

SUMMARY
CD40 has major roles in B cell development, activation, and germinal center responses. CD40 hypoactivity causes immunodeficiency whereas its overexpression causes autoimmunity and lymphomagenesis. To systematically identify B cell autonomous CD40 regulators, we use CRISPR/Cas9 genome-scale screens in Daudi B cells stimulated by multimeric CD40 ligand. These highlight known CD40 pathway components and reveal multiple additional mechanisms regulating CD40. The nuclear ubiquitin ligase FBXO11 supports CD40 expression by targeting repressors CTBP1 and BCL6. FBXO11 knockout decreases primary B cell CD40 abundance and impairs class-switch recombination, suggesting that frequent lymphoma monoallelic FBXO11 mutations may balance BCL6 increase with CD40 loss. At the mRNA level, CELF1 controls exon splicing critical for CD40 activity, while the N6-adenosine methyltransferase WTAP negatively regulates CD40 mRNA abundance. At the protein level, ESCRT negatively regulates activated CD40 levels while the negative feedback phosphatase DUSP10 limits downstream MAPK responses. These results serve as a resource for future studies and highlight potential therapeutic targets.
In Brief
CD40 is critical for B cell development, germinal center formation, somatic hypermutation, and class-switch recombination. Increased CD40 abundance is associated with autoimmunity and cancer, whereas CD40 hypoactivity causes immunodeficiency. Jiang et al. performed a genome-wide CRISPR/Cas9 screen to reveal key B cell factors that control CD40 abundance and that regulate CD40 responses.
Graphical Abstract
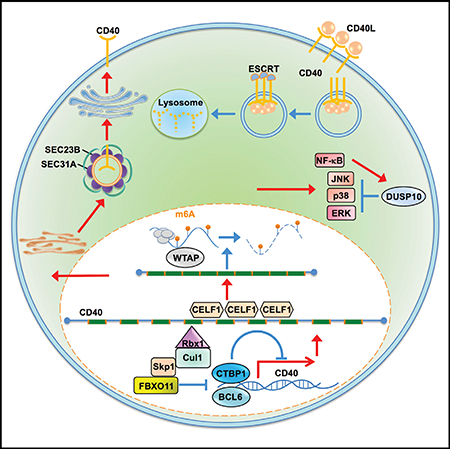
INTRODUCTION
Multiple signals are required to mount a successful humoral immune response. Together with B cell receptor activation by cognate antigen, the tumor necrosis factor receptor (TNFR) superfamily member CD40 (also called TNFRSF5) has obligatory roles in B cell activation, differentiation, survival, germinal center (GC) formation, and humoral responses (Bishop, 2009; Elgueta et al., 2009).
CD40 is activated by CD40-ligand (CD40L, also known as CD154), which is inducibly upregulated by T cells and multiple other cell types (Elgueta et al., 2009). Underscoring CD40 roles in humoral responses, congenital CD40L deficiency causes X-linked hyper-IgM (XHIGM) syndrome, with defective B cell function characterized by absence of memory, deficiency in class switch recombination (CSR) and somatic hypermutation, paucity of circulating isotype switched antibodies (Laman et al., 2017; van Kooten and Banchereau, 2000), and susceptibility to a broad range of pathogens (Johnson et al., 1993; Winkelstein et al., 2003). CD40 also has key roles in bidirectional communication between antigen-presenting cells and T cells.
CD40 is comprised of an extracellular ligand binding domain, a transmembrane domain, and a cytoplasmic tail. Activated CD40 recruits TNFR-associated factors (TRAFs) to three CD40 cytoplasmic tail domains to activate nuclear factor κB (NF-κB), mitogen activated kinase (MAPK), and phosphatidylinositol 3 kinase (PI3K) pathways (Bishop, 2004; Elgueta et al., 2009). Yet, negative regulators that down-modulate CD40 responses have not been characterized systematically.
CD40 upregulates multiple cytokines and cell surface molecules important for T cell activation, including the adhesion molecule ICAM1/CD54 and the costimulatory molecule B7–2/CD86 (Bishop, 2009; Elgueta et al., 2009; Hancock et al., 1996; Hennino et al., 2001; Liu et al., 1989; Tuscano et al., 1996). Interference with CD40/CD40L signaling collapses GC, which are secondary lymphoid organ structures necessary for key aspects of B cell development, differentiation somatic hypermutation, and class-switch recombination that underlie adaptive humoral responses (Han et al., 1995; Victora and Nussenzweig, 2012). Upregulation of the CD40 target Fas/CD95 is essential for GC B cell homeostasis (Hao et al., 2008).
CD40 levels must be tightly controlled to balance its essential roles in humoral responses with pathology that results from CD40 hyperactivity, but factors that control its plasma membrane abundance remain incompletely defined. Notably, a gain-of-function CD40 allele that increases its primary B cell plasma membrane abundance is associated with increased risk of rheumatoid arthritis (Li et al., 2013). Polymorphisms that elevate CD40 expression are associated with autoimmunity, including rheumatoid arthritis (Raychaudhuri et al., 2008), multiple sclerosis (Australia and New Zealand Multiple Sclerosis Genetics Consortium, 2009), Graves’ disease (Tomer et al., 2002), asthma (Park et al., 2007), Crohn’s disease (Blanco-Kelly et al., 2010), and systemic lupus erythematosus (Wakeland et al., 2001). Likewise, elevated CD40 abundance or signaling contributes to lymphomagenesis (Hatzivassiliou et al., 2007; Hömig-Hölzel et al., 2008; Huber et al., 2012; Nieters et al., 2011; Skibola et al., 2008).
Here, we use B cell CRISPR/Cas9 systematic genetic analysis to identify positive and negative regulators of CD40 responses.
RESULTS
Genome-wide CD40 CRISPR Screens
Daudi B cells with stable Cas9 expression were established for loss-of-function CD40 analysis, using CD40L-mediated upregulation of Fas plasma membrane (PM) abundance as a physiological readout of CD40 activity. To identify candidate CD40-positive and -negative regulators, we performed genome-wide pooled CRISPR screens, using the Avana single guide RNA (sgRNA) library (Doench et al., 2016). Five days post-transduction, B cells were stimulated with trimeric Mega-CD40L, and CD40-induced Fas levels were analyzed by fluorescence-activated cell sorting (FACS) (Holler et al., 2000). To identify candidate positive and negative regulators of CD40, populations of B cells with the lowest or highest three percent of plasma membrane (PM) Fas abundance were sorted, respectively (Figure 1A). As a control for general effects on PM protein levels, we gated out cells with diminished expression of an unrelated PM marker, CD37, whose B cell expression we found to be unaffected by CD40L stimulation. Genomic DNA was harvested from input versus sorted cell populations, and PCR-amplified sgRNA abundances were quantified by deep sequencing.
Figure 1. CRISPR/Cas9 Screens Identify Positive and Negative Regulators of CD40 Responses in B Cells.

(A) CRISPR/Cas9 screen workflow and screening strategy. Cas9+ Daudi B cells were transduced with the Avana sgRNA library at MOI 0.3, stimulated by multimerized CD40L at 50 ng/mL for 48 h and sorted for the 3% of cells with the lowest or highest Fas expression.
(B) Scatterplots showing the statistical significance of selected hits in the screen for CD40 positive regulators. −Log10(p value) for two biological screen replicates are shown. Statistical significance was quantitated by the STARS algorithm, using one biological screen replicate for each axis. Selected CD40 positive regulator screen hits are highlighted.
(C) Log2 sgRNA abundances in the indicated cell populations. sgRNA values in the input CRISPR Daudi cell library (red), in the sorted Faslow (blue) and Fashigh (green) populations are shown. Mean ± SD of two input libraries and four screen replicates are shown. ***p < 0.001.
(D) Scatterplots showing the −log10(p value) for two biological screen replicates. Selected CD40 negative regulator screen hits are highlighted.
(E) Schematic diagram of the CD40/NF-κB pathway, highlighting known components that scored in our screens. Positive regulator Faslow screen hits with p < 0.05 (red) or negative regulator Fashigh screen hit with p < 0.05 (light blue) are highlighted.
See also Figure S1.
The STARS algorithm was used to identify statistically significant hits, in which multiple independent sgRNAs against a human gene were enriched in either the sorted FasHi or FasLow populations versus the input population (Doench et al., 2016). At a multiple-hypothesis test adjusted q < 0.05 cutoff, 85 hits were identified as candidate positive regulators of CD40, whose knockout (KO) impaired CD40L-driven Fas upregulation (Figure 1B; Table S1). Encouragingly, FAS was the top hit in the sorted Faslow population, with anti-FAS sgRNAs ranking 3, 5, 6, and 14 out of the ~76,000 Avana sgRNAs. CD40 was also a top hit on this side of the screen, with anti-CD40 sgRNAs ranking 4, 10, 16, and 122 (Figure 1C). Likewise, Gene Set Enrichment Analysis (GSEA) identified CD40 and NF-κB pathways to be highly enriched among screen hits (Figure S1A). Multiple well-characterized CD40/NF-κB pathway components scored as important for CD40-mediated Fas upregulation, including the genes encoding TRAFs 5 and 6, TAK1 and its cofactor TAB2, IKKα, IKKβ, and IKKγ, the IκBα ubiquitin ligase β-TRCP, and the NF-κB transcription factor subunit p50 (Figure 1B; Table S1). These results suggest that CD40 canonical and non-canonical NF-κB pathways each contribute non-redundantly to B cell Fas upregulation.
Multiple hits had suspected roles in CD40 and/or Fas trafficking, without strongly perturbing control CD37 PM expression. These included five signal recognition particle (SRP) components and both SRP receptor subunits (Table S1), presumably due to roles in CD40 and/or Fas endoplasmic reticulum translocation. Genes encoding the COPII ER to Golgi trafficking pathway components SEC23B and SEC31A scored at the q < 0.05 cutoff (Figures S1B–S1G), suggesting probable roles in CD40 and/or Fas trafficking.
STARS analysis of sgRNAs enriched in the Fashigh population identified 57 candidate CD40 negative regulators at a q < 0.05 cutoff and 158 at a p < 0.05 cutoff (Figure 1D; Table S2). q < 0.05 hits included the NF-κB negative regulators IκBα (encoded by NFKBIA), IκBβ (encoded by NFKBIB), the deubiquitylase A20 (encoded by TNFAIP3), and its cofactor ABIN1 (encoded by TNIP1), denoting that each may play non-redundant roles in modulating B cell CD40 signaling (Figure 1E). Importantly, most other hits have not previously been implicated in CD40 or NF-κB responses, suggesting that CD40 signaling is subject to multiple additional layers of negative regulation.
The F-Box Protein FBXO11 Is a Dependency Factor for CD40 Expression
F-box proteins are substrate-recognition subunits of SKP1-cullin-1-F-box protein (SCF) E3 ligase complexes (O’Connell and Harper, 2007; Skaar et al., 2013). Unexpectedly, FBXO11 was a top hit in our CD40 positive regulator screen (Figure 1B) and was recently implicated as a regulator of GC reactions and B cell lymphomagenesis (Duan et al., 2012; Schneider et al., 2016). All four sgRNAs against FBXO11 were enriched in sorted Faslow but depleted in Fashigh populations (Figure 2A). Independent FBXO11 sgRNAs strongly depleted FBXO11 (Figure 2B) and significantly impaired CD40L-induced Fas expression (Figures 2C and 2D). By contrast, FBXO11 KO did not impair TNF-α-induced Fas induction (Figure S2A), consistent with the possibility of specific effects on a CD40 pathway.
Figure 2. FBXO11 Is a Critical Dependency Factor for B Cell CD40 Expression.

(A) Log2-normlized FBXO11 sgRNA abundances within the indicated CRISPR screen cell populations. CRISPR Daudi cell library (red), in the sorted Faslow (blue) and Fashigh (green) populations are shown. Mean ± SD of two input libraries and four screen replicates are shown.
(B) Immunoblot analysis of whole cell extracts (WCE) from Cas9+ Daudi B cells expressing the indicated control or independent FBXO11-targeting sgRNAs.
(C) FACS analysis of PM Fas levels in Cas9+ Daudi B cells expressing the indicated control or FBXO11 targeting sgRNA and stimulated by 50 ng/mL Mega-CD40L for 48 h as shown.
(D) FACS analysis of PM Fas median fluorescence intensity (MFI) as in (B) from n = 3 replicates.
(E) CD40 canonical NF-κB pathway IκBα and tubulin load control levels in WCE from control or FBXO11 sgRNA expressing Daudi B cells treated with 50 ng/mL Mega-CD40L for the indicated times. The ratios of IκBα to tubulin (tub) abundances are shown beneath.
(F) FACS analysis of PM CD40 MFI in Daudi B cells expressing the indicated sgRNA and stimulated by Mega-CD40L 50 ng/mL for 48 h, as indicated, using n = 3 replicates.
(G) Immunoblot analysis of WCE from Cas9+ Daudi B cells expressing the indicated control or FBOX11 sgRNA from a replicate shown in (F).
(H) FACS analysis of PM CD40 and Fas levels in primary spleen B cells from n = 3 wild type (WT) or FBXO11 KO mice stimulated by anti-CD40 agonist antibody (1 μg/mL) and recombinant mouse IL-4 (20 ng/mL) for 48 h.
(I) Percentages of IgG1 + B cells obtained from n = 5 WT or FBXO11 KO mice stimulated for the indicated days with anti-CD40 antibody (1 μg/mL) and recombinant mouse IL-4 (20 ng/mL).
All immunoblots were representative of at least n = 3 replicates. Mean + SD are shown in (D), (F), (H), and (I). *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001. Cas9+ Daudi B cells were used for (A)–(G).
See also Figures S2 and S3.
To gain insights into FBXO11 roles in CD40 regulation, we first examined effects of its KO on CD40/NF-κB pathways. FBXO11 loss strongly blocked CD40 canonical NF-κB driven IκBα turnover (Figure 2E) and non-canonical p100 processing (Figure S2B), raising the possibility of effects at the level of CD40 itself. To test this hypothesis, CD40 PM and whole cell level (WCL) were measured in control versus FBXO11 KO cells. FBXO11 sgRNAs significantly reduced CD40 PM in multiple B cell lines (Figures 2F, S2C, and S2D) and WCL CD40 levels (Figure 2G) without altering abundance of four other PM markers (Figures S2E and S2F).
FBXO11 KO Effects in Primary B Cells
To next test whether FBXO11 is a major regulator of primary B cell CD40 responses, we characterized B cells purified from control and conditional CD19/Cre-FBXO11fl/fl mice, in which CD19-driven Cre inactivated FBXO11 in B cells at an early stage in development (Figure S2G). PM CD40 levels and CD40-mediated Fas upregulation were significantly lower in FBXO11-deficient than in control B cells (Figure 2H). To investigate the functional significance of FBXO11 KO effects on a hallmark CD40-dependent B cell process, CSR was measured in activated B cells. Purified control and FBXO11 KO splenic B cells were treated with anti-CD40 agonist antibody and IL-4 for 96 h, and the percentage of IgG1 was monitored daily by FACS. By 72 h, FBXO11 KO cells exhibited significantly lower amounts of CSR (Figures 2I, S3A, and S3B). Collectively, these results suggest that FBXO11 is important for B cell CD40 responses.
Mechanistic Investigation of FBXO11 Effects on CD40 Expression
SCF ligases typically destabilize target proteins (Skaar et al., 2013), but in rare instances instead stabilize target proteins through monoubiquitination (Cepeda et al., 2013). To test whether FBXO11 stabilizes CD40, control versus FBXO11 KO cells were treated with the proteasome inhibitor bortezomib, and CD40 levels were measured by immunoblot. Proteasome inhibition failed to restore CD40 levels (Figure 3A), and FBXO11 loss did not appreciably alter CD40 half-life (Figure S3C). To test FBXO11 KO effects on CD40 mRNA steady-state levels, qPCR was performed and demonstrated a nearly 50% decrease in CD40 message in FBXO11 KO cells (Figures 3B and S3D), suggesting possible roles in CD40 transcription or mRNA stability.
Figure 3. FBXO11 Supports CD40 Expression by Targeting the Repressor CTBP1.

(A) Immunoblot analysis of CD40, poly-ubiquitin (poly-Ub), or control GAPDH levels in WCE from Cas9+ Daudi B cells that expressed the indicated sgRNA, treated with DMSO or the proteasome inhibitor bortezomib (200 nM) for 16 h. Increased poly-Ub signal indicates on-target bortezomib activity.
(B) qRT-PCR analysis of CD40 mRNA abundances relative to 18S control levels in Cas9+ Daudi B cells expressing the indicated sgRNA.
(C) FACS analysis of PM CD40 abundances in Cas9+ Daudi B cells expressing the indicated control or FBXO11 targeting sgRNA as well as the indicated control or CD40 cDNA construct.
(D) FACS analysis of Daudi B cell PM CD40 MFI as in (C) from n = 3 replicates.
(E) FACS analysis of PM Fas abundances in Cas9+ Daudi B cells expressing the indicated control or FBXO11 targeting sgRNA as well as the indicated control or CD40 cDNA rescue construct, stimulated by Mega-CD40L (50 ng/mL for 48 h), as indicated.
(F) Mean + SD Log2-normlized CTBP1 sgRNA abundances from both pre-FACS sort input libraries and from all four screen replicates are shown.
(G) Immunoblot analysis of CTBP1, FBXO11, and GAPDH control abundances in WCE from Cas9+ Daudi B cells expressing the indicated control, FBXO11, or either of two independent CTBP1 targeting sgRNAs.
(H) PM CD40 abundances in Cas9+ Daudi B cells expressing the indicated two sgRNAs.
Mean + SD levels from at least n = 3 experiments and shown in (B), (D), and (H). *p < 0.05, **p < 0.01, ***p < 0.001, ns, non-significant. Immunoblot results were representative of n = 3 experiments.
See also Figure S3.
To further test the hypothesis that FBXO11 controls CD40 transcription, we asked whether CD40 cDNA, driven by a heterologous promoter, rescued CD40 expression in FBXO11 KO cells. Interestingly, PM CD40 levels and CD40L-induced Fas induction were nearly completely rescued by lentivirus-driven stable CD40 cDNA expression (Figures 3C–3E). Because most characterized FBXO11 substrates are nuclear transcription factors including transcriptional repressors, our results suggested that FBXO11 may support CD40 expression by targeting a nuclear suppressor of CD40 transcription.
We hypothesized that putative FBXO11 target repressor(s) could be among the CD40 negative regulator FasHigh population screen hits (Table S2). We searched the Bioplex interactome and Biogrid databases and found that only one CD40 negative regulator hit, CTBP1, is a high-confidence FBXO11 interactor (Huttlin et al., 2017). Multiple Avana sgRNAs targeting CTBP1 were enriched in cells sorted for Fashigh expression and depleted in cells sorted for Faslow expression (Figure 3F). We confirmed that endogenous CTBP1 and FBXO11 associate in Daudi B cells by co-immunoprecipitation (Figure S3E) and that FBOX11 KO increased CTBP1 steady-state levels. Consistent with a CD40 repressor role, CTBP1 KO increased PM CD40 levels (Figures 3G and 3H).
Combined FBXO11 and CTBP1 KO resulted in control CD40 levels, suggesting that additional FBXO11 target(s) also regulate CD40 expression (Figure 3H). We therefore next examined whether increases in abundance of a well-characterized SCFFBXO11 target, the nuclear transcription repressor BCL6 (Basso and Dalla-Favera, 2010; Duan et al., 2012; Schneider et al., 2016), also contributed to FBXO11 KO effects on CD40 expression. Notably, CD40 signaling downregulates BCL6 expression (Saito et al., 2007), and we reasoned that BCL6 might in turn cross-regulate CD40 expression. Indeed, FBXO11 KO markedly increased Daudi cell BCL6 levels (Figure S3F). Although we were unable to obtain single cell BCL6 KO clones, given key BCL6 Burkitt B cell dependency factor roles (Ma et al., 2017; Schmitz et al., 2014), depletion of BCL6 increased Daudi cell steady-state CD40 levels (Figure S3G). Similarly, lentivirus-driven BCL6 overexpression decreased CD40 PM levels (Figures S3H and S3I). Taken together, these data support a model in which FBXO11 reinforces CD40 transcription by negatively regulating abundance of the nuclear repressors CTBP1 and BCL6.
CELF1 Is a Key Dependency Factor for CD40 Expression
CELF1 (also called CUG-binding protein 1 [CUGBP1]) binds to GU-rich elements and controls several steps of RNA metabolism, including splicing (Kalsotra et al., 2008; Philips et al., 1998; Tang et al., 2015), translation (Chaudhury et al., 2016), and mRNA turnover (Vlasova et al., 2008). While not previously implicated in CD40 biology, all four CELF1-targeting Avana sgRNAs scored in the CD40 positive regulator screen (Figures 1B and 4A; Table S1). CELF1 sgRNAs were enriched in Faslow population and depleted in the Fashigh population (Figure 4A). Independent CELF1 sgRNAs diminished CD40Linduced PM Fas abundance (Figures 4B, 4C, and S4A) and successfully depleted CELF1 levels (Figure 4D). CELF1 KO diminished CD40L-induced expression of a second CD40 target gene, ICAM-1 (Figures S4B and S4C), consistent with a CELF1 role in CD40 expression or signaling. Further suggesting on-target CRISPR effects, CELF1 cDNA rescue construct expression in a single-cell CELF1 KO clone restored CD40-driven Fas and ICAM-1 induction (Figures 4E, 4F, and S4D).
Figure 4. CELF1 Is a Critical Dependency Factor for B Cell CD40 Expression.

(A) Log2-normlized CELF1 sgRNA abundances of the indicated CRISPR screen cell populations. Mean + SD of two input libraries and four screen replicates are shown.
(B) FACS analysis of PM Fas levels in Cas9+ Daudi B cells expressing the indicated sgRNA and stimulated by 50 ng/mL Mega-CD40L for 48 h where indicated.
(C) PM MFI Fas abundances as in (B) from n = 3 experiments.
(D) Immunoblot of CELF1 or control GAPDH abundances using WCE from Cas9+ Daudi B cells expressing control or independent CELF1 targeting sgRNAs.
(E) Immunoblot of CELF1, V5 epitope-tagged CEFL1, or control GAPDH abundances in WCE from single-cell Cas9+ Daudi B cell control or CELF1 KO clones with stable V5-tagged CELF1 rescue (CELF1R) cDNA expression.
(F) FACS analysis of PM Fas abundances in Daudi B cell CELF1 KO expressing control or CELF1R cDNAs and stimulated by Mega-CD40L (50 ng/mL) for 48 h, as indicated.
(G) Immunoblot analysis of IκBα or tubulin load control abundances in WCE from Cas9+ Daudi B cells expressing control or CELF1 sgRNA and treated with 50 ng/mL Mega-CD40L for the indicated minutes (min). Ratios of IκBα/tubulin (tub) abundances are shown beneath each lane.
(H) Immunoblot analysis of p100 and p52 abundances in WCE from Cas9+ Daudi B cells expressing control or CELF1 sgRNA and treated with 50 ng/mL Mega-CD40L for the indicated hours (h). Ratios of p100:p52 abundances are shown beneath each lane. The immunoblot was representative of n = 3 experiments.
(I) FACS analysis of PM CD40 abundances in Cas9+ Daudi B cells expressing either control or one of two independent CELF1 sgRNAs.
(J) FACS analysis of Cas9+ Daudi B cell PM CD40 MFI as in (I) from n = 3 replicates
(K) Immunoblot analysis of CD40 or tubulin abundances in WCE from Cas9+ Daudi B cells expressing control or CELF1 sgRNAs.
(L) Immunoblot of CD40, Poly-Ub, or tubulin abundances in WCE from Cas9+ Daudi B cells that expressed the indicated sgRNA and that were treated with control DMSO or the proteasome inhibitor bortezomib (200 nM) for 16 h.
Mean + SD from n = 3 are shown in (C) and (J). **p < 0.01; ***p < 0.001. All immunoblots were representative of at least n = 3 replicates.
See also Figure S4.
We next sought to determine whether CELF1 regulated CD40 signaling versus CD40 abundance. CELF1 KO impaired both CD40 canonical and non-canonical NF-κB pathways (Figures 4G and 4H), suggesting a role at the level of CD40 itself. Consistent with this possibility, CELF1 CRISPR targeting did not impair TNF-α-induced PM Fas expression, despite similar CD40 and TNF-α downstream pathways (Figure S4E). Furthermore, independent CELF1 sgRNAs significantly reduced PM CD40 levels in Daudi B cells (Figure 4I–J) and in four additional B cell lines (Figure S4F) but did not significantly change expression of four other PM markers in unstimulated cells (Figures S4G and S4H). CELF1 sgRNAs also markedly diminished whole cell CD40 abundance (Figure 4K), yet treatment with the proteasome inhibitor bortezomib did not rescue CD40 levels in CELF1 KO cells (Figure 4L). Taken together, these results suggest that CELF1 regulates CD40 expression at the transcriptional and/or post-transcriptional level(s).
To investigate possible CELF1 roles on CD40 message stability, we measured steady-state CD40 mRNA levels in control versus CELF1 KO cells. While CELF1 typically binds to 5′ or 3′ untranslated regions (UTR) to destabilize target mRNAs (Vlasova et al., 2008), CELF1 KO instead significantly increased CD40 mRNA levels (Figure 5A). Yet, arguing against CELF1 roles at the CD40 5′ or 3′ UTR, CELF1 loss did not significantly change levels of luciferase reporters stably expressed from vectors in which either the 5′ or 3′ luciferase construct UTR was replaced with that of CD40 (Figure S5A). We therefore next tested CELF1 KO effects on CD40 splicing, using real-time PCR primers that targeted previously defined CD40 mRNA splice sites (Tone et al., 2001). Interestingly, whereas a single message was observed in control cells, we observed two truncated CD40 messages in CELF1 KO cells (Figures 5B and S5B). Sanger sequence analysis of reverse-transcribed CD40 messages identified loss of exon 6, or exons 5 and 6, in CELF1 KO cells (Figures 5C and S5C).
Figure 5. CELF1 Is Required for Proper CD40 mRNA Splicing.

(A) qRT-PCR analysis of 18S-rRNA normalized CD40 mRNA levels in Cas9+ Daudi B cells expressing the indicated control or CELF1 sgRNAs. Schematic maps of CD40 cDNA and primer sets used are shown below.
(B) Schematic diagram of CD40 exons and primers used for diagnostic analyses. Shown below is PCR analysis of CD40 mRNA species, amplified by the indicated primers by RT-PCR from Cas9+ Daudi B cell control, CELF1 KO pools, or a single-cell CELF1 KO clone.
(C) Schematic diagram of CD40 mRNA splicing in control versus CELF1 KO cells. Positions of aberrant splicing are indicated by thick red lines.
(D) FACS analysis of PM CD40 abundances in Daudi B cells expressing the indicated control or CELF1 sgRNAs as well as control or CD40 cDNA constructs.
(E) FACS analysis of PM CD40 abundances as in (D) from n = 3 replicates.
(F) FACS analysis of PM Fas MFI in Daudi B cells with the indicated sgRNA and cDNA expression, stimulated by 50 ng/mL Mega-CD40L for 48 h, as indicated.
(G) CEFL1 exon (in capitals) and intron (in small case) boundary sequences, with sequence encoding putative CELF1 GU-rich binding sites highlighted in red. Nucleotide number of human CD40 cDNA is indicated.
Mean + SD from n = 3 experiments are shown in (A) and (E), with *p < 0.05, **p < 0.01, ns, non-significant. (B), (D), and (F) are representative of at least n = 3 experiments.
See also Figure S5.
If CELF1 is required for CD40 splicing, we predicted that expression of an intron-less CD40 cDNA construct would be similar in control versus CELF1 KO cells. Indeed, CD40 cDNA expression restored PM CD40 abundance and CD40L-induced Fas levels in CELF1 KO cells (Figures 5D–5F and S5D). Notably, sequences encoding GU-rich elements, frequently present at CELF1 RNA binding sites, are present at the exon 5 and 6 boundaries (Figure 5G). Taken together, our results suggest that CELF1 is necessary for inclusion of CD40 exon 6 or 5/6, perhaps through effects on GU-rich exonic splicing enhancers.
An N6-Methyladenosine Writer Controls CD40 mRNA Levels
CD40 mRNA levels are tightly controlled to protect against autoimmunity and B cell lymphoma, though little has remained known about factors that negatively regulate its abundance. Notably, the N6-methyladenosine (m6A) writer WTAP components VIRMA/KIAA1429, METTL14, and METTL3 (Liu et al., 2014; Schwartz et al., 2014) scored as the second, sixth, and thirteenth strongest CD40 negative regulator screen hits, respectively (Figures 1D, 6A, and S6A; Table S2). The fourth component WTAP also scored within the q < 0.05 cutoff (Figure S6A). Similarly, the nuclear m6A reader YTHDF2, which regulates the stability of m6A-modified mRNA, also scored strongly as a candidate CD40 negative regulator (Figure S6A). METTL14 KO increased Fas and ICAM-1 induction by CD40L in B cell lines (Figures 6B and S6B–S6D), but did not significantly alter TNF-α-induced PM Fas expression (Figure S6E), suggesting effects at the level of CD40.
Figure 6. The m6A Writer METTL3-METTL14-WTAP and ESCRT Negatively Regulates CD40 Abundance.

(A) Log2-normlized CRISPR screen mean + SD abundances are shown for sgRNAs targeting the gene encoding METTL14. Mean + SD of two input libraries and four screen replicates are shown.
(B) FACS analysis of PM Fas abundances in Cas9+ Daudi B cells expressing control or METTL14-targeting sgRNAs and stimulated by 50 ng/mL Mega-CD40L for 48 h, as indicated.
(C) Immunoblots analysis of CD40, METTL14, or control tubulin abundances in WCE from Cas9+ Daudi B cells expressing the indicated sgRNAs.
(D) Immunoblots analysis of CD40, IκBα, or GAPDH abundances in Cas9+ Daudi B cells expressing the indicated sgRNAs pre-treated for 1 h with 5 μM Calbiochem IKK inhibitor VIII (IKK inh) and then treated with IKK inh together with 50 ng/mL Mega-CD40L for an additional 12 h, as indicated.
(E) RT-PCR analysis of 18S-rRNA normalized CD40 mRNA levels in Cas9+ Daudi B cells expressing the indicated sgRNA.
(F) RT-qPCR analysis of Daudi B cell CD40 mRNA immunopurified by control IgG versus IgG against WTAP component VIRMA, expressed as a percentage of WCE input CD40 RNA abundance.
(G) RT-qPCR analysis of CD40 abundances in control IgG versus anti-m6A mRNA immunoprecipitations from Cas9+ Daudi B cells that expressed control or METTL14 sgRNAs. Immunopurified CD40 mRNA abundances are expressed as a percentage of input CD40 mRNA levels.
(H) Schematic highlighting ESCRT component CD40 negative regulator screen hits. ESCRT-0, II, and III subunits hits included genes encoding HRS, VPS25,VPS36, CHMP5, and CHMP6.
(I) Log2-normalized CRISPR screen CHMP5 and VPS25 sgRNA abundances. Mean + SD of two input libraries and four screen replicates are shown.
(J) FACS analysis of PM Fas abundance in Cas9+ Daudi B cells with the indicated control, CHMP5, or VPS25 sgRNAs and stimulated with 50 ng/mL Mega-CD40L for 48 h, as indicated.
(K) FACS analysis of PM CD40 abundances in Cas9+ Daudi B cells with the indicated control, CHMP5, or VPS25 sgRNAs and stimulated with 50 ng/mL Mega-CD40L for 24 h, as indicated.
(L) PM MFI Fas abundances as in (K) from n = 3 experiments.
(M) Immunoblot analysis of WCE from Cas9+ Daudi B cells expressing the indicated control or either of two independent CHMP5 or VPS25 targeting sgRNAs and stimulated for 24 h with 50 ng/mL Mega-CD40L, as indicated.
Mean + SD from n = 3 are shown in (E)–(G), (J), and (L). *p < 0.05, **p < 0.01, ***p < 0.001. All immunoblots are representative of at least n = 3 experiments.
See also Figures S6 and S7.
Given that multiple screen hits were found to regulate CD40 abundance, we next examined effects of METTL14 KO on steady-state CD40 protein levels. Interestingly, METTL14 KO significantly increased CD40 abundance (Figure 6C), even in the presence of IKK blockade (Figure 6D), suggesting that METTL14 KO effects were not solely dependent on NF-κB-driven CD40 upregulation. On the mRNA level, METTL14 KO significantly increased CD40 steady-state mRNA abundance (Figures 6E, S6F, and S6G). To next examine whether WTAP associates with CD40 mRNA in Daudi cells, immunoprecipitation was performed using control versus anti-VIRMA/KIAA1429 IgG (Yue et al., 2018). Reverse transcription qPCR (RT-qPCR) analysis revealed that CD40 message was significantly enriched in WTAP pull-down (Figure 6F), with ~10% of CD40 input mRNA associated with VIRMA. In further support, m6A RNA-immunoprecipitation (RIP) followed by RT-qPCR analysis demonstrated that CRISPR targeting of METTL14 significantly diminished Daudi B cell CD40 mRNA m6A modification (Figure 6G). Taken together, these data strongly suggest that WTAP targets CD40 for m6A modification in B cells and supports the hypothesis that WTAP fine tunes CD40 responses by negatively regulating its mRNA levels.
ESCRT Negatively Regulates Activated CD40
Internalization activates CD40 signal transduction (Chen et al., 2006), but how internalized CD40 responses are terminated remains incompletely uncharacterized. Notably, CD40L stimulation together with IKK blockade, which blocks NF-κB driven CD40 re-synthesis, strongly decreased steady-state CD40 levels, suggesting that activated CD40 receptors are targeted for degradation in Daudi B cells (Figure 6D). Interestingly, five subunits of the endosomal-sorting complex required for transport (ESCRT) scored strongly in our screen for CD40 negative regulators (Figure 1D; Table S2). Hits included three of the four ESCRT pathway complexes. Notably, all four guides targeting top hits VPS25 and CHMP5 were enriched in the FasHigh screen population (Figures 6H, 6I, and S7A). CHMP5 or VPS25 sgRNAs increased CD40L-stimulated PM Fas upregulation, even though control PM CD37 levels were unchanged (Figures 6J, S7B, and S7C). ESCRT subunit KO likewise increased CD40L-induced ICAM-1 PM levels in Daudi and Akata B cells (Figures S7D–S7G). While CHMP5 or VPS25 KO did not alter basal PM CD40 levels, PM CD40 abundance was elevated in CD40L-stimulated cells in multiple B cell lines (Figures 6K, 6L, and S6H).
ESCRT downmodulates PM receptor expression by routing cargo through multivesicular bodies (MVB) for lysosomal degradation (Raiborg and Stenmark, 2009; Teis et al., 2010; Figure 6H). We therefore next investigated ESCRT component KO effects on total cell CD40 levels in unstimulated versus CD40L-treated cells. Independent CHMP5 or VPS25 sgRNAs increased CD40 WCL in stimulated, but not in resting cells (Figure 6M). Similar results were obtained in CD40L-stimulated cells treated with an IKK antagonist for 24 h, denoting that effects on downstream CD40/NF-κB signaling were not required for increased CD40 abundance (Figure S7I).
To further investigate CD40L effects on CD40 trafficking, we next used confocal immunofluorescence microscopy. In control sgRNA expressing Daudi cells stimulated by CD40L together with the lysosomal protease inhibitor leupeptin, CD40 co-localized with lysosomes, as judged by staining for CD40 and for the lysosomal marker LAMP1 (Figure S7J). By contrast, CHMP5 sgRNA expression diminished CD40 and LAMP1 colocalization in CD40L-stimulated and leupeptin-treated Daudi cells. Collectively, these results suggest that ESCRT downmodulates activated CD40 receptor abundance.
The Phosphatase DUSP10 Negatively Regulates CD40 MAPK Pathways
Phosphatases that negatively regulate CD40 responses have not been systematically studied. Notably, the dual specificity serine/threonine phosphatase DUSP10 was a top hit in our CD40 negative regulator screen (Figures 1D and 7A). We first confirmed that Avana sgRNAs depleted DUSP10 (Figure 7B) and found that in contrast to the other hits investigated above, DUSP10 KO significantly enhanced CD40L Fas induction (Figures 7C and 7D) without altering basal PM CD40 levels (Figure 7E), suggesting a distinct layer of CD40 regulation uncovered by the CRISPR screen. DUSP10 negatively regulates lipopolysaccharide-induced macrophage ERK signaling (Zhang et al., 2004) but has not been implicated in CD40 or B cell regulation.
Figure 7. The Phosphatase DUSP10 Is a CD40/MAPK Pathway Negative Feedback Regulator.

(A) Log2-normlized CRISPR screen mean + SD abundance values for DUSP10 sgRNAs.
(B) Immunoblots of DUSP10 or control GAPDH abundances in WCE from Cas9+ Daudi cells expressing control or independent DUSP10 sgRNAs.
(C) FACS analysis of PM Fas levels in a representative experiment with Cas9+ Daudi cells expressing the indicated control or DUSP10 sgRNA and stimulated by 50 ng/mL Mega-CD40L for 48 h as shown.
(D) FACS analysis of PM Fas MFI as in (C) from n = 3 experiments.
(E) FACS analysis of PM CD40 expression in Cas9+ Daudi B cells expressing control versus DUSP10 sgRNAs.
(F) Immunoblot analysis phosphorylated and total p38, ERK, and JNK MAPK abundances in WCE from Cas9+ Daudi B cells expressing the indicated sgRNAs and stimulated by 50 ng/mL Mega-CD40L for the indicated hours. Shown below are the ratios of phosphokinase to total kinase signal from a representative experiment. Ratios in unstimulated cells with control sgRNA expression were normalized to a value of 1.
(G) FACS analysis of PM Fas level in Cas9+ Daudi B cells treated with 10 ng/mL TNF-α for 24 h, as indicated.
(H) DUSP10 RNA-seq reads in primary human B cells stimulated with CD40L for 0, 1, or 18 h.
(I) Immunoblot analysis of DUSP10 versus loading control TBP abundances from primary human B cells stimulated with 50 ng/mL Mega-CD40L for 0, 1, 4, or 18 h. Immunoblots were representative of n = 3 experiments. Mean and SD values from three independent experiments are shown in (D) and (H). *p < 0.05, ***p < 0.001. Cas9+ Daudi B cells were used in (A)–(G).
See also Figure S7.
To investigate putative DUSP10 roles in CD40 MAPK regulation, we measured ERK, p38, and JNK activation loop phosphorylation levels in control versus DUSP10 KO cells. While DUSP10 depletion had little effect on basal MAPK phosphorylation levels, it significantly increased p38 and ERK phosphorylation levels at 4 h post-CD40L stimulation and JNK phosphorylation by 1 h (Figure 7F). Yet, DUSP10 KO did not significantly enhance Fas induction by TNF-α (Figure 7G), suggesting that DUSP10 may not be induced or activated by TNF-α stimulation. To further examine CD40 effects on DUSP10 expression, we measured CD40L-stimulation effects on primary human B cell DUSP10 mRNA and protein levels. DUSP10 mRNA and protein abundances were markedly increased by 1 and 4 h post-stimulation, respectively (Figures 7H and 7I). DUSP10 protein abundance remained elevated through 18 h post-stimulation (Figure 7I). These data suggest that DUSP10 is a major B cell negative feedback regulator of CD40/MAPK pathways.
DISCUSSION
CD40 signaling must be carefully balanced to allow GC formation, development of high-affinity class-switched antibodies, memory, and plasma cell differentiation (De Silva and Klein, 2015; Elgueta et al., 2009; Nonoyama et al., 1993; van Kooten and Banchereau, 2000; Victora and Nussenzweig, 2012), while limiting the establishment and pathogenesis of multiple autoimmune diseases and lymphomagenesis (Elgueta et al., 2009; Hatzivassiliou et al., 2007; Hömig-Hölzel et al., 2008; Peters et al., 2009). Yet, little has remained known about how B cell autonomous factors control CD40 levels to regulate immune responses. Systematic genetic analysis identified multiple layers that control CD40 abundance and responses.
We identified the tumor suppressor FBXO11 as an important regulator of B cell CD40 transcription. FBXO11 inactivating mutations are observed in ~6% of diffuse large B cell lymphomas (DLBCL) (Duan et al., 2012; Lohr et al., 2012; Pasqualucci et al., 2014), the most common type of non-Hodgkin lymphoma (Alizadeh et al., 2000; Basso and Dalla-Favera, 2015). Intriguingly, monoallelic FBXO11 mutations are the most common in primary tumors (Duan et al., 2012; Lohr et al., 2012). Monoallelic fbxo11 mutation also caused higher rates of lymphoproliferative disorder than biallelic fbxo11 KO in Cγ1 Cre-driven conditional B cell models, and DLBCL was only observed in fbxo11 heterozygotes (Schneider et al., 2016). Yet, oncogenic BCL6 expression was elevated to a greater extent in homozygous than in heterozygous FBXO11 KO mice (Schneider et al., 2016). It will be interesting to determine whether FBXO11 roles in support of CD40 abundance contribute to the mixed phenotype that favors monoallelic FBXO11 loss.
Primary GC B cells have extremely short lifespans ex vivo, necessitating our use of GC-derived B cell lymphoma cell lines for most of our studies. However, we validated that conditional B cell FBXO11 loss diminished primary B cell CD40 expression and impaired CSR, a hallmark of CD40 function. Notably, this phenotype may have been more evident in our CD19 Cre system mouse model, where conditional FBXO11 loss was induced early in B cell development, and all mature B cells are deleted. By contrast, the Cγ1 Cre used in the previous study presumably causes FBXO11 KO at a later stage, where many B cells had likely undergone IgG CSR prior to or concurrent with FBXO11 inactivation (Casola et al., 2006; Schneider et al., 2016).
CD40 signaling acts in particular stages of the GC reaction with key roles in light zone centrocyte survival (Basso et al., 2004). Thus, FBXO11 may play key roles in GC responses by supporting light zone CD40 expression. Mechanistically, FBXO11 supported CD40 expression by controlling levels of CTBP1 and BCL6, which are major transcription repressors. CTBP1 assembles polycomb complexes to silence target gene expression through hypermethylation. Because SCF ubiquitin ligases are typically gated by a phosphodegron switch (Skaar et al., 2013), an important future objective will be to define the B cell kinase(s) that control FBXO11 targeting of CTBP1 and to test whether their activity is controlled by CD40 or by other B cell receptors to dynamically regulate CD40 levels. Similarly, it will be of interest to determine whether FBXO11 targets DNA-bound CTBP1 complexes versus free CTBP1 and whether post-translational mechanisms regulate FBXO11 targeting of CTBP1 or BCL6 in germinal center dark versus light zones.
Little has been known about mechanisms that positively and negatively control CD40 expression at the RNA level. Yet, SNPs that alter CD40 abundance have been implicated in the pathogenesis of autoimmunity and cancer, underscoring the critical need to tightly control CD40 abundance. Furthermore, CD40 mRNA splicing is actively regulated, and congenital hyper-IgM syndrome can result from mutations that cis-acting exonic enhancer splice sites that promote inclusion of specific exon(s) (Ferrari et al., 2001). For instance, congenital mutation of a CD40 splice enhancer site causes CD40 exon5 skipping, resulting in absence of CD40 PM and autosomal recessive hyper-IgM syndrome (Ferrari et al., 2001). Our data implicated CELF1 as a key factor in CD40 biogenesis, suggesting an obligatory role CD40 exon 6 or exons 5/6 splicing. Additional screen hits, including the splicing factor SRSF11, may also have key roles in CD40 splicing. Further studies are required to determine if truncated CD40 resulting from lack of CELF1 activity may have dominant-negative effects, perhaps as soluble decoy receptors in the absence of exon 6.
These results also raise the intriguing hypothesis that signal-dependent CELF1 regulation may exert strong effects on B cell CD40 levels. Unfortunately, CELF1 KO results in frequent perinatally lethality, limiting opportunities to study effects on B cell responses in mouse models (Kress et al., 2007). However, in primary T cells, CELF1 is a major negative-regulator of RNA stability through interactions with 3′ UTR GU-rich element (GRE), including in the TNF receptor 1B message (Vlasova et al., 2008). Upon T cell activation, CELF1 phosphorylation downmodulates its GRE binding, stabilizing target RNAs (Beisang et al., 2012). Protein kinase C phosphorylation increases steady-state CELF1 levels (Kuyumcu-Martinez et al., 2007). Further, CD40L mRNA levels are regulated by signal-dependent mechanisms (Matus-Nicodemos et al., 2011). Thus, it will be of interest to determine how B cell networks control CELF1 activity and how CELF1 post-transcriptional regulation in turn alters CD40 splicing.
Systematic genetic analysis also revealed multiple layers of CD40 negative regulation. We newly implicate WTAP and m6A as key suppressors of CD40 abundance on the mRNA level. Although m6A mRNA modification is widespread (Schwartz et al., 2014), it has not previously been implicated in control of TNF receptor family signaling. We speculate that WTAP activity, possibly including differential effects on alternatively spliced CD40 messages, provides B cells a route of RNA level epigenetic regulation that fine tunes CD40 abundance. In an analogous manner, m6A has key roles in regulation the circadian clock speed and stability (Yue et al., 2015). WTAP has been implicated as a therapeutic target for cancer therapy (Vu et al., 2017), and enhanced immune activation may be an additional benefit to explore.
At the protein level, ESCRT downmodulated activated CD40 abundance, while DUSP10 restricted CD40/MAPK pathways. Neither has previously been implicated in CD40 regulation, although ESCRT restricts basal NF-κB signaling by limiting the population of ligand-free receptors at signaling endosomes (Mamińska et al., 2016). Our data suggest that ESCRT and DUSP10 are important regulators of CD40 signal strength and duration, likely together with IκBα, A20, and ABIN1. Physiologically, these mechanisms may be critical for limiting CD40 signaling as T cell stimulated centrocytes migrate to germinal center dark zones and undergo DNA editing (e.g., to protect against oncogenic transformation). Because ESCRT also controls exosome biogenesis, it will be interesting to determine whether ESCRT may also down-modulate CD40 responses through secretion into extracellular vesicles (Colombo et al., 2013).
Collectively, our results serve as a resource for ongoing studies of CD40 regulation. Given the critical role of CD40L/CD40 interactions on adaptive immune responses, multiple strategies are in clinical development to either increase or decrease these responses (Laman et al., 2017). CD40 agonists are in clinical development for tumor immunotherapy approaches, and these data suggest specific escape pathways that tumor cells may subvert to evade killing (e.g., by altering CD40 transcription or splicing). Likewise, manipulation of key CD40 regulators may lead to synergistic therapeutic strategies to downmodulate CD40 signaling in autoimmune diseases, perhaps together with CD40 blockade.
STAR★METHODS
LEAD CONTACT AND MATERIALS AVAILABILITY
Further information and requests for resources and reagents may be directed to Benjamin Gewurz (Lead Contact; bgewurz@bwh.harvard.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell lines and reagents
The EBV+ Burkitt lymphoma (BL) Daudi cell line (ATCC CCL-213) was used for CRISPR/Cas9 screens. For subsequent studies, GM12878, BJAB, KEM1, MUTU1, AKATA were used. GM12878 were obtained from Coriel Institute. The BL cell lines KEM I and MUTU I were gifts from Dr. Jeffery Sample. BJAB and Akata were gifts from Dr. Elliott Kieff. Cell lines with stable Streptococcus pyogenes Cas9 expression were generated by lentiviral transduction and blasticidin selection (see below). All cells used in this study were cultured in a humidified incubator at 37°C with 5% CO2. B cells were grown in GIBCO RPMI 1640 medium (Life Technologies) with 10% fetal calf serum (FCS). 293T were grown in Dulbecco’s Modified Eagle’s Medium (DMEM) with 10% FCS. For selection, puromycin was introduced to cells at the concentration of 3 μg/ml. Hygromycin was introduced at 200 μg/ml for the initial 4 days, and 100 μg/ml thereafter. Blasticidin was used at the concentration of 5 μg/ml. Cells were tested routinely to be mycoplasma free by the MycoAlert kit (Lonza) before used in experiments. MEGA-CD40L (ENZO) was used at 50 ng/ml. TNFα (R&D systems) was used at 10 ng/ml. Bortezomib (ApexBio) was used at 200 nM for 15 h. IKK inhibitor VIII (Calbiochem/Millipore) was used at 5 mM, a concentration at which it blocks IKKα and IKKβ activity. Leupeptin (Millipore) was used at 10 μM. Cells were pretreated with leupeptin for 1 hour prior to CD40L stimulation.
Lentivirus Preparation and Transduction
Lentiviruses were constructed by transfection of 293T (obtained from ATCC and used at low passage), plated at a density of 600,000 cells per well in 2 mL DMEM with 10% fetal calf serum 24 hr prior to transfection in a 6-well dish. For transfection, the TransIT-LT1 Transfection Reagent (Mirus) was used according to the manufacturer’s protocol. Two solutions were prepared for each transfection: a solution with 4 mL of LT1 diluted in 16 mL of Opti-MEM (Corning) and incubated for 5 minutes at room temperature, and a solution with 150 ng pCMV-VSVG (Addgene plasmid #8454), 400 ng psPAX2 (Addgene plasmid #12260) and 500 ng of the expression vector. The final volume was brought up to 20 uL with Opti-MEM. The two solutions were mixed and incubated at room temperature for 30 minutes. Thereafter, they were added dropwise to the tissue culture well and gently mixed. Plates were then returned to 37C incubator. 24 hours later, media was exchanged to RPMI with 10% fetal calf serum. Virus supernatant was harvested 48h post-transfection and filtered through a 0.45 uM sterile filter. Fresh media was replenished on the 293T producer cell and a second harvest was done 72h post-transfection and again sterile filtered. Virus supernatant was added to target cells at 48 and 72 hours post-transfection. Target cell selection was then begun 48 hr post-transduction.
Primary human B cell collection
Discarded, de-identified leukocyte fractions left over from platelet donations were obtained from the Brigham and Women’s Hospital Blood Bank. Blood cells were collected from platelet donors following institutional guidelines. Since these were de-identified samples, the gender was unknown. Our studies on primary human blood cells were approved by the Brigham & Women’s Hospital Institutional Review Board. Primary human B cells were isolated by negative selection using RosetteSep Human B Cell Enrichment and EasySep Human B cell enrichment kits (Stem Cell Technologies), according to the manufacturers’ protocols. B cell purity was confirmed by plasma membrane CD19 positivity through FACS. Cells were then cultured with RPMI 1640 with 10% FCS.
Mice
C57BL/6 mouse ES cells heterozygous for a targeted allele of FBXO11 were obtained from EUCOMM, the International Knockout mouse consortium, and microinjected into blastocysts derived from C57BL/6 mice after removal of the LacZ-neomycin cassette by Flipase-mediated recombination. The resulting FBXO11 conditional allele has two loxP sites into intronic regions upstream and downstream of exon 4, that allow disruption of the FBXO11 sequence after Cre-recombination. The FBXO11fl/fl mice were bred with CD19cretg/+ transgenic mice to specifically delete FBXO11 in CD19+ B cells. Male and female FBOX11 conditional knockout mice at 10 months of age were used in the experiments. A minimum of three mice for each experiment was used. No mice were excluded from the analysis and no randomization or blinding method was used. All mice were housed and maintained in the specific pathogen free (SPF) facility at Boston Children’s Hospital. Animal experiments were performed under protocols approved by the Institutional Animal Care and Use Committee (IACUC) of Boston Children’s Hospital (protocol #16–01-3093R). FBXO11 inactivation was confirmed by genotyping and western blot from purified B cells.
Mouse B cell purification
Untouched B cells were separated from total spleen cell suspensions using anti-CD43 magnetic microbeads (Miltenyi). The purity of isolated untouched B cell was typically around 96%–98%. Untouched B cells were cultured at a concentration of 5 × 105 cells/ml in RPMI medium supplemented with 15% FBS, penicillin-streptomycin (100 units/ml) and L-glutamine (2 mM).
METHODS DETAILS
Pooled genome-wide CRISPR screens
The Burkitt lymphoma (BL) cell line Daudi (ATCC CCL-213) was used for CRISPR/Cas9 screens. Daudi B cells have low-basal CD40 activation, but robustly respond to stimulation. The Broad Institute Avana sgRNA library was used to generate biological duplicate Daudi libraries for use in the FAS-FACS based screens (Doench et al., 2016). 130 million Cas9+ Daudi cells were infected with the Avana library at a multiplicity of infection (MOI) of 0.3 by spinoculation at 300 g for 2 hours, in the presence of 4 μg/ul polybrene. Plates were then returned to 37°C with 5% CO2 incubator and cells were cultured for 6 hr, followed by changing media into fresh RPMI. Transduced cells were selected by puromycin (3 μg/ml) after 2 day culturing. The libraries were then passaged every 72 hours, returning cell number to at least 40 million per library at each passage to maintain adequate complexity. After 5 days post puromycin selection, genomic DNA was extracted from 40 million cells per each screen replicate as input, using the QIAGEN Blood and Cell Culture DNA Maxi Kit. From the library, 160 million cells were stimulated with 50 ng/mL MEGACD40L for 48 h. Mega-CD40L (50 ng/mL) stimulated cells were then co-stained with Fas and CD37 antibodies and FACS sorted into two CD37+ positive bins: low Fas (bottom 3%) and high Fas (top 3%), using technical duplicates for each biological replicate. Pooled genome-wide CRISPR screen FACS sorting was performed at the Brigham and Women’s Hospital Human Immunology Center. Genomic DNA was then extracted from the two bins for per each screen replicate, using the QIAGEN DNeasy Blood & Cell Culture DNA Kit. sgRNAs were PCR-amplified from genomic DNA, as described (Ma et al., 2017). The abundance of the PCR product was quantified by Illumina Hiseq DNA deep sequencing.
Individual sgRNA CRISPR knockout analysis
Following the CRISPR screen, specific human genes were targeted by CRISPR/Cas9 engineering, using Avana library sgRNA sequences. sgRNA oligos were obtained from Integrated DNA Technologies and cloned into the pLentiGuide-Puro vector (Addgene plasmid #52963, a gift from Feng Zhang) or into pLenti Sp BsmBI sgRNA Hygro (Addgene plasmid # 62205, a gift from Rene Maehr). Cells were selected by puromycin (3 mg/ml) or hygromycin (200 ug/ml), added 48 hr post-transduction. Since CRISPR/Cas9 editing frequently does not result in significant loss of target gene mRNA, loss of target gene protein expression was assayed by immunoblotting.
Double sgRNA CRISPR knockout analysis
For double CRISPR/Cas9 knockout experiments, cells were sequentially transduced and selected. Cells were first selected for 1 week with hygromycin after the first sgRNA perturbation, and then transduced with lentivirus encoding the second sgRNA with puromycin resistance marker in the presence of hygromycin. Finally, cells were selected with puromycin before use. Loss of target gene expression was validated by immunoblot.
Immunoblot analysis
Whole cell extract samples were separated by SDS-PAGE, transferred onto nitrocellulose filters, and then probed with relevant primary antibodies at 4°C overnight, followed by secondary antibody incubation for 1 h at room temperature. Blots were then developed by incubation with ECL chemiluminescence (Western Lightening) for 1 min and images were captured on a Carestream workstation. All antibodies used in this study are listed in the Key Resources Table.
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse monoclonal anti-alpha Tubulin antibody Clone DM1A | Abcam | Cat# ab7291; RRID:AB_2241126 |
| Mouse monoclonal anti-alpha-Tubulin antibody | Sigma-Aldrich | Cat# T5168; RRID:AB_477579 |
| PE mouse monoclonal anti-Human CD54 antibody Clone HA58 | BD Biosciences | Cat# 555511; RRID:AB_395901 |
| FITC Mouse monoclonal Anti-Human CD40 antibody Clone 5C3 | BD Biosciences | Cat# 555588; RRID:AB_395963 |
| Rabbit polyclonal anti-FBXO11 antibody | Bethyl Laboratories | Cat# A301–177A-T; RRID:AB_2779727 |
| Rabbit polyclonal anti-CTBP1 antibody | Bethyl Laboratories | Cat# A300–338A-T; RRID:AB_2779159 |
| Rabbit polyclonal anti-CtBP antibody | Santa Cruz Biotechnology | Cat# sc-11390; RRID:AB_2086634 |
| Rabbit monoclonal anti-DUSP10 antibody Clone EPR9366 | Abcam | Cat# ab140123 |
| Mouse monoclonal anti-CD40 antibody Clone H10 | Santa Cruz Biotechnology | Cat# sc-13128; RRID:AB_627060 |
| Anti-rabbit IgG, HRP-linked Antibody | Cell Signaling Technology | Cat# 7074; RRID:AB_2099233 |
| Anti-mouse IgG, HRP-linked Antibody | Cell Signaling Technology | Cat# 7076; RRID:AB_330924 |
| Influenza hemagglutinin (HA) epitope (YPYDVPDYA) antibody | Covance Research Products Inc | Cat# MMS-101P; RRID:AB_2314672 |
| Rabbit polyclonal anti-TBP Antibody | Bethyl Laboratories | Cat# A301–229A-T; RRID:AB_890661 |
| APC Mouse monoclonal anti-human CD95 (Fas) Antibody Clone DX2 | Biolegend | Cat# 305612; RRID:AB_2782770 |
| Mouse monoclonal anti-GAPDH antibody | Abcam | Cat# ab8245; RRID:AB_2107448 |
| FITC Mouse monoclonal anti-human CD21 antibody Clone Bu32 | Biolegend | Cat# 354909; RRID:AB_2561574 |
| FITC Mouse monoclonal anti-human CD37 antibody Clone M-B371 | Biolegend | Cat# 356304; RRID:AB_2561837 |
| Mouse monoclonal Anti-NF-kappaB p52 antibody | Millipore | Cat# 05–361; RRID:CVCL_8H75 |
| Mouse monoclonal anti-Human CD19 Alexa Fluor 488 antibody | eBioscience | Cat# 53–0199–41; RRID:AB_1659679 |
| Rabbit polyclonal anti-NF-kappaB2 p100/p52 antibody | Cell Signaling Technology | Cat# 4882S; RRID:AB_10695537 |
| Rabbit monoclonal anti-ZIP7/SLC39A7 antibody Clone D103A | Cell Signaling Technology | Cat# 33176S; RRID:AB_2799032 |
| Rabbit monoclonal anti-phospho-p44/42 MAPK (Erk 1/2) (Thr202/Tyr204) Clone D13.14.4E | Cell Signaling Technology | Cat# 4370s; RRID:AB_2315112 |
| Rabbit polyclonal anti-p44/42 MAPK (Erk 1/2) antibody | Cell Signaling Technology | Cat# 9102S; RRID:AB_330744 |
| Rabbit monoclonal anti-phospho-p38 MAPK (Thr80/Tyr182) antibody Clone D3F9 | Cell Signaling Technology | Cat# 4511S; RRID:AB_2139682 |
| Rabbit monoclonal anti-p38 antibody Clone D13E1 | Cell Signaling Technology | Cat# 8690s; RRID:AB_10999090 |
| Rabbit polyclonal anti-phospho-SAPK/JNK (Thr183/Tyr185) antibody | Cell Signaling Technology | Cat# 9251s; RRID:AB_331659 |
| Rabbit polyclonal anti-SAPK/JNK antibody | Cell Signaling Technology | Cat# 9252s; RRID:AB_2250373 |
| Rabbit polyclonal anti-METTL14 antibody | Novus Biologicals | Cat# NBP1–81392; RRID:AB_11021780 |
| Mouse monoclonal anti-CUG-BP1 antibody Clone 3B1 | Santa Cruz Biotechnology | Cat# sc-20003; RRID:AB_627319 |
| Rabbit polyclonal V5 antibody | Bethyl | Cat# A190–120P; RRID:AB_162729 |
| Rabbit monoclonal anti-BCL6 antibody Clone D4I2V | Cell Signaling Technology | Cat# 14895; RRID:AB_2798638 |
| Rabbit monoclonal anti-TRAF5 antibody Clone D3E2R | Cell Signaling Technology | Cat# 41658; RRID:AB_2799205 |
| Mouse monoclonal anti-VPS25 antibody Clone B-4 | Santa Cruz Biotechnology | Cat# sc-271648; RRID:AB_10707989 |
| Mouse monoclonal anti-CHAMP5 antibody Clone F-7 | Santa Cruz Biotechnology | Cat# sc-374338; RRID:AB_10989738 |
| Peroxidase IgG Fraction Monoclonal mouse antiRabbit IgG, light chain specific | Jackson ImmunoResearch | Cat# 211–032–171; RRID:AB_2339149 |
| APC Mouse monoclonal anti-mouse CD95 (Fas) antibody Clone SA367H8 | Biolegend | Cat# 152604; RRID:AB_2632899 |
| FITC Rat monoclonal anti-mouse CD19 antibody Clone 1D3/CD19 | Biolegend | Cat# 152403; RRID:AB_152403 |
| APC Rat monoclonal anti-mouse CD40 antibody Clone 3/23 | Biolegend | Cat# 124612; RRID:AB_1134072 |
| PE Rat monoclonal anti-mouse IgG1 antibody Clone A85–1 | BD Biosciences | Cat# 550083; RRID:AB_393553 |
| APC Rat monoclonal anti-mouse CD45R/B220 antibody Clone RA3–6B2 | BD Biosciences | Cat# 561880; RRID:AB_10897020 |
| Armenian hamster monoclonal anti-Mouse/Rat CD40 antibody Clone HM40–3 | eBioscience | Cat# 14–0402–81; RRID:AB_467227 |
| Mouse Ubiquitin (P4D1) monoclonal antibody | Cell Signaling Technology | Cat# 3936S; RRID:AB_331292 |
| Rabbit kBa polyclonal antibody | Cell Signaling Technology | Cat# 9242S; RRID:AB_331623 |
| Rabbit monoclonal CD40 antibody | Abcam | Cat# ab224639 |
| Mouse monoclonal LAMP1 antibody | Cell Signaling Technology | Cat# 15665; RRID:AB_2798750 |
| Alexa Fluor 488-AffiniPure Goat Anti-Mouse IgG (H+L) | FISHER SCIENTIFIC | Cat# NC0260529; RRID:AB_2338840 |
| Alexa Fluor 594-AffiniPure Goat Anti-Rabbit IgG (H+L) | FISHER SCIENTIFIC | Cat# NC0414256; RRID:AB_2338059 |
| Rabbit polyclonal Virilizer (VIRMA) Antibody | Bethyl Laboratories | Cat# A302–124A; RRID:AB_1720422 |
| Bacterial and Virus Strains | ||
| B95.8 Epstein-Barr Virus | Dr. Elliott Kieff | N/A |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Dynabeads® Protein G | Thermo Scientific | Cat# 10003D |
| Agencourt AMPure XP SPRI beads | Beckman Coulter | Cat# A63880 |
| MEGA CD40L | Enzo life sciences | Cat# ALX-522- 110-C010 |
| CD40L (human) (multimeric) | Adipogen Life Sciences | Cat# AG-40B- 0010-C010 |
| Bortezomib | Apex bio | Cat# A2614 |
| polybrene | Sigma-Aldrich | Cat# TR-1003-G |
| Recombinant Human TNF-alpha Protein | R&D systems | Cat# 210-TA-005 |
| IKK-2 inhibitor VIII | Calbiochem | Cat# 401487 |
| Recombinant Murine IL-4 | PeproTech | Cat# 214–14 |
| InSolution Leupeptin | MilliporeSigma | Cat# 509281 |
| Cathepsin Inhibitor I | MilliporeSigma | Cat# 219415 |
| Hoechst | Molecular Probes | Cat# 33258 |
| 16% formaldehyde | Ted Pella Inc. | Cat# 9658705 |
| E-64 Protease Inhibitor | MilliporeSigma | Cat# 324890 |
| Normal goat serum | Invitrogen | Cat# 31873 |
| Cyclonheximide solution | MilliporeSigma | Cat# 66–81–9 |
| InSolution Pepstatin A | MilliporeSigma | Cat# 508437 |
| Critical Commercial Assays | ||
| RNeasy Mini Kit | QIAGEN | Cat# 74104 |
| miRNeasy Mini Kit | QIAGEN | Cat# 217004 |
| Power SYBR Green RNA-to-Ct 1-Step Kit | Applied Biosystems | Cat# 4389986 |
| QiAquick PCR Purification Kit | QIAGEN | Cat# 28106 |
| QIAquick Gel Extraction Kit | QIAGEN | Cat# 28704 |
| RNase-Free DNase Set | QIAGEN | Cat# 79254 |
| iScript Reverse Transcription Supermix for RT- qPCR | BIO-RAD | Cat# 1708841 |
| Power SYBR Green PCR Master Mix | Applied Biosystems | Cat# 4367659 |
| Gateway LR Clonase II Enzyme Mix | Invitrogen | Cat# 11789–020 |
| QIAprep Spin Miniprep Kit | QIAGEN | Cat# 27106 |
| Invitrogen PureLink Quick Plasmid Maxiprep Kit | Invitrogen | Cat# K210016 |
| Dead Cell Removal Kit | Miltenyi Biotec | Cat# 130–090–101 |
| NEBNext Poly(A) mRNA Magnetic Isolation Module | New England Biolabs | Cat# E7490 |
| NEBNext Ultra RNA Library Prep Kit for Illumina | New England Biolabs | Cat# E7530 |
| RosetteSep Human B Cell Enrichment Cocktail; Immunodensity isolation of untouched B cells | StemCell Technologies, Inc. | Cat# 15064 |
| EasySep Human B cell enrichment kit | StemCell Technologies, Inc. | Cat# 19054 |
| MycoAlert Mycoplasma Detection Kit | Lonza | Cat# LT07–218 |
| Blood and Cell Culture DNA Maxi Kit | QIAGEN | Cat# 13362 |
| DNeasy Blood& Tissue Kit | QIAGEN | Cat# 69504 |
| Magna RIP RNA-Binding Protein Immunoprecipitation Kit | MilliporeSigma | Cat# 17–700 |
| CD40 Gene expression Assays (Hs00386848_m1) | Thermo Fisher Scientific | Cat# 4448892 |
| TaqMan Fast Virus 1-Step Master Mix | TaqMan Fast Virus 1-Step Master Mix | Cat# 4444432 |
| Deposited Data | ||
| Resting and CD40L-stimulated primary B cell RNA-seq datasets Deposited into the NIH GEO omnibusRaw and analyzed data | Gewurz Laboratory | GSE101666 |
| Experimental Models: Cell Lines | ||
| GM12878 lymphoblastoid cell line (LCL) | Coriell Institute for Medical Research | N/A |
| EBV+ Burkitt lymphoma Daudi cell line | ATCC | CCL-213 |
| EBV- Burkitt lymphoma Akata cell line | Dr. Bo Zhao | N/A |
| P3HR1 clone 16 | Dr. Elliott Kieff | N/A |
| Burkitt lymphoma KEM I cell line | Dr. Jeff Sample | N/A |
| Burkitt lymphoma BJAB cell line | Dr. Bo Zhao | N/A |
| Burkitt lymphoma MUTU I cell line | Dr. Jeff Sample | N/A |
| GM12878-Cas9 | This paper | N/A |
| P3HR1-Cas9 | This paper | N/A |
| BJAB-Cas9 | This paper | N/A |
| MutuI-Cas9 | This paper | N/A |
| KemI-Cas9 | This paper | N/A |
| Akata-Cas9 | This paper | N/A |
| Daudi-Cas9 | This paper | N/A |
| HEK293T | ATCC | CRL-3216 |
| Experimental Models: Organisms/Strains | ||
| FBX011 f/f/C D19Cre+ mice | Dr. Roberto Chiarle | N/A |
| FBXO11f/f/CD19WT mice | Dr. Roberto Chiarle | N/A |
| Oligonucleotides | ||
| Table S3 | This paper | N/A |
| Table S4 | This paper | N/A |
| Recombinant DNA | ||
| pLX-TRC313 CELF1 (rescue cDNA) | This paper | N/A |
| pLX-TRC313 | Broad Institute | N/A |
| pLentiGuide-Puro | Addgene | Cat# 52963 |
| pLenti SpBsmBI sgRNA Hygro | Addgene | Cat# 62205 |
| pLentiCas9-Blast | Addgene | Cat# 52962 |
| pXPR-011 | John Doench | N/A |
| Avana CRISPR Lentivirus Library | Broad Institute | N/A |
| pGL3-p27Luc | Addgene | Cat# 23047 |
| pIS2 | Addgene | Cat# 12177 |
| CD40 (Homo sapiens) in pENTR223 | DNASU | Cat# HsCD00508711 |
| BCL6 (Homo sapiens) in pENTR223 | DNASU | Cat# HsCD00516212 |
| Software and Algorithms | ||
| STARS v1.2 | Doench et al., 2016 | http://portals.broadinstitute.org/gpp/public/software/stars |
| STAR2.5.2b | Dobin et al., 2013 | https://github.com/alexdobin/STAR |
| FlowJo_V10 | FLOWJO LLC | https://www.flowjo.com/ |
| DESeq2 v1.14.1 | Love et al., 2014 | https://bioconductor.org/packages/release/bioc/html/DESeq2.html |
| GraphPad Prism 7 | GraphPad Software | https://www.graphpad.com/scientific-software/prism/ |
| SRAMP: A sequence-based N6-methyladenosine (m6A) modification site predictor | Zhou et al., 2016 | http://www.cuilab.cn/sramp/ |
Mouse B-cell activation, and flow cytometry
Murine B cells cultured at a concentration of 5 × 105 cells/ml in RPMI medium supplemented with 15% FBS, penicillin-streptomycin (100 units/ml) and L-glutamine (2 mM) were treated with anti-CD40 antibody (1 μg/ml, eBioscience) and recombinant mouse IL-4 (20 ng/ml; PeproTech). Cells were collected at the indicated time points. Class switch recombination (CSR) was measured by staining with PE-labeled anti-mouse IgG1 (BD Biosciences) and APC-labeled anti-mouse CD45R/B220 (BD Biosciences). The expression of CD40 and FAS was measured by staining with APC-labeled anti-mouse CD40 (BioLegend) and APC-labeled anti-mouse CD95 (BioLegend), respectively. Data acquisition was performed using a FACSVerse flow cytometer (BD Biosciences) (Cheong et al., 2016).
Reverse transcriptase (RT)-PCR and quantitative real time (QRT)-PCR
Total RNA was harvested from cells using RNeasy Mini Kit (QIAGEN) and subjected to RNase-Free DNase Set (QIAGEN) for genomic DNA digestion. For RT-PCR, cDNA was generated using iScript Reverse Transcription kit. To compare CD40 mRNA products from cells with different sgRNA perturbations, the human CD40 gene was amplified by using primers binding to the exon sequences. The amplified fragments were then analyzed by agarose gel electrophoresis. Products were then extracted from agarose gels and purified prior to nucleic acid sequence analysis. GAPDH mRNA products was used as an internal control. Quantitative real-time RT-PCR was performed using Power SYBR Green RNA-to-CT 1-Step Kit (Applied Biosystems) on an CFX96 Touch Real-Time PCR Detection System (Bio-Rad), and data were normalized to internal control 18S rRNA. Relative expression was calculated using 2−ΔΔCt method. All samples were run in technical triplicates and at least three independent experiments were performed. The primer sequences were listed in the Key Resources Table.
cDNA rescue
CD40 cDNA construct in entry vector was purchased from DNASU and was subcloned into pLX-TRC313 vector (gift from John Doench) through Gateway cloning. Daudi Cas9 cells with stable C-terminal V5-tag CD40 cDNA expression were established by lentiviral transduction and hygromycin selection. CD40 cDNA expression was confirmed by immunoblot and FACS.
The design of rescue cDNA for CELF1is described as followed in the following table. sgRNA sequence is highlighted. The PAM sequence is underlined, and the PAM site mutation that abrogated CRISPR editing is shown in red. The rescue cDNA was synthesized by GenScript (Piscataway, NJ) and cloned into pLX-TRC313 vector.
| CELF1 KO and Rescue | |
|---|---|
| sgRNA | 5′ – GGACCTGATGGCCTGAGCCG – 3′ |
| Genomic DNA | 5′ – GGA CCT GAT GGC CTG AGC CGA GGT– 3′ |
| Rescue cDNA | 5′ – GGA CCT GAT GGC CTG AGC CGA GCT – 3′ |
| Rescue cDNA sequence surrounding the PAM site mutation (in red, sgRNA sequence in yellow) | GACATCCGAGTCATGTTCTCTTCGTTTGGACAGATTGAAGAATGCCGGATATTGCGGGGACCTGATGGCCTGAGCCGAGCTTGTGCATTTGTGACTTTTACAACAAGAGCCATGGCACAGACGGCTATCAAGGCAATGCACCAAGCACAGACCAT |
Luciferase reporter assay
The full-length CD40 3′UTR and CD40 5′UTR were synthesized (IDT) and subcloned into the pGL3-Luc plasmid to generate pGL3-CD40 3′UTR-Luc and pGL3-Luc-CD40 5′UTR reporter constructs. The sequence and orientation of the fragments inserted were confirmed by enzyme digestion and sequencing. The transient transfection was done by electroporation performed on Gene Pulser II (BIO-RAD). The luciferase reporter constructs were co-transfected into cells along with pIS2, a Renilla luciferase control reporter, serving as an internal control of the transfection efficiencies. After transfection, cells were cultured in 10% FCS containing RPMI medium for 48 h. Luciferase activity was then measured using Dual-Luciferase reporter assay system, according to manufacturer’s instructions (Promega). The SpectraMax (Molecular Devices) was used to perform the measurement.
RNA-seq
Primary human B cells were isolated from 3 donors independently and seeded at 1 million cells/mL, followed by MegaCD40L (50 ng/mL) stimulation for 0h, 4h and 18h respectively. Dead cells were then removed by Dead Cell Removal Kit (Miltenyi Biotec), following the manufacturer’s manual. Total RNAs were subsequently isolated using PureLink RNA Mini Kit (Thermo Fisher Scientific) following the manufacturer’s manual. An in-column DNA digestion step was included to remove any residual genomic DNA contamination. To construct RNA-seq libraries, 500 ng total RNA was used for polyA mRNA-selection, using the NEBNext Poly(A) mRNA Magnetic Isolation Module (New England Biolabs), followed by library construction via NEBNext Ultra RNA Library Prep Kit (New England Biolabs). Each experimental treatment was performed in triplicate. Libraries were multi-indexed, pooled and sequenced on an Illumina NextSeq 500 sequencer using single-end 75 bp reads (Illunima).
RNA immunoprecipitation
RNA immunoprecipitation (RIP) experiments were performed using the Magna RIP RNA-binding protein immunoprecipitation kit (17–700, Millipore, Billerica, MA) following the manual. Briefly, 2 × 107 of Daudi cells were lysed with lysis buffer supplemented with the RNase and protease inhibitors (which were included in the kit). Ten percent of total lysate was collected as input control. Anti-Virma antibody (A302–124A, Bethyl, Montgomery, TX) was used for immunoprecipitation of WTAP and WTAP-bound transcripts. Anti-Rabit IgG was used for negative control pulldowns. Immunopurified WTAP-bound mRNAs were subjected to RT-qPCR analysis, using a Taqman probe targeting the CD40 mRNA (Hs00386848_m1, ThermoFisher). RT-qPCR was then performed, using TaqMan Fast Virus 1-Step Master Mix (4444432, ThermoFisher), following the manufacturer instructions. Relative enrichment of RIP in each sample was visualized by normalizing the Cq value of the IP fragment to the Cq value of the corresponding 10% input.
CD40 mRNA Specific M6A QPCR Assay
The Magna MeRIP m6A kit (Millipore, Billerica, MA) was used to examine CD40 mRNA m6A modification, according to the manufacturer’s protocol. Briefly, 20 μg of total mRNA was sheared to approximately 100 nt in length by metal-ion induced fragmentation and was then purified. Ten percent of the fragmented RNA was collected as input control. The remaining fragmented RNA was incubated with mouse anti-m6A antibody (included in the kit) or control mouse IgG overnight at 4°C. Methylated RNAs were then bead immunoprecipitated, eluted by competition with free m6A, and recovered by miRNeasy mini kit (QIAGEN). Eluted RNAs were then subject to RT-PCR analysis, using the CD40-specific primers listed in Table S4. Relative enrichment of m6A in each sample was calculated by normalizing the Cq value of the m6a-IP sample to the Cq value of the corresponding 10% input. The CD40 mRNA m6A modification sites were predicted by SRAMP prediction server. RT-PCR primers were designed to flank the predicted m6A sites (Zhou et al., 2016).
Confocal Immunofluorescence Microscopy
Daudi cells were seeded on glass slides in phosphate buffered saline (PBS), allowed to air dry and then fixed with 4% paraformaldehyde (PFA) in PBS for 10 minutes. PFA was removed and fixed cells were permeabilized with 0.1% digitonin in PBS. Slides were then blocked with 20% newborn goat serum (NGS) in PBS for 1 hour at room temperature. Subsequently, they were incubated with primary antibodies against CD40 (1:500) or LAMP1 (1:100) in PBS containing 0.1% digitonin and 20% NGS for 1 hour at room temperature. Slides were then washed three times and then incubated with secondary antibodies (Alexa fluor 488-conjugated goat antimouse and Alex Fluor 594-conjugated goat anti-rabbit diluted 1:250 in PBS/0.1% digitonin/20%NGS) for 1 hour. Finally, slides were washed three times in PBS and incubated with 10 uL of Hoechst 33258 (10 mg/mL in PBS/0.1% digitonin) for 5 minutes to stain nuclear DNA. Cells were then washed and dehydrated sequentially from 70% (1 minute) to 90% (1 minute) to 100% ethanol (1 minute). ProLong Gold anti-fade reagent was applied to the slide, which was then sealed with a No. 1.5 coverslip. Image acquisition was performed at the Brigham and Women’s Hospital core facility with the Zeiss LSM 800 instrument. Image analysis was performed with the Zeiss ZEN Lite (Blue) software. Circular ROIs were drawn around the cells and diameters were automatically computed by ZEN Lite (Blue) software.
QUANTIFICATION AND STATISTICAL ANALYSIS
Unless otherwise indicated, all bar graphs and line graphs represent the arithmetic mean of three independent experiments (n = 3), with error bars denoting standard deviations. Data were analyzed using two-tailed paired Student t test or analysis of variance (ANOVA) with the appropriate post-test using GraphPad Prism7 software. P values correlate with symbols as follows, ns = not significant, p > 0.05; * p < 0.05; ** p < 0.01; *** p < 0.001. The statistical significance of CRISPR screen hits was calculated using the STARSs algorithm v1.0.0 (Doench et al., 2016). Multiple hypothesis testing was adjusted using the Benjamini-Hochberg method with false discovery rate < 0.05 to filter significant screen hits. STARs was used to compare sgRNA abundances in the Daudi B cell library prior to FACS sort with levels in either the Fas low or Fas high sorted populations. Thus, sgRNA abundances in the Fas low population were compared to the input library levels, and likewise sgRNA abundances in the Fas high population were compared to input levels. For RNaseq analysis, 1 million B cells (purified as described above) were used from each of three de-identified donors. For flow cytometry experiments using mouse B cells, 10,000 live cells were analyzed from each of at least n = 3 wild-type and FBXO11 knockout animals per figure (three mice per group were used for Figure 2H, five per group for Figure 2I, three per group for Figure S2G and five per group for Figures S3A and S3B). Mean + SD values were then plotted using GraphPad Prism7 software. Gene set enrichment analysis (GSEA) was performed using the GSEAPreranked module using the KEGG, BIOCARTA and REACTOME pathway databases. Default parameters of GSEAPreranked was used, with the exception that the Enrichment statistic was set as classic. Finally, false discovery rate reported by GSEA was used to filter significantly enriched pathways. GeneMANIA (https://genemania.org/) was used in Figure S7A to construct the protein-protein interaction map.
DATA AND CODE AVAILABILITY
The accession number for all RNA-seq datasets reported in this paper is GEO: GSE101666.
Supplementary Material
Highlights.
Genome-wide B cell CRISPR screen reveals factors controlling CD40 levels
FBXO11 targets repressors CTBP1 and BCL6 to support CD40 transcription
The RNA binding protein CELF1 controls CD40 through mRNA splicing roles
M6A writer WTAP, ESCRT machinery, and phosphatase DUSP10 suppress CD40
ACKNOWLEDGMENTS
We thank Adam Chicoine for assistance with flow cytometry and Emma Vaimberg for assistance with pooled screen PCR amplification and next-generation sequencing. We thank Drs. Katia Basso and Riccardo Dalla-Favera for helpful discussions and for supplying fixed tissues from control and FBXO11 deficient mice. We thank Dr. Jeff Sample for the KEM1 and MUTU1 cell lines and Michael Walsh for establishing multiple Cas9+ B-cell lines. This work was supported by a Burroughs Wellcome Career Award in Medical Sciences (to B.E.G.), NIAID (RO1 AI137337 to B.E.G. and RO1 AI123420 to B.Z.), an American Cancer Society Research Scholar Award (to B.E.G.), a Singapore Agency for Science, Technology and Research (A*STAR) pre-doctoral fellowship (to L.W.W.), a National Research Foundation of Korea (NRF) fellowship (to T.C.C.), Fondazione Veronesi (to C.P.), a Howard Hughes predoctoral fellowship (to S.J.), the Next Generation Fund at the Broad Institute (to J.G.D.), and a Boston Ellison Foundation (to R.C.).
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.celrep.2019.06.079.
REFERENCES
- Alizadeh AA, Eisen MB, Davis RE, Ma C, Lossos IS, Rosenwald A, Boldrick JC, Sabet H, Tran T, Yu X, et al. (2000). Distinct types of diffuse large B-cell lymphoma identified by gene expression profiling. Nature 403, 503–511. [DOI] [PubMed] [Google Scholar]
- Australia and New Zealand Multiple Sclerosis Genetics Consortium (2009). Genome-wide association study identifies new multiple sclerosis susceptibility loci on chromosomes 12 and 20. Nat. Genet 41, 824–828. [DOI] [PubMed] [Google Scholar]
- Basso K, and Dalla-Favera R (2010). BCL6: master regulator of the germinal center reaction and key oncogene in B cell lymphomagenesis. Adv. Immunol 105, 193–210. [DOI] [PubMed] [Google Scholar]
- Basso K, and Dalla-Favera R (2015). Germinal centres and B cell lymphomagenesis. Nat. Rev. Immunol. 15, 172–184. [DOI] [PubMed] [Google Scholar]
- Basso K, Klein U, Niu H, Stolovitzky GA, Tu Y, Califano A, Cattoretti G, and Dalla-Favera R (2004). Tracking CD40 signaling during germinal center development. Blood 104, 4088–4096. [DOI] [PubMed] [Google Scholar]
- Beisang D, Rattenbacher B, Vlasova-St Louis IA, and Bohjanen PR (2012). Regulation of CUG-binding protein 1 (CUGBP1) binding to target transcripts upon T cell activation. J. Biol. Chem 287, 950–960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bishop GA (2004). The multifaceted roles of TRAFs in the regulation of B-cell function. Nat. Rev. Immunol 4, 775–786. [DOI] [PubMed] [Google Scholar]
- Bishop GA (2009). The many faces of CD40: multiple roles in normal immunity and disease. Semin. Immunol 21, 255–256. [DOI] [PubMed] [Google Scholar]
- Blanco-Kelly F, Matesanz F, Alcina A, Teruel M, Díaz-Gallo LM, Gómez-García M, López-Nevot MA, Rodrigo L, Nieto A, Cardeña C, et al. (2010). CD40: novel association with Crohn’s disease and replication in multiple sclerosis susceptibility. PLoS ONE 5, e11520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casola S, Cattoretti G, Uyttersprot N, Koralov SB, Seagal J, Hao Z, Waisman A, Egert A, Ghitza D, and Rajewsky K (2006). Tracking germinal center B cells expressing germ-line immunoglobulin gamma1 transcripts by conditional gene targeting. Proc. Natl. Acad. Sci. USA 103, 7396–7401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cepeda D, Ng HF, Sharifi HR, Mahmoudi S, Cerrato VS, Fredlund E, Magnusson K, Nilsson H, Malyukova A, Rantala J, et al. (2013). CDK-mediated activation of the SCF(FBXO) (28) ubiquitin ligase promotes MYC-driven transcription and tumourigenesis and predicts poor survival in breast cancer. EMBO Mol. Med 5, 1067–1086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaudhury A, Cheema S, Fachini JM, Kongchan N, Lu G, Simon LM, Wang T, Mao S, Rosen DG, Ittmann MM, et al. (2016). CELF1 is a central node in post-transcriptional regulatory programmes underlying EMT. Nat. Commun 7, 13362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Chen J, Xiong Y, Da Q, Xu Y, Jiang X, and Tang H (2006). Internalization of CD40 regulates its signal transduction in vascular endothelial cells. Biochem. Biophys. Res. Commun 345, 106–117. [DOI] [PubMed] [Google Scholar]
- Cheong TC, Compagno M, and Chiarle R (2016). Editing of mouse and human immunoglobulin genes by CRISPR-Cas9 system. Nat. Commun 7, 10934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colombo M, Moita C, van Niel G, Kowal J, Vigneron J, Benaroch P, Manel N, Moita LF, Théry C, and Raposo G (2013). Analysis of ESCRT functions in exosome biogenesis, composition and secretion highlights the heterogeneity of extracellular vesicles. J. Cell Sci 126, 5553–5565. [DOI] [PubMed] [Google Scholar]
- De Silva NS, and Klein U (2015). Dynamics of B cells in germinal centres. Nat. Rev. Immunol 15, 137–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, and Gingeras TR (2013). STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doench JG, Fusi N, Sullender M, Hegde M, Vaimberg EW, Donovan KF, Smith I, Tothova Z, Wilen C, Orchard R, et al. (2016). Optimized sgRNA design to maximize activity and minimize off-target effects of CRISPR-Cas9. Nat. Biotechnol 34, 184–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duan S, Cermak L, Pagan JK, Rossi M, Martinengo C, di Celle PF, Chapuy B, Shipp M, Chiarle R, and Pagano M (2012). FBXO11 targets BCL6 for degradation and is inactivated in diffuse large B-cell lymphomas. Nature 481, 90–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elgueta R, Benson MJ, de Vries VC, Wasiuk A, Guo Y, and Noelle RJ (2009). Molecular mechanism and function of CD40/CD40L engagement in the immune system. Immunol. Rev 229, 152–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrari S, Giliani S, Insalaco A, Al-Ghonaium A, Soresina AR, Loubser M, Avanzini MA, Marconi M, Badolato R, Ugazio AG, et al. (2001). Mutations of CD40 gene cause an autosomal recessive form of immunodeficiency with hyper IgM. Proc. Natl. Acad. Sci. USA 98, 12614–12619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han S, Hathcock K, Zheng B, Kepler TB, Hodes R, and Kelsoe G (1995). Cellular interaction in germinal centers. Roles of CD40 ligand and B7–2 in established germinal centers. J. Immunol 155, 556–567. [PubMed] [Google Scholar]
- Hancock WW, Sayegh MH, Zheng XG, Peach R, Linsley PS, and Turka LA (1996). Costimulatory function and expression of CD40 ligand, CD80, and CD86 in vascularized murine cardiac allograft rejection. Proc. Natl. Acad. Sci. USA 93, 13967–13972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hao Z, Duncan GS, Seagal J, Su YW, Hong C, Haight J, Chen NJ, Elia A, Wakeham A, Li WY, et al. (2008). Fas receptor expression in germinal-center B cells is essential for T and B lymphocyte homeostasis. Immunity 29, 615–627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatzivassiliou EG, Kieff E, and Mosialos G (2007). Constitutive CD40 signaling phenocopies the transforming function of the Epstein-Barr virus oncoprotein LMP1 in vitro. Leuk. Res 31, 315–320. [DOI] [PubMed] [Google Scholar]
- Hennino A, Bérard M, Krammer PH, and Defrance T (2001). FLICE-inhibitory protein is a key regulator of germinal center B cell apoptosis. J. Exp. Med 193, 447–458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holler N, Kataoka T, Bodmer JL, Romero P, Romero J, Deperthes D, Engel J, Tschopp J, and Schneider P (2000). Development of improved soluble inhibitors of FasL and CD40L based on oligomerized receptors. J. Immunol. Methods 237, 159–173. [DOI] [PubMed] [Google Scholar]
- Hömig-Hölzel C, Hojer C, Rastelli J, Casola S, Strobl LJ, Müller W, Quintanilla-Martinez L, Gewies A, Ruland J, Rajewsky K, and Zimber-Strobl U (2008). Constitutive CD40 signaling in B cells selectively activates the noncanonical NF-kappaB pathway and promotes lymphomagenesis. J. Exp. Med 205, 1317–1329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huber AK, Finkelman FD, Li CW, Concepcion E, Smith E, Jacobson E, Latif R, Keddache M, Zhang W, and Tomer Y (2012). Genetically driven target tissue overexpression of CD40: a novel mechanism in autoimmune disease. J. Immunol 189, 3043–3053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huttlin EL, Bruckner RJ, Paulo JA, Cannon JR, Ting L, Baltier K, Colby G, Gebreab F, Gygi MP, Parzen H, et al. (2017). Architecture of the human interactome defines protein communities and disease networks. Nature 545, 505–509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson J, Filipovich AH, and Zhang K (1993). X-Linked Hyper IgM Syndrome In GeneReviews, Volume R, Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJH, Stephens K, and Amemiya A, eds. (University of Washington; ). [PubMed] [Google Scholar]
- Kalsotra A, Xiao X, Ward AJ, Castle JC, Johnson JM, Burge CB, and Cooper TA (2008). A postnatal switch of CELF and MBNL proteins reprograms alternative splicing in the developing heart. Proc. Natl. Acad. Sci. USA 105, 20333–20338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kress C, Gautier-Courteille C, Osborne HB, Babinet C, and Paillard L (2007). Inactivation of CUG-BP1/CELF1 causes growth, viability, and spermatogenesis defects in mice. Mol. Cell. Biol. 27, 1146–1157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuyumcu-Martinez NM, Wang GS, and Cooper TA (2007). Increased steady-state levels of CUGBP1 in myotonic dystrophy 1 are due to PKC-mediated hyperphosphorylation. Mol. Cell 28, 68–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laman JD, Claassen E, and Noelle RJ (2017). Functions of CD40 and Its Ligand, gp39 (CD40L). Crit. Rev. Immunol. 37, 371–420. [DOI] [PubMed] [Google Scholar]
- Li G, Diogo D, Wu D, Spoonamore J, Dancik V, Franke L, Kurreeman F, Rossin EJ, Duclos G, Hartland C, et al. ; Rheumatoid Arthritis Consortium International (RACI) (2013). Human genetics in rheumatoid arthritis guides a high-throughput drug screen of the CD40 signaling pathway. PLoS Genet. 9, e1003487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu YJ, Joshua DE, Williams GT, Smith CA, Gordon J, and MacLennan IC (1989). Mechanism of antigen-driven selection in germinal centres. Nature 342, 929–931. [DOI] [PubMed] [Google Scholar]
- Liu J, Yue Y, Han D, Wang X, Fu Y, Zhang L, Jia G, Yu M, Lu Z, Deng X, et al. (2014). A METTL3-METTL14 complex mediates mammalian nuclear RNA N6-adenosine methylation. Nat. Chem. Biol 10, 93–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lohr JG, Stojanov P, Lawrence MS, Auclair D, Chapuy B, Sougnez C, Cruz-Gordillo P, Knoechel B, Asmann YW, Slager SL, et al. (2012). Discovery and prioritization of somatic mutations in diffuse large B-cell lymphoma (DLBCL) by whole-exome sequencing. Proc. Natl. Acad. Sci. USA 109, 3879–3884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love MI, Huber W, and Anders S (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma Y, Walsh MJ, Bernhardt K, Ashbaugh CW, Trudeau SJ, Ashbaugh IY, Jiang S, Jiang C, Zhao B, Root DE, et al. (2017). CRISPR/Cas9 Screens Reveal Epstein-Barr Virus-Transformed B Cell Host Dependency Factors. Cell Host Microbe 21, 580–591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mamińska A, Bartosik A, Banach-Orłowska M, Pilecka I, Jastrzębski K, Zdżalik-Bielecka D, Castanon I, Poulain M, Neyen C, Wolińska-Nizioł L, et al. (2016). ESCRT proteins restrict constitutive NF-κB signaling by trafficking cytokine receptors. Sci. Signal. 9, ra8. [DOI] [PubMed] [Google Scholar]
- Matus-Nicodemos R, Vavassori S, Castro-Faix M, Valentin-Acevedo A, Singh K, Marcelli V, and Covey LR (2011). Polypyrimidine tract-binding protein is critical for the turnover and subcellular distribution of CD40 ligand mRNA in CD4+ T cells. J. Immunol. 186, 2164–2171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nieters A, Bracci PM, de Sanjosé S, Becker N, Maynadié M, Benavente Y, Foretova L, Cocco P, Staines A, Holly EA, et al. (2011). A functional TNFRSF5 polymorphism and risk of non-Hodgkin lymphoma, a pooled analysis. Int. J. Cancer 128, 1481–1485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nonoyama S, Hollenbaugh D, Aruffo A, Ledbetter JA, and Ochs HD (1993). B cell activation via CD40 is required for specific antibody production by antigen-stimulated human B cells. J. Exp. Med. 178, 1097–1102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Connell BC, and Harper JW (2007). Ubiquitin proteasome system (UPS): what can chromatin do for you? Curr. Opin. Cell Biol 19, 206–214. [DOI] [PubMed] [Google Scholar]
- Park JH, Chang HS, Park CS, Jang AS, Park BL, Rhim TY, Uh ST, Kim YH, Chung IY, and Shin HD (2007). Association analysis of CD40 polymorphisms with asthma and the level of serum total IgE. Am. J. Respir. Crit. Care Med 175, 775–782. [DOI] [PubMed] [Google Scholar]
- Pasqualucci L, Khiabanian H, Fangazio M, Vasishtha M, Messina M, Holmes AB, Ouillette P, Trifonov V, Rossi D, Tabbò F, et al. (2014). Genetics of follicular lymphoma transformation. Cell Rep. 6, 130–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters AL, Stunz LL, and Bishop GA (2009). CD40 and autoimmunity: the dark side of a great activator. Semin. Immunol 21, 293–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Philips AV, Timchenko LT, and Cooper TA (1998). Disruption of splicing regulated by a CUG-binding protein in myotonic dystrophy. Science 280, 737–741. [DOI] [PubMed] [Google Scholar]
- Raiborg C, and Stenmark H (2009). The ESCRT machinery in endosomal sorting of ubiquitylated membrane proteins. Nature 458, 445–452. [DOI] [PubMed] [Google Scholar]
- Raychaudhuri S, Remmers EF, Lee AT, Hackett R, Guiducci C, Burtt NP, Gianniny L, Korman BD, Padyukov L, Kurreeman FA, et al. (2008). Common variants at CD40 and other loci confer risk of rheumatoid arthritis. Nat. Genet 40, 1216–1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saito M, Gao J, Basso K, Kitagawa Y, Smith PM, Bhagat G, Pernis A, Pasqualucci L, and Dalla-Favera R (2007). A signaling pathway mediating downregulation of BCL6 in germinal center B cells is blocked by BCL6 gene alterations in B cell lymphoma. Cancer Cell 12, 280–292. [DOI] [PubMed] [Google Scholar]
- Schmitz R, Ceribelli M, Pittaluga S, Wright G, and Staudt LM (2014). Oncogenic mechanisms in Burkitt lymphoma. Cold Spring Harb. Perspect. Med 4, a014282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider C, Kon N, Amadori L, Shen Q, Schwartz FH, Tischler B, Bossennec M, Dominguez-Sola D, Bhagat G, Gu W, et al. (2016). FBXO11 inactivation leads to abnormal germinal-center formation and lymphoproliferative disease. Blood 128, 660–666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz S, Mumbach MR, Jovanovic M, Wang T, Maciag K, Bushkin GG, Mertins P, Ter-Ovanesyan D, Habib N, Cacchiarelli D, et al. (2014). Perturbation of m6A writers reveals two distinct classes of mRNA methylation at internal and 5′ sites. Cell Rep. 8, 284–296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skaar JR, Pagan JK, and Pagano M (2013). Mechanisms and function of substrate recruitment by F-box proteins. Nat. Rev. Mol. Cell Biol 14, 369–381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skibola CF, Nieters A, Bracci PM, Curry JD, Agana L, Skibola DR, Hubbard A, Becker N, Smith MT, and Holly EA (2008). A functional TNFRSF5 gene variant is associated with risk of lymphoma. Blood 111, 4348–4354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang Y, Wang H, Wei B, Guo Y, Gu L, Yang Z, Zhang Q, Wu Y, Yuan Q, Zhao G, and Ji G (2015). CUG-BP1 regulates RyR1 ASI alternative splicing in skeletal muscle atrophy. Sci. Rep 5, 16083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teis D, Saksena S, Judson BL, and Emr SD (2010). ESCRT-II coordinates the assembly of ESCRT-III filaments for cargo sorting and multivesicular body vesicle formation. EMBO J. 29, 871–883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomer Y, Concepcion E, and Greenberg DA (2002). A C/T single-nucleotide polymorphism in the region of the CD40 gene is associated with Graves’ disease. Thyroid 12, 1129–1135. [DOI] [PubMed] [Google Scholar]
- Tone M, Tone Y, Fairchild PJ, Wykes M, and Waldmann H (2001). Regulation of CD40 function by its isoforms generated through alternative splicing. Proc. Natl. Acad. Sci. USA 98, 1751–1756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuscano JM, Druey KM, Riva A, Pena J, Thompson CB, and Kehrl JH (1996). Bcl-x rather than Bcl-2 mediates CD40-dependent centrocyte survival in the germinal center. Blood 88, 1359–1364. [PubMed] [Google Scholar]
- van Kooten C, and Banchereau J (2000). CD40-CD40 ligand. J. Leukoc. Biol 67, 2–17. [DOI] [PubMed] [Google Scholar]
- Victora GD, and Nussenzweig MC (2012). Germinal centers. Annu. Rev. Immunol. 30, 429–457. [DOI] [PubMed] [Google Scholar]
- Vlasova IA, Tahoe NM, Fan D, Larsson O, Rattenbacher B, Sternjohn JR, Vasdewani J, Karypis G, Reilly CS, Bitterman PB, and Bohjanen PR (2008). Conserved GU-rich elements mediate mRNA decay by binding to CUG-binding protein 1. Mol. Cell 29, 263–270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vu LP, Pickering BF, Cheng Y, Zaccara S, Nguyen D, Minuesa G, Chou T, Chow A, Saletore Y, MacKay M, et al. (2017). The N6-methyladenosine (m6A)-forming enzyme METTL3 controls myeloid differentiation of normal hematopoietic and leukemia cells. Nat. Med. 23, 1369–1376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wakeland EK, Liu K, Graham RR, and Behrens TW (2001). Delineating the genetic basis of systemic lupus erythematosus. Immunity 15, 397–408. [DOI] [PubMed] [Google Scholar]
- Winkelstein JA, Marino MC, Ochs H, Fuleihan R, Scholl PR, Geha R, Stiehm ER, and Conley ME (2003). The X-linked hyper-IgM syndrome: clinical and immunologic features of 79 patients. Medicine (Baltimore) 82, 373–384. [DOI] [PubMed] [Google Scholar]
- Yue Y, Liu J, and He C (2015). RNA N6-methyladenosine methylation in post-transcriptional gene expression regulation. Genes Dev. 29, 1343–1355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yue Y, Liu J, Cui X, Cao J, Luo G, Zhang Z, Cheng T, Gao M, Shu X, Ma H, et al. (2018). VIRMA mediates preferential m6A mRNA methylation in 3’UTR and near stop codon and associates with alternative polyadenylation. Cell Discov. 4, 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Blattman JN, Kennedy NJ, Duong J, Nguyen T, Wang Y, Davis RJ, Greenberg PD, Flavell RA, and Dong C (2004). Regulation of innate and adaptive immune responses by MAP kinase phosphatase 5. Nature 430, 793–797. [DOI] [PubMed] [Google Scholar]
- Zhou Y, Zeng P, Li YH, Zhang Z, and Cui Q (2016). SRAMP: prediction of mammalian N6-methyladenosine (m6A) sites based on sequence-derived features. Nucleic Acids Res. 44, e91. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The accession number for all RNA-seq datasets reported in this paper is GEO: GSE101666.