

Abstract
Dendritic spines in the developing mammalian neocortex are initially overproduced and then eliminated during adolescence to achieve appropriate levels of excitation in mature networks. We show here that the L1 family cell adhesion molecule Close Homolog of L1 (CHL1) and secreted repellent ligand Semaphorin 3B (Sema3B) function together to induce dendritic spine pruning in developing cortical pyramidal neurons. Loss of CHL1 in null mutant mice in both genders resulted in increased spine density and a greater proportion of immature spines on apical dendrites in the prefrontal and visual cortex. Electron microscopy showed that excitatory spine synapses with postsynaptic densities were increased in the CHL1-null cortex, and electrophysiological recording in prefrontal slices from mutant mice revealed deficiencies in excitatory synaptic transmission. Mechanistically, Sema3B protein induced elimination of spines on apical dendrites of cortical neurons cultured from wild-type but not CHL1-null embryos. Sema3B was secreted by the cortical neuron cultures, and its levels increased when cells were treated with the GABA antagonist gabazine. In vivo CHL1 was coexpressed with Sema3B in pyramidal neuron subpopulations and formed a complex with Sema3B receptor subunits Neuropilin-2 and PlexinA4. CHL1 and NrCAM, a closely related L1 adhesion molecule, localized primarily to distinct spines and promoted spine elimination to Sema3B or Sema3F, respectively. These results support a new concept in which selective spine elimination is achieved through different secreted semaphorins and L1 family adhesion molecules to sculpt functional neural circuits during postnatal maturation.
SIGNIFICANCE STATEMENT Dendritic spines in the mammalian neocortex are initially overproduced and then pruned in adolescent life through unclear mechanisms to sculpt maturing cortical circuits. Here, we show that spine and excitatory synapse density of pyramidal neurons in the developing neocortex is regulated by the L1 adhesion molecule, Close Homolog of L1 (CHL1). CHL1 mediated spine pruning in response to the secreted repellent ligand Semaphorin 3B and associated with receptor subunits Neuropilin-2 and PlexinA4. CHL1 and related L1 adhesion molecule NrCAM localized to distinct spines, and promoted spine elimination to Semaphorin 3B and -3F, respectively. These results support a new concept in which selective elimination of individual spines and nascent synapses can be achieved through the action of distinct secreted semaphorins and L1 adhesion molecules.
Keywords: autism spectrum disorders, cell adhesion molecule, close homolog of L1, dendritic spine, Semaphorin 3B, spine pruning
Introduction
The vast majority of excitatory synapses in the mammalian brain are localized to dendritic spines of cortical pyramidal neurons (Shen and Cowan, 2010). During development spines are initially overproduced, eliminated in significant numbers during the transition through adolescence, and stabilized upon maturation (Huttenlocher, 1979; McAllister, 2007; Holtmaat and Svoboda, 2009; Petanjek et al., 2011). Although much is known about spine plasticity in the adult brain, the molecular mechanisms regulating dendritic spine reorganization in the juvenile-to-adult transition remain relatively obscure. Defining these mechanisms is vital for explaining how circuits arise during normal maturation, and may also help understand the elevated spine density that has been observed in autism spectrum disorders (ASDs) (Hutsler and Zhang, 2010; Tang et al., 2014) and cognitive disabilities (Forrest et al., 2018).
The L1 family cell adhesion molecules L1, Close Homolog of L1 (CHL1), NrCAM, and Neurofascin carry out diverse functions in the developing brain, and mutations in the genetic loci encoding these proteins are associated with neurodevelopmental diseases (Sytnyk et al., 2017). L1-CAMs have well studied roles as promoters of axon growth, but they also facilitate axon repulsion in response to class 3 semaphorins (Castellani et al., 2000; Wright et al., 2007). These secreted semaphorins (Sema3A-G) trigger axon repulsion and growth cone collapse through receptors comprising Neuropilins (Npn1/2) and PlexinA signaling subunits (PlexA1-4) (Koropouli and Kolodkin, 2014). During thalamocortical axon guidance, L1-CAMs function as obligate subunits of holoreceptor complexes for Sema3A (L1, CHL1) and Sema3F (NrCAM), which enable thalamic axon subpopulations to project to distinct neocortical areas through axon repulsion in the ventral telencephalon (Wright et al., 2007; Demyanenko et al., 2011a). The ability of L1-CAMs and Sema3s to promote axon repulsion, taken together with their robust expression in the postnatal neocortex, suggested that they might also have a repellent function in cortical pyramidal neurons. Consistent with this hypothesis, Sema3F induces spine retraction in cortical pyramidal neurons (Tran et al., 2009), and NrCAM serves as a functional subunit of the Sema3F receptor complex to limit the density of spine subpopulations in the prefrontal cortex (PFC) and primary visual cortex (V1) (Tran et al., 2009; Demyanenko et al., 2014; Mohan et al., 2019).
An emerging view is that maturing neuronal networks are sculpted by selective elimination of individual spines or synapses. Such pruning could give rise to local clustering of functionally related synaptic inputs, as observed in sensory cortical areas (Wilson et al., 2016; Iacaruso et al., 2017; Scholl et al., 2017). The molecular basis for selective spine pruning is not well understood but might involve mechanisms whereby different L1-CAMs promote spine elimination in response to distinct Sema3 ligands. Among L1 family members, CHL1 was considered to be a prominent candidate as it is expressed postnatally in cortical pyramidal neurons and contributes to apical dendrite orientation and neuronal positioning at early stages (Demyanenko et al., 2004; Ye et al., 2008). Moreover, CHL1 is important for proper development of cortical networks, as CHL1-null mice display impaired cortical functions of working memory, sociability, sensory gating, and attention (Pratte et al., 2003; Irintchev et al., 2004; Morellini et al., 2007; Kolata et al., 2008; Kleene et al., 2015).
Here we investigated an unexplored function for CHL1 in dendritic spine remodeling in cortical pyramidal neurons. We found that CHL1-deficient mice exhibit increased spine density and abnormal spine morphology in PFC and V1 during adolescence and adulthood, accompanied by altered regulation of excitatory synaptic strength. Strikingly, CHL1 promoted spine elimination on apical dendrites in response to Sema3B, a secreted Semaphorin with a previously unknown role in spine remodeling. CHL1 formed a complex with Semaphorin receptor subunits Npn2 and PlexA4, and colocalized with Sema3B on spine subpopulations. Notably, spines expressing CHL1 were selectively eliminated by Sema3B, whereas spines expressing NrCAM were eliminated by Sema3F. These results support the interpretation that L1-CAMs function within holoreceptor complexes as critical determinants of selective spine pruning to class 3 semaphorins in the maturing neocortex.
Materials and Methods
Mice.
Wild-type (WT) and CHL1-null mutant mice in the C57BL/6 genetic background have been described (Montag-Sallaz et al., 2003; Demyanenko et al., 2004). CHL1 mutant mice display no obvious neuroanatomical abnormalities but have aberrant hippocampal mossy fiber and olfactory sensory axon projections (Montag-Sallaz et al., 2003), as well as thalamocortical targeting defects (Wright et al., 2007; Demyanenko et al., 2011a). NexCre-ERT2 mice (C57BL/6J) (Agarwal et al., 2012) were crossed to the Ai9 reporter line Rosa-CAG-(LoxP-Stop-LoxP-tdTomato)-WPRE (C57BL/6) (Madisen et al., 2010) and treated with tamoxifen from P10-P13 to induce recombination in postmitotic, postmigratory pyramidal neurons as described previously (Mohan et al., 2019). Mice were maintained in the University of North Carolina Animal Facility and handled according to Institutional Animal Care and Use Committee policies in accordance with National Institutes of Health guidelines. Genotyping was done by PCR.
Analysis of spine density and dendritic arborization in cortical pyramidal neurons.
Mice at postnatal day 21 (P21, early adolescence) or P60 (young adulthood) were anesthetized with isoflurane and brains were rapidly frozen in isopentane and subjected to Golgi impregnation using the FD Rapid Golgi Stain Kit (FD NeuroTechnologies). Coronal sections (100 μm) containing the cingulate cortex 1 and 2 of the medial PFC, and V1 were cut on a cryostat and mounted on gelatin-coated microscope slides. Golgi-labeled neurons were imaged under bright-field illumination using a Zeiss AxioPlan2 microscope. Spine density and arborization of pyramidal cell dendrites were quantified using Neurolucida software (MBF Bioscience) by investigators blinded to genotype. Spines were scored along the first branch of the apical dendrite 100–150 μm from the soma. For basal dendrites, spines were scored along 35–50 μm segments of 3–5 secondary branches. Mean spine number per 10 μm of dendritic length (density) was calculated from measurements.
Dendritic arborization was measured by Sholl analysis of Golgi-labeled image stacks. The center of concentric circles was defined as the center of the cell body at a soma detector sensitivity of 1.5 μm, and the automatic tracing mode was used to seed and trace dendritic arbors. Images in DAT format were subjected to Sholl analysis using Neurolucida Explorer with a starting radius of 10 μm and radius increments of 10 μm ending at 300 μm. Data were analyzed using single-factor ANOVA.
Spine morphologies were scored as thin, stubby, or mushroom types on apical dendrite segments from WT and NrCAM null littermates (P21, P60) as described previously (Peters and Kaiserman-Abramof, 1970). To compare apical and basal dendrites across genotypes in PFC (layer 2/3) and V1 (layer 4), we applied multinomial logistic regression (Agresti, 2013). The method uses a pair of logistic regression models, because the response comprises three spine types: mushroom, stubby, and thin. Because thin spines constituted the smallest number of spine types in all regions and genotypes, we formulated the two models as log-odds of the outcome to be mushroom instead of thin, and log-odds of the outcome to be stubby instead of thin. In each logistic model, the main independent variable was genotype (WT or CHL1-null) and was coded as a binary indicator variable. To compare across genotypes in each region, for apical and basal dendrites, we employed the 'multinom' function from R 'nnet' package to estimate the multinomial logistic regression models. p-values were computed and shown as extended data in Fig. 2-1 and Fig. 2-2. Spine length and width measurements were made using Neurolucida as described previously (Sala et al., 2001; Hodges et al., 2014).
Electron microscopy.
WT and CHL1-null littermates (P21) were anesthetized and perfused transcardially with phosphate buffer (0.15 m sodium phosphate, pH 7.4) followed by 2% PFA and 2.5% glutaraldehyde in phosphate buffer. Brains were stored in fixative at 4°C for 2 d. Coronal vibratome sections (300 μm) were postfixed in 1% osmium tetroxide with 1.25% potassium ferrocyanide in phosphate buffer for 45 min, dehydrated in series of ethanol, and embedded in PolyBed 812 epoxy resin. Semithin sections (1 μm) were cut, stained with toluidine blue and used for orientation. Ultrathin (70 nm) sections of cortex were cut using a Leica Ultracut UCT microtome and mounted on 200 mesh copper grids. Ultrathin sections were contrasted with uranyl acetate and lead citrate and imaged on a JEOL JEM 1230 transmission electron microscope (JEOL USA) equipped with a Gatan Orius SC1000 CCD Digital Camera and Gatan Microscopy Suite version 3.0 software. The same magnification and size of electron micrograph were used to compare synaptic profiles in WT and CHL1 mutant PFC layer 2/3. Excitatory synaptic profiles on spines were identified by the presence of asymmetric postsynaptic densities (PSDs) aligned with a presynaptic terminal membrane containing clustered synaptic vesicles. In each micrograph, numerous spines were observed in cross-section, and those with asymmetric synaptic profiles were classified as containing one or multiple PSDs. In each micrograph, the numbers of synaptic profiles with PSDs and PSD lengths were counted on all spines. The number of PSDs on spines within a unit cross-sectional area of cortex was calculated from the magnification and image size, and expressed as mean number of PSDs per 100 μm2 cortical area ± SEM.
Whole-cell patch-clamp recording.
WT and CHL1-null mutant mice (P50–P60) were anesthetized with ketamine/xylazine (100/10 mg/kg) and decapitated. Coronal slices (350 μm) containing the anterior cingulate cortex portion of the PFC were prepared in ice-cold, oxygenated low-sodium artificial CSF containing the following (in mm): 135 N-methyl-D-glucamine, 20 choline chloride, 20 NaHCO3, 2.2 KCl, 0.5 CaCl2, 1.5 MgSO4, 1.2 NaH2PO4, and 10 D-glucose, pH 7.4, gassed with 95%O2/5% CO2. The slices were then incubated in extracellular artificial CSF (ACSF) recording solution containing the following (in mm): 125 NaCl, 2.5 KCl, 2 CaCl2, 1.3 MgSO4 20 D-glucose, 1.25 NaH2PO4, and 26 NaHCO3, pH 7.4, gassed with 95% O2/5% CO2 for 30 min at 34°C, followed by incubation for at least 30 min at room temperature. For recording, slices were transferred to a heated glass-bottom chamber on a Zeiss FS-2 microscope and viewed using oblique illumination. The slice was superfused with ACSF at ∼2 ml/min. All recordings were made at 34°C. mEPSCs were recorded from pyramidal cells in layer 2/3, in whole-cell voltage clamp with a recording pipette (KG-33, King Glass) containing the following (in mm): 130 CsMetSO3, 5 CsCl, 5 EGTA, 10 HEPES, 4 Mg2ATP, 0.3 Tris-GTP, 10 Tris-phosphocreatine, and 3 QX-314-Cl, pH adjusted to 7.2 with CsOH. Lucifer yellow (Invitrogen) was added to the internal solution at ∼0.5% to provide morphological identification of all recorded cells, and only results from pyramidal neurons were analyzed. During recording, the extracellular solution was supplemented with 1 μm tetrodotoxin (TTX) to block action potentials and 10 μm SR95531 to block GABAergic transmission. No series resistance compensation was used. All chemicals were obtained from Sigma-Aldrich, except SR95531 (Tocris Bioscience). Recordings were made with an AxoClamp 700A Amplifier (Molecular Devices) digitized with a National Instruments NI-6251 16-bit A-D card at 400 kHz and down-sampled to 10 kHz to reduce high-frequency noise. Data acquisition was controlled by ACQ4 (Campagnola et al., 2014), available at www.acq4.org.
The data for miniature synaptic currents were curated by excluding traces (10 s in duration) that exhibited large fluctuations, excess noise, or high-frequency bursts of events. Any linear trend in the baseline was subtracted, and the data then low-pass filtered at 2.8 kHz with a 4-pole Butterworth filter, subject to a bank of notch filters (Q = 20) at 60 Hz and any harmonics with significant energy up to 1.5 kHz, and an additional notch filter (Q = 20) at 4.0 kHz to remove a residual signal originating from the amplifier. Events were detected using a convolution-based algorithm (Pernía-Andrade et al., 2012). All cells were analyzed using a uniform threshold (2.75 * trace SD), and with a template defined by the following equation:
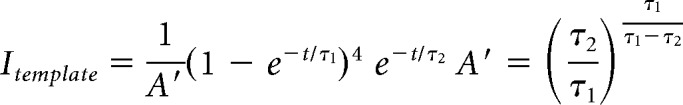 |
Where t is time, τ1 = 0.35 ms, and τ2 = 4.0 ms. This template has a sigmoidal rise and a single exponential decay, which more closely matches the shape of the recorded events than a simple exponential rise. For each cell, an average event was measured based on all of the events detected from that cell, and the cell was considered the experimental unit. Analysis was repeated using the template-based algorithm (using the same template) of Clements and Bekkers (1997) with similar results. All analysis of electrophysiology data was performed with programs written in Python (2.7.14; 3.6.5) and Cython (0.25.2).
Evoked EPSCs were measured in six female mice (three of each genotype), ages P66–P78. The extracellular solution was the same as for the mEPSC analysis, except that TTX was omitted. Likewise, the intracellular solution was the same as used for the mESPCs, except that Lucifer yellow was replaced by tetramethylrhodamine-biocytin (Thermo Fisher Scientific T12921, <0.5% by weight) for cell identification. A 25 μm inner diameter, 125 μm outer diameter concentric Pt/Ir, and stainless-steel electrode (CBABC75; Fredrick Haer) was used to stimulate locally in layer 3 between 150 and 300 μm dorsal or ventral to the recorded cell in the anterior cingulate cortex. Measurements were made in voltage-clamp, holding at −60 mV (−68 mV after correction for an estimated −8 mV junction potential) with the CsMetSO3-based intracellular electrode solution.
Measurements of the peak EPSC amplitude for input-output functions and paired-pulse facilitation were made after subtraction of a linear baseline to minimize the influence of the stimulus artifact on the measurements. The subtraction line was manually set to start just before the start of the EPSC, and ended 15–25 ms later, after the EPSC had decayed back to the holding current. All measures were made on an average of four repeats of the protocol for each cell. For the stimulus input-output functions, the mean uncompensated electrode series resistance (Rs) was 17.4 MΩ (SD = 6.29, n = 21 cells) and the average Rs compensation was 69.0% (SD = 2.7). For the paired-pulse measurements, the mean Rs was 17.2 MΩ (SD = 6.1, n = 36 cells), and the average Rs compensation was 68.2% (SD = 3.9).
To measure the NMDA:AMPA ratio, EPSCs were recorded while the membrane potential was stepped to different levels between −108 and +42 mV in 10 mV steps. The steps were 0.8 s in duration, presented once every 3 s and the EPSC was evoked 400 ms after the voltage step started. EPSC amplitudes were measured after subtracting the baseline current before stimulus, to remove the effects of residual outward currents through K+ channels at positive potentials. AMPA currents were measured at ∼−78 mV (corrected for the junction potential), close to the calculated Cl− reversal potential of −74.1 mV to minimize potential interference by GABAergic synaptic conductances. Recordings with unclamped slow inward currents (presumed calcium currents) were discarded. NMDA currents were measured 50 ms (averaging over a 0.5 ms window) following the stimulus, after AMPA (and any GABAergic) channels were closed, at +42 mV. All measures were made on an average of four repeats of the voltage protocol for each cell. The average uncompensated Rs for these measurements was 18.6 MΩ (SD = 6.6, n = 19 cells), and was corrected by 65.5% (SD = 11.0). For the evoked EPSC measures, all data acquisition and analyses were performed blinded to genotype.
Semaphorin-induced spine retraction in cortical neuronal cultures.
Neuronal cultures were prepared from brains of WT or CHL1-null mutant embryos (E15.5) as described previously (Demyanenko et al., 2014). Cells were cultured on chamber slides coated with laminin and poly-D-lysine and transfected at 11 d in vitro (DIV11) with pCAGG-IRES-mEGFP and indicated plasmids. At DIV14, the medium was changed and cultures were treated with recombinant Fc fusion proteins (5 nm) of Sema3A, Sema3B, Sema3C, Sema3D, and Sema3F; R&D Systems) and ephrinA5 (R&D Systems) or control Fc for 30 min before fixation in 4% paraformaldehyde (PFA). Cultures were permeablized with 0.3% Triton X-100, blocked in 10% serum, and labeled with anti-GFP (Abcam catalog #13970, RRID:AB_300798) and secondary Alexa Fluor 488 antibodies (Life Technologies catalog #A11039, RRID:AB_142924). Spine densities were measured from confocal images of at least 10 GFP-labeled neurons in each of four replicate cultures. Mean spine densities ± SEM were compared by the t test (two-tailed, unequal variances, with significance set at *p < 0.05).
For assaying Sema3B secretion after activity stimulation, neuronal cultures were treated on DIV12 with or without 20 μm gabazine without media change, and after 48 h conditioned medium was collected with the following additives: 200 μm sodium orthovanadate,10 mm NaF, and Protease Inhibitor Cocktail (Sigma-Aldrich). After concentration by Amicon Ultra 10K filtration, samples were diluted 1:1 in 2× RIPA with additives and Sema3B was immunoprecipitated and subjected to immunoblotting as described below. Calcium imaging of neuronal cultures using Oregon Green 488 BAPTA-1 confirmed that addition of 20 μm gabazine stimulated neuronal firing (data not shown). Cell lysates were prepared in RIPA with additives, and equal amounts of protein were immunoblotted for Sema3B directly.
Immunofluorescence localization.
Mice were anesthetized, perfused transcardially with 4% paraformaldehyde, and processed for staining as described previously (Demyanenko et al., 1999). Brains were removed and postfixed in 4% paraformaldehyde overnight at 4°C, and coronally sectioned using a Vibratome (100 μm). Sections were permeabilized in 0.3% Triton X-100 and blocked in 10% normal donkey serum in PBS for 1 h, and subjected to immunofluorescence staining for CHL1, Sema3B, Npn2, or NrCAM. Mouse cortical neuronal cultures transfected with pCAGG-IRES-mEGFP were fixed at DIV14 for immunofluorescence staining as described previously (Demyanenko et al., 2014). Sections or cultures were incubated overnight with goat polyclonal anti-CHL1 (R&D Systems catalog #AF2147, RRID:AB_2079332), rabbit polyclonal anti-NrCAM (Abcam catalog #24344, RRID:AB_448024), rabbit polyclonal anti-Sema3B (Abcam catalog #48197, RRID:AB_2185420), or chicken anti-GFP (Abcam catalog #13970, RRID:AB_300798). After washing, samples were incubated with anti-chicken Alexa Fluor 488 (Life Technologies catalog #A11039, RRID:AB_142924), anti-goat Alexa Fluor 647 (Life Technologies catalog #A21447, RRID:AB_2535864) or anti-rabbit Alexa Fluor 555 (Life Technologies catalog #A31572, RRID:AB_162543) (1:500) for 2 h before mounting and confocal imaging. The specificity of the CHL1 antibody, directed against the entire extracellular region of mouse CHL1, was confirmed previously (Demyanenko et al., 2004, 2010; Nikonenko et al., 2006; Wright et al., 2007). The antibody does not react with other L1-CAMs in mouse brain or transfected HEK293 cells and does not bind nonspecifically to CHL1-null mutant mouse brain (Nikonenko et al., 2006). The NrCAM antibody, also directed against the extracellular region, does not cross-react with other L1-CAMs, as previously characterized (Demyanenko et al., 2011b, 2014; Mohan et al., 2019). The Sema3B antibody was generated against a synthetic peptide (human Sema3B residues 100–200 in the Sema domain) and reacts specifically with mouse or human Sema3B, not with Sema3F (Nguyen et al., 2011).
Dendritic z-stacks were obtained by imaging on a confocal LSM700 microscope. Images were deconvolved using Autoquant version 3 software (Media Cybernetics) with default blind deconvolution settings. In Imaris (Bitplane) software, all images were subjected to same threshold settings. A mask was created for the green labeling, and the red and/or blue pixels outside the green were excluded from the analysis.
Immunoblotting and immunoprecipitation.
Mouse cortex (P7, P14, P21, and P80) was homogenized in Brij 98 lysis buffer (1% Brij 98, 10 mm Tris-Cl pH 7.0, 150 mm NaCl, 1 mm EDTA, 1 mm EGTA, 200 μm Na3VO4, 10 mm NaF, 1X protease inhibitors (Sigma-Aldrich). Lysates (50 μg) were subjected to SDS-PAGE and transferred to nitrocellulose. Filters were blocked in TBST containing 5% nonfat dried milk and incubated overnight in the following antibodies: anti-CHL1 (R&D Systems catalog #AF2147, RRID:AB_2079332, 1:500), anti-Sema3B (Abcam catalog #48197, RRID:AB_2185420, 1:500), or anti-actin (Millipore catalog #MAB1501, RRID:AB_2223041, 1:1000). Blots were washed and incubated in HRP-conjugated secondary antibodies (1:5000) for 1 h before being developed using Western Bright ECL Substrate (Advansta) and exposed to film for various times to obtain a linear response density. Bands were quantified by densitometric scanning using ImageJ. Synaptoneurosomes were prepared from P28 mouse brain as described previously (Villasana et al., 2006). Protein complexes were immunoprecipitated using goat anti-CHL1 antibodies and protein A/G Sepharose, separated on SDS PAGE gels, and immunoblotted with antibodies against Npn2 (CST catalog #3366s), Npn1 (Abcam catalog #ab25998, RRID:AB_448950), PlexA1 (ECM catalog #PM5351), PlexA2 (CST catalog #3994s), PlexA3 (Abcam catalog #ab41564, RRID:AB_944889), or PlexA4 (CST catalog #3816s) using Western Bright ECL Substrate. For assessing the interaction between Npn2 and PlexA4 in absence of CHL1, lysates were prepared from P60 cortex of WT and CHL1-null mice. Npn2 was immunoprecipitated from equal amounts of protein (500 μg) with Npn2 antibodies (R&D Systems catalog #AF567, RRID:AB_2155253) and immunoblotted for PlexA4 (CST catalog #3816s).
For assessing Sema3B binding to CHL1, Sema3B-Fc or Fc (10 nm) was incubated with HEK293T cells transfected with CHL1 for 30 min at 37C. Cells were washed extensively in OptiMEM, lysed in Brij 98 lysis buffer, and any Fc-proteins pulled down with Protein A/G-Agarose beads. Pull-downs were run by SDS-PAGE, and subjected to Western blotting with CHL1 antibodies.
Experimental design and statistical analysis.
All experiments were designed as described previously (Dupont and Plummer, 1990) to provide sufficient power (80–90%) to discriminate significant differences (α = 0.05) in means (± SEM) between independent controls and experimental subjects with unpaired t tests using equivalent samples sizes calculated with PS software (http://biostat.mc.vanderbilt.edu/PowerSampleSize). The type I error probability associated with these tests of the null hypothesis was set at 0.05. Student's t test (2- tailed, unequal variances) was used to compute p-values in experiments where only two groups were compared. For Sholl analysis, single-factor ANOVA was used to compare genotypes. To compare spine morphologies across genotypes in PFC (layer 2/3) and V1 (layer 4), we applied multinomial logistic regression analysis for computing the log-odds as described previously (Agresti, 2013). We used the 'multinom' function from the R 'nnet' package to estimate the multinomial logistic regression models and we report the resulting p-values in Fig. 2-1. For electrophysiology experiments, statistical analysis was performed with Python scripts using statsmodels (www.statsmodels.org) and scipy.stats (version 1.01; www.scipy.org), and in R (version 3.5.1, for Anderson-Darling tests and linear mixed effects models). Full details of the experimental design, sample sizes, and number of repetitions for each experiment are reported in the Results and in the figure legends.
Results
Increased spine density on apical dendrites of cortical pyramidal neurons in CHL1-null mice
To investigate a potential role for CHL1 in dendritic spine regulation, we focused on pyramidal neurons in layer 2/3 of the PFC due to the importance of this neocortical area in social and cognitive behaviors (Yizhar, 2012) and star pyramidal cells in layer 4 of the visual cortex (V1), where spine pruning is especially robust (Bian et al., 2015). Pyramidal neurons of WT and CHL1 homozygous null mutant mice (KO, knock-out) were analyzed during the peak stage of spine remodeling in early adolescence (P21) and after synaptic stabilization in young adults (P60) by Golgi staining. In layer 2/3 of the medial PFC, spine density on apical dendrites of pyramidal neurons was increased at P21 in CHL1 KO mice compared with WT (Fig. 1A,C). In contrast, spine density on basal dendrites of CHL1 KO pyramidal neurons did not differ from WT (Fig. 1B,C). In layer 4 of V1, star pyramidal neurons also displayed elevated spine density on apical dendrites in CHL1 KO mice, whereas spine density on basal dendrites was unaffected by CHL1 loss (Fig. 1D–F). To determine whether increased spine density on apical dendrites of CHL1 KO neurons persisted into adulthood, spine density was also quantified in WT and CHL1 mutant mice at P60. Apical dendrites of Golgi-labeled pyramidal neurons in both PFC and V1 continued to display elevated spine density in adult CHL1 KO mutant mice, and spine density on basal dendrites was unaffected in both areas (Fig. 1G–L). These results indicated that CHL1 loss resulted in increased spine density on apical rather than basal dendrites of pyramidal neurons during adolescence in both PFC and V1, and this phenotype persisted into adulthood.
Figure 1.

Loss of CHL1 increases spine density on apical dendrites of cortical pyramidal neurons in early adolescence and adulthood. A–F, Representative images of apical and basal dendrites from Golgi-labeled pyramidal neurons and quantification of spine density in PFC layer 2/3 and V1, layer 4 of adolescent WT and CHL1-null (KO) mice at P21. Mean spine density per neuron (±SEM) was significantly increased on apical but not basal dendrites of pyramidal neurons in CHL1-null mice compared with WT at P21 (*p = 1.70E-07 (PFC), 0.0377 (V1); two-tailed t test). Number of mice: 3 per genotype. Number of neurons scored: WT, n = 11 (apical), n = 10 (basal); CHL1-null, n = 10 (apical), n = 10 (basal). Scale bar, 10 μm. The total number of spines counted in each condition ranged from 280 to 563. G–L, Representative images of apical and basal dendrites from Golgi-labeled pyramidal neurons and quantification of spine density in PFC layer 2/3 and V1 layer 4 of adult WT and CHL1 KO mice at P60. Mean spine density per neuron (±SEM) was significantly increased on apical but not basal dendrites of pyramidal neurons in CHL1-null mice compared with WT at P60 (*p = 0.017 (PFC), 0.0176 (V1); two-tailed t test). Number of mice: 3 per genotype. Number of neurons scored: WT, n = 10 (apical), n = 10 (basal); CHL1-null, n = 10 (apical), n = 10 (basal). Scale bar, 10 μm. The total number of spines counted in each condition ranged from 450 to 863.
Dendritic protrusions acquire different morphologies and can be classified as mushroom, stubby, and thin or filopodial spines based on the relative size of the spine head and neck (Peters and Kaiserman-Abramof, 1970). The different types of spines are dynamically interchangeable, and comprise a continuum from immature thin spines, which can have synaptic structure and function, to mushroom spines with a mature synaptic contact (Bhatt et al., 2009; Holtmaat and Svoboda, 2009; Berry and Nedivi, 2017). In neurodevelopmental disorders such as Fragile X syndrome there is a relative increase in spines with the immature, thin morphology (Phillips and Pozzo-Miller, 2015). We quantified the percentage of spines with mushroom, stubby, and thin morphology on apical and basal dendrites in Golgi-labeled pyramidal neurons in PFC (layer 2/3) and V1 (layer 4) in adult WT and CHL1-null mice at P60 (Fig. 2A–C). In both cortical regions of CHL1-null mice, the percentage of thin spines on apical dendrites increased, whereas stubby and mushroom spines decreased compared with WT (Fig. 2). In contrast, spine morphologies on basal dendrites had a similar distribution in both genotypes. Multinomial logistic regression was used to compare the percentage of spine types between genotypes. p-values for testing the effect of genotype on spine morphology (Fig. 2-1) showed that for apical dendrites in the PFC or V1, there was a significant difference for spines to be mushroom instead of thin or stubby (Fig. 2C; *p < 0.001). There was not a significant difference in spine morphology on basal dendrites in either region between genotypes. Measurements of spine length and width showed that mean spine length in CHL1-null PFC (1.41 ± 0.04 μm; 235 spines) was significantly greater than WT (1.12 ± 0.03 μm; 219 spines; p = 9.6 e-09). In the CHL1-null V1, mean spine length (1.33 ± 0.01 μm; 203 spines) was also significantly greater than WT (1.20 ± 0.03 μm; p = 9.6 E-09; 216 spines). Spine widths were not significantly different between genotypes for either PFC or V1 (data not shown). Analysis of spine morphology in PFC (layer 2/3) and V1 (layer 4) of mice at P21 showed no significant difference for spines to be mushroom instead of thin or stubby in both PFC and V1 on apical or basal dendrites (Fig. 2-2). These results indicated that loss of CHL1 increases the proportion of immature, thin spines on apical dendrites in the adult PFC (layer 2/3) and V1 (layer 4).
Figure 2.

CHL1 regulates spine morphology on apical dendrites but has no effect on dendritic branching. A, B, Representative Golgi-labeled apical and basal dendrites in PFC and V1 of adult WT and CHL1 KO mice at P60. Arrows indicate spines with thin morphology. Scale bar, 10 μm. C, Percentage of spines of different morphological types (mushroom, stubby, thin) on apical and basal dendrites of Golgi-labeled pyramidal neurons in PFC and V1 at P60. Multinomial regression analysis tested the effect of genotype on spine morphology and yielded p-values that indicated a significant difference (*) in log-odds for spines to be mushroom instead of thin or stubby for apical but not basal dendrites of CHL1 KO mice in both regions, which was seen by an increase in the proportion of thin spines at the expense of stubby and mushroom spines (Figure 2-1; *p < 0.001). WT mice (3 mice, n = 20 neurons, 886 spines) and CHL1 KO mice (3 mice, n = 27 neurons, 1105 spines). D–G, Sholl analysis for dendrite arborization of pyramidal neurons in PFC and V1 of WT and CHL1-null mice at P60. Representative images are shown for Golgi-labeled pyramidal neurons in PFC (layer 2/3) and V1 (layer 4) of WT and CHL1-null mice at P60. Sholl analysis indicated no significant differences (N.S., not significant) (single-factor ANOVA) in branching of apical dendritic arbors at indicated distances (means ±SEM) within a 10–300 μm radius from the pyramidal cell body of WT (3 mice, n = 10 neurons) and CHL1-null mice (3 mice, n = 10 neurons) in (E) PFC L2/3 (p = 0.68) or (G) V1 L4 (p = 0.14). Scale bar, 100 μm. Figure 2-2 presents spine analysis at P21.
Multinomial logistic regression fits for spine morphology. Download Figure 2-1, DOCX file (14.7KB, docx)
Multinomial logistic regression fits for spine morphology at P21. Download Figure 2-2, DOCX file (14.9KB, docx)
To investigate whether loss of CHL1 affected arborization (branching) of apical dendrites, Sholl analysis was performed on the dendritic tree of Golgi-labeled pyramidal neurons in adult mice (P60). Dendritic arborization of CHL1 KO pyramidal neurons was not significantly different from WT in PFC (layer 2/3) or V1 (layer 4) despite a slight downward trend (Fig. 2D–G; single factor ANOVA). Sholl analysis of CHL1 KO and WT pyramidal neurons at P21 also indicated no significant differences (ANOVA) for PFC (layer 2/3; p = 0.27) or V1 (layer 4; p = 0.83). Apical dendrites of these neurons projected toward the apical surface and were not misoriented. Misoriented orientation of apical dendrites was not observed in these cortical regions, which was different from a subpopulation of layer 5 pyramidal cells in V1 that were displaced to layer 6 and had misoriented apical dendrites at P21–P28 (Demyanenko et al., 2004).
CHL1 loss increases excitatory synapse number and alters synaptic transmission
To determine if increased dendritic spine density in the CHL1-null cortex was associated with elevated numbers of excitatory synapses, ultrastructural examination by electron microscopy (EM) was performed in PFC (layer 2/3) and V1 (layer 4) in adult CHL1-null and WT mice (P80). In electron micrographs excitatory synapses were identified as asymmetric synaptic profiles with a PSD in close apposition to a presynaptic terminal containing synaptic vesicles (Fig. 3A–D). Asymmetric synaptic profiles with PSDs were scored in unit cross-sectional areas corresponding to 30 μm2 of cortex. The mean number of asymmetric synapses per 100 μm2 was calculated and found to be significantly increased in both PFC and V1 of CHL1-null mice (Fig. 3E). This analysis indicated that the elevated spine density and proportion of immature, thin spines on pyramidal neurons in the CHL1-null cortex were accompanied by an increase in excitatory spine synapses.
Figure 3.

Increased density of excitatory spine synapses in PFC and V1 of CHL1-null mice. A–D, Electron micrographs showing PSDs (arrowheads) opposed to presynaptic terminals containing synaptic vesicles, which characterize excitatory synapses in PFC, layer 2/3 and V1, layer 4 of WT and CHL1 KO mice at P60. Scale bar, 1 μm. E, Mean number of excitatory synaptic profiles with PSDs within a unit area (100 μm2; ±SEM) in PFC and V1 was increased in CHL1 KO compared with WT mice (two-tailed t test, *p = 0.022 (PFC), 0.002 (V1); n = 3 mice/genotype). The number of micrographs analyzed was as follows: for WT PFC, n = 29, WT V1, n = 51; CHL1-null PFC, n = 28; CHL1-null V1, n = 51.The total number of asymmetric synapses per genotype scored in each region was 118–211.
The structural results suggested that synaptic number and distribution in cortical pyramidal cells were altered in CHL1-null mice. To test whether this resulted in a functional difference in synaptic transmission, we recorded mEPSCs in the presence of TTX. The grand average mean currents for CHL1-null and WT mice are shown in Figure 4A. The frequency of detected events (Fig. 4B, mean intervals: CHL1-null: 124.9 (SD = 41.0), n = 10; WT: 115.6 (SD = 37.7), n = 10, t(17.9) = 0.500, p = 0.623) and mean amplitude of events (Fig. 4C, CHL1-null: U = 8.7 (SD = 2.9), n = 10; WT: U = 8.5 (SD = 1.7), n = 10, t(14.7) = 0.212, p = 0.835) were not different between genotypes. Rise-decay time constants of events are normally correlated because of the effects of electrotonic filtering (Rall et al., 1967). The same filtering is expected in voltage-clamp recordings in part due to imperfect control of the voltage at the synaptic site (Williams and Mitchell, 2008), as observed in hippocampal neurons (McBain and Dingledine, 1992). We found that the average event rise and decay times were correlated across WT cells (Fig. 4D; r2 = 0.574) but poorly correlated in cells from CHL1-null mice (r2 = 0.008). Similarly, there should be an inverse relationship between rise-time and amplitude. This was observed in the WT cells (Fig. 4E; r2 = 0.250) but the correlation was weak in CHL1-null cells (r2 = 0.055). We did not observe clear correlations for events in individual cells, likely because it was difficult to measure time constants accurately with the small-sized mEPSCs. As the effects of dendritic filtering on these correlations depend on the assumptions that all events have the same conductance time course and average amplitudes at their site of origin, it seems likely that at least one of these assumptions does not hold for excitatory synapses from CHL1-null mice.
Figure 4.

mEPSC measures in PFC layer 2/3 of CHL1-null and WT mice. A, Grand mean mEPSC waveforms for each genotype. The shaded areas represent 1 SD. mEPSCs have similar amplitudes and average shapes in each group (n = 10 cells from WT mice, 10 cells from CHL1-null mice). B–G, In these plots symbols show measures for individual cells, the box plots show the mean and 25–75 percentile points, and the box plot whiskers show the 5–95 percentiles of the distribution. Points with gray diamonds are marked as potential outliers that are >1.5× the interquartile range, but were not excluded from analysis. B, Event frequencies are not different between CHL1-null and WT mice. C, Mean event amplitudes are not different between the two genotypes. D, Plot of decay to 37% of peak amplitude against 10–90% rise time for the mean events between cells. There is a positive correlation between rise time and decay time in cells from WT mice; however, CHL1-null mice did not show a strong correlation. Shaded areas correspond to 1 SD (68% confidence interval) for the regression lines for the WT data only. E, Plot of amplitude against 10–90% rise time. In WT cells the amplitudes and rise times show the expected inverse relationship (dashed line and shaded area). However in CHL1-null cells there was not a strong correlation between these measures. Shaded areas are as in D. F, There is no difference in the 10–90% rise times between the two genotypes. G, Although there was no difference in the mean total event charge between genotypes, the variability of the event charge distribution was significantly larger in CHL1-null mice (Levene's test; p = 0.017). H, Measures of the skew of the mEPSC event amplitude distribution in WT mice. H1 and H2 show amplitude distributions for all events from two cells (legend indicates mouse number and recorded cell by letter, along with the measured skew). H3 summarizes the skew values for all WT cells; all values fall between 1.5 and 3.5. I, Measures of the skew of the mEPSC event amplitude distribution in CHL1 KO mice, in the same format as H1–H3. I3 shows that two of the CHL1 KO cells had a larger skew values than the rest of the cells from either WT or other CHL1 KO cells. J, Cumulative mEPSC amplitude distributions for cells from WT and CHL1 KO mice. Inset: Cumulative distribution of mEPSC amplitudes for all events from all cells of each genotype.
There were no differences in 10–90% rise times between the WT and CHL1-null mice (Fig. 4F, CHL1-null: 0.485 ms (SD = 0.139), n = 10; WT: 0.484 ms (SD = 0.066), n = 10; t(12.9) = 0.005, p = 0.996) or holding current needed to keep the cells at −72 mV (data not shown; mean CHL1-null: 182 pA (SD = 105), n = 10 WT: U = −149 pA (SD = 54), n = 10, t(13.5) = −0.834, p = 0.419). The total charge, which reflects changes in amplitude and time course of events, also did not differ (Fig. 4G, mean CHL1-null: 16.9 pA*ms (SD = 6.1), n = 10; WT: 16.1 (SD = 2.0), n = 10, t(10.9) = 0.364, p = 0.723). However, the variance of the charge measures differed significantly between two genotypes (Levene's test, W = 6.86, p = 0.017), suggesting altered regulation of excitatory synaptic strength in cells from CHL1-null mice.
Because of differences in variance and correlation between rise and decay times, we looked more closely at the event amplitude distribution for individual cells. Example distributions are shown for WT cells in Figure 4, H1 and H2, and for CHL1-null cells in Figure 4, I1 and I2. These distributions are plotted normalized against their median amplitude. The event amplitude distributions are not Gaussian, but have a tail toward larger amplitudes. To quantify this, we measured the skew of the amplitude distributions for each cell. These are summarized in Figure 4, H3 and I3. The skew values for all WT cells were positive and clustered between 1.5 and 3.5; however, two of the cells from CHL1 null mice had a larger positive skew (Fig. 4I3). As an exploratory analysis, we computed the Anderson-Darling k-sample statistic on the skew distributions (Fig. 4H3,I3), but the found that the distributions were not significantly different (d = 0.042, p = 0.33). We then computed the cumulative amplitude distributions across all events from each cell for each genotype (Fig. 4J). The Anderson–Darling k-sample test on the average amplitude on a per-cell basis indicated no significant difference (d = 0.052, p = 0.34) between the genotypes. The inset in Figure 4J shows the cumulative distribution of all event amplitudes pooled across all cells for each genotype (WT: n = 32,611 events; CHL1-null: n = 25,439 events). An Anderson–Darling k-sample on this pooled data showed a significant difference by genotype (d = 74.94, p < 10−6).
To further investigate how synaptic transmission might be altered in the CHL1-null mice, we examined evoked EPSCs in layer 2/3 pyramidal cells. Stimulus strength versus EPSC amplitude curves were not different between WT and CHL1-null cells (maximal EPSC at 300 μA: WT 1.44 nA (SD 1.17, n = 12 cells from 3 mice); CHL1-null 1.03 nA (SD 0.55, n = 9 cells from 3 mice), Welch's t test t = 1.247, p = 0.23), and the curves were not significantly different (linear mixed model, with genotype and current as factors; t = 1.022, p = 0.322). Paired-pulse facilitation was measured at 20, 50, 150, and 200 ms intervals (Fig. 5A,B). WT mice showed a modest average facilitation over all intervals. CHL1-null mice showed less facilitation, and sometimes slight depression, across all intervals. A linear mixed effects model, with genotype and interval as factors, revealed a significant difference between genotypes (t = −3.086, p = 0.0029), as well as an effect of interval (t = −7.38, p < 1e-6). Because the loss of CHL1 increased the proportion of thin, immature spines on apical dendrites of layer 2/3 cells, we also measured the NMDA to AMPA current ratios at −78 (to isolate the AMPA receptor mediated currents) and +42 mV (to activate the NMDA receptors) (Fig. 5C,D). The NMDA to AMPA ratios spanned a broad range (Fig. 5D) and there was no difference between the genotypes.
Figure 5.

Decreased paired-pulse facilitation in CHL1-null mice compared with WT mice, but no change in NMDA to AMPA ratio. A, Example traces for EPSCs at different intervals for pyramidal cells WT and CHL1-null. The first EPSC is marked with “P1”, and is plotted for each subsequent interval (20, 50, 200, 150, and 200 ms). B, Summary of paired-pulse ratio for all cells (n = 18 WT; n = 18 CHL1-null cells). There is a significant effect of genotype (p = 0.0029) in the paired-pulse ratio; the ratio is depressed for all intervals in the CHL1-null cells. C, Example traces for EPSCs at +42 and −78 mV to measure NMDA to AMPA ratios for WT and CHL1-null. Vertical markers indicate the times where the measurements of the AMPA (gray marker) and NMDA (black marker) are made. D, There is no difference in the NMDA to AMPA ratio between the two genotypes (Mann–Whitney U; t = 33.0, p = 0.174, WT: n = 9, CHL1-null: n = 10). ns, not significant.
In summary, differences in distributions for charge and amplitude skew, along with breakdown of the expected relationship between rise time and amplitude and the decrease in paired-pulse facilitation, indicate that the regulation of excitatory synaptic strength is disrupted in CHL1-null mice
Regulation of Sema3B-induced spine retraction by CHL1
CHL1 might mediate pruning of immature spines and synapses from apical dendrites during postnatal stages of maturation in response to secreted semaphorins. However, it was not known whether any class 3 semaphorin other than Sema3F had the capacity to elicit spine retraction in cortical pyramidal neurons. Several class 3 semaphorins were expressed postnatally in the mouse neocortex, as ascertained from in situ hybridization data in the Allen Mouse Brain Atlas: Sema3A, Sema3B, Sema3C, Sema3D, Sema3E, and Sema3F. We assessed the ability of purified Sema3-Fc fusion proteins to promote dendritic spine retraction in long-term cultures of primary cortical neurons from WT mice. Cortical neuron cultures from E15.5 mouse embryos were transfected on DIV11 with pCAGG-IRES-mEGFP and then treated at DIV14 with Fc-fusion proteins (5 nm) for 30 min, as described previously (Demyanenko et al., 2014). After fixation and immunostaining for EGFP, spine density was quantified on apical dendrites of neurons with pyramidal morphology in confocal z-stacks using Neurolucida software (Tran et al., 2009; Demyanenko et al., 2014; Mohan et al., 2019). Sema3B-Fc was found to induce a 40% decrease in spine density on apical dendrites compared with control Fc protein, whereas Sema3F-Fc caused an 81% decrease (Fig. 6A–C). Substantial spine density remained after Sema3B treatment, suggesting that a specific spine subpopulation is responsive to this ligand. In contrast Sema3A, Sema3C, Sema3D, and Sema3E fusion proteins had no effect on apical dendrite spine density. None of the Sema3-Fc proteins altered basal spine density. Because CHL1 can associate with the EphrinA5 receptor, EphA7 (Demyanenko et al., 2011a), we also assessed the ability of EphrinA5-Fc to induce spine retraction but found that it did not alter spine density (Fig. 6A). These findings revealed a novel role for Sema3B as a promoter of spine elimination in cortical neuron cultures.
Figure 6.

Sema3B promotes spine retraction on apical dendrites of cortical pyramidal neurons in culture from WT but not CHL1-null mice. A, Histogram depicts quantification of spine density (mean ±SEM) on apical dendrites of WT neurons (DIV14) treated with Fc, Sema3A-Fc, Sema3B-Fc, Sema3C-Fc, Sema3DFc, Sema3E-Fc, Sema3F-Fc, and EphrinA5 (5 nm, 30 min). Treatment with Sema3B-Fc and Sema3F-Fc led to significant decreases in spine density (*p = 8.64E-05 (Sema3B), 1.67E-05 (Sema3F), two-tailed t test; n = 10 neurons for each condition). The total number of spines counted in each condition ranged from 234 to 1149. B, Representative images of apical dendritic branches in Fc- and Sema3B-Fc-treated neuronal cultures from WT and CHL1 KO mice. Scale bar, 10 μm. C, Representative images of apical dendrites in Fc and Sema3F-Fc neuronal cultures from WT and CHL1 KO mice. Scale bar, 10 μm. D, Quantification of spine density (mean ±SEM) in WT and CHL1-null neuronal cultures treated with Fc, Sema3B-Fc, or Sema3F-Fc. Sema3B-Fc led to a significant decrease in mean spine density per neuron (±SEM) for WT but not CHL1 KO neurons (*p = 0.005, two-tailed t test). Sema3F-Fc led to a significant decrease in spine density for WT and CHL1 KO [*p = 1.40E-06 (WT), 6.48E-09 (CHL1 KO), two-tailed t test]. Number of neurons scored: WT, n = 10 (Fc), n = 10 (Sema3B-Fc), n = 14 (Fc), n = 14 (Sema3F-Fc); CHL1-null, n = 18 (Fc), n = 18 (Sema3B-Fc), n = 16 (Fc), n = 17 (Sema3F-Fc). The total number of spines counted in each condition ranged from 234 to 1098.
To investigate whether CHL1 was required for Sema3B- or Sema3F-induced spine retraction, cortical neuron cultures from CHL1-null mutant mice were transfected with pCAGGs-IRES-EGFP and then assayed for spine retraction in response to Fc fusion proteins at DIV14. Spine density on apical dendrites of untreated cortical neurons from WT and CHL1-null mice did not differ significantly (Fig. 6B–D) because there is little if any accumulation of Sema3B in culture supernatants as cultures were subjected to several media changes. Treatment with Sema3B-Fc decreased spine density in WT neurons by 32% but did not induce spine retraction in CHL1-null neurons. Treatment with Sema3F-Fc decreased spine density in WT neurons (50%) to approximately the same extent as in CHL1-null neurons (46%). Sema3F-Fc has been shown to induce spine retraction through NrCAM and fails to promote retraction in neurons from NrCAM null mice (Demyanenko et al., 2014). Taken together, these results showed that CHL1 mediates spine retraction on apical dendrites of cortical pyramidal neurons specifically in response to stimulation by Sema3B.
Expression of CHL1 and Sema3B in cortical pyramidal neurons and association of CHL1 with Sema3 receptor subunits Npn2 and PlexA4
To investigate whether CHL1 and Sema3B were coexpressed in cortical pyramidal neurons, double immunofluorescence staining of cortical sections was performed in tamoxifen-inducible Nex1-CreER: Ai9 mice in which fluorescent tdTomato marks postmitotic pyramidal neurons (Mohan et al., 2019). After tamoxifen induction (P10–P13), numerous cells in PFC layers 2/3 and V1 layer 4 expressed tdTomato by P21 (Fig. 7A,D left). In both cortical areas CHL1 labeling was prominent on tdTomato-positive pyramidal cell soma, which were readily seen at low magnification, and staining was also evident on processes between soma (Fig. 7A,D). Some CHL1-positive, tdTomato-negative cells likely corresponded to uninduced pyramidal neurons or other cell types. Sema3B labeling was clearly present on tdTomato-positive pyramidal cells in both areas (Fig. 7B,E). Neither CHL1 nor Sema3B exhibited colocalization with GABAergic cell marker Gad67 (data not shown). Merged images showed that CHL1 and Sema3B colocalized to a large population of pyramidal cells (Fig. 7C,F). Cell counts in PFC layer 2/3 (P21) indicated that CHL1 was expressed in ∼22% of tdTomato-positive neurons, whereas Sema3B was evident on 33% of the cell soma (186–220 cells scored in each of four images). Approximately half of CHL1-expressing pyramidal cells were also Sema3B-positive. At higher magnification CHL1 labeling on apical dendrites was visible on spines, as shown in PFC layer 2/3 (Fig. 7G,H). Sema3B immunolabeling was also present on spines (Fig. 7I), overlapping with CHL1 (Fig. 7J). Approximately 72% of spines showed colocalization of CHL1 and Sema3B.
Figure 7.
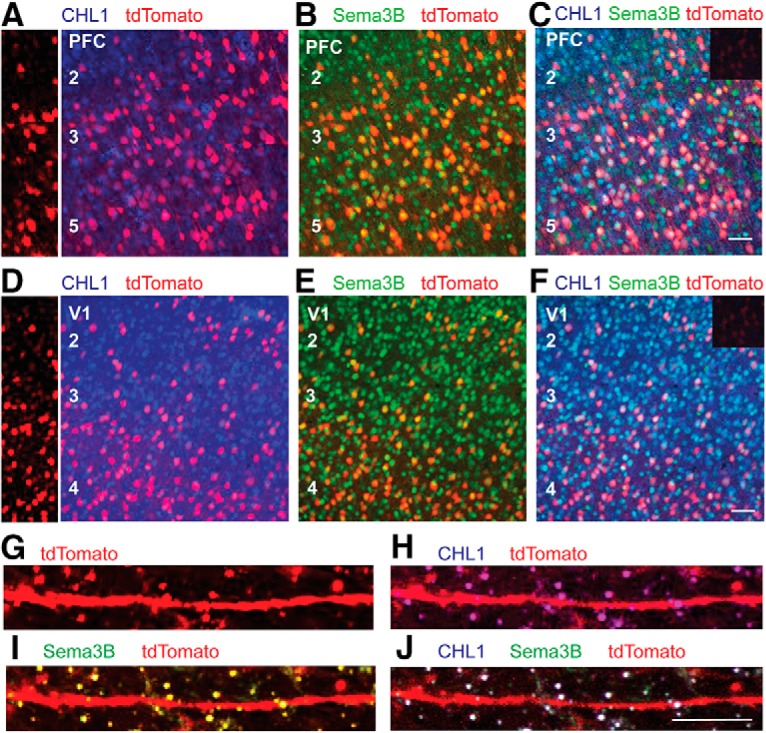
Localization of CHL1 and Sema3B in cortical pyramidal neurons in postnatal mouse neocortex. A–J, Nex1-CreERT2:Ai9 mice were induced to express tdTomato in postmitotic pyramidal neurons by tamoxifen injections (P10–P14), and brains were harvested at P21. Immunofluorescence staining was carried out for CHL1 and Sema3B in coronal sections of PFC and V1. Nonimmune IgG control is shown as an inset (C,F). Scale bar, 100 μm. A, D, Left panels show tdTomato labeling in unstained sections of PFC and V1. Larger right panels show localization of CHL1 (blue) in cell bodies of tdTomato-positive neurons (red), appearing as magenta. B, E, Localization of Sema3B (green) in cell bodies of tdTomato-positive neurons (red), appearing as yellow. C, F, Colocalization of CHL1 (blue) and Sema3B (green) in cell bodies of tdTomato- positive pyramidal neurons (red), appearing as white. G–J, Higher magnification of images of apical dendrites in the same sections shows co localization of CHL1 and Sema3B on numerous spines (white).
The developmental regulation of CHL1 and Sema3B was further assessed by immunoblotting of cortical lysates from postnatal and adult brain. CHL1 and Sema3B were coexpressed throughout early postnatal development (P7–P30) and then CHL1, but not Sema3B, declined to lower levels (relative to actin) in adulthood (Fig. 8A). Sema3B has been reported to be capable of binding to Npn1 or Npn2, which can associate constitutively with a PlexA1–4 subunit depending on cellular context to constitute a receptor complex (Sharma et al., 2012). Molecular constituents of the Sema3B receptor complex were analyzed in synaptoneurosomes isolated from P28 mouse brain. Synaptoneurosomes contain pinched-off presynaptic terminals (synaptosomes) attached to sealed postsynaptic sacs (neurosomes) and are enriched in PDZ domain-containing scaffold proteins of the PSD such as PSD95 and SAP102 (Villasana et al., 2006). CHL1, Npn1-2, and PlexA1-4 were each present in the synaptoneurosome fraction (input, Fig. 8B). CHL1 was immunoprecipitated from equal amounts of synaptoneurosomes and immunoblotted for associated proteins. CHL1 was found to coimmunoprecipitate preferentially with Npn2, and to a much lesser extent with Npn1 (Fig. 8B). CHL1 associated directly or indirectly with Npn2, as shown by coimmunoprecipitation from HEK293T cells transfected with CHL1 and Npn2 (Fig. 8C). The involvement of individual PlexAs in neuronal Sema3B receptor complexes has not been well established except for PlexA1, which promotes Sema3B guidance of commissural axons (Nawabi et al., 2010). PlexA4 was found to be a prominent component of CHL1 complexes immunoprecipitated from synaptoneurosomes, whereas PlexA1, PlexA2, or PlexA3 were not detectable (Fig. 8B). PlexA4 also functions as a component of the Sema3B receptor together with Npn1 or Npn2 in endothelial and glioblastoma cells (Sabag et al., 2014). Sema3B does not appear to bind CHL1 directly, as shown in a cell binding assay in which HEK293T cells expressing CHL1 were incubated with Sema3B-Fc or Fc (control). After washing, Fc proteins in cell lysates were pulled down with Protein A/G-Sepharose and probed for CHL1. CHL1 was not detected in Sema3B-Fc or Fc-treated cell lysates (Fig. 8D). To investigate the role of CHL1 in the binding between Npn2 and PlexA4, cortical lysates (500 μg) from CHL1-null and WT mice were immunoprecipitated with Npn2 antibodies and immunoblotted for PlexA4. PlexA4 coimmunoprecipitated with Npn2 from WT but not CHL1-null cortex (Fig. 8E), indicating that CHL1 promoted binding between Npn2 and PlexA4. These results are consistent with the interpretation that CHL1 engages Npn2, which associates with signaling subunit PlexA4 to comprise a holoreceptor complex for Sema3B in the postsynaptic membrane (Fig. 8F).
Figure 8.

Developmental regulation of CHL1 and Sema3B, and association of CHL1 with Sema3B receptor subunits. A, Expression of CHL1 and Sema3B in cortical lysates (equal amounts of protein) from WT mice during postnatal development (P7, P14, P30, and adult). Reprobing of the same blot for actin is shown below. Levels of expression of CHL1 and Sema3B relative to actin are calculated from densitometric scanning. B, Equal amounts of protein from the synaptoneurosome fraction of P28 mouse brain were immunoprecipitated with CHL1 antibodies (CHL1 IP), and associated Npn1/2 or PlexA1-4 were identified by immunoblotting. Equivalent exposures of immunoblots show that CHL1 coimmunoprecipitated with Npn2 to a much greater extent than with Npn1, and associated with PlexA4 but not detectably with PlexA1, PlexA2, or PlexA3. Input lanes demonstrate the presence of each protein in the synaptoneurosome fraction. This experiment was repeated 6 times with similar results. C, CHL1 associated with Npn2 as shown by coimmunoprecipitation from HEK293T cells transfected with CHL1 and Npn2. CHL1 was immunoprecipitated from cell lysates (500 μg) with CHL1 antibodies (CHL1 IP) or control normal IgG (nIg), and immunoprecipitated proteins or input samples (50 μg) were subjected to immunoblotting with Npn2 or CHL1 antibodies. D, Sema3B-Fc or Fc proteins (10 nm) were incubated with HEK293T cells expressing CHL1 for 30 min, then washed extensively, and lysed. Fc proteins were pulled down from equal amounts of lysates with Protein A/G-Sepharose, and immunoblotted for CHL1. Input lanes demonstrate equivalent levels of CHL1 expression in cell lysates. Results of prolonged exposure of blots show no direct binding of Sema3B-Fc or control Fc to cells expressing CHL1. E, PlexA4 coimmunoprecipitated with Npn2 from equal amounts (500 μg) of cortical lysates of WT but not CHL1-null (KO) mice. NIg, Normal IgG control. Blots were reprobed with antibodies to Npn2 and CHL1 in panels below. F, Schematic of the Sema3B holoreceptor complex containing CHL1, Npn2, and PlexA4 leading to Sema3B-induced spine retraction. CHL1 domains include Ig-like domains 1–5 and fibronectin III-like domains (FN). Rap-GAP (Rap1 GTPase-activating protein). This experiment was repeated three times with similar results. G, Cortical neuron cultures were treated with or without gabazine (20 μm) for 48 h. Conditioned medium was harvested from cultures at the same plating density and equivalent amounts were analyzed by immunoblotting with Sema3B antibodies. Cell lysates (equal protein) prepared from cultures from which conditioned media was removed were analyzed by immunoblotting for β-actin and Sema3B. H, Histogram depicts the fold change in Sema3B levels in conditioned medium relative to actin in the cell lysates (mean ±SEM; n = 3, *p = 0.029, t test).
To investigate whether Sema3B was secreted in response to neuronal activity (Orr et al., 2017), cortical neuron cultures were treated with the GABAA receptor antagonist gabazine (20 μm) to increase spontaneous network activity (Favuzzi et al., 2017). Sema3B was concentrated by immunoprecipitation from conditioned medium from untreated and gabazine-treated cultures after 48 h and then compared by immunoblotting. Gabazine was found to increase the amount of Sema3B protein (85, 83 kDa), evident as two forms due to proteolytic processing (Adams et al., 1997; Varshavsky et al., 2008) (Fig. 8G). The levels of Sema3B were significantly increased in conditioned medium from gabazine-treated cultures, as shown by the amount of secreted Sema3B relative to actin in the corresponding cell lysates (Fig. 8H). Gabazine treatment had no effect on the amount of Sema3B protein in cell lysates (Fig. 8G). These findings in cortical neuron cultures suggested that Sema3B, like Sema3F (Orr et al., 2017), can be secreted in response to excitatory activity.
Sema3B selectively eliminates CHL1-expressing spines on apical dendrites
Double immunofluorescence staining for CHL1 and NrCAM was carried out in DIV14 cortical neuron cultures expressing EGFP. Results showed that most spines of apical dendrites expressed either CHL1 (54%) or NrCAM (23%), whereas a minority of spines expressed both (15%) or neither (8%) (n = 459 spines, 12 images). To assess whether spines eliminated by Sema3B treatment were those specifically expressing CHL1, we assayed Sema3B-induced spine retraction in cortical neuron cultures followed by immunostaining for CHL1. EGFP-expressing cortical neurons were treated with 5 nm Sema3B-Fc or Fc for 30 min and then scored for the percentage of CHL1-positive spines labeled (red) by immunostaining, as well as density of total EGFP-labeled spines on apical dendrites. Remarkably, there was a significant decrease in percentage of CHL1-expressing spines in Sema3B-Fc-treated neurons compared with Fc control. This decrease correlated with a similar loss in total spine density (Fig. 9B,C). In merged images, the EGFP-labeled spines after Sema3B-Fc treatment were negative for CHL1 (Fig. 9A, arrows). Conversely, to assess whether spines eliminated by Sema3F treatment were those specifically expressing NrCAM, we similarly assayed Sema3F-induced spine retraction in cortical neuron cultures followed by immunostaining for NrCAM (red) (Fig. 9D). Results revealed a specific decrease in NrCAM-positive spines in Sema3F-Fc-treated neurons compared with Fc (Fig. 9E), correlating with loss of EGFP-labeled spines (Fig. 9F). In merged images, the EGFP-labeled spines remaining after Sema3F treatment were negative for NrCAM (Fig. 9D, arrows). To confirm these findings, we treated neurons with Sema3B-Fc and immunostained for NrCAM. The majority of the remaining spines were NrCAM-positive (Fig. 9G, arrows). Quantification showed a significant increase in the percentage of NrCAM-positive spines in Sema3B-treated neurons (Fig. 9I). Spine density was also reduced significantly (Fig. 9J). Similar results were found when Sema3F-treated neurons were immunostained for CHL1. There was a significant increase in the percentage of CHL1-positive spines in Sema3F-treated neurons compared with Fc (Fig. 9H,K, L). We also performed double staining for NrCAM and CHL1 on neurons treated with Sema3B-Fc or Fc. There was a significant increase in the percentage of NrCAM-positive spines and a reduction in the percentage of CHL1-positive spines in Sema3B-Fc-treated neurons. Approximately 20% of spines were immunoreactive for both NrCAM and CHL1 (Fig. 9M,N). CHL1 was localized to both apical and basal dendrites, whereas Npn2 was specifically localized to apical dendrites (Fig. 9O,P), consistent with previous findings (Tran et al., 2009). These results demonstrated a strong selectivity in Sema3-dependent mechanisms for eliminating dendritic spines expressing CHL1 or NrCAM.
Figure 9.

Selective elimination of spines by Sema3B and Sema3F. A, Apical dendrites from GFP-expressing cortical neurons (green) in DIV14 cultures were treated with either Fc or Sema3B-Fc (5 nm) for 30 min, then immunostained for CHL1 (red). CHL1-positive spines were selectively eliminated. Arrows point to residual CHL1-negative spines. Scale bar, 10 μm. B, Histogram depicting a significant decrease in the percentage of CHL1-positive spines in Sema3B-Fc-treated neurons (*p = 0.001, two-tailed t test, n = 10 neurons per condition). C, Histogram shows decreased spine density (mean ±SEM) in Sema3B-Fc-treated neurons versus Fc-treated neurons (*p = 3.8E-06, two-tailed t test, n = 10 neurons per condition). Total spines counted for each condition ranged from 171 to 343. D, Apical dendrites from GFP-expressing cortical neurons (green) in DIV14 cultures were treated with either Fc or Sema3F-Fc (5 nm) for 30 min, then immunostained for NrCAM (red). NrCAM-positive spines were selectively eliminated. Arrows point to residual NrCAM-negative spines. Scale bar, 10 μm. E, Histogram depicting a significant decrease in the percentage of NrCAM-positive spines in Sema3F-Fc-treated neurons (*p = 0.00013, two-tailed t test, n = 10 neurons per condition). F, Histogram showing decreased spine density (mean ±SEM) in Sema3F-Fc-treated neurons versus Fc-treated neurons (*p = 2.4E-05, t test, n = 10 neurons per condition). Total spines counted for each condition ranged from 76 to 204. G, Apical dendrites from GFP-expressing cortical neurons (green) in DIV14 cultures were treated with either Fc or Sema3B-Fc (5 nm) for 30 min and then immunostained for NrCAM (red). The majority of remaining spines after Sema3B-Fc treatment were NrCAM-positive (arrows). Scale bar, 10 μm. H, Apical dendrites from GFP-expressing cortical neurons (green) in DIV14 cultures were treated with either Fc or Sema3F-Fc (5 nm) for 30 min and then immunostained for CHL1 (red). Arrows point to residual CHL1-positive spines. Scale bar, 10 μm. I, Histogram depicting a significant increase in the percentage of NrCAM-positive spines in Sema3B-Fc-treated neurons (*p = 0.002, two-tailed t test, n = 10 neurons per condition). J, Histogram showing decreased spine density (mean ±SEM) in Sema3B-Fc-treated neurons versus Fc-treated controls (*p = 0.005, two-tailed t test, n = 5 neurons per condition). K, Histogram depicting a significant increase in the percentage of CHL1-positive spines in Sema3F-Fc-treated neurons (*p = 7.3E-06, two-tailed t test, n = 10 neurons per condition). L, Histogram showing decreased spine density (mean ±SEM) in Sema3B-Fc-treated neurons versus Fc-treated neurons (*p = 0.001, two-tailed t test, n = 5 neurons per condition). M, Apical dendrites from GFP-expressing cortical neurons (green) in DIV14 cultures were treated with either Fc or Sema3F-Fc (5 nm) for 30 min and then immunostained for CHL1 (blue) and NrCAM (red). Yellow spines are NrCAM-positive; cyan spines are CHL1-positive; and white spines are positive for both CHL1 and NrCAM. Arrows point to residual NrCAM-positive spines. Scale bar, 10 μm. N, Histogram depicts a significant increase in the percentage of NrCAM-positive spines in Sema3B-Fc-treated neurons (*p = 0.01, two-tailed t test, n = 10 neurons per condition) and a significant reduction in percentage of CHL1-positive spines (*p = 0.015, two-tailed t test, n = 10 neurons). O, Immunofluorescence staining of CHL1 in GFP-expressing cortical neuron in culture showing localization on both apical and basal dendrites of pyramidal-type neuron. P, Immunofluorescence staining of Npn2 in GFP-expressing cortical neuron cultures showing Npn2 localization to apical but not basal dendrites of pyramidal-type neuron.
Discussion
Here, we describe a novel molecular mechanism for elimination of dendritic spines on pyramidal neurons mediated by the neural adhesion molecule CHL1 and the class 3 secreted semaphorin Sema3B in the developing mouse cerebral cortex. CHL1 deletion in null mice increased the density of apical dendritic spines and excitatory synapses in early adolescence, which persisted through adulthood. Electrophysiological recordings showed altered regulation of excitatory synaptic strength in prefrontal slices from CHL1-null mutants. CHL1 and Sema3B were coexpressed in subpopulations of early postnatal pyramidal neurons and localized to dendritic spines. CHL1 formed a synaptoneurosome complex with Sema3 receptor subunits Npn2 and PlexA4, and mediated spine elimination on apical dendrites in cortical neuron cultures in response to Sema3B, which was secreted in an activity-dependent manner. Remarkably, Sema3B selectively eliminated spines expressing CHL1, whereas Sema3F eliminated a distinct cohort of spines expressing NrCAM. These results support a new concept in which developmental elimination of overproduced spines may be achieved selectively by secreted semaphorins acting locally through different L1-CAM containing receptor complexes. Such a mechanism could function at the level of individual synapses to determine which are culled from developing neural circuits in the maturing neocortex.
Our findings suggest that CHL1 limits spine density by mediating spine pruning on apical dendrites of cortical pyramidal neurons in response to secreted Sema3B as an essential subunit within a receptor complex comprising Npn2 and PlexA4. This function is consistent with the phenotype of Npn2 null mice, which display increased spine density only on apical dendrites in cortical pyramidal cell subpopulations (Tran et al., 2009). Also in accord, PlexA4 deficient mice display elevated spine density on granule cells of the dentate gyrus (Duan et al., 2014). CHL1 may play a role in formation or stabilization of the Npn2/PlexA3 complex, as suggested by coimmunoprecipitation from WT but not CHL1-null cortical lysates. The observation that CHL1 mutants exhibit proportionately more thin spines than mushroom or stubby spines compared with WT suggests that Sema3B may preferentially target thin, immature spines. Consistent with this notion, Sema3B-Fc treatment of WT cortical neuron cultures resulted in a decrease in thin spines, whereas the majority of remaining spines lacked CHL1.
These results add to the evidence that class 3 semaphorins bind holoreceptor complexes comprising different Npns, PlexAs, and L1-CAMs to serve as multifunctional regulators of spine and dendrite development. Notably, Sema3F binds a receptor complex consisting of NrCAM, Npn2, and PlexA3 to constrain spine density during adolescent development of cortical pyramidal neurons (Tran et al., 2009; Cheadle and Biederer, 2012; Mohan et al., 2019). Secreted Sema3F has an extended role in adult neurons in facilitating homeostatic downscaling of synaptic strength (Wang et al., 2017), which was also shown for Drosophila Sema2b (Orr et al., 2017). Sema3A promotes basal dendrite arborization through PlexA4 and Npn1 in cortical and hippocampal neurons (Tran et al., 2009; Cheadle and Biederer, 2012). In related cellular systems, Sema3B triggers morphological retraction of glioblastoma and endothelial cells through PlexA4 and PlexA2 with either Npn1 or Npn2 (Sabag et al., 2014), whereas PlexA4 and Npn2 mediate stereotyped pruning of corticospinal axons (Low et al., 2008).
Restriction of CHL1-dependent spine remodeling to apical dendrites of pyramidal cells may regulate the distribution of specific synaptic inputs. For example, apical and basal dendrites differ in the distribution of intracortical and thalamocortical inputs, as well as their mode of synaptic potentiation (Kawaguchi and Kubota, 1997; Gordon et al., 2006; Brzdak et al., 2019). Effects of CHL1 on remodeling of apical dendritic spines reflects the preferential localization of Npn2 to apical but not basal dendrites in cortical neurons, shown here and in Tran et al. (2009). In addition, Npn2 has a C-terminal PDZ binding motif (Ser-Glu-Ala) that could help position the Sema3B receptor complex, containing CHL1 and PlexA4, to the PSD (Prahst et al., 2008). PlexA4 also binds the AMPA receptor subunit GluA2 and localizes the complex to dendrites through Sema3A/Npn1/PlexA4 signaling (Yamashita et al., 2014), whereas for homeostatic scaling, GluA2 interacts with the Sema3F/Npn2/PlexA3 complex in cortical neurons (Wang et al., 2017). The spine remodeling function of CHL1 appears unrelated to its role in pyramidal neuron positioning and dendritic orientation, which occurs at earlier stages of development and is limited to a subset of layer 5/6 pyramidal cells (Demyanenko et al., 2004).
The electrophysiological evidence indicated that there is a disruption of normal excitatory synaptic function, with event amplitudes skewed toward larger sizes in slices from CHL1-null PFC (layer 2/3), but without detectable changes in mean amplitude or frequency. The CHL1 mutant showed changes in spine morphology that were limited to apical dendrites, whereas the electrophysiological measures sample from synapses that are biased toward the proximal regions of both basal and apical dendrites. With mixed populations of events that cannot be assigned to synapse location, it perhaps not surprising that differences in basic measures of event frequency and amplitude were not detected. In addition CHL1 was expressed in a subset of pyramidal neurons, possibly contributing to the wider range of event amplitudes, including larger events in some cells, as measured from the skew of the amplitude distributions. There are also limitations to the interpretation of mEPSC amplitudes, as the spine compartment is partially isolated from the dendritic shaft electrically (Jayant et al., 2017), precluding accurate measurements of synaptic conductance under voltage clamp (Beaulieu-Laroche and Harnett, 2018). The differences in event charge distributions could reflect differences in the electrical geometry of some spines (a decrease in spine neck resistance), differences in synaptic conductances (increased AMPA receptor conductance), or both. CHL1 loss increased the proportion of thin spines, which are characteristic of immature spines with nascent synapses (Berry and Nedivi, 2017). Such spines are thought to have a low NMDA to AMPA receptor ratio and should have a higher spine neck resistance. However, the measured NMDA to AMPA ratios did not indicate a difference between the genotypes. Considering that developmental synaptic pruning and long-term synaptic depression (LTD) share some common mechanisms (Piochon et al., 2016), it is possible that LTD and/or homeostatic downscaling at these synapses is impaired. Interfering with these mechanisms would upset the interplay of metaplasticity mechanisms needed to maintain a normal excitatory synaptic operating range (Hulme et al., 2013). Decreased paired-pulse facilitation is usually associated with an increased synaptic release probability, suggesting an elevated excitatory synaptic tone onto the CHL1-null cells. In summary, these data indicate that the regulation of excitatory synaptic strength is disrupted in the CHL1-null mice.
Spine formation and retraction contribute to remodeling and fine tuning of cortical networks in the developing and mature brain (Turrigiano, 2012; Stein and Zito, 2019). During postnatal development class 3 semaphorins and their respective L1-CAM/Npn/PlexA receptor complexes may function to refine nascent synaptic connections from specific inputs to sculpt neural circuits. Activity-dependent secretion of Sema3B in cortical neuron cultures suggested that Sema3B may be upregulated at active synapses and secreted locally to homeostatically eliminate inactive spine neighbors expressing CHL1, Npn2, and PlexA4. In this scenario, secreted Sema3B would spare spines expressing NrCAM, Npn2, and PlexA3, which would be subject instead to elimination by Sema3F. Class 3 semaphorins are able to associate with the extracellular matrix through their basic C terminal tail and in this way are in a position to prune nearby spines through autocrine/paracrine signaling (Hamm et al., 2016). A homeostatic mechanism of local competition has been demonstrated between stimulated spines and inactive neighboring spines in the hippocampus (Oh et al., 2015). Activity-dependent competition for adhesive cadherin/catenin complexes similarly leads to spine pruning in the developing somatosensory cortex (Bian et al., 2015). It will be interesting to determine if Sema3B is upregulated and released at active synapses to prune weaker neighboring spines and synapses in vivo. Genetic variations in the human CHL1/CALL gene (3p26.3) have been linked to cognitive dysfunction in ASD (Salyakina et al., 2011; Vega et al., 2017), schizophrenia (Sakurai et al., 2002; Chen et al., 2005; Chu and Liu, 2010), and intellectual disability (Angeloni et al., 1999; Frints et al., 2003; Cuoco et al., 2011). Thus, the present findings, which shed light on normal molecular mechanisms of developmental spine remodeling, may provide insight into spine dysgenesis in certain neurodevelopmental disorders.
Footnotes
This work was supported by US National Institutes of Mental Health grant R01 MH113280 (P.F.M.) and fellowship F32 MH111189 (C.S.), National Institute on Deafness and other Communication Disorders grant R01 DC004551 (P.B.M.), The University of North Carolina IDDRC Center grant NICHD U54HD079124 and support from the Li Kashing Foundation (M.S.). We thank Dr. Thomas Theis for providing CHL1 mutant mice, Dr. Soraia Barao for mutant brains, and Daniel Tidwell for assistance with graphics. We thank Victoria Madden for specimen preparation for electron microscopy and Dr. Pablo Ariel (Director) for helping in confocal image analysis at the Microscopy Services Laboratory, UNC Pathology and Laboratory Medicine (supported in part by P30 CA016086 Cancer Center Core Support Grant to the UNC Lineberger Comprehensive Cancer Center).
The authors declare no competing financial interests.
References
- Adams RH, Lohrum M, Klostermann A, Betz H, Püschel AW (1997) The chemorepulsive activity of secreted semaphorins is regulated by furin-dependent proteolytic processing. EMBO J 16:6077–6086. 10.1093/emboj/16.20.6077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agarwal A, Dibaj P, Kassmann CM, Goebbels S, Nave KA, Schwab MH (2012) In vivo imaging and noninvasive ablation of pyramidal neurons in adult NEX-CreERT2 mice. Cereb Cortex 22:1473–1486. 10.1093/cercor/bhr214 [DOI] [PubMed] [Google Scholar]
- Agresti A. (2013) Categorical data analysis, Ed 3 New York: Wiley. [Google Scholar]
- Angeloni D, Lindor NM, Pack S, Latif F, Wei MH, Lerman MI (1999) CALL gene is haploinsufficient in a 3p- syndrome patient. Am J Med Genet 86:482–485. 10.1002/(SICI)1096-8628(19991029)86:5<482::AID-AJMG15>3.0.CO;2-L [DOI] [PubMed] [Google Scholar]
- Beaulieu-Laroche L, Harnett MT (2018) Dendritic spines prevent synaptic voltage clamp. Neuron 97:75–82.e3. 10.1016/j.neuron.2017.11.016 [DOI] [PubMed] [Google Scholar]
- Berry KP, Nedivi E (2017) Spine dynamics: are they all the same? Neuron 96:43–55. 10.1016/j.neuron.2017.08.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhatt DH, Zhang S, Gan WB (2009) Dendritic spine dynamics. Annu Rev Physiol 71:261–282. 10.1146/annurev.physiol.010908.163140 [DOI] [PubMed] [Google Scholar]
- Bian WJ, Miao WY, He SJ, Qiu Z, Yu X (2015) Coordinated spine pruning and maturation mediated by inter-spine competition for Cadherin/Catenin complexes. Cell 162:808–822. 10.1016/j.cell.2015.07.018 [DOI] [PubMed] [Google Scholar]
- Brzdak P, Wójcicka O, Zareba-Koziol M, Minge D, Henneberger C, Wlodarczyk J, Mozrzymas JW, Wójtowicz T (2019) Synaptic potentiation at basal and apical dendrites of hippocampal pyramidal neurons involves activation of a distinct set of extracellular and intracellular molecular cues. Cereb Cortex 29:283–304. 10.1093/cercor/bhx324 [DOI] [PubMed] [Google Scholar]
- Campagnola L, Kratz MB, Manis PB (2014) ACQ4: an open-source software platform for data acquisition and analysis in neurophysiology research. Front Neuroinform 8:3. 10.3389/fninf.2014.00003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castellani V, Chédotal A, Schachner M, Faivre-Sarrailh C, Rougon G (2000) Analysis of the L1-deficient mouse phenotype reveals cross-talk between Sema3A and L1 signaling pathways in axonal guidance. Neuron 27:237–249. 10.1016/S0896-6273(00)00033-7 [DOI] [PubMed] [Google Scholar]
- Cheadle L, Biederer T (2012) The novel synaptogenic protein Farp1 links postsynaptic cytoskeletal dynamics and transsynaptic organization. J Cell Biol 199:985–1001. 10.1083/jcb.201205041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen QY, Chen Q, Feng GY, Lindpaintner K, Chen Y, Sun X, Chen Z, Gao Z, Tang J, He L (2005) Case-control association study of the close homologue of L1 (CHL1) gene and schizophrenia in the chinese population. Schizophr Res 73:269–274. 10.1016/j.schres.2004.06.001 [DOI] [PubMed] [Google Scholar]
- Chu TT, Liu Y (2010) An integrated genomic analysis of gene-function correlation on schizophrenia susceptibility genes. J Hum Genet 55:285–292. 10.1038/jhg.2010.24 [DOI] [PubMed] [Google Scholar]
- Clements JD, Bekkers JM (1997) Detection of spontaneous synaptic events with an optimally scaled template. Biophys J 73:220–229. 10.1016/S0006-3495(97)78062-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuoco C, Ronchetto P, Gimelli S, Béna F, Divizia MT, Lerone M, Mirabelli-Badenier M, Mascaretti M, Gimelli G (2011) Microarray based analysis of an inherited terminal 3p26.3 deletion, containing only the CHL1 gene, from a normal father to his two affected children. Orphanet J Rare Dis 6:12. 10.1186/1750-1172-6-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demyanenko GP, Schachner M, Anton E, Schmid R, Feng G, Sanes J, Maness PF (2004) Close homolog of L1 modulates area-specific neuronal positioning and dendrite orientation in the cerebral cortex. Neuron 44:423–437. 10.1016/j.neuron.2004.10.016 [DOI] [PubMed] [Google Scholar]
- Demyanenko GP, Halberstadt AI, Rao RS, Maness PF (2010) CHL1 cooperates with PAK1–3 to regulate morphological differentiation of embryonic cortical neurons. Neuroscience 165:107–115. 10.1016/j.neuroscience.2009.09.077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demyanenko GP, Siesser PF, Wright AG, Brennaman LH, Bartsch U, Schachner M, Maness PF (2011a) L1 and CHL1 cooperate in thalamocortical axon targeting. Cereb Cortex 21:401–412. 10.1093/cercor/bhq115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demyanenko GP, Riday TT, Tran TS, Dalal J, Darnell EP, Brennaman LH, Sakurai T, Grumet M, Philpot BD, Maness PF (2011b) NrCAM deletion causes topographic mistargeting of thalamocortical axons to the visual cortex and disrupts visual acuity. J Neurosci 31:1545–1558. 10.1523/JNEUROSCI.4467-10.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demyanenko GP, Mohan V, Zhang X, Brennaman LH, Dharbal KE, Tran TS, Manis PB, Maness PF (2014) Neural cell adhesion molecule NrCAM regulates semaphorin 3F-induced dendritic spine remodeling. J Neurosci 34:11274–11287. 10.1523/JNEUROSCI.1774-14.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demyanenko GP, Tsai AY, Maness PF (1999) Abnormalities in neuronal process extension, hippocampal development, and the ventricular system of L1 knockout mice. J Neurosci 19:4907–4920. 10.1523/JNEUROSCI.19-12-04907.1999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duan Y, Wang SH, Song J, Mironova Y, Ming GL, Kolodkin AL, Giger RJ (2014) Semaphorin 5A inhibits synaptogenesis in early postnatal- and adult-born hippocampal dentate granule cells. Elife. Advance online publication. Retrieved October 14, 2014. doi: 10.7554/eLife.04390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dupont WD, Plummer WD Jr (1990) Power and sample size calculations: a review and computer program. Control Clin Trials 11:116–128. 10.1016/0197-2456(90)90005-M [DOI] [PubMed] [Google Scholar]
- Favuzzi E, Marques-Smith A, Deogracias R, Winterflood CM, Sánchez-Aguilera A, Mantoan L, Maeso P, Fernandes C, Ewers H, Rico B (2017) Activity-dependent gating of parvalbumin interneuron function by the perineuronal net protein brevican. Neuron 95:639–655.e10. 10.1016/j.neuron.2017.06.028 [DOI] [PubMed] [Google Scholar]
- Forrest MP, Parnell E, Penzes P (2018) Dendritic structural plasticity and neuropsychiatric disease. Nat Rev Neurosci 19:215–234. 10.1038/nrn.2018.16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frints SG, Marynen P, Hartmann D, Fryns JP, Steyaert J, Schachner M, Rolf B, Craessaerts K, Snellinx A, Hollanders K, D'Hooge R, De Deyn PP, Froyen G (2003) CALL interrupted in a patient with nonspecific mental retardation: gene dosage-dependent alteration of murine brain development and behavior. Hum Mol Genet 12:1463–1474. 10.1093/hmg/ddg165 [DOI] [PubMed] [Google Scholar]
- Gordon U, Polsky A, Schiller J (2006) Plasticity compartments in basal dendrites of neocortical pyramidal neurons. J Neurosci 26:12717–12726. 10.1523/JNEUROSCI.3502-06.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamm MJ, Kirchmaier BC, Herzog W (2016) Sema3d controls collective endothelial cell migration by distinct mechanisms via Nrp1 and PlxnD1. J Cell Biol 215:415–430. 10.1083/jcb.201603100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodges JL, Vilchez SM, Asmussen H, Whitmore LA, Horwitz AR (2014) alpha-actinin-2 mediates spine morphology and assembly of the post-synaptic density in hippocampal neurons. PLoS One 9:e101770. 10.1371/journal.pone.0101770 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holtmaat A, Svoboda K (2009) Experience-dependent structural synaptic plasticity in the mammalian brain. Nat Rev Neurosci 10:647–658. 10.1038/nrn2699 [DOI] [PubMed] [Google Scholar]
- Hulme SR, Jones OD, Abraham WC (2013) Emerging roles of metaplasticity in behaviour and disease. Trends Neurosci 36:353–362. 10.1016/j.tins.2013.03.007 [DOI] [PubMed] [Google Scholar]
- Hutsler JJ, Zhang H (2010) Increased dendritic spine densities on cortical projection neurons in autism spectrum disorders. Brain Res 1309:83–94. 10.1016/j.brainres.2009.09.120 [DOI] [PubMed] [Google Scholar]
- Huttenlocher PR. (1979) Synaptic density in human frontal cortex - developmental changes and effects of aging. Brain Res 163:195–205. 10.1016/0006-8993(79)90349-4 [DOI] [PubMed] [Google Scholar]
- Iacaruso MF, Gasler IT, Hofer SB (2017) Synaptic organization of visual space in primary visual cortex. Nature 547:449–452. 10.1038/nature23019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irintchev A, Koch M, Needham LK, Maness P, Schachner M (2004) Impairment of sensorimotor gating in mice deficient in the cell adhesion molecule L1 or its close homologue, CHL1. Brain Res 1029:131–134. 10.1016/j.brainres.2004.09.042 [DOI] [PubMed] [Google Scholar]
- Jayant K, Hirtz JJ, Plante IJ, Tsai DM, De Boer WD, Semonche A, Peterka DS, Owen JS, Sahin O, Shepard KL, Yuste R (2017) Targeted intracellular voltage recordings from dendritic spines using quantum-dot-coated nanopipettes. Nat Nanotechnol 12:335–342. 10.1038/nnano.2016.268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawaguchi Y, Kubota Y (1997) GABAergic cell subtypes and their synaptic connections in rat frontal cortex. Cereb Cortex 7:476–486. 10.1093/cercor/7.6.476 [DOI] [PubMed] [Google Scholar]
- Kleene R, Chaudhary H, Karl N, Katic J, Kotarska A, Guitart K, Loers G, Schachner M (2015) Interaction between CHL1 and serotonin receptor 2c regulates signal transduction and behavior in mice. J Cell Sci 128:4642–4652. 10.1242/jcs.176941 [DOI] [PubMed] [Google Scholar]
- Kolata S, Wu J, Light K, Schachner M, Matzel LD (2008) Impaired working memory duration but normal learning abilities found in mice that are conditionally deficient in the close homolog of L1. J Neurosci 28:13505–13510. 10.1523/JNEUROSCI.2127-08.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koropouli E, Kolodkin AL (2014) Semaphorins and the dynamic regulation of synapse assembly, refinement, and function. Curr Opin Neurobiol 27:1–7. 10.1016/j.conb.2014.02.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Low LK, Liu XB, Faulkner RL, Coble J, Cheng HJ (2008) Plexin signaling selectively regulates the stereotyped pruning of corticospinal axons from visual cortex. Proc Natl Acad Sci U S A 105:8136–8141. 10.1073/pnas.0803849105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madisen L, Zwingman TA, Sunkin SM, Oh SW, Zariwala HA, Gu H, Ng LL, Palmiter RD, Hawrylycz MJ, Jones AR, Lein ES, Zeng H (2010) A robust and high-throughput cre reporting and characterization system for the whole mouse brain. Nat Neurosci 13:133–140. 10.1038/nn.2467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McAllister AK. (2007) Dynamic aspects of CNS synapse formation. Annu Rev Neurosci 30:425–450. 10.1146/annurev.neuro.29.051605.112830 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McBain C, Dingledine R (1992) Dual-component miniature excitatory synaptic currents in rat hippocampal CA3 pyramidal neurons. J Neurophysiol 68:16–27. 10.1152/jn.1992.68.1.16 [DOI] [PubMed] [Google Scholar]
- Mohan V, Sullivan CS, Guo J, Wade SD, Majumder S, Agarwal A, Anton ES, Temple BS, Maness PF (2019) Temporal regulation of dendritic spines through NrCAM-Semaphorin3F receptor signaling in developing cortical pyramidal neurons. Cereb Cortex 29:963–977. 10.1093/cercor/bhy004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montag-Sallaz M, Baarke A, Montag D (2003) Aberrant neuronal connectivity in CHL1-deficient mice is associated with altered information processing-related immediate early gene expression. J Neurobiol 57:67–80. 10.1002/neu.10254 [DOI] [PubMed] [Google Scholar]
- Morellini F, Lepsveridze E, Kähler B, Dityatev A, Schachner M (2007) Reduced reactivity to novelty, impaired social behavior, and enhanced basal synaptic excitatory activity in perforant path projections to the dentate gyrus in young adult mice deficient in the neural cell adhesion molecule CHL1. Mol Cell Neurosci 34:121–136. 10.1016/j.mcn.2006.10.006 [DOI] [PubMed] [Google Scholar]
- Nawabi H, Briançon-Marjollet A, Clark C, Sanyas I, Takamatsu H, Okuno T, Kumanogoh A, Bozon M, Takeshima K, Yoshida Y, Moret F, Abouzid K, Castellani V (2010) A midline switch of receptor processing regulates commissural axon guidance in vertebrates. Genes Dev 24:396–410. 10.1101/gad.542510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen H, Ivanova VS, Kavandi L, Rodriguez GC, Maxwell GL, Syed V (2011) Progesterone and 1,25-dihydroxyvitamin D(3) inhibit endometrial cancer cell growth by upregulating semaphorin 3B and semaphorin 3F. Mol Cancer Res 9:1479–1492. 10.1158/1541-7786.MCR-11-0213 [DOI] [PubMed] [Google Scholar]
- Nikonenko AG, Sun M, Lepsveridze E, Apostolova I, Petrova I, Irintchev A, Dityatev A, Schachner M (2006) Enhanced perisomatic inhibition and impaired long-term potentiation in the CA1 region of juvenile CHL1-deficient mice. Eur J Neurosci 23:1839–1852. 10.1111/j.1460-9568.2006.04710.x [DOI] [PubMed] [Google Scholar]
- Oh WC, Parajuli LK, Zito K (2015) Heterosynaptic structural plasticity on local dendritic segments of hippocampal CA1 neurons. Cell Rep 10:162–169. 10.1016/j.celrep.2014.12.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orr BO, Fetter RD, Davis GW (2017) Retrograde semaphorin-plexin signalling drives homeostatic synaptic plasticity. Nature 550:109–113. 10.1038/nature24017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pernía-Andrade AJ, Goswami SP, Stickler Y, Fröbe U, Schlögl A, Jonas P (2012) A deconvolution-based method with high sensitivity and temporal resolution for detection of spontaneous synaptic currents in vitro and in vivo. Biophys J 103:1429–1439. 10.1016/j.bpj.2012.08.039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petanjek Z, Judaš M, Šimic G, Rasin MR, Uylings HB, Rakic P, Kostovic I (2011) Extraordinary neoteny of synaptic spines in the human prefrontal cortex. Proc Natl Acad Sci U S A 108:13281–13286. 10.1073/pnas.1105108108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters A, Kaiserman-Abramof IR (1970) The small pyramidal neuron of the rat cerebral cortex. the perikaryon, dendrites and spines. Am J Anat 127:321–355. 10.1002/aja.1001270402 [DOI] [PubMed] [Google Scholar]
- Phillips M, Pozzo-Miller L (2015) Dendritic spine dysgenesis in autism related disorders. Neurosci Lett 601:30–40. 10.1016/j.neulet.2015.01.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piochon C, Kano M, Hansel C (2016) LTD-like molecular pathways in developmental synaptic pruning. Nat Neurosci 19:1299–1310. 10.1038/nn.4389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prahst C, Héroult M, Lanahan AA, Uziel N, Kessler O, Shraga-Heled N, Simons M, Neufeld G, Augustin HG (2008) Neuropilin-1-VEGFR-2 complexing requires the PDZ-binding domain of neuropilin-1. J Biol Chem 283:25110–25114. 10.1074/jbc.C800137200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pratte M, Rougon G, Schachner M, Jamon M (2003) Mice deficient for the close homologue of the neural adhesion cell L1 (CHL1) display alterations in emotional reactivity and motor coordination. Behav Brain Res 147:31–39. 10.1016/S0166-4328(03)00114-1 [DOI] [PubMed] [Google Scholar]
- Rall W, Burke RE, Smith TG, Nelson PG, Frank K (1967) Dendritic location of synapses and possible mechanisms for the monosynaptic EPSP in motoneurons. J Neurophysiol 30:1169–1193. 10.1152/jn.1967.30.5.1169 [DOI] [PubMed] [Google Scholar]
- Sabag AD, Smolkin T, Mumblat Y, Ueffing M, Kessler O, Gloeckner CJ, Neufeld G (2014) The role of the plexin-A2 receptor in Sema3A and Sema3B signal transduction. J Cell Sci 127:5240–5252. 10.1242/jcs.155960 [DOI] [PubMed] [Google Scholar]
- Sakurai K, Migita O, Toru M, Arinami T (2002) An association between a missense polymorphism in the close homologue of L1 (CHL1, CALL) gene and schizophrenia. Mol Psychiatry 7:412–415. 10.1038/sj.mp.4000973 [DOI] [PubMed] [Google Scholar]
- Sala C, Piëch V, Wilson NR, Passafaro M, Liu G, Sheng M (2001) Regulation of dendritic spine morphology and synaptic function by shank and homer. Neuron 31:115–130. 10.1016/S0896-6273(01)00339-7 [DOI] [PubMed] [Google Scholar]
- Salyakina D, Cukier HN, Lee JM, Sacharow S, Nations LD, Ma D, Jaworski JM, Konidari I, Whitehead PL, Wright HH, Abramson RK, Williams SM, Menon R, Haines JL, Gilbert JR, Cuccaro ML, Pericak-Vance MA (2011) Copy number variants in extended autism spectrum disorder families reveal candidates potentially involved in autism risk. PLoS One 6:e26049. 10.1371/journal.pone.0026049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scholl B, Wilson DE, Fitzpatrick D (2017) Local order within global disorder: synaptic architecture of visual space. Neuron 96:1127–1138.e4. 10.1016/j.neuron.2017.10.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma A, Verhaagen J, Harvey AR (2012) Receptor complexes for each of the class 3 semaphorins. Front Cell Neurosci 6:28. 10.3389/fncel.2012.00028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen K, Cowan CW (2010) Guidance molecules in synapse formation and plasticity. Cold Spring Harb Perspect Biol 2:a001842. 10.1101/cshperspect.a001842 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stein IS, Zito K (2019) Dendritic spine elimination: molecular mechanisms and implications. Neuroscientist 25:27–47. 10.1177/1073858418769644 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sytnyk V, Leshchyns'ka I, Schachner M (2017) Neural cell adhesion molecules of the immunoglobulin superfamily regulate synapse formation, maintenance, and function. Trends Neurosci 40:295–308. 10.1016/j.tins.2017.03.003 [DOI] [PubMed] [Google Scholar]
- Tang G, Gudsnuk K, Kuo SH, Cotrina ML, Rosoklija G, Sosunov A, Sonders MS, Kanter E, Castagna C, Yamamoto A, Yue Z, Arancio O, Peterson BS, Champagne F, Dwork AJ, Goldman J, Sulzer D (2014) Loss of mTOR-dependent macroautophagy causes autistic-like synaptic pruning deficits. Neuron 83:1131–1143. 10.1016/j.neuron.2014.07.040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tran TS, Rubio ME, Clem RL, Johnson D, Case L, Tessier-Lavigne M, Huganir RL, Ginty DD, Kolodkin AL (2009) Secreted semaphorins control spine distribution and morphogenesis in the postnatal CNS. Nature 462:1065–1069. 10.1038/nature08628 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turrigiano G. (2012) Homeostatic synaptic plasticity: local and global mechanisms for stabilizing neuronal function. Cold Spring Harb Perspect Biol 4:a005736. 10.1101/cshperspect.a005736 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varshavsky A, Kessler O, Abramovitch S, Kigel B, Zaffryar S, Akiri G, Neufeld G (2008) Semaphorin-3B is an angiogenesis inhibitor that is inactivated by furin-like pro-protein convertases. Cancer Res 68:6922–6931. 10.1158/0008-5472.CAN-07-5408 [DOI] [PubMed] [Google Scholar]
- Vega Y, Arias S, Paradisi I (2017) Most martin-bell syndrome (FMR1-related disorder) venezuelan patients did not show CGG expansion but instead display genetic heterogeneity. J Hum Genet 62:235–241. 10.1038/jhg.2016.114 [DOI] [PubMed] [Google Scholar]
- Villasana LE, Klann E, Tejada-Simon MV (2006) Rapid isolation of synaptoneurosomes and postsynaptic densities from adult mouse hippocampus. J Neurosci Methods 158:30–36. 10.1016/j.jneumeth.2006.05.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Q, Chiu SL, Koropouli E, Hong I, Mitchell S, Easwaran TP, Hamilton NR, Gustina AS, Zhu Q, Ginty DD, Huganir RL, Kolodkin AL (2017) Neuropilin-2/PlexinA3 receptors associate with GluA1 and mediate Sema3F-dependent homeostatic scaling in cortical neurons. Neuron 96:1084–1098.e7. 10.1016/j.neuron.2017.10.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams SR, Mitchell SJ (2008) Direct measurement of somatic voltage clamp errors in central neurons. Nat Neurosci 11:790–798. 10.1038/nn.2137 [DOI] [PubMed] [Google Scholar]
- Wilson DE, Whitney DE, Scholl B, Fitzpatrick D (2016) Orientation selectivity and the functional clustering of synaptic inputs in primary visual cortex. Nat Neurosci 19:1003–1009. 10.1038/nn.4323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright AG, Demyanenko GP, Powell A, Schachner M, Enriquez-Barreto L, Tran TS, Polleux F, Maness PF (2007) Close homolog of L1 and neuropilin 1 mediate guidance of thalamocortical axons at the ventral telencephalon. J Neurosci 27:13667–13679. 10.1523/JNEUROSCI.2888-07.2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamashita N, Usui H, Nakamura F, Chen S, Sasaki Y, Hida T, Suto F, Taniguchi M, Takei K, Goshima Y (2014) Plexin-A4-dependent retrograde semaphorin 3A signalling regulates the dendritic localization of GluA2-containing AMPA receptors. Nat Commun 5:3424. 10.1038/ncomms4424 [DOI] [PubMed] [Google Scholar]
- Ye H, Tan YL, Ponniah S, Takeda Y, Wang SQ, Schachner M, Watanabe K, Pallen CJ, Xiao ZC (2008) Neural recognition molecules CHL1 and NB-3 regulate apical dendrite orientation in the neocortex via PTP alpha. EMBO J 27:188–200. 10.1038/sj.emboj.7601939 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yizhar O. (2012) Optogenetic insights into social behavior function. Biol Psychiatry 71:1075–1080. 10.1016/j.biopsych.2011.12.029 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Multinomial logistic regression fits for spine morphology. Download Figure 2-1, DOCX file (14.7KB, docx)
Multinomial logistic regression fits for spine morphology at P21. Download Figure 2-2, DOCX file (14.9KB, docx)