

Abstract
Metal–superoxo species are typically proposed as key intermediates in the catalytic cycle of dioxygen activation by metalloenzymes involving different transition metal cofactors. In this regard, while a series of Fe–, Co–, and Ni–superoxo complexes have been reported to date, well-defined Mn–superoxo complexes remain rather rare. Herein, we report two mononuclear MnIII–superoxo species, Mn(BDPP)(O2•–) (2, H2BDPP = 2,6-bis((2-(S)-diphenylhydroxylmethyl-1-pyrrolidinyl)methyl)pyridine) and Mn(BDPBrP)(O2•–) (2′, H2BDPBrP = 2,6-bis((2-(S)-di(4-bromo)phenylhydroxyl-methyl-1-pyrrolidinyl)methyl)pyridine), synthesized by bubbling O2 into solutions of their MnII precursors, Mn(BDPP) (1) and Mn(BDPBrP) (1′), at −80 °C. A combined spectroscopic (resonance Raman and electron paramagnetic resonance (EPR) spectroscopy) and computational study evidence that both complexes contain a high-spin MnIII center (SMn = 2) antiferromagnetically coupled to a superoxo radical ligand (SOO• = 1/2), yielding an overall S = 3/2 ground state. Complexes 2 and 2′ were shown to be capable of abstracting a H atom from 2,2,6,6-tetramethyl-1-hydroxypiperidine (TEMPO-H) to form MnIII–hydroperoxo species, Mn(BDPP)(OOH) (5) and Mn(BDPBrP)(OOH) (5′). Complexes 5 and 5′ can be independently prepared by the reactions of the isolated MnIII-aqua complexes, [Mn(BDPP)(H2O)]OTf (6) and [Mn(BDPBrP)(H2O)]OTf (6′), with H2O2 in the presence of NEt3. The parallel-mode EPR measurements established a high-spin S = 2 ground state for 5 and 5′.
Short abstract
Two mononuclear MnIII−superoxo complexes, synthesized by bubbling O2 into solutions of their MnII precursors at −80 °C and well-characterized spectroscopically and computationally, were shown to be capable of abstracting a H atom from TEMPO-H to form MnIII−hydroperoxo species.
Introduction
Functionalization of organic compounds using O2, a green oxidant, represents an essential process in nature.1 Despite having favorable thermodynamic driving forces, direct substrate oxygenations usually have to traverse high kinetic barriers arising from the triplet-to-singlet spin inversion of O2.1 Nature has developed a diverse array of enzymes that contain transition metal cofactors to activate O2; thus, metalloenzyme-catalyzed reactions typically proceed with unrivaled efficiencies under ambient conditions. The majority of O2-activating wild-type enzymes utilize Fe and Cu in their active sites, whereas a few of them feature Mn and Ni. To the best of our knowledge, none of them incorporates Co. Catechol dioxygenases play a pivotal role in the global carbon cycle because they catalyze cleavage of aromatic rings and hence reclaim large quantities of carbon sequestered in ubiquitous aromatic compounds in nature.2 Two forms of homoprotocatechuate 2,3-dioxygenases with the respective active-site metal ions being FeII and MnII have been isolated, namely, Fe-HPCD from Brevibacterium fuscum(3) and Mn-MndD from Arthrobacter globiformis.4 Surprisingly, the native enzymes, Fe-HPCD and Mn-MndD, and their artificial variants, Mn-HPCD, Fe-MndD, and Co-HPCD, generated by reconstituting the native enzymes with nonphysiological metal ions, all exhibit comparable catalytic activity.5 Therefore, it has long been believed that transition-metal-catalyzed oxidative transformations share common mechanistic features, in which O2 initially binds to a reduced metal center to form an oxidized metal–superoxo species, which functions as the reagent to perform subsequent chemistry.6 Isopenicillin N synthase (IPNS), an iron-dependent nonheme enzyme, which catalyzes formation of isopenicillin N en route to a number of important antibacterial drugs,7 serves as a prototypical example for the proposed mechanism. O2 association at the FeII center in IPNS has been experimentally shown to yield a FeIII–superoxo intermediate, which initiates hydrogen atom abstraction (HAA) to generate presumably a hydroperoxo species (Scheme 1a).8
Scheme 1. Mechanism of Dioxygen Activation by (a) IPNS, (b) Mn-HPCD, and (c) Model Complexes Supported by a BDPP2– or BDPBrP2– Ligand.

Notably, FeIII– and CoIII–superoxo species were detected in the His200Asn mutant of Fe-HPCD and Co-HPCD in the turnover of an electron-poor substrate, 4-nitocatechol.9 Upon mixing of O2 to Mn-HPCD, two intermediates were trapped and characterized by electron paramagnetic resonance (EPR) spectroscopy. They were assigned to a MnIII ion coordinated by an unidentified radical, assumed to be a superoxo, and a MnII–alkylperoxo species (Scheme 1b), but the conclusive evidence remains elusive.10 Furthermore, MnIII–superoxo intermediates have been invoked in a series of Mn-catalyzed chemical transformations on the basis of the measurements using mass spectrometry,11 and the existence of the putative Mn–superoxo species was postulated by investigations using low-temperature absorption spectroscopy12 and reactivity studies.13
In addition to several synthetic nonheme FeIII– and CoIII–superoxo models,14−18 a MnIII–superoxo complex, [MnIII(O2)(OH2)L]2+ (L = [5,11,17,23-tetrakis(trimethylammonium)-25,26,27,28-tetrahydroxylcalix[4]arene)]), has been crystallographically characterized to feature an unusual linear Mn–O2 arrangement.19 Its electronic structure has been probed by EPR and resonance Raman (rR) spectroscopy, and [MnIII(O2)(OH2)L]2+ was shown to exhibit catalytic reactivity toward alkene epoxidation. Unexpectedly, the determined MnIII–O2 bond distance (2.444 Å) is considerably longer than that of the trans Mn–OH2 bond (2.210 Å). Notably, the MnIII–O bond lengths found for Mn–(O2), Mn–OOR, and Mn–OR (R = H, Me, Ph, Ph-pNO2) species range from 1.832 to 1.901 Å (Table S1). Therefore, more work on well-defined MnIII–superoxo complexes remains highly desirable due to the important roles played by such species in chemistry and biology.
Our earlier work reported synthesis and reactivity of nonheme FeIII– and CoIII–superoxo complexes, Fe(BDPP)(O2•–) (3) and Co(BDPP)(O2•–) (4) (H2BDPP = 2,6-bis((2-(S)-diphenylhydroxylmethyl-1-pyrrolidinyl)methyl)pyridine).14,16 Complexes 3 and 4 were synthesized by oxygenating the corresponding divalent metal precursors and found to be capable of performing HAA reactions (Scheme 1c). The aforementioned mechanistic hypothesis for O2 activation prompted us to test the accessibility of the MnIII–superoxo congeners via the same reaction route. To this end, we herein present the synthesis of two well-defined MnIII–superoxo species and their spectroscopic characterizations as well as reactivity studies.
Results and Disscussion
Synthesis and Characterization of MnIII–Superoxo Complexes
Bubbling O2 into THF or CH2Cl2 solutions of Mn(BDPP) (1) and Mn(BDPBrP) (1′) (H2BDPBrP = 2,6-bis((2-(S)-di(4-bromo)phenylhydroxyl-methyl-1-pyrrolidinyl)methyl)pyridine) at −80 °C results in a color change from pale yellow to deep yellow, due to the growth of three absorption bands at 370, 450, and 740 nm (Figures S4 and 1). This observation hints at the formation of Mn–O2 adducts, Mn(BDPP)(O2•–) (2) and Mn(BDPBrP)(O2•–) (2′). Our repeated attempts to obtain single crystals of 2 and 2′ were foiled due to their gradual decomposition even at −80 °C.
Figure 1.
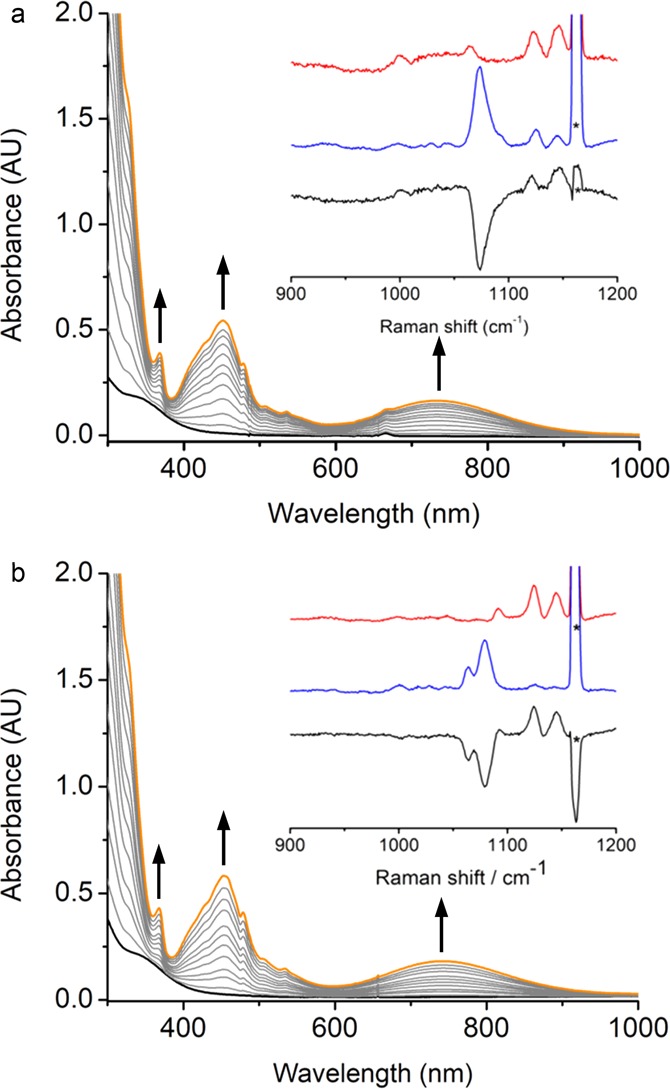
UV–vis spectra change for formation of (a) 2 and (b) 2′. Complex 2 or 2′ (orange) from a reaction of 1 or 1′ (black, 0.4 mM) with O2 in THF at −80 °C. Inset: rR spectra of 2 or 2′ (7.5 mM) in CH2Cl2 (λex 457 nm, 10 mW, 77 K; red line: 2-16O or 2′-16O, blue line: 2-18O or 2′-18O, black line: difference spectrum 2-16O – 2-18O or 2′-16O – 2′-18O). Asterisks denote solvent peaks.
The rR spectra (λex at 457 nm) of 2-16O in CH2Cl2 reveal a Fermi doublet at 1125 and 1145 cm–1 (1135 cm–1 on average; see the inset in Figure 1a), which shifts to 1073 cm–1 upon 18O substitution (99 atom %). The signals at 1125 and 1145 cm–1 in the 2-18O rR spectrum originate from trace 16O2 contamination presumably coming from air during the sample preparation. The observed isotope shifts (62 cm–1) are close to the value for a diatomic O–O bond stretching mode (νO–O) estimated by Hooke’s law. In the case of 2′, its 16O rR spectrum (the inset in Figure 1b) also shows a doublet with exactly the same frequencies as those detected for 2-16O. Interestingly, its 18O rR spectrum also demonstrates a different doublet at 1064 and 1078 cm–1. The relative intensities of the two peaks of a given doublet measured for 2′-16O or 2′-18O are different, which essentially rules out the possibility that there exists two isomers for 2′. On the basis of these observations, we attributed the two peaks observed for 2′-16O and 2′-18O to Fermi doublets. For the related Fe– and Cu–superoxo complexes, Fermi doublets arising from O–O stretching vibrations were typically found for either 16O or 18O samples,15b,20a but detection of both 16O and 18O Fermi doublets has been reported for a structurally characterized diiron(III)-μ-1,2-peroxo complex.20b The O–O stretching frequencies detected for 2 and 2′ closely match those reported for well-characterized metal–superoxo complexes (Table 1), thereby evidencing that 2 and 2′ accommodate a superoxo ligand.
Table 1. O–O Stretching Frequencies of Mononuclear Metal Superoxo Complexesa.
| complex | ν(16O–16O), cm–1 | ν(18O–18O), cm–1 | Δν(O–O), cm–1 | ref |
|---|---|---|---|---|
| 2 | 1125, 1145 | 1073 | 62 | this work |
| 2′ | 1125, 1145 | 1064, 1078 | 61 | this work |
| [MnIIIL(O2•–)(H2O)](PF6)2 | 1124 | 1035 | 89 | (19) |
| 3 | 1125 | 1062 | 63 | (14) |
| 4 | 1135 | 1070 | 65 | (16) |
| [FeIII(TAML)(η2-O2•–)]2– | 1260 | 1183 | 77 | (15) |
| FeIII(TpMe2)(LPh)(O2•–) | 1168 | 1090 | 78 | (17) |
| CoIII(TpMe2)(LPh)(η1-O2•–) | 1150 | 1090 | 60 | (17) |
| CoIII(salen)(py)(O2•–) | 1144 | 1082 | 62 | (18) |
| [CuII(TMG3tren)(η1-O2•–)]+ | 1117 | 1059 | 58 | (21) |
| [CuII(6-pivTPA)(O2•–)]+ | 1130 | 1067 | 63 | (22) |
| [CrIII (14-TMC)(η1-O2•–)(Cl)]+ | 1170 | 1104 | 66 | (23) |
L = 5,11,17,23-tetrakis(trimethylammonium)-25,26,27,28-tetrahydroxycalix[4]arene; TAML = 3,3,6,6,9,9-hexametyl-2,5,7,10-tetraoxo-3,5,6,7,9,10-hexahydro-2H-benzo[e][1,4,7,10]tetraazacyclotridecine-1,4,8,11-tetraide; TpMe2 = hydro-tris(3,5-dimethylpyrazolyl)borate; LPh = bis(2-N-methylimizadolyl)methylphenylborate; salen = N,N′-ethylene-bis(salicylideneiminato); TMG3tren = 1,1,1-tris(1-tetramethylguanidino)]ethyl)amine); 6-pivTPA = (6-pivaloyl-amidopyridyl-2-methyl)bis(6-methylpyridyl-2-methyl)amine; 14-TMC = 1,4,8,11-tetramethyl-1,4,8,11-tetraazacyclo-tetradecane.
In the following, we focus
our discussion on 2′ because it has higher stability
and solubility than those of 2 in MeTHF, and we summarize
similar results of 2 in the Supporting Information. The X-band EPR spectrum of 2′ in MeTHF at 6.3 K (Figure 2) is typical for near-axial quartet systems with a sizable
axial zero-field splitting D and marginal rhombicity E/D (|2D| ≫ 0.3
cm–1, and E/D ≈
0). Specifically, the resonances of geff,⊥ ≈ 4 and geff,|| ≈ 2 originate
from the transition within its MS ≈
± 1/2 Kramers doublet. Consequently, the weak sextet around 115
mT can be attributed to the edge of the intra-Kramers doublet spectrum
(geff,|| ≈ 6 and geff,⊥ → 0) for the MS ≈ ± 3/2 levels. The nicely resolved six lines
at the geff,|| ≈ 2 and 6 and geff,⊥ ≈ 4 regions are due to the
hyperfine coupling with 55 Mn (I = 5/2).
From the relative intensity of the geff ≈ 4 and 6 signals, the effective splitting 2D′ (D′ = 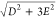 of the quartet ground state was estimated
to be −10.4(5) cm–1 (see the inset in Figure 2).
of the quartet ground state was estimated
to be −10.4(5) cm–1 (see the inset in Figure 2).
Figure 2.
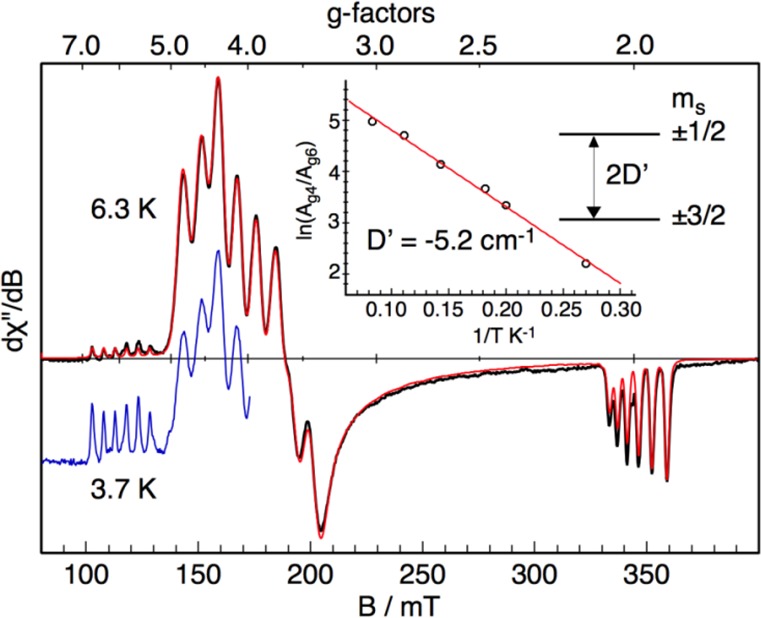
X-band EPR spectra of 2′ (2 mM) recorded at 6.3 K (black) and at 3.7 K (blue), and the simulation for 6.3 K (red). The simulation yields the following parameters: giso = 1.997(10), D = −5.2(5) cm–1, E/D = 0.03(1), and |Ax,y,z| = 234(10), 225(10), and 142(10) MHz. Note that the sign of the A values cannot determined experimentally. The inset shows the logarithm of the intensity ratio of the derivative signals geff ≈ 4 and geff ≈ 6 as a function of inverse temperature and a fit with the corresponding Boltzmann function (red line). Frequency 9.63595 GHz, power 0.2 mW, modulation 100 kHz/0.75 mT.
The experimental EPR spectra of 2′ can be nicely fitted by using a usual spin-Hamiltonian for S = 3/2:
in which g is the intrinsic g matrix and A is the magnetic hyperfine coupling matrix. Double integration of the spectrum revealed nearly quantitative conversion of 1′ to 2′ in a yield of 96 (10)%.
The spectroscopic investigations suggest that 2′ is best formulated as a high-spin MnIII ion (SMn = 2) bound to a O2•− radical (SOO• = 1/2) in an antiferromagnetic fashion yielding an overall quartet ground state (S = 3/2). As we will elaborate below, the exchange interaction in 2′ is in the strong coupling regime. The intrinsic DMn and AMn values of the MnIII center in 2′ can be obtained from the effective D and A values determined experimentally for the total spin S = 3/2 ground state by using the relation D = (7/5)DMn and A = (6/5)AMn.24 The resulting intrinsic values, DMn = −3.7 cm–1 and AMn.iso = 167 MHz, fall into the range typically measured for distorted octahedral MnIII complexes.25 In contrast, an alternative formulation of 2′ as a high-spin MnIV complex (SMn = 3/2) can be safely ruled out, because well-characterized six-coordinate S = 3/2 MnIV compounds rarely feature negative D values and the magnitude of D never exceed 3 cm–1, whereas negative D values are rather common for high-spin MnIII complexes in elongated octahedral environments.26 Experimentally, no signals that can be attributed to S = 5/2 have been observed up to 40 K, above which the fast relaxation induces significant line broadening of the EPR spectrum. Therefore, the exchange coupling constant between the high-spin MnIII center and the superoxo ligand cannot be determined accurately.
Conversion of MnIII–Superoxo Complexes to MnIII–Hydroperoxo Complexes
Treating 2 and 2′ with 10 equiv of TEMPO-H in THF at −90 °C resulted in an exponential decay of their signature bands (Figures S7 and 3) and nearly quantitative yields of TEMPO radical (86% for 2, Figure S8; 93% for 2′, Figure S9). Our previous work showed that the reaction of the related FeIII- and CoIII–superoxo complexes (3 and 4) with H-donor, DHA or TEMPO-H, produced the corresponding hydroperoxo species;14,16 we thus surmised the products of the similar reactions with 2 and 2′ to be MnIII–hydroperoxo species, Mn(BDPP)(OOH) (5) and Mn(BDPBrP)(OOH) (5′). Treating the resulting solutions containing 5 and 5′ with HOTf followed by NaI allowed us to determine the yields of H2O2 to be 83% and 81%, respectively (Figures S11b and S12b).27 The final products (Figures S11a and S12a) obtained by the acidified solution of 5 and 5′ were identified as MnIII-aqua complexes, [Mn(BDPP)(H2O)]OTf (6) and [Mn(BDPBrP)(H2O)]OTf (6′), which were characterized by X-ray crystallography (Figure 4). More importantly, complexes 5 and 5′ can be directly synthesized from the reactions of 6 and 6′ with an excess amount of a 2:1 mixture of H2O2 and NEt3 in THF at −90 °C as monitored by electronic spectroscopy. As shown in Figures S13 and 5, upon addition of the mixture of H2O2 and NEt3, the characteristic absorptions of 6 and 6′ at 460 and 600 nm gradually vanished along with the steady growth of those of 5 and 5′. Our repeated attempts to obtain the O–O vibrational frequencies of 5 and 5′ from rR measurements failed, largely because 5 and 5′ have no intense chromophores in the usual UV–vis region (Figure 3) and the intensity of the O–O stretching signal is too low to be readily detected.
Figure 3.
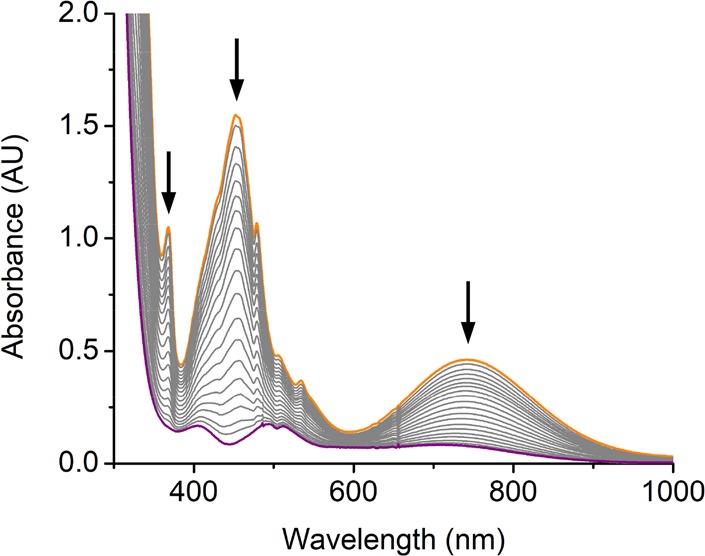
UV–vis spectra change monitored from the reaction of 2′ (orange line, 1 mM) with 10 equiv of TEMPO-H in THF at −90 °C. The purple line represents Mn(BDPBrP)(OOH) (5′) with four absorption bands at 410, 495, 515, and 720 nm.
Figure 4.
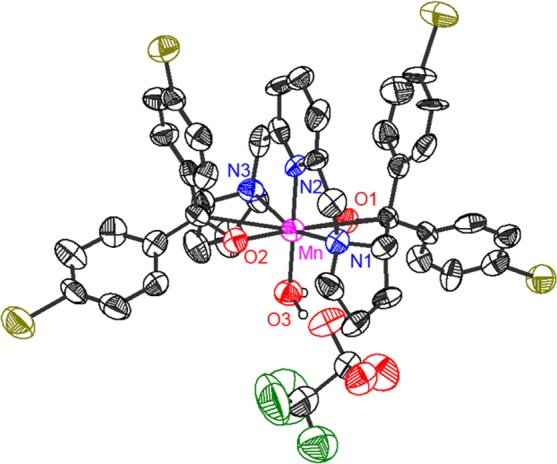
ORTEP presentation of [Mn(BDPBrP)(H2O)]OTf (6′). Hydrogen atoms are omitted for clarity. The MnIII center of 6′ possesses an octahedral coordination environment. The important bond distances and angles of 6′ are as follows: Mn–N1 = 2.248(7) Å, Mn–N2 = 2.069(7) Å, Mn–N3 = 2.255(7) Å, Mn–O1 = 1.861(5) Å, Mn–O2 = 1.881(5) Å, Mn–O3 = 2.023(5) Å; ∠N1–Mn–N2 = 77.7(3)°, ∠N2–Mn–N3 = 79.1(3)°, ∠N1–Mn–O3 = 101.5(3)°, ∠N3–Mn–O3 = 101.7(3)°, ∠O1–Mn–N2 = 90.7(3)°, ∠O2–Mn–N2 = 91.3(2)°, ∠O1–Mn–O3 = 90.5(2)°, ∠O2–Mn–O3 = 87.5(2)°.
Figure 5.
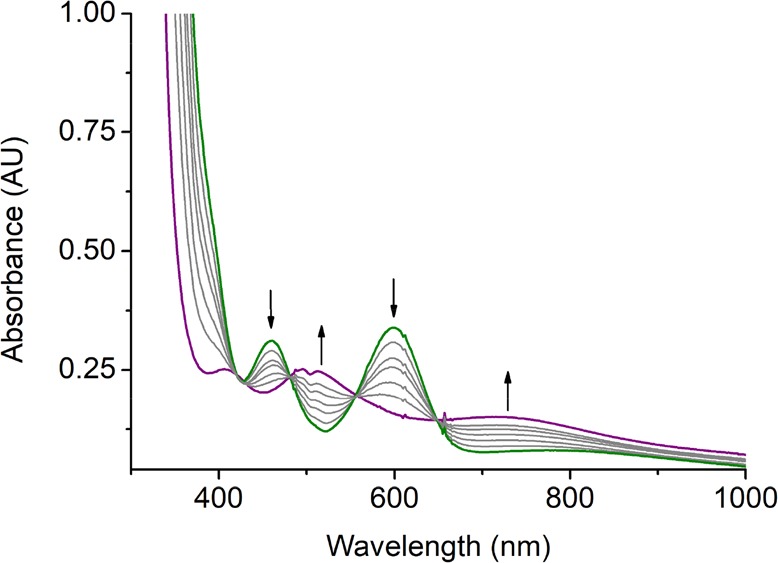
UV–vis spectra change monitored from the reaction of [Mn(BDPBrP)(H2O)]OTf (6′) (green line, 1 mM) with an excess amount of a 2:1 mixture of H2O2 and NEt3 in THF at −90 °C. The purple line represents Mn(BDPBrP)(OOH) (5′).
Figure 6 depicts a parallel-mode X-band EPR spectrum
of 5′ in MeTHF at 10 K. Complex 5′ shows a six-line hyperfine pattern at g ∼
8 that stems from the transition within the |2±⟩
non-Kramers doublet of an S = 2 spin manifold, thus
indicating that 5′ contains a high-spin MnIII center. Its intensity declines with the temperature; hence, 5′ possesses a negative D value. The
fit of the intensity variation as a function of temperature gives D = −3.6(5) cm–1. The resonance
condition for this parallel-mode signal is ℏν =  , where δ±2 ≈
, where δ±2 ≈ 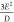 , ℏν is the microwave quanta, and θ is the angle between the magnetic
field (B) and the z-axis of the D tensor. Moreover, its intensity is proportional to
, ℏν is the microwave quanta, and θ is the angle between the magnetic
field (B) and the z-axis of the D tensor. Moreover, its intensity is proportional to 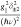 . Thus, one cannot simultaneously determine g|| and δ±2 values accurately. During
the simulation of the EPR spectrum, we therefore only allowed E/D to vary while fixing D to −3.6
cm–1 and giso to 2,
the typical g value often found for related high-spin
MnIII complexes.28 A satisfactory
simulation yields the following parameters, E/D =
0.09(1) and |Az| = 119(10)
MHz. The broad underlying feature starting from ca. 60 mT is tentatively
assigned to a high-spin MnIII decay product since its relative
intensity is dependent on the preparation. The onset of the signal
at ca. 150 mT originates from small amount of O2 in the
solution.
. Thus, one cannot simultaneously determine g|| and δ±2 values accurately. During
the simulation of the EPR spectrum, we therefore only allowed E/D to vary while fixing D to −3.6
cm–1 and giso to 2,
the typical g value often found for related high-spin
MnIII complexes.28 A satisfactory
simulation yields the following parameters, E/D =
0.09(1) and |Az| = 119(10)
MHz. The broad underlying feature starting from ca. 60 mT is tentatively
assigned to a high-spin MnIII decay product since its relative
intensity is dependent on the preparation. The onset of the signal
at ca. 150 mT originates from small amount of O2 in the
solution.
Figure 6.
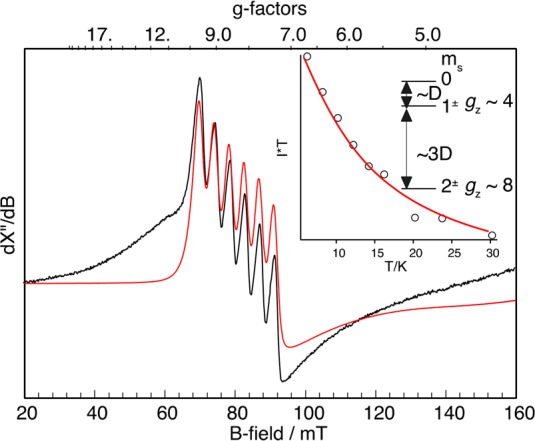
Parallel mode X-band EPR spectrum of 5′ (7.5 mM) recoded at 10 K (black) and the simulation (red). The inset shows signal intensity times temperature versus temperature of g ∼ 8 signal (dots) and a fit with the corresponding Boltzmann function (red line). Frequency 9.396 GHz, power 0.2 mW, and modulation 100 kHz/0.75 mT.
Remarkably, the D and Az values measured for complex 5′ are nearly identical to the intrinsic values of the MnIII center found for 2′ (DMn = −3.7 cm–1 and AMn,z = 118 MHz), thereby suggesting that the MnIII centers in both complexes feature a similar electronic structure and are ligated in an analogous coordination environment. All these observations confirm that 5 and 5′ are indeed high-spin MnIII–hydroperoxo species. Unlike 5 and 5′, 6 and 6′ were found to be EPR-silent in both normal and parallel modes (Figure S10). This finding likely reflects a considerable change in the electronic structure induced by replacing an anionic hydroperoxo ligand with H2O.
DFT Calculations of 2′ and 5′
In order to better understand the nature of 2′ and 5′, detailed computational studies on 2′ and 5′ were undertaken. Complex 2′ was predicted to have a quartet ground state, and the lowest-energy sextet state in which the high-spin MnIII center is ferromagnetically coupled to the superoxo ligand is situated at ∼1300 cm–1 higher in energy. The optimized quartet structure of 2′ features elongated octahedral coordination geometry. The two Mn–Npyrrolidine bonds along the z direction are substantially longer than all of the metal–ligand bond distances in the equatorial plane (Figure 7). The different metal–ligand bond lengths simply reflect the fact that the equatorial plane consists of three negatively charged O donors and a rigidly chelated Npyridine atom, whereas there are two neutral Npyrrolidine donors in the axial direction. In keeping with this reasoning, the MnIII center features an electron configuration of (dyz)1(dxz)1(dxy)1(dz2)1(dx2–y2)0, in which the MnIII dx2–y2 based molecular orbital is vacant. The calculated O–O distance (1.294 Å) of the O2 moiety agrees with those found in structurally characterized metal–superoxo complexes (Table S2). Furthermore, one of the two O2 π* orbitals is doubly occupied, and the other is resided by a β-electron. Such an orbital occupation pattern defines a high-spin MnIII ion (SMn = 2) that is antiferromagnetically coupled to a superoxo radical (SOO• = 1/2).
Figure 7.
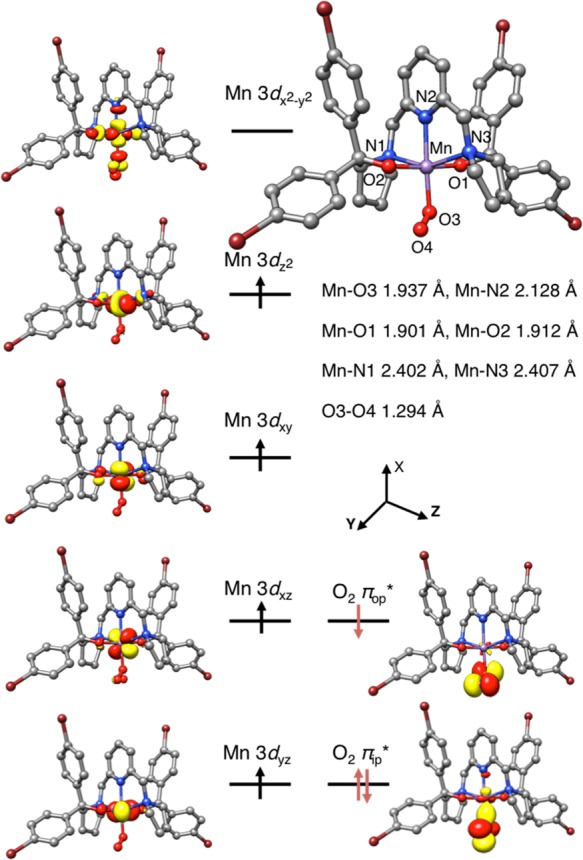
Computed structure of 2′ with key geometric parameters and its schematic molecular orbital diagram.
Complex 5′ also features an elongated octahedral coordination arrangement with the two considerably longer Mn–Npyrrolidine bonds, similar to that of 2′ (Figure 8). Consequently, the MnIII centers in both complexes possess the same electron configuration. Relative to 2′, the estimated O–O distance of the OOH unit in 5′ is significantly lengthened to 1.449 Å, a typical O–O bond length observed for metal–hydroperoxo species.16 Furthermore, the hydroperoxo ligand forms a hydrogen bond with one of the O atom of BDPBrP, analogous to hydrogen bonding observed in the crystal structure of Co(BDPP)(OOH). Our theoretical results revealed that such interaction stabilizes 5′ by 5.3 kcal/mol relative to another isomer, in which the H atom in the OOH ligand points out of the O2 atom of BDPBrP. Moreover, the computed spectroscopic parameters for 2′ and 5′ and the O–O stretching frequency for 2′ (1162 cm–1) match the experimental data within the uncertainty range of the calculations, which further lends credence to the proposed geometric and electronic structures for them (Table S3).
Figure 8.
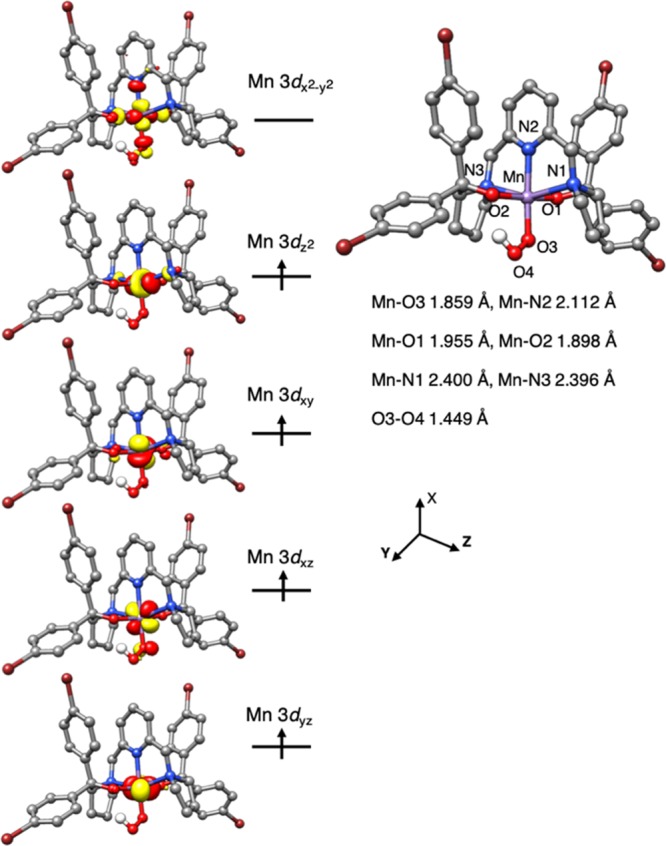
Computed structure of 5′ with key geometric parameters and its schematic molecular orbital diagram.
The weak axial coordination found for the MnIII centers in complexes 2′ and 5′ is consistent with the observed negative D values and the relative magnitude of the three A components (|Ax| ≈ |Ay| < |Az|) (for details, see the Supporting Information). In fact, [MnIII(O2)(OH2)L]2+ features the same coordination geometry as that of 2′, but the distortion was found along the O2–Mn–OH2 axis,19 because the bowl-shaped calix[4]arene ligand with four phenolate groups predefines an equatorial plane and forces the superoxo ligand to coordinate the MnIII center in the axial position.
Reactivity Comparisons of O2 Activations by Mn, Fe, and Co Counterparts
Our findings reveal that 2 and 2′ can be prepared by using the same synthetic route as that for their Fe and Co counterparts, 3 and 4.14,16 However, under the same experimental conditions, formation of 4 (∼0.26 min–1 in THF, even slower rate in CH2Cl2) is at least 10-fold slower than that of 2′ and 3, a situation similar to that observed for O2 binding to Co- and Fe-HPCD.9 Unexpectedly, the relative formation rates of 2′, 3, and 4 do not follow the trend of the redox potentials of their divalent precursors ([MnII(BDPP)]0/+, −0.555 V; [FeII(BDPP)]0/+, −0.328 V; [CoII(BDPP)]0/+, −0.476 V vs Fc/Fc+). Thus, we reason that the difference mainly results from the unavoidable spin transition from a high-spin CoII in Co(BDPP) to a low-spin CoIII in 4, whereas such processes are not required for the generation of 2, 2′, and 3. Bubbling N2 into a THF solution of 3 at −80 °C leads to O2 dissociation,14 whereas analogous interconversion processes have not been found for 2, 2′ (Figure S5), and 4. This observation suggests that the metal–superoxo bonding is weaker in 3 than that in 2, 2′, and 4, consistent with the Fe–O2•– distance computed for 3 being considerably longer than those for 2, 2′, and 4 (Table S4).
Complex 3 is capable of performing HAA toward dihydroanthracene14 (DHA, DC–H 78 kcal/mol29), whereas no reaction was observed when mixing 2, 2′, and 4 with DHA. Instead, 4 was shown to convert 2,2,6,6-tetramethyl-1-hydroxypiperidine (TEMPO-H), a more potent H atom donor (DO–H 69.7 kcal/mol29), to TEMPO radical via HAA.16 Relative to 4, superior HAA efficacy is found for 2′, which breaks the O–H bond at least 500-fold faster. Owing to the rapid reaction rate of 2′ with a large excess of TEMPO-H, where pseudo-first-order reaction kinetics was beyond the temporal resolution of our apparatus, we then performed a kinetic titration experiment with n equiv of TEMPO-H (n ∼ 0.8–2.0). Second-order reaction kinetics was confirmed by the excellent agreement of experimental data and the model below, as shown in Figure 9:
where [A]0 and [A]t represent the concentrations of 2′ before and at a given time t after the addition of TEMPO-H, respectively. The ratio [A]t/[A]0 was obtained from the normalized transient absorption signal of 2′. An initial concentration of 2′ ([A]0 = 1.86 × 10–4 M, 93% yield from the EPR data) was applied in the nonlinear least-squared fitting, and the exact n value (for details, see the Supporting Information) and the rate constant, k, were obtained. The second-order rate constant of this reaction, k2 = 500 ± 60 M–1 s–1, was deduced from the average of 4 independent measurements. Deuterated TEMPO-D was examined similarly. Linear plots (inset of Figure 9) were derived according to the following kinetic equation for both TEMPO-H and TEMPO-D at the concentration around 1.2 equiv to 2′.
A KIE value of 1.5 was obtained, suggesting that the homolytic O–H bond cleavage does not make the major contribution to the reaction coordinate of the rate-determining step for the conversion of MnIII-superoxo 2′ to MnIII-hydroperoxo 5′.
Figure 9.
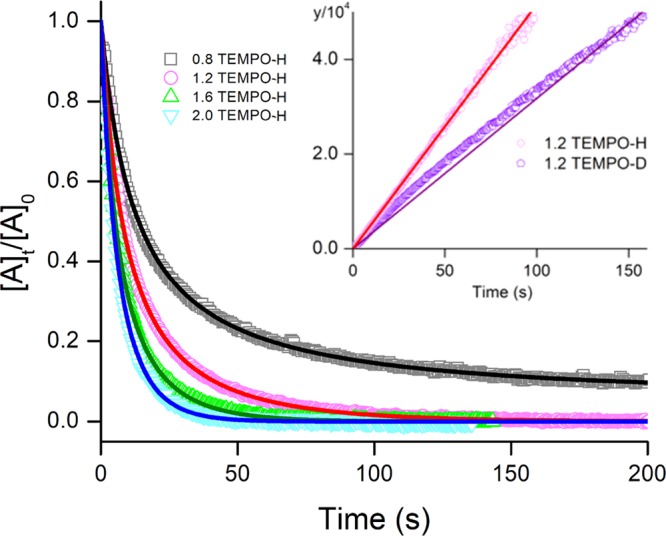
Nonlinear least-squared fitting for the reaction traces of 2′ in THF with various equiv of TEMPO-H monitored by UV–vis spectroscopy. The inset shows the linear plots of TEMPO-H and TEMPO-D following kinetic equation at the concentration around 1.2 equiv to 2′.
Obviously, complexes 2′, 3, and 4 exhibit differential HAA activity due to the nature of the individual metal center. After adjustment for differences in temperature and substrate DC–H values, it appears that 2′ has a rate for HAA toward TEMPO-H much faster than those of Co(BDPP)(O2•–) (4, k2 = 0.97 M–1 s–1 at −90 °C), [L1CuII(O2•–)]PF6 (L1 = 1-(2-phenethyl)-5-[2-(2-pyridyl)ethyl]-1,5-diazacyclooctane, k2 = 2.4 M–1 s–1 at −85 °C),30 [L2CuII(μ-1,2-O2•–)]BPh4 (L2 = tacn/pyrazolate hybrid ligand, k2 = 0.254 M–1 s–1 at −10 °C),31 and [K(Krypt)][L3Cu(O2•–)] (L2 = a bis(arylcarboxamido)pyridine ligand, k2 = 34.9 M–1 s–1 at −80 °C).32
Conclusion
Mononuclear MnIII–superoxo complexes 2 and 2′ were synthesized by reacting O2 with precursors 1 and 1′ at −80 °C. Our combined spectroscopic and computational study reveals that 2 and 2′ are best formulated as a high-spin MnIII center antiferromagnetically coupled to a superoxo ligand yielding an overall quartet ground state. Complexes 2 and 2′ were shown to activate a weak O–H bond of TEMPO-H to generate MnIII–hydroperoxo complexes 5 and 5′. Notably, complex 2′ cleaves the O–H bond in TEMPO-H much faster than the reported Co- and Cu–superoxo complexes.30−32 Acidification of 5 and 5′ produces nearly quantitative H2O2 and MnIII-aqua complexes 6 and 6′. Conversely, complexes 5 and 5′ can be directly prepared from the reactions of 6 and 6′ with an excess amount of a 2:1 mixture of H2O2 and NEt3 in THF at −90 °C.
Our earlier work showed that similar to 2 and 2′ their FeIII and CoIII congeners (3 and 4) can be prepared in the same way and that the resulting trivalent metal–superoxo complexes can initiate HAA reactions and produce trivalent metal–hydroperoxo complexes. All these findings provide direct experimental support for the proposed mechanism for O2 activation by distinct transition metals. Despite this, the three metal centers display different O2 bonding rates, and O2 adducts 2′, 3, and 4 exhibit distinct metal–superoxo bonding strengths and HAA activities. The observations warrant further systematic investigations devoting to pinpoint key features responsible for the metal specificity.
Experimental Section
Materials and Methods
All manipulations are performed under N2 conditions using glovebox or Schlenk techniques. Acetonitrile was purified by the MBraun Solvent Purification System (MB-SPS). Diethyl ether, THF, and pentane were dried by sodium/benzophenone and distilled prior to use. Dichloromethane were dried by CaH2 and distilled prior to use. 2,6-Bis(((S)-2-diphenylhydroxymethyl-1-pyrolidinyl)methyl)pyridine (H2BDPP), 2,6-bis(((S)-2(bis(4-bromophenyl)hydroxyl-methyl-1-pyrolidinyl)methyl)pyridine (H2BDPBrP), and 2,2,6,6-tetramethyl-1-hydroxypiperidine (TEMPO-H) were synthesized according to the literature methods.33 All the other chemical reagents were obtained from commercial sources and used without purification. UV–vis spectra were recorded with Agilent 8453 spectrophotometer equipped with cryostat from Unisoku Scientific Instruments, Osaka, Japan. Elemental analyses for C, H, and N were performed on an elementar Vario EL cube analyzer at the Instrumentation Center in National Taiwan University.
X-ray Data Collection and Structure Determination
X-ray diffraction data of crystals of 1, 1′, and 6′ were collected on a Bruker Kappa APEX II CCD diffractometer employing Mo Kα radiation (λ = 0.7107 Å) at 200 K and with a θ–2θ scan mode. The space groups for 1, 1′, and 6′ were determined on the basis of systematic absences and intensity statistics. Their structures were solved by direct methods using SIR92 or SIR97, and refined using SHELXL-97 with anisotropic displacement factors for all non-hydrogen atoms. For X-ray structures of 1′ and 6′, half or one CH2Cl2 solvent molecule in 1 and one Et2O solvent molecule in 6′ per unit cell are squeezed by PLATON program. The detailed crystallographic data of 1, 1′, and 6′ were provided in their crystallographic information files.
EPR Measurements
X-band cw-EPR measurements were performed on a Bruker E500 ELEXSYS spectrometer equipped with the Bruker dual-mode cavity (ER4116DM) and an Oxford Instruments helium flow cryostat (ESR 900). The microwave bridge was the high-sensitivity bridge Super-X from Bruker (ER-049X) with integrated microwave frequency counter. The magnetic field controller (ER032T) was externally calibrated with a Bruker NMR field probe (ER035M). Spectral analysis and simulations were handled by using the EasySpin program.34
Resonance Raman Collections
Resonance Raman spectra were recorded on a TriVista 555 triple monochromator equipped with a liquid-nitrogen-cooled Roper Scientific 400BR Excelon CCD camera. An excitation wavelength of 457 nm and 10 mW from a cobalt solid-state laser was used. The solution of a sample was packed into a quartz EPR tube and measured in an EPR quartz finger Dewar which was cooled with liquid nitrogen. The Raman signal was collected in a 180° backscattering geometry with a Thorlabs MPD249H–P01 off-axis parabolic mirror and focused onto the entrance slit of a spectrograph with a 100 mm diameter f/4 lens. The scattered light was dispersed with a grating of 1800 mm–1 and further with two filter prestages of 900 mm–1. The slit widths at the first and third stages amounted to 100 μm, thus providing a spectral resolution at the CCD camera of about 0.8 cm–1. Spectra were collected for about 30 min for a given wavelength and spectral window. Calibration of the Raman shifts has been achieved to an accuracy of 1 cm–1 by using Na2SO4 as well as the solvent signals as references.
Synthesis of Mn(BDPP) (1) and Mn(BDPBrP) (1′)
Mn(BDPP) (1) was synthesized by reacting Mn(OTf)2 (141.2 mg, 0.4 mmol) with H2BDPP (244.0 mg, 0.4 mmol) and NaH (24.0 mg, 1.0 mmol) in 15 mL of CH3CN in a 50 mL Schlenk flask. The solution was stirred for 3 h at ambient temperature and then vacuumed. The pale yellow residue was dissolved in CH2Cl2 (20.0 mL) and filtered. The filtrate was concentrated under vacuum and recrystallized by slow diffusion of Et2O into the concentrated CH2Cl2 filtrate at ambient temperature. Yellow crystals of 1 were obtained in 55% yield over 1 day. The structure of 1 features a distorted square pyramidal geometry (τ5 = 0.45). UV–vis (THF): 338 nm (450 M–1 cm–1). Anal. Calcd for C41H41MnN3O2 (F.W. = 662.71): C, 69.35; H, 5.92; N, 5.82. Found: C, 69.509; H, 6.176; N, 6.121. The synthesis of Mn(BDPBrP) (1′) is similar to the procedures for that of 1 (Mn(OTf)2: 0.1412 g, 0.4 mmol; H2BDPBrP: 0.3770 g, 0.4 mmol; NaH: 0.024 g, 1.0 mmol). Slow diffusion of pentane into the THF solution of 1′ at ambient temperature was performed for recrystallization. Yellow crystals of 1′ were obtained in 50% yield over 1 day. The structure of 1′ features a distorted square pyramidal geometry (τ5 = 0.34). UV–vis (THF): 350 nm (322 M–1 cm–1). Anal. Calcd for C41H37Br4MnN3O2·THF (F.W. = 1050.43): C, 51.45; H, 4.32; N, 4.00. Found: C, 51.587; H, 4.463; N, 3.801.
Formation of Mn(BDPP)(O2•–) (2) and Mn(BDPBrP)(O2•–) (2′)
Complexes 2 and 2′ were generated by bubbling O2 into THF or CH2Cl2 solutions of 1 and 1′ at −80 °C from an oxygen balloon for 2 min. Formation of 2 and 2′ was monitored by UV–vis spectroscopy on characteristic absorption bands at 370, 450, and 740 nm. We found that the volume of O2 in the balloon and the elasticity of the balloon have a large influence on the formation rate of 2 and 2′, a situation similar to that encountered for Fe(BDPP)(O2•) (3), indicating that the formation of 2 and 2′ is rapid and close to the diffusion rate of O2 in the working solutions.
Production of MnIII(BDPP)(H2O)(OTf) (6) and MnIII(BDPBrP)(H2O)(OTf) (6′) from Protonation of 5 and 5′
Complex 6 was produced from the reaction of 5 (0.2 mmol) with 1 equiv of HOTf at −90 °C then warming up to room temperature. After 3 h, the resulting green solution was evaporated. The crude powder was redissolved in mixed THF/CH3CN (10:1). Pentane was then added to the mixing THF/CH3CN solution to obtain a green precipitate of 6 which was isolated in a yield of 116.2 mg (70%). Complex 6′ was prepared with the same procedure. Crystallization of 6′ gave a yield of 169.5 mg (74%). Anal. Calcd for C42 H39 Br4 F3 Mn N3 O6 S·0.5C4H8O (F.W. = 1182.46): C, 44.69; H, 3.75; N, 3.55; S, 2.71. Found: C, 44.50; H, 3.91; N, 3.47; S, 2.52.
Preparation of Mn(BDPP)(OOH) (5) and Mn(BDPBrP)(OOH) (5′) from Reactions of 6 and 6′ with a Mixture of H2O2/NEt3
A solution of basic hydrogen peroxide (10 equiv of H2O2 with 5 equiv of NEt3, 100 μL) was added to a THF solution of 6 (1.0 mM, 3.0 mL) at −90 °C. The color of the reaction solution changed from green to pale purple exhibiting the formation of 5. Meanwhile, the absorption bands of 6 at 460 and 600 nm declined, and the bands of 5 at 510 and 720 nm grew as monitored by the UV–vis spectroscopy. Complex 5′ was prepared by the same procedure.
Kinetic Study of 2′ toward TEMPO-H
TEMPO-H (1.0, 1.5, 2.0, and 2.5 equiv) was added to a THF solution of 2′ at −90 °C. The reacting solution was monitored by UV–vis spectroscopy, and a rapid decline of the absorption band at 450 nm was recorded. Each condition was examined three times. Reaction of 2′ with 1.5 equiv of TEMPO-D was also performed, and the rate was obtained to derive the kinetic isotope effect (KIE) value.
Computational Details
Geometry optimizations and frequency calculations were carried out with the B3LYP density functional.35 The triple-ζ quality basis set, def2-TZVP,36 was used for Mn and the atoms in the first coordination sphere, while the remaining atoms were treated with def2-SVP37 basis set. The calculations were accelerated by using RI (resolution of the identity) approximation,38,39 for which the auxiliary coulomb-fitting basis set def2-TZVP/J38 was used. All geometry optimizations were performed incorporating solvation effect through conductor-like polarizable continuum model (CPCM)40 with the solvent THF (ε = 7.25). The noncovalent interactions were accounted for through atom-pairwise dispersion corrections with Becke–Johnson (D3BJ)41 damping.
Spectroscopic parameters were computed by using the TPSSh density functional42 in combination with the CP(PPP) basis set for Mn,43 the def2-TZVP basis set for the atoms in the first coordination sphere, and the def2-SVP for the remaining atoms. The magnetic hyperfine coupling matrix, A, of the 55Mn center was calculated by taking the isotropic Fermi contact term, the first-order traceless dipolar contribution, and the second-order nontraceless spin–orbit contribution into account. The Fermi-contact contributions were scaled by a factor of 1.49.44 Spin–orbit contributions to the hyperfine tensors were calculated as second order properties employing the coupled perturbed (CP) Kohn–Sham theory.45 The Mn magnetic hyperfine coupling constants of the “genuine” antiferromagnetic state AiAF were calculated from the magnetic hyperfine coupling constants of the corresponding broken symmetry state Ai by conversion into “site values” and multiplication with the spin projection coefficients Ci:46
The contributions of spin orbit coupling (SOC) to the zero-field splitting (ZFS)24 were calculated by linear response theory.47
The spin–spin coupling contributions to ZFSs are calculated from the equation of McWeeny and Mizuno.48
in which spin density matrix Pα–β was obtained on the basis of the spin-unrestricted natural orbital (UNO) determinant.49 All calculations were performed by using the ORCA quantum chemical program package.50
Acknowledgments
We thank Mr. Dennis Skerra at the Max-Planck Institute for Chemical Energy Conversion for resonance Raman measurements. We are grateful for the financial support from the Ministry of Science and Technology of Taiwan (MOST 105-2113-M-003-005-MY3 to W.-Z.L.) and the Max-Planck Society, in particular, the joint work space between MPI-CEC and MPI-KOFO. W.-Z.L. and S.Y. also acknowledge the MOST-DAAD Project-Based Personnel Exchange Program (MOST 107-2911-I-003-502 and DAAD 57320810).
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.inorgchem.9b00767.
UV–vis, NMR and HRMS spectra of 1–3, EPR spectra of 1–2, and computational result of 1 (PDF)
Accession Codes
CCDC 1898662–1898664 contain the supplementary crystallographic data for this paper. These data can be obtained free of charge via www.ccdc.cam.ac.uk/data_request/cif, or by emailing data_request@ccdc.cam.ac.uk, or by contacting The Cambridge Crystallographic Data Centre, 12 Union Road, Cambridge CB2 1EZ, UK; fax: +44 1223 336033.
The authors declare no competing financial interest.
Supplementary Material
References
- Bertini I.; Gray H. B.; Lippard S. J.; Valentine J. S.. Dioxygen Reactions. In Bioinorganic Chemistry; University Science Books: Mill Valley, CA, 1994; pp 253–313. [Google Scholar]
- a Costas M.; Mehn M. P.; Jensen M. P.; Que L. DiOxygen Activation at Mononuclear Non-Heme Iron Active Sites: Enzymes, Models, and Intermediates. Chem. Rev. 2004, 104, 939–986. 10.1021/cr020628n. [DOI] [PubMed] [Google Scholar]; b Vaillancourt F. H.; Bolin J. T.; Eltis L. D. The Ins and Outs of Ring-Cleaving Dioxygenases. Crit. Rev. Biochem. Mol. Biol. 2006, 41, 241–267. 10.1080/10409230600817422. [DOI] [PubMed] [Google Scholar]; c Kovaleva E. G.; Lipscomb J. D. Versatility of Biological Non-heme Fe(II) Centers in Oxygen Activation Reactions. Nat. Chem. Biol. 2008, 4, 186–193. 10.1038/nchembio.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller M. A.; Lipscomb J. D. Homoprotocatechuate 2,3-Dioxygenase from Brevibacterium fuscum. J. Biol. Chem. 1996, 271, 5524–5535. 10.1074/jbc.271.10.5524. [DOI] [PubMed] [Google Scholar]
- Whiting A. K.; Boldt Y. R.; Hendrich M. P.; Wackett L. P.; Que L. Manganese(II)-Dependent Extradiol-Cleaving Catechol Dioxygenase from Arthrobacter Globiformis CM-2. Biochemistry 1996, 35, 160–170. 10.1021/bi951979h. [DOI] [PubMed] [Google Scholar]
- a Emerson J. P.; Kovaleva E. G.; Farquhar E. R.; Lipscomb J. D.; Que L. Jr. Swapping Metals in Fe- and Mn-Dependent Dioxygenases: Evidence for Oxygen Activation without a Change in Metal Redox State. Proc. Natl. Acad. Sci. U. S. A. 2008, 105, 7347–7352. 10.1073/pnas.0711179105. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Fielding A. J.; Kovaleva E. G.; Farquhar E. R.; Lipscomb J. D.; Que L. Jr. A Hyperactive Cobalt-substituted Extradiol-cleaving Catechol Dioxygenase. JBIC, J. Biol. Inorg. Chem. 2011, 16, 341–355. 10.1007/s00775-010-0732-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Solomon E. I.; Brunold T. C.; Davis M. I.; Kemsley J. N.; Lee S.-K.; Lehnert N.; Neese F.; Skulan A. J.; Yang Y.-S.; Zhou J. Geometric and Electronic Structure/Function Correlations in Non-Heme Iron Enzymes. Chem. Rev. 2000, 100, 235–349. 10.1021/cr9900275. [DOI] [PubMed] [Google Scholar]; b Huang X.; Groves J. T. Beyond Ferryl-Mediated Hydroxylation: 40 Years of the Rebound Mechanism and C–H Activation. JBIC, J. Biol. Inorg. Chem. 2017, 22, 185–207. 10.1007/s00775-016-1414-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baldwin J. E.; Bradley M. Isopenicillin N Synthase: Mechanistic Studies. Chem. Rev. 1990, 90, 1079. 10.1021/cr00105a001. [DOI] [Google Scholar]
- Tamanaha E.; Zhang B.; Guo Y.; Chang W.-c.; Barr E. W.; Xing G.; St. Clair J.; Ye S.; Neese F.; Bollinger J. M.; Krebs C. Spectroscopic Evidence for the Two C–H-Cleaving Intermediates of Aspergillus nidulans Isopenicillin N Synthase. J. Am. Chem. Soc. 2016, 138, 8862–8874. 10.1021/jacs.6b04065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fielding A. J.; Lipscomb J. D.; Que L. Jr. Characterization of an O2 Adduct of an Active Cobalt-Substituted Extradiol-Cleaving Catechol Dioxygenase. J. Am. Chem. Soc. 2012, 134, 796–799. 10.1021/ja2095365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gunderson W. A.; Zatsman A. I.; Emerson J. P.; Farquhar E. R.; Que L.; Lipscomb J. D.; Hendrich M. P. Electron Paramagnetic Resonance Detection of Intermediates in the Enzymatic Cycle of an Extradiol Dioxygenase. J. Am. Chem. Soc. 2008, 130, 14465–14467. 10.1021/ja8052255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deville C.; Padamati S. K.; Sundberg J.; McKee V.; Browne W. R.; McKenzie C. J. O2 Activation and Double C-H Oxidation by a Mononuclear Manganese(II) Complex. Angew. Chem., Int. Ed. 2016, 55, 545–549. 10.1002/anie.201508372. [DOI] [PubMed] [Google Scholar]
- Coggins M. K.; Sun X.; Kwak Y.; Solomon E. I.; Rybak-Akimova E.; Kovacs J. A. Characterization of Metastable Intermediates Formed in the Reaction between a Mn(II) Complex and Dioxygen, Including a Crystallographic Structure of a Binuclear Mn(III)–Peroxo Species. J. Am. Chem. Soc. 2013, 135, 5631–5640. 10.1021/ja311166u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Shook R. L.; Peterson S. M.; Greaves J.; Moore C.; Rheingold A. L.; Borovik A. S. Catalytic Reduction of Dioxygen to Water with a Monomeric Manganese Complex at Room Temperature. J. Am. Chem. Soc. 2011, 133, 5810–5817. 10.1021/ja106564a. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Guo M.; Lee Y. M.; Gupta R.; Seo M. S.; Ohta T.; Wang H. H.; Liu H. Y.; Dhuri S. N.; Sarangi R.; Fukuzumi S.; Nam W. Dioxygen Activation and O–O Bond Formation Reactions by Manganese Corroles. J. Am. Chem. Soc. 2017, 139, 15858–15867. 10.1021/jacs.7b08678. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Sankaralingam M.; Lee Y. M.; Jeon S. H.; Seo M. S.; Cho K. B.; Nam W. A Mononuclear Manganese(III)–Hydroperoxo Complex: Synthesis by Activating Dioxygen and Reactivity in Electrophilc and Nucleophilic Reactions. Chem. Commun. 2018, 54, 1209–1212. 10.1039/C7CC09492B. [DOI] [PubMed] [Google Scholar]
- Chiang C. W.; Kleespies S. T.; Stout H. D.; Meier K. K.; Li P. Y.; Bominaar E. L.; Que L. Jr.; Münck E.; Lee W. Z. Characterization of a Paramagnetic Mononuclear Nonheme Iron-Superoxo Complex. J. Am. Chem. Soc. 2014, 136, 10846–10849. 10.1021/ja504410s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Hong S.; Sutherlin K. D.; Park J.; Kwon E.; Siegler M. A.; Solomon E. I.; Nam W. Crystallographic and Spectroscopic Characterization and Reactivities of a Mononuclear Non-Haem Iron(III)-Superoxo Complex. Nat. Commun. 2014, 5, 5440–5547. 10.1038/ncomms6440. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Blakely M. N.; Dedushko M. A.; Yan Poon P. C.; Villar-Acevedo G.; Kovacs J. A. Formation of a Reactive, Alkyl Thiolate-Ligated FeIII-Superoxo Intermediate Derived from Dioxygen. J. Am. Chem. Soc. 2019, 141, 1867. 10.1021/jacs.8b12670. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Noh H.; Cho J. Synthesis, Characterization and Reactivity of Non-Heme 1st Row Transition Metal-Superoxo Intermediates. Coord. Chem. Rev. 2019, 382, 126–144. 10.1016/j.ccr.2018.12.006. [DOI] [Google Scholar]
- Wang C. C.; Chang H. C.; Lai Y. C.; Fang H.; Li C. C.; Hsu H. K.; Li Z. Y.; Lin T. S.; Kuo T. S.; Neese F.; Ye S.; Chiang Y. W.; Tsai M. L.; Liaw W. F.; Lee W. Z. Structurally Characterized Nonheme Cobalt–Hydroperoxo Complex Derived from Its Superoxo Intermediate via Hydrogen Atom Abstraction. J. Am. Chem. Soc. 2016, 138, 14186–14189. 10.1021/jacs.6b08642. [DOI] [PubMed] [Google Scholar]
- Oddon F.; Chiba Y.; Nakazawa J.; Ohta T.; Ogura T.; Hikichi S. Characterization of Mononuclear Non-heme Iron(III)-Superoxo Complex with a Five-Azole Ligand Set. Angew. Chem., Int. Ed. 2015, 54, 7336–7339. 10.1002/anie.201502367. [DOI] [PubMed] [Google Scholar]
- a Sacramento J. J. D.; Goldberg D. P. The hydrogen atom transfer reactivity of a porphyrinoid cobalt superoxide complex. Chem. Commun. 2019, 55, 913–916. 10.1039/C8CC08453J. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Bajdor K.; Nakamoto K.; Kanatomi H.; Murase I. Resonance Raman Spectra of Molecular Oxygen Adducts of Co(salen) and Its Derivatives in Solution. Inorg. Chim. Acta 1984, 82, 207–210. 10.1016/S0020-1693(00)82493-7. [DOI] [Google Scholar]
- Liu L. L.; Li H. X.; Wan L. M.; Ren Z. G.; Wang H. F.; Lang J. P. Mn(III)–Superoxo Complex of a Zwitterionic Calix[4]arene with an Unprecedented Linear End-On Mn(III)–O2 Arrangement and Good Catalytic Performance for Alkene Epoxidation. Chem. Commun. 2011, 47, 11146–11148. 10.1039/c1cc14262c. [DOI] [PubMed] [Google Scholar]
- a Woertink J. S.; Tian L.; Maiti D.; Lucas H. R.; Himes R. A.; Karlin K. D.; Neese F.; Würtele C.; Holthausen M. C.; Bill E.; Sundermeyer J.; Schindler S.; Solomon E. I. Spectroscopic and Computational Studies of an End-on Bound Superoxo-Cu(II) Complex: Geometric and Electronic Factors That Determine the Ground State. Inorg. Chem. 2010, 49, 9450–9459. 10.1021/ic101138u. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Zhang X.; Furutachi H.; Fujinami S.; Nagatomo S.; Maeda Y.; Watanabe Y.; Kitagawa T.; Suzuki M. Structural and Spectroscopic Characterization of (μ-Hydroxo or μ-Oxo)(μ-peroxo) diiron(III) Complexes: Models for Peroxo Intermediates of Non-Heme Diiron Proteins. J. Am. Chem. Soc. 2005, 127, 826–827. 10.1021/ja045594a. [DOI] [PubMed] [Google Scholar]
- Schatz M.; Raab V.; Foxon S. P.; Brehm G.; Schneider S.; Reiher M.; Holthausen M. C.; Sundermeyer J.; Schindler S. Combined Spectroscopic and Theoretical Evidence for a Persistent End-On Copper Superoxo Complex. Angew. Chem., Int. Ed. 2004, 43, 4360–4363. 10.1002/anie.200454125. [DOI] [PubMed] [Google Scholar]
- Peterson R. L.; Himes R. A.; Kotani H.; Suenobu T.; Tian L.; Siegler M. A.; Solomon E. I.; Fukuzumi S.; Karlin K. D. Cupric Superoxo-Mediated Intermolecular C-H Activation Chemistry. J. Am. Chem. Soc. 2011, 133, 1702–1705. 10.1021/ja110466q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho J.; Woo J.; Nam W. An “End-On” Chromium(III)-Superoxo Complex: Crystallographic and Spectroscopic Characterization and Reactivity in C-H Bond Activation of Hydrocarbons. J. Am. Chem. Soc. 2010, 132, 5958–5959. 10.1021/ja1015926. [DOI] [PubMed] [Google Scholar]
- Bencini A.; Gatteschi D.. Electron Paramagnetic Resonance of Exchange Coupled Systems; Sringer-Verlag: Berlin, 1990. [Google Scholar]
- a Krzystek J.; Ozarowski A.; Telser J. Multi-Frequency, High-Field EPR as a Powerful Tool to Accurately Determine Zero-Field Splitting in High-Spin Transition Metal Coordination Complexes. Coord. Chem. Rev. 2006, 250, 2308–2324. 10.1016/j.ccr.2006.03.016. [DOI] [Google Scholar]; b Orio M.; Pantazis D. A.; Petrenko T.; Neese F. Magnetic and Spectroscopic Properties of Mixed Valence Manganese(III,IV) Dimers: A Systematic Study Using Broken Symmetry Density Functional Theory. Inorg. Chem. 2009, 48, 7251. 10.1021/ic9005899. [DOI] [PubMed] [Google Scholar]
- Duboc C. Determination and Prediction of the Magnetic Anisotropy of Mn Ions. Chem. Soc. Rev. 2016, 45, 5834–5847. 10.1039/C5CS00898K. [DOI] [PubMed] [Google Scholar]
- a Mair R. D.; Graupner A. J. Determination of Organic Peroxides by Iodine Liberation Procedures. Anal. Chem. 1964, 36, 194–201. 10.1021/ac60207a061. [DOI] [Google Scholar]; b Das D.; Lee Y.-M.; Ohkubo K.; Nam W.; Karlin K. D.; Fukuzumi S. Acid-Induced Mechanism Change and Overpotential Decrease in Dioxygen Reduction Catalysis with a Dinuclear Copper Complex. J. Am. Chem. Soc. 2013, 135, 4018–4026. 10.1021/ja312256u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta R.; Taguchi T.; Borovik A. S.; Hendrich M. P. Characterization of Monomeric MnII/III/IV–Hydroxo Complexes from X- and Q-Band Dual Mode Electron Paramagnetic Resonance (EPR) Spectroscopy. Inorg. Chem. 2013, 52, 12568–12575. 10.1021/ic401681r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo Y.-R.Handbook of Bond Dissociation Energies in Organic Compounds; CRC Press: Boca Raton, FL, 2002. [Google Scholar]
- Tano T.; Okubo Y.; Kunishita A.; Kubo M.; Sugimoto H.; Fujieda N.; Ogura T.; Itoh S. Redox Properties of a Mononuclear Copper(II)-Superoxide Complex. Inorg. Chem. 2013, 52, 10431–10437. 10.1021/ic401261z. [DOI] [PubMed] [Google Scholar]
- Kindermann N.; Gunes C. J.; Dechert S.; Meyer F. Hydrogen Atom Abstraction Thermodynamics of a μ-1,2-Superoxo Dicopper(II) Complex. J. Am. Chem. Soc. 2017, 139, 9831–9834. 10.1021/jacs.7b05722. [DOI] [PubMed] [Google Scholar]
- Bailey W. D.; Dhar D.; Cramblitt A. C.; Tolman W. B. Mechanistic Dichotomy in Proton-Coupled Electron-Transfer Reactions of Phenols with a Copper Superoxide Complex. J. Am. Chem. Soc. 2019, 141, 5470–5480. 10.1021/jacs.9b00466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Zhang Y.-X.; Du D.-M.; Chen X.; Lü S.-F.; Hua W.-T. Enantiospecific Synthesis of Pyridinylmethyl Pyrrolidinemethanols and Catalytic Asymmetric Borane Reduction of Prochiral Ketones. Tetrahedron: Asymmetry 2004, 15, 177–182. 10.1016/j.tetasy.2003.10.037. [DOI] [Google Scholar]; b Mader E. A.; Larsen A. S.; Mayer J. M. Hydrogen Atom Transfer from Iron(II)-Tris[2,2′-bi(tetrahydropyrimidine)] to TEMPO: A Negative Enthalpy of Activation Predicted by the Marcus Equation. J. Am. Chem. Soc. 2004, 126, 8066–8067. 10.1021/ja049246k. [DOI] [PubMed] [Google Scholar]; c Park J. K.; Lee H. G.; Bolm C.; Kim B. M. Asymmetric Diethyl- and Diphenylzinc Additions to Aldehydes by Using a Fluorine-Containing Chiral Amino Alcohol: A Striking Temperature Effect on the Enantioselectivity, a Minimal Amino Alcohol Loading, and an Efficient Recycling of the Amino. Chem. - Eur. J. 2005, 11, 945–950. 10.1002/chem.200400703. [DOI] [PubMed] [Google Scholar]
- Stoll S.; Schweiger A. EasySpin, a Comprehensive Software Package for Spectral Simulation and Analysis in EPR. J. Magn. Reson. 2006, 178, 42–55. 10.1016/j.jmr.2005.08.013. [DOI] [PubMed] [Google Scholar]
- a Becke A. D. Density-Fnnctional Exchange-Energy Approximation with Correct Asymptotic Behavior. Phys. Rev. A: At., Mol., Opt. Phys. 1988, 38, 3098–3100. 10.1103/PhysRevA.38.3098. [DOI] [PubMed] [Google Scholar]; b Lee C.; Yang W.; Parr R. G. Development of the Colic-Salvetti Correlation-Energy Formula into a Functional of the Electron Density. Phys. Rev. B: Condens. Matter Mater. Phys. 1988, 37, 785–789. 10.1103/PhysRevB.37.785. [DOI] [PubMed] [Google Scholar]
- Schäfer A.; Huber C.; Ahlrichs R. Fully Optimized Contracted Gaussian Basis Sets of Triple Zeta Valence Quality for Atoms Li to Kr. J. Chem. Phys. 1994, 100, 5829–5835. 10.1063/1.467146. [DOI] [Google Scholar]
- Schäfer A.; Horn H.; Ahlrichs R. Fully Optimized Contracted Gaussian Basis Sets for Atoms Li to Kr. J. Chem. Phys. 1992, 97, 2571–2577. 10.1063/1.463096. [DOI] [Google Scholar]
- Eichkorn K.; Weigend F.; Treutler O.; Ahlrichs R. Auxiliary Basis Sets for Main Row Atoms and Transition Metals and Their Use to Approximate Coulomb Potentials. Theor. Chem. Acc. 1997, 97, 119–124. 10.1007/s002140050244. [DOI] [Google Scholar]
- Eichkorn K.; Treutler O.; Öhm H.; Häser M.; Ahlrichs R. Auxiliary Basis Sets to Approximate Coulomb Potentials. Chem. Phys. Lett. 1995, 240, 283–290. 10.1016/0009-2614(95)00621-A. [DOI] [Google Scholar]
- Tomasi J.; Mennucci B.; Cammi R. Quantum Mechanical Continuum Solvation Models. Chem. Rev. 2005, 105, 2999–3093. 10.1021/cr9904009. [DOI] [PubMed] [Google Scholar]
- Grimme S.; Ehrlich S.; Goerigk L. Effect of the Damping Function in Dispersion Corrected Density Functional Theory. J. Comput. Chem. 2011, 32, 1456–1465. 10.1002/jcc.21759. [DOI] [PubMed] [Google Scholar]
- Staroverov V. N.; Scuseria G. E.; Tao J.; Perdew J. P. Comparative Assessment of a New Nonempirical Density Functional: Molecules and Hydrogen-Bonded Complexes. J. Chem. Phys. 2003, 119, 12129–12137. 10.1063/1.1626543. [DOI] [Google Scholar]
- Neese F. Prediction and Interpretation of the 57Fe Isomer Shift in Mössbauer Spectra by Density Functional Theory. Inorg. Chim. Acta 2002, 337, 181–192. 10.1016/S0020-1693(02)01031-9. [DOI] [Google Scholar]
- Orio M.; Pantazis D. A.; Petrenko T.; Neese F. Magnetic and Spectroscopic Properties of Mixed Valence Manganese(III,IV) Dimers: A Systematic Study Using Broken Symmetry Density Functional Theory. Inorg. Chem. 2009, 48, 7251–7260. 10.1021/ic9005899. [DOI] [PubMed] [Google Scholar]
- Neese F.; Solomon E. I. Calculation of zero-field splittings, g-values, and the relativistic nephelauxetic effect in transition metal complexes. Application to high-spin ferric complexes. Inorg. Chem. 1998, 37, 6568–6582. 10.1021/ic980948i. [DOI] [PubMed] [Google Scholar]
- Sinnecker S.; Neese F.; Noodleman L.; Lubitz W. Calculating the Electron Paramagnetic Resonance Parameters of Exchange Coupled Transition Metal Complexes Using Broken Symmetry Density Functional Theory: Application to a MnIII/MnIV Model Compound. J. Am. Chem. Soc. 2004, 126, 2613–2622. 10.1021/ja0390202. [DOI] [PubMed] [Google Scholar]
- Neese F. Calculation of the Zero-Field Splitting Tensor on the Basis of Hybrid Density Functional and Hartree-Fock Theory. J. Chem. Phys. 2007, 127, 164112. 10.1063/1.2772857. [DOI] [PubMed] [Google Scholar]
- Mcweeny R.; Mizuno Y. The Density Matrix in Many-Electron Quantum Mechanics. II. Separation of Space and Spin Variables; Spin Coupling Problems. Proc. R. Soc. London A 1961, 259, 554–577. 10.1098/rspa.1961.0008. [DOI] [Google Scholar]
- Sinnecker S.; Neese F. Spin-Spin Contributions to the Zero-Field Splitting Tensor in Organic Triplets, Carbenes and Biradical-A Density Functional and Ab Initio Study. J. Phys. Chem. A 2006, 110, 12267–12275. 10.1021/jp0643303. [DOI] [PubMed] [Google Scholar]
- a Neese F. The ORCA program system. WIREs Comput. Mol. Sci. 2012, 2, 73–78. 10.1002/wcms.81. [DOI] [Google Scholar]; b Neese F. Software update: the ORCA program system, version 4.0. WIREs Comput. Mol. Sci. 2018, 8, e1327. 10.1002/wcms.1327. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.