

Summary
By releasing glutamate, astrocytes actively regulate synaptic transmission and also contribute to excitotoxicity in neurological diseases. However, the mechanisms of astrocytic glutamate release have been debated. Here, we report non-vesicular release of glutamate through the glutamate-permeable volume-regulated anion channel (VRAC). Both cell swelling and receptor stimulation activated astrocytic VRAC, which requires its only obligatory subunit, Swell1. Astrocyte-specific Swell1 knockout mice exhibited impaired glutamatergic transmission due to the decreases in presynaptic release probability and ambient glutamate level. Consistently, the mutant mice displayed hippocampal-dependent learning and memory deficits. During pathological cell swelling, deletion of astrocytic Swell1 attenuated glutamate-dependent neuronal excitability and protected mice from brain damage after ischemic stroke. Our identification of a new molecular mechanism for channel-mediated glutamate release establishes a role for astrocyte-neuron interactions in both synaptic transmission and brain ischemia. It provides a rationale for targeting VRAC for the treatment of stroke and other neurological diseases associated with excitotoxicity.
eTOC Blurb
Whether and how astrocytes release glutamate to regulate neuronal function are debated. Yang et al. show that Swell1 volume-regulated anion channel is a glutamate-releasing channel in astrocytes, which regulates basal synaptic transmission and contributes to excitotoxicity in ischemic stroke.
Graphical Abstract
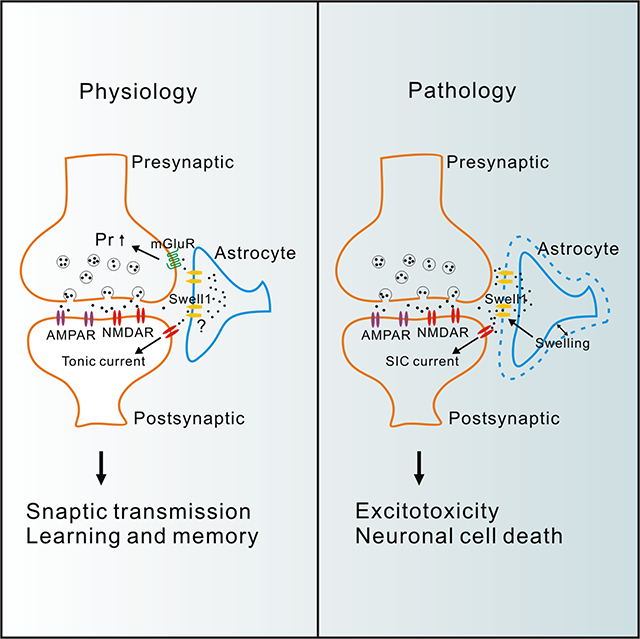
Introduction
Glutamate is the principal excitatory neurotransmitter in the central nervous system. Neurons release glutamate through Ca2+-dependent exocytosis which underlies fast synaptic transmission. Astrocytes may also release glutamate and actively modulate neuronal excitability, synaptic transmission and plasticity (Allen and Eroglu, 2017; Araque et al., 2014; Gundersen et al., 2015). However, there has been considerable controversy in the field over the molecular mechanisms of astrocytic glutamate release (Hamilton and Attwell, 2010). Some studies suggest that similar to neurons, astrocytes release glutamate through Ca2+-dependent vesicular exocytosis (Savtchouk and Volterra, 2018). Others argue that the various methods for manipulating astrocyte Ca2+ level and subsequent glutamate release are not specific and that astrocytes lack sufficient expression of vesicular release machinery (Barres, 2008; Fiacco and McCarthy, 2018). Therefore, astrocytes likely possess additional glutamate-releasing mechanisms other than vesicular exocytosis (Gundersen et al., 2015; Hamilton and Attwell, 2010).
Cytosolic glutamate concentrations in astrocytes are high (0.1–5 mM) (Attwell et al., 1993), whereas extracellular levels are in the sub-micromolar range (Cavelier and Attwell, 2005). This creates a steep concentration gradient favoring glutamate release through ion channels permeable to relatively large anions. Indeed, non-selective large pore-forming hemichannels and purinergic P2X7 receptor were shown to mediate glutamate release in a Ca2+-independent manner likely under pathological conditions (Duan et al., 2003; Ye et al., 2003). Ca2+-activated bestrophin-1 anion channel and K+-selective TREK-1 channel were also suggested to mediate astrocytic glutamate release upon G protein-coupled receptor (GPCR) activation (Woo et al., 2012), although the glutamate permeability of bestrophin-1 needs further investigation (Kane Dickson et al., 2014; Vaisey et al., 2016). Furthermore, the significance of these astrocytic channel candidates in glutamate release for normal neuronal function and animal cognitive behavior still remains elusive (Gundersen et al., 2015).
VRAC is ubiquitously expressed in mammalian cells and plays an important role in cell volume regulation (Osei-Owusu et al., 2018). Extracellular hypotonicity or intracellular hypertonicity, both inducing water influx and cell swelling, is widely used to activate the prominent Ca2+-independent VRAC currents. Accumulating evidence suggests VRAC as an astrocytic glutamate-releasing channel candidate (Hyzinski-Garcia et al., 2014; Mongin, 2016). Cell swelling activates VRAC, which mediates efflux of Cl− and organic osmolytes, such as glutamate from astrocytes, thus facilitating regulatory cell volume decrease (Osei-Owusu et al., 2018). Swollen astrocytes are the pathological feature associated with many brain diseases, including ischemic stroke, traumatic brain injury, and epilepsy (Mongin, 2016). VRAC activation may mediate the excessive release of glutamate from swollen astrocytes, which over-activates neuronal glutamate receptors and causes the excitotoxic neuronal death (Feustel et al., 2004; Fiacco et al., 2007). Astrocytic VRAC was also shown to be activated by ATP under isotonic conditions and mediate glutamate release (Takano et al., 2005), indicating a potential physiological role for this channel. However, due to the lack of molecular identity, the evidence supporting VRAC as an astrocytic glutamate-releasing channel was indirect and largely based on nonspecific pharmacological inhibitors, which affect the activity of many other membrane proteins, including those directly involved in glutamate transport (Bowens et al., 2013). Additionally, whether astrocytic VRAC regulates neuronal function and animal cognitive behavior under physiological conditions is still unknown. Recently, we and others identified SWELL1 (LRRC8A, a member of the leucine-rich repeat containing family 8 proteins) as the only essential VRAC subunit (Qiu et al., 2014; Voss et al., 2014). SWELL1 forms hetero-hexameric VRAC channels with its four homologs (LRRC8B-E), composition of which determines channel biophysical properties and substrate selectivity (Planells-Cases et al., 2015; Syeda et al., 2016; Voss et al., 2014). In contrast to bestrophin-1 and TREK-1 channel (Lolicato et al., 2017; Vaisey et al., 2016), structural analysis of VRAC revealed a wider selectivity filter which is compatible with the permeability of large anions (Deneka et al., 2018; Kasuya et al., 2018; Kefauver et al., 2018; Kern et al., 2019).
In this study, we generated a mouse model in which the only obligatory VRAC subunit Swell1 is disrupted specifically in astrocytes. We demonstrate that Swell1-dependent VRAC is a glutamate-permeable channel which mediates both tonic and cell swelling-induced glutamate release from astrocytes in the hippocampus. Deletion of astrocytic VRAC led to defective glutamatergic transmission due to a reduction in presynaptic release probability, and learning and memory defects. Furthermore, mice lacking astrocytic VRAC were protected from ischemic stroke-induced brain damage. Our study establishes astrocytic Swell1 channel as an important mechanism for astrocyte-neuron communication in both physiological and pathological states.
Results
Swell1 is required for VRAC activity induced by hypotonicity and ATP in astrocytes
To determine whether Swell1 is essential for VRAC activity in astrocytes, we generated Swell1 floxed mice (Swell1F/F) with two loxP sites flanking the major coding exon (Figure S1A), and then crossed with Nestin-cre line (Figure S1B), which induces potent and widespread recombination in precursors of neurons and astrocytes during embryonic development. We isolated and cultured primary cortical astrocytes from the knockout (KO) neonatal pups and their control littermates (McCarthy and de Vellis, 1980). The loss of Swell1 protein in KO astrocytes was confirmed by western blot (Figure 1A). To examine whether the basic electrophysiological properties of the astrocytes were altered during the culture process (Foo et al., 2011), we performed whole-cell patch-clamp recordings with KCl-based internal solution. Both control and KO cells exhibited highly negative resting membrane potential (~ −75 mV) and low membrane resistance (< 20 MΩ) with high basal K+ conductance (Figure S2A–S2E), consistent with the basic properties of native astrocytes. To isolate VRAC currents, we used CsCl-based isotonic internal solution. Perfusion of hypotonic solution quickly elicited cell swelling-activated currents in control astrocytes, which increased to very high levels after 5 min because of the constant osmolality gradient across the cell membrane under the recording conditions (Figure 1B). These currents exhibited characteristic features of VRAC: inhibition by channel inhibitor 4-(2-butyl-6, 7-dichloro-2-cyclopentyl-indan-1-on-5-yl) oxobutyric acid (DCPIB) (Figure 1B) and mild outward rectification (Figure 1C). Strikingly, the response of Swell1 KO astrocytes to hypotonic stimulus was completely abolished (Figure 1B–1D), indicating that Swell1 is an essential subunit of VRAC in astrocytes.
Figure 1. Swell1 is required for astrocytic VRAC activated by cell swelling and ATP.

(A) Representative western blots (n = 3 mice for each genotype) showing the loss of Swell1 protein from cultured Swell1 KO astrocytes. Gapdh was used as the loading control.
(B) Time course of whole-cell current densities at +100 mV induced by hypotonic solution (200 mOsm/kg, HYPO) for control and Swell1 KO astrocytes. DCPIB (50 μM) is a VRAC blocker.
(C) Representative whole-cell currents recorded by voltage step (left) and ramp (right) protocols in HYPO solution for cultured control and Swell1 KO astrocytes.
(D) Quantification of current densities at +100 mV evoked by HYPO for control (n = 14 cells) and Swell1 KO (n = 10 cells) astrocytes. Mann-Whitney test, *** p < 0.001.
(E) Left, time course of whole-cell currents at ±100 mV induced by ATP (100 μM) for control and Swell1 KO astrocytes. DCPIB was added as indicated. Right, whole-cell currents recorded by ramp protocol from −100 to +100 mV.
(F) Quantification of current densities at +100 mV activated by ATP. Numbers in bars correspond to experimental “n” of cells. Mann-Whitney test, ** p < 0.01.
Data are reported as mean ± SEM. Cultured astrocytes were independently isolated from 3 mice for each genotype.
Besides cell swelling, astrocytic VRAC can also be activated under isotonic conditions by other physiologically relevant stimuli associated with neuronal activity, such as ATP, glutamate, and bradykinin (Akita and Okada, 2014). Indeed, bath application of 100 μM ATP in isotonic solution induced the development of a DCPIB-sensitive Cl− current with mild outwardly rectification in control astrocytes (Figure 1E). Compared to cell swelling-induced currents, ATP-induced VRAC currents took a longer time (5–10 min) to develop and were much smaller in amplitude, suggesting alternative mode of channel activation and the potential involvement of second messenger systems (Akita and Okada, 2014). VRAC activity induced by ATP was also eliminated in Swell1 KO astrocytes (Figure 1E and 1F). Thus, both cell swelling and receptor stimulation activate Swell1-dependent VRAC in primary astrocytes.
Swell1-dependent VRAC is a glutamate-permeable channel
In addition to conducting Cl−, one of the key features of VRAC is its ability to permeate organic osmolytes, such as glutamate from astrocytes (Osei-Owusu et al., 2018). To determine the glutamate permeability of astrocytic VRAC, we replaced intracellular Cl− with equimolar glutamate as the only permeant anion and activated VRAC by hypotonic bath solution. The negatively charged glutamate carried considerable inward currents representing glutamate efflux from control astrocytes (Figure 2A). The leftward shifts in reversal potential indicated that the glutamate permeability of VRAC is significant, although less than that of Cl− (Pglutamate/PCl: ~ 0.3) (Figure 2B). The inward current was abolished in Swell1 KO astrocytes suggesting that Swell1 is also required for the glutamate permeability of VRAC (Figure 2C).
Figure 2. Swell1-dependent VRAC is a glutamate-releasing channel.

(A) Representative whole-cell currents recorded by ramp protocol from −100 to +100 mV in HYPO solution from control and Swell1 KO astrocytes with Cl−-based (133 mM CsCl) or glutamate-based (133 mM Cs-glutamate) pipette solution. Arrows indicate the reversal potentials.
(B) Quantification of the reversal potentials of control astrocytes. n = 11 cells for each group.
(C) Quantification of current densities at −100 mV evoked by HYPO from control and Swell1 KO astrocytes with Cl−-based or glutamate-based pipette solution. n = 9 cells for each group. Student’s t tests, *** p < 0.001.
(D) Representative images (top) and schematic illustration (below) of sniffer-patch technique. The small GFP positive cell is the HEK293T sensor cell expressing GluR1-L497Y. Astrocyte is whole-cell patched with hypertonic pipette solution (400 mOsm/kg, containing 1 or 5 mM glutamate). Scale bar, 20 μm.
(E-G) Representative current traces recorded at holding potential of −60 mV from control astrocytes (E and F), Swell1 KO astrocytes (G) and the sensor cells (red). The inward currents in the astrocytes indicate VRAC activation. The inward currents in the sensor cells indicate the detection of glutamate release from the nearby astrocytes. After the experiment, full current activation in the sensor cells (inset) was recorded by bath application of 5 mM glutamate (green line).
(H) Quantification of the percentage of full activation in control (n = 8 cells) and Swell1 KO astrocytes (n = 6 cells).
Data are reported as mean ± SEM. Cultured astrocytes were independently isolated from 2 mice for each genotype.
To determine whether VRAC-mediated glutamate efflux from astrocytes can be detected by neighboring cells, we adopted the sniffer-patch technique as a sensitive functional bioassay for glutamate (Lee et al., 2007). As illustrated in Figure 2D, we performed double whole-cell patch-clamp recordings in which astrocytes as the source cell, and the adjacent HEK293T cell transfected with GluR1-L497Y, a non-desensitizing α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptor mutant, as the sensor cell. The GluR1-L497Y has a high glutamate affinity, allowing detection of sub-micromole glutamate release. Intracellular hypertonic solution (containing 1 or 5 mM glutamate,) activated VRAC in the control source cells as indicated by the developing inward current at −60 mV (Figure 2E and 2F). At the same time, we observed an inward current in the sensor cells which can be washed away or blocked by DNQX, an AMPA receptor antagonist (Figure 2E and 2F and data not shown), indicating that glutamate efflux via astrocytic VRAC can be detected by the neighboring sensor cells. To quantify the amount of astrocytic glutamate release, we normalized the GluR1-L497Y activation to the maximal receptor activation by direct bath application of 5 mM glutamate (Figure 2E–2G). Intracellular solution containing 5 mM glutamate in control astrocytes generated more glutamate release than that with 1 mM glutamate (Figure 2H). As expected, intracellular hypertonicity-induced VRAC activity was absent in Swell1 KO source astrocytes (Figure 2G). Remarkably, glutamate efflux from these KO cells was also completely abolished (Figure 2G and 2H), demonstrating that astrocytic glutamate is released through Swell1-dependent VRAC channel. Similar results were also obtained in control and SWELL1 KO HeLa cells (Figure S3). Consistent with Swell1 siRNA knockdown and radiotracer D-aspartate (a non-metabolizable analogue for glutamate) efflux assays (Hyzinski-Garcia et al., 2014), these data provide direct evidence that glutamate permeates through Swell1 channel to yield glutamate release from astrocytes.
Reduced mEPSC frequency and impaired synaptic plasticity in astrocyte-specific Swell1 cKO mice
Astrocytes modulate neuronal function by releasing glutamate (Gundersen et al., 2015). To test whether astrocytic VRAC channel participates in the regulation of basal synaptic transmission and synaptic plasticity, we generated astrocyte-specific Swell1 knockout mice (cKO) by crossing Swell1F/F mice with mGFAP-cre (line 77.6), in which Cre expression is under the control of the mouse glial fibrillary acidic protein (mGFAP) promoter and restricted to astrocytes in the postnatal brain (Tao et al., 2011). To verify its specificity, mGFAP-cre mice were crossed with a stop-floxed-tdTomato reporter line (Ai9) so that the Cre positive cells are labeled with fluorescent tdTomato. As expected, the co-localization of tdTomato with astrocyte specific marker GFAP was nearly complete in the hippocampus (Figure S1C and S1E). In contrast, its co-localization with neuronal marker NeuN was very rare (~ 1.5%) (Figure S1D and S1F). In situ hybridization analysis revealed similar Swell1 mRNA levels in the hippocampal neurons and astrocytes in control mice, and confirmed the specific loss of Swell1 expression in astrocytes in cKO mice (Figure 3A and 3B). Consistently, western blot revealed a significant reduction of Swell1 protein in the mutant brains (Figure S1G and S1H). We then performed whole-cell patch-clamping recordings of astrocytes in stratum radiatum (SR) of the hippocampal CA1 region in brain slice preparations. Perfusion of hypotonic solution activated the typical DCPIB-sensitive outwardly-rectifying VRAC currents in control slices (Figure 3C–3E). Due to the low membrane resistance, the current amplitude of astrocytes in slices was underestimated and smaller than that of cultured astrocytes. Consistently, the swelling-activated VRAC currents were absent in astrocytes of mutant slices (Figure 3C–3E). These data demonstrate the specific loss of Swell1 and VRAC activity in astrocytes of Swell1 cKO mice.
Figure 3. Astrocytic Swell1 regulates glutamatergic synaptic transmission.

(A) Representative images of Swell1 RNAscope in situ hybridization and GFAP co-staining in hippocampal CA1 region from control and Swell1 cKO mice. Arrows indicate representative GFAP+ astrocytes with a clear cell body and not in the pyramidal cell layer (Pyr) or near blood vessel. SR: stratum radiatum. Scale bar, 20 μm.
(B) Quantification of Swell1 mRNA in situ hybridization signals. n = 197 neurons and 155 astrocytes from 4 mice for control; n = 207 neurons and 120 astrocytes from 4 mice for Swell1 cKO. Mann-Whitney test.
(C) Time course of whole-cell currents at ±80 mV induced by hypotonic aCSF (HYPO) for control and Swell1 cKO astrocytes in brain slices. DCPIB: 20 μM.
(D) Representative baseline subtracted whole-cell VRAC currents of astrocytes in slices recorded by ramp protocol from −80 to +80 mV.
(E) Quantification of baseline subtracted whole-cell currents at ±80 mV of astrocytes in slices. n = 13 cells from 6 mice for control; n = 10 cells from 4 mice for Swell1 cKO.
(F) Representative traces of mEPSCs recorded from hippocampal CA1 neurons of control and Swell1 cKO slices.
(G and H) Quantification of mEPSC frequency (G) and amplitude (H). n = 23 cells from 6 mice for control; n = 27 cells from 6 mice for Swell1 cKO. Student’s t tests, *** p < 0.001.
(I) Depressed I/O curves in the hippocampus of Swell1 cKO mice. n = 13–15 slices from 4 mice for each genotype. Two-way ANOVA, Bonferroni post hoc test, * p < 0.05, ** p < 0.01.
(J) TBS-induced LTP at SC-CA1 synapses in control and Swell1 cKO mice. Normalized fEPSP slopes were plotted every 1 min. Arrow indicates LTP induction. Sample traces represent fEPSPs taken before (1) and 50 min after TBS stimulation (2). Scale bars represent 0.5 mV (vertical), 5 ms (horizontal).
(K) Summary of experiments shown in (J). The amplitude of fEPSP slopes is averaged during 55–60 min after the stimulation. n = 13 slices from 5 mice for control; n = 12 slices from 5 mice for Swell1 cKO. Student’s t tests, ** p < 0.01.
Data are reported as mean ± SEM.
Swell1 cKO mice were viable and appeared grossly normal compared to their control littermates. To examine their glutamatergic synaptic transmission, we recorded, in whole-cell configuration, miniature excitatory postsynaptic currents (mEPSCs) of pyramidal neurons in the CA1 region of the hippocampus. mEPSC frequency was significantly reduced in the mutant hippocampus compared with controls, while no change was observed for mEPSC amplitude (Figure 3F–3H). Furthermore, there was a downward shift of input-output curves of field evoked postsynaptic potentials (fEPSPs) slope as the function of stimulus intensity at Schaffer collateral-CA1 synapses in hippocampal slices of Swell1 cKO mice (Figure 3I), indicating an impaired basal synaptic transmission. Furthermore, long-term potentiation (LTP) induced by theta burst stimulation (TBS) in the mutant hippocampus was partially, but significantly reduced (Figure 3J and 3K). To exclude the possibility that the defects we observed in Swell1 cKO mice are due to unspecific deletion of Swell1 in very rare neurons, we generated neuron-specific Swell1 knockout mice using NEX-cre line, which induces efficient recombination in pyramidal neurons in the neocortex and hippocampus (Goebbels et al., 2006) (Figure S4A–S4D). Both mEPSC (frequency and amplitude) of CA1 pyramidal neurons and hippocampal LTP were normal in Swell1F/F; NEX-cre mice compared with controls (Figure S4E–S4I). Thus, Swell1 in astrocytes is important for normal glutamatergic synaptic transmission and synaptic plasticity.
To explore the underlying mechanisms responsible for the defects in synaptic transmission and plasticity upon the deletion of Swell1 in astrocytes, we first characterized the cell number and morphology of astrocytes in Swell1 cKO mice and did not observe any difference compared to controls (Figure S2F–S2I), indicating that VRAC is not required for astrocyte proliferation and development in vivo. Astrocytes express an abundance of passive K+ channels which contribute to the hyperpolarized resting membrane potential and mediate K+ buffering function. We recorded hippocampal astrocytes in slices from control and Swell1 cKO mice and found they exhibited similar current-voltage relationships of K+ conductance and resting membrane potentials (Figure S2J–S2L), suggesting that the basic electrophysiological properties of astrocytes are normal without Swell1. Since astrocytes are implicated in synaptogenesis and synapse elimination during early development (Allen and Eroglu, 2017), the impairment of synaptic transmission and plasticity may be due to changes in neuron morphology and spine number. However, no defect in the laminar structure of hippocampus or in neuronal number was detected in Swell1 cKO mice (Figure S5A and S5B). Dendritic arborization and spine density in hippocampal CA1 pyramidal neurons were also comparable between control and mutant mice (Figure S5C–S5G). In addition, we did not observe any significant difference in the resting membrane potential, action potential threshold and amplitude (Figure S5H–S5J). We next examined the intrinsic excitability of CA1 pyramidal neurons by injecting a series of currents in the presence of blockers that inhibit excitatory and inhibitory inputs to the neurons. Again, no difference in neuronal firing frequency was observed (Figure S5K and S5L). These data suggest that deletion of Swell1 in astrocytes does not affect neuronal development and intrinsic electrophysiological properties.
Given that the spine number of CA1 neurons was normal (Figure S5E–S5G), a reduction of presynaptic release probability might be responsible for the impaired synaptic transmission in Swell1 cKO mice. To determine whether loss of astrocytic Swell1 affects excitatory vesicle release, we performed two independent experiments. First, we measured the paired-pulse ratios (PPRs) of evoked EPSCs (eEPSCs) in hippocampal CA1 pyramidal neurons in response to two consecutive stimulations at different short intervals. The second pulse generates a larger eEPSC at excitatory synapses due to the residual calcium from the first pulse in the presynaptic terminals (Zucker and Regehr, 2002) (Figure 4A). PPRs, inversely correlating with the release probability, were increased in Swell1 cKO mice compared to those of controls (Figure 4B), suggesting a reduction of presynaptic release probability in the mutant hippocampus. Second, we characterized EPSCs in CA1 pyramidal neurons evoked by minimal stimulation that activates single or very few presynaptic fibers (Stevens and Wang, 1995). The rate of successful synaptic response (probability of release) in Swell1 cKO mice was significantly lower compared with controls (Figure 4C and 4D), while the synaptic potency (mean amplitude of successful eEPSCs) was the same (Figure 4E). Accordingly, synaptic efficacy (mean amplitude of all EPSCs including failed responses) was decreased (Figure 4F). Both increased PPR and reduced successful rate of EPSCs by minimal stimulation suggest that the decreased mEPSC frequency in Swell1 cKO mice is due to the diminished probability of excitatory vesicle release from presynaptic terminals.
Figure 4. Reduced excitatory vesicle release probability in the absence of astrocytic Swell1.

(A) Representative paired-pulse traces with 25 and 200 ms intervals from hippocampal CA1 neurons of control and Swell1 cKO mice.
(B) Paired-pulse ratios (PPRs) with different inter-stimulus intervals. n = 13 cells from 6 mice for control; n = 15 cells from 6 mice for Swell1 cKO. Two-way ANOVA, Bonferroni post hoc test, ** p < 0.01, *** p < 0.001.
(C) Representative responses (top) and plot of EPSC amplitudes (bottom) evoked by minimal stimulation (20 consecutive stimuli) from hippocampal CA1 neurons of control (left) and Swell1 cKO (right) mice.
(D-F) Quantification of release probability (D), synaptic potency (E) and synaptic efficacy (F). n = 18 cells from 5 mice for control; n = 19 cells from 5 mice for Swell1 cKO. Student’s t tests, ** p < 0.01, *** p < 0.001.
Data are reported as mean ± SEM.
Swell1 channel mediates tonic glutamate release from astrocytes
In the brain, glutamate is released not only phasically through synaptic vesicle exocytosis but also tonically from non-synaptic sources of likely astrocytic origin (Cavelier and Attwell, 2005; Sah et al., 1989). Because of its sustained nature, tonic activation of extra-synaptic N-methyl-D-aspartate (NMDA) receptors (NMDAR) by ambient concentration of glutamate plays an important role in controlling neuronal excitability (Sah et al., 1989). However, the mechanisms underlying the tonic release of glutamate remain unknown (Cavelier and Attwell, 2005). To address whether ambient glutamate release occurs in an astrocytic VRAC-dependent manner, we performed whole-cell recordings on hippocampal CA1 pyramidal neurons at a holding potential of +40 mV and in the presence of extracellular Mg2+, tetrodotoxin (TTX) and picrotoxin to isolate NMDAR-mediated currents. Consistent with the previous findings (Cavelier and Attwell, 2005; Sah et al., 1989), application of NMDAR antagonist D-AP5 revealed a tonic NMDAR current in CA1 pyramidal neurons (Figure 5A). Interestingly, the tonic NMDAR currents were significantly reduced in Swell1 cKO hippocampus (Figure 5A and 5B). DCPIB treatment resulted in a similar reduction in the tonic NMDAR currents in control slices, while no further decrease was observed in Swell1 cKO hippocampus (Figure 5A and 5B). These data suggest that Swell1 channel is a major contributor to tonic glutamate release from astrocytes. The source of the remaining tonic NMDAR activation requires further investigation. To examine the effect of diminished tonic glutamate release on neuronal excitability, we injected a series of depolarizing current pulses to CA1 pyramidal neurons without blocking neuronal excitatory and inhibitory inputs and observed that neurons of mutant mice fired fewer action potentials than those of controls, indicating a decreased neuronal excitability in Swell1 cKO mice (Figure 5C and 5D).
Figure 5. Modulation of ambient glutamate level and neuronal excitability by astrocytic Swell1.

(A) Tonic NMDAR current recordings from CA1 pyramidal neurons in control and Swell1 cKO mice. Dashed lines indicate the changes of baseline induced by application of the NMDAR antagonist D-AP5 (50 μM). DCPIB, 25 μM.
(B) Quantification of D-AP5-sensitive tonic NMDAR currents. aCSF: n = 14 cells from 6 mice for each genotype. DCPIB: n = 12–14 cells from 4 mice for each genotype. One-way ANOVA, Bonferroni post hoc test, *** p < 0.001.
(C) Representative traces of action potentials in CA1 pyramidal neurons evoked by depolarizing current injection.
(D) Summarized results of firing rate under increasing step currents. n = 23 cells from 6 mice for each genotype. Two-way ANOVA, Bonferroni post hoc test, * p < 0.05, ** p < 0.01, *** p < 0.001.
(E) Representative traces of mEPSCs recorded from hippocampal CA1 neurons of Swell1 cKO mice before and after brief DHPG (50 μM) treatment.
(F and G) Quantification of mEPSCs frequency (F) and amplitude (G). n = 14 cells from 6 mice for each genotype. Paired Student’s t-test for (F), ** p < 0.001; Wilcoxon matched-pairs signed rank test for (G).
(H) Representative infrared differential interference contrast (DIC) image of paired recorded CA1 pyramidal neurons and the stimulation electrode in hippocampal slices. Sti, stimulation pipette; ND, neuronal depolarization. Scale bar, 20 μm.
(I) EPSC amplitudes from a representative synapse evoked by minimal stimulation in control (left) and Swell1 cKO (right) mice. Dot line at zero time indicates the application of neuronal depolarization.
(J) The fraction of paired recording experiments with an increase (>10%) in the neurotransmitter release probability before and after neuronal depolarization in control (left) and Swell1 cKO (right) mice. n = 20 cells from 6 mice for each genotype. Due to data overlapping, the data point is less than the n number. Note the basal release probability is lower in CA1 neurons of the cKO mice, consistent with Figure 4D.
Data are reported as mean ± SEM.
Activation of presynaptic group I metabotropic glutamate receptors (mGluR5/1) by glutamate released from astrocytes is a major mechanism for potentiating synaptic vesicle release probability in the hippocampus (Fiacco and McCarthy, 2004; Perea and Araque, 2007). Indeed, group I mGluR5/1 antagonists, 2-methyl-6-(phenylethynyl)-pyridine (MPEP) and (+)-2-methyl-4-carboxyphenylglycine (LY367385), significantly reduced mEPSC frequency of control CA1 pyramidal neurons without affecting mEPSC amplitude (Figure S6A and S6B). However, these antagonists failed to further decrease mEPSC frequency in Swell1 cKO hippocampus (Figure S6C and S6D), suggesting the involvement of mGluR hypofunction and reduced ambient glutamate levels in the regulation of synaptic vesicle release in the mutant mice. Short-term application (< 5 min) of mGluR5/1 agonist DHPG stimulates basal synaptic transmission by a presynaptic mechanism (Martin et al., 2015; Rodriguez-Moreno et al., 1998). Consistently, brief DHPG treatment rescued the phenotype of the reduced mEPSC frequency in Swell1 mutant mice without changing its amplitude (Figure 5E–5G). DHPG does not activate NMDA receptors and its application failed to rescue the depressed input-output curve of fEPSPs slope in the mutant hippocampal slices (data not shown). Therefore, the pharmacological study supports an mGluR-dependent presynaptic mechanism in the reduction of synaptic release probability in Swell1 cKO mice.
Astrocytes are also known to response to neuronal depolarization and release glutamate following intracellular Ca2+ elevation. This type of glutamate release has been demonstrated to rapidly and transiently increase the probability of neurotransmitter release at nearby CA3-CA1 synapses in a presynaptic mGluR1-depdendent manner (Navarrete and Araque, 2010). To test whether Swell1 channel is also required for this form of neuron-astrocyte signaling, we performed paired recordings in hippocampal slices by depolarizing one CA1 pyramidal neuron and monitoring synaptic transmission at synapses in an adjacent neuron with minimal stimulation technique (Navarrete and Araque, 2010) (Figure 5H). Neuronal depolarization evoked a transient increase of the neurotransmitter release probability in a similar fraction of paired recording experiments in Swell1 cKO mice compared to that in controls (Figure 5I and 5J). Thus, unlike the basal glutamatergic synaptic transmission, the phasic regulation of synaptic transmission by astrocytic glutamate release linked to the specific neuronal activities appeared to occur through a different mechanism from VRAC-mediated glutamate release.
Learning and memory deficits in astrocyte-specific Swell1 cKO mice
Normal animal behavior is dependent on precise synaptic transmission, whose dysregulation has been implicated in various neurological and psychiatric disorders. To determine whether the defective synaptic transmission and plasticity in slices lead to any abnormal animal behaviors, we first performed the open field test to evaluate the locomotor activity of Swell1 cKO mice. After 30 min free moving in the open field, the mutant mice had the same total travel distance as controls (Figure 6A). The number of vertical rearing was also comparable between cKO and control mice (Figure 6B). The anxiety level of mice can be determined by calculating the percentage of time mice spend in the center region of the open field. Compared with control mice, Swell1 mutant mice spent similar amount of time in the center (Figure 6C and 6D). These results indicate normal locomotor function and anxiety level in Swell1 cKO mice.
Figure 6. Swell1 cKO mice show impaired learning and memory.

(A-D) Quantification of total traveled distance (A), rearing number (B), distance traveled in the center (C), and distance traveled in the periphery (D) in 30 min open field test from control (n = 16 mice) and Swell1 cKO mice (n = 13 mice). Student’s t-test.
(E) No difference in spatial learning curves measured as the latency to find the hidden platform during MWM training between the two genotypes. n = 16 mice for control and 20 mice for Swell1 cKO. Two-way ANOVA, no significant difference was detected.
(F) Representative traces during the probe test of Morris water maze.
(G) Swell1 cKO mice spend significant less time in the target quadrant than control mice during the probe trials. n = 16 mice for control and 20 mice for cKO. Two-way ANOVA, Bonferroni post hoc test, ** p < 0.01.
(H) Trace fear conditioning elicits similar freezing behavior during the training trials in control and Swell1 cKO mice. n = 15 mice for control and 14 mice for Swell1 cKO (the same number of mice for I and J). Two-way ANOVA, no significant difference was detected.
(I) Swell1 cKO mice exhibit less freezing time during the contextual freezing test, but showed normal freezing in a novel context. Two-way ANOVA, Bonferroni post hoc test, ** p < 0.01.
(J) Swell1 cKO mice exhibit similar freezing time during tone presentation. Two-way ANOVA, no significance was detected.
Data are reported as mean ± SEM.
To test whether Swell1 deletion in astrocytes causes learning and memory deficits, we performed the Morris water maze (MWM) assay, a hippocampal-dependent spatial memory test (Morris, 1984). Mice were trained to locate a hidden platform in the water maze with four visual cues outside of the pool. As a control, when exposed to a visible platform in the same pool, the swimming latency and distance to the platform of Swell1 mutant mice were similar as those of controls (data not shown), indicating their visual perception and swimming capability are normal. During the training sessions, the escape latency for Swell1 cKO mice to locate the hidden platform was similar to that of control mice (Figure 6E), suggesting that they have a normal learning ability. To test spatial memory, we conducted the probe test 3 days after the final training session in which mice were exposed to the same pool but the platform was removed. Compared to controls, Swell1 cKO mice spent significantly less time in the target quadrant (Figure 6F and 6G), indicating that their spatial memory is impaired.
To further investigate the learning and memory impairment in Swell1 mutant mice, we perform trace fear conditioning, another classical behavior paradigm for studying learning and memory. In this task, mice were first trained to associate a conditional stimulus (tone) with an aversive unconditional stimulus (shock) separated by a 20 s trace period. During the training session, freezing behavior of both control and Swell1 cKO mice increased quickly and similarly (Figure 6H). 24 hours later, mice were placed back to the training context and their freezing time was measured. Compared to controls, Swell1 mutant mice exhibited significantly decreased context-evoked freezing behavior (Figure 6I). In contrast, there was no difference in the baseline freezing in a new context and no difference in the elevated freezing due to the tone presentation (Figure 6I and 6J), indicating that the learning deficit of Swell1 mutant mice is specific to hippocampal-dependent contextual fear conditioning. Consistent with the impairment of synaptic transmission and plasticity in Swell1 mutant hippocampus, these data demonstrate a role of astrocytic Swell1 for spatial and contextual memory.
Deletion of astrocytic Swell1 provides neuroprotection from stroke
Astrocytes are prone to pathological swelling in many neurological diseases, such as stroke and traumatic brain injury. Activation of VRAC channel in swollen astrocytes is thought to mediate the excessive release of glutamate, which then leads to over-activation of NMDA receptors and excitotoxic neuronal death (Mongin, 2016). One of the early effects of osmotic cell swelling on excitability of hippocampal CA1 pyramidal neurons in brain slices is the generation of slow inward currents (SICs) (Fiacco et al., 2007; Lauderdale et al., 2015). Similar to tonic glutamate currents, SICs are also mediated by the activation of extra-synaptic NMDA receptors and independent of neuronal vesicular glutamate release (Fellin et al., 2004). Application of hypotonic artificial cerebrospinal fluid (aCSF) stimulated NMDAR-mediated SICs with a characteristic slow kinetics and large amplitude (compared to mEPSCs) in hippocampal CA1 pyramidal neurons (Figure 7A). Their frequency peaked approximately two minutes after hypotonic solution perfusion (Figure 7B), correlating with the time course of rapid swelling of intact astrocytes in slices (Lauderdale et al., 2015). Interestingly, peak frequency of SICs was significantly blunted in neurons of Swell1 cKO mice compared to those of controls (Figure 7B), leading to the overall reduction in SIC frequency evoked by hypotonic stimulus (Figure 7C). These data suggest that astrocytic VRAC plays an important role in cell swelling-induced glutamate release in hippocampal slices.
Figure 7. Swell1 cKO mice are protected in ischemic stroke.

(A) Representative traces of slow inward current (SIC) recording from CA1 pyramidal neurons evoked by hypotonic aCSF (220 mOsm/kg, HYPO). Insets are examples of SIC. Scale bar: 40 pA (vertical) and 500 ms (horizontal).
(B) Quantification of the distribution of SICs analyzed every two minutes before and after HYPO application. Swell1 cKO mice (n = 13 cells from 5 mice) have significant less SICs than control mice (n = 14 cells from 5 mice) during the first 2 min of HYPO perfusion. Student’s t-test, *** p < 0.001.
(C) Summary histogram of SIC frequency per min for control (n = 13 cells from 5 mice) and Swell1 cKO (n = 14 cells from 5 mice) mice before and after HYPO application. Two-way ANOVA, Bonferroni post hoc test, *** p < 0.001.
(D) Representative traces of whole-cell current-clamp recording of neuronal action potentials (APs) evoked by HYPO application (220 mOsm/kg). HYPO-evoked APs are blocked by NMDAR antagonist D-AP5 (left middle panel). Stars and arrowheads indicate the burst firing and single AP firing, respectively. The dotted boxed areas are enlarged on the right.
(E and F) Quantification of HYPO-evoked burst number (E) and AP frequency (F). n = 14 cells from 5 mice for control; n = 13 cells from 5 mice for Swell1 cKO. Student’s t-test for (E) and Mann-Whitney test for (F), ** p < 0.01.
(G) Representative images of triphenyltetrazolium chloride (TTC) staining 1 day after transient middle cerebral artery occlusion (tMCAO).
(H) Quantification of infarct volume for each coronal section. n = 10 mice for control and 11 mice for Swell1 cKO. Two-way ANOVA, Bonferroni post hoc test, ** p < 0.01, *** p < 0.001.
(I) Quantification of total infarct area volume. n = 10 mice for control; n = 11 mice for Swell1 cKO. Student’s t-test, *** p < 0.001.
(J) Quantification of neurological score 1 day after tMCAO. n = 10 mice for control; n = 11 mice for Swell1 cKO. Mann-Whitney test, ** p < 0.01.
Data are reported as mean ± SEM.
Next, we examined the effect of osmotic cell swelling on neuronal excitability by current clamp recordings in the presence of DNQX to exclude the influence of AMPA receptors. Since AMPA receptors-mediated membrane depolarization is important for removing Mg2+ block from NMDAR, recordings were performed in Mg2+-free solution. Consistent with the slow kinetics and large amplitude of SICs, hypotonic aCSF evoked NMDAR-dependent both burst and single action potential firings in CA1 pyramidal neurons of control hippocampus (Figure 7D). However, both burst number and action potential frequency were significantly reduced in neurons of Swell1 cKO mice (Figure 7E and 7F), indicating a critical role for astrocytic VRAC in osmotic cell swelling-induced neuronal excitability. Together with the data in Figure 5, these results suggest that Swell1-dependent VRAC in astrocytes releases glutamate both tonically and during pathological cell swelling conditions, which contributes to the enhancement of neuronal excitability.
VRAC inhibitors exhibited significant neuroprotective effects in rodent ischemic stroke models, suggesting a beneficial role of blocking VRAC in stroke (Feustel et al., 2004; Zhang et al., 2008). However, the existing VRAC inhibitors are poorly selective and interfere with functions of other cellular components (Mongin, 2016). To specifically test whether astrocytic VRAC is involved in excitotoxicity in vivo, we performed the temporal middle cerebral artery occlusion (tMCAO) stroke model and induced ischemic brain injury by the occlusion of the common carotid artery with an intraluminal monofilament in Swell1 cKO mice and their control littermates (Engel et al., 2011). Swell1 cKO mice displayed significantly smaller infarct volumes one day after tMCAO across most coronal sections and brain regions compared to those in control mice (Figure 7G–7I). Accordingly, Swell1 cKO mice also had markedly improved behavioral scores (Figure 7J). Thus, Swell1 channel in astrocytes plays an important role in the pathogenesis of ischemic brain injury in vivo.
Discussion
Our study identified Swell1-containing volume-regulated anion channel (VRAC) as an astrocytic glutamate-releasing channel and uncovered a new molecular mechanism for astrocyte-neuron interactions regulating diverse processes from neuronal excitability and synaptic transmission, to cognitive behavior and the pathogenesis of stroke. Recent molecular identification of Swell1 as an essential VRAC subunit made it possible to perform genetic studies, which circumvent the limitation of nonspecific VRAC inhibitors used in the field in the last two decades (Mongin, 2016). We provided direct evidence that glutamate permeates through Swell1 channel in astrocytes, which can be detected by the neighboring cells. Tonic glutamate release from astrocytes into the extracellular space occurs throughout the brain. Yet, the mechanism of non-vesicular release has been a mystery (Cavelier and Attwell, 2005). In addition to its suspected role in excitotoxic glutamate release during ischemic stroke, we surprisingly discovered that Swell1-dependent VRAC channel in astrocytes also significantly contributes to tonic glutamate release in the normal brain. Tonic activation of NMDA and metabotropic glutamate receptors by ambient glutamate increases neuronal excitability and modulates presynaptic release of neurotransmitter in the hippocampus (Dalby and Mody, 2003; McBain et al., 1994; Sah et al., 1989). Indeed, astrocyte-specific Swell1 knockout mice exhibited reduced mEPSC frequency, as a result of decreased presynaptic release probability, which can be rescued by activation of group I mGluR. Thus, Swell1 channel-mediated tonic glutamate release appears to exert a global and homeostatic enhancement of neuronal excitability and synaptic plasticity.
Our study also revealed the complexity of neuron-astrocyte communication with different, and possibly complimentary, mechanisms underlying astrocytic glutamate release. While the non-inactivating nature of VRAC currents is consistent with sustained glutamate efflux contributing to the ambient glutamate levels, it appears not to fit with the occasional occurrence of NMDAR-mediated SICs. Actually, what exactly explains the SIC kinetics is still unknown. The remaining stimulatory effect of hypotonic aCSF on SIC frequency in Swell1 cKO slices (Figure 7A–7C) suggest that glutamate release underlying SICs may be mediated by VRAC together with other phasic glutamate-releasing pathways which may have a fast kinetics. Indeed, Ca2+-dependent SICs in CA1 pyramidal neurons evoked by protease-activated receptor 1 (PAR-1) activation in astrocytes were normal in Swell1 mutant mice (Figure S7). These data indicate the existence of VRAC-independent glutamate-releasing and SIC-generating mechanisms (Gomez-Gonzalo et al., 2018), which may include other channel candidates, such as bestrophin-1 and hemichannels. Besides glutamate release, the concomitant reduction of the extracellular space during cell swelling markedly increases the effective glutamate concentration which could also contribute to SIC generation (Lauderdale et al., 2015).
How VRAC in astrocytes is activated under physiological conditions is still unknown. There are several possibilities. First, signaling molecules, such as ATP and glutamate, released from the presynaptic neuron during synaptic transmission activate small but significant VRAC currents in cultured astrocytes (Akita and Okada, 2014). This mode of activation may involve signaling transduction pathways, such as protein phosphorylation by protein kinase C (Rudkouskaya et al., 2008). In addition, ATP stimulation is associated with a small transient increase (~5%) in astrocyte cell volume, which may contribute to VRAC activation (Takano et al., 2005). Second, bursts of neuronal activity lead to a transient accumulation of extracellular K+, which is absorbed by astrocytes through the highly expressed inward-rectifying K+ channels. This buffering function is critical for keeping extracellular K+ at levels that do not interfere with normal action potential propagation. The net accumulation of K+ causes physiological astrocyte swelling which may then stimulate VRAC (Larsen et al., 2014). Third, although difficult to detect by electrophysiological recordings, small basal activity of VRAC could exist in the absence of a hypotonic challenge or agonist exposure. Such basal activity may be responsible for the import of the antibiotic blasticidin S (larger than glutamate) under isotonic cell culture conditions (Lee et al., 2014). Therefore, VRAC as a glutamate-releasing channel represents a new mechanism by which astrocytes modulate neuronal function even in the absence of Ca2+ elevation. Future study will elucidate the exact mechanisms of VRAC activation under physiological conditions.
VRAC-dependent cell volume regulation has been implicated in cell proliferation, apoptosis and migration in cell lines (Akita and Okada, 2014). However, it is not essential for the development, morphology and overall function of astrocytes in vivo, possibly because cells possess other redundant cell volume regulatory mechanisms. Rather, we found that Swell1-containing VRAC plays a key role in astrocyte-neuron interactions by releasing glutamate. Cell-cell/cell-environment communications have emerged as a major function of VRAC based on its unique ability as a conducting channel for large (and not necessarily negatively charged) compounds with various degrees of permeability, such as neurotransmitters/neuromodulators (glutamate, taurine, GABA, D-serine, and even ATP) and cancer drugs (Lutter et al., 2017; Planells-Cases et al., 2015). Because glutamate is the brain’s most abundant amino acid and a highly permeable anion for VRAC, we focused on glutamate release in the current study. The permeability of VRAC to other neutral neurotransmitters, such as GABA and D-serine, is detectable but very low in cell lines based on radiotracer efflux assays (Lutter et al., 2017), which will require further examination in native brain cells. The substrate selectivity of astrocytic Swell1 channel is determined by the composition of non-obligatory LRRC8 subunits: for example, LRRC8D critical for taurine efflux, and LRRC8C/8E for the release of D-aspartate (Schober et al., 2017). Thus, in addition to their intracellular concentration, the expression ratio of LRRC8 channel subunits in heterogeneous astrocyte populations may influence the relative permeability and release of different neurotransmitters. This could provide a complex yet elegant mechanism for astrocytes to differentially regulate neuronal activity. By cell/region-specific disruption of individual LRRC8 subunits, future investigation will address their specific role in fine-tuning astrocyte-neuron communication.
Astrocyte-specific deletion of Swell1 protected mouse brain against damage in an experimental stroke model. This directly validates the proposed role of glutamate-releasing VRAC in excitotoxic neuronal death in vivo, resulting from over-activation of NMDA receptors by extracellular glutamate accumulation (Feustel et al., 2004). Furthermore, it highlights the critical involvement of astrocytes in the pathogenesis of ischemic brain injury. Given the pivotal role of NMDAR in excitotoxicity, blocking NMDAR was a major therapeutic approach for treating stroke. However, NMDAR antagonists failed to show efficacy in clinical trials of stroke. One of the major reasons is believed to be that inhibition of NMDAR also blocks its normal synaptic functions thus hindering neuronal survival (Lai et al., 2014). Our study suggests that targeting glutamate-releasing VRAC channel might represent a better therapeutic strategy than the current one of blocking NMDAR and its downstream signaling pathways, which are essential for neuronal function and survival. Furthermore, it may also avoid a dramatic impairment of the beneficial phasic regulation of synaptic transmission by astrocytic glutamate (Figure 5H–5J). In addition to ischemic stroke, this strategy could be relevant for glutamate-mediated excitotoxicity in other disorders, such as traumatic brain injury, epilepsy, and neurodegenerative diseases.
STAR METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Zhaozhu Qiu (zhaozhu@jhmi.edu)
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Animals
All procedures related to animal care and treatment were approved by the Johns Hopkins University Animal Care and Use Committee and met the guidelines of the National Institute of Health Guide for the Care and Use of Laboratory Animals. All animals were group housed in a standard 12 hours light/ 12 hours dark cycle with ad libitum access to food and water. Male and female animals were used for all experiments. Mouse embryonic stem (ES) cells (on C57BL6 background) with floxed Swell1 allele were obtained from the European Conditional Mouse Mutagenesis Program (EUCOMM) and were used to generate chimeric mice by blastocyst injection (Figure S1A). Germline-transmissions were achieved and selection cassette was removed by breeding with FLP delete mice. mGFAP-cre (B6. Cg-Tg(Gfap-cre)77.6Mvs/2J), Nestin-cre (B6. Cg-Tg(Nes-cre)1Kln/J), and Ai9 (B6. Cg-Gt(ROSA)26Sortm9(CAG-tdTomato)Hze/J) were purchased from The Jackson Laboratory. NEX-cre was a gift from Klaus-Armin Nave (Goebbels et al., 2006).
Cell culture and transfection
HEK293T cells and HeLa cells were purchased from ATCC and not further authenticated. Cells were maintained in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum (FBS) and 1% penicillin/streptomycin (P/S) at 37°C in humidified 95% CO2 incubator. One day before the sniffer patch recording, cells were digested in 0.25% trypsin and plated onto Poly-D-lysine coated 12-mm coverslips. HEK293T cells were co-transfected with EGFP and GluR1-L497Y plasmids by Lipofectamine 2000 (Invitrogen) following the manufacturer’s instructions. On the day of sniffer patch, HEK293T cells expressing GluR1-L497Y were dissociated, triturated, and added onto the coverslips with primary astrocytes or HeLa cells. Cells were not cultured past 20 passages.
To culture primary astrocytes, cortices from P0-P1 newborn pups of both male and female were dissected in ice-cold HBSS and digested in 0.25% trypsin at 37°C for 20 min. Cells were dissociated by triturating 15–20 times in the culture media (MEM supplemented with 10% FBS and 1% P/S) and plated into a culture flask for 7–10 days. The purity of astrocyte cultures was >95% as routinely confirmed with GFAP immunostaining. For astrocyte recording, cells were digested and plated onto Poly-D-lysine coated coverslips for at least 24 hours before recording.
SWELL1 knockout HeLa cells were generated by CRISPR/Cas9-mediated gene deletion. Guide RNA was cloned into PX458-mCherry, a modification of the plasmid pSpCas9(BB)-2A-GFP (PX458), which was a gift from Feng Zhang (Addgene plasmid # 48138). Two days after guide RNA transfection, single fluorescent cells were FACS-sorted into 96-well plates containing DMEM with 20% FBS. Single colonies were isolated and expanded for genotyping analysis of frameshift mutations by target-site-specific PCR and TA cloning followed by Sanger sequencing. The loss of SWELL1 was further validated by VRAC current recordings.
METHOD DETAILS
Immunostaining
Anesthetized mice were perfused transcardially with phosphate-buffered saline (PBS), followed by 4% paraformaldehyde (PFA) in PBS. Brains were removed and post-fixed in 4% PFA at 4°C overnight. After dehydration by 30% sucrose, brains were embedded in OCT (Tissue-Tek) and cut into 35-μm-thick sections on cryostat microtome (Leica). Sections were permeabilized with 0.2% Triton X-100 and 1% BSA in PBS for 45 min at room temperature (RT), washed with PBS three times, blocked in 10% BSA, and incubated with primary antibodies at 4°C overnight. Primary antibody concentrations: anti-GFAP (1:500; eBioscience), anti-NeuN (1:200; Millipore). After washing with PBS 3 times, samples were incubated with Alexa Fluor-conjugated secondary antibodies (1:500; Invitrogen) for 1h at RT. Images were taken under Zeiss LSM780 confocal microscope.
RNAscope In situ hybridization
Fixed brains were embedded in OCT (Tissue-Tek) and sectioned at a thickness of 14 μm. RNAscope Multiplex Fluorescent Reagent Kit v2 (ACD, Advanced Cell Diagnostics) was used following the manufacturer’s manual for the fixed frozen tissues. Probe targeting Swell1 (#458371) was purchased from ACD. TSA Plus fluorescein (#NEL741E001KT) were used for developing the fluorescence signal. After in situ hybridization, sections were performed immunostaining as described above. Images were collected by a Zeiss LSM 880 confocal microscope.
Biocytin labelling
To reconstruct the neuronal morphology of recorded CA1 pyramidal neurons, the recording pipettes were filled with 0.1% biocytin (Sigma). After the recording, the pipettes were removed slowly and the slices were fixed overnight with 4% PFA at 4 °C, permeabilized with 0.2% Triton X-100 and 1% BSA in PBS for 1 h at RT, and incubated with the fluorophore-conjugated streptavidin (1:1000; Invitrogen) for 2 h. After washing with PBS three times, samples were imaged with Zeiss LSM780 confocal microscope.
Cell number and morphological analysis
For the astrocyte number counting, cell counts were performed by selecting a 100 × 100 μm area in the stratum radiatum region of hippocampal CA1. For the neuron number counting, the counts were performed by selecting a 50 × 50 μm area in the CA1 and CA3 regions. To examine the morphology of astrocyte and neuron, Z-stack images were taken using the Zeiss 780 confocal microscope. Astrocyte and neuron morphological complexity was analyzed via Sholl analysis by counting the number of intersections between cellular processes and concentric circles emanating from the center of the soma. The analysis was performed using the Sholl analysis plugin in FIJI. The Sholl parameters included: 1) starting Sholl radii at 10 μm from the line start, and 2) Sholl radii at 5 μm increments (for astrocytes), and 20 μm increments (for neurons). Dendritic spines were counted using FIJI. Primary and second dendrites were counted from a region of 15 μm length.
Western blot
Homogenates of brain tissues and cultured astrocytes were prepared in lysis Buffer containing (in mM): 20 Tris-HCl, pH 7.5, 150 NaCl, 1% triton and 1% protease inhibitors cocktails. Samples were resolved on SDS/PAGE and transferred to nitrocellulose membranes, which were incubated in the TBS buffer containing 0.1% Tween-20 and 5% milk for 1h at RT before the addition of primary antibody (anti-Swell1; 1:1000; from a rabbit immunized with Swell1 C-terminal peptide antigen) for incubation overnight at 4°C. After wash, the membranes were incubated with HRP-conjugated secondary antibody (Thermo Scientific) in the same TBS buffer for 1 hour at RT. Immunoreactive bands were visualized using enhanced chemiluminescence. Films were scanned with a scanner and analyzed with Image J (NIH). Band density of interested proteins was normalized in relation to loading control.
Astrocyte electrophysiology
When cultured astrocytes were confluent in the T25 flask, cells were re-plated onto coverslips for recoding. For hypotonicity-activated VRAC current recordings, astrocytes were whole-cell patched in isotonic bath solution containing (in mM): 90 NMDG-Cl, 2 KCl, 1 MgCl2, 2 CaCl2, 10 HEPES, 10 glucose, 100 mannitol (pH adjusted to pH 7.3 with NMDG and osmolality adjusted to 310 mOsm/kg), then hypotonic solution which has the same ionic composition but without mannitol were applied. Recording electrodes (2–4 MΩ) were filled with a standard internal solution containing (mM):133 CsCl, 10 HEPES, 4 Mg-ATP, 0.5 Na3-GTP, 2 CaCl2, 5 EGTA (pH adjusted to 7.2 with CsOH and osmolality was 290–300 mOsm/kg). For ATP- or glutamate-activated VRAC current recordings, the bath solution contained (in mM): 145 NMDG-Cl, 2 KCl, 1 MgCl2, 2 CaCl2, 10 HEPES, 10 glucose (pH adjusted to pH 7.3 with NMDG and osmolality adjusted to 300–310 mOsm/kg). For the VRAC time course, constant voltage ramps (5 s interval, 500 ms duration) were applied from a holding potential of 0 to ± 100 mV. For the step protocol, cells were held at −60 mV and voltage step pulses (3 s interval, 500 ms duration) were applied from −100 to +100 mV in 20 mV increments. The ramp protocol was used with a holding potential of 0 and depolarized from −100 to +100 mV. To determine the glutamate permeability of astrocytic VRAC, the pipette solution was made by replacing CsCl with equimolar amounts of Cs-glutamate. The reversal potentials were determined using the ramp protocol.
For recordings of basic electrophysiological properties of astrocytes in cultured and hippocampal slices, the pipettes were filled with an internal solution containing (in mM): 130 KCl, 2 MgCl2, 10 HEPES, 5 EGTA, 2 Mg-ATP, 0.5 CaCl2, with pH adjusted to 7.2, osmolality 290–300 mOsm/kg. For astrocytes in slice, cells were held at −80 mV and voltage step pulses were applied from −120 mV to +40 mV in 20 mV increments. For cultured astrocytes, cells were held at −60 mV and voltage step pulses were applied from −100 mV to +100 mV in 20 mV increments.
To record astrocyte VRAC currents in slices, recording electrodes (5–7 MΩ) were filled with an internal solution containing (mM): 133 CsCl, 10 HEPES, 4 Mg-ATP, 0.5 Na3-GTP, 2 CaCl2, 5 EGTA (pH adjusted to 7.2 with CsOH and osmolality was 290–300 mOsm/kg). The isotonic aCSF containing (in mM): 75 NaCl, 1 MgCl2, 2 CaCl2, 1.3 NaH2PO4, 26 NaHCO3, 10 glucose, 100 mannitol. Hypotonic aCSF had the same ionic composition but without mannitol. Cells were held at 0 mV and a ramp protocol from −80 mV to +80 mV was applied every 5 s to monitor the VRAC currents developing over time. pCLAMP10.7 software and a MultiClamp 700B amplifier was used for electrophysiology (Axon Instruments).
Sniffer patch recording
Primary astrocytes or HeLa cells were used as the source cells, and HEK293T cells co-transfected with GluR1-L497Y and EGFP were the sensor cells. 12–18 h after transfection, HEK293T cells were reseeded onto source cells for recording. To activate VRAC in astrocytes or HeLa cells, recording electrodes (2–4 MΩ) were filled with a hypertonic internal solution containing (mM): 133 CsCl, 10 HEPES, 4 Mg-ATP, 0.5 Na3-GTP, 2 CaCl2, 5 EGTA, 100 mannitol, and various concentrations of glutamate (pH adjusted to 7.2 with CsOH and osmolality was 400–410 mOsm/kg). For HEK293T cells recording, the same internal solution was used except there was no mannitol or glutamate. External solution contained (in mM): 145 NaCl, 10 HEPES, 2 KCl, 2 CaCl2, 1 MgCl2, and 10 glucose (pH adjusted to pH 7.3 with NaOH and osmolality adjusted to 300–310 mOsm/kg). All the cells were held at −60 mV using Multiclamp 700B amplifier, and data was acquired with pClamp 10.7 software (Molecular Devices).
Neuron electrophysiology
To prepare acute hippocampal slices, mice (P20-P30) were anesthetized with the inhalation anesthetic isoflurane, and then perfused with ice-cold oxygenated cutting solution. The brain was removed rapidly and immersed in ice-cold choline-based cutting solution containing (in mM): 110 choline chloride, 7 MgCl2, 2.5 KCl, 0.5 CaCl2, 1.3 NaH2PO4, 25 NaHCO3, 20 glucose, saturated with 95% O2 and 5% CO2. Transversal hippocampal slices (300 μm) were cut in the cutting solution using a vibratome (VT-1200S, Leica) and transferred to artificial cerebrospinal fluid (aCSF) containing (in mM): 125 NaCl, 2.5 KCl, 2.5 CaCl2, 1.3 MgCl2, 1.3 NaH2PO4, 26 NaHCO3, 10 glucose, saturated with 95% O2 and 5% CO2. The slices were allowed to recover for 1 h at 32 °C and then at RT for at least 1 h before recoding. All recordings were made at RT in a submerged recording chamber with constant aCSF perfusion. Whole-cell recordings from CA1 pyramidal neurons were visualized under an upright microscope (BX51WI, Olympus) with infrared optics. Recording pipettes were pulled by a micropipette puller (P-1000, Sutter instrument) and had a resistance of 3–5 MΩ when filled with an internal solution containing (in mM): 125 K-gluconate, 15 KCl, 10 HEPES, 1 MgCl2, 4 Mg-ATP, 0.3 Na3-GTP, 10 phosphocreatine, and 0.2 EGTA (pH 7.2, osmolality 290–300 mOsm/kg). Recordings were made with MultiClamp 700B amplifier and 1550B digitizer (Molecular Device). Data acquisition were performed with pClamp 10.7 software (Molecular Device), filtered at 1 kHz and digitized at 10 kHz. In all experiments, the series resistance (Rs) was monitored throughout the recording and controlled below 20 MΩ with no compensation. Data was discarded when the series resistance varied by ≥ 20%. To block inhibitory synaptic transmission, picrotoxin (100 μM) was added to the bath in all recordings.
For AMPAR mEPSCs recording, cells were held at −70 mV and TTX (1 μM) was added to the bath. In pharmacological experiments, after 10–15 min baseline recording, MPEP and LY367385 or DHPG was bath applied. For tonic NMDAR-mediated currents recording, neurons were held at +40 mV in normal aCSF with an internal solution containing (in mM): 110 Cs methylsulfate, 20 TEA-Cl, 15 CsCl, 4 ATP-Mg, 0.3 Na3-GTP, 0.5 EGTA, 10 HEPES, 4.0 QX-314, and 1.0 spermine, pH adjusted to 7.2 with CsOH, osmolality 290–300 mOsm/kg with sucrose. Recoding was performed in the present of 1 μM TTX and 20 μM DNQX. After baseline was stable, tonic NMDAR current was observed by bath applying D-AP5.
For PPR recording, CA1 pyramidal neurons were patch clamped at a holding potential of −70 mV to record Schaffer collateral (SC)-evoked EPSCs. A succession of paired pulses stimulation separated by intervals of 25, 50, 100, and 200 ms were given every 15 s. Paired-pulse ratio was quantified as 2nd EPSC/1st EPSC. To examine the intrinsic excitability of CA1 pyramidal neurons, a series of step currents (from −40 to 240 pA at a step of 20 pA, 500 ms duration) were injected to the cells under current-clamp configuration in the presence of 20 μM DNQX, 50 μM D-AP5 and 10 μM bicuculline. To investigate the effect of tonic NMDAR currents on neuronal excitability (Figure 5C), recordings were done without any blockers. Spike properties were calculated from the very first action potentials.
For minimal stimulation recording, a glass microelectrode (3–5 MΩ) filled with aCSF was placed in the stratum radiatum using visual guidance to stimulate SC afferents. CA1 pyramidal neurons were clamped at −70 mV. Single pulse (100 μs) was delivered at 0.1 Hz by a STG 400 (Multichannel system) stimulator. The stimulus intensity (6–15 μA) was adjusted to fulfill the criteria that putatively stimulate single presynaptic fibers. Responses that did not meet these criteria were rejected. Release probability (Pr, number of successes as percentage of total number of stimulation), synaptic efficacy (average amplitude of all responses including failures), and synaptic potency (average amplitude of successes) were calculated. EPSC amplitude was measured as the peak current (2–6 ms after stimulus) relative to the mean baseline current which was taken 10–20 ms before stimulus artifact. For paired CA1 pyramidal neurons recording, two neurons within 100 μm distance were chosen. After the baseline recording (20 consecutive stimuli at 0.1 Hz), one pyramidal neuron was depolarized to 0 mV for 5 s and then another 20 consecutive responses were recorded after neuronal depolarization. Release probability changed more than 10 % between before and after the neuronal depolarization was considered as potentiation or depression.
For hypotonicity-induced slow inward current recordings, neurons were held at −70 mV in the isotonic solution containing (mM): 80 NaCl, 2.5 KCl, 2.5 CaCl2, 0 MgCl2, 1.3 NaH2PO4, 26 NaHCO3, 10 glucose, 90 mannitol and in the presence of 1 μM TTX, 100 μM picrotoxin, 20 μM DNQX and 10 D-serine. After a 10 min baseline, hypotonic aCSF solution which had the same ionic composition but without mannitol was applied. For hypotonic aCSF-evoked action potential recording, neurons were recorded under the current clamp configuration in the presence of 0 Mg2+ and 20 μM DNQX. For PAR-1 agonist TFLLR-induced slow inward current recording, neurons were held at −70 mV in the presence of 1 μM TTX, 100 μM picrotoxin, 20 μM DNQX.
Field recording
For LTP recording, juvenile mice (3–4 weeks) were anesthetized with isoflurane and transverse hippocampal slices (350 μm) were cut in ice-cold, choline-based cutting solution (saturated with 95% O2 and 5% CO2). The slices were allowed to recover for 40 min at 32 °C and then at room temperature for at least 2h before recoding. Picrotoxin (100 μM) was added to block inhibitory transmission. A cut between CA3 and CA1 was made to minimize recurrent activity during recording. Slices were transferred to the recording chamber, which were perfused continuously with aCSF (125 NaCl, 2.5 KCl, 2.5 CaCl2, 1 MgCl2, 1.3 NaH2PO4, 26 NaHCO3, 10 glucose, saturated with 95% O2 and 5% CO2, flow rate at 2–3 ml/min) at 30 °C. A concentric bipolar electrode (World Precision Instruments) was placed in the middle of CA1stratum radiatum to stimulate Schaffer collateral. Field EPSPs (fEPSPs) from the CA1 neurons were recorded with a glass pipette (2–3 MΩ) filled with aCSF. Constant current pulses (70–120 μA, 100 μs) were delivered at 0.033 Hz by a STG 400 stimulator. The stimulus intensity was adjusted to evoke 40–50% of the maximal response. LTP was induced by theta burst stimulation (TBS) consisting of a single train of 5 bursts at 5 Hz, and each burst contained 4 pulses at 100 Hz.
Behavioral analysis
Mice were handled by investigators for five days before any behavioral test. Locomotor activity was carried out with adult (3–5 month old) male mice by investigators blinded to their genotype. The animals were placed in a chamber (18” × 18”) (Photobeam activity system-San Diego Instruments) and monitored for movement by using horizontal and vertical photobeams. Beam breaks were converted to directionally specific movements and summated at 5-minute intervals over 30 minutes. Ambulatory activity was measured as total horizontal photobeam breaks, rearing was evaluated as total vertical beam breaks.
Morris water maze test was performed using adult (3–5 month old) male mice. The arena consisted of a circular pool (diameter of 120 cm) filled with water that was at 24°C and made opaque with non-toxic white tempera paint. A circular, plexiglass platform (length of 10 cm) was submerged 1 cm below the surface of the water and four local cues were provided to allow spatial map generation. Mice were trained for a total of 20 trials over 5 days, with 4 trials per session and 1 session per day. Prior to the first training trial, mice were given a single habituation trial without the platform to assess any spatial bias. Trials were 60 s and mice that did not find the platform within that time were guided to the platform by the experimenter. Once on top of the platform, mice were left for an additional 10 s before being removed. Start locations (north, south, east and west) were pseudo-randomized so that each start location was used once per session and the sequence of start locations in any session was never used twice. Two probe trials were performed 24 and 72 hours after the last training. The probe trials were 60s long and the platform was removed. Tracking and analysis of animal movement was done using the ANY-maze tracking system (SD instruments). Data were analyzed by comparing quadrant preferences and escape latencies averaged across animals within groups (control or cKO).
Trace fear conditioning was performed using adult (3–5 months old) male mice as previously described with some modifications (Makuch et al., 2011). Briefly, percent of time spent freezing was quantified using automated motion detection software (FreezeScan by CleverSys, Inc.). On day 1 mice were habituated to the training chamber for 10 min. Training occurred on day 2 as follows: mice were allowed to acclimate to the chamber for 2 min prior to the onset of four consecutive training blocks, each consisting of a 20 s baseline, followed by a 20 s, 2 KHz, 80 dB tone, followed by an 18 s trace interval of silence, followed by a 2 s scrambled 0.5 mA foot shock, followed by a 20 s inter-trial interval (ITI). On day 3 mice were tested. Mice were first placed in the training chamber for 3 min to assess contextual fear conditioning, after which they were returned to their home cage. Testing for trace fear conditioning took place in a novel chamber, which was distinct from the training chamber. Mice were allowed to acclimate to the novel chamber for 2 min prior to tone presentation to assess % freezing in the novel chamber. Mice were presented with three testing blocks consisting of a 20 s baseline followed by a 20 s 2 KHz, 80 dB tone followed by a 20s ITI.
Transient middle cerebral artery occlusion (tMCAO)
Male Swell1 cKO mice and control littermates (6 weeks, ~20 g) were anesthetized with isoflurane and body temperature was maintained during surgery. Ischemia was induced by introducing a 0.19 mm silicone-coated monofilament (7–0; Doccol) from the external carotid artery into the internal carotid artery and advancing it into MCA origin. Reperfusion was performed 1 hour after occlusion by removing the occluding suture from the MCA. The mortality rate immediately after surgery is ~10%, and these mice were excluded from further analysis. 24 hours after tMCAO, neurological function was evaluated by an observer blinded to the genotype of the mice with a scale of 0–5: 0, no neurological deficit; 1, failed to extend right forepaw; 2, circled to the right; 3, fell to the right; 4, could not walk spontaneously and; 5, death.
After neurological evaluation, brains were removed and cut into six 1-mm serial coronal sections using a vibratome (VT-1200S, Leica). The sections were stained with 1.5% 2,3,5-triphenyltetrazolium chloride (TTC) in PBS for 5 min at 37°C, followed by fixation in 4% paraformaldehyde for 15 min. The TTC stained sections were scanned and digitized. Using Image J, the areas of TTC negative and the ipsilateral hemisphere were measured. The percent infarct area per slice was calculated as: (area of infarct/area of ipsilateral hemisphere). The percent volume of infarct was calculated as: (the sum area of infarct/the sum area of ipsilateral hemisphere).
QUANTIFICATION AND STATISTICAL ANALYSIS
GraphPad Prism 6.0 software was used for all statistical analyses. Images were analyzed with ImageJ (US National Institutes of Health) and ZEN software (ZEISS). All the behavior assays, stroke model and image quantifications were performed by investigators blinded to mouse genotype. Before each test, Gaussian distribution of the data was assessed using Shapiro-Wilk normality test to determine whether the data were normally distributed. Parametric tests (paired or unpaired Student’s t test for two groups) were used for normally distributed data while non-parametric tests (Wilcoxon matched-pairs signed rank test for paired groups, Mann-Whitney for unpaired two groups, and Kruskal-Wallis test followed by Dunn’s post hoc test for three or more groups) were used for data not normally distributed. Two-way ANOVA followed by Bonferroni’s post hoc test was used to analyze studies which have two factors. Data are reported as mean ± SEM. The significance level was set at p < 0.05. When the p values were greater than 0.05, it is stated as a precise value. Number (n) of cells and animals for each experiment are shown in each figure and described in each figure legend.
Supplementary Material
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse anti-NeuN (clone A60) | Millipore | Cat# MAB377; RRID: AB_4339490 |
| Mouse anti-GFAP (clone GA5) | Thermo Fisher Scientific | Cat# 14–9892-82;RRID: AB_4339490 |
| Rabbit anti-Swell1 | This paper | N/A |
| Rabbit anti-GAPDH (clone EPR16891) | Abcam | Cat# ab181602; RRID: AB_21135 |
| Alexa Fluor 546 Goat anti-mouse | Thermo Fisher Scientific | Cat# A11018; RRID: AB_1944229 |
| Alexa Fluor 488 Goat anti-mouse | Thermo Fisher Scientific | Cat# A11017; RRID: AB_2107298 |
| Cy3-conjugated Streptavidin | Jackson ImmunoResearch | Cat# 016–160-084; RRID: AB_130355 |
| TSA Plus Fluorescein Evaluation Kit | PerkinElmer | Cat #NEL741; RRID: AB_2490275 |
| RNAscope Multiplex Fluorescent Reagent Kit v.2 | Advanced Cell Diagnostics | Cat# 323100 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| TTX | Tocris | Cat# 1069 |
| DL-APV | Sigma-Aldrich | Cat# A8054 |
| DNQX | Sigma-Aldrich | Cat# D0540 |
| Picrotoxin | Sigma-Aldrich | Cat# P1675 |
| Bicuculine | Sigma-Aldrich | Cat# 14340 |
| DCPIB | Tocris | Cat# 1540 |
| D-serine | Sigma-Aldrich | Cat# S4250 |
| MPEP | Tocris | Cat# 1212 |
| LY367385 | Tocris | Cat# 1237 |
| Biocytin | Sigma-Aldrich | Cat# B4261 |
| TFLLR | Sigma-Aldrich | Cat# T7830 |
| DHPG | Sigma-Aldrich | Cat# D3689 |
| L-glutamic acid | Sigma-Aldrich | Cat# G1251 |
| ATP | Sigma-Aldrich | Cat# A2383 |
| 2,3,5-Triphenyltetrazolium chloride | Sigma-Aldrich | Cat# T8877 |
| QX-314 | Tocris | Cat# 2313 |
| Experimental Models: Cell Lines | ||
| Human: Human Embryonic Kidney (HEK) 293T cells | ATCC | Cat# CRL-3216 |
| Human: HeLa cells | ATCC | Cat# CCL-2 |
| Experimental Models: Organisms/Strains | ||
| Mouse: mGFAP-cre; (B6. Cg-Tg(Gfapcre)77.6Mvs/2J) | The Jackson Laboratory | RRID: IMSR_JAX:024098 |
| Mouse: Nestin-cre; (B6.Cg-Tg(Nes-cre)1Kln/J) | The Jackson Laboratory | RRID: IMSR_JAX:003771 |
| Mouse: NEX-cre | Goebbels et al., 2006 | N/A |
| Mouse: Swell1flox | This paper | N/A |
| Mouse: Ai9flox; (B6.Cg-Gt(ROSA)26Sortm9(CAG-tdTomato)Hze/J) | The Jackson Laboratory | RRID: IMSR_JAX:007909 |
| Oligonucleotides | ||
| RNAscope probe: Mm-Lrrc8a | Advanced Cell Diagnostics | Cat# 458371 |
| Recombinant DNA | ||
| GluR1-L497Y cDNA | Lee et al., 2007 | N/A |
| pSpCas9(BB)-2A-GFP (PX458) | Addgene | Cat# 48138 |
| Software and Algorithms | ||
| pCLAMP 10.7 | Molecular Devices | http://www.moleculardevices.com/products/software/pclamp.html; RRID:SCR_011323 |
| Clampfit 10.7 | Molecular Devices | N/A |
| CorelDraw11 | Corel Corporation | https://www.coreldraw.com/en/;RRID:SCR_014235 |
| ImageJ | NIH | https://imagej.nih.gov/ij/download.html; RRID: SCR_003070 |
| GraphPad Prism | GraphPad Software | http://www.graphpad.com/; RRID:SCR_002798 |
| ZEN 2.3 | ZEISS | http://www.zeiss.com/microscopy/en_us/products/microscopesoftware/zen.html#introduction;RRID:SCR_013672 |
Highlights.
Swell1 is essential for the glutamate-permeable VRAC channel in astrocytes
Swell1 channel mediates both tonic and cell swelling-induced glutamate release
Astrocytic Swell1 regulates synaptic transmission and neuronal excitability
Knockout of astrocytic Swell1 provides neuroprotection from ischemic stroke
Acknowledgements
We thank Ardem Patapoutian for his invaluable support of the generation of conditional Swell1 mice; Woo-Ping Ge for helpful discussion; Xiang Shi, Siqi Zhao, Feng Yan, and Sai Yang for assistance with stroke model, Senthilkumar Karuppagounder for assistance with glutamate measurement, Stephen Traynelis for GluR1-L497Y plasmid, Marie Hardwick for NEX-cre mice from Klaus-Armin Nave’s laboratory. This work was supported by Johns Hopkins University School of Medicine and a grant from the NIH (R35 GM124824 to Z.Q.). This paper is dedicated to the memory of our dear friend and wonderful colleague, Maria del Carmen Vitery, who recently passed away.
Footnotes
Declaration of Interests
The authors declare no competing interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Akita T, and Okada Y (2014). Characteristics and roles of the volume-sensitive outwardly rectifying (VSOR) anion channel in the central nervous system. Neuroscience 275, 211–231. [DOI] [PubMed] [Google Scholar]
- Allen NJ, and Eroglu C (2017). Cell Biology of Astrocyte-Synapse Interactions. Neuron 96, 697–708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Araque A, Carmignoto G, Haydon PG, Oliet SH, Robitaille R, and Volterra A (2014). Gliotransmitters travel in time and space. Neuron 81, 728–739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Attwell D, Barbour B, and Szatkowski M (1993). Nonvesicular release of neurotransmitter. Neuron 11, 401–407. [DOI] [PubMed] [Google Scholar]
- Barres BA (2008). The mystery and magic of glia: a perspective on their roles in health and disease. Neuron 60, 430–440. [DOI] [PubMed] [Google Scholar]
- Bowens NH, Dohare P, Kuo YH, and Mongin AA (2013). DCPIB, the proposed selective blocker of volume-regulated anion channels, inhibits several glutamate transport pathways in glial cells. Molecular pharmacology 83, 22–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cavelier P, and Attwell D (2005). Tonic release of glutamate by a DIDS-sensitive mechanism in rat hippocampal slices. The Journal of physiology 564, 397–410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dalby NO, and Mody I (2003). Activation of NMDA receptors in rat dentate gyrus granule cells by spontaneous and evoked transmitter release. J Neurophysiol 90, 786–797. [DOI] [PubMed] [Google Scholar]
- Deneka D, Sawicka M, Lam AKM, Paulino C, and Dutzler R (2018). Structure of a volume-regulated anion channel of the LRRC8 family. Nature. [DOI] [PubMed] [Google Scholar]
- Duan S, Anderson CM, Keung EC, Chen Y, Chen Y, and Swanson RA (2003). P2X7 receptor-mediated release of excitatory amino acids from astrocytes. The Journal of neuroscience : the official journal of the Society for Neuroscience 23, 1320–1328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engel O, Kolodziej S, Dirnagl U, and Prinz V (2011). Modeling stroke in mice - middle cerebral artery occlusion with the filament model. Journal of visualized experiments : JoVE. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fellin T, Pascual O, Gobbo S, Pozzan T, Haydon PG, and Carmignoto G (2004). Neuronal synchrony mediated by astrocytic glutamate through activation of extrasynaptic NMDA receptors. Neuron 43, 729–743. [DOI] [PubMed] [Google Scholar]
- Feustel PJ, Jin Y, and Kimelberg HK (2004). Volume-regulated anion channels are the predominant contributors to release of excitatory amino acids in the ischemic cortical penumbra. Stroke 35, 1164–1168. [DOI] [PubMed] [Google Scholar]
- Fiacco TA, Agulhon C, Taves SR, Petravicz J, Casper KB, Dong X, Chen J, and McCarthy KD (2007). Selective stimulation of astrocyte calcium in situ does not affect neuronal excitatory synaptic activity. Neuron 54, 611–626. [DOI] [PubMed] [Google Scholar]
- Fiacco TA, and McCarthy KD (2004). Intracellular astrocyte calcium waves in situ increase the frequency of spontaneous AMPA receptor currents in CA1 pyramidal neurons. The Journal of neuroscience : the official journal of the Society for Neuroscience 24, 722–732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiacco TA, and McCarthy KD (2018). Multiple Lines of Evidence Indicate That Gliotransmission Does Not Occur under Physiological Conditions. The Journal of neuroscience : the official journal of the Society for Neuroscience 38, 3–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foo LC, Allen NJ, Bushong EA, Ventura PB, Chung WS, Zhou L, Cahoy JD, Daneman R, Zong H, Ellisman MH, et al. (2011). Development of a method for the purification and culture of rodent astrocytes. Neuron 71, 799–811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goebbels S, Bormuth I, Bode U, Hermanson O, Schwab MH, and Nave KA (2006). Genetic targeting of principal neurons in neocortex and hippocampus of NEX-Cre mice. Genesis 44, 611–621. [DOI] [PubMed] [Google Scholar]
- Gomez-Gonzalo M, Zehnder T, Requie LM, Bezzi P, and Carmignoto G (2018). Insights into the release mechanism of astrocytic glutamate evoking in neurons NMDA receptor-mediated slow depolarizing inward currents. Glia 66, 2188–2199. [DOI] [PubMed] [Google Scholar]
- Gundersen V, Storm-Mathisen J, and Bergersen LH (2015). Neuroglial Transmission. Physiological reviews 95, 695–726. [DOI] [PubMed] [Google Scholar]
- Hamilton NB, and Attwell D (2010). Do astrocytes really exocytose neurotransmitters? Nature reviews Neuroscience 11, 227–238. [DOI] [PubMed] [Google Scholar]
- Hyzinski-Garcia MC, Rudkouskaya A, and Mongin AA (2014). LRRC8A protein is indispensable for swelling-activated and ATP-induced release of excitatory amino acids in rat astrocytes. The Journal of physiology 592, 4855–4862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kane Dickson V, Pedi L, and Long SB (2014). Structure and insights into the function of a Ca(2+)-activated Cl(−) channel. Nature 516, 213–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kasuya G, Nakane T, Yokoyama T, Jia Y, Inoue M, Watanabe K, Nakamura R, Nishizawa T, Kusakizako T, Tsutsumi A, et al. (2018). Cryo-EM structures of the human volume-regulated anion channel LRRC8. Nat Struct Mol Biol 25, 797–804. [DOI] [PubMed] [Google Scholar]
- Kefauver JM, Saotome K, Dubin AE, Pallesen J, Cottrell CA, Cahalan SM, Qiu Z, Hong G, Crowley CS, Whitwam T, et al. (2018). Structure of the human volume regulated anion channel. Elife 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kern DM, Oh S, Hite RK, and Brohawn SG (2019). Cryo-EM structures of the DCPIB-inhibited volume-regulated anion channel LRRC8A in lipid nanodiscs. Elife 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai TW, Zhang S, and Wang YT (2014). Excitotoxicity and stroke: identifying novel targets for neuroprotection. Progress in neurobiology 115, 157–188. [DOI] [PubMed] [Google Scholar]
- Larsen BR, Assentoft M, Cotrina ML, Hua SZ, Nedergaard M, Kaila K, Voipio J, and MacAulay N (2014). Contributions of the Na(+)/K(+)-ATPase, NKCC1, and Kir4.1 to hippocampal K(+) clearance and volume responses. Glia 62, 608–622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lauderdale K, Murphy T, Tung T, Davila D, Binder DK, and Fiacco TA (2015). Osmotic Edema Rapidly Increases Neuronal Excitability Through Activation of NMDA Receptor-Dependent Slow Inward Currents in Juvenile and Adult Hippocampus. ASN Neuro 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee CC, Freinkman E, Sabatini DM, and Ploegh HL (2014). The protein synthesis inhibitor blasticidin s enters mammalian cells via leucine-rich repeat-containing protein 8D. The Journal of biological chemistry 289, 17124–17131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee CJ, Mannaioni G, Yuan H, Woo DH, Gingrich MB, and Traynelis SF (2007). Astrocytic control of synaptic NMDA receptors. The Journal of physiology 581, 1057–1081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lolicato M, Arrigoni C, Mori T, Sekioka Y, Bryant C, Clark KA, and Minor DL Jr. (2017). K2P2.1 (TREK-1)-activator complexes reveal a cryptic selectivity filter binding site. Nature 547, 364–368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lutter D, Ullrich F, Lueck JC, Kempa S, and Jentsch TJ (2017). Selective transport of neurotransmitters and modulators by distinct volume-regulated LRRC8 anion channels. J Cell Sci 130, 1122–1133. [DOI] [PubMed] [Google Scholar]
- Makuch L, Volk L, Anggono V, Johnson RC, Yu Y, Duning K, Kremerskothen J, Xia J, Takamiya K, and Huganir RL (2011). Regulation of AMPA receptor function by the human memory-associated gene KIBRA. Neuron 71, 1022–1029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin R, Bajo-Graneras R, Moratalla R, Perea G, and Araque A (2015). Circuit-specific signaling in astrocyte-neuron networks in basal ganglia pathways. Science 349, 730–734. [DOI] [PubMed] [Google Scholar]
- McBain CJ, DiChiara TJ, and Kauer JA (1994). Activation of metabotropic glutamate receptors differentially affects two classes of hippocampal interneurons and potentiates excitatory synaptic transmission. The Journal of neuroscience : the official journal of the Society for Neuroscience 14, 4433–4445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCarthy KD, and de Vellis J (1980). Preparation of separate astroglial and oligodendroglial cell cultures from rat cerebral tissue. J Cell Biol 85, 890–902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mongin AA (2016). Volume-regulated anion channel--a frenemy within the brain. Pflugers Archiv : European journal of physiology 468, 421–441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris R (1984). Developments of a water-maze procedure for studying spatial learning in the rat. J Neurosci Methods 11, 47–60. [DOI] [PubMed] [Google Scholar]
- Navarrete M, and Araque A (2010). Endocannabinoids potentiate synaptic transmission through stimulation of astrocytes. Neuron 68, 113–126. [DOI] [PubMed] [Google Scholar]
- Osei-Owusu J, Yang J, Vitery MDC, and Qiu Z (2018). Molecular Biology and Physiology of Volume-Regulated Anion Channel (VRAC). Curr Top Membr 81, 177–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perea G, and Araque A (2007). Astrocytes potentiate transmitter release at single hippocampal synapses. Science 317, 1083–1086. [DOI] [PubMed] [Google Scholar]
- Planells-Cases R, Lutter D, Guyader C, Gerhards NM, Ullrich F, Elger DA, Kucukosmanoglu A, Xu G, Voss FK, Reincke SM, et al. (2015). Subunit composition of VRAC channels determines substrate specificity and cellular resistance to Pt-based anti-cancer drugs. The EMBO journal 34, 2993–3008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu Z, Dubin AE, Mathur J, Tu B, Reddy K, Miraglia LJ, Reinhardt J, Orth AP, and Patapoutian A (2014). SWELL1, a plasma membrane protein, is an essential component of volume-regulated anion channel. Cell 157, 447–458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez-Moreno A, Sistiaga A, Lerma J, and Sanchez-Prieto J (1998). Switch from facilitation to inhibition of excitatory synaptic transmission by group I mGluR desensitization. Neuron 21, 1477–1486. [DOI] [PubMed] [Google Scholar]
- Rudkouskaya A, Chernoguz A, Haskew-Layton RE, and Mongin AA (2008). Two conventional protein kinase C isoforms, alpha and beta I, are involved in the ATP-induced activation of volume-regulated anion channel and glutamate release in cultured astrocytes. Journal of neurochemistry 105, 2260–2270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sah P, Hestrin S, and Nicoll RA (1989). Tonic activation of NMDA receptors by ambient glutamate enhances excitability of neurons. Science 246, 815–818. [DOI] [PubMed] [Google Scholar]
- Savtchouk I, and Volterra A (2018). Gliotransmission: Beyond Black-and-White. The Journal of neuroscience : the official journal of the Society for Neuroscience 38, 14–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schober AL, Wilson CS, and Mongin AA (2017). Molecular composition and heterogeneity of the LRRC8-containing swelling-activated osmolyte channels in primary rat astrocytes. The Journal of physiology 595, 6939–6951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stevens CF, and Wang Y (1995). Facilitation and depression at single central synapses. Neuron 14, 795–802. [DOI] [PubMed] [Google Scholar]
- Syeda R, Qiu Z, Dubin AE, Murthy SE, Florendo MN, Mason DE, Mathur J, Cahalan SM, Peters EC, Montal M, et al. (2016). LRRC8 Proteins Form Volume-Regulated Anion Channels that Sense Ionic Strength. Cell 164, 499–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takano T, Kang J, Jaiswal JK, Simon SM, Lin JH, Yu Y, Li Y, Yang J, Dienel G, Zielke HR, et al. (2005). Receptor-mediated glutamate release from volume sensitive channels in astrocytes. Proceedings of the National Academy of Sciences of the United States of America 102, 16466–16471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tao J, Wu H, Lin Q, Wei W, Lu XH, Cantle JP, Ao Y, Olsen RW, Yang XW, Mody I, et al. (2011). Deletion of astroglial Dicer causes non-cell-autonomous neuronal dysfunction and degeneration. The Journal of neuroscience : the official journal of the Society for Neuroscience 31, 8306–8319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaisey G, Miller AN, and Long SB (2016). Distinct regions that control ion selectivity and calcium-dependent activation in the bestrophin ion channel. Proceedings of the National Academy of Sciences of the United States of America 113, E7399–E7408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voss FK, Ullrich F, Munch J, Lazarow K, Lutter D, Mah N, Andrade-Navarro MA, von Kries JP, Stauber T, and Jentsch TJ (2014). Identification of LRRC8 heteromers as an essential component of the volume-regulated anion channel VRAC. Science 344, 634–638. [DOI] [PubMed] [Google Scholar]
- Woo DH, Han KS, Shim JW, Yoon BE, Kim E, Bae JY, Oh SJ, Hwang EM, Marmorstein AD, Bae YC, et al. (2012). TREK-1 and Best1 channels mediate fast and slow glutamate release in astrocytes upon GPCR activation. Cell 151, 25–40. [DOI] [PubMed] [Google Scholar]
- Ye ZC, Wyeth MS, Baltan-Tekkok S, and Ransom BR (2003). Functional hemichannels in astrocytes: a novel mechanism of glutamate release. The Journal of neuroscience : the official journal of the Society for Neuroscience 23, 3588–3596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Zhang H, Feustel PJ, and Kimelberg HK (2008). DCPIB, a specific inhibitor of volume regulated anion channels (VRACs), reduces infarct size in MCAo and the release of glutamate in the ischemic cortical penumbra. Experimental neurology 210, 514–520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zucker RS, and Regehr WG (2002). Short-term synaptic plasticity. Annual review of physiology 64, 355–405 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.