

SUMMARY
Direct cellular reprogramming provides a powerful platform to study cell plasticity and dissect mechanisms underlying cell fate determination. Here we report a single cell transcriptomic study of human cardiac (hiCM) reprogramming that utilizes an analysis pipeline incorporating current data normalization methods, multiple trajectory prediction algorithms, and a Cell Fate Index calculation we developed to measure reprogramming progression. These analyses revealed hiCM reprogramming-specific features and a decision point at which cells either embark on reprogramming or regress toward their original fibroblast state. In combination with functional screening, we found immune response-associated DNA methylation is required for hiCM induction and validated several downstream targets of reprogramming factors as necessary for productive hiCM reprograming. Collectively this single cell transcriptomics study provides detailed datasets that reveal molecular features underlying hiCM determination and rigorous analytical pipelines for predicting cell fate conversion.
In brief:
Zhou et al. performed single-cell RNA-seq to unravel molecular features of human cardiac reprogramming. They identified a “decision” point where cells either reprogram or regress to initial fate. Further, progression of reprogramming was quantitatively assessed by their developed “cell fate index”, which could be used for studying other biological processes.
Graphical Abstract
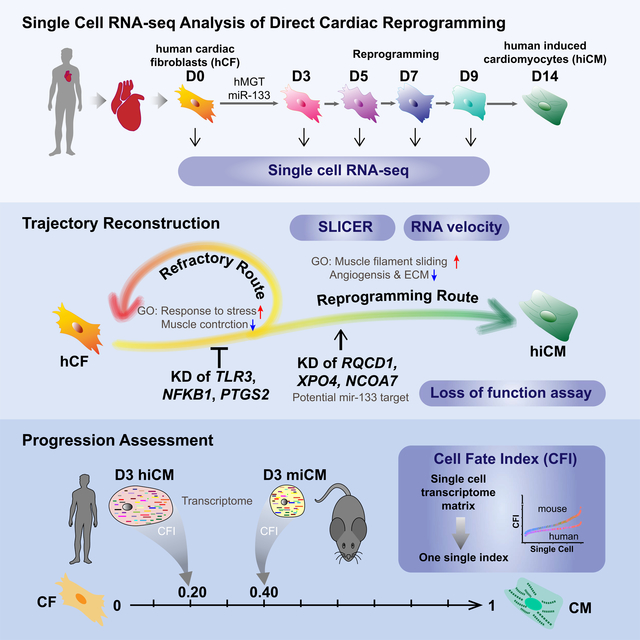
INTRODUCTION
Cardiac reprogramming that converts cardiac fibroblasts into induced cardiomyocytes (iCMs) has emerged as a promising avenue to regenerate damaged hearts (Ieda et al., 2010; Qian et al., 2012; Song et al., 2012). Despite many recent advances in mouse iCM (miCM) reprogramming (see reviews Kojima and Ieda, 2017; Vaseghi et al., 2017), human iCM (hiCM) reprogramming remains a great challenge. hiCM reprogramming could only be achieved with more complex cocktails, suffers from lower efficiency and requires longer reprogramming time (Christoforou et al., 2017; Fu et al., 2013; Mohamed et al., 2016; Nam et al., 2013; Wada et al., 2013). The difficulties in hiCM generation suggest species differences in the gene regulatory networks in controlling CM fate and fibroblast plasticity, understanding of which might be the key for next step clinical application.
Single-cell RNA sequencing (scRNA-seq) has become a powerful technology to dissect cellular dynamics, uncover differential cellular responses to stimuli and delineate molecular state transitions in biological processes (Lein et al., 2017; Stubbington et al., 2017). scRNA-seq is particularly well-suited to circumvent the hurdles imposed by the heterogeneous and asynchronous nature of cellular reprogramming (Biddy et al., 2018; Liu et al., 2017b; Treutlein et al., 2016).
In this study, we performed scRNA-seq to study the molecular and cellular dynamics of hiCM reprogramming. Consistent with a “decision point” uncovered by RNA velocity (La Manno et al., 2018), SLICER based trajectory reconstruction (Welch et al., 2016) identified a bifurcation event that led to acquisition of hiCM fate or regression toward fibroblast fate, with each of the fate choice being associated with distinct hiCM specific biological pathways and signaling molecules. Combining single cell analyses with functional studies, we further demonstrated that immune response associated DNA methylation is involved in hiCM induction and silencing several previously uncharacterized downstream targets of miR-133 could replace its indispensable function in hiCM induction. Additionally, we developed Cell Fate Index (CFI) algorithm to quantitatively assess reprogramming progression, and found a slower progression of hiCM reprogramming than miCM reprogramming. Collectively, our scRNA-seq study provides valuable resources to delineate gene networks underlying hiCM reprogramming at a much higher resolution. We also envision that the CFI algorithm can be applied to quantitatively compute progression of cell fate transition for other biological processes.
RESULTS
Optimized platform for hiCM reprogramming
Our previous study demonstrated that the miCM factors, when delivered as a single polycistronic transgene in the splicing order of Mef2c, Gata4 and Tbx5, drastically enhanced both the quality and the quantity of miCM induction (Wang et al., 2015a). To achieve efficient and reproducible hiCM reprogramming, we generated human polycistronic MEF2C, GATA4 and TBX5 (hMGT for short), and transduced human fibroblasts with hMGT in combination with other factors from previously published cocktails (Fu et al., 2013; Nam et al., 2013; Wada et al., 2013). hMGT plus microRNA miR-133 (hereafter referred to as hMGT133) was found to be the most efficient minimal hiCM reprogramming cocktail (Figures 1A and 1B). We then further optimized hiCM reprogramming by using SureCoat as coating substrate, performing puromycin selection at early stage and culturing the cells in low serum hiCM media at late stage of reprogramming (Figures 1C, S1A and S1B). Under the optimized condition, approximately 40% and 60% of the reprogramming human cardiac fibroblasts (hCF1 see Methods) were cTnT+ and Į-Actinin+ at 2 weeks post infection, respectively (Figures 1D, 1E and S1C). Furthermore, these hiCMs established CM molecular signature, exhibited periodic Ca2+ oscillations (Figures 1F–1H, Movie S1), and initiated contraction after co-culture with mouse neonatal CMs (Movie S2). Similar reprogramming efficiency was achieved when H9Fs (Fu et al., 2013) and another previously established human primary cardiac fibroblast resource (hCF2, see Methods) were used for reprogramming (Figures 1I–1L). For consistent and reproducible hiCM induction, we used this optimized hMGT133 reprogramming platform for the entire study.
Figure 1. Optimizing hiCM reprogramming platform.

(A, B) Representative flow plots (A) and quantification (B) for cTnT+ cells 2-week post transduction of the indicated cocktails into H9Fs.
(C) Schematic of our optimized hiCM reprogramming platform.
(D) Flow analysis for cTnT+ cells in hMGT133 infected hCF1 at day 14.
(E) Representative ICC images and quantification for expression of cTnT and a-actinin 14 days after transduction of hMGT133 in hCF1. Scale bars, 100 μm.
(F) Heatmap of the relative expression of a set of CM and CF marker genes determined by RT-qPCR in hCF1 at different time points as indicated after hMGT133 infection.
(G, H) Representative images (G) and quantification (H) of hiCMs exhibiting calcium oscillation indicated by Calcium sensor GCamp5. Scale bar, 200 μm. RFUs, relative fluorescence units. (I, J) Representative flow plots (I) and quantification (J) for cTnT+ hiCMs from H9F and hCF2 at reprogramming day 14.
(K, L) ICC images with quantification for hiCMs expressing cTnT, a-Actinin and Cx43 derived from H9F (K) and hCF2 (L). Scale bars, 100 μm.
Error bars indicate mean ± SEM, N=3 in (B, D, J), N=20 in (K, L). See also Figure S1.
Single-cell RNA-seq of day 3 reprogramming hiCM
scRNA-seq study of miCM demonstrated that day 3 reprogramming culture comprises a wide range of cells transitioning from fibroblast to iCM (Liu et al., 2017b), we thus performed pilot scRNA-seq on day 3 hMGT133-transduced hCFs (d3hMGT) and three controls, namely, primary hCFs (d0hCF), day 3 uninfected hCFs (d3hU) and day 3 hCFs infected with DsRed plus empty vector (d3hR). We captured single cells with Fluidigm microfluidic system and generated single-cell full-length transcriptomes using SMART-Seq as we previously reported (Figure 2A) (Liu et al., 2017b). A series of quality controls were performed to exclude low-quality cells and non-expressing genes (Figures S1D and S1E, Methods). To estimate the sensitivity and accuracy of our scRNA-seq, we plotted the expression levels of external RNA spike-ins measured by RNA-seq against their theoretical numbers of molecules/chamber (bottom panels of Figure S1F, Table S1). This analysis revealed sequencing sensitivity at single-molecule level, and a robust correlation between the RNA-seq measured expression level and the theoretical molecule number of spike-ins across the dynamic range (~105) of ERCC concentration. Additionally, we found that the concentration of spike-ins covers the full range of human gene expression levels (top histograms of Figure S1F), indicating high fidelity of our scRNA-seq data across a wide range of human gene expression levels. We then applied SCnorm, a newly developed scRNA-seq normalization method (Bacher et al., 2017), to adjust for sequencing depth and other sample specific features. This normalization approach allows for appropriate comparison of a gene’s expression across samples (Figures S1G–S1H and Methods), and as indicated by the principal component analysis (PCA), the cells were better distributed on the 3D PCA plots generated post SCnorm than post DESeq (Figures S1I and S1J). After normalization, a total of 13381 human genes were detected (normalized expression > 1 counts per million, CPM) in 329 high-quality control and d3 reprogramming cells for subsequent analysis.
Figure 2. scRNA-seq on day 3 reprogramming hCFs revealed the cell cycle status during hiCM induction.

(A) Schematic of experimental design for scRNA-seq (see details in Methods).
(B, C) 3D PCA score plot (B) and 2D loading plot (C) calculated with the top 400 genes in all single cells.
(D) Heatmap showing identification of a cell-cycle related gene cluster by HC of all single cells with top 400 PCA genes. CCA, cell-cycle active. CCI, cell-cycle inactive. Rep Genes, representative genes.
(E) PCA of all single cells color-coded by CCA/CCI state.
(F) Each dot represents a cell and the cell cycle status of each cell was determined based on its mean score calculated by reCAT.
(G) PCA of all single cells color-coded by cell-cycle states from (F).
(H) Violin plot showing the distribution of expression of cyclin and cyclin-dependent kinases in d0hCF cells and the rest single cells. Box plots were overlaid.
(I-K) HC and PCA of day 3 control and reprogramming cells were calculated with top 400 PCA genes. (I) Heatmap showing HC results and relevant GO terms. Pos., positive; reg., regulation; neg., negative; rep., replication. PCA score (J) and loading (K) plots showing the five cell groups identified by HC.
(L) Comparison of the CCA:CCI ratio in hiFib and hiCMs.
(M) Cell number count of hMGT133 or LacZ-EV infected hCF along first 3 days after infection. Error bars indicate mean ± SEM, N= 3 in (M), *p < 0.05. See also Figures S1, S2 and Table S1.
To obtain a general picture of the relationship among single cells, we performed unbiased dimensionality reduction using PCA with the top 400 genes showing highest loading values (weight) in the first three principal components (PC) and visualized the sample relationships by 3D PCA score plot (Figures 2B and S1K). The 3D PCA plot resembled a tripod shape with d3hU localized in the center and three ramifying branches that were mainly composed of d0hCF, d3hR, and d3hMGT cells, possibly representing three biological processes (Figures 2B and S1L). Indeed, we found that the PCA loadings (genes) in the direction of d0hCF cells were highly associated with cell cycle regulation, while immune response genes were enriched in the direction of d3hR cells and cardiac structural genes were enriched in the direction of d3hMGT cells (Figure 2C; also see below for more details).
Analyses of cell cycle status in reprogramming hiCM
Hierarchical clustering (HC) analysis of the top 400 PCA genes revealed that the largest identified gene cluster was enriched in cell cycle-related genes (Figure 2D). Based on this result, we grouped the cells into cell cycle-active (CCA) and cell-cycle-inactive (CCI) populations. A similar segregation of CCA and CCI populations was evident in PCA plot (Figure 2E), which also demonstrated that most CCA cells were from the d0hCF samples. We then applied reCAT (Liu et al., 2017c), a recently developed algorithm to recover cell cycle along time for unsynchronized single-cell transcriptome data, to determine cell cycle status of individual cell. reCAT analysis identified distinct G2/M and G0/G1/S populations that exhibited similar distributions on the PCA plot as the CCA and CCI populations, respectively (Figures 2F and S2A, Figure 2G compared to Figure 2E). This observation was further supported by the difference in the expression of representative cyclin and cyclin dependent kinase genes in individual cell between d0hCF and the rest cell populations (Figure 2H), suggesting human cardiac fibroblasts lost their highly proliferative capacity along in vitro reprogramming.
We next performed unsupervised HC and PCA analyses only on the day 3 reprogramming and control samples (Figures 2I–2K, S2B and S2C). These analyses identified five major cell populations: uninfected hCFs (hU), control hCFs expressing low/high level of DsRed (hRlow/hRhigh), hMGT133-infected hCFs that retained fibroblast features (hiFib), and hMGT133-transduced hCFs that gained CM gene signatures (hiCM) (Figures 2I, S2B and S2C). On the PCA plots, the hRlow/hRhigh cells and hiFib/hiCM were distributed along two bifurcating branches ramifying from uninfected hCFs (Figures 2J and 2K). Interestingly, while about 8% of hiFib cells also belonged to the proliferative CCA group, none of hiCMs were actively proliferating (Figure 2L), suggesting that cell-cycle exit might be essential for successful hiCM reprogramming. Indeed, upon hMGT133 transduction, expression of the cell proliferation marker Ki67 was dramatically reduced (Figure S2D). Cell growth curve further demonstrated that transduction of hMGT133 suppressed hCF proliferation compared with control treatment (Figure 2M). These observations are consistent with previous findings that cell cycle exit is prerequisite for successful fate conversion of murine iCM (Bektik et al., 2018; Liu et al., 2017b; Zhou et al., 2016).
The role of immune response in hiCM fate transition
We observed an enrichment of immune response genes in d3hR control samples (Figures 2B and 2C). We speculated that this upregulation was simply an antiviral response. Analyses of single cell data from day 3 control hCFs allowed us to identify three cell populations: uninfected hCFs (hU), hCFs with low expression of DsRed (hRlow) and hCFs with high expression level of DsRed (hRhigh). The three populations were associated with highly expressed genes representing fibroblast markers, antiviral response and immune response, respectively (Figures 3A–3C and S3A–S3C). Since the cell population grouping was significantly correlated with the expression level of DsRed (Figure 3D), we checked whether the upregulation of immune genes was due to DsRed viruses per se or a high titer of viruses. We determined the expression of representative immune response genes in both hCFs infected with DsRed at a gradient of dosages and with viruses expressing LacZ, GFP, or Td-Tomato (Figure S3D). Adjusting the DsRed viral titer to the same level as other types of viruses diminished the activation of immune genes (Figure 3E), while transduction of overdosed LacZ viruses produced similar activation of immune response genes as from DsRed-infected cells (Figure 3F). Similar changes of the same immune genes were also observed in virus infected H9F cells, excluding the possibility of cell type specific effects (Figure S3E). While substantial increase in the expression of immune-related genes was observed in DsRed control cells, hMGT133 cells demonstrated a moderate yet significant upregulation of the immune response genes, including factors in the interferon (IFN) signaling pathway (Figures S3F and S3G). It has been reported that the viral induced innate immunity is required for efficient reprogramming of induced pluripotent stem cell (iPSC) (Lee et al., 2012; Sayed et al., 2017). Our data showed similar activation of innate immune response, however it is unknown whether this activation is required for hiCM reprogramming.
Figure 3. Activation of innate immunity is required for hiCM generation.

(A-C) HC and PCA of d3hU and d3hR cells were calculated with top 400 PCA genes. (A) Heatmap showing HC-identified clusters (left) and GO terms (right). Neg., negative; reg., regulation. PCA score (B) and loading (C) plot showing the three cell groups identified by HC. (D) Expression levels of DsRed in the three cell groups. *, p < 0.05; ***, p < 0.001.
(E-F) Heatmap of the relative expression of immune-related genes determined by RT-qPCR in hCF 3 days after infection of hMGT133 together with indicated control viruses. Less amount of DsRed led to reduced immune response (E). Overdose of LacZ showed over activation of immune genes (F).
(G-I) Flow cytometry (G), immunostaining images (H) and quantification (I) of cTnT+ cells 14 days post-coinfection of hMGT133 and indicated shRNAs. EV, empty vector; shTubo,control. #1 and #2 are two individual shRNA oligos of each gene.
(J) RT-qPCR of marker gene expression at day 14 after knockdown of the indicated genes in hiCMs. Fibr, fibroblast genes.
(K) Histogram of normalized Z score of % cTnT+ cells after infection of shRNA lentiviruses targeting epigenetic regulators in TLR3 depleted hiCMs.
(L) Flow cytometry for cTnT+ hiCMs in TLR3 knockdown cells and TLR3/TET1 double knockdown cells.
(M) Bisulfite sequencing results showing DNA methylation at MYH6 and MYL7 loci. Open circle, unmethylated CpGs; closed circle, methylated CpGs.
Error bars indicate mean ± SEM, N=10 in (I), N= 3 in (G and J), **p < 0.01. See also Figure S3 and Table S2.
Thus, we sought to study the role of immune response in hiCM reprogramming by knocking down candidate immune regulatory genes, including toll-like receptor 3 (TLR3, reported in iPSC reprogramming (Lee et al., 2012)) and its target, nuclear factor kappa B subunit 1 (NFKB1), and prostaglandin-endoperoxide synthase 2 (PTGS2 or COX-2), the well-known target of nonsteroidal anti-inflammatory drugs (VANE, 1971). shRNA-mediated knockdown of these three genes suppressed their respective upregulation in hMGT133a cells (Figures S3H and S3I), and led to a profound reduction in hiCM induction (Figures 3G–3J). Importantly, knockdown of these genes had negligible effect on the expression of exogenous reprogramming factors (Figures 3J and S3J–3L), suggesting that inhibition of innate immunity had minimal effect on gene delivery and that compromised hiCM reprogramming was not due to failed upregulation of reprogramming factors. It has been shown that the effect of innate immunity on iPSC reprogramming is through altered chromatin remodeling (Lee et al., 2012). Hence, we performed a shRNA loss-of-function screen to identify epigenetic modifiers that mediated the effect of immune response on hiCM reprogramming (Table S2). Notably, Tet methylcytosine dioxygenase 1(TET1) knockdown resulted in a 4-fold increase in percentage of hiCM compared to control treatment (Figure 3K). More importantly, knockdown of TET1 partially rescued the reduction of reprogramming efficiency caused by TLR3 knockdown (Figure 3L). Given the critical role of TET1 in DNA methylation, we speculated that changes in DNA methylation could underpin the TET1 knockdown mediated reprogramming recovery in TLR3 depleted cells. Indeed, bisulfite sequencing results in double knockdown cells showed reduced methylation rates at the promoter regions of cardiac genes (Figure 3M). These data suggest that immune response is critical for myocyte fate acquisition during hiCM reprogramming, possibly through impacting DNA methylation status of cardiac loci.
Dynamics of hCF to hiCM fate conversion revealed by SLICER and RNA velocity
Using SLICER (Welch et al., 2016), we constructed the early reprogramming trajectory and calculated the relative position (pseudotime) of each cell on the route of fate switch from hCF to hiCM (Figures S4A and S4B). Built upon SLICER trajectory, we further applied RNA velocity analysis (La Manno et al., 2018) to assess the dynamics of each reprogramming cell. By calculating the ratio between spliced and unspliced reads for each gene in a given cell, RNA velocity predicts whether the gene is currently being turned on or off, and how rapidly this is occurring. Accordingly, it is possible to estimate gene-specific RNA velocity values for each cell using a kinetic model of transcription, in which the appearance of unspliced pre-mRNA precedes spliced mature mRNA as a particular gene is turned on (La Manno et al., 2018). We calculated RNA velocity for each cell used in the trajectory analysis, then projected the vector field onto the SLICER trajectory (Figures 4A and 4B).
Figure 4. Identification of cell fate decision point by RNA velocity and downstream gene networks regulated by reprogramming factors.

(A, B) Vector field of RNA velocity projected onto the SLICER trajectory. Arrows indicate the direction and “speed” for each cell (A) or the average velocity at a grid of points (B) along the trajectory.
(C-G) Potential TBX5 and miR-133 target genes whose expression significantly correlated (P < 0.05) to TBX5 (C, D) or miR-133 (E, F) expression. (C, E) Heatmaps showing Pearson correlation coefficient and representative GO terms enriched in positively- or negatively-correlated target genes. Genes were ordered the same as (D) and (F). Reg., regulation; ECM, extracellular matrix. (D, F) Heatmaps showing inter-correlation of TBX5 (D) or miR-133 (F) target genes with co-expressed genes highlighted with boxes a-c. Genes were ordered by hierarchical clustering. Representative (rep.) genes in each box are listed to the right of the heatmap. (G) Venn diagram showing the number of target genes significantly correlated with miR-133 (red) or TBX5 (purple) expression.
(H) Time course analysis of relative expression of miR-133 candidate targets in hMGT-infected or hMGT133 infected hCFs. Expression value of each gene in D0 cells was set as 1.
(I-J) Flow cytometry plots (I) and quantification (J) showing % cTnT+ cells in hMGT-infected hCFs with addition of indicated shRNA to replace miR-133 for reprogramming.
Error bars indicate mean ± SEM, N= 3 in (H and J). See also Figure S4 and Table S2.
The velocity analysis suggests several interesting properties of hiCM reprogramming. The most salient feature of the velocity plot is the largely opposing directions of the velocity vectors for Fib/iFib and iCM cells (Figures 4A and 4B). That is, unexpectedly, many of the Fib/iFib cells appear to be regressing toward the fibroblast fate. This suggests that there may be a “decision point” after introduction of the reprogramming factors, in which a cell either responds to the hMGT133 cocktail and begins proceeding toward an iCM fate or drops back toward a fibroblast fate. However, intriguingly, not all of the Fib/iFib cells are proceeding in the same direction (inset in Figure 4A). Instead, a subset of the Fib/iFib cells have velocity vectors that point toward the iCM fate, suggesting that these cells have not yet reached the decision point and may be “late responders” still reacting to the introduction of the reprogramming cocktail. Thus, the Fib/iFib portion of the trajectory may consist of “opposing flows” (Figure 4A right panel), rather than a population of cells uniformly proceeding toward either iCM or fibroblast fate. This prediction was further supported experimentally by our time-course scRNA-seq (see below), suggesting the great value of using RNA velocity in combination with SLICER for prediction of the “direction” and “speed” at which a particular cell traverses high-dimensional gene expression space when the reprogramming is initiated.
Transcription factors and microRNA coordinate for hiCM reprogramming
To understand the contribution of individual reprogramming factors to transcriptome changes during reprogramming, we first navigated the relationship between TBX5 expression and its downstream targets in each single cell due to the availability of other datasets (He et al., 2011; Waldron et al., 2016). We calculated the Pearson correlation coefficient between TBX5 expression and the expression of its predicted downstream targets within each reprogramming cell (Figure 4C), and performed GO analysis (Figures S4C and S4D). We also calculated the inter-correlation of TBX5 target genes to determine their co-expression patterns (Figure 4D). GO results and co-expression patterns suggest that TBX5 acts by promoting cardiac structure- and function-related genes and by suppressing fibroblast-related function and non-cardiomyocyte lineages (Figure 4C).
Because miR-133 is an indispensable factor unique for hiCM reprogramming, we focused on analyzing the gene network and co-expression patterns regulated by miR-133 by calculating Pearson correlation coefficient between expression of miR-133 and its predicted targets (Agarwal et al., 2015) (Figures 4E, 4F, S4E and S4F). Global GO analysis of positively correlated miR-133 target genes revealed enrichment of GO terms related to general cellular function (Figures 4E, S4E and S4F). Further examination of target genes that were strongly positively correlated with miR-133 expression (R > 0.4) and also strongly inter-correlated (box a in Figure 4F) identified a few genes that were cardiac related (rep. genes of box a). These genes might be critical mediators of miR-133 during reprogramming. GO analysis of negatively-correlated miR-133 target genes revealed enrichment of ribosome and cell-cycle related genes (Figure 4E). Consistent with the GO analyses, the inter-correlation matrix also identified cell cycle (box b) and translation (box c) related genes based on their co-expression patterns (Figure 4F).
Moreover, we found minimal overlap in GO terms enriched in TBX5 and miR-133 downstream targets during hiCM reprogramming (Figures 4C and 4E). Consistent with different GO terms, comparison of the lists of miR-133 and TBX5 target genes revealed limited overlap (10% of miR-133 target genes, Figure 4G), suggesting TBX5 and miR-133 contribute to hiCM reprogramming through distinct pathways. Next we attempted to identify the functional targets of miR-133 during hiCM reprogramming. We selected 24 potential miR-133 targets based on the prediction score provided by miRDB (Wong and Wang, 2015) and the Pearson correlation coefficient calculated between expression of miR-133 and its target genes in MEF2C highly expressed cells (expression value > 200) (Figure S4G). Among these selected targets, CNN2 and TAGLN2 are previously confirmed functional targets of miR133 in smooth muscle cells (Liu et al., 2008; Torella et al., 2011). We then determined if majority of these genes were downregulated by addition of miR-133 to hMGT-infected hCFs (Figures 4H and S4H). In parallel, we functionally knocked down candidate targets of miR-133 in hMGT-infected hCFs. FACS analyses showed that silencing several genes including NCOA7 (Nuclear receptor coactivator 7), XPO4 (Exportin-4) and RQCD1 (RCD1 required for cell differentiation1 homolog, also known as CNOT9) resulted in various degrees of enhancement in hiCM induction, suggesting that miR 133 promotes hiCM reprogramming partly through silencing these target genes (Figures 4I and 4J). It is important to note that NCOA7 functions as a transcription coactivator (Shao et al., 2002), XPO4 is a bidirectional nuclear transport receptor (Lipowsky, 2002), and RQCD1 belongs to CCR4-NOT complex that is one of the major cellular mRNA deadenylase (Mathys et al., 2014). It will be of great interest to further explore their roles in cardiomyocyte fate determination and fibroblast plasticity.
Cell fate index (CFI) to quantitatively assess reprogramming progression
Although the molecular mechanisms underlying cardiac fate acquisition are largely conserved between mouse and human, differences do exist including considerable species-specific genetic and epigenetic modulators. Given the availability of scRNA-seq data sets we recently generated for miCM reprogramming, we investigated the transcriptomic differences between hiCM and miCM reprogramming. We first analyzed the heterogeneity of reprogramming hCFs by calculating intercellular transcriptome variance. We found that the gene expression variance across hMGT133-infected cells is significantly less than that across murine MGT infected CFs (Figure S5A). As a control, DsRed plus EV introduced more intercellular variance in gene expression, possibly resulting from the over-activation of antiviral immune response as aforementioned (Figures 3E–3F, S3D–S3E). Intriguingly, in sharp contrast to miCM induction but similar to the initiation of iN reprogramming, the transcriptome variance in hMGT133 fibroblasts was much smaller than uninfected control hCFs, suggesting a possible synchronized response upon hMGT133 transduction (Liu et al., 2017b; Treutlein et al., 2016). Notably, we found that the number of genes whose expression change significantly at day 3 of reprogramming in human was only 1/3 of that in mouse and 57% changed genes in human were also changed in mouse (Figure S5B), suggesting that human cardiac programming might progress at a slower rate than the mouse one.
We then sought to develop an approach to quantitatively measure the reprogramming progression at single cell level. By defining the starting fibroblast cellular state as 0 and the targeting myocyte state as 1, all reprogramming cells can be positioned mathematically along this fate path and given a calculated index, which we call “cell fate index” (Figure 5A). With CFI, we evaluated the distance a reprogramming cell departed from fibroblast fate and how much the cell progressed toward myocyte fate. We first calculated CFIs of all mouse and human single cells using cardiomyocyte fate- and function-related genes obtained by comparison of bulk transcriptome profiles of de novo CFs and CMs (Figures S5C, S5D and Table S3). The results revealed a continuous acquisition of cardiomyocyte fate for both mouse and human reprogramming fibroblasts, the order of which was well aligned with the HC/PCA grouping of cell states (Figures 5B–5D). To further dissect the progression of CM fate acquisition during reprogramming, we calculated CFIs of both mouse and human cells using subgroups of CM fate- and function-related genes (Figure 5E). Mouse cells consistently progressed further toward the target cell type than human cells in all gene categories: cardiac structural protein, cell junction, contractility, and ion channel. In addition to target cell fate acquisition, suppression of starting cell fate is equivalently important in reprogramming. Therefore, we also calculated CFIs using genes that are expressed significantly higher in the starting cell compared to the target cell (Figure S5E, S5F and Table S3). The results revealed a continuous suppression of the starting cell fate in both mouse and human cells with the order of cells well-matched to HC/PCA grouping of cell states (Figures 5F and 5G). Similar to CM fate acquisition, the suppression of starting cell fate in human cells is less prominent than that in mouse cells, further confirming a slower rate of cardiac reprogramming in human cells than in murine cells (Figures 5F, S5E–S5F). Moreover, human-specific mRNA-splicing, transcription and translation related reduction of cell fate index was determined along human reprogramming (Figures 5G, S5E–S5F), consistent with our early findings in mouse.
Figure 5. Quantitative assessment of mouse and human iCM progression.

(A) Schematic for the calculation of cell fate index (CFI). See Methods for details.
(B-D) CFIs of day 3 mouse and human single cells undergoing reprogramming. (B) Overall CFIs calculated with cardiac fate- and function-related genes. Trendlines were show in grey (mouse) and lavender (human). Violin plots showing distribution of CFI in subpopulations of cells during hiCM (C) or miCM (D) reprogramming. Center yellow dots indicate the median value. mpiCM, mouse pre-iCM.
(E-G) CFIs calculated with subgroups of cardiac genes (E) or starting cell fate-related genes (F, G). ECM, extracellular matrix.
(H) Quantile-quantile plot of mouse and human CFIs shown in (B). The solid line indicates hypothetical data if speed of mouse equals to speed of human.
(I) Formula for calculating instantaneous speed of reprogramming. See Figure S5G and S5H for details about CFIcubic.
(J) Speed of hiCM and miCM reprogramming calculated based on (I). Trendline was calculated by fitting cubic smoothing spline and confidence bands were shown in grey. Two-sided t tests were performed, α= 0.05.
(K, L) Kinetics of miCM (K) and hiCM (L) reprogramming plotted by relative percentage of marker positive cells determined by flow cytometry along reprogramming. The end point of reprogramming was set as 1.
Error bars indicate mean ± SEM, N=3. See also Figure S5 and Table S3
Using CFI, we calculated the speed of hiCM and miCM reprogramming. We first determined the overall speed of these two reprogramming processes by considering the quantile-quantile plot (QQ-plot) using 600 equally-spaced quantiles from the CFIs for both human and mouse (Figure 5B). Linear regression estimated the slope of mouse’s quantiles over human’s quantiles to be 1.559, suggesting that mouse fibroblasts progressed significantly faster (P < 1e−16) toward the target cardiomyocyte fate, at a speed that is about 1.6 times faster compared to human cells (Figure 5H). This result is also consistent with the highest CFIs achieved by mouse and human cells, with the fastest miCM arriving at CFI of 0.45 and the fastest hiCM arriving at 0.25 (Figure 5B). We also calculated the speed of reprogramming at each moment along the process by fitting cubic smoothing splines to CFIs first (Figures S5G and S5H) and then calculating the derivative using first order finite difference approximation (Figure 5I). The results showed that instantaneous rate of miCM reprogramming was higher than that of hiCM reprogramming at any given time (Figure 5J, mean speed difference = 0.1944, p value = 0.0002), consistent with the overall rate estimated from QQ-plot. Interestingly, even though differing in absolute values, the rate of miCM and hiCM reprogramming demonstrated similar trend of progression over “time” featuring three consecutive phases. The first phase is characterized by initially high yet decreasing rate of reprogramming, which is followed by a second phase with steady and lower reprogramming rate and the last phase of higher and accelerating reprogramming rate (Figure 5J). Further analysis of the reprogramming rate confirmed an overall higher progression rate of miCM reprogramming than hiCM reprogramming, with the mean speed differences being 0.1633 (initial phase), 0.0875 (middle phase) and 0.4434 (late phase) and the p values smaller than 0.05. To experimentally verify this calculation, we performed a pairwise comparison of the kinetics of hiCM and miCM reprogramming. Results from flow cytometry (Figures S5I and S5J) and the calculation of reprogramming efficiency demonstrated distinct kinetics of miCM (Figure 5K) and hiCM reprogramming (Figure 5L). We found that cTnT+ cells emerged on day 4 in mouse and day 8 in human cells, demonstrating a latency period of 2 days and 6 days accordingly (Figures 5K–5L, S5I–S5J). Collectively, our results suggest that cardiac reprogramming in human cells progressed more slowly than in mouse cells, at least using the cocktails and platforms we described here.
Time-course scRNA-seq identified a refractory route along hiCM reprogramming
Because day 3-reprogramming human CFs lagged behind toward CM fate when compared to day 3-reprogramming mouse CFs, we decided to perform scRNA-seq at later time points along hiCM reprogramming. In contrast to its expression in day 3 miCMs, the cardiac marker cTnT was expressed at lower level in day 3 hiCMs, but demonstrated significant level of expression from day 5 to day 9 (Figure S6A). We thus collected single cells of hMGT133- or TdTomato+EV-infected hCFs at post-infection day 3, 5, 7 and 9 using the same Fluidigm C1 platform followed by Illumina sequencing as aforementioned (Figures 6A and S6A). Similarly, quality control and normalization were performed to exclude low quality reads and cells, yielding 13479 genes detected in 323 high-quality control and reprogramming cells for further analysis (Figures S6B–S6G and Methods). Interestingly, we found that all control TdTomato+EV-infected cells were clustered at the top-left corner of the PCA plots, while the hMGT133-infected cells bifurcated into two branches (Figures 6B–6C). The first branch highlighted by pink circle along PC1 was associated with CM fate acquisition since PC1 accounts for the variations of cardiac genes (Figures 6B–6C and S6H–S6I). In contrast, the second branch highlighted by the green circle along PC2 featured fibroblast markers and genes in the interferon pathway (Figures 6B–6C and S6I). Intriguingly, within the second population, the day 3 and day 5 reprogramming cell clusters appeared to be further separated from the control cells compared to the day 7 and day 9 samples (Figure 6C). Similar clustering pattern was demonstrated by t-SNE (Figures 6D and S6J) and HC analyses (Figures 6E and S6K). Using the major groups of gene cluster generated by these two analyses, we classified the cells into three major groups: the control cells, the cells that assumed cardiac fate (hereinafter referred as reprogramming cells), and the cells that retained fibroblast molecular signature (referred as refractory cells) (Figure S6K). Similarly, two subgroups of refractory cells corresponding to their duration of reprogramming (D3/D5 and D7/D9) were clearly distinguishable on the plot. The D3/D5 refractory cells were associated with stress response genes, while D7/D9 refractory cells more resembled control fibroblasts and were also characterized by upregulation of antiviral response related genes (Figure S6K).
Figure 6. Time-course scRNA-seq of hiCM reprogramming identified a refractory route.

(A) Schematic of experimental design for time-course scRNA-seq of hiCM reprogramming (see details in Methods).
(B-E) PCA and tSNE of control and reprogramming cells from day 0, 3, 5, 7, and 9 were calculated with top 400 PCA genes. Cells were color-coded by treatment (B-D) or clusters defined by HC (E) and shown in 3D PCA (B, E), 2D PCA (C), or tSNE (D) plots.
(F-H) Reprogramming trajectories constructed by SLICER shown in 3D with cells color-coded by treatment as indicated in (B) (F and H) or HC clusters (G). (H) Cells at each time point were highlighted by coloring cells from other time points in gray.
(I-J) Six gene clusters that are significantly related to and show similar trends along the reprogramming (Rep) (I) or refractory (Ref) (J) trajectory were identified. GO analysis was performed for each gene cluster and listed in the bottom. The number of genes in each cluster is shown in parentheses.
As the hMGT133-infected cells were distributed on bifurcating branches, we speculated that these cells could take alternative routes to either adopt cardiac fate or enter the refractory state and subsequently assume original fibroblast fate. Interestingly, our RNA velocity analysis on day 3 hiCMs (Figures 4A–4B) revealed a “decision point” at which the cells proceed toward an iCM fate or fall back toward their original fate. To test our hypothesis, we performed SLICER analysis with the entire scRNA-seq dataset generated from the samples at day 3 time point onward to reconstruct the reprogramming trajectory and calculate the pseudotime (Figures 6F–6H). Consistently with HC grouping, the trajectory was comprised of two separate paths: one path (pink) progressing from control fibroblast to hiCM fate, and the other path (green) looping back to control fibroblast fate (Figure 6F), thus we defined the pink path as a reprogramming route (branches 1 and 3) and the green path as a refractory route (branches 1 and 2, Figures 6G and S6L). Plotting the cells at real time points on the trajectory, we further demonstrated the segregation of two cell populations (Figure 6H), and the relative distribution of which on the routes was consistent with the reprogramming efficiency determined by ICC and flow analyses (Figure 1D). These data strongly support the prediction from day 3 RNA velocity analysis.
We next performed nonparametric regression and k-medoids clustering to delineate the underlying regulatory networks. Six clusters of genes based on their dynamic expression patterns were identified along pseudotime of reprogramming (Rep) or refractory (Ref) route (Figures 6I–6J and S6M–S6O, Table S4). As expected, the upregulated genes in cluster Rep1 were associated with cardiac function (Figure 6I, left panel), while fibroblast function related genes in cluster Rep2 were downregulated within reprogramming route (Figure 6I, middle panel). Notably, the cluster Rep3 showed an up-then-down-regulation in the expression of the genes related to “ATP biosynthetic process” and “mitotic cell cycle” (Figure 6I, right panel), consistent with our finding that cell cycle was inhibited once reprogramming initiated (Figure 2).
The three gene clusters identified for refractory route were enriched in completely different molecular pathways and biological processes (Figures 6J and S6O). In particular, the characteristic GO terms enriched in the up-regulated cluster Ref1 were “NIK/NF-kappaB signaling”, “response to stress” and “viral process”, suggesting upregulated viral and stress response in cells distributed along refractory route (Figure 6J, left panel). Meanwhile, the downregulated genes were highly involved in “viral transcription”, “muscle contraction” and “response to mechanical stimulus” (Figure 6J, middle panel). More interestingly, “regulation of mRNA stability” and “protein folding” were enriched in cluster 3 genes, with their expression peaking at the middle point of refractory pseudotime (Figure 6J, right panel). Therefore, molecular pathway analysis along pseudotime of refractory route provided candidate genes and pathways for future studies.
Comparative analyses between reprogramming and refractory routes
Given the separation of hMGT133-infected cells into either reprogramming or refractory subpopulations with time, we next aimed to identify the underlying molecular differences between these two routes. We first examined the expression of reprogramming factors. As shown on the tSNE plot, we found that the expression of M, G, T, and miR133 in the reprogramming cell population was higher than that in the refractory population (Figure S7A), suggesting that the level of reprogramming factors could influence cell fate decision. We then applied CFI to further evaluate the progression of each route in different gene categories related to cardiac or fibroblast cell fate. The CFIs of cardiac structural proteins or contractility genes showed that the cells following reprogramming route underwent more progression into cardiac cell fate over time than those following refractory route (Figures 7A and S7B). Fibroblast-fate- related CFIs were decreased in a time dependent manner in reprogramming cells but regressed in refractory cells (Figures 7B top panels and S7B). The CFI of mRNA splicing, which has been identified as barriers in mouse iCM reprogramming (Liu et al., 2017b), showed similar decrease in reprogramming cells but little alteration in refractory cells (Figure 7B bottom panels). More importantly, two categories of cardiac CFIs indicated by cell junction and ion channel genes failed to show differences in progression along either reprogramming or refractory route (Figure 7C). This data pointed to a promising future direction whereby re-activation of genes in these two categories might be a viable approach to promote the efficiency of hiCM induction.
Figure 7. Comparative CFI, DEG and RNA velocity analyses between reprogramming and refractory cells.

(A-C) CFIs of cells on reprogramming (left) or refractory (right) route. Indexes calculated with subgroups of cardiac genes (A, C) or starting cell fate-related genes (B). Cells ordered based on their indexes.
(D) Differentially expressed genes (DEG) between cells on reprogramming verses refractory route at each time point were identified by one-way ANOVA followed by pairwise comparisons (adjusted p value < 0.05, number of DEG shown in parenthesis). Left panels: volcano plots showing DEGs with more than 2-fold change in red. Right panels: GO plot showing enriched terms for DEGs highly expressed in refractory (green) or reprogramming cells (red).
(E-F) Violin plots showing distribution of the expression of potential positive (E) and negative (F) selection markers of reprogramming.
(G-H) RNA velocity analysis showing vector field of RNA velocity projected onto the SLICER trajectory. Arrows indicate the direction and “speed” for each cell along the trajectory. Cells colored by treatment (G) or HC clusters (H).
To identify the different molecular signatures between reprogramming and refractory cell populations, we examined DEGs at each time point by one-way ANOVA and GO analysis (Table S5). In agreement with the above findings, the GO terms consistently enriched for highly expressed genes in reprogramming cells at each time point were cardiac cell fate or function related terms (Figure 7D). In contrast, we found that genes involved in “protein folding”, “response to endoplasmic reticulum stress” and “regulation of autophagy/mitophagy” (Figure 7D. see D3 and D5), were upregulated in refractory cells, suggesting that these cells activated stress response pathways upon transduction of reprogramming factor.
To identify the potential selection markers for reprogramming or refractory populations, we focused on those genes repeatedly ranked at the top of the DEG list (p value < 0.05 at each time point, Figure 7D, highlight in red dots, Table S4). Among the genes highly expressed in reprogramming cells (Figure 7E), we found several makers already known in heart, like cardiomyopathy associated 5 (CMYA5) encoding MYOSPRYN (Benson et al., 2017; Durham et al., 2006), crystallin alpha B (CRYAB, or HSPB5) and Heat Shock Protein Family B (Small) Member 2 (HSPB2) (Sugiyama et al., 2000). Transforming growth factor beta 3 (TGFB3), another potential marker for reprogramming cells, exhibited increasing level of expression along reprogramming route (Bujak and Frangogiannis, 2007). The other two potential positive markers junction plakoglobin (JUP) and fibrinogen-like protein 2 (FGL2) have unknown function in heart development or regeneration. We also identified five negative markers for reprogramming that were highly expressed in control and refractory cells but barely detectable in reprograming cells (Figure 7F). It is interesting to identify one immune cytokine TNF alpha induced protein 6 (TNFAIP6), and one metabolic enzyme aldo-keto reductase family 1 member C1 (AKR1C1), with which small molecular inhibitors can be discovered and applied to eliminate the undesired refractory population. Moreover, fibroblast activation protein alpha (FAP) is a cell surface serine protease, which can be a promising surface marker to select against the refractory cells for enriching reprogramming hiCMs.
Lastly, we calculated RNA velocity of cells at all examined time points and projected that onto the entire time course-reconstructed trajectory (Figures 7G–7H and S7C–7D). It is interesting to notice that the arrows (velocity) of cells on the reprogramming route is unidirectional, always pointing to cells at the next time point on the reprogramming route. However, some of the D3 and the majority of the D5, D7 and D9 cells on the refractory route regressed toward the starting fibroblast cells (Figures 7G and 7H), which is consistent with our finding in Figures 4A and 4B, indicating that the “decision point” is where the cells take the bifurcating reprogramming or refractory route. Additionally, the vector field plot of RNA velocity suggests that the early initiation of cell fate conversion progressed more rapidly than the late determination of hiCMs (Figures S7C and S7D). Therefore, the time course scRNA-seq analysis demonstrated the early separation of reprogramming and non-reprogramming cells and provided more detailed molecular differences and potential biomarkers to distinguish these two populations.
DISCUSSION
In this study, we established a stable and reproducible platform for hiCM reprogramming. Using this optimized platform, we performed scRNA-Seq on reprogramming fibroblasts at multiple time points in the reprogramming process. In-depth scRNA-seq analyses demonstrated several salient properties of hiCM reprogramming. First, RNA velocity revealed a “decision point” at which a cell begins progressing toward a hiCM fate or regress toward a fibroblast fate. This observation is consistent with SLICER trajectory that an hMGT133-infected fibroblast could take alternative routes to either acquire hiCM fate or regress toward fibroblast fate. Additionally, these two separate routes are associated with distinct hiCM specific signaling molecules and factors that warrant in-depth future studies. Second, by performing cell fate index to quantitatively assess reprogramming progression, we found that hiCM reprogramming progresses at a much slower rate toward acquiring cardiac fate than mouse cardiac reprogramming. This is likely due to the fact that the instantaneous rate of hiCM reprogramming was lower than that of mouse cardiac reprogramming at any given time. Third, through further scRNA-seq analysis and follow-up functional studies, we demonstrated that immune response associated DNA methylation regulation is required for hiCM induction and silencing several previously uncharacterized downstream targets of reprogramming factor enhanced hiCM induction. Collectively, our scRNA-seq based approach revealed previously unrecognized molecular features and regulatory mechanisms of human cardiac reprogramming.
Our finding that depletion of immune genes represses iCM reprogramming, as well as previous reports of critical role of innate immunity in other reprogramming systems (Hodgkinson et al., 2018; Lee et al., 2012; Sayed et al., 2015, 2017) highlighted a critical role of innate immune signaling pathway in cell fate determination and maintenance. It is interesting to notice that activation of immune gene mainly occurs at the early stage of iCM reprogramming (Figure S6K) and is correlated with alterations in stress response genes (Figure 6I see Cluster Rep2), suggesting that innate immunity is required for and associated with initial iCM fate conversion rather than late maturation. It is also reasonable to believe that the activation of innate immune response is at least partially associated with the collateral stress/damage induced by the initiation of reprogramming. Mechanistically, we and others have shown that alteration of immune response during reprogramming is associated with epigenetic changes on histone modification and/or DNA methylation (Lee et al., 2012). Yet it is still largely unknown how regulators of immune response acts on epigenetic modifiers to allow histone and DNA modification changes. Future research on this topic will inform us more on how a cell senses environmental stimuli and then relates to its epigenetic re-patterning to ultimately alter its cell status or identity.
The cell fate index approach that we developed allows a proof of principle comparison of cellular processes across species and conditions, and we anticipate that it will be useful for a variety of biological processes. We found that both hiCM and miCM reprogramming occurs with the “three phase” of differential rate of progression (Figure 5J). There are two turning points on each trendline that change the speed of reprogramming. The high yet decreasing speed in the initial phase seems to be consistent with a relative fast and uniform initial response to transcription factors and may suggest barriers impeding the progress of reprogramming around the turning point. In contrast, the steep burst in the late phase suggests a committed cardiomyocyte fate and certain critical regulators around the turning point might be important for this abrupt acceleration of speed. Follow-up studies about potential predicting functions of CFI and its derivatives will be of great interest.
Ultimately, interdisciplinary approaches combining biological experiments with statistical analyses and computational modeling will open opportunities for novel biological discoveries. Our studies incorporating the latest algorithms such as SCnorm, reCAT, RNA velocity, and our own SLICER and CFI analyses represent such an example and may inspire next crucial steps toward understanding the nature of cell identity and translating this knowledge into regenerative therapies.
STAR METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for reagents should be directed to and will be fulfilled by the Lead Contact, Li Qian (li_qian@med.unc.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Human fibroblasts culture
The human cardiac tissue was provided by the Duke Human Heart Repository (DHHR) with IRB protocol # Pro00005621. The patient is male at age of 63, Caucasian and non-Hispanic. Human cardiac fibroblasts (hCF1) were derived from heart tissue by explant culture. The heart tissue was firstly minced into small aggregates, then plated onto one gelatin coated 10 cm dish for explant culture in HCF medium (20% FBS/IMDM/1×P/S). After 7 days of migration, fibroblasts were trypsinized and dissociated into single cells. These cells were ready for reprogramming or frozen for future use. hCF2 cells were non-myocytes derived in Cheng Lab as described previously (Cheng et al., 2012). H9F cells were fibroblasts differentiated from aMHC-mCherry transgenic human embryonic stem cell H9 as previously described (Fu et al., 2013). Briefly, embryonic bodies were generated by suspension culture of H9 cells for 7 days, and plated onto gelatin-coated dished in H9F medium (20% FBS/DMEM) for additional 2 weeks.
Mouse fibroblasts culture
Fresh isolation of CFs was performed according to the protocols described previously (Wang et al., 2015b). Briefly, hearts were dissected from postnatal 1.5 (P1.5) ĮMHC-GFP transgenic mice (CD1 background), rinsed with cold PBS and cut into small pieces with a sterile blade. Then, heart tissues were digested with 0.05% Trypsin at 37 °C for 10 minutes and 0.2% collagenase type II/HBSS (Life Technologies) at 37°C for 5 minutes followed by 1 minute of vortexing for 5 times. Each time, supernatant containing single cells was filtered through 40 μm cell strainer (BD) and neutralized in equal volume of FB media. Red cells were removed using red cell lysis buffer (150 ml NH4Cl, 10 mM KHCO3, and 0.1 mM EDTA) for 1 minute on ice. Then, magnetic-activated cell sorting (MACS) was performed to enrich Thy1.2 positive fibroblasts. Cells were incubated with 10 μl of biotin anti-Thy1.2 antibody (Biolegend) in FACS buffer (DPBS/2% FBS/2 mM EDTA) for 30 minutes at 4 °C and then with 10 μl of Anti-Biotin Microbeads (Miltenyi Biotec.) in MACS buffer (DPBS/0.5% BSA/2 mM EDTA) at 4 °C for 30 minutes. After that, cells were washed and resuspended in MACS buffer and applied to calibrated LS column (Miltenyi Biotec.). Thy1.2 positive cells (CF) were flushed out and seeded for reprogramming.
METHOD DETAILS
Plasmids
Retroviral vectors encoding human MEF2C, GATA4, TBX5, ESRRG, MESP1 and MYOCD in pMXs backbone vectors used for Fu (GMTEMsp) and Wada (GMTMyMsp) cocktail were described previously (Fu et al., 2013; Wada et al., 2013). Retroviral vectors encoding human GATA4, HAND2, MEF2C, TBX5, MYOCD, MESP1, miR-1 and miR-133 were gifts from Nam (for GMTMyMsp-miR-1/133 cocktail) (Nam et al., 2013). To generate polycistronic hMGT construct, human MEF2C, GATA4, TBX5 were PCR amplified and inserted into pGEMT-P2A-T2A vector as previously described (Wang et al., 2015a) and the hMGT cassette was sequentially cloned into pMXs-puro vector. pMXs-puro-LacZ was purchased from Cell Biolabs. pMXs-puro-dsRed, pMXs-GFP and pMXs-tdTomato were described previously (Liu et al., 2017d; Wang et al., 2015a). TroponinT-GCaMP5-Zeo was a gift from John Gearhart (Addgene plasmid #46027) (Addis et al., 2013). All the lentiviral shRNA plasmids were purchased from Sigma and listed in Table S2.
Virus packaging
To obtain retroviruses for human cell infection, retroviral DNA plasmids along with pCMV-VSV-G (Addgene #8454) and pUMVC (Addgene #8449) were transfected by NanoFect (ALSTEM) into 293T cells. While, we used PlatE cells to package retroviruses for mouse cell infection. One day before transfection, 4–5 million 293T or PlatE cells were seeded onto poly-l-lysine (Sigma) coated 10 cm dish with 293T media (DMEM supplemented with 10% fetal bovine serum (FBS), 0.1 mM non-essential amino acids (NEAA) and 1% penicillin/streptomycin (P/S)). Fresh 293T media without P/S was changed prior to transfection. For 293T cell transfection, DMEM mixture with 45 μl of NanoFect was added into DMEM mixture with 8 μg of retroviral DNA plasmid, 8 μg of pUMVC and 1 μg of pCMV-VSV-G and vortexed. For PlatE cell transfection, 20 μg of retroviral DNA plasmid was used instead. After 15 minutes of incubation at room temperature, total mixture was added dropwise to 293T or PlatE cells. Supernatant containing retroviruses was collected 48 and 72 hours after transfection, filtered through a 0.45 μm filter (Thermo Scientific) and incubated with 8% of PEG6000 (Sigma) at 4 °C overnight. Viral particles were pelleted with centrifuge at 3900 rpm for 30 minutes at 4 °C. Viruses from each 10 cm dish were resuspended in 100 μl of DMEM and ready for future use. For lentiviral packaging in 293T cells, same protocol with Nanofect was used, except the different viral DNA mixture, which contained 10 μg of lentiviral shRNA plasmids cotranformed with 7 μg of psPAX2 (Addgene #12260) and 3 μg of MD2.G (Addgene #12259).
Human fibroblasts cardiac reprogramming
To test different combinations of transcription factors, 2 × 104 of H9Fs per well were seeded onto gelatin-coated 24 well plate, and infected with 10 μl of each indicated viruses in iCM media (10% FBS/20% M199/DMEM) supplemented with 8 μg/ml polybrene. Media were changed every 4 days thereafter. At reprogramming day 14, cells were trypsinized and analyzed by flow cytometry. The optimized protocol of cardiac reprogramming was performed as following. Human fibroblasts were seeded onto SureCoat (Celltron) pre-coated wells of 24 well plate at a cell density of 2 × 104 per well one day before infection. Cells were infected with 10 μl of puro-hMGT and 10 μl of miR-133 with iCM media supplemented with 8 μg/ml polybrene. Puromycin selection was performed from day 3 to day 7. The media were replaced with hiCM (RPMI1640/2% B27/2%FBS/0.05%BSA/50 μg/ml Ascorbic Acid/0.2 mM GlutaMAX, 1× NEAA) media at reprogramming day 10. Reprogramming cells were harvested at indicated time point for immunostaining, RT-qPCR and flow cytometry.
Co-culture of hiCM with mouse neonatal CM
H9Fs were infected with retroviral hMGT133 for iCM reprogramming and lentiviral GFP to indicate the human cells. After 2 weeks of infection, the infected cells were trypsinized and re-plated onto neonatal mouse cardiomyocytes at a ratio of 1:10 with DMEM/M199/10% FBS medium. Neonatal mouse cardiomyocytes were isolated from P3.5 mouse pups by using the neonatal cardiomyocytes isolation system (Worthington Biochemical Corporation) except that all enzymes were used at a quarter of the recommended concentration to increase cell viability. After a 1.5 h of pre-plating on an uncoated surface to remove attached non-cardiomyocytes, the unattached cardiomyocytes were counted and used for seeding with GFP labeled reprogramming cells together.
Mouse fibrobroblasts cardiac reprogramming
Freshly isolated neonatal mouse cardiac CF (Thy1.2+) seeded for reprogramming at a density of 2 × 104 per well onto gelatin-coated 24 well plate. iCM media with 10 μl of retroviral puro-MGT and 4 μg/ml polybrene were replaced for FB media at reprogramming day 0. iCM media with 1 μg/ml puromycin were used at day 3 and replaced by regular iCM media at day 6. Reprogramming cells were collected at indicated time point in TRIzol for RNA extraction or fixed with 4% paraformaldehyde (PFA) for immunostaining or flow cytometry.
Flow cytometry
For flow cytometry, cells were trypsinized with 0.05% Trypsin/EDTA (Life Technologies), fixed with Fixation/Permeabilization Solution (BD Biosciences) for 30 minutes at 4 °C. 1× Perm/Wash Solution (BD Bioscience) was used for wash between each step. Cells were incubated with primary antibodies (GFP, 1:500, Invitrogen; cTnT, 1:400, Thermo Scientific) diluted in BD Perm/Wash Solution for 30 minutes at 4°C and Alex Fluor 488-or 647-conjugated secondary antibodies (1:500, Jackson ImmnoResearch Inc.) for 30 minutes at 4°C. Cells were run on Beckman Coulter CyAn ADP flow cytometer. Data analyses was performed by FlowJo software (Tree Star).
Imaging Calcium oscillation
Calcium signals were indicated with GFP fluorescence from cTnT driven GCamp5. H9Fs were infected with TroponinT-GCaMP5-Zeo lentiviruses and seed to perform hiCM reprograming 2 days postinfection. At reprogramming day 20, calcium flux was observed and videos of calcium oscillation were recorded using an image acquisition software liteCam HD (RSUPPORT) and an EVOS microscope system (Invitrogen). The calcium signals were quantified via ImageJ (Fiji version, NIH). The relative fluorescence unit (RFU) of GFP was normalized to the background fluorescence intensity.
Immunofluorescence staining
Cells were fixed with 4% PFA, permeablized with 0.1% Triton-X100 for 20 minutes, blocked by 5% BSA for 30 minutes at room temperature, and then incubated with primary antibody (GFP, 1:500, Invitrogen; aActinin, 1:500, Sigma-Aldrich, Connexin 43, 1:400, Abcam) at 4°C overnight and Alex Fluor 488- or 647-conjugated secondary antibodies (1:500, Jackson ImmnoResearch Inc.) for 1 hour at room temperature. Finally, Hoechst 33342 (Life Technologies) was used to label nuclei. PBS was used for wash between each step. Images were captured using EVOS® FL Auto Cell Imaging System (Life Technologies). For the quantification of ICC, 10 images were randomly acquired under 20× magnification at the same exposure setting. Then the indicated cells were counted manually.
RNA extraction and qRT-PCR
According to the manufacturer’s instruction, cells lysate in TRIzol reagent (Invitrogen) were separated with chloroform. RNA in the aqueous phase was precipitated with isopropanol, pelleted with centrifuge, washed with ethanol and eluted in DNase- and RNase-free water. Purified RNA was quantified by Nanodrop (Thermo Scientific) and reverse-transcribed into cDNA using SuperScript III Reverse Transcriptase (Invitrogen). qPCR was performed using Power SYBR Green PCR Master Mix (Applied Biosystems) on the ABI ViiA 7 Real-Time PCR system (Applied Biosystems). Additional primer sequences for RT-qPCR are provide in Supplemental Table S2.
Bisulfite sequencing
H9Fs were infected with lentiviral shRNAs targeting TLR3 and selected with puromycin for 2 days. Then TLR3 knocked down H9F were seed at density of 40,000 and infected with hMGT133 with shNT or shTET1 the next day after seeding to perform reprogramming. Fresh iCM medium was replaced post-transduction day 2 and cells were harvested at day 6 by trypsinization, and genomic DNA was extracted using DNeasy Blood & Tissue Kits (Qiagen) and bisulfite converted (~ 500 ng/reaction) using the EZ DNA Methylation-Gold Kit (Zymo Research). 1 ȝl of the bisulfite converted DNA was used for PCR amplification of the promoter regions of MYH6 and MYL7 genes using previously described primers (Fu et al., 2013) and the EpiMark Hot Start Taq DNA Polymerase (NEB). The PCR products were then gel purified and TA-cloned into pGEMT vector (Promega). Ten clones in each sample were successfully sequenced. Bisulfite Sequencing DNA Methylation Analysis (BISMA) was used to analyze the bisulfite sequencing DNA methylation data (Rohde et al., 2010).
Single cell capture and cDNA preparation
Single cells were captured using the Fluidigm C1 system with the capacity of up to 96 single cells per experiment. A total of eight individual single-cell experiments were performed (see Fig. 2A and Fig. 7A for experimental design). One plate (D0hCF) contained uninfected d0 human primary fibroblasts. Two plates (D3M1 and D3M2) contained d3 MGT+miR-133-transduced cells. The fourth plate (D3UN) contained d3 DsRed-transduced cells (DsRed+ by FACS) mixed with d3 uninfected cells at 3:1 ratio. The rest four plates contained MGT+miR-133-transduced cells mixed with Tdtomato-transduced cells (Tdtomato+ by FACS) at 3:1 ratio, which were collected on d3 (D3M3), d5 (D5M1), d7 (D7M1), and d9 (D9M1), respectively. The cells were collected by trypsinization, stained with NearIR live/dead dye (Thermo Fisher Scientific), and FACS-sorted for live cells (negative for the live/dead dye). For plates D3M3, D5M1, D7M1 and D9M1, the MGT+miR-133-transduced cells were also stained with the green Carboxyfluorescein succinimidyl ester (CFSE) dye as described previously(Liu et al., 2017b) and only CFSE+ cells by FACS were collected and used. Pilot experiments showed that hCF had a diameter of 22.5 μm and a buoyancy of 7.5 (cells): 2.5 (buoyancy buffer). Therefore, the sorted single cell suspension (~2000 cells μl−1) was loaded on a large-sized (17–25 μm) microfluidic RNA-seq chip and single cells were captured with the C1 system. Bright field and/or green (CFSE)/red (DsRed or Tdtomato) fluorescence images were taken for each capture site. For the next step, cDNAs were prepared on chip using the SMARTer Ultra Low RNA kit from Illumina (Clontech). Briefly, control RNA spike-ins were added to the plate before cell lysis and processed in parallel to cellular RNA. The experiment D3M1 used the Ambion Array Control spike-ins (AM1780) that were included in the SMARTer kit (spike 1, 4, and 7) according to Fluidigm’s protocol. For the other experiments, D0hCF, D3M2/3, D3UN, D5M1, D7M1, and D9M1, the External RNA Controls Consortium (ERCC) RNA spike-in Mix 1 (Ambion, Life Technologies), were added after a 80,000-fold dilution. After the addition of spike-ins, cell lysis, reverse transcription, and cDNA pre-amplification were performed on the chip according to Fluidigm’s standard protocol.
Illumina library preparation and sequencing
After in situ cDNA library preparation, the bright field and/or fluorescence images of each nest on the chip were carefully examined and only single and healthy cells were processed further. This step removed 64 empty nests, nests with two or more cells, or nests containing morphologically unhealthy cells from a total of 768 capture sites in the eight chips, resulting 704 single-cell cDNA libraries for further processing. Size distribution and quality of each of these cDNA libraries were assessed and confirmed on Agilent Bioanalyzer 2100 by UNC LCCC Genomics Core. Two negative control libraries from empty nests on the D3UN plate and the D9M1 plate was included and processed in parallel with other single healthy cells. Next, the 704 high-quality cDNA libraries were sent to UNC HTSF Core and Illumina libraries were prepared using the Nextera XT DNA Sample Preparation kit according to Fluidigm’s standard protocol except that 13 cycles of amplification was used instead of 12 cycles. The barcoded single-cell Illumina libraries of each experiment were pooled and sequenced for 50 base pairs (bp) single-end reads on one lane of Illumina HiSeq 2500. Previous studies showed that 0.5−1 million reads per cell were sufficient to detect most genes expressed by single cells (Treutlein et al., 2016; Wu et al., 2013) and we sequenced about 1–5×106 reads per cell in this study. Raw reads were re-assigned to each single cell by their unique Nextera barcode and sequencing reads removed of barcodes were received from UNC HTSF Core in .fastq format.
QUANTIFICATION AND STATISTICAL ANALYSIS
QC, alignment, and counting scRNA-seq data
Quality of sequencing results was first checked by FASTQC and no trimming was performed because of high-quality reads. Therefore the raw reads were mapped to a merged genome of hg19, ERCC, E.coli K12, pMXs-DsRed, pMXs-Tdtomato, and pBabe-miR-133 with Tophat2 using default settings (allowed mismatch = 2, maximum hits = 1). Information about the number of total reads and the percentages of reads mapped to spike-in or human genome for each single cell were detailed in the Table S1. Outliers showing high ratios of % reads mapped to spike-in to % reads mapped to human genome were removed (Fig. S2A and Fig. S7B). This step removed 29 outliers from the 704 sequenced single cells. After normalization (see next paragraph), additional outliers were removed at the default outlier removal step during analysis using the “SINGuLAR Analysis Toolset” R package (see “Analysis of single cell RNA-seq data”). A total of 23 additional outliers were removed based on median gene expression (Fig. S7E) or PCA (Fig. S7F), resulting 652 high-quality single cells for analysis including 85 original hCFs (D0hCF), 14 uninfected hCFs (D3UN), 54 day 3 DsRed- and EV-transduced hCFs (D3UN), 14, 21, 18, and 18 Tdtomato and EV-transduced hCFs collected on day 3 (D3M3), 5 (D5M1), 7 (D7M1), and 9 (D9M1), and 248, 54, 64, and 62 hMGT- and miR-133-transduced hCFs collected at day 3 (D3M1, D3M2, and D3M3), 5 (D5M1), 7 (D7M1) and 9 (D9M1, Table S3). Gene expression was counted with Htseq-count using the union mode against the UCSC hg19 annotation file (Anders et al., 2015) (http://www-huber.embl.de/users/anders/HTSeq). No-feature and ambiguous counts were excluded for downstream analysis.
Normalization of scRNA-seq data
The human mRNA raw counts were then normalized using SCnorm (Bacher et al., 2017) to correct the dependence of read counts on sequencing depth (FilterCellProportion = .1, 1 condition). Plate D0hCF, D3M1, D3M2, and D3UN were normalized together (329 cells, K = 7, Fig. 2D and Fig. S2C) and plate D3M3, D5M1, D7M1, and D9M1 were normalized together (346 cells, K=9, Fig. S7C,D). The count-depth plot (Fig. 2D and Fig. S7C) clearly documented the shift of slope (indicating the dependence of gene counts on sequencing depth of each cell and each gene) from 1 before normalization to 0 after normalization, suggesting the successful removal of count-depth dependence. PCA analyses suggest that normalization with SCnorm outperformed our previous normalization strategy (Liu et al., 2017b) using DEseq (Bacher et al., 2017) and SCnorm outperformed our previous strategy judging from PCA (Fig. 2E, F). After normalization, total number of counts per cell is about 1 million (Fig. S2C, S7D). Therefore the unit of gene expression reported here is counts per million (CPM). Based on the normalized DsRed counts, cells in D3UN were classified as DsRed-infected (R, expressing high levels of DsRed), or uninfected cells (U, low DsRed expression, Fig. S2E). Similarly, cells in D3M3, D5M1, D7M1, and D9M1 were classified as Tdtomato-infected or MGTmiR133-infected based on Tdtomato versus MGTmiR133 counts (Fig. S7G).
Analysis of scRNA-seq data
Dimension reduction and correlation analysis
Single cell transcriptomes normalized with SCnorm were analyzed in two batches. D0hCF, D3M1, D3M2, and D3UN were analyzed together (Fig. 2–6, S2–S6, 329 cells in total). D3M3, D5M1, D7M1, and D9M1 (323 cells) were analyzed together with the 14 day 3 uninfected control cells from plate D3UN after sequencing depth adjustment (Fig. 6–7 and Fig. S7–S9, 337 cells in total). First, genes expressed at a level below limit of detection (average normalized expression = 1 CPM) were excluded, resulting in 13381 and 13479 detected genes for the two batches of analyses. Then a logarithm transformation (log2)is applied to all single-cell data to avoid non-Gaussianity. Outlier removal, PCA, HC, tSNE, ANOVA, and the generation of volcano and violin plots were performed with the “SINGuLAR Analysis Toolset” package (Fluidigm) in R. Expression of the reprogramming factors M, G, T, and miR133 was excluded before analysis. Next, top 400 PCA genes were selected by largest weight (loading) contribution to principle component 1, 2, or 3. Then HC was performed with these 400 genes and cells were grouped (grouping information in Table S3). tSNE was also performed with these genes. One-way ANOVA was performed on all detected genes using cell clusters determined by HC and pairwise comparisons of cells on reprogramming route vs. refractory route were performed for each time point. The expression of a total of 4333 genes were considered statistically significant by showing p value < 0.05 in one or more of the four pairwise comparisons (Table S5). Genes showing p < 0.05 and fold change > 2 between cells on reprogramming vs. refractory route at all four time points were selected as potential positive or negative markers of reprogramming. SLICER analysis, pseudotime calculation and identification of genes significantly related to reprogramming progress were performed as previously described (Liu et al., 2017b) except that SLICER auto-selected genes were used. For the calculation of Fig. 7I, cells on branch 1 and 3 (Fig. S8E) were included. For the calculation of Fig. 7J, cells on branch 1 and 2 (Fig. S8E) were included. Analysis of cell cycle status with reCAT (Liu et al., 2017c) was performed with R package available at https://github.com/tinglab/reCAT. GO analysis was performed using the DAVID functional annotation tool version 6.8 (https://david.ncifcrf.gov/). The long list of GO terms then were categorized and visualized by REVIGO at http://revigo.irb.hr (Supek et al., 2011). For correlation analysis, only reprogramming cells were included. Potential miR-133 target genes were determined using the TargetScan tool (Agarwal et al., 2015); precomputed predictions were downloaded from http://www.targetscan.org/vert_71. Potential TBX5 targets were obtained as previously reported (Liu et al., 2017b). Because all three reprogramming factors were expressed from one polycistronic viral vector, expression counts of all three factors were averaged and used to calculate correlation between TBX5 and its targets. The associated p-values were obtained by considering a t distribution with degrees of freedom being equal to the sample size minus 2, under the null hypothesis. To identify the potential targets of miR-133, we ranked the Pearson correlation coefficient between expression of miR-133 and other genes in MEF2C highly expressed cells (expression value > 200), and picked up the hits with negative Pearson’s R (< −0.2) and high prediction score (> 60) provided by miRDB (http://mirdb.org) (Wong and Wang, 2015) as candidates for further functional validation.
Transcriptome heterogeneity
In order to estimate intercellular heterogeneity of d0hCF and compare heterogeneity in human v.s. mouse fibroblasts receiving different treatments, intercellular transcriptome variance was calculated. Single cell gene expression profiles of mESC (2iLif), hESC, day 0 MEF, and five primary hGMB cells were obtained from previous studies (Patel et al., 2014). Htseq counts of uninfected, DsRed-infected, and reprogramming mouse fibroblasts from 513 single cells in our previous study (Liu et al., 2017b) were normalized by SCnorm first in order to be compared with human cells. To ensure that gene expression calculated from different pipelines are comparable, linear expression of all single cell datasets was scaled so that the median total mRNA counts of each cell populations equal to 1 million. Then each expression dataset was filtered by removing genes with average expression level < 1. Log2(Exp+1) was calculated and intercellular variance of each gene was calculated. The distribution of variances for all genes per cell type/treatment condition was then plotted as box plots and statistics were calculated with ANOVA followed by Bonferroni correction.
Cell Fate Index
The cell-fate index of each single cell was calculated in three steps: calculation of differentially expressed genes (DEG) between starting and target cell types from bulk transcriptome data, filtering DEG to obtain genelists representing the starting and target cell fates, and calculation of cell-fate index using the transcriptome of single cells. To calculate DEG between cardiac fibroblast (starting cell type) and cardiomyocytes (target cell type), bulk RNA-seq data in previously published studies were downloaded from the Gene Expression Omnibus (GEO) website: mouse neonatal cardiac fibroblasts (mCF, GSM1223642/3) and cardiomyocytes (mCM, GSM1223646/7) (Giudice et al., 2014), human ESC (H1, GSM2264850/1, H9, GSM2264858/9 (Liu et al., 2017a) and human ESC-derived cardiomyocyte (H1, GSM2264856/7, H9, GSM2264864/5 (Liu et al., 2017a), H7, GSM1536176/7/8 (Kuppusamy et al., 2015). Quality of sequencing results was first checked by FASTQC and low-quality bases were trimmed off by trimmomatic/0.36 (trim until the average score of adjacent 4 bases is above 30). Processed reads were then mapped to mm10 or hg19 with Tophat2. Gene expression was counted with Htseq-count using the union mode (Anders et al., 2015) (http://www-huber.embl.de/users/anders/HTSeq). DEG between mCF and mCM were calculated with DESeq2 (Love et al., 2014). Due to lack of human cardiac fibroblast bulk RNA-seq data, we used assembly of our day 3 uninfected hCF single cell RNA-seq data for the calculation of DEG between hCF and hESC-CM (H7, referred to as hCM hereafter). Briefly, the 14 d3hU cells were randomly divided into two groups (7 cells per group) and average gene expression was calculated for each group based on normalized single-cell gene counts from SCnorm. The two groups mimic a duplicated bulk RNA-seq sample. Gene expression from the two groups were then scaled up (about 30 fold) to the same sequencing depth as hESC-CM because differences in sequencing depth would lead to miscalculation of DEG by DESeq2. After the scale-up, gene expression values were rounded up because DESeq2 requires raw counts (integer) as input data.
To obtain gene lists for the calculation of cell-fate index, the following filtering criteria were applied to DEGs calculated between mCF and mCM, and between hCF and hCM. For mouse, only DEGs with p < 0.05, average normalized expression (counts per million, CPM) > 5, and fold change (FC) of expression in CF versus CM (either direction) > 1.25 were kept. Then DEGs with 0.01< p < 0.05 and 1.25 < FC < 1.5 were further excluded, resulting in 2428 genes expressed higher in mCF and 2965 genes expressed higher in mCM. For human, first, only DEGs with p < 0.05, CPM > 1, and FC > 1.25 were kept. Second, in order to exclude carry-over ESC genes in hESC-CM, DEGs between hESC-H1 and hESC-CM-H1 and those between hESC-H9 and hESC-CM-H9 were calculated with DESeq2 and overlapping genes between H1 and H9 that were expressed at the same level in ESC and CM (p > 0.05) or expressed higher in ESC than CM (p < 0.05) were excluded. Third, DEGs with 1 < CPM < 5 and 0.001 < p < 0.05, and DEGs with 5 < CPM < 50, 0.01< p < 0.05 and 1.25 < FC < 1.5 were further excluded, resulting in 2866 genes expressed higher in hCF and 2713 genes expressed higher in hCM. Next, GO analyses were performed with the four selected gene lists and enriched GO terms were grouped based on their related biological function. Then genes in each GO term group were combined and used for the calculation of cell-fate index.
To calculate cell-fate index for each gene list, we first defined the average of the beginning 10 cells in pseudotime (smallest pseudotime value, Fig. 3G for human and Fig. 1e (Liu et al., 2017b) for mouse) as the start point (CF), and the average of mCM or hCM bulk transcriptome as the end point (CM). Next, for each gene j in each single cell i, we calculated relative expression of that gene as rij = (gij – gCF) / (gCM – gCF) for gene lists expressed higher in CM than CF, and as rij = (gij – gCM) / (gCF – gCM) for gene lists expressed higher in CF than CM, where g denotes normalized gene expression from SCnorm. The value of rij was set to 0 or 1 when it’s < 0 or > 1. Then for each single cell i and each gene list containing m genes, we calculated the cell fate index (CFIi) as the weighted sum of relative gene expression across different genes: CFIi = <MI>wj ×rij, where the weight of each gene wj is proportional to its expression level at the end point (gCM) such that the sum of wj adds up to 1. The use of both relative gene expression rij and weight wj is to ensure that all expression changes are on the same scale of 0~1. For gene lists expressed higher in CF than CM, wj is proportional to its expression level at the start point (gCF). The plots in Fig. 4 were generated by sorting the cell fate indexes for all cells and then plotting on the y-axis with x-axis being the cell fraction time between 0 and 1. Day 3 cells on branch 1 and 2 (Fig. S4C) were included for human and non-proliferating cells (Fig. 1e in ref (Liu et al., 2017b) were used for mouse).
To compare the “reprogramming rate” in mouse and human (Fig. 7A), we consider the quantile-quantile plot (QQ-plot) by using 600 equally-spaced quantiles from the cell fate indexes of both human and mouse. We then fit a linear regression of mouse’s quantiles over human’s quantiles. The slope parameter estimate is 1.559 with standard error 0.005. This result suggests that the mouse progression rate is 1.56 times faster than that of human. The rate estimate is also highly significant p-value < 1e-16, which suggests that the speed of progression in mouse cells is statistically significantly higher than that of human cells. For the estimation of the smoothed CFI curve and its derivative (Figure S5E–F, and 7B–C), we adopt the cubic smoothing spline method, which is a nonparametric statistical approach that has been widely used for estimation and smoothing of functions (Gu, 2013; Wahba, 1990) Compared to parametric methods (e.g., linear regression), smoothing spline provides a wide choice of shapes to characterize the behavior of CFI over time, which is extremely useful in our situation since the CFI curve and its derivatives can be highly nonlinear as the study time varies. In addition, smoothing splines effectively deals with the over-fitting problem by shrinking the coefficients of the estimated function (in terms of its basis expansion). The main idea behind cubic smoothing spline is very simple and intuitive the underlying smooth function (e.g., CFI curve) is approximated by a linear combination of polynomials. This idea allows easy computations of the derivative and is theoretically well-supported based on the results from the approximation theory for polynomials.
RNA velocity
We calculated RNA velocity using the velocyto.R package (https://github.com/velocyto-team/velocyto.R). Following the examples in the notebooks included with the package, we estimated gene-relative velocity using a gamma fit based on extreme quantiles. We did not employ knn pooling, as this might remove the effects of “opposing flows”, which we observed in Fib/iFib cells. To produce the plots shown in Fig. 4I–J, we projected the vector field onto the SLICER trajectory using the show.velocity.on.embedding.cor function with the same settings as in the sample notebook.
Statistical analyses
Where appropriate, values are presented as the mean ± SEM of replicate experiments. Statistical analyses were performed with Student t test or one-way ANOVA followed by post hoc tests. Generally, * P < 0.05 was considered statistically significant, *** P < 0.01 was considered highly significant and *** P < 0.001 was considered very highly significant. All data are representative of multiple repeated experiments. Statistical analyses were performed in Prism or R 3.3.2, which is freely available at https://www.r-project.org/
DATA AND SOFTWARE AVAILABILITY
Software
All software is freely or commercially available and is listed in the STAR Methods description and Key Resources Table.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse monoclonal to cardiac troponin T | ICC: abcam clone 1F11 Flow: Thermo Fisher clone: 3–11 |
ICC: ab10214 Flow: MS-295-p |
| Mouse monoclonal to a-Actinin (clone EA-53) | sigma | A7811–2ml |
| Rabbit polyclonal to Connexin 43 polyclonal | abcam | Ab11370 |
| Rabbit polyclonal to GFP | Thermo Fisher | A11122 |
| Donkey anti-mouse IgG(H+L), AlexaFluor647 conjugated | Jackson Lab | 715–695-151 |
| Donkey anti-rabbit IgG(H+L), AlexaFluor488 conjugated | Jackson Lab | 711–545-152 |
| Biotin anti-Thy1.2 | Invitrogen | 13–0903-85 |
| Bacterial and Virus Strains | ||
| E.coli: HB101 | Promega | |
| Biological Samples | ||
| Human cardiac tissue from a male patient | Duke Human Heart Repository | https://sites.duke.edu/dhhr/ |
| Neonatal P1.5 mouse hearts | This paper | |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Poly-l-lysine | Electron microscopy science (EMS) | 19321-B |
| DMEM | Corning | 10–013-CV |
| FBS | Millipore | TMS-013-B |
| Non-essential amino acids | Corning | 25–025-CI |
| Penicillin/streptomycin | Corning | 30–002-CI |
| PEG6000 | Sigma | 81255–2.5g |
| 0.05% Trypsin | Corning | 25–052-CI |
| M199 | Corning | 10–060-CV |
| Polybrene | Millipore | TR-1003-G |
| SurCoat | Celltron | SC-9035 |
| RPMI1640 | gibco | 11875–093 |
| B27 supplement | gibco | 17504–044 |
| BSA | Hyclone | SH3057402 |
| GlutaMAX | gibco | 35050–061 |
| Gelatin | sigma | G1393–100ml |
| Collagenase type II | Worthington | LS004176 |
| HBSS | Corning | 21–022-CV |
| EDTA | sigma | E5134 |
| Anti-Biotin Microbeads | Miltenyi Biotec | 130–090-485 |
| TRIzol | Life technologies | 15596018 |
| 32% paraformaldehyde | EMS | 15714 |
| Triton X-100 | Acros | 21568–2500 CAS 900293–1 |
| Hoechst 33342 | Life Technologies | H3570 |
| Isopropanol | Fisher | M-15805 |
| Puromycin | Thermo Fisher | A1113803 |
| Critical Commercial Assays | ||
| NanoFect transfection Reagent | Alstem | NF101 |
| neonatal cardiomyocytes isolation system | Worthington Biochemical Corporation | 130–101-372 |
| Fixation/Permeabilization Solution | BD Biosciences | 51–2090KZ |
| Perm/Wash Solution | BD Biosciences | 51–2091KZ |
| SuperScript III Reverse Transcriptase kit | Invitrogen | 18080093 |
| Power SYBR Green PCR Master Mix | Applied Biosystems | 1710515 |
| DNeasy Blood & Tissue Kits | Qiagen | 69504 |
| EZ DNA Methylation-Gold Kit | Zymo Research | D5005 |
| EpiMark Hot Start Taq DNA Polymerase | NEB | M0490S |
| NearIR live/dead dye | Thermo Fisher Scientific | L10119 |
| Carboxyfluorescein succinimidyl ester (CFSE) dye | Thermo Fisher Scientific | 65–0850-84 |
| Ambion Array Control spike-ins | Thermo Fisher | AM1780 |
| External RNA Controls Consortium (ERCC) RNA spike-in Mix 1 | Ambion | 4456740 |
| C1™ Single-Cell mRNA Seq IFC, 17–25 µm—10 IFCs | Fluidigm | 100–6042 |
| C1™ Single-Cell Reagent Kit for mRNA Seq | Fluidigm | 100–6201 |
| Deposited Data | ||
| Raw and analyzed data | This paper | GEO: GSE106888 |
| Human reference genome NCBI build 37, GRCh37 | Genome Reference Consortium | http://www.ncbi.nlm.nih.gov/projects/genome/assembly/grc/human/ |
| Experimental Models: Cell Lines | ||
| Human: PlatE | Cell Biolabs | RV-101 |
| Human: 293LTV Cell Line | Cell Biolabs | LTV-100 |
| Human: ESC H9 derived fibroblasts: H9F | Fu et al., 2013 | |
| Human: primary cardiac fibroblasts: HCF1 | This paper | |
| Human: primary cardiac fibroblasts: HCF2 | Cheng et al., 2012 | |
| Experimental Models: Organisms/Strains | ||
| Mouse: aMHC-GFP transgenic CD1 | Ieda et al., 2010 | |
| Oligonucleotides | ||
| See primer sequences for RT-PCR in Table S2 | This paper | |
| Primers for bisulfite sequencing | Fu et al., 2013 | |
| Recombinant DNA | ||
| pMXs-MEF2C | Fu et al., 2013 | |
| pMXs-GATA4 | Fu et al., 2013 | |
| pMXs-TBX5 | Fu et al., 2013 | |
| pMXs-ESRRG | Fu et al., 2013 | |
| pMXs-MESP1 | Fu et al., 2013 | |
| pMXs-MYOCD | Fu et al., 2013 | |
| pBabe-GATA4 | Nam et al., 2013 | |
| pBabe-HAND2 | Nam et al., 2013 | |
| pBabe-MEF2C | Nam et al., 2013 | |
| pBabe-TBX5 | Nam et al., 2013 | |
| pBabe-MYOCD | Nam et al., 2013 | |
| pBabe-MESP1 | Nam et al., 2013 | |
| pBabe-miR-1 | Nam et al., 2013 | |
| pBabe-miR-133 | Nam et al., 2013 | |
| pMXs-puro-hMGT | This paper | |
| pMXs-puro-LacZ | Cell Biolabs | |
| pMXs-puro-dsRed | Wang et al., 2015a | |
| pMXs-GFP | Wang et al., 2015a | |
| pMXs-tdTomato | Ziqing Liu et al. | |
| TroponinT-GCaMP5-Zeo | Addgene #46027 | |
| pCMV-VSV-G | Addgene #8454 | |
| pUMVC | Addgene #8449 | |
| psPAX2 | Addgene #12260 | |
| MG2.G | Addgene #12259 | |
| pGEMT | Promega | |
| See list of shRNA cloned in pLKO.1 vector in Table S2. | Sigma | |
| Software and Algorithms | ||
| ImageJ (Fiji version) | NIH | https://imagej.net/Fiji/Downloads |
| FlowJo | ||
| GraphPad Prism | ||
| liteCam HD | RSUPPORT | |
| EVOS microscope system | Invitrogen | |
| Bisulfite Sequencing DNA Methylation Analysis (BISMA) | Rohde et al., 2010 | |
| R 3.3.2 | https://www.r-project.org/ | |
| SLICER | Welch et al., 2016 | https://github.com/jw156605/SLICER |
| SCnorm | Bacher et al., 2017 | https://bioconductor.org/packages/release/bioc/html/SCnorm.html |
| SINGuLAR Analysis Toolset | Fluidigm | https://www.fluidigm.com/software/ |
| reCAT | Zehua Liu et al., 2017 | https://github.com/tinglab/reCAT |
| DESeq2 | Love et al., 2014 | |
| TargetScan tool | Agarwal et al., 2015 | http://www.targetscan.org/vert_71 |
| miRDB | Wong and Wang, 2015 | http://mirdb.org |
| DAVID GO | https://david.ncifcrf.gov/ | |
| REVIGO | Supek et al., 2011 | http://revigo.irb.hr |
| HTSeq | Anders et al., 2015 | http://www.huber.embl.de/users/anders/HTSeq |
| velocyto.R | La Manno et al., 2018 | https://github.com/velocytoteam/velocyto.R)) |
| Other | ||
Data Resources
The accession number for the scRNA-seq data reported in this study is GEO: GSE106888.
Supplementary Material
Movie S1. Related to Figure 1. Calcium fluxes indicated by GCamp5 GFP fluorescence.
Movie S2. Related to Figure 1. Contractile hiCMs co-cultured with mouse neonatal CMs.
Table S1. Related to Figure 2, 6, S1, and S6. Sample information for all single cells (mapping and counting summaries, treatment, and HC cluster assignment).
Table S2. Related to Figures 3 and 4, and Methods. shRNA oligoes used for RNAi loss of function assay and primers used for qRT-PCR.
Table S3. Related to Figure 5. Gene lists for the calculation of cell fate index.
Table S4. Related to Figure 6 and 7. Gene list of the 6 clusters in Fig 6I–6J and gene list of positive and negative markers for hiCMs.
Table S5. Related to Figure 7. ANOVA and pairwise comparison results between cells on reprogramming vs. refractory routes.
Highlights:
Single-cell transcriptomics reveals a bifurcated trajectory of hiCM reprogramming
Discovery of a decision point for a cell to enter reprogramming or refractory route
Identification of hiCM specific features when compared to miCM
Development of cell fate index to evaluate the progression of cell fate conversion
ACKNOWLEDGEMENTS
We thank UNC AA Core, HTSF Core, and Flow Cytometry Core for technical support. This study was supported by NIH HG06272 to J.F.P., NIH BD2K Fellowship (T32 CA201159) and NIH F31 Fellowship (HG008912) to J.D.W., UNC Integrative Vascular Biology Training Grant (NIH T32 HL069768, PI: C. Mack) to B.K., NIH/NHLBI R01HL139880 to J.L., AHA 18TPA34180058, NIH/NHLBI R01HL128331, R01HL144551 to L.Q., and generous gifts from Dr. Hugh “Chip” McAllister and Cecil Sewell.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Addis RC, Ifkovits JL, Pinto F, Kellam LD, Esteso P, Rentschler S, Christoforou N, Epstein J. a., and Gearhart JD (2013). Optimization of direct fibroblast reprogramming to cardiomyocytes using calcium activity as a functional measure of success. J. Mol. Cell. Cardiol 60, 97–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agarwal V, Bell GW, Nam J-W, and Bartel DP (2015). Predicting effective microRNA target sites in mammalian mRNAs. Elife 4, e05005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anders S, Pyl PT, and Huber W (2015). HTSeq-A Python framework to work with high-throughput sequencing data. Bioinformatics 31, 166–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bacher R, Chu L-F, Leng N, Gasch AP, Thomson JA, Stewart RM, Newton M, and Kendziorski C (2017). SCnorm: robust normalization of single-cell RNA-seq data. Nat. Methods 14, 584–586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bektik E, Dennis A, Pawlowski G, Zhou C, Maleski D, Takahashi S, Laurita K, Deschênes I, Fu J-D, Bektik E, et al. (2018). S-phase Synchronization Facilitates the Early Progression of Induced-Cardiomyocyte Reprogramming through Enhanced Cell-Cycle Exit. Int. J. Mol. Sci 19, 1364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benson MA, Tinsley CL, Waite AJ, Carlisle FA, Sweet SMM, Ehler E, George CH, Lai FA, Martin-Rendon E, and Blake DJ (2017). Ryanodine receptors are part of the myospryn complex in cardiac muscle. Sci. Rep 7, 6312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biddy BA, Kong W, Kamimoto K, Guo C, Waye SE, Sun T, and Morris SA (2018). Single-cell mapping of lineage and identity in direct reprogramming. Nature 564, 219–224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bujak M, and Frangogiannis NG (2007). The role of TGF-ȕ signaling in myocardial infarction and cardiac remodeling. Cardiovasc. Res [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng K, Blusztajn A, Shen D, Li TS, Sun B, Galang G, Zarembinski TI, Prestwich GD, Marbán E, Smith RR, et al. (2012). Functional performance of human cardiosphere-derived cells delivered in an in situ polymerizable hyaluronan-gelatin hydrogel. Biomaterials 33, 5317–5324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christoforou N, Chakraborty S, Kirkton RD, Adler AF, Addis RC, and Leong KW (2017). Core Transcription Factors, MicroRNAs, and Small Molecules Drive Transdifferentiation of Human Fibroblasts Towards The Cardiac Cell Lineage. Sci. Rep 7, 40285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durham JT, Brand OM, Arnold M, Reynolds JG, Muthukumar L, Weiler H, Richardson JA, and Naya FJ (2006). Myospryn is a direct transcriptional target for MEF2A that encodes a striated muscle, Į-actinin-interacting, costamere-localized protein. J. Biol. Chem [DOI] [PubMed] [Google Scholar]
- Fu JD, Stone NR, Liu L, Spencer CI, Qian L, Hayashi Y, Delgado-Olguin P, Ding S, Bruneau BG, and Srivastava D (2013). Direct reprogramming of human fibroblasts toward a cardiomyocyte-like state. Stem Cell Reports 1, 235–247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giudice J, Xia Z, Wang ET, Scavuzzo M. a, Ward AJ, Kalsotra A, Wang W, Wehrens XHT, Burge CB, Li W, et al. (2014). Alternative splicing regulates vesicular trafficking genes in cardiomyocytes during postnatal heart development. Nat. Commun 5, 3603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu C (2013). Smoothing Spline ANOVA Models (New York, NY: Springer New York; ). [Google Scholar]
- He A, Kong SW, Ma Q, and Pu WT (2011). Co-occupancy by multiple cardiac transcription factors identifies transcriptional enhancers active in heart. Proc. Natl. Acad. Sci. U. S. A 108, 5632–5637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodgkinson CP, Pratt RE, Kirste I, Dal-Pra S, Cooke JP, and Dzau VJ (2018). Cardiomyocyte Maturation Requires TLR3 Activated Nuclear Factor Kappa B. Stem Cells [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ieda M, Fu JD, Delgado-Olguin P, Vedantham V, Hayashi Y, Bruneau BG, and Srivastava D (2010). Direct reprogramming of fibroblasts into functional cardiomyocytes by defined factors. Cell 142, 375–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kojima H, and Ieda M (2017). Discovery and progress of direct cardiac reprogramming. Cell. Mol. Life Sci 74, 2203–2215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuppusamy KT, Jones DC, Sperber H, Madan A, Fischer KA, Rodriguez ML, Pabon L, Zhu W-Z, Tulloch NL, Yang X, et al. (2015). Let-7 family of microRNA is required for maturation and adult-like metabolism in stem cell-derived cardiomyocytes. Proc. Natl. Acad. Sci 112, E2785–E2794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J, Sayed N, Hunter A, Au KF, Wong WH, Mocarski ES, Pera RR, Yakubov E, and Cooke JP (2012). Activation of innate immunity is required for efficient nuclear reprogramming. Cell 151, 547–558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lein E, Borm LE, and Linnarsson S (2017). The promise of spatial transcriptomics for neuroscience in the era of molecular cell typing. Science (80-. ) 358, 64–69. [DOI] [PubMed] [Google Scholar]
- Lipowsky G (2002). Exportin 4: a mediator of a novel nuclear export pathway in higher eukaryotes. EMBO J [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu N, Bezprozvannaya S, Williams AH, Qi X, Richardson JA, Bassel-Duby R, and Olson EN (2008). microRNA-133a regulates cardiomyocyte proliferation and suppresses smooth muscle gene expression in the heart. Genes Dev 22, 3242–3254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Q, Jiang C, Xu J, Zhao M-T, Van Bortle K, Cheng X, Wang G, Chang HY, Wu JC, and Snyder MP (2017a). Genome-Wide Temporal Profiling of Transcriptome and Open Chromatin of Early Cardiomyocyte Differentiation Derived From hiPSCs and hESCsNovelty and Significance. Circ. Res 121, 376–391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Z, Wang L, Welch JD, Ma H, Zhou Y, Vaseghi HR, Yu S, Wall JB, Alimohamadi S, Zheng M, et al. (2017b). Single-cell transcriptomics reconstructs fate conversion from fibroblast to cardiomyocyte. Nature 551, 100–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Z, Lou H, Xie K, Wang H, Chen N, Aparicio OM, Zhang MQ, Jiang R, and Chen T (2017c). Reconstructing cell cycle pseudo time-series via single-cell transcriptome data. Nat. Commun 8, 22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Z, Chen O, Wall JBJ, Zheng M, Zhou Y, Wang L, Ruth Vaseghi H, Qian L, and Liu J (2017d). Systematic comparison of 2A peptides for cloning multi-genes in a polycistronic vector. Sci. Rep 7, 2193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love MI, Huber W, and Anders S (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15, 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- La Manno G, Soldatov R, Zeisel A, Braun E, Hochgerner H, Petukhov V, Lidschreiber K, Kastriti ME, Lönnerberg P, Furlan A, et al. (2018). RNA velocity of single cells. Nature 560, 494–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathys H, Basquin JÔ, Ozgur S, Czarnocki-Cieciura M, Bonneau F, Aartse A, Dziembowski A, Nowotny M, Conti E, and Filipowicz W (2014). Structural and Biochemical Insights to the Role of the CCR4-NOT Complex and DDX6 ATPase in MicroRNA Repression. Mol. Cell [DOI] [PubMed] [Google Scholar]
- Mohamed TMA, Stone NR, Berry EC, Radzinsky E, Huang Y, Pratt K, Ang Y-S, Yu P, Wang H, Tang S, et al. (2016). Chemical Enhancement of In Vitro and In Vivo Direct Cardiac Reprogramming. Circulation [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nam Y-J, Song K, Luo X, Daniel E, Lambeth K, West K, Hill J. a, DiMaio JM, Baker L. a, Bassel-Duby R, et al. (2013). Reprogramming of human fibroblasts toward a cardiac fate. Proc. Natl. Acad. Sci. U. S. A 110, 5588–5593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel AP, Tirosh I, Trombetta JJ, Shalek AK, Gillespie SM, Wakimoto H, Cahill DP, Nahed BV, Curry WT, Martuza RL, et al. (2014). Single-cell RNA-seq highlights intratumoral heterogeneity in primary glioblastoma. Science (80-. ) 344, 1396–1401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qian L, Huang Y, Spencer CI, Foley A, Vedantham V, Liu L, Conway SJ, Fu J, and Srivastava D (2012). In vivo reprogramming of murine cardiac fibroblasts into induced cardiomyocytes. Nature 485, 593–598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohde C, Zhang Y, Reinhardt R, and Jeltsch A (2010). BISMA - Fast and accurate bisulfite sequencing data analysis of individual clones from unique and repetitive sequences. BMC Bioinformatics [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sayed N, Wong WT, Ospino F, Meng S, Lee J, Jha A, Dexheimer P, Aronow BJ, and Cooke JP (2015). Transdifferentiation of human fibroblasts to endothelial cells: role of innate immunity. Circulation 131, 300–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sayed N, Ospino F, Himmati F, Lee J, Chanda P, Mocarski ES, and Cooke JP (2017). Retinoic Acid Inducible Gene 1 Protein (RIG1)-Like Receptor Pathway Is Required for Efficient Nuclear Reprogramming. Stem Cells [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shao W, Halachmi S, and Brown M (2002). ERAP140, a Conserved Tissue-Specific Nuclear Receptor Coactivator. Mol. Cell. Biol [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song K, Nam Y-J, Luo X, Qi X, Tan W, Huang GN, Acharya A, Smith CL, Tallquist MD, Neilson EG, et al. (2012). Heart repair by reprogramming non-myocytes with cardiac transcription factors. Nature 485, 599–604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stubbington MJT, Rozenblatt-Rosen O, Regev A, and Teichmann SA (2017). Single-cell transcriptomics to explore the immune system in health and disease. Science (80-. ) 358, 58–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugiyama Y, Suzuki A, Kishikawa M, Akutsu R, Hirose T, Waye MMY, Tsui SKW, Yoshida S, and Ohno S (2000). Muscle develops a specific form of small heat shock protein complex composed of MKBP/HSPB2 and HSPB3 during myogenic differentiation. J. Biol. Chem [DOI] [PubMed] [Google Scholar]
- Supek F, Bošnjak M, Škunca N, and Šmuc T (2011). Revigo summarizes and visualizes long lists of gene ontology terms. PLoS One [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torella D, Iaconetti C, Catalucci D, Ellison GM, Leone A, Waring CD, Bochicchio A, Vicinanza C, Aquila I, Curcio A, et al. (2011). MicroRNA-133 controls vascular smooth muscle cell phenotypic switch in vitro and vascular remodeling in vivo. Circ. Res [DOI] [PubMed] [Google Scholar]
- Treutlein B, Lee QY, Camp JG, Mall M, Koh W, Shariati SAM, Sim S, Neff NF, Skotheim JM, Wernig M, et al. (2016). Dissecting direct reprogramming from fibroblast to neuron using single-cell RNA-seq. Nature 534, 391–395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- VANE JR (1971). Inhibition of Prostaglandin Synthesis as a Mechanism of Action for Aspirin-like Drugs. Nat. New Biol [DOI] [PubMed] [Google Scholar]
- Vaseghi H, Liu J, and Qian L (2017). Molecular barriers to direct cardiac reprogramming. Protein Cell [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wada R, Muraoka N, Inagawa K, Yamakawa H, Miyamoto K, Sadahiro T, Umei T, Kaneda R, Suzuki T, Kamiya K, et al. (2013). Induction of human cardiomyocyte-like cells from fibroblasts by defined factors. Proc. Natl. Acad. Sci. U. S. A 110, 12667–12672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wahba G (1990). Spline Models for Observational Data (Society for Industrial and Applied Mathematics)
- Waldron L, Steimle JD, Greco TM, Gomez NC, Dorr KM, Kweon J, Temple B, Yang XH, Wilczewski CM, Davis IJ, et al. (2016). The Cardiac TBX5 Interactome Reveals a Chromatin Remodeling Network Essential for Cardiac Septation. Dev. Cell [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Liu Z, Yin C, Asfour H, Chen O, Li Y, Bursac N, Liu J, and Qian L (2015a). Stoichiometry of Gata4, Mef2c, and Tbx5 influences the efficiency and quality of induced cardiac myocyte reprogramming. Circ. Res 116, 237–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Liu Z, Yin C, Zhou Y, Liu J, and Qian L (2015b). Improved Generation of Induced Cardiomyocytes Using a Polycistronic Construct Expressing Optimal Ratio of Gata4, Mef2c and Tbx5. J. Vis. Exp e53426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welch JD, Hartemink AJ, and Prins JF (2016). SLICER: inferring branched, nonlinear cellular trajectories from single cell RNA-seq data. Genome Biol 17, 106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong N, and Wang X (2015). miRDB: An online resource for microRNA target prediction and functional annotations. Nucleic Acids Res [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu AR, Neff NF, Kalisky T, Dalerba P, Treutlein B, Rothenberg ME, Mburu FM, Mantalas GL, Sim S, Clarke MF, et al. (2013). Quantitative assessment of single-cell RNA-sequencing methods. Nat. Methods 11, 41–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y, Wang L, Vaseghi HR, Liu Z, Lu R, Alimohamadi S, Yin C, Fu J-D, Wang GG, Liu J, et al. (2016). Bmi1 Is a Key Epigenetic Barrier to Direct Cardiac Reprogramming. Cell Stem Cell 18, 382–395. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Movie S1. Related to Figure 1. Calcium fluxes indicated by GCamp5 GFP fluorescence.
Movie S2. Related to Figure 1. Contractile hiCMs co-cultured with mouse neonatal CMs.
Table S1. Related to Figure 2, 6, S1, and S6. Sample information for all single cells (mapping and counting summaries, treatment, and HC cluster assignment).
Table S2. Related to Figures 3 and 4, and Methods. shRNA oligoes used for RNAi loss of function assay and primers used for qRT-PCR.
Table S3. Related to Figure 5. Gene lists for the calculation of cell fate index.
Table S4. Related to Figure 6 and 7. Gene list of the 6 clusters in Fig 6I–6J and gene list of positive and negative markers for hiCMs.
Table S5. Related to Figure 7. ANOVA and pairwise comparison results between cells on reprogramming vs. refractory routes.
Data Availability Statement
Software
All software is freely or commercially available and is listed in the STAR Methods description and Key Resources Table.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse monoclonal to cardiac troponin T | ICC: abcam clone 1F11 Flow: Thermo Fisher clone: 3–11 |
ICC: ab10214 Flow: MS-295-p |
| Mouse monoclonal to a-Actinin (clone EA-53) | sigma | A7811–2ml |
| Rabbit polyclonal to Connexin 43 polyclonal | abcam | Ab11370 |
| Rabbit polyclonal to GFP | Thermo Fisher | A11122 |
| Donkey anti-mouse IgG(H+L), AlexaFluor647 conjugated | Jackson Lab | 715–695-151 |
| Donkey anti-rabbit IgG(H+L), AlexaFluor488 conjugated | Jackson Lab | 711–545-152 |
| Biotin anti-Thy1.2 | Invitrogen | 13–0903-85 |
| Bacterial and Virus Strains | ||
| E.coli: HB101 | Promega | |
| Biological Samples | ||
| Human cardiac tissue from a male patient | Duke Human Heart Repository | https://sites.duke.edu/dhhr/ |
| Neonatal P1.5 mouse hearts | This paper | |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Poly-l-lysine | Electron microscopy science (EMS) | 19321-B |
| DMEM | Corning | 10–013-CV |
| FBS | Millipore | TMS-013-B |
| Non-essential amino acids | Corning | 25–025-CI |
| Penicillin/streptomycin | Corning | 30–002-CI |
| PEG6000 | Sigma | 81255–2.5g |
| 0.05% Trypsin | Corning | 25–052-CI |
| M199 | Corning | 10–060-CV |
| Polybrene | Millipore | TR-1003-G |
| SurCoat | Celltron | SC-9035 |
| RPMI1640 | gibco | 11875–093 |
| B27 supplement | gibco | 17504–044 |
| BSA | Hyclone | SH3057402 |
| GlutaMAX | gibco | 35050–061 |
| Gelatin | sigma | G1393–100ml |
| Collagenase type II | Worthington | LS004176 |
| HBSS | Corning | 21–022-CV |
| EDTA | sigma | E5134 |
| Anti-Biotin Microbeads | Miltenyi Biotec | 130–090-485 |
| TRIzol | Life technologies | 15596018 |
| 32% paraformaldehyde | EMS | 15714 |
| Triton X-100 | Acros | 21568–2500 CAS 900293–1 |
| Hoechst 33342 | Life Technologies | H3570 |
| Isopropanol | Fisher | M-15805 |
| Puromycin | Thermo Fisher | A1113803 |
| Critical Commercial Assays | ||
| NanoFect transfection Reagent | Alstem | NF101 |
| neonatal cardiomyocytes isolation system | Worthington Biochemical Corporation | 130–101-372 |
| Fixation/Permeabilization Solution | BD Biosciences | 51–2090KZ |
| Perm/Wash Solution | BD Biosciences | 51–2091KZ |
| SuperScript III Reverse Transcriptase kit | Invitrogen | 18080093 |
| Power SYBR Green PCR Master Mix | Applied Biosystems | 1710515 |
| DNeasy Blood & Tissue Kits | Qiagen | 69504 |
| EZ DNA Methylation-Gold Kit | Zymo Research | D5005 |
| EpiMark Hot Start Taq DNA Polymerase | NEB | M0490S |
| NearIR live/dead dye | Thermo Fisher Scientific | L10119 |
| Carboxyfluorescein succinimidyl ester (CFSE) dye | Thermo Fisher Scientific | 65–0850-84 |
| Ambion Array Control spike-ins | Thermo Fisher | AM1780 |
| External RNA Controls Consortium (ERCC) RNA spike-in Mix 1 | Ambion | 4456740 |
| C1™ Single-Cell mRNA Seq IFC, 17–25 µm—10 IFCs | Fluidigm | 100–6042 |
| C1™ Single-Cell Reagent Kit for mRNA Seq | Fluidigm | 100–6201 |
| Deposited Data | ||
| Raw and analyzed data | This paper | GEO: GSE106888 |
| Human reference genome NCBI build 37, GRCh37 | Genome Reference Consortium | http://www.ncbi.nlm.nih.gov/projects/genome/assembly/grc/human/ |
| Experimental Models: Cell Lines | ||
| Human: PlatE | Cell Biolabs | RV-101 |
| Human: 293LTV Cell Line | Cell Biolabs | LTV-100 |
| Human: ESC H9 derived fibroblasts: H9F | Fu et al., 2013 | |
| Human: primary cardiac fibroblasts: HCF1 | This paper | |
| Human: primary cardiac fibroblasts: HCF2 | Cheng et al., 2012 | |
| Experimental Models: Organisms/Strains | ||
| Mouse: aMHC-GFP transgenic CD1 | Ieda et al., 2010 | |
| Oligonucleotides | ||
| See primer sequences for RT-PCR in Table S2 | This paper | |
| Primers for bisulfite sequencing | Fu et al., 2013 | |
| Recombinant DNA | ||
| pMXs-MEF2C | Fu et al., 2013 | |
| pMXs-GATA4 | Fu et al., 2013 | |
| pMXs-TBX5 | Fu et al., 2013 | |
| pMXs-ESRRG | Fu et al., 2013 | |
| pMXs-MESP1 | Fu et al., 2013 | |
| pMXs-MYOCD | Fu et al., 2013 | |
| pBabe-GATA4 | Nam et al., 2013 | |
| pBabe-HAND2 | Nam et al., 2013 | |
| pBabe-MEF2C | Nam et al., 2013 | |
| pBabe-TBX5 | Nam et al., 2013 | |
| pBabe-MYOCD | Nam et al., 2013 | |
| pBabe-MESP1 | Nam et al., 2013 | |
| pBabe-miR-1 | Nam et al., 2013 | |
| pBabe-miR-133 | Nam et al., 2013 | |
| pMXs-puro-hMGT | This paper | |
| pMXs-puro-LacZ | Cell Biolabs | |
| pMXs-puro-dsRed | Wang et al., 2015a | |
| pMXs-GFP | Wang et al., 2015a | |
| pMXs-tdTomato | Ziqing Liu et al. | |
| TroponinT-GCaMP5-Zeo | Addgene #46027 | |
| pCMV-VSV-G | Addgene #8454 | |
| pUMVC | Addgene #8449 | |
| psPAX2 | Addgene #12260 | |
| MG2.G | Addgene #12259 | |
| pGEMT | Promega | |
| See list of shRNA cloned in pLKO.1 vector in Table S2. | Sigma | |
| Software and Algorithms | ||
| ImageJ (Fiji version) | NIH | https://imagej.net/Fiji/Downloads |
| FlowJo | ||
| GraphPad Prism | ||
| liteCam HD | RSUPPORT | |
| EVOS microscope system | Invitrogen | |
| Bisulfite Sequencing DNA Methylation Analysis (BISMA) | Rohde et al., 2010 | |
| R 3.3.2 | https://www.r-project.org/ | |
| SLICER | Welch et al., 2016 | https://github.com/jw156605/SLICER |
| SCnorm | Bacher et al., 2017 | https://bioconductor.org/packages/release/bioc/html/SCnorm.html |
| SINGuLAR Analysis Toolset | Fluidigm | https://www.fluidigm.com/software/ |
| reCAT | Zehua Liu et al., 2017 | https://github.com/tinglab/reCAT |
| DESeq2 | Love et al., 2014 | |
| TargetScan tool | Agarwal et al., 2015 | http://www.targetscan.org/vert_71 |
| miRDB | Wong and Wang, 2015 | http://mirdb.org |
| DAVID GO | https://david.ncifcrf.gov/ | |
| REVIGO | Supek et al., 2011 | http://revigo.irb.hr |
| HTSeq | Anders et al., 2015 | http://www.huber.embl.de/users/anders/HTSeq |
| velocyto.R | La Manno et al., 2018 | https://github.com/velocytoteam/velocyto.R)) |
| Other | ||
Data Resources
The accession number for the scRNA-seq data reported in this study is GEO: GSE106888.