on freshly purified blood monocytes from the trio (Supp. Methods). We first validated the specificity of the human ACF1 (hACF1) antibody in Saos‐2 cells using western blot (Fig. 2D). We then performed qrtPCR on the immunoprecipitated DNA to determine the occupancy of ACF1 on CYP24A1 promoter. The results demonstrated that the patient's monocytes have significantly less ACF1 occupancy on the CYP24A1 promoter as compared with the parents (P value = 0.03) (Fig. 2D). These results indicate that the mutated ACF1 has a decreased ability to bind and repress CYP24A1, leading to an increase in CYP24A1 expression.
RNA Sequencing of the Trio
To investigate whether the de novo variant has an impact on genes involved in other pathways, we performed global RNA‐seq on polyA+ RNA extracted from blood samples from the trio family, producing 60–90 million mapped sequence reads per sample. In order to identify DEGs in the patient compared with the parents, and to minimize false‐positive findings, the two R programs DESeq [Anders and Huber, 2010] and EdgeR [Robinson et al., 2010] were used and only genes identified as differentially expressed by both programs were further used in our analysis. Using a stringent P value of <0.0001, 823 genes were determined to be differentially expressed using this strategy (Supp. Table S2). The RNA‐seq results also confirmed the previously identified upregulation of CYP24A1 in the patient (fold change 2.38).
A GO analysis of the DEGs resulted in 108 significantly enriched nonredundant GO categories (Supp. Table S3). In line with the role of ACF1 as a suppressor of the VD3 pathway, we note that the GO category “response to vitamin D” is significantly enriched in the analysis (adjusted P value = 0.018). We then compared the genes differentially expressed in our analysis with a more detailed list of genes known to be regulated by vitamin D [Ramagopalan et al., 2010]. Again, a significant over‐representation of vitamin D‐regulated genes was observed among the DEGs (P value = 0.0016, Fisher's exact test). In a previous study, four genes in the vitamin D pathway have been experimentally shown to be regulated by ACF1 [Ewing et al., 2007]. Of these four, we found two to be differentially expressed in the patient (CYP24A1 and IGFBP3), whereas the other two genes were expressed at very low levels in blood in the trio samples (TNFSF11 and IGF1).
The GO analysis of DEGs also yielded significant enrichment for both the canonical and noncanonical Wnt signaling pathways (P value = 9.0E‐5 and 9.7E‐5, respectively). Consistent with these findings, a previous study in Drosophila melanogaster revealed that ACF1 is involved in the basal repression of Wnt target genes in the absence of active Wnt signaling [Liu et al., 2008]. To further pursue the enrichment of the Wnt signaling pathway genes, we created a list of genes previously identified as targets of the Wnt pathway (n = 76, http://web.stanford.edu/group/nusselab/cgi‐bin/wnt/target_genes) and compared these genes with the list of DEGs resulting from our RNA‐seq analysis. Of the 76 genes, 14 genes showed differential expression in the patient (Supp. Table S4) yielding a significant over‐representation of Wnt target genes among the DEGs identified (P value = 3.63E‐7, Fisher's exact test). To investigate the possibility that this is an effect of the age difference between the patient and his parents, the number of DEGs overlapping with the Wnt target genes was counted for a second unrelated trio family with healthy parents and a child with mild mental retardation. This test gave a nonsignificant result, indicating that the differential expression in Wnt target genes in the patient is not an influence of age difference. To further investigate the impact on Wnt pathway target genes, the expression fold change was measured for the genes in the Wnt pathway GO categories as well as the Wnt target genes. The results showed a stronger response in fold change of the Wnt target genes as compared with the Wnt pathway genes (see Fig. 3).
Figure 3.
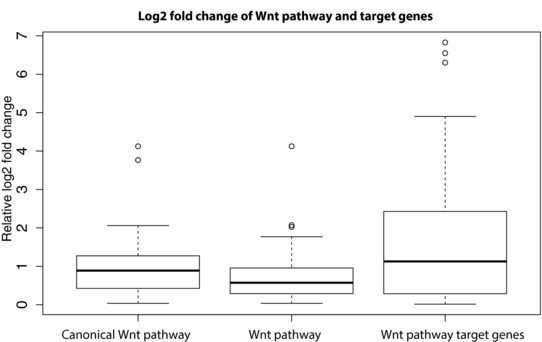
The log2 fold change of the genes in the GO categories Wnt pathway (n = 123), canonical Wnt pathway (n = 45), and Wnt target genes (n = 44). Genes having no reads aligned in the RNA sequencing experiments were not counted.
Considering the phenotype of the patient, we note that additional significantly enriched GO categories include axon guidance and nervous system development. To find specific genes relevant to the phenotype of the patient, we searched our list of DEGs for genes known to be involved in ID [Veltman and Brunner, 2012] and found that 25 of the DEGs have previously been shown to cause ID. The most significant differential expression among these ID genes was found for SYNGAP1 [OMIM 603384] and SMARCA4 [OMIM 603254]. SYNGAP1 is known to cause both epilepsy and ID [Berryer et al., 2013; Carvill et al., 2013]. We identified SYNGAP1 to be overexpressed in the patient compared with his parents. Overexpression of SYNGAP1 has been previously shown to significantly affect neuronal signaling and synapse function [Rumbaugh et al., 2006]. SMARCA4, a member of SWI/SNF chromatin‐remodeling complex previously linked to Coffin‐Siris syndrome, was also overexpressed in the patient. To experimentally validate the differential expression of SYNGAP1 and SMARCA4, we performed qrtPCR on the trio samples, confirming overexpression of both genes in the patient compared with the parents (fold change 3 and 1.5, respectively) (Fig. 4A and B). To exclude the possibility that the differential expression is age related, we also measured relative expression of the two genes in an independent control trio family with two healthy parents and a child of similar age suffering from mental retardation and epilepsy. The results showed no differential expression between the child and the parents in the unaffected trio (Fig. 4A and B).
Figure 4.
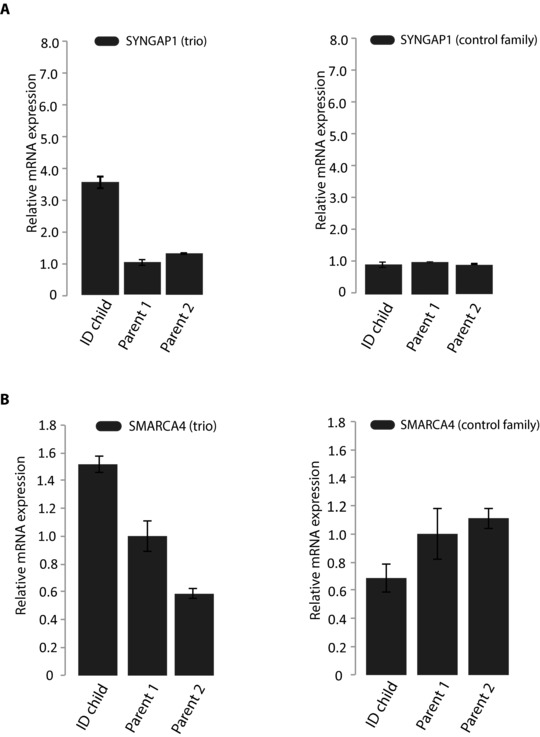
The relative expression of SYNGAP1 and SMARCA4 in the trio. A: qrtPCR validation of the differential expression of SYNGAP1 between the patient and the parents (left). To the right are the expression levels of SYNGAP1 in a control trio family, indicating no age‐related differential expression. B: qrtPCR validation of the differential expression of SMARCA4 between the patient and the parents (left). To the right are the expression levels of SMARCA4 in a control trio family showed no age‐related differential expression. All qrtPCR results were normalized to the level of B‐actin in each respective sample. Expression levels represent mean values of the biological and technical replicates and error bars are ±SD.
The Role of ACF1 in Regulation of Gene Expression
To date, there is limited knowledge about the general function of BAZ1A. Therefore, we next sought to investigate the role of ACF1 in gene expression regulation. We performed siRNA‐mediated knockdown of BAZ1A in Saos‐2 cells (Fig. 5A) followed by polyA+ RNA‐seq on Saos‐2 cells with BAZ1A knockdown and cells transfected with control siRNA. RNA‐seq analysis indicated a large number of genes to be differentially expressed upon knockdown (n = 1,256, data not shown). GO analysis of the DEGs showed enrichment of GO categories related to several biological pathways including cell signaling and system development. The top 10 GO categories are shown in Supp. Table S5. Consistent with previously published BAZ1A loss‐of‐function data [Ewing et al., 2007], our RNA‐seq data showed no differential expression of CYP24A1 in Saos‐2 cells with BAZ1A knockdown. However, in the same study, Ewing et al. (2007) demonstrated that overexpression of ACF1 lacking the C terminus, where the de novo variant is located, in cells resulted in increased levels CYP24A1. These data suggest that the de novo variant in the patient does not cause loss‐of‐function of BAZ1A. It may rather reflect a dominant negative gain‐of‐function, at least for the transcriptional regulation of CYP24A1.
Figure 5.
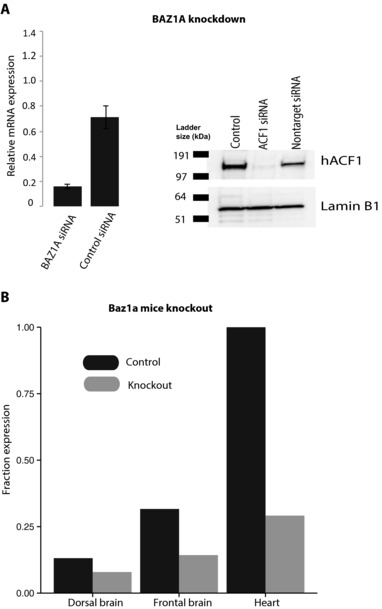
Validation of BAZ1A knockdown in Saos‐2 cells and Baz1a knockout in mouse. A: Left, expression levels of BAZ1A in Saos‐2 cells transfected with BAZ1A siRNA and Saos‐2 with control siRNA. All expression values were normalized to the level of B‐actin in each respective sample. Expression levels represent mean values of the biological and technical replicates and error bars are ±SD. Right, protein expression of ACF1 in Saos‐2 cells transfected with BAZ1A siRNA, Saos‐2 with control siRNA and untransfected cells. B: Bar plot showing the fraction of Baz1a expression in each mouse tissue as compared with the tissue with the highest Baz1a expression (heart).
We also investigated the role of ACF1 on gene expression in vivo using polyA+ RNA‐seq on frontal brain, dorsal brain, and heart tissues from four Baz1a knockout mice and four wild‐type litter mate controls (two males and two females for each condition). Even though the highest expression of Baz1a was observed in the heart compared with frontal and dorsal brain (Fig. 5B), we found that the frontal brain harbored the highest number of DEGs between knockout and control mice (DEG = 287) compared with heart (DEG = 44). No DEGs were found in the dorsal brain, where expression of Baz1a was very low. These results indicate a larger influence of ACF1 on gene expression regulation in the frontal brain compared with the dorsal brain and heart. The results also suggest that ACF1 carries out distinctive functions in different tissues. GO analysis of the DEGs of the knockout mice revealed enrichment for several GO categories including synaptic transmission (P value = 4.6E‐10), neurological system process (P value = 3.6E‐7), and ensheathment of neurons (P value = 5.5E‐6) in the frontal brain (most significant GO terms are listed in Supp. Table S6). These results indicate an important role of ACF1 in regulation of gene expression in the nervous system. On the other hand, the heart showed enrichment for categories associated with cardiovascular system development and extracellular matrix. Most significant GO categories enriched in the heart are listed in Supp. Table S7. It is obvious in our data that DEGs and GO categories show weak overlap between the different tissues, highlighting that ACF1 may be involved in different regulatory networks in different tissues. Nevertheless, it is interesting to note that ACF1 influences the expression of genes related to development in mouse frontal brain and heart, and in Saos‐2 cells, indicating a role for ACF1 in development across several tissues. The weak overlap between the different tissues is also evident from the low number of DEGs that overlap between the different RNA‐seq analyses (Fig. 6). We do note that the SYNGAP1 gene, which was found as differentially expressed in the patient blood (compared with parents), and has previously been linked to ID and epilepsy, was also identified as a DEG in the frontal cortex of knockout mice.
Figure 6.
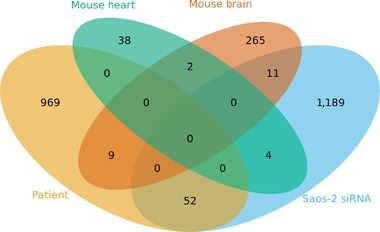
Venn diagram showing the number of DEGs in mouse heart (green), mouse frontal brain (brown), patient blood (yellow) and Saos‐2 cells (blue), and the overlap of the DEGs between the different tissues.
A recent study reported the genomic locations of ACF1 binding in the nucleus accumbens in the brain of control and depression mouse models using ChIP‐sequencing (ChIP‐seq) [Sun et al., 2015]. We investigated whether the genes showing differential expression (P value < 0.05) in the knockout mice overlap with these genomic‐binding locations. As expected, the genomic locations of ACF1 bindings overlapped more with the DEGs in the brain of the knockout mice (n = 14) compared with the heart (n = 3) (P value = 0.048, Fisher's exact test). The overlapping genes are shown in Supp. Tables S8 and S9. We also note that the ChIP‐seq experiment revealed ACF1 binding on the promoter region of SMARCA4, which was found as differentially expressed in the patient blood. These results further support a role of ACF1 in the regulation of gene expression in the brain.
Although ACF1 has been mainly reported as a transcriptional repressor, we observed several downregulated genes in the RNA‐seq data from the Baz1a null mice. Similarly, we found downregulated genes in Saos‐2 cells with BAZ1A knockdown and in the patient among the DEGs using a P value cut‐off of 0.0001. The number of upregulated and downregulated genes in RNA‐seq data from all samples is shown in Supp. Table S10. These results indicate that ACF1 is also involved in functions that could indirectly lead to transcriptional activation.
Discussion
ACF1 is a multidomain protein, with 1,556 amino acids, containing several conserved motifs for interaction with DNA, histones, and the other member of the ACF complex (Fig. 1). In this study, we identified a de novo p.Phe1348Cys variant in BAZ1A in a child with ID from a trio family. Since the general function of BAZ1A is not well characterized, we further investigated the role of BAZ1A in a cell line, and in mice brain and heart. The variant we identified in the patient is located in a conserved region in the linker motif between the PHD finger and the Brd domains. The PHD finger interacts with the central domain of the core histone and is known to be involved in chromatin‐mediated transcriptional regulation [Aasland et al., 1995; Eberharter et al., 2004]. Brd domains bind to acetylated histone H4 and H3 N‐termini to promote the interaction of chromatin‐remodeling factors to the DNA and facilitate the function of chromatin‐remodeling machinery [Jenuwein and Allis, 2001; Marmorstein and Berger, 2001]. Moreover, in agreement with our results, recent evidence implicated a role for Brd‐containing proteins in the initiation and development of ID and other neurodevelopmental diseases [Li et al., 2013]. In addition, cooperation of Brds together with PHD fingers has been implicated in chromatin‐mediated transcriptional regulation [Aasland et al., 1995; Schultz et al., 2001; Zeng et al., 2008]. Linker regions in multidomain proteins have been suggested to bridge flanking domains and support interdomain communication. Their conformation and structure is critical for the function of the flanking domains [Bhaskara et al., 2013]. The exchange of the phenylalanine (nonpolar, hydrophobic) to cysteine (polar, uncharged) in a highly conserved motif will likely impact the folding of the linker region and affect the flanking domains, that is, binding of the PHD and Brd to the histones, especially in the presence of other cysteines upstream the mutation. In fact, it has been shown that cysteine‐rich domains create disulfide‐bonded cysteine loops that influence the function of a protein [Roseman, 1988; Koticha et al., 2002]. Furthermore, analysis of post‐translational modification previously reported in ACF1 revealed large numbers of phosphorylation sites in the vicinity of the mutation [Hornbeck et al., 2012], which further supports the potential impact of the mutation on protein function. In line with this, a previous study has reported that deletion of the C‐terminus of ACF1, spanning the PHD and the bromodomain, influenced the function of ACF1 in vitamin D pathway and relieved repression of the VDR‐regulated genes [Ewing et al., 2007]. Depletion of hACF1 in human cells causes delay in cell cycle progression [Collins et al., 2002]. In Drosophila, disruption of BAZ1A compromises fly viability [Chioda et al., 2010]. To date, there is only one publication on Baz1a knockout mice, showing that male mice suffer from reduced fertility due to global misregulation of gene expression in sperm cells [Dowdle et al., 2013]. In addition to male infertility, a number of more modest phenotypes are described by the International Mouse Phenotyping Consortium (measures with P value < 0.005) and these include hypoactivity, decreased body mass and length, decreased body fat, increased mean platelet volume, and increased blood urea nitrogen levels. Interestingly, the mice also display abnormal bone mineralization, potentially linked to the established role of ACF1 in VD3 metabolism. BAZ1B, a BAZ1A homolog, has been previously implicated in Williams syndrome, a well‐known neurodevelopmental disorder [Lu et al., 1998]. Baz1b‐deficient mice die at the neonatal stage [Yoshimura et al., 2009].
The specific variant we identified is not present in >60,000 unrelated individuals accessible through the Exome Aggregation Consortium database [Exome Aggregation Consortium (ExAC), 2015]. Deletions encompassing BAZ1A have been described in the literature, but no clear phenotypic associations have been made to date. In a comprehensive study of copy‐number variants identified in patients with developmental delay and healthy controls [Coe et al., 2014], heterozygous deletions spanning BAZ1A have been identified in six patients and no controls (P value = 0.045), whereas duplications have been identified in four patients and five controls. We further note that BAZ1A, along with several other chromatin remodelers in the SWI/SNF complex, have been implicated in carcinosarcomas [Jones et al., 2014]. Interestingly, the mutations detected in BAZ1A in patients with carcinosarcoma accumulate in the same part of the protein (linker region and flanking domains) where the de novo mutation in the present study is located.
A large number of chromatin‐remodeling genes have been implicated in ID syndromes [Iwase et al., 2011; Harakalova et al., 2012; Kosho et al., 2013]. The majority of these genes are components of the SWI/SNF chromatin‐remodeling complex. In all cases, mutations in these genes give rise to a range of symptoms in addition to ID. The patient in the present study shares several clinical manifestations with patients carrying mutations in components of the SWI/SNF complex [Kosho et al., 2013]. Shared symptoms include severe ID, autism, profound speech impairment (in agreement with patients with mutations in any component of the SWI/SNF complex), seizures (mutations in SMARCA2 and SMARCB1), hypotonia (mutations in SMARCB1, SMARCA2, and SMARCA4), hypoplastic finger nails (SMARCA4 mutations), and distal phalanges (SMARCB1, SMARCA4, and ARID1B mutations). The most striking clinical overlap is found with patients with Coffin–Siris syndrome, caused by mutations in SMARCA4. The fact that we also find significant changes in the expression of SMARCA4 in our patient raises the possibility that there is an interaction between the ISWI and SWI/SNF complexes.
Since we are not aware of any other patients with similar phenotype with mutation in BAZ1A, we prioritized functional analysis of the mutation, and of BAZ1A in general. We initially focused on the known involvement of BAZ1A in the VD3 pathway in Saos‐2 cells. Targeted qrtPCR revealed overexpression of the CYP24A1 in the patient compared with the parents, which was also confirmed in Saos‐2 cells transfected with the mutated ACF1 (mut‐ACF1) transcript, verifying the effect of the mutation on the expression of CYP24A1. The ChIP‐qPCR analysis revealed this effect was caused by the reduced efficiency of the mut‐ACF1 to bind and repress the expression of CYP24A1. CYP24A1 was previously shown to control vitamin D levels in the cell in a negative feedback process. In the absence of VD3, CYP24A1 expression is suppressed by ACF1 and the N‐CoR protein [Vaisanen et al., 2005; Ewing et al., 2007]. The overexpression of CYP24A1 could therefore explain the vitamin D insufficiency detected in the patient. On the other hand, individuals with ID and epilepsy commonly suffer from vitamin D deficiency [Knudtzon et al., 1987], and the deficiency may be further exacerbated by anticonvulsant drug treatment [Silver et al., 1974; Harijan et al., 2013]. However, to our knowledge, no direct link to changes in expression of factors in vitamin D metabolism, as we find for CYP24A1, has previously been established in this patient group. VD3 deficiency has been linked to a range of clinical symptoms, including effects on the developing brain. Mice with VD3 deficiency suffer from profound brain structural alterations [Eyles et al., 2003]. Our results show that the role of BAZ1A in VD3 regulation is similar to that of BAZ1B [Fujiki et al., 2005], a homolog to BAZ1A known to be involved in the causation of Williams syndrome [Lu et al., 1998].
Consistent with the function of ACF1 as a chromatin‐remodeling factor, our RNA‐seq data revealed many genes in the patient to be differentially expressed, most notably, genes involved in the Wnt signaling pathway. The link between chromatin‐remodeling factors and Wnt/β‐catenin signaling pathway has been highlighted in several studies [Barker et al., 2001; Griffin et al., 2011; Holik et al., 2014]. Wnt signaling is a highly conserved cascade known to play key roles in many developmental processes including the central nervous system [Salinas, 2012]. Mutations in genes encoding members of the Wnt signaling pathway have previously been implicated in human cognitive disorders such as ID and autism [de Ligt et al., 2012; O'Roak et al., 2012]. Our observation is supported by previous studies of the role of ACF1 in the basal repression of Wnt target genes in Drosophila [Liu et al., 2008]. In the absence of Wnt signaling, ACF1 binds to target genes and antagonizes the acetylation of histone H4 causing transcriptional repression. Consistent with the role of the C terminus of ACF1 in binding histones H3 and H4, the mutation in the patient could therefore explain the misregulation of Wnt target genes observed in the RNA‐seq data. Similarly, ARID1B, a chromatin‐remodeling factor, was recently shown to repress Wnt/β‐catenin signaling. Moreover, ID patients with loss‐of‐function mutations an ARIDB1, show upregulation Wnt/β‐catenin target genes [Vasileiou et al., 2015]. The influence of ACF1 on Wnt signaling further adds to the growing evidence of a link between chromatin‐remodeling factors and cancer. Additionally, studies in cultured cells and animal models revealed a crosstalk between VDR and Wnt signaling where VD3 antagonizes the activity of Wnt signaling [Larriba et al., 2013]. Therefore, the reduced levels of VD3 in the patient could further contribute to the differential expression of Wnt targets in the patient.
Our analysis also revealed DEGs involved in synaptic function, including overexpression of SYNGAP1 encoding synaptic GTPase‐activating protein. SYNGAP1 is a major member of the postsynaptic density [Chen et al., 1998] and is essential for postsynaptic signaling [Komiyama et al., 2002]. Mutations in this gene have previously been associated with ID [Hamdan et al., 2009] and epilepsy [Carvill et al., 2013]. Moreover, SYNGAP1 overexpression was reported to hinder axon outgrowth in granule cells [Tomoda et al., 2004]. SMARCA4, a member of SWI/SNF chromatin‐remodeling complex, was also overexpressed in the patient. As most chromatin remodelers, SMARCA4 is known to have broad range of functions including roles in the development of the heart and the nervous system in a dosage‐dependent manner [Lessard et al., 2007; Wu et al., 2007; Takeuchi et al., 2011; Smith‐Roe and Bultman, 2013]. Mice lacking SMARCA4 die in early development [Bultman et al., 2000]. Targeted depletion of SMARCA4 in neural stem/progenitor cells result in reduced numbers of both neurons and glia, and reduced brain size and the absence of the cerebellum in mouse embryos [Lessard et al., 2007]. In line with this, mutations in SMARCA4 have been linked to Coffin–Siris syndrome [Tsurusaki et al., 2012; Kosho et al., 2013]. It is also interesting to note that both SYNGAP1 and SMARCA4 are interconnected with the Wnt signaling pathway [Griffin et al., 2011; Krumm et al., 2014].
We also investigated the role of ACF1 on regulating gene expression using RNA‐seq. Upon BAZ1A knockdown in Saos‐2 cells, we found large numbers of gene to be differentially expressed including genes important for cell signaling and development. In agreement with our results, ACF1 was reported to be involved in chromatin remodeling during development and in several signaling processes in human and drosophila, especially in hormone responsiveness and the signaling necessary for DNA damage repair [Ewing et al., 2007; Sanchez‐Molina et al., 2011]. In Baz1a knockout mice, we demonstrate that the frontal brain harbors the largest number of DEGs as compared with heart and dorsal brain. Interestingly, we observed that many DEGs in the brain are important for neurological processes and synaptic function, highlighting a previously unknown function of ACF1 in nervous system. We also found that ACF1 function is tissue specific, obvious from the weak overlap between the DEGs in different tissues. Similarly, a recent study demonstrated that overexpression of Baz1a specifically in the nucleus accumbens contributes to susceptibility to stress‐induced depressive‐like behaviors [Sun et al., 2015]. Nevertheless, we observed that DEGs in the different tissues are enriched in pathways important for development, accentuating a general role for ACF1 in tissue development. Although ACF1 is generally recognized as a transcription repressor, we show several genes to be upregulated upon BAZ1A depletion, highlighting new role of ACF1 in transcription regulation.
In this study, we report, for the first time, a de novo mutation in BAZ1A, in a patient with a syndromic form of ID. Consistent with the function of ACF1 as a chromatin‐remodeling factor, the de novo mutation affects the expression of genes involved in several biological pathways. Our data support a role for BAZ1A on three distinct pathways: vitamin D regulation, Wnt signaling, and postsynaptic signaling. The diverse roles of BAZ1A could explain several syndromic clinical phenotype of our patient, similar to the symptoms identified in other patients with mutations in chromatin‐remodeling factors. We particularly demonstrate that the differential expression of CYP24A1, SYNGAP1, and SMARCA4 correlates with clinical findings in the patient, raising the possibility that the de novo mutation in BAZ1A may contribute to the phenotypes of the patient. However, further functional studies are needed to prove unambiguously the pathogenicity of this de novo variant in ID. We also report new roles of BAZ1A in nervous system and neurological process. Our findings add to the growing evidence for a role of chromatin‐remodeling factors in cognition and in syndrome related to neurodevelopment.
Disclosure statement: The authors declare no conflict of interest.
Supporting information
Disclaimer: Supplementary materials have been peer‐reviewed but not copyedited.
Supp. Figure S1: Baz1a knockout mice validation. Expression levels of Baz1a in control and knockout mice tissues analyzed using qrtPCR. All expression values were normalized to the levels of Gapdh in in each respective sample. Expression levels represent mean values and error bars are ± sd.
Supp. Table S1: Primer sequences
Supp. Table S2: All list of differentially expressed genes between the patient and parents, with a p‐value <0.0001
Supp. Table S3: Gene Ontology (GO) categories for the differentially expressed genes in the patient
Supp. Table S4: Expression of Wnt pathway target genes in the patient
Supp. Table S5: Top ten Gene Ontology (GO) categories for the differentially expressed genes in Saos‐2 cell after BAZ1A knockdown
Supp. Table S6: Gene Ontology (GO) categories for the differentially expressed genes in frontal brain tissue of Baz1a knockout mice
Supp. Table S7: Gene Ontology (GO) categories for the differentially expressed genes in heart tissue from Baz1a knockout mice
Supp. Table S8: DEGs in the knockout mice brain overlapping with the genomic location of ACF1 binding detected in (Sun et al., 2015)
Supp. Table S9: DEGs in the knockout mice heart overlapping with the genomic location of ACF1 binding detected in (Sun et al., 2015)
Supp. Table S10: Differentially expressed genes upon BAZ1A depletion
Supp. Methods
Supp. References
Acknowledgments
We thank Patrick Varga‐Weisz for the polyclonal anti‐hACF1 antibody and for the original ACF1 plasmid. We would also like to thank Sabrina Kutschan, Di Yu, and Pernilla Martinsson for assistance with the qrtPCR experiment, monocyte extraction, and western blot. We are grateful to the Uppsala Genome Center for technical support with library preparation and sequencing experiments. Computational analyses were performed on resources provided by SNIC through Uppsala Multidisciplinary Center for Advanced Computational Science (UPPMAX).
Contract grant sponsors: Hans von Kantzow Foundation; Swedish Research Council (Dnr. 521‐2014‐3093, Dnr. 521‐2012‐2639); Swedish Cancer Society (2013/404); European Research Council (ERC starting grant agreement no. 282330).
Communicated by Mark H. Paalman
References
- Aasland R, Gibson TJ, Stewart AF. 1995. The PHD finger: implications for chromatin‐mediated transcriptional regulation. Trends Biochem Sci 20:56–59. [DOI] [PubMed] [Google Scholar]
- Alvarez‐Saavedra M, De Repentigny Y, Lagali PS, Raghu Ram EV, Yan K, Hashem E, Ivanochko D, Huh MS, Yang D, Mears AJ, Todd MA, Corcoran CP, et al. 2014. Snf2h‐mediated chromatin organization and histone H1 dynamics govern cerebellar morphogenesis and neural maturation. Nat Commun 5:4181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anders S, Huber W. 2010. Differential expression analysis for sequence count data. Genome Biol 11:R106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banting GS, Barak O, Ames TM, Burnham AC, Kardel MD, Cooch NS, Davidson CE, Godbout R, Mcdermid HE, Shiekhattar R. 2005. CECR2, a protein involved in neurulation, forms a novel chromatin remodeling complex with SNF2L. Hum Mol Genet 14:513–524. [DOI] [PubMed] [Google Scholar]
- Barker N, Hurlstone A, Musisi H, Miles A, Bienz M, Clevers H. 2001. The chromatin remodelling factor Brg‐1 interacts with beta‐catenin to promote target gene activation. EMBO J 20:4935–4943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becker PB, Horz W. 2002. ATP‐dependent nucleosome remodeling. Annu Rev Biochem 71:247–273. [DOI] [PubMed] [Google Scholar]
- Berryer MH, Hamdan FF, Klitten LL, Moller RS, Carmant L, Schwartzentruber J, Patry L, Dobrzeniecka S, Rochefort D, Neugnot‐Cerioli M, Lacaille JC, Niu Z, et al. 2013. Mutations in SYNGAP1 cause intellectual disability, autism, and a specific form of epilepsy by inducing haploinsufficiency. Hum Mutat 34:385–394. [DOI] [PubMed] [Google Scholar]
- Bhaskara RM, De Brevern AG, Srinivasan N. 2013. Understanding the role of domain‐domain linkers in the spatial orientation of domains in multi‐domain proteins. J Biomol Struct Dyn 31:1467–1480. [DOI] [PubMed] [Google Scholar]
- Bradley A, Anastassiadis K, Ayadi A, Battey JF, Bell C, Birling MC, Bottomley J, Brown SD, Burger A, Bult CJ, Bushell W, Collins FS, et al. 2012. The mammalian gene function resource: the International Knockout Mouse Consortium. Mamm Genome 23:580–586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buganim Y, Goldstein I, Lipson D, Milyavsky M, Polak‐Charcon S, Mardoukh C, Solomon H, Kalo E, Madar S, Brosh R, Perelman M, Navon R, et al. 2010. A novel translocation breakpoint within the BPTF gene is associated with a pre‐malignant phenotype. PLoS One 5:e9657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bultman S, Gebuhr T, Yee D, La Mantia C, Nicholson J, Gilliam A, Randazzo F, Metzger D, Chambon P, Crabtree G, Magnuson T. 2000. A Brg1 null mutation in the mouse reveals functional differences among mammalian SWI/SNF complexes. Mol Cell 6:1287–1295. [DOI] [PubMed] [Google Scholar]
- Carvill GL, Heavin SB, Yendle SC, Mcmahon JM, O'roak BJ, Cook J, Khan A, Dorschner MO, Weaver M, Calvert S, Malone S, Wallace G, et al. 2013. Targeted resequencing in epileptic encephalopathies identifies de novo mutations in CHD2 and SYNGAP1. Nat Genet 45:825–830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen HJ, Rojas‐Soto M, Oguni A, Kennedy MB. 1998. A synaptic Ras‐GTPase activating protein (p135 SynGAP) inhibited by CaM kinase II. Neuron 20:895–904. [DOI] [PubMed] [Google Scholar]
- Cheng J, Randall A, Baldi P. 2006. Prediction of protein stability changes for single‐site mutations using support vector machines. Proteins 62:1125–1132. [DOI] [PubMed] [Google Scholar]
- Chioda M, Vengadasalam S, Kremmer E, Eberharter A, Becker PB. 2010. Developmental role for ACF1‐containing nucleosome remodellers in chromatin organisation. Development 137:3513–3522. [DOI] [PubMed] [Google Scholar]
- Coe BP, Witherspoon K, Rosenfeld JA, Van Bon BW, Vulto‐Van Silfhout AT, Bosco P, Friend KL, Baker C, Buono S, Vissers LE, Schuurs‐Hoeijmakers JH, Hoischen A, et al. 2014. Refining analyses of copy number variation identifies specific genes associated with developmental delay. Nat Genet 46:1063–1071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins N, Poot RA, Kukimoto I, Garcia‐Jimenez C, Dellaire G, Varga‐Weisz PD. 2002. An ACF1‐ISWI chromatin‐remodeling complex is required for DNA replication through heterochromatin. Nat Genet 32:627–632. [DOI] [PubMed] [Google Scholar]
- Day JJ, Sweatt JD. 2011. Epigenetic mechanisms in cognition. Neuron 70:813–829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Ligt J, Willemsen MH, Van Bon BW, Kleefstra T, Yntema HG, Kroes T, Vulto‐Van Silfhout AT, Koolen DA, De Vries P, Gilissen C, Del Rosario M, Hoischen A, et al. 2012. Diagnostic exome sequencing in persons with severe intellectual disability. N Engl J Med 367:1921–1929. [DOI] [PubMed] [Google Scholar]
- Dirscherl SS, Henry JJ, Krebs JE. 2005. Neural and eye‐specific defects associated with loss of the imitation switch (ISWI) chromatin remodeler in Xenopus laevis. Mech Dev 122:1157–1170. [DOI] [PubMed] [Google Scholar]
- Dowdle JA, Mehta M, Kass EM, Vuong BQ, Inagaki A, Egli D, Jasin M, Keeney S. 2013. Mouse BAZ1A (ACF1) is dispensable for double‐strand break repair but is essential for averting improper gene expression during spermatogenesis. PLoS Genet 9:e1003945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eberharter A, Becker PB. 2004. ATP‐dependent nucleosome remodelling: factors and functions. J Cell Sci 117:3707–3711. [DOI] [PubMed] [Google Scholar]
- Eberharter A, Vetter I, Ferreira R, Becker PB. 2004. ACF1 improves the effectiveness of nucleosome mobilization by ISWI through PHD‐histone contacts. EMBO J 23:4029–4039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ewing AK, Attner M, Chakravarti D. 2007. Novel regulatory role for human Acf1 in transcriptional repression of vitamin D3 receptor‐regulated genes. Mol Endocrinol 21:1791–1806. [DOI] [PubMed] [Google Scholar]
- Exome Aggregation Consortium (ExAC), C., MA . 2015. Exome Aggregation Consortium (ExAC), Cambridge, MA.Available at: http://exac.broadinstitute.org).
- Eyles D, Brown J, Mackay‐Sim A, Mcgrath J, Feron F. 2003. Vitamin D3 and brain development. Neuroscience 118:641–653. [DOI] [PubMed] [Google Scholar]
- Flaus A, Martin DM, Barton GJ, Owen‐Hughes T. 2006. Identification of multiple distinct Snf2 subfamilies with conserved structural motifs. Nucleic Acids Res 34:2887–2905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujiki R, Kim MS, Sasaki Y, Yoshimura K, Kitagawa H, Kato S. 2005. Ligand‐induced transrepression by VDR through association of WSTF with acetylated histones. EMBO J 24:3881–3894. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Fyodorov DV, Kadonaga JT. 2002. Binding of Acf1 to DNA involves a WAC motif and is important for ACF‐mediated chromatin assembly. Mol Cell Biol 22:6344–6353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffin CT, Curtis CD, Davis RB, Muthukumar V, Magnuson T. 2011. The chromatin‐remodeling enzyme BRG1 modulates vascular Wnt signaling at two levels. Proc Natl Acad Sci U S A 108:2282–2287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamdan FF, Gauthier J, Spiegelman D, Noreau A, Yang Y, Pellerin S, Dobrzeniecka S, Cote M, Perreau‐Linck E, Carmant L, D'anjou G, Fombonne E, et al. 2009. Mutations in SYNGAP1 in autosomal nonsyndromic mental retardation. N Engl J Med 360:599–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harakalova M, Van Den Boogaard MJ, Sinke R, Van Lieshout S, Van Tuil MC, Duran K, Renkens I, Terhal PA, De Kovel C, Nijman IJ, Van Haelst M, Knoers NV, Van Haaften G, Kloosterman W, Hennekam RC, Cuppen E, Ploos Van Amstel HK. 2012. X‐exome sequencing identifies a HDAC8 variant in a large pedigree with X‐linked intellectual disability, truncal obesity, gynaecomastia, hypogonadism and unusual face. J Med Genet 49:539–543. [DOI] [PubMed] [Google Scholar]
- Hargreaves DC, Crabtree GR. 2011. ATP‐dependent chromatin remodeling: genetics, genomics and mechanisms. Cell Res 21:396–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harijan P, Khan A, Hussain N. 2013. Vitamin D deficiency in children with epilepsy: do we need to detect and treat it? J Pediatr Neurosci 8:5–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He X, Fan HY, Garlick JD, Kingston RE. 2008. Diverse regulation of SNF2h chromatin remodeling by noncatalytic subunits. Biochemistry 47:7025–7033. [DOI] [PubMed] [Google Scholar]
- Ho L, Jothi R, Ronan JL, Cui K, Zhao K, Crabtree GR. 2009. An embryonic stem cell chromatin remodeling complex, esBAF, is an essential component of the core pluripotency transcriptional network. Proc Natl Acad Sci USA 106:5187–5191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holik AZ, Young M, Krzystyniak J, Williams GT, Metzger D, Shorning BY, Clarke AR. 2014. Brg1 loss attenuates aberrant wnt‐signalling and prevents wnt‐dependent tumourigenesis in the murine small intestine. PLoS Genet 10:e1004453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hornbeck PV, Kornhauser JM, Tkachev S, Zhang B, Skrzypek E, Murray B, Latham V, Sullivan M. 2012. PhosphoSitePlus: a comprehensive resource for investigating the structure and function of experimentally determined post‐translational modifications in man and mouse. Nucleic Acids Res 40:D261–D270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito T, Levenstein ME, Fyodorov DV, Kutach AK, Kobayashi R, Kadonaga JT. 1999. ACF consists of two subunits, Acf1 and ISWI, that function cooperatively in the ATP‐dependent catalysis of chromatin assembly. Genes Dev 13:1529–1539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwase S, Xiang B, Ghosh S, Ren T, Lewis PW, Cochrane JC, Allis CD, Picketts DJ, Patel DJ, Li H, Shi Y. 2011. ATRX ADD domain links an atypical histone methylation recognition mechanism to human mental‐retardation syndrome. Nat Struct Mol Biol 18:769–776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jenuwein T, Allis CD. 2001. Translating the histone code. Science 293:1074–1080. [DOI] [PubMed] [Google Scholar]
- Jones G, Prosser DE, Kaufmann M. 2012. 25‐Hydroxyvitamin D‐24‐hydroxylase (CYP24A1): its important role in the degradation of vitamin D. Arch Biochem Biophys 523:9–18. [DOI] [PubMed] [Google Scholar]
- Jones MH, Hamana N, Nezu J, Shimane M. 2000. A novel family of bromodomain genes. Genomics 63:40–45. [DOI] [PubMed] [Google Scholar]
- Jones S, Stransky N, Mccord CL, Cerami E, Lagowski J, Kelly D, Angiuoli SV, Sausen M, Kann L, Shukla M, Makar R, Wood LD, Diaz LA, Jr , Lengauer C, Velculescu VE. 2014. Genomic analyses of gynaecologic carcinosarcomas reveal frequent mutations in chromatin remodelling genes. Nat Commun 5:5006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kircher M, Witten DM, Jain P, O'roak BJ, Cooper GM, Shendure J. 2014. A general framework for estimating the relative pathogenicity of human genetic variants. Nat Genet 46:310–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knudtzon J, Aksnes L, Akslen LA, Aarskog D. 1987. Elevated 1,25‐dihydroxyvitamin D and normocalcaemia in presumed familial Williams syndrome. Clin Genet 32:369–374. [DOI] [PubMed] [Google Scholar]
- Komiyama NH, Watabe AM, Carlisle HJ, Porter K, Charlesworth P, Monti J, Strathdee DJ, O'carroll CM, Martin SJ, Morris RG, O'dell TJ, Grant SG. 2002. SynGAP regulates ERK/MAPK signaling, synaptic plasticity, and learning in the complex with postsynaptic density 95 and NMDA receptor. J Neurosci 22:9721–9732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kosho T, Okamoto N, Ohashi H, Tsurusaki Y, Imai Y, Hibi‐Ko Y, Kawame H, Homma T, Tanabe S, Kato M, Hiraki Y, Yamagata T, et al. 2013. Clinical correlations of mutations affecting six components of the SWI/SNF complex: detailed description of 21 patients and a review of the literature. Am J Med Genet A 161A:1221–1237. [DOI] [PubMed] [Google Scholar]
- Koticha DK, Mccarthy EE, Baldini G. 2002. Plasma membrane targeting of SNAP‐25 increases its local concentration and is necessary for SNARE complex formation and regulated exocytosis. J Cell Sci 115:3341–3351. [DOI] [PubMed] [Google Scholar]
- Krumm N, O'roak BJ, Shendure J, Eichler EE. 2014. A de novo convergence of autism genetics and molecular neuroscience. Trends Neurosci 37:95–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lan L, Ui A, Nakajima S, Hatakeyama K, Hoshi M, Watanabe R, Janicki SM, Ogiwara H, Kohno T, Kanno S, Yasui A. 2010. The ACF1 complex is required for DNA double‐strand break repair in human cells. Mol Cell 40:976–987. [DOI] [PubMed] [Google Scholar]
- Landry J, Sharov AA, Piao Y, Sharova LV, Xiao H, Southon E, Matta J, Tessarollo L, Zhang YE, Ko MS, Kuehn MR, Yamaguchi TP, Wu C. 2008. Essential role of chromatin remodeling protein Bptf in early mouse embryos and embryonic stem cells. PLoS Genet 4:e1000241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langst G, Becker PB. 2001. Nucleosome mobilization and positioning by ISWI‐containing chromatin‐remodeling factors. J Cell Sci 114:2561–2568. [DOI] [PubMed] [Google Scholar]
- Langst G, Manelyte L. 2015. Chromatin remodelers: from function to dysfunction. Genes (Basel) 6:299–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larriba MJ, Gonzalez‐Sancho JM, Barbachano A, Niell N, Ferrer‐Mayorga G, Munoz A. 2013. Vitamin D is a multilevel repressor of Wnt/b‐catenin signaling in cancer cells. Cancers (Basel) 5:1242–1260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leonard H, Wen X. 2002. The epidemiology of mental retardation: challenges and opportunities in the new millennium. Ment Retard Dev Disabil Res Rev 8:117–134. [DOI] [PubMed] [Google Scholar]
- Lessard J, Wu JI, Ranish JA, Wan M, Winslow MM, Staahl BT, Wu H, Aebersold R, Graef IA, Crabtree GR. 2007. An essential switch in subunit composition of a chromatin remodeling complex during neural development. Neuron 55:201–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li G, Reinberg D. 2011. Chromatin higher‐order structures and gene regulation. Curr Opin Genet Dev 21:175–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Zhao G, Gao X. 2013. Development of neurodevelopmental disorders: a regulatory mechanism involving bromodomain‐containing proteins. J Neurodev Disord 5:4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu YI, Chang MV, Li HE, Barolo S, Chang JL, Blauwkamp TA, Cadigan KM. 2008. The chromatin remodelers ISWI and ACF1 directly repress Wingless transcriptional targets. Dev Biol 323:41–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez AJ, Wood MA. 2015. Role of nucleosome remodeling in neurodevelopmental and intellectual disability disorders. Front Behav Neurosci 9:100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love MI, Huber W, Anders S. 2014. Moderated estimation of fold change and dispersion for RNA‐seq data with DESeq2. Genome Biol 15:550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu X, Meng X, Morris CA, Keating MT. 1998. A novel human gene, WSTF, is deleted in Williams syndrome. Genomics 54:241–249. [DOI] [PubMed] [Google Scholar]
- Luo W, Karpf AR, Deeb KK, Muindi JR, Morrison CD, Johnson CS, Trump DL. 2010. Epigenetic regulation of vitamin D 24‐hydroxylase/CYP24A1 in human prostate cancer. Cancer Res 70:5953–5962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marmorstein R, Berger SL. 2001. Structure and function of bromodomains in chromatin‐regulating complexes. Gene 272:1–9. [DOI] [PubMed] [Google Scholar]
- Mohrmann L, Verrijzer CP. 2005. Composition and functional specificity of SWI2/SNF2 class chromatin remodeling complexes. Biochim Biophys Acta 1681:59–73. [DOI] [PubMed] [Google Scholar]
- Narlikar GJ, Fan HY, Kingston RE. 2002. Cooperation between complexes that regulate chromatin structure and transcription. Cell 108:475–487. [DOI] [PubMed] [Google Scholar]
- O'roak BJ, Vives L, Girirajan S, Karakoc E, Krumm N, Coe BP, Levy R, Ko A, Lee C, Smith JD, Turner EH, Stanaway IB, et al. 2012. Sporadic autism exomes reveal a highly interconnected protein network of de novo mutations. Nature 485:246–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez Jurado AL. 2003. Williams‐Beuren syndrome: a model of recurrent genomic mutation. Horm Res 59 Suppl 1:106–113. [DOI] [PubMed] [Google Scholar]
- Ramagopalan SV, Heger A, Berlanga AJ, Maugeri NJ, Lincoln MR, Burrell A, Handunnetthi L, Handel AE, Disanto G, Orton SM, Watson CT, Morahan JM, et al. 2010. A ChIP‐seq defined genome‐wide map of vitamin D receptor binding: associations with disease and evolution. Genome Res 20:1352–1360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson MD, Mccarthy DJ, Smyth GK. 2010. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26:139–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ronan JL, Wu W, Crabtree GR. 2013. From neural development to cognition: unexpected roles for chromatin. Nat Rev Genet 14:347–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roseman MA. 1988. Hydrophilicity of polar amino acid side‐chains is markedly reduced by flanking peptide bonds. J Mol Biol 200:513–522. [DOI] [PubMed] [Google Scholar]
- Rumbaugh G, Adams JP, Kim JH, Huganir RL. 2006. SynGAP regulates synaptic strength and mitogen‐activated protein kinases in cultured neurons. Proc Natl Acad Sci USA 103:4344–4351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salinas PC. 2012. Wnt signaling in the vertebrate central nervous system: from axon guidance to synaptic function. Cold Spring Harb Perspect Biol 4:pii:a008003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez‐Molina S, Mortusewicz O, Bieber B, Auer S, Eckey M, Leonhardt H, Friedl AA, Becker PB. 2011. Role for hACF1 in the G2/M damage checkpoint. Nucleic Acids Res 39:8445–8456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santen GW, Aten E, Vulto‐Van Silfhout AT, Pottinger C, Van Bon BW, Van Minderhout IJ, Snowdowne R, Van Der Lans CA, Boogaard M, Linssen MM, Vijfhuizen L, Van Der Wielen MJ, et al. 2013. Coffin‐Siris syndrome and the BAF complex: genotype‐phenotype study in 63 patients. Hum Mutat 34:1519–1528. [DOI] [PubMed] [Google Scholar]
- Santen GW, Kriek M, Van Attikum H. 2012. SWI/SNF complex in disorder: SWItching from malignancies to intellectual disability. Epigenetics 7:1219–1224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schultz DC, Friedman JR, Rauscher FJ, 3rd . 2001. Targeting histone deacetylase complexes via KRAB‐zinc finger proteins: the PHD and bromodomains of KAP‐1 form a cooperative unit that recruits a novel isoform of the Mi‐2alpha subunit of NuRD. Genes Dev 15:428–443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwarz JM, Rodelsperger C, Schuelke M, Seelow D. 2010. MutationTaster evaluates disease‐causing potential of sequence alterations. Nat Methods 7:575–576. [DOI] [PubMed] [Google Scholar]
- Sherry ST, Ward MH, Kholodov M, Baker J, Phan L, Smigielski EM, Sirotkin K. 2001. dbSNP: the NCBI database of genetic variation. Nucleic Acids Res 29:308–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shevell M, Ashwal S, Donley D, Flint J, Gingold M, Hirtz D, Majnemer A, Noetzel M, Sheth RD, Quality Standards Subcommittee of the American Academy of Neurology; Practice Committee of the Child Neurology Society . 2003. Practice parameter: evaluation of the child with global developmental delay: report of the Quality Standards Subcommittee of the American Academy of Neurology and The Practice Committee of the Child Neurology Society. Neurology 60:367–380. [DOI] [PubMed] [Google Scholar]
- Silver J, Davies TJ, Kupersmitt E, Orme M, Petrie A, Vajda F. 1974. Prevalence and treatment of vitamin D deficiency in children on anticonvulsant drugs. Arch Dis Child 49:344–350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skarnes WC, Rosen B, West AP, Koutsourakis M, Bushell W, Iyer V, Mujica AO, Thomas M, Harrow J, Cox T, Jackson D, Severin J, et al. 2011. A conditional knockout resource for the genome‐wide study of mouse gene function. Nature 474:337–342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith‐Roe SL, Bultman SJ. 2013. Combined gene dosage requirement for SWI/SNF catalytic subunits during early mammalian development. Mamm Genome 24:21–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stopka T, Skoultchi AI. 2003. The ISWI ATPase Snf2h is required for early mouse development. Proc Natl Acad Sci USA 100:14097–14102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strohner R, Nemeth A, Jansa P, Hofmann‐Rohrer U, Santoro R, Langst G, Grummt I. 2001. NoRC—a novel member of mammalian ISWI‐containing chromatin remodeling machines. EMBO J 20:4892–4900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun H, Damez‐Werno DM, Scobie KN, Shao NY, Dias C, Rabkin J, Koo JW, Korb E, Bagot RC, Ahn FH, Cahill ME, Labonte B, et al. 2015. ACF chromatin‐remodeling complex mediates stress‐induced depressive‐like behavior. Nat Med 21:1146–1153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Supek F, Bosnjak M, Skunca N, Smuc T. 2011. REVIGO summarizes and visualizes long lists of gene ontology terms. PLoS One 6:e21800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeuchi JK, Lou X, Alexander JM, Sugizaki H, Delgado‐Olguin P, Holloway AK, Mori AD, Wylie JN, Munson C, Zhu Y, Zhou YQ, Yeh RF, et al. 2011. Chromatin remodelling complex dosage modulates transcription factor function in heart development. Nat Commun 2:187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomoda T, Kim JH, Zhan C, Hatten ME. 2004. Role of Unc51.1 and its binding partners in CNS axon outgrowth. Genes Dev 18:541–558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsurusaki Y, Okamoto N, Ohashi H, Kosho T, Imai Y, Hibi‐Ko Y, Kaname T, Naritomi K, Kawame H, Wakui, K , Fukushima Y, Homma T, et al. 2012. Mutations affecting components of the SWI/SNF complex cause Coffin‐Siris syndrome. Nat Genet 4:376–378. [DOI] [PubMed] [Google Scholar]
- Vaisanen S, Dunlop TW, Sinkkonen L, Frank C, Carlberg C. 2005. Spatio‐temporal activation of chromatin on the human CYP24 gene promoter in the presence of 1alpha,25‐dihydroxyvitamin D3. J Mol Biol 350:65–77. [DOI] [PubMed] [Google Scholar]
- Vasileiou G, Ekici AB, Uebe S, Zweier C, Hoyer J, Engels H, Behrens J, Reis A, Hadjihannas MV. 2015. Chromatin‐remodeling‐factor ARID1B represses Wnt/beta‐catenin signaling. Am J Hum Genet 97:445–456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veltman JA, Brunner HG. 2012. De novo mutations in human genetic disease. Nat Rev Genet 13:565–575. [DOI] [PubMed] [Google Scholar]
- Wang K, Li M, Hakonarson H. 2010. ANNOVAR: functional annotation of genetic variants from high‐throughput sequencing data. Nucleic Acids Res 38:e164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson BG, Roberts CW. 2011. SWI/SNF nucleosome remodellers and cancer. Nat Rev Cancer 11:481–492. [DOI] [PubMed] [Google Scholar]
- Wu JI, Lessard J, Olave IA, Qiu Z, Ghosh A, Graef IA, Crabtree GR. 2007. Regulation of dendritic development by neuron‐specific chromatin remodeling complexes. Neuron 56:94–108. [DOI] [PubMed] [Google Scholar]
- Ye Y, Xiao Y, Wang W, Wang Q, Yearsley K, Wani AA, Yan Q, Gao JX, Shetuni BS, Barsky SH. 2009. Inhibition of expression of the chromatin remodeling gene, SNF2L, selectively leads to DNA damage, growth inhibition, and cancer cell death. Mol Cancer Res 7:1984–1999. [DOI] [PubMed] [Google Scholar]
- Yoshimura K, Kitagawa H, Fujiki R, Tanabe M, Takezawa S, Takada I, Yamaoka I, Yonezawa M, Kondo T, Furutani Y, Yagi H, Yoshinaga S, et al. 2009. Distinct function of 2 chromatin remodeling complexes that share a common subunit, Williams syndrome transcription factor (WSTF). Proc Natl Acad Sci USA 106:9280–9285. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Zeng L, Yap KL, Ivanov AV, Wang X, Mujtaba S, Plotnikova O, Rauscher FJ, 3rd, Zhou MM. 2008. Structural insights into human KAP1 PHD finger‐bromodomain and its role in gene silencing. Nat Struct Mol Biol 15:626–633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zentner GE, Scacheri PC. 2012. The chromatin fingerprint of gene enhancer elements. J Biol Chem 287:30888–30896. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Disclaimer: Supplementary materials have been peer‐reviewed but not copyedited.
Supp. Figure S1: Baz1a knockout mice validation. Expression levels of Baz1a in control and knockout mice tissues analyzed using qrtPCR. All expression values were normalized to the levels of Gapdh in in each respective sample. Expression levels represent mean values and error bars are ± sd.
Supp. Table S1: Primer sequences
Supp. Table S2: All list of differentially expressed genes between the patient and parents, with a p‐value <0.0001
Supp. Table S3: Gene Ontology (GO) categories for the differentially expressed genes in the patient
Supp. Table S4: Expression of Wnt pathway target genes in the patient
Supp. Table S5: Top ten Gene Ontology (GO) categories for the differentially expressed genes in Saos‐2 cell after BAZ1A knockdown
Supp. Table S6: Gene Ontology (GO) categories for the differentially expressed genes in frontal brain tissue of Baz1a knockout mice
Supp. Table S7: Gene Ontology (GO) categories for the differentially expressed genes in heart tissue from Baz1a knockout mice
Supp. Table S8: DEGs in the knockout mice brain overlapping with the genomic location of ACF1 binding detected in (Sun et al., 2015)
Supp. Table S9: DEGs in the knockout mice heart overlapping with the genomic location of ACF1 binding detected in (Sun et al., 2015)
Supp. Table S10: Differentially expressed genes upon BAZ1A depletion
Supp. Methods
Supp. References