

Abstract
The coassembly of different building blocks into supramolecular copolymers provides a promising avenue to control their properties and to thereby expand the potential of supramolecular polymers in applications. However, contrary to covalent copolymerization which nowadays can be well controlled, the control over sequence, polymer length, and morphology in supramolecular copolymers is to date less developed, and their structures are more determined by the delicate balance in binding free energies between the distinct building blocks than by kinetics. Consequently, to rationalize the structures of supramolecular copolymers, a thorough understanding of their thermodynamic behavior is needed. Though this is well established for single-component assemblies and over the past years several models have been proposed for specific copolymerization cases, a generally applicable model for supramolecular cooperative copolymers is still lacking. Here, we provide a generalization of our earlier mass-balance models for supramolecular copolymerizations that encompasses all our earlier models. In this model, the binding free energies of each pair of monomer types in each aggregate type can be set independently. We provide scripts to solve the model numerically for any (co)polymerization of one or two types of monomer into an arbitrary number of distinct aggregate types. We illustrate the applicability of the model on data from literature as well as on new experimental data of triarylamine triamide-based copolymers in three distinct solvents. We show that apart from common properties such as the degree of polymerization and length distributions, our approach also allows us to investigate properties such as the copolymer microstructure, that is, the internal ordering of monomers within the copolymers. Moreover, we show that in some cases, also intriguing analytical approximations can be derived from the mass balances.
1. Introduction
Supramolecular polymers are one-dimensional assemblies of monomeric units held together via moderately strong, reversible, and often highly directional noncovalent interactions.1−4 The dynamic nature of these noncovalent interactions makes such supramolecular polymers versatile systems with high potential for use as adaptive materials that incorporate a variety of interesting mechanical, optical, electronic, or biological functionalities.5−10 The coassembly of multiple building blocks provides a promising avenue to further expand their potential in applications and to control the properties of the produced supramolecular polymers by changing the stoichiometry or feed ratio of the distinct components,11 whereas for covalent copolymers, the molecular structure can be predicted via the classical copolymer equation12 in combination with advanced controlled polymerization techniques13−19 and highly depends via relative reactivities on kinetics; for supramolecular polymers, control over sequence, polymer length, and morphology is yet less developed, and the structure is determined by the ratio of the binding free energies between the distinct building blocks rather than on kinetics. It is well established that supramolecular polymerizations can also form out-of-equilibrium,20 that is, in a dissipative state21−24 or in a kinetically trapped state, which has been exploited to control the degree of polymerization and form supramolecular block copolymers via living supramolecular polymerization.25−29 However, to understand the degree of polymerization, polydispersity, distribution over aggregate types, and internal structure (e.g., random, alternating, and blocked ordering) in the majority of supramolecular (co)polymerizations, a thorough understanding of the thermodynamic equilibrium is needed.
A theoretical foundation of supramolecular polymerization thermodynamics30−41 and kinetics42−53 is now well established. An important notion, for instance, is that of isodesmic versus (anti-)cooperative self-assembly, depending on whether the binding free energies in all monomer association steps are equal (isodesmic), that up to a certain critical nucleus size the binding free energies are smaller (cooperative), or that small aggregates are most stable and the binding free energies for monomer association steps to larger aggregates are smaller (anticooperative). These models show how thermodynamic parameters determine the degree of polymerization, the average lengths of the polymers, and their length–distribution profile, as well as their inclination to assemble/disassemble in response to temperature. More recently, these models have been extended to describe supramolecular polymerization with competing aggregation types.50,54−58
Following models of discrete coassemblies59−66 and initiator-based reversible copolymerization,67−69 theory on supramolecular copolymerization has been developed as well.70 Weller et al.71 and Evstigneev et al.,72,73 for instance, considered the case of noncooperative indefinite molecular heteroassembly. Moreover, van der Schoot et al. extended their original modeling for cooperative homopolymers74 to sergeants and soldiers,75 majority rules,76,77 blocky,78 and most recently, alternating79 copolymerizations. While their approach is based on Ising models and the transfer matrix method, we proposed a more direct approach, based on expressing the equilibrium concentrations of all aggregates in terms of the monomer equilibrium concentrations and on analytical expressions for the infinite sums in the resulting mass-balance equations.80,81 We initially focused on majority rule-based chiral amplification in the copolymerization of two enantiomers. Later, we extended this approach to multiple monomer82 and aggregate types,83 and competition between cooperative and isodesmic aggregates.84 Finally, extending the description that the free energy changes upon monomer associations may not only depend on the type of the monomer and the type of aggregate it binds to but also on the monomer at the end of the aggregate, we could investigate how the balance of interaction energies not only influences the size distribution but also the polymer microstructure, that is, from self-sorting to blocky, random, and alternating internal order.85
Here, we present the extension of this latter model to multiple aggregate types. We show that this model comprises all our earlier mass-balance models56,80−85 and can be used to describe cooperative as well as noncooperative polymerization, homoassembly as well as two-component coassembly, and the aggregation into a single aggregate type as well as competition between multiple aggregate types. In the next section, we describe this copolymer model, the derivation of the corresponding mass-balance equations, and the consequent limits on the equilibrium monomer concentrations. Moreover, we show how to calculate monomer/polymer concentrations and length distributions as well as other properties such as the sizes of blocks of repeating monomer units making up the polymer microstructure. In Section 3, we demonstrate how the scripts provided in the Supporting Information to numerically solve our general copolymerization model can be applied to not only reproduce our earlier model results but also to properly describe sergeants and soldiers data from the literature86 as well as some new data on solvent-dependent triarylamine triamide-based supramolecular block copolymerizations. After this illustration of the general applicability of our model, which for most cases must be solved numerically, we show in Section 4 that in several special cases, it can also provide interesting analytical approximations, such as (i) for a threshold on the heterointeraction above which polymers elongate rather than shorten upon mixing in small amounts of a comonomer, (ii) for the fraction of homo- and heterobonds as a function of the heterointeraction upon mixing two distinct highly polymerized homopolymers, and (iii) for the free monomer concentrations and polymerized fraction in the case of purely alternating copolymers.
2. Copolymerization Model
2.1. Copolymer Types
We consider the coassembly of two types of monomers, which we denote as A and B, into one or more types of supramolecular polymers. We assume that the aggregates have an intrinsic direction, that is, from a bottom element to a top element, and that the binding strength between two neighboring monomers in an aggregate is determined by the type of this aggregate as well as the types and order of the two monomers. The formation of aggregates of one type can then be described by four dimer formation reactions and four elongation reactions as schematically illustrated in Figure 1a. In the case of multiple possible copolymer types, for example, differing in helicity or morphology, we assume that copolymers of distinct types have no direct interaction. They do interact indirectly, as schematically illustrated in Figure 1b, because they share the same monomer pool. In the following subsections, we will first derive the mass-balance equations for the case of only one copolymer type. Also, the computation of various properties of copolymers, such as block lengths or amounts of bonds, will first be given for the case of a single-aggregate type. The step to systems with several distinct copolymer types will be made in Section 2.11.
Figure 1.
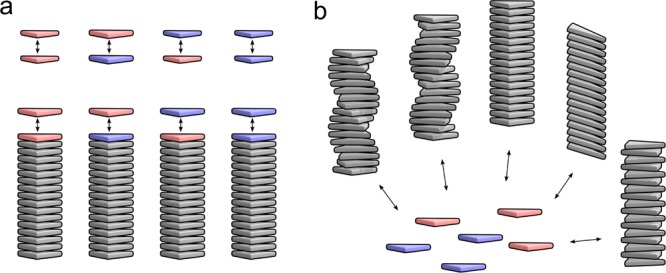
Schematic illustration of the considered supramolecular copolymerization. (a) Equilibrium constants of elongation of a supramolecular copolymer with a monomer may depend on the type of the monomer added and the type of the monomer at top of the copolymer, and they may differ from the equilibrium constants for the four possible dimerization reactions. Monomers that are not on the top of the copolymer do not influence the equilibrium constants of elongation and are drawn gray. (b) In the case of formation of multiple copolymer types, they compete for the same monomers and each aggregate type may have its own equilibrium constants for the eight reactions as illustrated in part (a).
2.2. Equilibrium Reactions
The copolymers will be written as sequences, with bottom left and top right. AABAB thus represents a copolymer of length five with an A monomer at the bottom and a B monomer at the top, which differs from BABAA with bottom B and top A. When needed, the length of a copolymer will be indicated by a subscript, that is, Pi denotes a copolymer of length i, and PiA is a copolymer of length i + 1 with top monomer A. Without the subscript, P represents a copolymer of arbitrary length.
Homopolymers of A can be generated by nucleation elongation reactions of the form
| 1 |
| 2 |
where KA–A and K*A–A are the equilibrium constants of nucleation and elongation, respectively, and PA represents an arbitrary polymer with top A and length at least 2. The corresponding cooperativity parameter is σA = KA–A/K*A–A. Note that elongation reaction 2 describes only growth at the top of the polymer. Growth at the bottom is described by the additional reaction
| 3 |
A consequence of adding growth at the bottom is that different ways arise to generate the same polymer. Polymer AAA, for instance, can be generated by the initial formation of dimer AA and subsequent association of an A monomer either at its top or at its bottom. As the equilibrium concentrations should satisfy according to the first route, [AAA] = K*A–A[AA][A] and according to the second route, [AAA] = KA–A*[AA][A], elongation of the homopolymer at the top or bottom must occur with the same equilibrium constant, that is, KA–A* = K*A–A. This is a so-called detailed balance condition. In general, the detailed balance condition requires that at thermodynamic equilibrium, the product of the equilibrium constants along two different routes to generate a (co)polymer must be equal.
Homopolymers of B can be generated by an analogous reaction scheme with nucleation equilibrium constant KB–B, elongation equilibrium constant K*B–B, and cooperativity parameter σB = KB–B/K*B–B. If growth at the bottom is also possible, the detailed balance condition gives again that the corresponding equilibrium constant KB–B* equals K*B–B.
Because of the assumption that the binding strength between two monomers is determined by the types of those two monomers, the generation of mixed copolymers is described by the additional reactions
| 4 |
| 5 |
| 6 |
| 7 |
Note that now in the reactions 6 and 7, but also in the reactions 2 and 3, P may be an arbitrary copolymer but with the same sequence at both sides of the reaction. These copolymers may also elongate at their bottom, but we do not require explicit description of those reactions as these reactions do not result in aggregates that cannot be made via the already given reactions. Their equilibrium constants are thus fixed via detailed balance relations, and their presence or absence does not influence the equilibrium concentrations.
2.3. Detailed Balance Conditions
The copolymer AAB can be generated in two ways, namely, initial AA dimer formation followed by association of B at the top
| 8 |
and initial AB dimer formation followed by association of A at the bottom
| 9 |
Detailed balance now implies that KA–AK*A–B = KA–BKA–A*, and because KA–A* = K*A–A, we obtain
| 10 |
Analogously, the two possible routes for construction of BBA lead to
| 11 |
Hence, from the four equilibrium constants KA–B, KB–A, K*A–B, K*B–A in the copolymerization reactions 4–7, only two are independent. The total set of reactions to construct all possible copolymers can thus be described with just six independent parameters: K*A–A, σA (for the A homopolymers), K*B–B, σB (for the B homopolymers), and K*A–B and K*B–A (for the heterointeraction between A and B).
Note that eqs 10 and 11 do not imply that KA–B = KB–A. Hence, the concentrations of the two dimers AB and BA will in general not be equal. If, by symmetry considerations, [AB] = [BA] is required, the additional symmetry conditionKA–B = KB–A may be imposed. Then, K*B–A = σAK*A–B/σB, and the A–B interaction is described by only a single parameter K*A–B. In Section SI-1 of the Supporting Information, we show that in this case the concentration of a copolymer and its reversed version are equal. In the sequel, we shall not impose this additional symmetry condition. Of course, all results for the general case also hold for this symmetric case.
2.4. Copolymer Concentration
To derive the mass-balance equations for general A- and B-based copolymers described above, we start with an iteration process for the concentration of polymers of given length. As the equilibrium constants for elongation of a copolymer with a given monomer depend on the top of the copolymer, it is useful to distinguish copolymers with top A and top B. Hence, we define cnA and cn as the concentration of all copolymers of length n with top elements A and B, that is, Pn–1A and Pn–1B, respectively. For the monomer concentrations, we write a = [A] and b = [B].
For a copolymer Pn with top element A, that is, a copolymer of form Pn = Pn–1A, we know that [PnA] = [Pn–1AA] = K*A–Aa[Pn–1A]. Also, for copolymer Pn with top element B, that is, a copolymer of form Pn = Pn–1B, the relation [PnA] = [Pn–1BA] = K*B–Aa[Pn–1B] holds. Because each copolymer Pn has either top A or top B, we obtain
| 12 |
For the copolymers with top B, we obtain in this way
| 13 |
Consequently, the concentrations of all possible copolymers are given by an iteration process, which can be written in the matrix form as
| 14 |
where the 2 × 2 matrix Mc is given by
| 15 |
As the equilibrium concentrations of the shortest (co)-polymers, that is the dimers, are coupled to the equilibrium monomer concentration, equation 14 implies that once the monomer concentrations are known, the concentrations of all copolymers are known, as well.
2.5. Equivalent Concentrations
For the mass-balance equations, knowledge of the concentration of the copolymers alone is not sufficient. We also need the amount of A and B monomers that is present in those copolymers. Therefore, we introduce the notion of equivalent concentration of a part X of a copolymer P (in computing science terms: a substring X of a string P). The part X can be a monomer (A or B) or a bond (A–A, A–B, B–A, B–B). The equivalent concentration of X in polymer P is the product of the number of occurrences of X in P and the concentration of P. It will be written as [P]X. For instance, if P = AAABAB, then [P]A = 4[P], [P]A–A = 2[P] and [P]A–B = 2[P]. Note that the equivalent A (B) concentration in copolymer P equals the concentration of free A (B) monomers that results if all copolymers P would be broken down to monomers.
We now describe how equivalent concentrations can be computed. Let X be one of the possible parts mentioned above. Define fnA and fnB to be the equivalent X concentration in copolymers of length n with top A and B, respectively. To derive recurrence relations for these variables, we study first what happens with the equivalent X concentration if copolymer PA or PB is elongated at its top with an A monomer. Consider first the case that PA is elongated with an A monomer. Suppose that PA contains k occurrences of X, so [PA]X = k[PA]. All these k occurrences of X will also be present in PAA. Moreover, it is possible that the elongation of PA with an additional A monomer has introduced one additional X. Let δAA be the number of additional parts X that is introduced in this step. Then PAA contains k + δAA parts X. The equivalent X concentration now becomes
| 16 |
| 17 |
This formula gives the equivalent X concentration in PAA, expressed in the equivalent X concentration of (shorter) copolymer PA and the (ordinary) concentration of PA. Note that δAA is simply 0 or 1, depending on whether the elongation of PA with an A on its top leads to a new X part.
Analogously, when copolymer PB containing k occurrences of X is elongated with an A monomer, the elongated copolymer PBA contains k + δBA occurrences of X, with δBA, the number (0 or 1) of additional parts X introduced in this elongation step. For the equivalent X concentration, this means
| 18 |
| 19 |
This formula gives the equivalent X concentration in PBA, expressed in the equivalent X concentration of (shorter) copolymer PB and the (ordinary) concentration of PB.
Because each polymer with top element A is either of the form PAA or of the form PBA, we obtain that the equivalent X concentrations in copolymers PA with length n + 1 are given by
| 20 |
Similarly a recurrence relation for the equivalent X concentration in copolymers of form PB can be derived as
| 21 |
The iteration processes described in eqs 14, 20, and 21 can be combined. In terms of vector un = (cnA, cnB, fnA, fnB)T where superscript T indicates transposing the vector to make it a column vector again; this process is written as
| 22 |
with Mc given in eq 15, and the 2 × 2 matrix MX defined by
| 23 |
The matrix MX describes the number of additional X parts in a copolymer for the four possible ways a copolymer can be elongated. For instance, for X = A, δAA = δBA = 1 and δAB = δBB = 0, because only the elongation of PA and PB with an A monomer leads to an extra A monomer. For the case X = B–A, δBA = 1 and δAA = δAB = δBB = 0, only the elongation of PB with an A monomer leads to an extra B–A bond. Hence,
| 24 |
The iteration process can be started from n = 1 with vector u1 = (σAa, σBb, f1A, f1B)T. The initial values for f1A and f1B depend on the considered part X. For X = A, we find that f1A = σAa and f1B = 0, as the only “copolymer” with length 1 and top A is monomer A (the additional σA comes from the nucleation step). If X is one of the four bond types, the initial values are always f1A = f1B = 0, as the “copolymers” of length 1 do not contain bonds. For more details, see the Supporting Information, Section SI-2. The equivalent X concentrations for an arbitrary copolymer length can now be computed from
| 25 |
For the mass-balance equations, we need the sum of the equivalent A and B concentrations over all copolymer lengths. To obtain sum PX of the equivalent X concentration over all copolymer lengths, n = 2, 3, ... corresponding to free monomer concentrations a and b, we proceed as follows
| 26 |
where U3 and U4 are the third and fourth components of the (4-dim.) vector U given by
| 27 |
In this formula, I is the 4 × 4 identity matrix, and we used the matrix identity ∑n=1∞Mn = (I – M)−1·M, which only holds if the absolute eigenvalues of M are all smaller than 1. This condition holds because the total equivalent X concentration PX, as defined in eq 26, is finite in any chemically relevant system.
This method, with correct matrices MX and initial values for f1A and f1B, can be used to compute the equivalent X concentration for all mentioned X parts. The resulting sum of equivalent X concentration will be called PA, PB, PA–A, PA–B, PB–A, and PB–B, respectively. Sometimes, we will write PA(a,b), and so forth to stress that all these equivalent X concentrations depend on the used values for the free monomer concentrations a and b.
Finally, note that the sum of all copolymer concentrations Ctot can easily be computed from
| 28 |
2.6. Mass-Balance Equations
The mass-balance equations state that each monomer is either a free monomer or occurs in some copolymer. In the case of one copolymer type, they read
| 29 |
| 30 |
where a and b are the unknown free monomer concentrations, and atot and btot are the given total monomer concentrations. The solution of these equations, with absolute eigenvalues of matrix M smaller than 1, are the free monomer concentrations a and b in thermodynamic equilibrium. The mass-balance equations form a system of two nonlinear algebraic equations that in general can only be solved by numerical methods. In Section SI-8 of the Supporting Information, a MATLAB function to solve the mass-balance equations is described.
2.7. Limits on Equilibrium Monomer Concentrations: The Allowed Region
The solution of the mass balance equations is only useful if it corresponds to a situation where the total amount of the material in the copolymers is finite. Hence, only solutions, for which all absolute eigenvalues of matrix M are smaller than 1, are chemically relevant. For each X (monomer or bond), the eigenvalues of M are the same as those of Mc. Hence, the free monomer concentrations a and b must be in the part of the a, b plane, where the absolute eigenvalues of Mc are smaller than 1 (and a ≥ 0, b ≥ 0), which we shall denote as the allowed region.
The degree of polymerization depends on the largest absolute eigenvalue λ1 of M. If λ1 ≪ 1, then the vectors un tend to 0 very fast for increasing n, which means there is hardly any material in the copolymers. If λ1 ≈ 1, then vectors un tend to 0 very slowly for increasing n, which corresponds with a high degree of polymerization. The part of the boundary of the allowed region where a > 0 and b > 0 is the critical curve, that is, the curve where λ1 = 1. By setting the determinant of Mc – I equal to 0, this curve is found to satisfy
| 31 |
with D = K*A−AK*B−B − K*A−BK*B−A.
In Figure 2, the allowed regions for weak and strong interaction between the A and B monomers are shown. The shape of the allowed region depends on the value of D. If D > 0, the homopolymer interactions are stronger than the heteropolymer interactions, and the allowed region is convex (Figure 2a). If D < 0, the heterointeractions are the strongest and the allowed region is nonconvex (Figure 2b). If D = 0 the (right-upper) boundary is a straight line. Some more examples of allowed regions can be found in the Supporting Information, Section SI-3.
Figure 2.
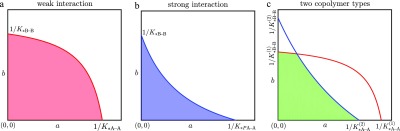
Allowed regions for the monomer concentrations a, b. (a) For weak A–B interaction (D = K*A–AK*B–B – K*A–BK*B–A > 0), the allowed region, that is the colored part under the critical curve, is convex. (b) For strong A–B interaction (D < 0), the allowed region is nonconvex. (c) For multiple copolymer types, their critical curves (red and blue) may intersect, and the allowed region is the (green) area that is below all critical curves.
Consider a point (atot, btot) that lies outside the allowed region. Because the corresponding free monomer point (a, b) must always lie inside the allowed region, some degree of polymerization must be present. If the point (atot, btot) lies inside the allowed region, that argument does not hold. Therefore, a point (atot, btot) on the critical curve is called a critical concentration pair. Note that there is no fixed critical concentration for atot and btot individually. Only the pair (atot, btot) can be considered critical, if it satisfies eq 31. In the case of a titration experiment (at fixed temperature), the point (atot, btot) changes and therefore may pass the critical curve. At that point, the degree of copolymerization will change drastically.
2.8. Elongation Temperatures
The equilibrium constants (K) are often related to an enthalpy difference (ΔH) and entropy difference (ΔS) by K = exp(−(ΔH – TΔS)/RT), where R is the gas constant and T is the absolute temperature, see also Section SI-6 of the Supporting Information. Consequently, in experiments at fixed temperature (e.g., titration experiments), the equilibrium constants do not change, whereas in cooling experiments, the equilibrium constants do change. As usually ΔH < 0, the equilibrium constants increase during cooling, which means that the allowed region shrinks. Thus, a point (atot, btot) can be inside the allowed region for high temperatures and outside the (smaller) allowed region for low temperatures. The temperature where the point (atot, btot) is on the critical curve (i.e., it is a critical concentration pair for this temperature) is called the elongation temperatureTe for the copolymers. Hence, in a cooling experiment, Te is the temperature at which the degree of (co)polymerization must start to grow. More details on the computation of the elongation temperature and a MATLAB implementation are given in Sections SI-6 and SI-8 of the Supporting Information.
2.9. Copolymer Properties
Although the solution of the mass balance equations as described above consists of the free monomer concentrations a and b, more information can easily be obtained. Because PA(a,b) and PB(a,b) are the total amount of A and B monomers occurring in copolymers corresponding to the free monomer concentrations a and b, the total amount of the material in the copolymers is given by Ptot = PA + PB, where to simplify the notation, we omit the (a, b) arguments here. The degree of polymerization can then be computed by ϕ = Ptot/(atot + btot). The fractions of A and B monomers in the copolymers is given by PA/Ptot and PB/Ptot, respectively. These notions might be useful in cases, where the experimental observables of a copolymer depend on the number of occurring A and B monomers, but the contribution per A or B monomer is different. Also, the equivalent bond concentrations PA–A, PA–B, PB–A and PB–B can easily be found from eq 26 once the equilibrium concentrations are known. Hence, the fraction A–A bonds is given by PA–A/(PA–A + PA–B + PB–A + PB–B), and similar expressions hold for other bond fractions. The average concentration-weighted copolymer length is given by
| 32 |
where the sum of all copolymer concentrations Ctot is given in eq 28. For the computation of the average mass-weighted copolymer length and the corresponding MATLAB function, see Sections SI-4 and SI-8 in the Supporting Information.
2.10. Block Lengths
The calculation of some other properties, such as, for example, the average block lengths, requires some more work. Blocks are defined as specific series of monomers occurring in a copolymer. We distinguish A blocks, B blocks, and AB (=alternating) blocks. All blocks are assumed to be as long as possible; therefore, in copolymer BAAAABAAA there is one A block of length 4 and one A block of length 3. AB blocks can start and terminate with A or B, as long as the A and B monomers inside the block are alternating. In the copolymer above, there is thus one AB block of length 2 and one AB block of length 3. Formally blocks can also have length 1, as the two B blocks of length 1 in the copolymer above. It is clear that block lengths give additional information with respect to bond fractions. The copolymers BAAAABAAA and BAAAAAABA, for instance, have the same number of A and B monomers and the same bonds, but their A block lengths differ.
To quantify the amount of blocks in the copolymers, we use the notion of equivalent block concentration. If in copolymer P, a certain block occurs k times, the equivalent block concentration in that copolymer is defined as k[P], that is, k times the concentration of P. Therefore, for P = BBBABBB, the equivalent concentration of B blocks of length 3 is 2[P], as P contains two B blocks of length 3. Clearly, equivalent block concentrations (of blocks with the same length) occurring in different copolymers can be added, yielding the total amount of blocks of that length.
We first describe the computation of A blocks. In the computation of the previously considered equivalent concentrations, we used an iteration process over length n of the copolymers, thereby making a distinction between copolymers with top A and top B. Here, the situation is more complicated. First of all, there are blocks of different lengths. Moreover, the effect of elongation with a new A monomer on an A block depends on the position of that A block. Only A blocks that run until the top of the copolymer become longer by an elongation with A. Consider as example the copolymer P = AAAABAA. This copolymer contains an A block of length 4 and an A block of length 2. Only the length of this last A block grows by 1 due to elongation with an A. Hence, it is necessary to distinguish between closed A blocks and open A blocks. Closed A blocks are always terminated by a B monomer. Open A blocks run until the top of the copolymer.
To formulate the iteration process for the A block lengths, we define for copolymers of length n and
Of course, copolymers with top B cannot contain open A blocks. Next, we explain the recurrence relations, to make the step from length n to n + 1.
Adding A on the top of a copolymer with top A or B does not lead to new closed A blocks. Each closed A block in the original copolymer also occurs in the elongated copolymer. Because the concentrations of the original and elongated copolymer differ by factor K*A–Aa (original with top A) or K*B–Aa (original with top B), this leads to
| 33 |
for k = 1 ..., n − 1.
Further, open A blocks in the elongated copolymer can only occur if A is added. Each open A block with length k ≥ 1 in the original copolymer leads to an open A block of length k + 1 (≥2) in the elongated copolymer. Moreover, adding an A monomer to copolymers with top B leads to new open A blocks with length 1. These copolymers, with top BA, have concentration cnBK*B–Aa. Hence,
| 34 |
for k = 2, ..., n + 1, and
| 35 |
If a B monomer is added on the top of a copolymer with top B, all closed A blocks in the original copolymer also occur in the elongated copolymer. The same happens if the original copolymer has A as top. However, in this latter case, open A blocks at the end of the original copolymer become closed as well because they are now followed by the newly added B. Again, a factor occurs due to the different concentrations of the original and elongated copolymers. This results in
| 36 |
The relations in eqs 33–36 allow to compute the equivalent concentrations of open and closed A blocks in copolymers of any length n. This iteration proces can be started at n = 2, with C2B(1) = [AB] = KA–Bab, O2(1) = [BA] = KB–Aab, and O2A(2) = [AA] = KA–Aa2.
The total equivalent concentration 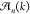 of an (arbitrary) A block of length k in copolymers of length n is now given
by
of an (arbitrary) A block of length k in copolymers of length n is now given
by
Hence, in this way, we can compute the distribution
of A block
lengths in copolymers of length n. The total equivalent
concentration 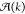 of an (arbitrary) A block of length k in any copolymer is now given by
of an (arbitrary) A block of length k in any copolymer is now given by
This summation can be compared with the summation
in eq 26 in the computation
of the equivalent
X concentrations. In that case, the actual summation over all copolymer
lengths n could be avoided by using standard summation
formulas based on the formulas for the sum of geometrical series.
In the case of block lengths, we did not find a simple way to avoid
performing the actual iteration process of eqs 33–36 and computing
the 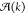 during that process. In practice, the iteration
process can be stopped if the amount of the material in copolymers
of length more than n can be neglected. Depending
on the average copolymer length, the running time of the block length
computation for a whole cool curve with 100 points is on a standard
PC between seconds and a few minutes. The average A block length can
now be found from
during that process. In practice, the iteration
process can be stopped if the amount of the material in copolymers
of length more than n can be neglected. Depending
on the average copolymer length, the running time of the block length
computation for a whole cool curve with 100 points is on a standard
PC between seconds and a few minutes. The average A block length can
now be found from
| 37 |
The computation of (average) B block lengths and also the (alternating) AB block lengths can be done in a similar way. In Section SI-8 of the Supporting Information, we describe a MATLAB script that computes the average length of A, B, and alternating AB blocks. The used method is not restricted to block lengths. Other more complicated properties of copolymers, such as the total concentration of copolymers with a given number of (for instance) B monomers can be computed by a similar iteration process.
2.11. Multiple Copolymer Types
So far, we have described copolymerization with one copolymer type. Here, we consider the case that there are multiple copolymer types; for instance, copolymers that show a different morphology or helicity. Each aggregate type may have its own equilibrium constants and thus matrices, Mc and MX. For given a and b, the equivalent monomer concentrations in each aggregate type, which we denote as PA,i and PB,i, respectively, can be calculated. The amount of monomers that occur in all copolymers is now found by adding the equivalent monomer concentrations of all copolymer types. For p copolymer types, the resulting mass-balance equations are
| 38 |
| 39 |
The solution of the mass-balance equation gives again the free monomer concentrations a and b in thermodynamic equilibrium. Because the amount of material in each copolymer type must be finite, the free monomer point (a, b) must lie in the intersection of the allowed regions of all individual copolymer types, see Figure 2c for an example. As the degree of polymerization in a cooperative aggregate type is only high if the free monomer point (a, b) is close to the corresponding critical curve, coexistence of multiple aggregate types will correspond to free monomer concentrations a and b close to the intersection of critical curves. Properties such as the degree of polymerization, average copolymer lengths, equivalent bond concentrations, and average block lengths can be computed straightforwardly per copolymer type. In Section SI-8 in the Supporting Information, we provide MATLAB scripts to solve the mass-balance equations for multiple copolymer types as well as for the computation of their equivalent bond concentrations, average block lengths, and so on.
3. Examples of Model Application
In previous papers,56,80−85 we used several dedicated mass-balance models, up to one component with four polymer types and two components with three copolymer types. The equilibrium copolymerization model presented in the previous section encompasses all these earlier mass-balance models and can be used to reproduce the earlier reported results. In Section SI-8 of the Supporting Information scripts for a variety of examples thereof are provided, illustrating how the current model can be used to produce titration curves, cool curves, and speciation plots in order to elucidate diverse copolymerization phenomena. In the remainder of this section, we will illustrate the applicability of the model also on two new examples.
3.1. Sergeants and Soldiers
The first new application of our model concerns the copolymerization of N,N′,N″-trialkyl-benzene-1,3,5-tricarboxamides (BTAs) equipped with achiral and (R)-chiral aliphatic side chains (Figure 3a). These molecules form helical aggregates in apolar alkane solvents, where in the absence of the chiral (R)-1 monomers left (M)- and right (P)-handed helices will be equally abundant. The addition of small amounts of (chiral) sergeant (R)-1 to (achiral) soldier (A)-2, while keeping the total concentration constant, results in an excess helical sense corresponding to the preferred sense of (R)-1, that is, the so-called sergeants and soldiers effect.87 In ref (80), we modeled this system using stochastic simulations88 as the mass balance model presented there for the copolymerization of two enantiomers could not describe the copolymerization of two monomers that individually form homopolymers with distinct cooperativity. However, it can also be described by the general copolymerization model described here, which leads to much more efficient computation than using stochastic simulations.
Figure 3.

Sergeants and soldiers. (a) Structure of BTAs equipped with achiral and (R)-chiral aliphatic side chains. (b) Schematic view of the P-type and M-type copolymer types and the different thermodynamic parameters which here are assumed to only depend on the monomer added and aggregate type.
To describe this sergeants and soldiers system, we consider two monomer types, that is, A for achiral (A)-2 monomers and B for chiral (R)-1 monomers, and two copolymer types, which will be denoted as P and M. As described in Section 2, there are in principle six independent equilibrium constants per aggregate type and some of those will be the same. For instance, for reasons of symmetry, the equilibrium constant for elongation as well as the cooperativity factor for the achiral molecules should be equal for both P- and M-type aggregates. To limit the number of parameters further and to reuse the thermodynamic parameters as found with the stochastic simulations in ref (80), we assume in this case that the equilibrium constants only depend on the monomer that is added, and thus independent of the top of the aggregate, as schematically depicted in Figure 3b. The equilibrium constants (K) are given by the corresponding enthalpy (ΔH) and entropy (ΔS) changes under standard conditions, that is K = e(−ΔH+TΔS)/RT, where R is the gas constant, and T the absolute temperature. For A monomers, ΔHA = −75 kJ mol–1, ΔSA = −0.1255 kJ mol–1 K–1, and a cooperativity parameter σA = eNPA/RT with nucleation penalty NPA = −27 kJ mol–1 were obtained in ref (80) by fitting multiple UV coolcurves of homopolymerizations at different concentrations. As A monomers are achiral, these values hold for both P-type and M-type copolymers. For the chiral B monomers that only form homopolymers with P-type helicity, fitting CD cool curves yielded enthalpy change ΔHBP = −66 kJ mol–1, entropy change ΔSB = −0.1015 kJ mol–1 K–1, and cooperativity parameter σB = eNPB/RT with nucleation penalty NPB = −35 kJ mol–1 for those P-type aggregates.80 Because of the choice that the equilibrium constants only depend on the monomer that is added, for P-type copolymers holds that K*A–B = K*B–BP and also K*B–A = K*A–AP. Analogous relations with the superscripts P replaced by M hold for the M-type copolymers. The only remaining parameters then are for the B monomers in M-type aggregates. Following once more ref (80), these were chosen the same as for the P-type aggregates except for an enthalpy penalty MMP = −0.5 kJ mol–1 for incorporation in their unpreferred aggregates, that is, ΔHB = ΔHBP – MMP.
Now for a given temperature and total amounts of achiral and chiral monomers, the mass-balance eqs 38 and 39 with p = 2 can be solved, giving the equilibrium values of the free monomer concentrations a and b. As an example, we computed for a total concentration of 21 μM and for a series of eight temperatures, the helical excess Ptot – Mtot as a function of the sergeant fraction, where Ptot = PA + PB and Mtot = MA + MB are the total amounts of material in P- and M-type copolymers, respectively. The results are, together with the experimental CD data,86 shown in Figure 4a and correspond with Figure 6a from ref (80). To demonstrate the various possible properties that can be computed using the scripts provided in Section SI-8 of the Supporting Information, we computed for T = 337 K a speciation plot with Ptot and Mtot (i.e., the total equivalent monomer concentrations in P-type and M-type copolymers, respectively), the contribution of the A and B monomers to those equivalent concentrations and both the free monomer concentrations. The result is shown in Figure 4b and is again fully consistent with stochastic simulation results in Figure 6c in ref (80). Furthermore, Figure 5a shows the average copolymer length, and part (b) shows the equivalent concentration of the four bond types. Although the stochastic simulation method in ref (80) is consistent with the mass-balance approach in this paper, the computing times are very different. While the computation of one point on a cooling curve, that is, one computation of the equilibrium state, takes on a standard PC about 90 min with the stochastic simulation method, it takes less than a second with the mass-balance method. This makes the mass-balance approach suitable for fitting of experimental data to find thermodynamic parameters.
Figure 4.

Sergeants and soldiers titration curves. (a) Model computed Ptot – Mtot (lines) and experimental CD data from ref (86) (*) for eight temperatures. (b) Speciation plot of the model results for T = 337 K.
Figure 5.

Sergeants and soldiers (a) Average length of P-type and M-type copolymers. (b) Equivalent concentrations of the four possible bond types in P-type copolymers (the equivalent concentration of a bond type in copolymer P is the concentration of P, multiplied by the number of occurrences of that bond type in P; see also the paragraph on equivalent concentrations in Section 2).
3.2. Solvent-Dependent Copolymerizations
The second new application of our model comprises the copolymerization of tripyridylamine triamide (1) and triphenylamine triamide (2), both with a chiral (S)-3,7-dimethyloctyl chain (S-1, S-2), see also Figure 6a. In ref (85), the copolymerization of these triarylamine triamide-based monomers, with chiral and achiral chains, was studied in decalin. Here, we study the effect of different solvents on copolymerization. Precisely, the copolymerization is studied in pure apolar solvents, decalin and methylcyclohexane (MCH) and in a solvent mixture of decalin and 1,2-dichloroethane (DCE, decalin/DCE v/v = 97/3) in order to evaluate the effect of an increased polarity of the environment on the block lengths. The S-1 monomers will be denoted by A and the S-2 monomers by B. Because of the (S) configuration of the stereocenters, these triarylamine triamide-based monomers form helical aggregates with one preferred handedness. Therefore, we now use the model from Section 2 with a single aggregate type and assume that, contrary to the above sergeants and soldiers example, the equilibrium constants depend on the top monomer in the aggregate as well, as schematically depicted in Figure 6b.
Figure 6.

(a) Chemical structure of the tripyridylamine triamide- and triphenylamine triamide-based monomers. (b) Schematic of the copolymerization reactions and the corresponding (elongation) equilibrium constants dependent on both the added monomer and top of the copolymer.
The first step in modeling the copolymerization is to find the properties of the homopolymers. Fitting the results of cooling experiments with the one-component model software81 yields the thermodynamic parameters of the homopolymers shown in Table 1. To avoid the complexity related to the interaction of the supramolecular polymers with the codissolved water89 occurring below 30 °C, we restricted the fitting of cooling curves to temperatures above 40 °C, see Section SI-7 of the Supporting Information for more details.
Table 1. Thermodynamic Parameters for Triarylamine Triamide-Based Homopolymers in Three Distinct Solvents.
| monomer/solvent | ΔH [kJ mol–1] | ΔS [kJ mol–1 K–1] | NP [kJ mol–1] |
|---|---|---|---|
| A in decalin | –85.4 | –0.1520 | –25.05 |
| B in decalin | –71.3 | –0.127 | –17.6 |
| A in decalin/DCE | –81.8 | –0.148 | –20.0 |
| B in decalin/DCE | –58.2 | –0.0931 | –15.4 |
| A in MCH | –92.0 | –0.168 | –29.5 |
| B in MCH | –87.5 | –0.173 | –14.0 |
The only remaining parameters for the copolymerization of A and B are the heteroassociation equilibrium constants K*A–B and K*B–A. To reduce the number of unknown parameters, we assume the symmetry condition (see Section 2.3), retaining K*A–B as the only remaining parameter. The temperature dependence of K*A–B is again given by an enthalpy term ΔHA–B and an entropy term ΔSA–B. These parameters were found by fitting CD data of cooling curves of an A–B mixture, with concentrations atot = 15 μM and btot = 15 μM against the model predictions. The resulting parameters for the three solvents are given in Table 2. The fit for the decalin solvent is shown in Figure 7a and those for the other two solvents in Figure S6 in the Supporting Information. These figures also contain a “no interaction” line that gives the model result in case that there is no copolymerization at all. In this way, the effect of copolymerization can easily be seen. In the absence of mixing, Figure 7a shows that two individual polymerizations can be discerned, each with its own elongation temperature. In the case with copolymerization, the cooling curve below the elongation temperature is much more linear, as is the case for the experimental cooling curve.
Table 2. Thermodynamic Parameters for the Copolymerization of Tripyridylamine Triamide- and Triphenylamine Triamide-Based Monomers in Three Distinct Solvents.
| solvent | ΔHA–B [kJ mol–1] | ΔSA–B [kJ mol–1 K–1] |
|---|---|---|
| in decalin | –93.4 | –0.178 |
| in decalin/DCE | –66.7 | –0.108 |
| in MCH | –98.0 | –0.177 |
Figure 7.

(a) Fit of the CD data of the mixture in decalin with the results of the copolymerization model and a “no interaction” model result, (b) ΔGcopol as function of the temperature for the three solvents.
To investigate the differences between the three solvents, we introduce the net free copolymerization energy ΔGcopol, defined as ΔGcopol = ΔGA–A + ΔGB–B – ΔGA–B – ΔGB–A, where the free energy terms have their usual definition (ΔG = ΔH – TΔS). Then, clearly,
which means that ΔGcopol < 0 corresponds with the situation, where the homopolymer interaction is the strongest and ΔGcopol > 0, where the heteropolymer interactions dominates. In fact, ΔGcopol < 0 corresponds with a convex allowed region (D > 0), and ΔGcopol > 0 corresponds with a nonconvex allowed region (D < 0). In Figure 7b, we show ΔGcopol as function of the considered temperatures for the three solvents. For temperatures below the elongation temperatures of the copolymers, ΔGcopol is in the range from −4 to −2 kJ mol–1 for all three solvents. In all cases, ΔGcopol < 0, which means that the homopolymer bonds are stronger than the heteropolymer bonds independent of the solvent.
In Figure 8, the average block lengths of the copolymers for all three solvents are shown. All three graphs show a strong increase in A block length during cooling, which is consistent with the fact that the elongation temperature for the A homopolymers is higher than that of the B homopolymers. For lower temperatures, more B monomers will occur in the copolymers, which leads to a decrease of A block length and a relative growth of B and alternating A–B block lengths. Remarkably, we observe in decalin/DCE a smaller growth of A block lengths below the elongation temperature but a somewhat larger A block length at low temperatures compared to the other two solvents. Finally, the equivalent bond concentrations for the three solvents are shown in Figure 9. In all cases, the A–A bond occurs most often for all temperatures, which is consistent with the longest block length for A blocks. Note that for the decalin/DCE solvent, the fraction of A–A bonds at low temperatures is higher than for the other solvents, which agrees with the somewhat longer A block lengths in this case. This result confirms the hypothesis previously reported by the Meijer group,85 where the authors, based on spectroscopic and microscopy analysis, speculate that the presence of denaturant agents in solution favors the homointeraction over the heterointeraction.
Figure 8.
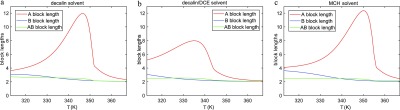
Average block lengths for (a) decalin, (b) decalin/DCE, and (c) MCH solvent.
Figure 9.
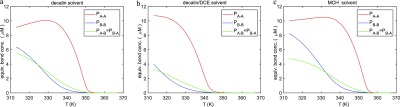
Equivalent bond concentrations for (a) decalin, (b) decalin/DCE, and (c) MCH solvent.
4. Special Cases
Usually the mass-balance equations must, in the absence of an analytical solution, be solved numerically, as illustrated in the previous section. However, in some cases, an analytical approximation of the solution can be found. The advantage of such analytical solutions is that they provide more insights into the way the system properties depend on the parameters, that is, the equilibrium constants and total monomer concentrations. Here, we will consider three examples where an analytical solution can be derived from the mass-balance equations, where we restrict ourselves to a single aggregate type and the symmetric case, that is, we assume in this section that KA–B = KB–A.
4.1. Mixing in Small Amounts of Comonomers
The first example we consider is the situation where a small amount of B monomers is added to a system of, initially pure, A monomers, and we study the effect on the degree of polymerization and the length of the copolymers. We assume that the initial system is highly polymerized, so K*A–Aatot ≫ 1, and that the B monomers mix into those polymers rather than forming homopolymers of their own. To simplify the formulas, we therefore put K*B–B = 0.
In the described situation, the equivalent A and B concentration in the copolymers are approximately related by
| 40 |
see the Supporting Information, Section SI-5, for derivation. Using this relation, the mass-balance eqs 29 and 30 can be rewritten as
| 41 |
To find a second relation between a and b, note that the system is assumed to be highly polymerized, which means that the free monomer point (a, b) must lie inside the allowed region, very close to the critical curve. This means that also eq 31 must hold approximately (with K*B–B = 0). Combining this with eq 41 gives a system of two equations for the free monomer concentrations a and b that can easily be solved, leading to
| 42 |
| 43 |
Hence, with increasing btot, (i) b and PB = btot – b both grow linearly, (ii) a decreases, and (iii) PA = atot – a increases. This means that the total material in the copolymers PA + PB also increases with increasing btot. Note that this does not imply that the degree of polymerization ϕ = (PA + PB)/(atot + btot) also increases because the denominator of this quotient grows as well.
To study
the effect of the addition of B monomers on the degree
of polymerization ϕ and the average copolymer length ⟨n⟩, we first introduce the dimensionless parameter
γ by setting btot = γatot. Keeping all other parameters fixed, we
consider all (equivalent) concentrations, the average copolymer length
⟨n⟩, and the degree of polymerization
ϕ as function of γ. The derivatives of these notions with
respect to γ in γ = 0 indicate what happens if B monomers
are added to a pure A homopolymer solution. The derivatives will be
written with a prime, like a′, b′, Ptot′, and ϕ′ and can be computed
with the standard calculus rules, starting from eqs 42 and 43. The resulting
expressions for ϕ′ and ⟨n⟩′
are given in the Supporting Information, Section SI-5. It turns out that upon addition of B to a pure A
system, the degree of polymerization ϕ decreases for small values
of the heterointeraction constant K*A–B and increases for large values of K*A–B. This behavior for small K*A–B is not surprising. For very small K*A–B, hardly any of the added B monomers will be included in a polymer,
which means that the total amount of material in copolymers Ptot will almost not increase. Because the total
amount atot + btot does increase, the quotient ϕ decreases. For larger values
of K*A–B, this argument does not
hold and ϕ increases. The turning point 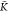 *A–B can be found by solving
the equation ϕ′ = 0 for K*A–B, see Section SI-5 of the Supporting Information for details. This results in
*A–B can be found by solving
the equation ϕ′ = 0 for K*A–B, see Section SI-5 of the Supporting Information for details. This results in
where α
= K*A–Aatot. Therefore, for K*A–B < 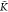 *A–B (K*A–B >
*A–B (K*A–B > 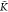 *A–B), the degree of copolymerization
decreases (increases) upon addition of B monomers.
*A–B), the degree of copolymerization
decreases (increases) upon addition of B monomers.
The average
copolymer length shows a similar behavior. Also, here,
there is a turning point 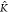 *A–B given by
*A–B given by
such that the
addition of B to a pure A system
leads to a decrease (increase) of the average copolymer length for K*A–B < 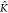 *A–B (K*A–B >
*A–B (K*A–B > 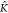 *A–B). Again, for small
values of K*A–B, the addition of
B monomers will lead to a very small increase of Ptot. The total concentration of the copolymers Ctot grows much faster, which leads to a decreasing
copolymer length.
*A–B). Again, for small
values of K*A–B, the addition of
B monomers will lead to a very small increase of Ptot. The total concentration of the copolymers Ctot grows much faster, which leads to a decreasing
copolymer length.
In the example in Figure 10a, the degree of polymerization ϕ
(left) is shown as
a function of btot/atot for three values of β = K*A–B/K*A–A, namely, for β =
0.6, the turning point value 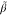 =
= 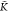 *A–B/K*A–A = 0.487 and for β = 0.4. Figure 10b gives the corresponding
copolymer length ⟨n⟩ as a function
of btot/atot, for β = 0.4, the turning point value
*A–B/K*A–A = 0.487 and for β = 0.4. Figure 10b gives the corresponding
copolymer length ⟨n⟩ as a function
of btot/atot, for β = 0.4, the turning point value 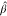 ^ =
^ = 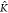 *A–B/K*A–A = 0.321 and for β = 0.3. Indeed ϕ
and ⟨n⟩ decrease upon addition of B
monomers for values of β below the turning points
*A–B/K*A–A = 0.321 and for β = 0.3. Indeed ϕ
and ⟨n⟩ decrease upon addition of B
monomers for values of β below the turning points 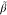 ~ and
~ and 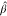 respectively.
Note that in this case for
β = 0.4, the addition of B monomers leads to a decrease of ϕ
but an increase of ⟨n⟩.
respectively.
Note that in this case for
β = 0.4, the addition of B monomers leads to a decrease of ϕ
but an increase of ⟨n⟩.
Figure 10.

Mixing in comonomers example. (a) Degree of polymerization ϕ and (b) average copolymer length ⟨n⟩ and their slopes for three values of β = K*A–B/K*A–A. Parameters: atot = 10–4 M, K*A–A = 105 M–1, K*B–B = 0 M–1, σA = 10–4, and σB = 5 × 10–5.
4.2. Mixing of Two Highly Polymerized Monomer Types
In this second case, we are interested in the dependence of the fraction of homo- and heterobonds on the heteroassociation K*A–B under conditions where both monomer types individually would be highly polymerized, that is, K*A–Aatot ≫ 1 and K*B–Bbtot ≫ 1. It can be shown in all cases, where K*A–AK*B–B ≠ 0 (see Supporting Information, Section SI-5) that
| 44 |
Because we restrict ourselves here to the symmetric case, PA–B = PB–A. Moreover, PA–A + PA–B ≈ PA ≈ atot. The first approximation holds because each A in a copolymer, except A at the top, is either followed by another A or by B. For long copolymers, the relative error we make by omitting A monomers at the top of a copolymer is very small. The second approximation holds because we assume a high degree of copolymerization; hence, almost all A monomers occur in copolymers. Substitution of PA–A = atot – PA–B and similarly PB–B = btot – PA–B in eq 44 gives a quadratic equation for PA–B with as only positive solution
| 45 |
In this way, for this special case, the amount of A–B (and B–A) bonds and hence also the remaining A–A and B–B bonds can be computed as function of the interaction parameters, without solving the mass-balance equations. It is easily verified that PA–B = 0 for K*A–B = 0. To describe the results for large heterointeraction, assume that atot > btot. Then, for K*A–B becoming very large, it is readily inferred from eq 45 that PA–B → btot and hence PA–A → atot – btot and PB–B → 0. Therefore, for large heterointeraction, all B–B bonds disappear and the amount of A–A bonds equals the excess of the total amount of A monomers over the total amount of B monomers.
In Figure 11, we give an example. In part (a) of that figure, the equivalent bond concentrations computed by the approximation are compared with the bond concentrations obtained by solving the mass-balance equation. Part (b) shows the block lengths, computed as described before. Note the very sharp decrease of the length of the A blocks and B blocks for small positive interaction parameter K*A–B.
Figure 11.

Mixing highly polymerized monomer types example. (a) Approximated and exact equivalent bond concentrations. (b) Corresponding block lengths. Parameters: atot = 2 × 10–4 M, btot = 1.5 × 10–4 M, K*A–A = 1.2 × 105 M–1, K*B–B = 1.5 × 105 M–1, σA = 5 × 10–5, and σB = 7 × 10–5.
4.3. Purely Alternating Copolymers
The final special case we consider is that where all copolymers are alternating, that is, where K*A–A = 0 and K*B–B = 0, and consequently no A–A and B–B bonds can be formed. This case, which was also recently considered by van Buel et al.79 with a dedicated model, is also captured by our general model. For long copolymers, the absence of homo bonds implies that PA and PB are almost equal. The mass balance in eqs 29 and 30 now implies that
If K*A–B is so large that most monomers occur in some (alternating) copolymer, the free monomer point (a, b) must lie inside the allowed region, but very close to the critical curve. Hence, eq 31 yields for this case that
| 46 |
From the last two equations, the free monomer concentrations can be solved, which gives the following approximations
| 47 |
| 48 |
| 49 |
In this way, for this case of purely alternating polymers, the free monomer concentrations, the amount of A and B monomers in the copolymers, and the amount of A–B (and B–A) bonds can be computed as a function of the interaction parameters, without solving the mass-balance equations. To describe the results for large hetero interaction, assume that atot > btot. Then, as K*A–B (and hence as K*B–A) becomes very large, a → atot – btot, b → 0 and PA–B, PA and PB all tend to btot. Therefore, all B monomers occur in the copolymers and the excess of A monomers occur as free monomers.
The example in Figure 12 shows that for K*A–B > 0.9 × 104 M–1, the approximations for the free monomer concentrations and the material in copolymers are very good. For smaller values of K*A–B, where the degree of copolymerization is not yet large enough and eq 46 hence does not hold, the monomer concentration is limited to the total monomer concentration.
Figure 12.

Example for purely alternating copolymers: (a) exact and approximated monomer concentrations. (b) PA and PB and their approximation. The lines of PA and PB almost coincide; therefore, PA is not visible. Parameters: atot = 2 × 10–4 M, btot = 1.5 × 10–4 M, K*A–A = K*B–B = 0 M–1, σA = 5 × 10–5, and σB = 7 × 10–5.
5. Discussion and Conclusions
The mass-balance model described in this work allows a fast computation of the thermodynamic equilibrium state in a large number of supramolecular copolymerizations, where two monomer types can aggregate into an arbitrary number of copolymer types. The homopolymers corresponding to the monomers can have arbitrary and different cooperativity. The bonds between two monomers in a copolymer may depend on both monomers, their order, and the copolymer type. The solution of the mass-balance equations gives both monomer concentrations, the amount of monomers of each type occurring in each copolymer type, and the average length of the copolymers. This information gives already some insight in the microstructure of the copolymers. Once the thermodynamic equilibrium state is known, information on the occurring bond types and the block lengths of homogeneous and alternating blocks can be computed, which provides additional insight in the structure of the copolymers. The latter are often difficult or impossible to obtain via experimental methods. The model encompasses all our earlier mass-balance models56,80−85 and the MATLAB scripts provided in the Supporting Information are able to reproduce the earlier reported results. Here, we used the mass-balance model and the MATLAB scripts to model the sergeants and soldiers system, that we previously described with stochastic simulations, and for the copolymerization of triarylamine triamide-based monomers in three different solvents.
Note that our method computes the thermodynamical equilibrium state, that is, it does not give a good description of for instance kinetically trapped systems. For those systems kinetic models are needed, like the stochastic approach to copolymerization in ref (90). However, stochastic simulations are computationally much more expensive than our method. Hence, to compute the thermodynamical equilibrium state, and also in cases where only a part of the system is in equilibrium,82 our method is preferable.
The model can straightforwardly be generalized to 3 or more monomer types. In the case of m monomer types, the iteration matrix Mc given in eq 15 becomes a m × m matrix. Also, the assumption that the equilibrium constant of an elongation at the top (or the bottom) of a copolymer depends only on the added monomer and the top (respectively bottom) element of the copolymer and of course on the copolymer type, may be relaxed. All methods described in this paper can be extended to the case where an elongation equilibrium constant depends on the added monomer and the upper k (or lower k) elements of the copolymer, for k = 1, 2, 3, .... Note, however, that both these generalizations lead to many more unknown parameters that must be given a useful value.
The detailed balance condition, the assumption that the copolymers can grow at top and bottom, and the assumption that the equilibrium constants for elongation at the top or bottom of a copolymer depend only on the two monomers forming a new bond are essential for deriving the relations 10 and 11 between the nucleation and elongation equilibrium constants. In fact, the detailed balance condition states that the total gain in free energy when constructing a molecule along two different routes must be the same. This condition ensures that the free energy is a function of the state of the system, and not, for instance, depends on its history. However, in principle, the used method to formulate and solve the mass-balance equations and also the computation of bonds and block lengths can easily be adapted to a case where the relations 10 and 11 do not hold.
The method we used to compute the equivalent monomer concentrations in the copolymers and also the methods to compute equivalent bond concentrations and average block lengths always considered elongation of the copolymers at their top element. Of course, it is equally well possible to compute these notions by elongation of copolymers at their bottom or even by a fragmentation and coagulation mechanism. As long as the equilibrium constants of used equilibrium reactions satisfy the detailed balance condition, the resulting concentrations of copolymers and so forth are always the same. Although the kinetics of the reactions may change drastically by the possibility of the additional reactions, the equilibrium state does not.
The general copolymerization model and the MATLAB scripts given here form a powerful tool to unravel supramolecular copolymerizations and hereby make a next step in controlling supramolecular copolymerizations. They provide tools to obtain a better insight into the microstructure of supramolecular copolymers which is experimentally difficult to attain. Ultimately, this will expand the potential of supramolecular copolymer applications.
Acknowledgments
This work was funded by the Dutch Ministry of Education, Culture and Science (Gravity program FMS, 024.001.035). We acknowledge E. W. (Bert) Meijer for the stimulating discussions during this work.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.jpcb.9b04373.
Notes on the consequences of the symmetry condition on heterodimer concentrations, initial values for the recursive relations, computing mass weighted copolymer lengths, the derivation of analytical expressions for the special cases, computing elongation temperatures, additional data for solvent dependent copolymerizations, and description of the provided MATLAB scripts (PDF)
MATLAB scripts for solving mass-balance equations, computing equivalent bond concentrations, block lengths and elongation temperatures (ZIP)
Author Present Address
∥ PASTEUR, Département de Chimie, École Normale Supérieure, PSL University, Sor-bonne Université, CNRS, 75005 Paris, France.
The authors declare no competing financial interest.
Supplementary Material
References
- Brunsveld L.; Folmer B. J. B.; Meijer E. W.; Sijbesma R. P. Supramolecular polymers. Chem. Rev. 2001, 101, 4071–4098. 10.1021/cr990125q. [DOI] [PubMed] [Google Scholar]
- De Greef T. F. A.; Smulders M. M. J.; Wolffs M.; Schenning A. P. H. J.; Sijbesma R. P.; Meijer E. W. Supramolecular polymerization. Chem. Rev. 2009, 109, 5687–5754. 10.1021/cr900181u. [DOI] [PubMed] [Google Scholar]
- Yang L.; Tan X.; Wang Z.; Zhang X. Supramolecular polymers: historical development, preparation, characterization, and functions. Chem. Rev. 2015, 115, 7196–7239. 10.1021/cr500633b. [DOI] [PubMed] [Google Scholar]
- Krieg E.; Bastings M. M. C.; Besenius P.; Rybtchinski B. Supramolecular polymers in aqueous media. Chem. Rev. 2016, 116, 2414–2477. 10.1021/acs.chemrev.5b00369. [DOI] [PubMed] [Google Scholar]
- Yan X.; Wang F.; Zheng B.; Huang F. Stimuli-responsive supramolecular polymeric materials. Chem. Soc. Rev. 2012, 41, 6042–6065. 10.1039/c2cs35091b. [DOI] [PubMed] [Google Scholar]
- Aida T.; Meijer E. W.; Stupp S. I. Functional supramolecular polymers. Science 2012, 335, 813–817. 10.1126/science.1205962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehn J.-M. Perspectives in Chemistry-Aspects of Adaptive Chemistry and Materials. Angew. Chem., Int. Ed. 2015, 54, 3276–3289. 10.1002/anie.201409399. [DOI] [PubMed] [Google Scholar]
- Bakker M. H.; Lee C. C.; Meijer E. W.; Dankers P. Y. W.; Albertazzi L. Multicomponent supramolecular polymers as a modular platform for intracellular delivery. ACS Nano 2016, 10, 1845–1852. 10.1021/acsnano.5b05383. [DOI] [PubMed] [Google Scholar]
- Mann J. L.; Yu A. C.; Agmon G.; Appel E. A. Supramolecular polymeric biomaterials. Biomater. Sci. 2018, 6, 10–37. 10.1039/c7bm00780a. [DOI] [PubMed] [Google Scholar]
- Kitamoto Y.; Aratsu K.; Yagai S. In Photoactive Functional Soft Materials: Preparation, Properties, and Applications; Li Q., Ed.; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2019; Chapter 2, pp 45–90. [Google Scholar]
- Besenius P. Controlling supramolecular polymerization through multicomponent self-assembly. J. Polym. Sci., Part A: Polym. Chem. 2017, 55, 34–78. 10.1002/pola.28385. [DOI] [Google Scholar]
- Odian G.Principles of Polymerization; John Wiley & Sons, 2004. [Google Scholar]
- Hawker C. J. ″Living″ Free Radical Polymerization: A Unique Technique for the Preparation of Controlled Macromolecular Architectures. Acc. Chem. Res. 1997, 30, 373–382. 10.1021/ar960248m. [DOI] [Google Scholar]
- Braunecker W. A.; Matyjaszewski K. Controlled/living radical polymerization: Features, developments, and perspectives. Prog. Polym. Sci. 2007, 32, 93–146. 10.1016/j.progpolymsci.2006.11.002. [DOI] [Google Scholar]
- Moad G.; Rizzardo E.; Thang S. H. Toward living radical polymerization. Acc. Chem. Res. 2008, 41, 1133–1142. 10.1021/ar800075n. [DOI] [PubMed] [Google Scholar]
- Bielawski C. W.; Grubbs R. H. Living ring-opening metathesis polymerization. Prog. Polym. Sci. 2007, 32, 1–29. 10.1016/j.progpolymsci.2006.08.006. [DOI] [Google Scholar]
- Bates F. S.; Hillmyer M. A.; Lodge T. P.; Bates C. M.; Delaney K. T.; Fredrickson G. H. Multiblock Polymers: Panacea or Pandora’s Box?. Science 2012, 336, 434–440. 10.1126/science.1215368. [DOI] [PubMed] [Google Scholar]
- Ouchi M.; Terashima T.; Sawamoto M. Transition metal-catalyzed living radical polymerization: toward perfection in catalysis and precision polymer synthesis. Chem. Rev. 2009, 109, 4963–5050. 10.1021/cr900234b. [DOI] [PubMed] [Google Scholar]
- Martens S.; Van den Begin J.; Madder A.; Du Prez F. E.; Espeel P. Automated synthesis of monodisperse oligomers, featuring sequence control and tailored functionalization. J. Am. Chem. Soc. 2016, 138, 14182–14185. 10.1021/jacs.6b07120. [DOI] [PubMed] [Google Scholar]
- Sorrenti A.; Leira-Iglesias J.; Markvoort A. J.; de Greef T. F. A.; Hermans T. M. Non-equilibrium supramolecular polymerization. Chem. Soc. Rev. 2017, 46, 5476–5490. 10.1039/c7cs00121e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boekhoven J.; Brizard A. M.; Kowlgi K. N. K.; Koper G. J. M.; Eelkema R.; van Esch J. H. Dissipative self-assembly of a molecular gelator by using a chemical fuel. Angew. Chem., Int. Ed. 2010, 49, 4825–4828. 10.1002/anie.201001511. [DOI] [PubMed] [Google Scholar]
- Boekhoven J.; Hendriksen W. E.; Koper G. J. M.; Eelkema R.; van Esch J. H. Transient assembly of active materials fueled by a chemical reaction. Science 2015, 349, 1075–1079. 10.1126/science.aac6103. [DOI] [PubMed] [Google Scholar]
- Debnath S.; Roy S.; Ulijn R. V. Peptide nanofibers with dynamic instability through nonequilibrium biocatalytic assembly. J. Am. Chem. Soc. 2013, 135, 16789–16792. 10.1021/ja4086353. [DOI] [PubMed] [Google Scholar]
- Leira-Iglesias J.; Sorrenti A.; Sato A.; Dunne P. A.; Hermans T. M. Supramolecular pathway selection of perylenediimides mediated by chemical fuels. Chem. Commun. 2016, 52, 9009–9012. 10.1039/c6cc01192f. [DOI] [PubMed] [Google Scholar]
- Ogi S.; Sugiyasu K.; Manna S.; Samitsu S.; Takeuchi M. Living supramolecular polymerization realized through a biomimetic approach. Nat. Chem. 2014, 6, 188. 10.1038/nchem.1849. [DOI] [PubMed] [Google Scholar]
- Ogi S.; Stepanenko V.; Sugiyasu K.; Takeuchi M.; Würthner F. Mechanism of self-assembly process and seeded supramolecular polymerization of perylene bisimide organogelator. J. Am. Chem. Soc. 2015, 137, 3300–3307. 10.1021/ja511952c. [DOI] [PubMed] [Google Scholar]
- Kang J.; Miyajima D.; Mori T.; Inoue Y.; Itoh Y.; Aida T. A rational strategy for the realization of chain-growth supramolecular polymerization. Science 2015, 347, 646–651. 10.1126/science.aaa4249. [DOI] [PubMed] [Google Scholar]
- Mukhopadhyay R. D.; Ajayaghosh A. Living supramolecular polymerization. Science 2015, 349, 241–242. 10.1126/science.aac7422. [DOI] [PubMed] [Google Scholar]
- Görl D.; Zhang X.; Stepanenko V.; Würthner F. Supramolecular block copolymers by kinetically controlled co-self-assembly of planar and core-twisted perylene bisimides. Nat. Commun. 2015, 6, 7009. 10.1038/ncomms8009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oosawa F.; Kasai M. A theory of linear and helical aggregations of macromolecules. J. Mol. Biol. 1962, 4, 10–21. 10.1016/s0022-2836(62)80112-0. [DOI] [PubMed] [Google Scholar]
- Martin R. B. Comparisons of indefinite self-association models. Chem. Rev. 1996, 96, 3043–3064. 10.1021/cr960037v. [DOI] [PubMed] [Google Scholar]
- Goldstein R. F.; Stryer L. Cooperative polymerization reactions. Analytical approximations, numerical examples, and experimental strategy. Biophys. J. 1986, 50, 583–599. 10.1016/s0006-3495(86)83498-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao D.; Moore J. S. Nucleation-elongation: a mechanism for cooperative supramolecular polymerization. Org. Biomol. Chem. 2003, 1, 3471–3491. 10.1039/b308788c. [DOI] [PubMed] [Google Scholar]
- Ercolani G. Assessment of cooperativity in self-assembly. J. Am. Chem. Soc. 2003, 125, 16097–16103. 10.1021/ja038396c. [DOI] [PubMed] [Google Scholar]
- Ciferri A.Supramolecular Polymers; CRC Press, 2005. [Google Scholar]
- Hamacek J.; Borkovec M.; Piguet C. Simple thermodynamics for unravelling sophisticated self-assembly processes. Dalton Trans. 2006, 1473–1490. 10.1039/b518461d. [DOI] [PubMed] [Google Scholar]
- Douglas J. F.; Dudowicz J.; Freed K. F. Lattice model of equilibrium polymerization. VII. Understanding the role of ″cooperativity″ in self-assembly. J. Chem. Phys. 2008, 128, 224901. 10.1063/1.2909195. [DOI] [PubMed] [Google Scholar]
- Hunter C. A.; Anderson H. L. What is cooperativity?. Angew. Chem., Int. Ed. 2009, 48, 7488–7499. 10.1002/anie.200902490. [DOI] [PubMed] [Google Scholar]
- Fernández G.; Stolte M.; Stepanenko V.; Würthner F. Cooperative Supramolecular Polymerization: Comparison of Different Models Applied on the Self-Assembly of Bis(merocyanine) Dyes. Chem.—Eur. J. 2013, 19, 206–217. 10.1002/chem.201202679. [DOI] [PubMed] [Google Scholar]
- Smulders M. M.; Nieuwenhuizen M. M.; de Greef T. F.; van der Schoot P.; Schenning A. P.; Meijer E. How to distinguish isodesmic from cooperative supramolecular polymerisation. Chem.—Eur. J 2010, 16, 362–367. 10.1002/chem.200902415. [DOI] [PubMed] [Google Scholar]
- Gershberg J.; Fennel F.; Rehm T. H.; Lochbrunner S.; Würthner F. Anti-cooperative supramolecular polymerization: a new K2-K model applied to the self-assembly of perylene bisimide dye proceeding via well-defined hydrogen-bonded dimers. Chem. Sci. 2016, 7, 1729–1737. 10.1039/c5sc03759j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frieden C.; Goddette D. W. Polymerization of actin and actin-like systems: evaluation of the time course of polymerization in relation to the mechanism. Biochemistry 1983, 22, 5836–5843. 10.1021/bi00294a023. [DOI] [PubMed] [Google Scholar]
- Ferrone F. A.; Hofrichter J.; Eaton W. A. Kinetics of sickle hemoglobin polymerization. J. Mol. Biol. 1985, 183, 611–631. 10.1016/0022-2836(85)90175-5. [DOI] [PubMed] [Google Scholar]
- Ferrone F.Methods in Enzymology; Elsevier, 1999; Vol. 309, pp 256–274. [DOI] [PubMed] [Google Scholar]
- Powers E. T.; Powers D. L. The Kinetics of Nucleated Polymerizations at High Concentrations: Amyloid Fibril Formation Near and Above the ″Supercritical Concentration″. Biophys. J. 2006, 91, 122–132. 10.1529/biophysj.105.073767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Powers E. T.; Powers D. L. Mechanisms of protein fibril formation: nucleated polymerization with competing off-pathway aggregation. Biophys. J 2008, 94, 379–391. 10.1529/biophysj.107.117168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue W.-F.; Homans S. W.; Radford S. E. Systematic analysis of nucleation-dependent polymerization reveals new insights into the mechanism of amyloid self-assembly. Proc. Natl. Acad. Sci. U.S.A. 2008, 105, 8926–8931. 10.1073/pnas.0711664105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris A. M.; Watzky M. A.; Finke R. G. Protein aggregation kinetics, mechanism, and curve-fitting: a review of the literature. Biochim. Biophys. Acta, Proteins Proteomics 2009, 1794, 375–397. 10.1016/j.bbapap.2008.10.016. [DOI] [PubMed] [Google Scholar]
- Knowles T. P. J.; Waudby C. A.; Devlin G. L.; Cohen S. I. A.; Aguzzi A.; Vendruscolo M.; Terentjev E. M.; Welland M. E.; Dobson C. M. An analytical solution to the kinetics of breakable filament assembly. Science 2009, 326, 1533–1537. 10.1126/science.1178250. [DOI] [PubMed] [Google Scholar]
- Korevaar P. A.; George S. J.; Markvoort A. J.; Smulders M. M. J.; Hilbers P. A. J.; Schenning A. P. H. J.; De Greef T. F. A.; Meijer E. W. Pathway complexity in supramolecular polymerization. Nature 2012, 481, 492. 10.1038/nature10720. [DOI] [PubMed] [Google Scholar]
- Cohen S. I.; Vendruscolo M.; Welland M. E.; Dobson C. M.; Terentjev E. M.; Knowles T. P. Nucleated polymerization with secondary pathways. I. Time evolution of the principal moments. J. Chem. Phys. 2011, 135, 08B615. 10.1063/1.3608916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meisl G.; Kirkegaard J. B.; Arosio P.; Michaels T. C. T.; Vendruscolo M.; Dobson C. M.; Linse S.; Knowles T. P. J. Molecular mechanisms of protein aggregation from global fitting of kinetic models. Nat. Protoc. 2016, 11, 252. 10.1038/nprot.2016.010. [DOI] [PubMed] [Google Scholar]
- Michaels T. C. T.; Liu L. X.; Meisl G.; Knowles T. P. J. Physical principles of filamentous protein self-assembly kinetics. J. Phys.: Condens. Matter 2017, 29, 153002. 10.1088/1361-648x/aa5f10. [DOI] [PubMed] [Google Scholar]
- Fennel F.; Wolter S.; Xie Z.; Plötz P.-A.; Kühn O.; Würthner F.; Lochbrunner S. Biphasic self-assembly pathways and size-dependent photophysical properties of perylene bisimide dye aggregates. J. Am. Chem. Soc. 2013, 135, 18722–18725. 10.1021/ja409597x. [DOI] [PubMed] [Google Scholar]
- Cai K.; Xie J.; Zhang D.; Shi W.; Yan Q.; Zhao D. Concurrent cooperative J-aggregates and anticooperative H-aggregates. J. Am. Chem. Soc. 2018, 140, 5764–5773. 10.1021/jacs.8b01463. [DOI] [PubMed] [Google Scholar]
- Mabesoone M. F. J.; Markvoort A. J.; Banno M.; Yamaguchi T.; Helmich F.; Naito Y.; Yashima E.; Palmans A. R. A.; Meijer E. W. Competing interactions in hierarchical porphyrin self-assembly introduce robustness in pathway complexity. J. Am. Chem. Soc. 2018, 140, 7810–7819. 10.1021/jacs.8b02388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y.; Zhang Y.; Fennel F.; Wagner W.; Würthner F.; Chen Y.; Chen Z. Coupled cooperative supramolecular polymerization: A new model applied to the competing aggregation pathways of an amphiphilic aza-BODIPY dye into spherical and rod-like aggregates. Chem.—Eur. J. 2018, 24, 16388–16394. 10.1002/chem.201803336. [DOI] [PubMed] [Google Scholar]
- van der Zwaag D.; Pieters P. A.; Korevaar P. A.; Markvoort A. J.; Spiering A. J. H.; de Greef T. F. A.; Meijer E. W. Kinetic analysis as a tool to distinguish pathway complexity in molecular assembly: an unexpected outcome of structures in competition. J. Am. Chem. Soc. 2015, 137, 12677–12688. 10.1021/jacs.5b08138. [DOI] [PubMed] [Google Scholar]
- Wu A.; Isaacs L. Self-sorting: the exception or the rule?. J. Am. Chem. Soc. 2003, 125, 4831–4835. 10.1021/ja028913b. [DOI] [PubMed] [Google Scholar]
- Sun W.-Y.; Yoshizawa M.; Kusukawa T.; Fujita M. Multicomponent metal-ligand self-assembly. Curr. Opin. Chem. Biol. 2002, 6, 757–764. 10.1016/s1367-5931(02)00358-7. [DOI] [PubMed] [Google Scholar]
- Prins L. J.; Timmerman P.; Reinhoudt D. N. Amplification of Chirality: The ″Sergeants and Soldiers″ Principle Applied to Dynamic Hydrogen-Bonded Assemblies†. J. Am. Chem. Soc. 2001, 123, 10153–10163. 10.1021/ja010610e. [DOI] [PubMed] [Google Scholar]
- Mateos-Timoneda M. A.; Crego-Calama M.; Reinhoudt D. N. Controlling the amplification of chirality in hydrogen-bonded assemblies. Supramol. Chem. 2005, 17, 67–79. 10.1080/10610270412331328826. [DOI] [PubMed] [Google Scholar]
- Ballester P.; Oliva A. I.; Costa A.; Deyà P. M.; Frontera A.; Gomila R. M.; Hunter C. A. DABCO-induced self-assembly of a trisporphyrin double-decker cage: thermodynamic characterization and guest recognition. J. Am. Chem. Soc. 2006, 128, 5560–5569. 10.1021/ja060608t. [DOI] [PubMed] [Google Scholar]
- Lombardo T. G.; Stillinger F. H.; Debenedetti P. G. Thermodynamic mechanism for solution phase chiral amplification via a lattice model. Proc. Natl. Acad. Sci. U.S.A. 2009, 106, 15131–15135. 10.1073/pnas.0812867106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castilla A. M.; Miller M. A.; Nitschke J. R.; Smulders M. M. J. Quantification of Stereochemical Communication in Metal-Organic Assemblies. Angew. Chem., Int. Ed. 2016, 55, 10616–10620. 10.1002/anie.201602968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y.; Fang H.; Tranca I.; Qu H.; Wang X.; Markvoort A. J.; Tian Z.; Cao X. Elucidation of the origin of chiral amplification in discrete molecular polyhedra. Nat. Commun. 2018, 9, 488. 10.1038/s41467-017-02605-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tobolsky A. V.; Owen G. D. T. A general treatment of equilibrium copolymerization. J. Polym. Sci. 1962, 59, 329–337. 10.1002/pol.1962.1205916811. [DOI] [Google Scholar]
- Szwarc M.; Perrin C. L. General treatment of equilibrium copolymerization of two or more comonomers deduced from the initial state of the system. Macromolecules 1985, 18, 528–533. 10.1021/ma00145a038. [DOI] [Google Scholar]
- Szymański R. Reversible copolymerization at equilibrium. Makromol. Chem. 1986, 187, 1109–1114. 10.1002/macp.1986.021870507. [DOI] [Google Scholar]
- Buchelnikov A. S.; Evstigneev V. P.; Evstigneev M. P. The hetero-association models of non-covalent molecular complexation. Phys. Chem. Chem. Phys. 2019, 21, 7717. 10.1039/c8cp03183e. [DOI] [PubMed] [Google Scholar]
- Weller K.; Schütz H.; Petri I. Thermodynamical model of indefinite mixed association of two components and NMR data analysis for caffeine-AMP interaction. Biophys. Chem. 1984, 19, 289–298. 10.1016/0301-4622(84)87012-x. [DOI] [PubMed] [Google Scholar]
- Evstigneev V. P.; Mosunov A. A.; Buchelnikov A. S.; Hernandez Santiago A. A.; Evstigneev M. P. Complete solution of the problem of one-dimensional non-covalent non-cooperative self-assembly in two-component systems. J. Chem. Phys. 2011, 134, 194902. 10.1063/1.3589899. [DOI] [PubMed] [Google Scholar]
- Buchelnikov A. S.; Evstigneev V. P.; Evstigneev M. P. General statistical-thermodynamical treatment of one-dimensional multicomponent molecular hetero-assembly in solution. Chem. Phys. 2013, 421, 77–83. 10.1016/j.chemphys.2013.06.006. [DOI] [Google Scholar]
- Jonkheijm P.; van der Schoot P.; Schenning A. P. H. J.; Meijer E. W. Probing the solvent-assisted nucleation pathway in chemical self-assembly. Science 2006, 313, 80–83. 10.1126/science.1127884. [DOI] [PubMed] [Google Scholar]
- Gestel J. v.; van der Schoot P.; Michels M. A. J. Amplification of chirality in helical supramolecular polymers beyond the long-chain limit. J. Chem. Phys. 2004, 120, 8253–8261. 10.1063/1.1689645. [DOI] [PubMed] [Google Scholar]
- van Gestel J. Amplification of chirality in helical supramolecular polymers: the majority-rules principle. Macromolecules 2004, 37, 3894–3898. 10.1021/ma030480p. [DOI] [Google Scholar]
- Smulders M. M. J.; Filot I. A. W.; Leenders J. M. A.; van der Schoot P.; Palmans A. R. A.; Schenning A. P. H. J.; Meijer E. W. Tuning the extent of chiral amplification by temperature in a dynamic supramolecular polymer. J. Am. Chem. Soc. 2010, 132, 611–619. 10.1021/ja908053d. [DOI] [PubMed] [Google Scholar]
- Jabbari-Farouji S.; van der Schoot P. Theory of supramolecular co-polymerization in a two-component system. J. Chem. Phys. 2012, 137, 064906. 10.1063/1.4742192. [DOI] [PubMed] [Google Scholar]
- van Buel R.; Spitzer D.; van der Schoot P.; Besenius P.; Jabbari-Farouji S.; et al. Supramolecular copolymers predominated by alternating order: Theory and application. J. Chem. Phys. 2019, 151, 014902. 10.1063/1.5097577. [DOI] [PubMed] [Google Scholar]
- Markvoort A. J.; Eikelder H. M. T.; Hilbers P. A. J.; De Greef T. F. A.; Meijer E. W. Theoretical models of nonlinear effects in two-component cooeperative supramolecular copolymerizations. Nat. Commun. 2011, 2, 509. 10.1038/ncomms1517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ten Eikelder H. M. M.; Markvoort A. J.; De Greef T. F. A.; Hilbers P. A. J. An equilibrium model for chiral amplification in supramolecular polymers. J. Phys. Chem. B 2012, 116, 5291–5301. 10.1021/jp300622m. [DOI] [PubMed] [Google Scholar]
- Cantekin S.; Ten Eikelder H. M. M.; Markvoort A. J.; Veld M. A. J.; Korevaar P. A.; Green M. M.; Palmans A. R. A.; Meijer E. W. Consequences of cooperativity in racemizing supramolecular systems. Angew. Chem., Int. Ed. 2012, 124, 6532–6537. 10.1002/ange.201201701. [DOI] [PubMed] [Google Scholar]
- Nakano Y.; Markvoort A. J.; Cantekin S.; Filot I. A. W.; ten Eikelder H. M. M.; Meijer E. W.; Palmans A. R. A. Conformational analysis of chiral supramolecular aggregates: modeling the subtle difference between hydrogen and deuterium. J. Am. Chem. Soc. 2013, 135, 16497–16506. 10.1021/ja4073645. [DOI] [PubMed] [Google Scholar]
- Das A.; Vantomme G.; Markvoort A. J.; ten Eikelder H. M. M.; Garcia-Iglesias M.; Palmans A. R. A.; Meijer E. W. Supramolecular copolymers: structure and composition revealed by theoretical modeling. J. Am. Chem. Soc. 2017, 139, 7036–7044. 10.1021/jacs.7b02835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adelizzi B.; Aloi A.; Markvoort A. J.; Ten Eikelder H. M. M.; Voets I. K.; Palmans A. R. A.; Meijer E. W. Supramolecular block copolymers under thermodynamic control. J. Am. Chem. Soc. 2018, 140, 7168–7175. 10.1021/jacs.8b02706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smulders M. M. J.; Schenning A. P. H. J.; Meijer E. W. Insight into the Mechanisms of Cooperative Self-Assembly: The ″Sergeants-and-Soldiers″ Principle of Chiral and Achiral C3-Symmetrical Discotic Triamides. J. Am. Chem. Soc. 2008, 130, 606–611. 10.1021/ja075987k. [DOI] [PubMed] [Google Scholar]
- Green M. M.; Reidy M. P.; Johnson R. D.; Darling G.; O’Leary D. J.; Willson G. Macromolecular stereochemistry: the out-of-proportion influence of optically active comonomers on the conformational characteristics of polyisocyanates. The sergeants and soldiers experiment. J. Am. Chem. Soc. 1989, 111, 6452–6454. 10.1021/ja00198a084. [DOI] [Google Scholar]
- Gillespie D. T. Stochastic simulation of chemical kinetics. Annu. Rev. Phys. Chem. 2007, 58, 35–55. 10.1146/annurev.physchem.58.032806.104637. [DOI] [PubMed] [Google Scholar]
- Van Zee N. J.; Adelizzi B.; Mabesoone M. F. J.; Meng X.; Aloi A.; Zha R. H.; Lutz M.; Filot I. A. W.; Palmans A. R. A.; Meijer E. W. Potential enthalpic energy of water in oils exploited to control supramolecular structure. Nature 2018, 558, 100–103. 10.1038/s41586-018-0169-0. [DOI] [PubMed] [Google Scholar]
- Markvoort A. J.; ten Eikelder H. M. M.; Hilbers P. A. J.; de Greef T. F. A. Fragmentation and coagulation in supramolecular (co)polymerization kinetics. ACS Cent. Sci. 2016, 2, 232–241. 10.1021/acscentsci.6b00009. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.