

Abstract
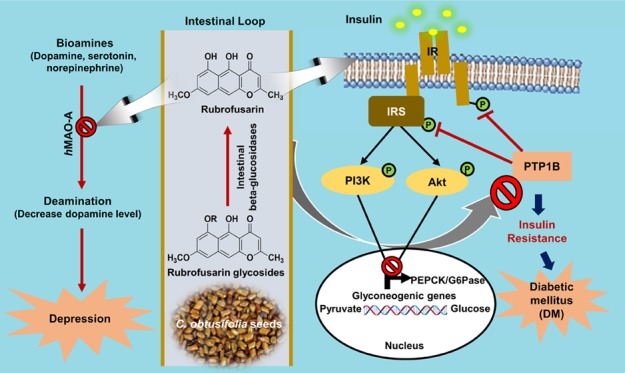
A number of nature-derived biologically active compounds comprise glycosides. In some cases, the glycosidic residue is needed for bioactivity; however, in other cases, glycosylation just improves some pharmacokinetic/dynamic parameters. The patterns of protein tyrosine phosphatase 1B (PTP1B) and human monoamine oxidase A (hMAO-A) inhibition by rubrofusarin 6-O-β-d-glucopyranoside (1), rubrofusarin 6-O-β-d-gentiobioside (2), rubrofusarin triglucoside (3), and cassiaside B2 (4) were compared with the aglycone, rubrofusarin, isolated from Cassia obtusifolia seeds. Rubrofusarin showed potent inhibition against the PTP1B enzyme (IC50; 16.95 ± 0.49 μM), and its glycosides reduced activity (IC50; 87.36 ± 1.08 μM for 1 and >100 μM for 2–4) than did the reference drug, ursolic acid (IC50; 2.29 ± 0.04 μM). Similarly, in hMAO-A inhibition, rubrofusarin displayed the most potent activity with an IC50 value of 5.90 ± 0.99 μM, which was twice better than the reference drug, deprenyl HCl (IC50; 10.23 ± 0.82 μM). An enzyme kinetic and molecular docking study revealed rubrofusarin to be a mixed-competitive inhibitor of both these enzymes. In a western blot analysis, rubrofusarin increased glucose uptake significantly and decreased the PTP1B expression in a dose-dependent manner in insulin-resistant HepG2 cells, increased the expression of phosphorylated protein kinase B (p-Akt) and phosphorylated insulin receptor substrate-1 (p-IRS1) (Tyr 895), and decreased the expression of glucose-6-phosphatase (G6Pase) and phosphoenol pyruvate carboxykinase (PEPCK), key enzymes of gluconeogenesis. Our overall results show that glycosylation retards activity; however, it reduces toxicity. Thus, Cassia seed as functional food and rubrofusarin as a base can be used for the development of therapeutic agents against comorbid diabetes and depression.
Introduction
A number of glycosides derived from natural sources are biologically active. Glycosylation, in some cases, is essential for activity; however, in other cases, glycosidic residue merely improves some pharmacokinetic parameters. Recently, molecular glycobiology has contributed to a better understanding of the activities of aglycone and glycoside. The significant difference in activity between aglycone and glycosides has created a new platform to develop more potent and/or more effective glycodrugs.1 Despite that, it is nearly impossible to generalize how the specific pattern of biological activities of the glycosides differs from that of aglycones.2
The structural diversity and function of natural products have been the recent focus of drug research and drug discovery with a routine evaluation of absorption, distribution, metabolism, and excretion-toxicity (ADMET) and drug-likeness of secondary metabolites from natural sources. Interestingly, glycosylation has been found to modulate these properties. Furthermore, glycosylation of natural products alters the water solubility, bioavailability, and pharmacological activities, which provides a challenge as well as passion in developing and applying glycosylated natural products in modern medicine.3 Previous studies have reported that the amino sugars must have a natural α-glycoside linkage with the aglycone for antitumor activity, which the β-analogs lacked.4 Similarly, another study on some vitamins found more advantages from glycosylated vitamins than from the aglycone ones. Glycosylation increased water solubility (especially the lipophilic ones), increased stability against heat, ultraviolet light, and oxidation, masked bitter taste and odor (e.g., thiamine), and improved resistance to enzymatic action.2
Among various natural glycosides, flavonoid glycosides have gained much attention. In a comparative study of the effect of icaritin and its glycosides (icariin and icariside II) on the differentiation and proliferation of osteoblasts in vitro, aglycones demonstrated more potent activity than glycoside derivatives.5 Although icariin and icariside II showed activity on streptozotocin-induced diabetic retinopathy in vivo,6 a recent study by Kim et al.7 stated that the activity of two metabolites, icariside II and icaritin, is responsible for the overall in vivo effects of icariin because, in the small intestine, icariin is hydrolyzed to icariside II and icaritin by human intestinal microflora.8 In our recent study on how natural naringenin derivatives affect protein tyrosine phosphate 1B (PTP1B) inhibition and glucose uptake, a single-glucose-containing flavanone glycoside (prunin) showed the most potent activity among other glycosides and an aglycone, naringenin.9 Sugars attached to specific positions with definite orientations on the aglycone core of natural compounds are the eminent source of structural biodiversity within natural products. Generally, these sugars play a vital role in the molecular recognition of its cellular target.4 The nature, its position, and orientation of the sugar residue may increase the absorption of particular compounds in the small intestine. However, many studies have reported aglycones to have greater biological effects than the glycosides.10
Type 2 diabetes mellitus (T2DM) is a chronic disease resulting from insulin resistance, in which, although enough insulin is produced, cells fail to respond to it; so, glucose uptake in tissues declines and the blood glucose level elevates.11 PTP1B is a negative regulator of insulin signaling that catalyzes the dephosphorylation of the insulin receptor/insulin receptor substrate. Further, studies on molecular biology have demonstrated inhibition on tyrosine phosphorylation of insulin receptor (IR) and insulin receptor substrate-1 (IRS-1) due to PTP1B overexpression, thereby enhancing serine phosphorylation and finally leading to insulin resistance. In addition, insulin sensitivity was enhanced via PTP1B knockout from liver, muscles, and fat tissues.12 Therefore, PTP1B is considered a target for insulin-sensitizing drugs in preventing and treating T2DM. Similarly, the liver is a center for hepatic gluconeogenesis catalyzed by two key enzymes, glucose-6-phosphatase (G6Pase) and phosphoenol pyruvate carboxykinase (PEPCK); insulin inhibits the activity of these enzymes and finally blocks gluconeogenesis,13,14 controlling the blood glucose level. Therefore, either increasing the sensitivity of insulin-resistant cells toward insulin to increase glucose uptake or inhibiting the enzyme activities of PEPCK and G6Pase or both could be a better way to treat T2DM and associated complications.
Human monoamine oxidase (hMAO) is a metalloenzyme that catalyzes the oxidative deamination of various bioamines, and its inhibitors were the first developed antidepressants. Since the major depressive disorder is estimated to be the second leading cause of diseases worldwide, the discovery of inhibitors of two isozymes of MAO (MAO-A and MAO-B), which differ in sensitivity toward inhibitors and substrate specificity,15 creates a sense of urgency. At present, the association between T2DM and depression has been a subject of focus. Some studies argue that there is a solid association between depression and incidence of T2DM and a weak alliance between diabetes and risk of depression,16,17 and some stick with a conclusion that diabetics have a 2–4-fold greater risk of depression.18 On the same note, some authors have concluded the relationship between diabetes and depression as bidirectional.19 So the discovery of dual hMAO-A and PTP1B inhibitors from natural sources might be a promising strategy to treat comorbid diabetes and depression.
Cassia obtusifolia Linn seed is one of the most popular traditional Chinese medicines. It has been used for decades because of its inherent antifungal, antioxidant, antimutagenic, antigenotoxic, antiglycation, and antihepatotoxic properties.20−22 Recent in vivo experiments in streptozotocin-induced diabetic mice demonstrated that the ethanolic extract of Cassia seed caused significant reduction in blood glucose, total cholesterol, triglyceride, phospholipids, and free fatty acid,23 whereas the n-butanol fraction showed a beneficial effect on postprandial blood glucose control.24 In our previous study, we evaluated the anti-Alzheimer’s disease activity25,26 and antidiabetic potentials27 of major anthraquinones, naphthopyrones, and naphthalene glycosides from C. obtusifolia seeds. Recently, we have reported the hepatoprotective property of anthraquinone and naphthopyrone glycosides from the n-BuOH fraction28 as well as the EtOAc fraction29 of Cassia seed in a t-BHP-treated HepG2 cell model. In this study, we aimed to evaluate the inhibitory activity of rubrofusarin against PTP1B and hMAO-A, set forth its enzyme kinetics, and simulate a molecular docking study. In addition, glucose uptake activity was assessed in insulin-resistant HepG2 cells, and the mechanism of action was explored via cell signaling. We also made an attempt to explore how PTP1B and hMAO-A are inhibited by glycosylation by naturally occurring naphthopyrone glycosides from the seeds of C. obtusifolia and discuss the effect of glycosylation.
Results
Protein Tyrosine Phosphatase 1B (PTP1B) and Human Monoamine Oxidase A (hMAO-A) Inhibition
The methanol extract of C. obtusifolia seeds and its components were evaluated for their inhibitory potential against PTP1B and hMAO-A in vitro. As shown in Table 1, the methanol extract potently inhibited PTP1B with an IC50 value of 14.79 ± 0.31 μg/mL.27 Among rubrofusarin and its glycosides (Figure 1), rubrofusarin showed potent inhibition of the PTP1B enzyme with an IC50 of value 16.95 ± 0.49 μM. In contrast, glycosides showed much less inhibition. The 50% inhibition concentration for the PTP1B enzyme was 87.36 ± 1.08 μM for 1 as compared with the reference compound, ursolic acid (IC50; 2.29 ± 0.04 μM). However, an increase in the number of glucose molecules retarded the activity significantly (IC50; >100 μM) in compounds 2–4 containing two, three, and four glucose molecules, respectively.
Table 1. Protein Tyrosine Phosphatases 1B (PTP1B) and Human Monoamine Oxidase A (hMAO-A) Inhibitory Activity of Rubrofusarin and Its Glycosidesa.
| PTP1B
inhibition |
hMAO-A inhibition |
|||||
|---|---|---|---|---|---|---|
| compounds | IC50 (μM)b | Ki value (μM) | inhibition typee | IC50 (μM)b | Ki value (μM) | inhibition typee |
| methanol extractf | 14.79 ± 0.31 | 86.89 ± 3.80 | ||||
| rubrofusarin | 16.95 ± 0.49 | 6.99c/18.31d | mixed-competitive | 5.90 ± 0.99 | 4.92c/5.96d | mixed-competitive |
| 1 | 87.36 ± 1.08 | NT | >100 | NT | ||
| 2 | >100 | NT | >100 | NT | ||
| 3 | >100 | NT | 85.50 ± 1.92 | NT | ||
| 4 | >100 | NT | 40.57 ± 0.75 | NT | ||
| ursolic acidg | 2.29 ± 0.04 | NT | NT | |||
| deprenyl HClg | 10.23 ± 0.82 | |||||
NT not tested.
The 50% inhibition concentration (μM) was calculated from a log-dose inhibition curve and is expressed as the mean ± standard deviation of the triplicate experiments.
Binding constant of the inhibitor with the free enzyme (Kic).
Binding constant of the inhibitor with the enzyme–substrate complex (Kiu) calculated from secondary plots.
Calculated from the Lineweaver–Burk plot.
IC50 values are expressed in μg/mL.
Used as reference controls.
Figure 1.

Chemical structures of rubrofusarin and its glycosides.
Similarly, in hMAO-A inhibition, the MeOH extract displayed moderate inhibition with an IC50 value of 14.79 ± 0.31 μg/mL. Rubrofusarin displayed the most potent activity with an IC50 value of 5.90 ± 0.99 μM, which was twice better than the reference drug, deprenyl HCl (IC50; 10.23 ± 0.82 μM). 1 and 2 bearing single and double glucose moieties did not show notable effect up to 100 μM. However, further increase in the number of glucose moiety displayed a mild activity by 3 (IC50; 85.50 ± 1.92 μM) and a moderate one by 4 (IC50; 40.57 ± 0.75 μM). In both enzymes, rubrofusarin showed potent inhibition; however, inhibition by glycosides showed a reversed pattern in these enzymes.
Enzyme Kinetics in PTP1B and hMAO-A Inhibition
To find out the mode of enzyme inhibition and the enzyme inhibition constant (Ki) of the enzyme–inhibitor complex, we carried out a kinetic study of rubrofusarin against PTP1B and hMAO-A enzymes using Lineweaver–Burk plots and secondary plots (Supporting Information Figure S1). The results showed a mixed-competitive mode of PTP1B (Kic: 6.99 μM; Kiu: 18.31 μM) and hMAO-A (Kic: 4.92 μM; Kiu: 5.96 μM) inhibition for rubrofusarin (Table 1).
Molecular Docking Simulation in PTP1B and hMAO-A Inhibition
Based on the molecular docking studies, we evaluated the binding-site-directed PTP1B and hMAO-A inhibition by rubrofusarin using AutoDock 4.2 (Figure 2). As shown in Table 2, binding energies for rubrofusarin to the catalytic and allosteric sites of PTP1B were −5.47 and −6.41 kcal/mol, respectively. Similarly, rubrofusarin shared the same hydrophobic interacting residues, Tyr46 via π–π stacked interaction, and Ala217 via π-alkyl interaction with the catalytic inhibitor, 3-({5-[(N-acetyl-3-{4-[(carboxycarbonyl)(2-carboxyphenyl)amino]-1-naphthyl}-l-alanyl)amino]pentyl}oxy)-2-naphthoic acid (compound 23) (Figure 2A-a2). At the allosteric site, it shared two common residues, Ala189 and Leu192, via π–alkyl interaction with the allosteric inhibitor, 3-(3,5-dibromo-4-hydroxy-benzoyl)-2-ethyl-benzofuran-6-sulfonic acid (4-sulfamoyl-phenyl)-amide (compound 2) (Figure 2A-a3). Furthermore, the common H-bond interacting residues for rubrofusarin and reference inhibitors were Asn193 at the allosteric site and Asp48 and Lys120 at the catalytic site.
Figure 2.

Molecular docking studies. (A) Rubrofusarin at the binding sites of PTP1B; (a2) and (a3) represent a close-up of the 3D binding pose of rubrofusarin in the predicted catalytic and allosteric sites of PTP1B, respectively. (B) Rubrofusarin at the binding sites of human monoamine oxidase A (2z5x); (b2) and (b3) represent a close-up of the 3D binding pose of rubrofusarin in the predicted allosteric and catalytic sites of hMAO-A, respectively. The different color dotted lines represent specific interactions; deep pink, π–π stacked and π–π T-shaped; pale pink, π–alkyl; purple, π–σ; green, H-bond; O–H; and pale green, H-bond, C–H.
Table 2. Docking Affinity Scores and Possible H-Bond Formation to PTP1B (1t49) and hMAO A (2z5x) Active Sites by Rubrofusarin Along with Reported Inhibitors.
| compounds | binding score (kcal/mol)a | no. of H-bonds | H-bond interacting residuesb | hydrophobic interacting residuesb | others |
|---|---|---|---|---|---|
| PTP1B (1t49) | |||||
| compound 23c | –10.18 | 11 | Lys116, Lys120 (2), Asp48 (4), Asp181, Gln262 | π–π stacked: Tyr46, π–alkyl: Met258, Ala217 | π–sulfur: Met258 |
| compound 2c | –10.98 | 2 | Asn193, Glu276, Lys279, Phe280 | π–π stacked: Phe280, alkyl: Ile280, π–alkyl: Phe196, Phe280, Leu192, Ala189 | |
| rubrofusarin (catalytic mode) | –5.47 | 6 | Asp48, Lys120, Cys215, Gln262 (3) | π–π stacked: Tyr46, alkyl: Lys120, π–alkyl: Ala217 | |
| rubrofusarin (allosteric mode) | –6.41 | 1 | Asn193, Gly277 | π–σ: Leu192, π–π stacked: Phe280, π–alkyl: Ala189, Leu192 | |
| hMAO-A (2z5x) | |||||
| harmined | –9.18 | 3 | Phe208, Gln215, Tyr407 | π–alkyl; Ile180, Ile335, Leu337, Tyr407, Tyr444, π–π stacked: Tyr407, π–σ: Ile335 | π–sulfur: Cys323, van der Waals: FAD |
| rubrofusarin | |||||
| (catalytic mode) | –9.67 | 6 | Tyr444, Asn181 (2), Gly443, Tyr407 (2) | π–σ: Tyr444, π–π stacked: Tyr407, π–π T-shaped: FAD, alkyl: Ile335, Leu337, π–alkyl: Ile180, Ile335 | van der Waals: FAD |
| rubrofusarin | |||||
| (allosteric mode) | –5.60 | 5 | Gly404 (2), Gln296, Leu298, Met300 | π–alkyl: Ala302, Pro299 | |
Estimated binding free energy of the ligand–receptor complex.
All amino acid residues located 5 Å from the original enzyme/compound complex in the AutoDock 4.2 program.
Compound 23, 3-({5-[(N-acetyl-3-{4-[(carboxycarbonyl)(2-carboxyphenyl)amino]-1-naphthyl}-l-alanyl)amino]pentyl}oxy)-2-naphthoic acid, and compound 2, 3-(3,5-dibromo-4-hydroxy-benzoyl)-2-ethyl-benzofuran-6-sulfonic acid (4-sulfamoyl-phenyl)-amide, were used as reference ligands for PTP1B.
Reference catalytic ligand for hMAO-A.
Rubrofusarin bound to a catalytic gorge of hMAO-A (−9.67 kcal/mol) resulting from prime hydrophobic interactions such as π–alkyl with Ile180 and Ile335, π–σ with Tyr444, π–π stacked with Tyr407, and π–π T-shaped along with van der Waals interactions with FAD (Figure 2B-b3). At the allosteric site (Figure 2B-b2), rubrofusarin bound with −5.60 kcal/mol energy, forming five H-bonds with Gly404, Gln296, Leu298, and Met300. Besides this, π–alkyl interactions with Ala302 and Pro299 were also observed at the allosteric site. Though the mode of inhibition was identified as a mixed one, secondary plots and docking results specified it as a mixed-competitive inhibition. The overall results of the enzyme kinetic study revealed rubrofusarin as a mixed-competitive-dual inhibitor of PTP1B and hMAO-A.
Effect of Glucose Moiety on Cytotoxicity in HepG2 Cells
To find out the effect of glycosylation on the cytotoxicity in HepG2 cells, we evaluated cell viability using an MTT assay. As shown in Figure 3A, all glycosides depicted no toxicity up to 100 μM. However, rubrofusarin showed toxicity at 100 μM.
Figure 3.

Effect of rubrofusarin and its glycosides, rubrofusarin 6-O-β-d-glucopyranoside (1), rubrofusarin 6-O-β-d-gentiobioside (2), rubrofusarin triglucoside (3), and cassiaside B2 (4), on cell viability in HepG2 cells, and *p < 0.05 indicates significant differences from the control group (A). Effect of rubrofusarin on insulin-stimulated glucose uptake in insulin-resistant HepG2 cells, and ###p < 0.001 indicates significant differences from the control group; **p < 0.01 and ***p < 0.001 indicate significant differences from the 10–6 M insulin-treated control group (B). Data shown represent means ± standard deviation of triplicate experiments.
Effects of Rubrofusarin on Glucose Uptake in Insulin-Resistant HepG2 Cells
A 2-[N-(7-nitrobenz-2-oxa-1,3-diazol-4-yl)amino]-2-deoxyglucose (2-NBDG) test was used to measure the glucose uptake in insulin-resistant HepG2 cells. Rubrofusarin increased insulin-stimulated 2-NBDG uptake in insulin-resistant HepG2 cells significantly in a dose-dependent manner (Figure 3B). The relative glucose uptake percentages of rubrofusarin at 12.5, 25, and 50 μM concentrations were 68.54, 81.27, and 98.47%, respectively. Rosiglitazone, which was used as a reference drug, increased insulin-stimulated glucose uptake significantly to 118.52% on average at 10 μM concentration. Since our PTP1B enzyme assay showed negligible inhibition by glycosides, we did not perform a glucose uptake assay for those glycosides.
Effect of Rubrofusarin on the PTP1B Expression Level in Insulin-Resistant HepG2 Cells
PTP1B is a negative regulator of insulin signaling, and its high expression is implicated in insulin resistance. Therefore, to confirm whether rubrofusarin increases insulin sensitivity by inhibiting PTP1B expression, we evaluated the expression in insulin-resistant HepG2 cells by western blot. As shown in Figure 4A, rubrofusarin inhibited PTP1B expression in a dose-dependent manner, disclosing its insulin-sensitizing potential.
Figure 4.

Effect of rubrofusarin on protein tyrosine phosphatase 1B (PTP1B) expression in insulin-resistant HepG2 cells (A). Western blotting was performed, and protein band intensities were quantified by densitometric analysis. The upper panels display representative blots. Equal protein loading was ensured and normalized against β-actin levels. Values are the mean ± standard deviation of three independent experiments; ###p < 0.001 indicates significant differences from the control group; **p < 0.01 and ***p < 0.001 indicate significant differences from the 10–6 M insulin-treated control group. Similarly, (B) represents the effects of rubrofusarin on the levels of total and phosphorylated insulin receptor substrate-1 (IRS-1), phosphatidylinositol-3-kinase (PI3K), and protein kinase B (Akt) as well as on two key enzymes for gluconeogenesis, glucose-6-phosphatase (G6Pase) and phosphoenol pyruvate carboxykinase (PEPCK), in insulin-resistant HepG2 cells.
Effects of Rubrofusarin on the IRS-1/PI3K/Akt Signaling Pathway in Insulin-Resistant HepG2 Cells
The expression levels of proteins involved in insulin signaling were measured by western blotting to explore the molecular mechanism underlying the effects of rubrofusarin. As shown in Figure 4B, rubrofusarin dose-dependently increased the expression of total IRS-1 and p-IRS-1 (Tyr 895) in insulin-resistant HepG2 cells, and the levels were like those observed in normal control cells sensitive to insulin. Furthermore, rubrofusarin significantly in a dose-dependent manner increased the relative abundances of p-PI3K and p-Akt. Rosiglitazone was used as a reference drug in our experiment. These findings indicate that rubrofusarin increased insulin sensitivity in insulin-resistant HepG2 cells via upregulating the expressions of total IRS-1 and p-IRS-1 of tyrosine residue and activating the PI3K/Akt signaling pathway.
Effect of Rubrofusarin on PEPCK and G6Pase Expression Levels in Insulin-Resistant HepG2 Cells
To investigate the effect of rubrofusarin on the expression of genes of gluconeogenesis, we evaluated the expression levels of two key enzymes of gluconeogenesis, PEPCK, and G6Pase, in insulin-resistant HepG2 cells. The western blot result (Figure 4B) showed that rubrofusarin was able to downregulate the expressions of PEPCK and G6Pase, implying that rubrofusarin mimics the ability of insulin in regulating these enzymes.
Drug-likeness and ADME Prediction
Drug-likeness was predicted for rubrofusarin and its predominant glycoside 2, which suggested that rubrofusarin has good druglike properties, as it followed the MDDR and Lipinski’s rule (Supporting Information Table 1). In the case of 2, though it showed a druglike property according to the MDDR-like rule,30 it did not obey Lipinski’s rule.31 ADME prediction results revealed a good percentage of plasma protein binding (85.78%), a high percentage of human intestinal absorption (HIA) (93.22%), and good lipophilicity with log Po/w values of 2.89. The blood–brain barrier (BBB) penetration value ([brain]/[blood]) of rubrofusarin was 0.64, whereas the value for 2 was too low (0.03). Since human epithelial colorectal adenocarcinoma cells (Caco-2) and Madin–Darby canine kidney cells are commonly used in vitro methods to assess intestinal permeability in early drug discovery, we also predicted the permeabilites of rubrofusarin and 2 in these cell models. Results showed higher permeability for rubrofusarin than 2. All of these predicted results will be helpful for the optimization of druglike properties.
Discussion
These days, for the treatment of T2DM, several approaches to enzyme inhibition, enhanced insulin secretion, and sensitivity along with gluconeogenesis inhibition have been considered.32 By continuous search for natural PTP1B inhibitors from natural resources, about 300 inhibitors have been discovered.33 However, along with concerns about the development of PTP1B inhibitors, high structural conservation and toxicity have become a barrier.34 Most of the natural compounds exist as aglycone and glycoside conjugates simultaneously, and despite the high activity of aglycones, their toxicity at certain concentrations has forced advances in glycoscience.
Glycosylation is a natural route for the development of new therapeutic carriers for site-specific drug delivery because of its reduced toxicity and high absorption cum bioavailability.35 To compare the toxicity of aglycone to that of its glycosides, we performed a cytotoxicity assay in HepG2 cells. Glycosides reduced the cytotoxicity as well as PTP1B enzyme inhibition. Rubrofusarin was nontoxic up to a 50 μM concentration; however, its glycosides showed no toxicity even at 100 μM. Most of the natural glycosides are hydrolyzed in the human intestine by the β-glucosidase enzyme, cleaving the glycosidic bond and resulting aglycone moiety. The role of intracellular-glycosidase is difficult to assess, but we suspect that rubrofusarin glycosides, when taken orally, are deglycosylated/hydrolyzed by intestinal enzymes, such as β-glucosidase, in the small intestine, into rubrofusarin. In C. obtusifolia seeds, rubrofusarin gentiobioside is present abundantly, and it can be provided in various forms, such as foods and drinks, seasonings, alcoholic beverages, functional foods, and medicinal products for oral consumption.36 Being a dominant component in Cassia seeds, rubrofusarin gentiobioside, because of some of its inherent properties, like hydrophilicity, high stability to pH and light, and lack of toxicity at relatively higher concentration, could be useful as a medicinal entity. High hydrophilicity of glycosides makes them satisfactorily soluble in the gastrointestinal juice, which are hydrolyzed into absorbable aglycones. To optimize pharmacokinetic properties in a drug discovery program especially in light of typical constraints on in vivo resources, in vitro screens should be utilized, which could serve as an effective tool to triage and prioritize compounds for in vivo pharmacokinetic studies. Furthermore, characterization of ADME properties and evaluation of drug-likeness are the first steps in drug discovery. Therefore, we predicted the ADME properties and drug-likeness of our active compound, rubrofusarin, and compared it with its glycoside, 2. As in our result, HIA of rubrofusarin was much higher than its glycoside, 2 (93.22 vs 13.23%). Although 2 was predicted to be druglike from the MDDR-like rule, it violated Lipinski’s rule and also the ADME properties were not satisfactory. However, rubrofusarin was predicted to be druglike with improved ADME properties, as shown in Supporting Information Table 1. Similarly, in the PTP1B enzyme assay of this study, glycosides showed marked reduction in activity. Regardless of their reduced activity, we presume they are active in vivo because these glycosides might be metabolized to the active aglycone in the gut/intestine. However, whether the intake of Cassia seed leads to blood or tissue concentration range of IC50 value of our study is yet to be explored. Our ADME prediction result obtained from PreADMET demonstrated good results for rubrofusarin (PPB: 85.78%; BBB: 0.64) unlike 2 (PPB: 34.48%; BBB: 0.03). Recently, a clinical trial conducted to evaluate safety and beneficial effects of Cassia seed supplementation (330 mg × 3 times a day) in healthy adults demonstrated no change in biological safety markers.37 So it is worth performing in vivo studies of Cassia seed extract and determine blood/tissue concentration of rubrofusarin as well as a high but safe dose of its glycosides to compare them with the activity of aglycone.
The pattern of PTP1B and hMAO-A inhibition was quite interesting in our study. Rubrofusarin showed potent effect against these enzymes, but glycosylation reduced the activity. Unlike the reduced activity of glycosides on PTP1B, highly glycosylated compounds 3 and 4 showed reoccurrence of activity on the hMAO-A enzyme. Whether the activities of 3 and 4 are real indeed or it is the result of the masking effect of long sugar chain remains unexplored. Since only rubrofusarin showed potent PTP1B and hMAO-A inhibition, we worked more on understanding the kinetics and molecular docking. The cell signaling mechanism for PTP1B inhibition and glucose uptake in insulin-resistant HepG2 cell was also explored. Enzyme kinetics studies have become an essential component of investigating drug action or characterizing inhibitors. To characterize the mode of PTP1B and hMAO-A enzyme inhibition, the double reciprocal Lineweaver–Burk plots and secondary plots were used. From the kinetic results, rubrofusarin showed the mixed mode of inhibition because increasing the concentration of the compound resulted in a family of lines with different slopes and intercepts, but they intersected one another in the second quadrant. This behavior shows that rubrofusarin can bind not only with the free enzyme but also with the enzyme–substrate complex. In enzymatic reactions and, definitely, for their inhibition, ligand binding is the key step. Hence, understanding the interactions between small molecules and proteins in detail may form a base for rational drug design. A thorough understanding of the mechanisms of enzyme inhibition is of crucial importance in biomedical research.38 Molecular docking has been a good tool to elucidate the mechanistic pathway. To confirm the kinetics result, and to investigate the mechanisms of interaction between the inhibitor and the active sites of PTP1B and hMAO-A, we performed a molecular docking study. Rubrofusarin shared common hydrophobic interacting residues Tyr46 and Ala217 with compound 23 at the catalytic site via π–π stacked and π–alkyl interactions, respectively. Similarly, sharing the same residues with compound 2 through π–π stacked, π–alkyl, and π–σ interactions, rubrofusarin displayed allosteric inhibition of PTP1B. The docking results of the PTP1B–rubrofusarin complex revealed that rubrofusarin was stably positioned in both the catalytic and allosteric domains of the PTP1B residue, with most of the shared interacting residues being the same as in the reference ligands; however, in comparison, the shared number of hydrophobic interacting residues at the allosteric site was greater.
For hMAO-A inhibition, rubrofusarin bound to the enzyme targets via π–alkyl interactions with Ile180 and Ile335, π–π stacking with Tyr407, and π–π T-shaped and van der Waals interactions with FAD at the catalytic site to provide further stability to the complexes. In addition, the modeling predicted π–alkyl interaction with Ala302 and H-bond interaction with Met300 as a principal mechanism for allosteric inhibition. Furthermore, binding affinity toward the catalytic site overweighed the allosteric site as indicated by the binding energy (−9.67 kcal/mol for the catalytic site and −5.60 kcal/mol for the allosteric site), which denotes that the inhibition was mixed but toward the catalytic site of the hMAO-A enzyme.
Inhibition of PTP1B is a recognized therapeutic approach for the management of T2DM and its associated complications. Protein tyrosine phosphatases downregulate the cellular signal transduction mediated by receptor tyrosine kinases, such as the insulin receptor and the epidermal growth factor receptor.39 PTP1B is thought to function as a negative regulator of insulin signal transduction and has been implicated in the negative regulation of IR and IRS-1 within the insulin-stimulated signal-transduction pathway.40,41 Thus, drug entities with PTP1B inhibition have insulin-sensitizing potential and prolong insulin action for the management of T2DM.
Insulin-resistant cells, compared with normal cells, are better in an vitro model to investigate type 2 diabetes because, to use normal cells, many parameters related to glucose metabolism must be altered. In our study, we used insulin-resistant HepG2 to evaluate the glucose uptake potential of rubrofusarin along with its role in the insulin transduction pathway. Increased glucose uptake in insulin-resistant cells treated with samples, as compared with the untreated insulin-resistant group, confirms the improvement of insulin sensitivity. The result of a 2-NBDG assay showed that rubrofusarin could increase the insulin-stimulated glucose uptake significantly in a dose-dependent manner. Similarly, in western blot analysis for PTP1B expression, rubrofusarin at 50 μM concentration significantly reduced the expression.
Binding of insulin to its receptor results in the phosphorylation of IRS-1 at the tyrosine residue and finally activates PI3K. IRS-1 is the first receptor protein that is cloned and plays a vital role in insulin-stimulated cell signaling.42 Activation of PI3K/Akt upon binding insulin to its receptor increases the transportation of glucose from the blood to cells. However, if there is insulin resistance, the phosphorylation of IRS-1 at the tyrosine residue is downregulated, causing blockade of PI3K/Akt signaling. The expression and activity of PTP1B elevate in hyperglycemic liver, which impairs IR/IRS1-mediated insulin signaling by increasing the association of IR with this phosphatase. The result from our western blot analysis showed greater expressions of IRS-1, p-IRS-1 (Tyr 895), p-Akt, and p-PI3K in insulin-resistant HepG2 upon rubrofusarin treatment, leading to the activation of the PI3K/Akt pathway. These modulations in insulin-signaling pathways restored hepatic IR/IRS1-associated PI3K activity in response to insulin and markedly improved insulin sensitivity and glucose uptake. Taken together, these results demonstrate that hyperglycemia impairs insulin signaling via increasing the expression level and activity of PTP1B and that inhibition of PTP1B activity by the specific PTP1B inhibitor rubrofusarin may provide a potential effective therapeutic strategy for rescuing the impaired insulin signaling under diabetic condition.
The rate of gluconeogenesis in type 2 diabetic patients is higher than in nondiabetic ones.43 G6Pase and PEPCK are the two key enzymes responsible for hepatic gluconeogenesis. Therefore, inhibiting the activity of these two enzymes definitely halts or reduces glucose formation in diabetic patients. Insulin is the drug that suppresses hepatic gluconeogenesis.44 When the cells are resistant to insulin, the rate of hepatic gluconeogenesis elevates. Therefore, suppression of hepatic glucose production leads to the management of diabetes. In this regard, we evaluated the effect of rubrofusarin on these two enzymes, and western blot result showed dose-dependent suppression by rubrofusarin of the expressions of G6Pase and PEPCK.
Diabetic people frequently have depressive disorders along with significant levels of depressive symptoms.45 A recent study by Sakimura et al.46 explored the reduced level of serotonin in spontaneously diabetic Torii fatty rat, concluding this animal as an appropriate model for diabetes with comorbid depression associated with impaired neurotransmitter. A number of pieces of evidence suggest a deficit in the serotonergic system as one underlying pathophysiology of depression. In addition, the mechanism of antidepressant action is generally based on MAO-A inhibition and consequently the ability to counter the reduced serotonin level. Even though the American Psychiatric Association and the British Association for Psychopharmacology consider MAO inhibitors as a second choice following serotonin selective reuptake inhibitors,47 discovery of natural dual MAO-A/PTP1B inhibitors will be an asset for drug discovery against comorbid diabetes and depression. Our overall result indicates that rubrofusarin is a dual inhibitor of PTP1B and hMAO-A, stimulates glucose uptake, reduces the expression of hepatic enzymes of gluconeogenesis, decreases PTP1B expression, and activates the insulin-signaling pathway in insulin-resistant HepG2 cells, demonstrating the utmost importance of in vivo studies to determine the blood/tissue concentration of rubrofusarin and a high but safe dose of its glycosides to compare with the activity of aglycone.
In summary, our study extends the knowledge on rubrofusarin showing its potential in inhibiting hMAO-A and PTP1B, promoting glucose uptake in insulin-resistant HepG2 cells, and activating PI3K/Akt through p-IRS-1 tyrosine 895 phosphorylation in insulin-signaling and -suppressing key enzymes of gluconeogenesis in vitro. In addition, glycosylation of rubrofusarin reduces toxicity in HepG2 cells. Most importantly, our findings introduce Cassia seeds as a functional food and rubrofusarin as a new class of phytochemicals with hMAO-A-inhibiting and insulin-sensitizing activity. Therefore, these compounds may have potential in treating insulin resistance and T2DM with comorbid depression. However, further in vivo studies must be conducted for a comparative study of rubrofusarin and its glycosides along with the determination of blood/tissue concentration of rubrofusarin after ingestion of Cassia seed for the validation of our findings.
Materials and Methods
General Experimental Procedures
All of the spectral data including proton (1H-) and carbon (13C-) NMR spectra were measured using dimethyl sulfoxide-d6 (DMSO-d6) or methanol-d3 (MeOH-d3) as the solvent by a JNM ECP-400 spectrometer (JEOL, Tokyo, Japan) at 600 MHz for 1H NMR and 150 MHz for 13C NMR. Chemical shift values were referenced versus appropriate residual solvent peaks (2.50 ppm for 1H NMR and 39.52 ppm for 13C NMR). Silica (Si) gel 60 (70-230 mesh, Merck, Darmstadt, Germany) was used to perform column chromatography. Thin-layer chromatography was employed on precoated Merck Kieselgel 60 F254 plates (20 × 20 cm2, 0.25 mm, Merck, Darmstadt, Germany) sprayed with 50% H2SO4. All solvents used for column chromatography were of reagent grade and were acquired from commercial sources (E. Merck, Fluka and Sigma-Aldrich Co).
Chemicals, Reagents, and Cell Line
Ethylenediaminetetraacetic acid (EDTA), p-nitrophenyl phosphate (pNPP), rosiglitazone, and DMSO were purchased from Sigma-Aldrich Co. (St. Louis, MO). PTP1B (human recombinant) was purchased from Biomol International LP (Plymouth Meeting, PA). Fetal bovine serum (FBS), minimum essential medium (MEM), sodium pyruvate, penicillin–streptomycin, and nonessential amino acids were purchased from Gibco-BRL Life Technologies (Grand Island, NY). The fluorescent d-glucose analogue and glucose tracer 2-[N-(7-nitrobenz-2-oxa-1,3-diazol-4-yl)amino]-2-deoxy-d-glucose (2-NBDG) was purchased from Life Technologies (Carlsbad, CA). Human insulin was purchased from Eli Lilly (Fegersheim, France). Phospho-Akt (Serine 473) (D9E) rabbit monoclonal antibody, p-IRS-1 (Tyr 895), and PEPCK antibody were obtained from Cell Signaling Technology (Danvers, MA). PTP1B (D-4), IRS-1 (C-20) antibody, Akt1 (C-20), PI3-kinase p110 (D-4), p-PI3-kinase p85α (Tyr 508), G6Pase-α (H-4), β-Actin (C4), and all secondary antibodies were obtained from Santa Cruz Biotechnology (Dallas, TX). The human hepatocarcinoma (HepG2) cell line was purchased from the American Type Culture Collection (HB-8065; Manassas, VA). All of the other chemicals and solvents used were of reagent grade and were acquired from commercial sources.
Plant Material
Raw seeds of C. obtusifolia were purchased (Omni Herb Co.) and identified by Prof. J.-H. Lee (Dongguk University, Gyeongju). A voucher specimen (No. 20160302) was registered and deposited in the Prof. J. S. Choi laboratory (Pukyong National University, Busan).
Extraction, Fractionation, and Isolation
The MeOH extract (209.92 g), obtained by refluxing 2.0 kg of dried seeds of C. obtusifolia in MeOH for 3 h (3 × 5 L), was suspended in distilled water and successively partitioned with dichloromethane (CH2Cl2), ethyl acetate (EtOAc), and n-butanol (n-BuOH) to yield CH2Cl2 (33.21 g), EtOAc (14.72 g), and n-BuOH (63.36 g) fractions, respectively, and a final H2O residue (94.28 g). The n-BuOH fraction was subjected to Diaion HP 20 resin column chromatography using H2O/MeOH (100:0 → 0:100 gradient) and finally acetone to obtain five subfractions. The 60% MeOH subfraction (25.77 g) was then subjected to Si gel column chromatography to yield 11 subfractions (M1–M11). Subfraction M6 (1.17 g) was further chromatographed on a Si gel column (3 × 80 cm2) employing EtOAc–MeOH–H2O (30:2:1) as an eluent, and eight subfractions were obtained. Subfraction 8 was further chromatographed on a reverse phase column using 30% acetone as the eluent to yield compound 1 (11.0 mg). In addition, M8, which had the highest yield (9.00 g), was chromatographed over a Si gel column (3 × 80 cm2) using EtOAc–MeOH–H2O (24:3:2) as the eluent to obtain 11 subfractions. Subfraction 10 gave a yellow precipitate that recrystallized into yellow crystals, compound 2 (250 mg). Similarly, M9 (0.78 g) was chromatographed over a Si gel column using EtOAc–MeOH–H2O (24:3:2) as the eluent to obtain 80 subfractions. Subfraction 80, upon purification on a reverse phase column using 50% methanol as the eluent, gave compound 3 (17.0 mg). Finally, subfraction 20 of M11 (1.02 g) obtained using EtOAc–MeOH–H2O (21:5:3) gave a light-green precipitate that, upon washing with methanol, gave compound 4 (14.8 mg). All four compounds isolated were identified spectroscopically, which included the use of 1H- and 13C NMR, by comparing spectral data with published values.26,48−51 The four compounds 1, 2, 3, and 4 were identified as rubrofusarin 6-O-β-d-glucopyranoside, rubrofusarin 6-O-β-d-gentiobioside, rubrofusarin triglucoside, and cassiaside B2, respectively. The EtOAc fraction was subjected to Si gel column chromatography using CH2Cl2–MeOH–H2O (10:1:0.1) to yield 10 subfractions. Subfraction 1 (1.5 g) gave a precipitate in methanol, which was filtered and further purified on a reverse phase column using 80% methanol to give rubrofusarin (8.9 mg), as identified by comparing the spectral data with our previous report.52 The purities of these compounds were considered to be >98% as evidenced by proton and carbon NMR spectra.
Assay for PTP1B and hMAO-A Inhibition
The inhibitory activity of the methanol extract and isolated compounds against PTP1B was measured using pNPP following the procedure reported by Sun et al.34 with slight modifications.53 To each well of a 96-well plate (final volume 100 μL), 40 μL of PTP1B enzyme (0.5 unit diluted with the PTP1B reaction buffer containing 50 mM citrate [pH 6.0], 0.1 M NaCl, 1 mM EDTA, and 1 mM DTT) was added with or without test samples (10 μL). The plate was preincubated at 37 °C for 10 min, and then 50 μL of 2 mM pNPP in the PTP1B reaction buffer was added. After incubation at 37 °C for 20 min, the reaction was terminated by the addition of 10 M NaOH. The amount of p-nitrophenyl produced by enzymatic dephosphorylation of pNPP was estimated by measuring the absorbance at 405 nm using a microplate spectrophotometer (Molecular Devices, Sunnyvale, CA). Ursolic acid was used as a positive control.
Similarly, for hMAO-A inhibition, a chemiluminescent assay was performed in a white opaque 96-well plate using the MAO-Glo kit (Promega, Madison, WI). All of the experimental conditions and procedures followed herein were similar to those reported in our previous paper.54 The percent of inhibition (%) was obtained by the following equation: % inhibition = (Ac – As)/Ac × 100, where Ac is the absorbance of the control and As is the absorbance of the sample.
Kinetic Parameters in PTP1B and hMAO-A Inhibition: Dixon and Lineweaver–Burk Plots
Two kinetic methods, Lineweaver–Burk and Dixon plots, were used to find out the kinetic mechanism.55,56 The reaction mixture consisted of three different concentrations of substrate (pNPP for PTP1B and MAO substrate for hMAO-A) in the presence or absence of test compounds. The Michaelis–Menten constant (Km) and maximum velocity (Vmax) for each enzyme inhibition were measured by Lineweaver–Burk plots, and the inhibition constant (Ki) was calculated from secondary plots using SigmaPlot 12.0 TM software (SPCC, Inc., Chicago, IL).
Molecular Docking
The docking of the target enzyme and rubrofusarin was successfully simulated by using the AutoDock 4.2 program.57,58 X-ray crystallography of the PTP1B-allosteric ligand compound 2 complex (PDB ID: 1T49) and the hMAO A–harmine complex (PDB ID: 2Z5X) was obtained from the RCSB Protein Data Bank (PDB) website (http://www.rcsb.org/) with resolutions of 1.90 and 2.2 Å, respectively.59,60 The 3D structure of rubrofusarin was obtained from PubChem Compound (NCBI), with compound CIDs of 72537. Automated docking simulation was performed using AutoDockTools (ADT) to assess the appropriate binding orientations and conformations of the ligand molecules with different protein inhibitors. For the docking calculations, Gasteiger charges were added by default, the rotatable bonds were set by the ADT, and all torsions were allowed to rotate. The grid maps were generated by the AutoGrid. The docking protocol for rigid and flexible ligand docking consisted of 10 independent genetic algorithms; the other parameters used were the ADT defaults. The reported catalytic inhibitor compound 23 (PDB ID: 1NNY) was used to compare the interaction residues and aspect. The results were visualized and analyzed using Discovery Studio (v17.2, Accelrys, San Diego, CA).
Cell Culture, MTT Assay, and Insulin-Resistance Induction
The HepG2 cells were subcultured in 10% FBS MEM, and the medium was replaced with fresh FBS MEM every 48 h until the plate was confluent enough. The confluent cells were seeded in a 96-well plate, and cytotoxicity was evaluated by the MTT assay.61 Similarly, for the induction of insulin resistance in HepG2 cells, we followed the previously reported method.62 Overall procedures, experimental conditions, and setup have been described in detail in our previous report.63
Glucose Uptake Assay
The glucose uptake potential of rubrofusarin was evaluated fluorimetrically by using fluorescent d-glucose analog, 2-NBDG (Life Technologies, Carlsbad, CA), and a Synergy HT microplate reader (PerkinElmer, MA).53 The experiment was performed in triplicates, and the result was validated using rosiglitazone (10 μM) as a reference drug.
Preparation of Cell Lysates and Western Blot Analysis
In brief, insulin-resistant HepG2 cells were treated with rubrofusarin or rosiglitazone for 24 h and stimulated with 100 nM insulin for 30 min. Cells were then washed with ice-cold PBS, harvested, and lysed with a sample buffer to extract protein. Then, aliquots of protein were resolved by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and transferred onto a nitrocellulose membrane, followed by blocking the membrane with 10% nonfat milk (w/v) in TBST. After that, the membrane was incubated overnight in primary antibodies and in secondary antibody for 1 h and visualized with the Supersignal West Pico chemiluminescence substrate (Pierce, Rockford, IL) on X-ray films (Kodak, Rochester, NY) and quantified using CS analyzer software (Atto Corp., Tokyo, Japan).
Drug-likeness and ADME Prediction
Drug-likeness prediction was carried out using PreADMET. This web-based server is used to calculate absorption, distribution, metabolism, and excretion (ADME) data and build the druglike library using an in silico method.64
Statistical Analysis
A one-way analysis of variance and Student’s t-test (Systat Inc., Evanston, IL) were used to calculate the statistical significance. A p-value <0.05 was considered to be significant. All of the results are presented as the mean ± standard error of the mean.
Acknowledgments
This research was supported by the Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Science, ICT & Future Planning (2015R1D1A1A01057156) and the Ministry of Education (2019R1I1A3A01049380).
Glossary
Abbreviations
- PTP1B
protein tyrosine phosphatase 1B
- hMAO-A
human monoamine oxidase A
- p-Akt
phosphorylated protein kinase B
- p-IRS1
phosphorylated insulin receptor substrate-1
- G6Pase
glucose-6-phosphatase
- PEPCK
phosphoenol pyruvate carboxykinase
- CH2Cl2
dichloromethane
- EtOAc
ethyl acetate
- DS
discovery studio
- IC50
half-maximal inhibitory concentration
- NMR
nuclear magnetic resonance
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsomega.9b01433.
Lineweaver–Burk plots and secondary plots for PTP1B and hMAO-A inhibition kinetics along with drug-likeness and ADME characteristics of rubrofusarin (PDF)
(PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Křen V.Glycoside vs. aglycon: the role of glycosidic residue in biological activity. In Glycoscience; Fraser-Reid B. O.; Kuniaki T., Eds.; Springer: Berlin, 2008; pp 2589–2644. ISBN 978-3-540-30429-6. [Google Scholar]
- Kren V.; Martinkova L. Glycosides in medicine: The role of glycosidic residue in biological activity. Curr. Med. Chem. 2001, 8, 1303–1328. 10.2174/0929867013372193. [DOI] [PubMed] [Google Scholar]
- Huang G.; Lv M.; Hu J.; Huang K.; Xu H. Glycosylation and activities of natural products. Mini-Rev. Med. Chem. 2016, 16, 1013–1016. 10.2174/138955751612160727164559. [DOI] [PubMed] [Google Scholar]
- Weymouth-Wilson A. C. The role of carbohydrates in biologically active natural products. Nat. Prod. Rep. 1997, 14, 99–110. 10.1039/np9971400099. [DOI] [PubMed] [Google Scholar]
- Li Y.; Sun S.; Chang Q.; Zhang L.; Wang G.; Chen W.; Miao X.; Zheng Y. A strategy for the improvement of the bioavailability and antiosteoporosis activity of BCS IV flavonoid glycosides through the formulation of their lipophilic aglycone into nanocrystals. Mol. Pharm. 2013, 10, 2534–2542. 10.1021/mp300688t. [DOI] [PubMed] [Google Scholar]
- Xin H.; Zhou F.; Liu T.; Li G.-Y.; Liu J.; Gao Z.-Z.; Bai G.-Y.; Lu H.; Xin Z.-C. Icariin ameliorates streptozotocin-induced diabetic retinopathy in vitro and in vivo. Int. J. Mol. Sci. 2012, 13, 866–878. 10.3390/ijms13010866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D. H.; Jung H. A.; Sohn H. S.; Kim J. W.; Choi J. S. Potential of icariin metabolites from Epimedium koreanum nakai as antidiabetic therapeutic agents. Molecules 2017, 22, 986 10.3390/molecules22060986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu H.; Kim M.; Han J. Icariin metabolism by human intestinal microflora. Molecules 2016, 21, 1158 10.3390/molecules21091158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung H. A.; Paudel P.; Seong S. H.; Min B. S.; Choi J. S. Structure-related protein tyrosine phosphatase 1B inhibition by naringenin derivatives. Bioorg. Med. Chem. Lett. 2017, 27, 2274–2280. 10.1016/j.bmcl.2017.04.054. [DOI] [PubMed] [Google Scholar]
- Williamson G.; Plumb G. W.; Uda Y.; Price K. R.; Rhodes M. J. Dietary quercetin glycosides: antioxidant activity and induction of the anticarcinogenic phase II marker enzyme quinone reductase in Hepalclc7 cells. Carcinogenesis 1996, 17, 2385–2387. 10.1093/carcin/17.11.2385. [DOI] [PubMed] [Google Scholar]
- Wilcox G. Insulin and insulin resistance. Clin. Biochem. Rev. 2005, 26, 19–39. [PMC free article] [PubMed] [Google Scholar]
- Klaman L. D.; Boss O.; Peroni O. D.; Kim J. K.; Martino J. L.; Zabolotny J. M.; Moghal N.; Lubkin M.; Kim Y.-B.; Sharpe A. H. Increased energy expenditure, decreased adiposity, and tissue-specific insulin sensitivity in protein-tyrosine phosphatase 1B-deficient mice. Mol. Cell. Biol. 2000, 20, 5479–5489. 10.1128/MCB.20.15.5479-5489.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pessin J. E.; Saltiel A. R. Signaling pathways in insulin action: molecular targets of insulin resistance. J. Clin. Invest. 2000, 106, 165–169. 10.1172/JCI10582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yabaluri N.; Bashyam M. D. Hormonal regulation of gluconeogenic gene transcription in the liver. J. Biosci. 2010, 35, 473–484. 10.1007/s12038-010-0052-0. [DOI] [PubMed] [Google Scholar]
- Youdim M. B. H.; Edmondson D.; Tipton K. F. The therapeutic potential of monoamine oxidase inhibitors. Nat. Rev. Neurosci. 2006, 7, 295–309. 10.1038/nrn1883. [DOI] [PubMed] [Google Scholar]
- Schmitz N.; Deschênes S. S.; Burns R. J.; Smith K. J.; Lesage A.; Strychar I.; Rabasa-Lhoret R.; Freitas C.; Graham E.; Awadalla P.; Wang J. L. Depression and risk of type 2 diabetes: the potential role of metabolic factors. Mol. Psychiatry 2016, 21, 1726–1732. 10.1038/mp.2016.7. [DOI] [PubMed] [Google Scholar]
- Mezuk B.; Eaton W. W.; Albrecht S.; Golden S. H. Depression and type 2 diabetes over the lifespan: a meta-analysis. Diabetes Care 2008, 31, 2383–2390. 10.2337/dc08-0985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bădescu S. V.; Tătaru C.; Kobylinska L.; Georgescu E. L.; Zahiu D. M.; Zăgrean A. M.; Zăgrean L. The association between Diabetes mellitus and depression. J. Med. Life 2016, 9, 120–125. [PMC free article] [PubMed] [Google Scholar]
- Pan A.; Lucas M.; Sun Q.; van Dam R. M.; Franco O. H.; Manson J. E.; Willett W. C.; Ascherio A.; Hu F. B. Bidirectional association between depression and type 2 diabetes in women. Arch. Intern. Med. 2010, 170, 1884–1891. 10.1001/archinternmed.2010.356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong S. M.; Wong M. M.; Seligmann O.; Wagner H. New antihepatotoxic naphtho-pyrone glycosides from the seeds of Cassia tora. Planta Med. 1989, 55, 276–280. 10.1055/s-2006-962003. [DOI] [PubMed] [Google Scholar]
- Park Y. B.; Kim S. B. Isolation and identification of antitumor promoters from the seeds of Cassia tora. J. Microbiol. Biotechnol. 2011, 21, 1043–1048. 10.4014/jmb.1103.03040. [DOI] [PubMed] [Google Scholar]
- Mondal A. Phenolic constituents and traditional uses of Cassia (Fabaceae) plants: An update. J. Org. Biomol. Chem. 2014, 3, 93–141. [Google Scholar]
- Kumar V.; Singh R.; Mahdi F.; Mahdi A. A.; Singh R. K. Experimental validation of antidiabetic and antioxidant potential of Cassia tora (L.): An indigenous medicinal plant. Indian J. Clin. Biochem. 2017, 32, 323–328. 10.1007/s12291-016-0608-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nam J.; Choi H. Effect of butanol fraction from Cassia tora L. seeds on glycemic control and insulin secretion in diabetic rats. Nutr. Res. Pract. 2008, 2, 240–246. 10.4162/nrp.2008.2.4.240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung H. A.; Ali M. Y.; Jung H. J.; Jeong H. O.; Chung H. Y.; Choi J. S. Inhibitory activities of major anthraquinones and other constituents from Cassia obtusifolia against β-secretase and cholinesterases. J. Ethnopharmacol. 2016, 191, 152–160. 10.1016/j.jep.2016.06.037. [DOI] [PubMed] [Google Scholar]
- Shrestha S.; Seong S. H.; Paudel P.; Jung H. A.; Choi J. S. Structure related inhibition of enzyme systems in cholinesterases and BACE1 in vitro by naturally occurring naphthopyrone and its glycosides isolated from Cassia obtusifolia. Molecules 2018, 23, 69. 10.3390/molecules23010069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung H. A.; Ali M. Y.; Choi J. S. Promising inhibitory effects of anthraquinones, naphthopyrone, and naphthalene glycosides, from Cassia obtusifolia on α-glucosidase and human protein tyrosine phosphatases 1B. Molecules 2017, 22, 28. 10.3390/molecules22010028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paudel P.; Jung H. A.; Choi J. S. Anthraquinone and naphthopyrone glycosides from Cassia obtusifolia seeds mediate hepatoprotection via Nrf2-mediated HO-1 activation and MAPK modulation. Arch. Pharm. Res. 2018, 41, 677–689. 10.1007/s12272-018-1040-4. [DOI] [PubMed] [Google Scholar]
- Ali M. Y.; Jannat S.; Jung H. A.; Min B. S.; Paudel P.; Choi J. S. Hepatoprotective effect of Cassia obtusifolia seed extract and constituents against oxidative damage induced by tert-butyl hydroperoxide in human hepatic HepG2 cells. J. Food Biochem. 2018, 42, e12439 10.1111/jfbc.12439. [DOI] [Google Scholar]
- Oprea T. I. Property distribution of drug-related chemical databases. J. Comput.-Aided Mol. Des. 2000, 14, 251–264. 10.1023/A:1008130001697. [DOI] [PubMed] [Google Scholar]
- Lipinski C. A.; Lombardo F.; Dominy B. W.; Feeney P. J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Delivery Rev. 1997, 23, 3–25. 10.1016/S0169-409X(96)00423-1. [DOI] [PubMed] [Google Scholar]
- Bloomgarden Z. T. Approaches to treatment of type 2 diabetes. Diabetes Care 2008, 31, 1697–1703. 10.2337/dc08-zb08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao B. T.; Nguyen D. H.; Le D. D.; Choi J. S.; Min B. S.; Woo M. H. Protein tyrosine phosphatase 1B inhibitors from natural sources. Arch. Pharm. Res. 2018, 41, 130–161. 10.1007/s12272-017-0997-8. [DOI] [PubMed] [Google Scholar]
- Sun J.; Wang Y.; Fu X.; Chen Y.; Wang D.; Li W.; Xing S.; Li G. Magnolia officinalis extract contains potent inhibitors against PTP1B and attenuates hyperglycemia in db/db mice. Biomed. Res. Int. 2015, 2015, 139451 10.1155/2015/139451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jain K.; Kesharwani P.; Gupta U.; Jain N. K. A review of glycosylated carriers for drug delivery. Biomaterials 2012, 33, 4166–4186. 10.1016/j.biomaterials.2012.02.033. [DOI] [PubMed] [Google Scholar]
- Asami S.Rubrofusarin glycoside-containing composition. U.S. Patent Application No. US10/516,288, 2008.
- Kambalachenu H. R.; Reddy T. M.; Rao S. D.; Dorababu K.; Reddy K. K.; Sarma K. V. S. A randomized, double blind, placebo controlled, crossover study to assess the safety and beneficial effects of Cassia tora supplementation in healthy adults. Rev. Recent Clin. Trials 2018, 13, 69–78. 10.2174/1574887112666171120094539. [DOI] [PubMed] [Google Scholar]
- de Ruyck J.; Brysbaert G.; Blossey R.; Lensink M. F. Molecular docking as a popular tool in drug design, an in silico travel. Adv. Appl. Bioinform. Chem. 2016, 9, 1–11. 10.2147/AABC.S105289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burke T. R. Jr.; Zhang Z. Y. Protein-tyrosine phosphatases: structure, mechanism, and inhibitor discovery. Biopolymers 1998, 47, 225–241. . [DOI] [PubMed] [Google Scholar]
- Boucher J.; Kleinridders A.; Kahn C. R. Insulin receptor signaling in normal and insulin-resistant states. Cold Spring Harbor Perspect. Biol. 2014, 6, a009191 10.1101/cshperspect.a009191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao B. T.; Le D. D.; Nguyen P. H.; Ali M. Y.; Choi J. S.; Min B. S.; Shin H. M.; Rhee H. I.; Woo M. H. PTP1B, α-glucosidase, and DPP-IV inhibitory effects for chromene derivatives from the leaves of Smilax china L. Chem.-Biol. Interact. 2016, 253, 27–37. 10.1016/j.cbi.2016.04.012. [DOI] [PubMed] [Google Scholar]
- Hu X.; Wang S.; Xu J.; Wang D.-B.; Chen Y.; Yang G.-Z. Triterpenoid saponins from Stauntonia chinensis ameliorate insulin resistance via the AMP-activated protein kinase and IR/IRS-1/PI3K/Akt pathways in insulin-resistant HepG2 cells. Int. J. Mol. Sci. 2014, 15, 10446–10458. 10.3390/ijms150610446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boden G.; Chen X.; Stein T. P. Gluconeogenesis in moderately and severely hyperglycemic patients with type 2 diabetes mellitus. Am. J. Physiol. Endocrinol. Metab. 2001, 280, E23–E30. 10.1152/ajpendo.2001.280.1.E23. [DOI] [PubMed] [Google Scholar]
- Puigserver P.; Rhee J.; Donovan J.; Walkey C. J.; et al. Insulin-regulated hepatic gluconeogenesis through FOXO1-PGC-1α interaction. Nature 2003, 423, 550. 10.1038/nature01667. [DOI] [PubMed] [Google Scholar]
- Lloyd C.; Nouwen A.; Sartorius N.; Ahmed H.; Alvarez A.; Bahendeka S.; Basangwa D.; Bobrov A.; Boden S.; Bulgari V.; et al. Prevalence and correlates of depressive disorders in people with Ttype 2 diabetes: results from the International Prevalence and Treatment of Diabetes and Depression (INTERPRET-DD) study, a collaborative study carried out in 14 countries. Diabetic Med. 2018, 35, 760–769. 10.1111/dme.13611. [DOI] [PubMed] [Google Scholar]
- Sakimura K.; Maekawa T.; Sasagawa K.; Ishii Y.; Kume S.; Ohta T. Depression-related behavioural and neuroendocrine changes in the spontaneously diabetic torii (SDT) fatty rat, an animal model of type 2 diabetes mellitus. Clin. Exp. Pharmacol. Physiol. 2018, 45, 927–933. 10.1111/1440-1681.12965. [DOI] [PubMed] [Google Scholar]
- Bortolato M.; Chen K.; Shih J. C. Monoamine oxidase inactivation: from pathophysiology to therapeutics. Adv. Drug Delivery Rev. 2008, 60, 1527–1533. 10.1016/j.addr.2008.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatano T.; Uebayashi H.; Ito H.; Shiota S.; Tsuchiya T.; Yoshida T. Phenolic constituents of Cassia seeds and antibacterial effect of some naphthalenes and anthraquinones on methicillin-resistant Staphylococcus aureus. Chem. Pharm. Bull. 1999, 47, 1121–1127. 10.1248/cpb.47.1121. [DOI] [PubMed] [Google Scholar]
- Choi J. S.; Lee H. J.; Kang S. S. Alaternin, cassiaside and rubrofusarin gentiobioside, radical scavenging principles from the seeds of Cassia tora on 1,1-diphenyl-2-picrylhydrazyl (DPPH) radical. Arch. Pharm. Res. 1994, 17, 462–466. 10.1007/BF02979126. [DOI] [PubMed] [Google Scholar]
- Jing Y.; Yang J.; Wu L.; Zhang Z.; Fang L. Rubrofusarin glucosides of Berchemia polyphylla var. leioclada and their scavenging activities for DPPH radical. Zhongguo Zhong Yao Za Zhi 2011, 36, 2084–2087. [PubMed] [Google Scholar]
- Shrestha S.; Paudel P.; Seong S. H.; Min B. S.; Seo E. K.; Jung H. A.; Choi J. S. Two new naphthalenic lactone glycosides from Cassia obtusifolia L. seeds. Arch. Pharm. Res. 2018, 41, 737–742. 10.1007/s12272-018-1044-0. [DOI] [PubMed] [Google Scholar]
- Lee H. J.; Park J. C.; Choi J. S. The 13C-NMR assignment of nor-rubrofusarin having strong radical scavenging effect on 1,1-diphenyl-2picrylhydrazyl radical. Nat. Prod. Sci. 1998, 4, 95–99. [Google Scholar]
- Paudel P.; Yu T.; Seong S. H.; Kuk E. B.; Jung H. A.; Choi J. S. Protein tyrosine phosphatase 1B inhibition and glucose uptake potentials of mulberrofuran G, albanol B, and kuwanon G from root bark of Morus alba L. in insulin-resistant HepG2 cells: An in vitro and in silico study. Int. J. Mol. Sci. 2018, 19, 1542 10.3390/ijms19051542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung H. A.; Roy A.; Choi J. S. In vitro monoamine oxidase A and B inhibitory activity and molecular docking simulations of fucoxanthin. Fish. Sci. 2017, 83, 123–132. 10.1007/s12562-016-1036-2. [DOI] [Google Scholar]
- Cornish-Bowden A. A simple graphical method for determining the inhibition constants of mixed, uncompetitive and non-competitive inhibitors. Biochem. J. 1974, 137, 143–144. 10.1042/bj1370143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lineweaver H.; Burk D. The determination of enzyme dissociation constants. J. Am. Chem. Soc. 1934, 56, 658–666. 10.1021/ja01318a036. [DOI] [Google Scholar]
- Goodsell D. S.; Morris G. M.; Olson A. J. Automated docking of flexible ligands: applications of AutoDock. J. Mol. Recognit. 1996, 9, 1–5. . [DOI] [PubMed] [Google Scholar]
- Li D.; Chi B.; Wang W.-W.; Gao J.-M.; Wan J. Exploring the possible binding mode of trisubstituted benzimidazoles analogues in silico for novel drug designtargeting Mtb FtsZ. Med. Chem. Res. 2017, 26, 153–169. 10.1007/s00044-016-1734-4. [DOI] [Google Scholar]
- Wiesmann C.; Barr K. J.; Kung J.; Zhu J.; Erlanson D. A.; Shen W.; Fahr B. J.; Zhong M.; Taylor L.; Randal M.; et al. Allosteric inhibition of protein tyrosine phosphatase 1B. Nat. Struct. Mol. Biol. 2004, 11, 730–737. 10.1038/nsmb803. [DOI] [PubMed] [Google Scholar]
- Son S.-Y.; Ma J.; Kondou Y.; Yoshimura M.; Yamashita E.; Tsukihara T. Structure of human monoamine oxidase A at 2.2-Å resolution: the control of opening the entry for substrates/inhibitors. Proc. Natl. Acad. Sci. U.S.A. 2008, 105, 5739–5744. 10.1073/pnas.0710626105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mosmann T. Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays. J. Immunol. Methods 1983, 65, 55–63. 10.1016/0022-1759(83)90303-4. [DOI] [PubMed] [Google Scholar]
- Liu Z. Q.; Liu T.; Chen C.; Li M. Y.; Wang Z. Y.; Chen R. S.; Wei G. X.; Wang X. Y.; Luo D. Q. Fumosorinone, a novel PTP1B inhibitor, activates insulin signaling in insulin-resistance HepG2 cells and shows anti-diabetic effect in diabetic KKAy mice. Toxicol. Appl. Pharmacol. 2015, 285, 61–70. 10.1016/j.taap.2015.03.011. [DOI] [PubMed] [Google Scholar]
- Jung H. A.; Ali M. Y.; Bhakta H. K.; Min B. S.; Choi J. S. Prunin is a highly potent flavonoid from Prunus davidiana stems that inhibits protein tyrosine phosphatase 1B and stimulates glucose uptake in insulin-resistant HepG2 cells. Arch. Pharm. Res. 2017, 40, 37–48. 10.1007/s12272-016-0852-3. [DOI] [PubMed] [Google Scholar]
- Kumar A.; Srivastava G.; Srivastava S.; Verma S.; Negi A. S.; Sharma A. Investigation of naphthofuran moiety as potential dual inhibitor against BACE-1 and GSK-3β: molecular dynamics simulations, binding energy, and network analysis to identify first-in-class dual inhibitors against Alzheimer’s disease. J. Mol. Model. 2017, 23, 239. 10.1007/s00894-017-3396-7. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.