

Abstract
Thirty years of research on the biology of posttraumatic stress disorder now provides a foundation for hypotheses related to the mechanisms underlying the pharmacotherapy of this disorder. Only two medications, sertraline and paroxetine, are approved by the U.S. Food and Drug Administration for the treatment of PTSD. While these medications are somewhat effective, other treatment mechanisms must be explored to address the unmet need for effective treatment. This article provides a concise summary of advances in our understanding of the neurobiology of PTSD that suggest novel approaches to pharmacotherapy.
Keywords: PTSD, noradrenergic, serotonin, GABA, glutamate, pharmacology
Introduction
Lifetime prevalence of posttraumatic stress disorder (PTSD) in the general population ranges between 6.4 and 7.8% (Pietrzak, Goldstein, Southwick, & Grant, 2011). Rates are much higher among military veterans, with upwards of 20% of combat-exposed veterans meeting criteria for PTSD during their lifetime (Seal et al., 2009). Although there have been significant advances in the understanding of the pathophysiology of PTSD and clinical psychopharmacology, the U.S. Food and Drug Administration (FDA)-approved treatment options are limited. Paroxetine and sertraline, both selective serotonin reuptake inhibitors (SSRIs), are the only FDA-approved pharmacotherapies for PTSD. Unfortunately, there have been no new FDA-approved medications for PTSD for more than 15 years. While response rates as high as 60% have been reported, only about 30% of PTSD patients achieve clinical remission when administered paroxetine or sertraline monotherapy (Alexander, 2012; Berger et al., 2009). Given the suboptimal response rates of first-line pharmacotherapy for PTSD, research is needed to investigate the basic mechanisms and underlying pathways associated with the pathophysiology of this disorder and novel drug development is critical. This view is consistent with current clinical practice. In one study of veterans with long-standing antidepressant-resistant symptoms of PTSD, the average patient entering the study was already receiving, on average, approximately three psychotropic medications (Krystal et al., 2011).
Although numerous neurobiological mechanisms are implicated in the pathogenesis of PTSD, we do not yet fully understand how these mechanisms interact with individual patients. We hypothesize that these risk mechanisms are present to varying degrees in different patients and that they interact in complex ways to facilitate or reduce PTSD risk and recovery, as well as to influence vulnerability to a variety of comorbid conditions. The current paper provides a concise review of the neurobiological factors implicated in PTSD. To highlight factors relevant to pharmacotherapy – which are almost exclusively focused on modulation of neurotransmitter systems – the emphasis is placed on the monoaminergic, glutamatergic, and gamma-amino-butyric acid (GABA)-ergic systems.
Brain Function and Neurotransmitter Response Following Stress
Structural and functional abnormalities have consistently revealed a general imbalance between frontal lobe, amygdala, hippocampus and anterior cingulate cortex (ACC) activation in PTSD (Pitman et al., 2012). Imbalance in distinct neural systems overlaps with the structural and functional brain abnormalities and contributes to the expression of fear and anxiety observed in PTSD. Specifically, the amygdala by virtue of being involved in emotional processing and regulation of fear, including fear conditioning, generalization, and extinction learning serves as a connecting hub of the neural circuits implicated in PTSD (Shin, Rauch, & Pitman, 2006). The amygdala activates brainstem monoaminergic inputs to the PFC; thus, it is likely that elevated amygdalar activity leads to increased input to the frontal lobe regions (Mahan & Ressler, 2012). In order to deal with increased input from the amygdala, an increased feedback inhibition is observed that may explain a decrease in activity in the PFC, which together may be responsible for some of the core behavioral patters observed in PTSD – exaggerated fear response, hypervigilance, and failure to extinguish maladaptive fear response (Shin & Liberzon, 2010; Shin et al., 2006). More specifically, during mild stress, arousal increases transmission of catecholamines [norepinephrine (NE), serotonin (5-HT), and dopamine (DA)], input to the frontal lobe, which enables the PFC to inhibit the amygdala via neuronal glutamatergic outputs that in turn stimulate intra-amygdala GABA-ergic inhibitory interneurons (Pitman et al., 2012). Sensory characteristics of the environment and relevant experiential memories are coded in the hippocampus, which modulates both PFC and amygdala activation (Shin et al., 2006). During high levels of stress or specific threat, monoamine release is increased further to engage noradrenergic (NA) alpha1, dopaminergic (DA1), and serotonergic (5-HT2A) receptors that interfere with PFC-mediated executive function and inhibition of the amygdala (Robbins & Arnsten, 2009). In addition to modifying PFC function, trauma and stress lead to changes in amygdala circuitry that promote defensive responding; for example, chronic stress and fear conditioning diminish GABA mediated activity within the amygdala (Mahan & Ressler, 2012). During intense stress, 5-HT2A receptors are also upregulated in the amygdala, as discussed below.
Monoaminergic System
Serotonergic Neurobiology
Studies suggest that the 5-HT system impacts both PTSD risk and symptom severity (Pitman et al., 2012). The cell bodies of the 5-HT system are located in brainstem’s median and dorsal raphe nuclei, which project widely in the brain, including to key fear circuitry loci within the amygdala, hippocampus, and ventromedial prefrontal cortex (vmPFC), and primarily targets GABA-ergic inhibitory neurons (Neumeister et al., 2013). Numerous preclinical studies have reported heightened 5-HT release, and increased 5-HT synthesis and turnover in response to acute stress (Krystal & Neumeister, 2009). Some alterations in 5HT systems have been described in PTSD using positron emission tomography (PET). Sullivan et al. reported higher density of ligand binding to 5HT1A receptors in both brainstem and forebrain regions (Sullivan et al., 2013). Since 5HT1A receptors are somatic autoreceptors for 5HT neurons, this might suggest that 5HT neurons see increased feedback inhibition. However, there are signs that 5HT nerve terminals may have increased efficacy in that both 5HT1B receptors, terminal autoreceptors, and 5HT transporters are downregulated among patients with PTSD (Murrough et al., 2011; Pietrzak et al., 2013). Together, these findings might suggest that enhanced synaptic 5HT transmission at the level of the nerve terminals might contribute to symptoms of PTSD. This hypothesis was first tested using the 5HT partial agonist, meta-chlorophenylpiperzine (mCPP), which stimulates 5HT2C receptors, among other effects on 5HT signaling. When administered to PTSD patients, this drug evokes anxiety, panic attacks, and PTSD symptoms, including flashbacks (Southwick et al., 1997). The limited efficacy of SSRIs in the treatment of PTSD must be understood in terms of variable individual genetic profile, specific symptom clusters, and other medications typically co-administered with SSRIs and not serve as a clue to abandon our understanding of the 5-HT signaling. (See Figure 1 for a more detailed exposition of the role of the 5-HT system in PTSD symptoms)
Figure 1.
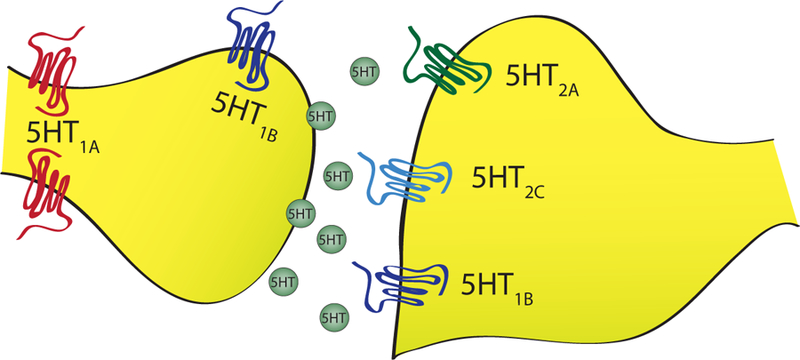
Schematic of serotonin system.
Serotoninergic Pharmacology
Two SSRIs, paroxetine and sertraline, have received FDA- approval for the treatment of PTSD. Randomized controlled trials (RCTs) have demonstrated that paroxetine and sertraline improve PTSD symptomatology in contrast to placebo, and produce remission in about 30% of study participants (Alexander, 2012); although no statistically significant difference has been established in favor of any single SSRI (Benedek DM, 2009). SSRIs have been shown to have a broad spectrum of action and are effective for managing re-experiencing, avoidance, numbing, and hyperarousal symptoms. While multiple RCTs have significantly favored SSRIs in comparison with placebo, there have been numerous studies that have failed to show any difference compared to placebo (Brady et al., 2005; Friedman, Marmar, Baker, Sikes, & Farfel, 2007; Shalev et al., 2012; Tucker et al., 2001). A recent systematic review conducted by the Institute of Medicine questioned the use of SSRIs for veterans with chronic course of combat-related PTSD, reporting that this subgroup responds rather poorly to SSRIs. Interestingly, this refractory group also responds more poorly to psychotherapeutic interventions (Benedek DM, 2009). The issue of SSRIs’ efficacy among combat-exposed veterans vs civil trauma-related PTSD is still under debate and requires further evaluation. The heterogeneity of PTSD may also contribute to the inconsistencies across SSRI RCTs. Furthermore, it is possible that a 6 to 12 week clinical trial does not allot an adequate time to separate the magnitude of the benefit. In an open-label study, when the treatment with sertraline was extended from 12 weeks to 36 weeks, the remission rates increased from 30 to 55% (Londborg et al., 2001). This separation of the magnitude of the benefit of pharmacological treatment vs placebo is also demonstrated in double-blind, placebo-controlled trials (J. Davidson, Baldwin, et al., 2006; J. Davidson et al., 2001).
In addition to SSRI monotherapy, a few small studies have tested whether combining SSRIs with cognitive-behavioral therapy (CBT) might benefit partial responders. One study randomized patients with PTSD with partial response to prolonged exposure (PE) (after eight sessions) to PE continuation with paroxetine augmentation vs placebo and found no benefit to the addition of paroxetine (Simon et al., 2008). On the other hand, two small studies found that the reverse design was effective, PTSD patients who had inadequately responded to sertraline monotherapy showed significant improvement when sertraline treatment was augmented with PE (Otto et al., 2003; Rothbaum et al., 2006). Further research is needed to clarify if and how SSRIs and CBT should be combined to optimize treatment gains.
Other Antidepressants:
Besides SSRIs, a number of other antidepressant medications – most of which affect multiple neurotransmitter systems – have been investigated for the treatment of PTSD. Two multicenter RCTs, one 12 weeks (J. Davidson, Rothbaum, et al., 2006) and the other 6 months (J. Davidson, Baldwin, et al., 2006), showed that venlafaxine – a serotonin and NE reuptake inhibitor (SNRI) – is superior to placebo for the treatment of PTSD. Participants who received venlafaxine reported greater overall symptom reduction and greater rates of remission than those who received placebo. In fact, response rates to venlafaxine were about 10% higher than those reported in previous SSRI trails (J. Davidson, Baldwin, et al., 2006).
One study has shown that mirtazapine, another antidepressant with a mixed mechanism of action (most potent at blocking histamine 1 (H1) receptor, but also exhibits lower potency blockade of postsynaptic 5-HT2 and 5-HT3 receptors and presynaptic noradrenergic alpha-2A/C receptors), was as effective as sertraline (Chung et al., 2004). However, another RCT found no statistically significant differences between mirtazapine and placebo (J. R. Davidson et al., 2003). It is likely that mirtazapine primarily assists with sleep as an antihistamine and is recommended as second-line agent for PTSD-related sleep disturbance (Forbes et al., 2010). However, a recent review concluded that only prazosin, an adrenergic inhibiting agent (see section below), has had its efficacy established in multiple RCTs, and that antidepressants such as sertraline, venlafaxine and mirtazapine, and benzodiazepines and non-benzodiazepine hypnotics such as zolpidem appear ineffective in treating PTSD-related sleep disturbance (Lipinska, Baldwin, & Thomas, 2016).
Tricyclic antidepressants, which block presynaptic reuptake of serotonin and NE, such as amitriptyline (J. Davidson et al., 1990), imipramine (Kosten, Frank, Dan, McDougle, & Giller, 1991) have demonstrated to be superior to placebo for the treatment of PTSD; although there was also a negative RCT with desipramine (Reist et al., 1989). In another study, paroxetine did not show statistical superiority to desipramine, but desipramine was superior to paroxetine with respect to study retention and alcohol use outcomes (Petrakis et al., 2012). An RCT with a monoamine oxidase inhibitor (MAOI), phenelzine, which blocks the intraneuronal metabolic breakdown of 5-HT, NE, and DA, has shown to successfully reduce re-experiencing and arousal symptoms (Kosten et al., 1991). Despite some promising results, tricyclics and MAOIs are typically considered after several failed trials of other SRIs due to the potentially serious side effects associated with tricyclics and MAOIs.
An atypical serotonin agent, 3,4-methylenedioxy-methamphetamine (MDMA), also identified as the street drug “ecstasy”, has been shown to facilitate fear extinction in a preclinical model of PTSD (Young, Andero, Ressler, & Howell, 2015). It induces the release of presynaptic serotonin, activating 5-HT1A, 5-HT1B and 5-HT2A receptors (Liechti & Vollenweider, 2001). MDMA also stimulates the release of DA and NE, producing psychostimulant effects, which may increase motivation to engage in psychotherapy (Hysek et al., 2011).
A pilot clinical study reported long-lasting improvements in symptoms of PTSD following exposure-based psychotherapy sessions paired with MDMA (Mithoefer, Wagner, Mithoefer, Jerome, & Doblin, 2011; Mithoefer et al., 2013). Pilot studies suggest it may be possible to safely administer MDMA to patients with PTSD, and that MDMA assisted-therapy appears to facilitate the emergence of fearful memories that can then be reprocessed in therapy, (Mithoefer et al., 2011). Another MDMA-assisted psychotherapy double-blind active-placebo study (comparing low dose vs high dose of MDMA) found no statistically significant differences in PTSD severity between the two treatment groups at post-treatment but the severity scores improved further at one-year follow-up (Oehen, Traber, Widmer, & Schnyder, 2013). Pilot studies suggest that it may be possible to safely administer MDMA to patients with PTSD. However, negative mood states, distress reactivity, and substantial abuse liability have been associated with MDMA (Mithoefer et al., 2013). Further research is required to explore both the positive and negative effects of MDMA in the context of PTSD treatment.
Noradrenergic Neurobiology
The prominence of hyperadrenergic symptoms in PTSD (e.g. hyperarousal, re-experiencing, and anxiety) naturally made the noradrenergic system a compelling point of investigation (Bailey, Cordell, Sobin, & Neumeister, 2013; van der Kolk, Greenberg, Boyd, & Krystal, 1985). Evidence for noradrenergic hyperactivity in PTSD derives from 1) studies showing increased levels of norepinephrine and its metabolites in cerebrospinal fluid (CSF), blood, and urine at rest or following exposure to trauma reminders, (Delahanty, Raimonde, Spoonster, & Cullado, 2003; Strawn, Ekhator, Horn, Baker, & Geracioti, 2004), and 2) increased catecholamine, behavioral, and neural responses to the alpha2 adrenergic antagonist, yohimbine (Bremner et al., 1997). Early studies found abnormally high levels of catecholamines and their metabolites in the plasma and urine of individuals undergoing severe stress, as well as patients with PTSD, suggesting increased levels of catecholamines may be responsible for some PTSD symptoms (Southwick et al., 1999). Factors that may contribute to noradrenergic hyperactivity in response to traumatic stress or conditioned reminders of stress include decreased expression, affinity, or function of alpha-2-adrenergic autoreceptors (see Figure 2) and genetic or stress-induced decrements in neuropeptide Y (NPY), which inhibits norepinephrine release (A. M. Rasmusson et al., 2000).
Figure 2.
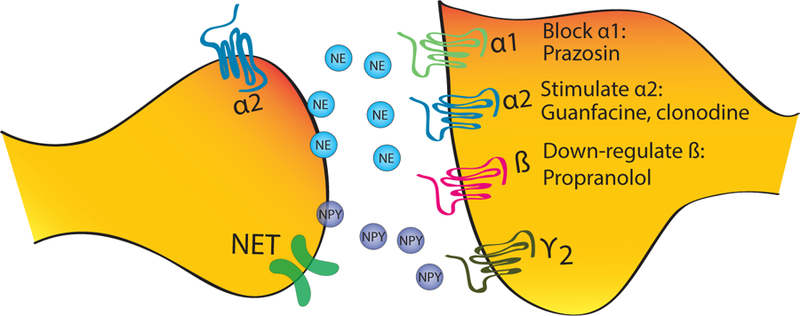
Schematic of noradrenergic system.
There are increased NE and metabolite levels in blood at rest and after reminder of trauma. By process of increased inhibition, not surprisingly, early studies examining the number of alpha-2-adrenergic receptor sites on blood elements (platelets) have shown that both combat veterans and children diagnosed with PTSD had fewer alpha-2-adrenergic receptor binding sites than healthy controls (Perry, Giller, & Southwick, 1987; Pitman et al., 2012). Reduced alpha-2 receptor function conveys increased response to administration of yohimbine, an alpha-2-adrenergic antagonist which increases synaptic norepinephrine by blocking autoinhibitory feedback, associated with symptoms of hyperarousal and re-experiencing in veterans with PTSD and alterations in cortical metabolism (decreased activity in the PFC) (Southwick et al., 1997) (Bremner et al., 1997)Pre-clinical studies have shown that yohimbine facilitates not only the formation and recall of aversive memories in rodents, but also extinction of cue and contextual fear (Mueller, Olivera-Figueroa, Pine, & Quirk, 2009). Although clinical trials are currently underway (Wangelin et al., 2013), there is not yet evidence that yohimbine can be used to augment exposure-based therapies for PTSD.
NPY is released in response to intense psychological stress or pain and binds to NPY-Y1 receptors to decreased release of norepinephrine (A. M. Rasmusson, Southwick, Hauger, & Charney, 1998). NPY is implicated in arousal and the assignment of emotional valences to stimuli and memory (Silva, Xapelli, Grouzmann, & Cavadas, 2005). Not surprisingly, stress-induced reductions in NPY are implicated in overall increased release of NE in PTSD (Neumeister et al., 2013; Perry et al., 1987). Human research suggests NPY-Y1 receptor agonist in the amygdala exerts anxiolytic effects (Pitman et al., 2012) and is also involved in stress resilience (Morgan et al., 2002). Soldiers in the Special Forces who produced higher plasma levels of NPY levels during highly stressful military training were found to have better performance during training and lower stress-induced dissociation and distress (Morgan et al., 2000). Low baseline plasma NPY levels and blunted NPY response to increased NE levels by administration of yohimbine have been found in male veterans with PTSD (A. M. Rasmusson et al., 2000). Pretreatment with intranasal (before exposure to stress paradigm) NPY has been shown to attenuate the development of PTSD-like symptoms in rodent models of PTSD (Sabban, Alaluf, & Serova, 2015). However, efforts to develop pharmacological agents harnessing NPY-receptor-mediated effects have thus far been unsuccessful (Pitman et al., 2012).
Reduction in the norepinephrine transporter (NET) expression enhances synaptic availability of NE. It is likely that the dyregulation of NET expression may result from ongoing elevated NE levels, as preclinical studies show that endogenous DA and norepinephrine stimulate NET expression (Krystal & Neumeister, 2009). NET acts as norepinephrine plasma membrane transporter and maintains NE presynaptic storage. Chronic stress leads to a reduction of NET availability in locus ceruleus (LC), while in the PFC there is an increase in NET expression, suggesting this may represent an attempt to maintain normal availability, and consequently normal function of NE (Bailey et al., 2013). These preclinical findings were recently replicated in a human PET study demonstrating that PTSD is associated with significantly decreased NET availability in the LC, which might, in turn, result in the exaggerated synaptic availability of norepinephrine in projection areas, such as the PFC (Pietrzak et al., 2013). Despite these informative preclinical models and clinical evidence, the role of the antidepressants with a high-affinity for NETs (e.g., venlafaxine) in the treatment of PTSD remains unclear.
Noradrenergic Pharmacology
Normalization of hyperadrenergic activity, which is suggested to contribute to hyperarousal symptoms, has been attempted by 1) blocking alpha-1-adrenoreceptor with prazosin (a post-synaptic alpha-1-adrenoreceptor antagonist), 2) stimulating alpha-2-adrenoreceptor with guanfacine and clonidine (both alpha-2-adrenoreceptor agonists with slight differential affinity for different alpha-2 receptors), and 3) blocking beta-adrenergic receptor with the antagonist propranolol. Two small RCTs have shown prazosin to be more effective than placebo at reducing traumatic nightmares for patients with PTSD (Raskind et al., 2007; Taylor et al., 2006). A recent RCT with combat-exposed veterans also showed that prazosin effectively reduced overall PTSD symptom severity (Raskind et al., 2013). However, a recent double-blind placebo-controlled study showed that prazosin had no effect in reducing PTSD symptoms or improving sleep in a 96 patient with PTSD and comorbid alcohol dependence (Petrakis et al., 2016). Findings thus far suggest that prazosin may be effective at treating sleep disturbances in PTSD; however, results may vary based on comorbid conditions. Both guanfacine and clonidine have been failed to demonstrate efficacy in the treatment of PTSD in open trials (Kolb, 1984; Neylan et al., 2006). The role of propranolol has been investigated as monotherapy, as prophylactic following the immediate aftermath of trauma, and in conjunction with therapy as an aid to disrupt reconsolidation of traumatic memories in patients with PTSD; however, the results have been inconsistent and unpromising (Pitman et al., 2012). Despite the promising results in preventing consolidation of aversive memories in preclinical studies, propranolol has been shown to be ineffective for the prevention of PTSD (Pitman et al., 2012; Stein, Kerridge, Dimsdale, & Hoyt, 2007). Although propranolol may not be effective as monotherapy for treatment of PTSD, there has been a recent resurgence of interest in using it in conjunction with exposure-based therapies.
This highlights the importance of conducting studies in a ‘real-world’ patients with PTSD (Petrakis et al., 2016).
Dopaminergic Neurobiology
Less is known about the involvement of the dopaminergic system in PTSD because, unlike 5-HT and NE peripheral measures of dopamine function do not correlate well with the central measures (A. M. Rasmusson, Riddle, M. A., Leckman, J. F., Anderson, G. M., & Cohen, D. J., 1990). However, the dopaminergic system is implicated in the pathophysiology of this disorder by virtue of being one of the most important neurotransmitters in reward signal processing, a process that appears to be downregulated in PTSD (Elman et al., 2009). Further evidence for the involvement of the dopaminergic system in PTSD comes from rodent studies showing that DA release in the amygdala promotes modulation of unconditional and conditional stress responding.
Dopaminergic Pharmacology
Atypical antipsychotics, primarily effect the dopaminergic system, have largely been considered as adjunctive medications for partial responders to antidepressants. A series of small RCTs, involving olanzapine and risperidone, suggest that they might be useful as SSRI adjunctive medications for PTSD (Pitman et al., 2012). However, these findings are limited by small to moderate sample sizes. A recent large, multisite RCT investigated adjunctive risperidone in 247 veterans who were partial responders to antidepressants and found that risperidone augmentation was no better than placebo (Krystal et al., 2011). To date, the evidence for the use of atypical antipsychotics is insufficient to determine efficacy in the treatment of PTSD.
Glutamatergic System
Neurobiology
The main projections from the PFC to the amygdala and hippocampus are glutamatergic in nature; more specifically, the PFC can modulate the activity of these areas either directly or indirectly by acting on DA and acetylcholine neurons (Del Arco & Mora, 2009). The deficient top-down control from the PFC to the amygdala, as described above, implies that the glutamatergic pathways are potentially involved in the pathophysiology of PTSD. Furthermore, glutamatergic projections from the PFC, specifically vmPFC, hippocampus and amygdala are crucial in the modulation of fear learning, extinction and contextual fear conditioning (Morrison & Ressler, 2014), a firmly established finding in the literature.
Glutamate, the most common and main excitatory neurotransmitter in the central nervous system (CNS), binds to a variety of receptors most generally classified into two categories: 1) ionotropic (ion bearing channel) glutamatergic receptors consists of N-methyl-D-aspartate receptor (NMDA), α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA), and kainite, and 2) metabotropic glutamatergic receptors (mGluRs) – which can be eight different types (mGluR1 – mGluR8) – operate pre- and postsynaptically to increase or decrease ionotropic activity. Ionotropic receptors are crucial for synaptic plasticity and, as such, most forms of learning and memory. Not surprising, aberrant glutamatergic transmission is implicated in the pathophysiology of PTSD via essential role the NMDA receptors play in fear conditioning and extinction (Bailey et al., 2013). This makes ionotropic receptors critically important for the development and treatment of PTSD. Although metabotropic glutamate receptors have also been implicated in stress-related pathology, much of the work is isolated to preclinical models of anxiety and more conclusive work remains to be done in human studies.
NMDA receptor activation plays an essential role in fear learning; whereas blockade of NMDA receptors with an antagonist blocks the memory consolidation processes (Morrison & Ressler, 2014). Paradoxically anesthetic doses of ketamine, NMDA receptor antagonist, in case series report among burn victims receiving surgery has shown to reduce posttraumatic stress symptoms (McGhee, Maani, Garza, Gaylord, & Black, 2008). Conversely, peritraumatic administration of anesthetic ketamine before surgery was associated with a worsening of acute posttraumatic stress symptoms among accident victims (Schonenberg, Reichwald, Domes, Badke, & Hautzinger, 2005, 2008). Furthermore dissociative symptoms, including emotional numbing, reduction in awareness of one’s environment, depersonalization, and memory impairment, frequently seen in acute trauma have all been observed in ketamine intoxication (Krystal et al., 1994). Individuals who dissociate following a trauma are more likely to develop PTSD than those who do not (Chambers et al., 1999). Ketamine administration in the hours following trauma may make the development of PTSD more likely (Schonenberg et al., 2005, 2008), suggesting an important role of intact NMDA receptor for healthy trauma response and providing further evidence that NMDA receptor dysfunction contributes to PTSD symptomatology.
Pharmacological Interventions
The above-mentioned studies suggest that anesthetic doses of ketamine might potentiate memory if administered shortly following a traumatic event and may reduce symptomatology if administered well after the traumatic event. A recent case report suggested that a single intravenous subanesthetic dose of ketamine may temporarily reduce posttraumatic stress symptoms (D’Andrea & Andrew Sewell, 2013). More recent study administered ketamine and midazolam to forty-one patients with a primary diagnosis of PTSD in a randomized, double-blind, crossover trial, found that single ketamine infusion provided a temporary reduction in symptoms (Feder et al., 2014). Both drugs were associated with rapid reductions in posttraumatic stress symptoms, but ketamine outperformed midazolam; however, no inactive placebo was used in this study.
D-Cycloserine (DCS) is a partial NMDA receptor agonist that has been tested as a monotherapy and treatment augmentation agent for PTSD. The DCS clinical studies have mostly failed to demonstrate efficacy in the treatment of PSD. There is minimal evidence that DCS is efficacious as monotherapy (Heresco-Levy et al., 2002) or as an add-on for standard pharmacological treatments for PTSD (Attari, Rajabi, & Maracy, 2014). There is weak evidence that DCS can enhance the efficacy of exposure therapy for PTSD. Only one RCT has found that PTSD patients who receive DCS and exposure therapy fare better than their counterparts who receive exposure therapy with a placebo pill (Difede et al., 2014). However, two other RCTs failed to show therapeutic exposure enhancing effects of DCS (de Kleine, Hendriks, Kusters, Broekman, & van Minnen, 2012; Rothbaum et al., 2014). One study reported that DCS was detrimental to extinction learning (Litz et al., 2012). The only study to report positive effects of DCS used a larger dose (100 mg) than those studies that reported negative findings (50 mg) (Difede et al., 2014), raising the possibility that larger doses are required in PTSD to significantly enhance exposure therapy.
GABA
Neurobiology
The deficits in GABA signaling demonstrated in PTSD may contribute more broadly to risk for PTSD symptoms (Goddard et al., 2001; Pollack, Jensen, Simon, Kaufman, & Renshaw, 2008; Rosso et al., 2014). For example, deficits in the GABA-ergic inhibition may explain why the administration of mCPP exacerbates PTSD symptoms in patients but not in healthy controls, but mCPP can produce panic attacks and dissociative symptoms in healthy controls when pretreated with iomazenil, inverse GABA agonist {D’Souza, 2006 #274}. GABA-ergic inhibitory control seems to be critically involved in fine-tuning regulation of expression, consolidation, and extinction of fear conditioning (Mahan & Ressler, 2012) (Bustos, Maldonado, & Molina, 2006). Not surprisingly, studies have shown significantly lower GABA levels among individuals exposed to trauma who developed PTSD as compared to those who did not (Meyerhoff, Mon, Metzler, & Neylan, 2014; Rosso et al., 2014) and reduced GABA levels have are strongly correlated with severity of insomnia symptoms among patients with PTSD (Meyerhoff et al., 2014). Decreased GABA levels in PTSD are consistent with previous finding of low cortical and subcortical benzodiazepine receptor binding in patients with PTSD and panic disorder (Bremner, Southwick, Darnell, & Charney, 1996; Charney, 2004), and suggest that the GABAergic system may be a potential therapeutic target.
Pharmacological Interventions
Treatment of PTSD-related symptoms has long included benzodiazepines, GABA receptor agonists. However, the results from RCTs indicate that this class of medications has not been helpful in the treatment of PTSD, despite their widespread use in the treatment for PTSD. Though recently their use has been in decline (Lund, Bernardy, Alexander, & Friedman, 2012) due to low risk/benefit ratio because of addictive potential, sedation in older adults, and the possibility that they might interfere with psychological processes needed to benefit from CBT. Therefore, the 2010 VA/DoD Clinical Practice Guideline discouraged the use of benzodiazepines for the treatment of both acute stress disorder and PTSD (Forbes et al., 2010). Eszopiclone – a high affinity GABA-A receptor agonist – has been shown to significantly improve sleep disturbance associated with PTSD (Pollack et al., 2008). GABAergic anticonvulsants are also often prescribed for PTSD-related symptoms, but results have not been uniform. One large placebo-controlled study of tiagabine found no significant effect of the drug on PTSD, depression, or functional impairment (J. R. Davidson, Brady, Mellman, Stein, & Pollack, 2007). Divalproex was similarly shown to have no significant effect on PTSD symptoms (Hamner et al., 2009). In a small international trial, topiramate was reported to significantly reduce PTSD symptoms (Yeh et al., 2011), but these results have yet to be replicated. In summary, GABAergic drugs are widely prescribed but incompletely understood, and clinical trials have thus far yielded underwhelming results. Each comes with potential side effects, tolerance issues, and addictive potential, as well as possible effects on neurocognitive functioning.
Future Directions
Pharmacological trials for PTSD, so far, have utilized agents with established efficacy for other psychiatric disorders. A weakness of the current clinical research is the lack of focus on how treatments help different PTSD symptom clusters. In order to identify effective treatments for PTSD, research could focus on identifying which disruptions in neural circuitry and neurotransmitter systems relate to specific symptom clusters. This approach will then lead to developing medications designed address various dimensions of PTSD symptomatology.
One promising approach to improving the treatment for PTSD is to combine pharmacological agents with behavioral interventions that specifically target maladaptive fear processing and learning. For example, neuroimaging studies over the past two decades have repeatedly revealed specific brain activation in response to fearful and trauma-related stimuli in patients with PTSD. This robust neural signal has been a useful biological marker of PTSD and has guided the development of improved exposure-based therapies for PTSD by targeting dysfunction in this primary regulatory control network that drives exaggerated fear response and dysregulated affect (Morrison & Ressler, 2014). Although, as mentioned earlier, propranolol as a monotherapy for treatment of PTSD has demonstrated mixed results, at best - a recent resurgence of interest in using propranolol in conjunction with psychotherapy is gaining research traction. In addition, MDMA has recently been shown to facilitate fear extinction in a preclinical model of PTSD and preliminary evidence indicates that MDMA may be effective for assisting psychotherapeutic interventions for PTSD. In this context, the administration of MDMA is hypothesized to reduce the fear response to the emotional threat, allowing patients to revisit traumatic memories in a therapeutic setting, without feeling overwhelmed (Mithoefer et al., 2011). Along these same lines of research, agents targeting the cannabinoid system (for a more thorough review see Berardi et al., 2016) have shown preliminary support for management anxious and dysphoric symptoms and sleep disturbances in PTSD (Neumeister, Seidel, Ragen, & Pietrzak, 2015) (Berardi, Schelling, & Campolongo, 2016). Oxytocin has also received considerable attention recently in treatment of psychological disorders. Oxytocin is known for its pro-social effects. The effect of oxytocin on anxiety and fear extinction and expression provides a possibility as an adjunct treatment for patients with PTSD. Human studies have provided evidence that oxytocin administration may attenuate fear expression and facilitate fear extinction but the timing of administration appears to be critical in whether it can facilitate fear extinction (Bailey et al., 2013). Whether oxytocin can be used a prophylactic, monotherapy, or in conjunction with exposure-based therapies remains unclear. However, while these small studies are of benefit, they do not carry the same weight as RCTs leaving the degree of potential benefit in question.
Conclusion
Currently, FDA-approved treatments have demonstrated mild to moderate success. The research reviewed in this article reveals that, while much knowledge has been generated towards the underlying pathophysiology of PTSD and many putative pharmacological treatments have emerged, very few studies have resulted in treatments that are novel, effective, and specific to PTSD symptoms. Most novel treatments have insufficient evidence to draw meaningful conclusions about their efficacy. A promising direction in the field is the use of brain plasticity inducers as monotherapies or in conjunction with learning-based psychotherapies. Another possible treatment approach for this complex disorder may be the use of multiple drugs simultaneously. The development of treatment algorithms to guide such combinatorial treatments will be a necessary next step.
Supplementary Material
References
- Alexander W (2012). Pharmacotherapy for Post-traumatic Stress Disorder In Combat Veterans: Focus on Antidepressants and Atypical Antipsychotic Agents. P T, 37(1), 32–38. [PMC free article] [PubMed] [Google Scholar]
- Attari A, Rajabi F, & Maracy MR (2014). D-cycloserine for treatment of numbing and avoidance in chronic post traumatic stress disorder: A randomized, double blind, clinical trial. J Res Med Sci, 19(7), 592–598. [PMC free article] [PubMed] [Google Scholar]
- Bailey CR, Cordell E, Sobin SM, & Neumeister A (2013). Recent progress in understanding the pathophysiology of post-traumatic stress disorder: implications for targeted pharmacological treatment. CNS Drugs, 27(3), 221–232. doi: 10.1007/s40263-013-0051-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benedek DM FM, Zatzick D, Ursano RJ. (2009). Guideline watcha (March 2009): Practice guideline for the treatment of patients with acute stress disorder and posttraumatic stress disorder. J Lifelong Learning(7), 204–2013. [Google Scholar]
- Berardi A, Schelling G, & Campolongo P (2016). The endocannabinoid system and Post Traumatic Stress Disorder (PTSD): From preclinical findings to innovative therapeutic approaches in clinical settings. Pharmacol Res, 111, 668–678. doi: 10.1016/j.phrs.2016.07.024 [DOI] [PubMed] [Google Scholar]
- Berger W, Mendlowicz MV, Marques-Portella C, Kinrys G, Fontenelle LF, Marmar CR, & Figueira I (2009). Pharmacologic alternatives to antidepressants in posttraumatic stress disorder: a systematic review. Prog Neuropsychopharmacol Biol Psychiatry, 33(2), 169–180. doi: 10.1016/j.pnpbp.2008.12.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brady KT, Sonne S, Anton RF, Randall CL, Back SE, & Simpson K (2005). Sertraline in the treatment of co-occurring alcohol dependence and posttraumatic stress disorder. Alcohol Clin Exp Res, 29(3), 395–401. [DOI] [PubMed] [Google Scholar]
- Bremner JD, Innis RB, Ng CK, Staib LH, Salomon RM, Bronen RA, … Charney DS (1997). Positron emission tomography measurement of cerebral metabolic correlates of yohimbine administration in combat-related posttraumatic stress disorder. Arch Gen Psychiatry, 54(3), 246–254. [DOI] [PubMed] [Google Scholar]
- Bremner JD, Southwick SM, Darnell A, & Charney DS (1996). Chronic PTSD in Vietnam combat veterans: course of illness and substance abuse. Am J Psychiatry, 153(3), 369–375. doi: 10.1176/ajp.153.3.369 [DOI] [PubMed] [Google Scholar]
- Bustos SG, Maldonado H, & Molina VA (2006). Midazolam disrupts fear memory reconsolidation. Neuroscience, 139(3), 831–842. doi: 10.1016/j.neuroscience.2005.12.064 [DOI] [PubMed] [Google Scholar]
- Chambers RA, Bremner JD, Moghaddam B, Southwick SM, Charney DS, & Krystal JH (1999). Glutamate and post-traumatic stress disorder: toward a psychobiology of dissociation. Semin Clin Neuropsychiatry, 4(4), 274–281. doi: 10.153/SCNP00400274 [DOI] [PubMed] [Google Scholar]
- Charney DS (2004). Psychobiological mechanisms of resilience and vulnerability: implications for successful adaptation to extreme stress. Am J Psychiatry, 161(2), 195–216. doi: 10.1176/appi.ajp.161.2.195 [DOI] [PubMed] [Google Scholar]
- Chung MY, Min KH, Jun YJ, Kim SS, Kim WC, & Jun EM (2004). Efficacy and tolerability of mirtazapine and sertraline in Korean veterans with posttraumatic stress disorder: a randomized open label trial. Hum Psychopharmacol, 19(7), 489–494. doi: 10.1002/hup.615 [DOI] [PubMed] [Google Scholar]
- D’Andrea D, & Andrew Sewell R (2013). Transient resolution of treatment-resistant posttraumatic stress disorder following ketamine infusion. Biol Psychiatry, 74(9), e13–14. doi: 10.1016/j.biopsych.2013.04.019 [DOI] [PubMed] [Google Scholar]
- Davidson J, Baldwin D, Stein DJ, Kuper E, Benattia I, Ahmed S, … Musgnung J (2006). Treatment of posttraumatic stress disorder with venlafaxine extended release: a 6-month randomized controlled trial. Arch Gen Psychiatry, 63(10), 1158–1165. doi: 10.1001/archpsyc.63.10.1158 [DOI] [PubMed] [Google Scholar]
- Davidson J, Kudler H, Smith R, Mahorney SL, Lipper S, Hammett E, … Cavenar JO Jr. (1990). Treatment of posttraumatic stress disorder with amitriptyline and placebo. Arch Gen Psychiatry, 47(3), 259–266. [DOI] [PubMed] [Google Scholar]
- Davidson J, Pearlstein T, Londborg P, Brady KT, Rothbaum B, Bell J, … Farfel G (2001). Efficacy of sertraline in preventing relapse of posttraumatic stress disorder: results of a 28-week double-blind, placebo-controlled study. Am J Psychiatry, 158(12), 1974–1981. doi: 10.1176/appi.ajp.158.12.1974 [DOI] [PubMed] [Google Scholar]
- Davidson J, Rothbaum BO, Tucker P, Asnis G, Benattia I, & Musgnung JJ (2006). Venlafaxine extended release in posttraumatic stress disorder: a sertraline- and placebo-controlled study. J Clin Psychopharmacol, 26(3), 259–267. doi: 10.1097/01.jcp.0000222514.71390.c1 [DOI] [PubMed] [Google Scholar]
- Davidson JR, Brady K, Mellman TA, Stein MB, & Pollack MH (2007). The efficacy and tolerability of tiagabine in adult patients with post-traumatic stress disorder. J Clin Psychopharmacol, 27(1), 85–88. doi: 10.1097/JCP.0b013e31802e5115 [DOI] [PubMed] [Google Scholar]
- Davidson JR, Weisler RH, Butterfield MI, Casat CD, Connor KM, Barnett S, & van Meter S (2003). Mirtazapine vs. placebo in posttraumatic stress disorder: a pilot trial. Biol Psychiatry, 53(2), 188–191. [DOI] [PubMed] [Google Scholar]
- de Kleine RA, Hendriks GJ, Kusters WJ, Broekman TG, & van Minnen A (2012). A randomized placebo-controlled trial of D-cycloserine to enhance exposure therapy for posttraumatic stress disorder. Biol Psychiatry, 71(11), 962–968. doi: 10.1016/j.biopsych.2012.02.033 [DOI] [PubMed] [Google Scholar]
- Del Arco A, & Mora F (2009). Neurotransmitters and prefrontal cortex-limbic system interactions: implications for plasticity and psychiatric disorders. J Neural Transm (Vienna), 116(8), 941–952. doi: 10.1007/s00702-009-0243-8 [DOI] [PubMed] [Google Scholar]
- Delahanty DL, Raimonde AJ, Spoonster E, & Cullado M (2003). Injury severity, prior trauma history, urinary cortisol levels, and acute PTSD in motor vehicle accident victims. J Anxiety Disord, 17(2), 149–164. [DOI] [PubMed] [Google Scholar]
- Difede J, Cukor J, Wyka K, Olden M, Hoffman H, Lee FS, & Altemus M (2014). D-cycloserine augmentation of exposure therapy for post-traumatic stress disorder: a pilot randomized clinical trial. Neuropsychopharmacology, 39(5), 1052–1058. doi: 10.1038/npp.2013.317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elman I, Lowen S, Frederick BB, Chi W, Becerra L, & Pitman RK (2009). Functional neuroimaging of reward circuitry responsivity to monetary gains and losses in posttraumatic stress disorder. Biol Psychiatry, 66(12), 1083–1090. doi: 10.1016/j.biopsych.2009.06.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feder A, Parides MK, Murrough JW, Perez AM, Morgan JE, Saxena S, … Charney DS (2014). Efficacy of intravenous ketamine for treatment of chronic posttraumatic stress disorder: a randomized clinical trial. JAMA Psychiatry, 71(6), 681–688. doi: 10.1001/jamapsychiatry.2014.62 [DOI] [PubMed] [Google Scholar]
- Forbes D, Creamer M, Bisson JI, Cohen JA, Crow BE, Foa EB, … Ursano RJ (2010). A guide to guidelines for the treatment of PTSD and related conditions. J Trauma Stress, 23(5), 537–552. doi: 10.1002/jts.20565 [DOI] [PubMed] [Google Scholar]
- Friedman MJ, Marmar CR, Baker DG, Sikes CR, & Farfel GM (2007). Randomized, double-blind comparison of sertraline and placebo for posttraumatic stress disorder in a Department of Veterans Affairs setting. J Clin Psychiatry, 68(5), 711–720. [DOI] [PubMed] [Google Scholar]
- Goddard AW, Mason GF, Almai A, Rothman DL, Behar KL, Petroff OA, … Krystal JH (2001). Reductions in occipital cortex GABA levels in panic disorder detected with 1h-magnetic resonance spectroscopy. Arch Gen Psychiatry, 58(6), 556–561. [DOI] [PubMed] [Google Scholar]
- Hamner MB, Faldowski RA, Robert S, Ulmer HG, Horner MD, & Lorberbaum JP (2009). A preliminary controlled trial of divalproex in posttraumatic stress disorder. Ann Clin Psychiatry, 21(2), 89–94. [PubMed] [Google Scholar]
- Heresco-Levy U, Kremer I, Javitt DC, Goichman R, Reshef A, Blanaru M, & Cohen T (2002). Pilot-controlled trial of D-cycloserine for the treatment of post-traumatic stress disorder. Int J Neuropsychopharmacol, 5(4), 301–307. doi: 10.1017/S1461145702003061 [DOI] [PubMed] [Google Scholar]
- Hysek CM, Simmler LD, Ineichen M, Grouzmann E, Hoener MC, Brenneisen R, … Liechti ME (2011). The norepinephrine transporter inhibitor reboxetine reduces stimulant effects of MDMA (“ecstasy”) in humans. Clin Pharmacol Ther, 90(2), 246–255. doi: 10.1038/clpt.2011.78 [DOI] [PubMed] [Google Scholar]
- Kolb LC, Burris BC, & Griffiths S (1984). Propranolol and clonidine in the treatment of chronic post-traumatic stress disorders of war. In van der Kolk BA (Ed.), Post-traumatic stress disorder: Psychological and biological sequelae Washington, D.C.: American Psychiatric Press. [Google Scholar]
- Kosten TR, Frank JB, Dan E, McDougle CJ, & Giller EL Jr. (1991). Pharmacotherapy for posttraumatic stress disorder using phenelzine or imipramine. J Nerv Ment Dis, 179(6), 366–370. [DOI] [PubMed] [Google Scholar]
- Krystal JH, Karper LP, Seibyl JP, Freeman GK, Delaney R, Bremner JD, … Charney DS (1994). Subanesthetic effects of the noncompetitive NMDA antagonist, ketamine, in humans. Psychotomimetic, perceptual, cognitive, and neuroendocrine responses. Arch Gen Psychiatry, 51(3), 199–214. [DOI] [PubMed] [Google Scholar]
- Krystal JH, & Neumeister A (2009). Noradrenergic and serotonergic mechanisms in the neurobiology of posttraumatic stress disorder and resilience. Brain Res, 1293, 13–23. doi: 10.1016/j.brainres.2009.03.044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krystal JH, Rosenheck RA, Cramer JA, Vessicchio JC, Jones KM, Vertrees JE, … Veterans Affairs Cooperative Study No, G. (2011). Adjunctive risperidone treatment for antidepressant-resistant symptoms of chronic military service-related PTSD: a randomized trial. JAMA, 306(5), 493–502. doi: 10.1001/jama.2011.1080 [DOI] [PubMed] [Google Scholar]
- Liechti ME, & Vollenweider FX (2001). Which neuroreceptors mediate the subjective effects of MDMA in humans? A summary of mechanistic studies. Hum Psychopharmacol, 16(8), 589–598. doi: 10.1002/hup.348 [DOI] [PubMed] [Google Scholar]
- Lipinska G, Baldwin DS, & Thomas KG (2016). Pharmacology for sleep disturbance in PTSD. Hum Psychopharmacol, 31(2), 156–163. doi: 10.1002/hup.2522 [DOI] [PubMed] [Google Scholar]
- Litz BT, Salters-Pedneault K, Steenkamp MM, Hermos JA, Bryant RA, Otto MW, & Hofmann SG (2012). A randomized placebo-controlled trial of D-cycloserine and exposure therapy for posttraumatic stress disorder. J Psychiatr Res, 46(9), 1184–1190. doi: 10.1016/j.jpsychires.2012.05.006 [DOI] [PubMed] [Google Scholar]
- Londborg PD, Hegel MT, Goldstein S, Goldstein D, Himmelhoch JM, Maddock R, … Farfel GM (2001). Sertraline treatment of posttraumatic stress disorder: results of 24 weeks of open-label continuation treatment. J Clin Psychiatry, 62(5), 325–331. [DOI] [PubMed] [Google Scholar]
- Lund BC, Bernardy NC, Alexander B, & Friedman MJ (2012). Declining benzodiazepine use in veterans with posttraumatic stress disorder. J Clin Psychiatry, 73(3), 292–296. doi: 10.4088/JCP.10m06775 [DOI] [PubMed] [Google Scholar]
- Mahan AL, & Ressler KJ (2012). Fear conditioning, synaptic plasticity and the amygdala: implications for posttraumatic stress disorder. Trends Neurosci, 35(1), 24–35. doi: 10.1016/j.tins.2011.06.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGhee LL, Maani CV, Garza TH, Gaylord KM, & Black IH (2008). The correlation between ketamine and posttraumatic stress disorder in burned service members. J Trauma, 64(2 Suppl), S195–198; Discussion S197–198. doi: 10.1097/TA.0b013e318160ba1d [DOI] [PubMed] [Google Scholar]
- Meyerhoff DJ, Mon A, Metzler T, & Neylan TC (2014). Cortical gamma-aminobutyric acid and glutamate in posttraumatic stress disorder and their relationships to self-reported sleep quality. Sleep, 37(5), 893–900. doi: 10.5665/sleep.3654 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mithoefer MC, Wagner MT, Mithoefer AT, Jerome L, & Doblin R (2011). The safety and efficacy of {+/−}3,4-methylenedioxymethamphetamine-assisted psychotherapy in subjects with chronic, treatment-resistant posttraumatic stress disorder: the first randomized controlled pilot study. J Psychopharmacol, 25(4), 439–452. doi: 10.1177/0269881110378371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mithoefer MC, Wagner MT, Mithoefer AT, Jerome L, Martin SF, Yazar-Klosinski B, … Doblin R (2013). Durability of improvement in post-traumatic stress disorder symptoms and absence of harmful effects or drug dependency after 3,4-methylenedioxymethamphetamine-assisted psychotherapy: a prospective long-term follow-up study. J Psychopharmacol, 27(1), 28–39. doi: 10.1177/0269881112456611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan CA 3rd, Rasmusson AM, Wang S, Hoyt G, Hauger RL, & Hazlett G (2002). Neuropeptide-Y, cortisol, and subjective distress in humans exposed to acute stress: replication and extension of previous report. Biol Psychiatry, 52(2), 136–142. [DOI] [PubMed] [Google Scholar]
- Morgan CA 3rd, Wang S, Southwick SM, Rasmusson A, Hazlett G, Hauger RL, & Charney DS (2000). Plasma neuropeptide-Y concentrations in humans exposed to military survival training. Biol Psychiatry, 47(10), 902–909. [DOI] [PubMed] [Google Scholar]
- Morrison FG, & Ressler KJ (2014). From the neurobiology of extinction to improved clinical treatments. Depress Anxiety, 31(4), 279–290. doi: 10.1002/da.22214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mueller D, Olivera-Figueroa LA, Pine DS, & Quirk GJ (2009). The effects of yohimbine and amphetamine on fear expression and extinction in rats. Psychopharmacology (Berl), 204(4), 599–606. doi: 10.1007/s00213-009-1491-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murrough JW, Czermak C, Henry S, Nabulsi N, Gallezot JD, Gueorguieva R, … Neumeister A (2011). The effect of early trauma exposure on serotonin type 1B receptor expression revealed by reduced selective radioligand binding. Arch Gen Psychiatry, 68(9), 892–900. doi: 10.1001/archgenpsychiatry.2011.91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neumeister A, Normandin MD, Pietrzak RH, Piomelli D, Zheng MQ, Gujarro-Anton A, … Huang Y (2013). Elevated brain cannabinoid CB1 receptor availability in post-traumatic stress disorder: a positron emission tomography study. Mol Psychiatry, 18(9), 1034–1040. doi: 10.1038/mp.2013.61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neumeister A, Seidel J, Ragen BJ, & Pietrzak RH (2015). Translational evidence for a role of endocannabinoids in the etiology and treatment of posttraumatic stress disorder. Psychoneuroendocrinology, 51, 577–584. doi: 10.1016/j.psyneuen.2014.10.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neylan TC, Lenoci M, Samuelson KW, Metzler TJ, Henn-Haase C, Hierholzer RW, … Marmar CR (2006). No improvement of posttraumatic stress disorder symptoms with guanfacine treatment. Am J Psychiatry, 163(12), 2186–2188. doi: 10.1176/appi.ajp.163.12.2186 [DOI] [PubMed] [Google Scholar]
- Oehen P, Traber R, Widmer V, & Schnyder U (2013). A randomized, controlled pilot study of MDMA (+/− 3,4-Methylenedioxymethamphetamine)-assisted psychotherapy for treatment of resistant, chronic Post-Traumatic Stress Disorder (PTSD). J Psychopharmacol, 27(1), 40–52. doi: 10.1177/0269881112464827 [DOI] [PubMed] [Google Scholar]
- Otto MW, Hinton D, Korbly NB, Chea A, Ba P, Gershuny BS, & Pollack MH (2003). Treatment of pharmacotherapy-refractory posttraumatic stress disorder among Cambodian refugees: a pilot study of combination treatment with cognitive-behavior therapy vs sertraline alone. Behav Res Ther, 41(11), 1271–1276. [DOI] [PubMed] [Google Scholar]
- Perry BD, Giller EL Jr., & Southwick SM (1987). Altered platelet alpha 2-adrenergic binding sites in posttraumatic stress disorder. Am J Psychiatry, 144(11), 1511–1512. [DOI] [PubMed] [Google Scholar]
- Petrakis IL, Desai N, Gueorguieva R, Arias A, O’Brien E, Jane JS, … Ralevski E (2016). Prazosin for Veterans with Posttraumatic Stress Disorder and Comorbid Alcohol Dependence: A Clinical Trial. Alcohol Clin Exp Res, 40(1), 178–186. doi: 10.1111/acer.12926 [DOI] [PubMed] [Google Scholar]
- Petrakis IL, Ralevski E, Desai N, Trevisan L, Gueorguieva R, Rounsaville B, & Krystal JH (2012). Noradrenergic vs serotonergic antidepressant with or without naltrexone for veterans with PTSD and comorbid alcohol dependence. Neuropsychopharmacology, 37(4), 996–1004. doi: 10.1038/npp.2011.283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pietrzak RH, Gallezot JD, Ding YS, Henry S, Potenza MN, Southwick SM, … Neumeister A (2013). Association of posttraumatic stress disorder with reduced in vivo norepinephrine transporter availability in the locus coeruleus. JAMA Psychiatry, 70(11), 1199–1205. doi: 10.1001/jamapsychiatry.2013.399 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Pietrzak RH, Goldstein RB, Southwick SM, & Grant BF (2011). Prevalence and Axis I comorbidity of full and partial posttraumatic stress disorder in the United States: results from Wave 2 of the National Epidemiologic Survey on Alcohol and Related Conditions. J Anxiety Disord, 25(3), 456–465. doi: 10.1016/j.janxdis.2010.11.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pitman RK, Rasmusson AM, Koenen KC, Shin LM, Orr SP, Gilbertson MW, … Liberzon I (2012). Biological studies of post-traumatic stress disorder. Nat Rev Neurosci, 13(11), 769–787. doi: 10.1038/nrn3339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pollack MH, Jensen JE, Simon NM, Kaufman RE, & Renshaw PF (2008). High-field MRS study of GABA, glutamate and glutamine in social anxiety disorder: response to treatment with levetiracetam. Prog Neuropsychopharmacol Biol Psychiatry, 32(3), 739–743. doi: 10.1016/j.pnpbp.2007.11.023 [DOI] [PubMed] [Google Scholar]
- Raskind MA, Peskind ER, Hoff DJ, Hart KL, Holmes HA, Warren D, … McFall ME (2007). A parallel group placebo controlled study of prazosin for trauma nightmares and sleep disturbance in combat veterans with post-traumatic stress disorder. Biol Psychiatry, 61(8), 928–934. doi: 10.1016/j.biopsych.2006.06.032 [DOI] [PubMed] [Google Scholar]
- Raskind MA, Peterson K, Williams T, Hoff DJ, Hart K, Holmes H, … Peskind ER (2013). A trial of prazosin for combat trauma PTSD with nightmares in active-duty soldiers returned from Iraq and Afghanistan. Am J Psychiatry, 170(9), 1003–1010. doi: 10.1176/appi.ajp.2013.12081133 [DOI] [PubMed] [Google Scholar]
- Rasmusson AM, Hauger RL, Morgan CA, Bremner JD, Charney DS, & Southwick SM (2000). Low baseline and yohimbine-stimulated plasma neuropeptide Y (NPY) levels in combat-related PTSD. Biol Psychiatry, 47(6), 526–539. [DOI] [PubMed] [Google Scholar]
- Rasmusson AM, Riddle MA, Leckman JF, Anderson GM, & Cohen DJ (1990). Neurotrasmitter assessment in neuropsychiatric disorders of childhood New York: Plenum Press. [Google Scholar]
- Rasmusson AM, Southwick SM, Hauger RL, & Charney DS (1998). Plasma neuropeptide Y (NPY) increases in humans in response to the alpha 2 antagonist yohimbine. Neuropsychopharmacology, 19(1), 95–98. doi: 10.1016/S0893-133X(97)00199-1 [DOI] [PubMed] [Google Scholar]
- Reist C, Kauffmann CD, Haier RJ, Sangdahl C, DeMet EM, Chicz-DeMet A, & Nelson JN (1989). A controlled trial of desipramine in 18 men with posttraumatic stress disorder. Am J Psychiatry, 146(4), 513–516. doi: 10.1176/ajp.146.4.513 [DOI] [PubMed] [Google Scholar]
- Robbins TW, & Arnsten AF (2009). The neuropsychopharmacology of fronto-executive function: monoaminergic modulation. Annu Rev Neurosci, 32, 267–287. doi: 10.1146/annurev.neuro.051508.135535 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosso IM, Weiner MR, Crowley DJ, Silveri MM, Rauch SL, & Jensen JE (2014). Insula and anterior cingulate GABA levels in posttraumatic stress disorder: preliminary findings using magnetic resonance spectroscopy. Depress Anxiety, 31(2), 115–123. doi: 10.1002/da.22155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothbaum BO, Cahill SP, Foa EB, Davidson JR, Compton J, Connor KM, … Hahn CG (2006). Augmentation of sertraline with prolonged exposure in the treatment of posttraumatic stress disorder. J Trauma Stress, 19(5), 625–638. doi: 10.1002/jts.20170 [DOI] [PubMed] [Google Scholar]
- Rothbaum BO, Price M, Jovanovic T, Norrholm SD, Gerardi M, Dunlop B, … Ressler KJ (2014). A randomized, double-blind evaluation of D-cycloserine or alprazolam combined with virtual reality exposure therapy for posttraumatic stress disorder in Iraq and Afghanistan War veterans. Am J Psychiatry, 171(6), 640–648. doi: 10.1176/appi.ajp.2014.13121625 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabban EL, Alaluf LG, & Serova LI (2015). Potential of neuropeptide Y for preventing or treating post-traumatic stress disorder. Neuropeptides doi: 10.1016/j.npep.2015.11.004 [DOI] [PubMed] [Google Scholar]
- Schonenberg M, Reichwald U, Domes G, Badke A, & Hautzinger M (2005). Effects of peritraumatic ketamine medication on early and sustained posttraumatic stress symptoms in moderately injured accident victims. Psychopharmacology (Berl), 182(3), 420–425. doi: 10.1007/s00213-005-0094-4 [DOI] [PubMed] [Google Scholar]
- Schonenberg M, Reichwald U, Domes G, Badke A, & Hautzinger M (2008). Ketamine aggravates symptoms of acute stress disorder in a naturalistic sample of accident victims. J Psychopharmacol, 22(5), 493–497. doi: 10.1177/0269881107082481 [DOI] [PubMed] [Google Scholar]
- Seal KH, Metzler TJ, Gima KS, Bertenthal D, Maguen S, & Marmar CR (2009). Trends and risk factors for mental health diagnoses among Iraq and Afghanistan veterans using Department of Veterans Affairs health care, 2002–2008. Am J Public Health, 99(9), 1651–1658. doi: 10.2105/AJPH.2008.150284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shalev AY, Ankri Y, Israeli-Shalev Y, Peleg T, Adessky R, & Freedman S (2012). Prevention of posttraumatic stress disorder by early treatment: results from the Jerusalem Trauma Outreach And Prevention study. Arch Gen Psychiatry, 69(2), 166–176. doi: 10.1001/archgenpsychiatry.2011.127 [DOI] [PubMed] [Google Scholar]
- Shin LM, & Liberzon I (2010). The neurocircuitry of fear, stress, and anxiety disorders. Neuropsychopharmacology, 35(1), 169–191. doi: 10.1038/npp.2009.83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin LM, Rauch SL, & Pitman RK (2006). Amygdala, medial prefrontal cortex, and hippocampal function in PTSD. Ann N Y Acad Sci, 1071, 67–79. doi: 10.1196/annals.1364.007 [DOI] [PubMed] [Google Scholar]
- Silva AP, Xapelli S, Grouzmann E, & Cavadas C (2005). The putative neuroprotective role of neuropeptide Y in the central nervous system. Curr Drug Targets CNS Neurol Disord, 4(4), 331–347. [DOI] [PubMed] [Google Scholar]
- Simon NM, Connor KM, Lang AJ, Rauch S, Krulewicz S, LeBeau RT, … Pollack MH (2008). Paroxetine CR augmentation for posttraumatic stress disorder refractory to prolonged exposure therapy. J Clin Psychiatry, 69(3), 400–405. [DOI] [PubMed] [Google Scholar]
- Southwick SM, Bremner JD, Rasmusson A, Morgan CA 3rd, Arnsten A, & Charney DS (1999). Role of norepinephrine in the pathophysiology and treatment of posttraumatic stress disorder. Biol Psychiatry, 46(9), 1192–1204. [DOI] [PubMed] [Google Scholar]
- Southwick SM, Krystal JH, Bremner JD, Morgan CA 3rd, Nicolaou AL, Nagy LM, … Charney DS (1997). Noradrenergic and serotonergic function in posttraumatic stress disorder. Arch Gen Psychiatry, 54(8), 749–758. [DOI] [PubMed] [Google Scholar]
- Stein MB, Kerridge C, Dimsdale JE, & Hoyt DB (2007). Pharmacotherapy to prevent PTSD: Results from a randomized controlled proof-of-concept trial in physically injured patients. J Trauma Stress, 20(6), 923–932. doi: 10.1002/jts.20270 [DOI] [PubMed] [Google Scholar]
- Strawn JR, Ekhator NN, Horn PS, Baker DG, & Geracioti TD Jr. (2004). Blood pressure and cerebrospinal fluid norepinephrine in combat-related posttraumatic stress disorder. Psychosom Med, 66(5), 757–759. doi: 10.1097/01.psy.0000138133.72365.45 [DOI] [PubMed] [Google Scholar]
- Sullivan GM, Ogden RT, Huang YY, Oquendo MA, Mann JJ, & Parsey RV (2013). Higher in vivo serotonin-1a binding in posttraumatic stress disorder: a PET study with [11C]WAY-100635. Depress Anxiety, 30(3), 197–206. doi: 10.1002/da.22019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor FB, Lowe K, Thompson C, McFall MM, Peskind ER, Kanter ED, … Raskind MA (2006). Daytime prazosin reduces psychological distress to trauma specific cues in civilian trauma posttraumatic stress disorder. Biol Psychiatry, 59(7), 577–581. doi: 10.1016/j.biopsych.2005.09.023 [DOI] [PubMed] [Google Scholar]
- Tucker P, Zaninelli R, Yehuda R, Ruggiero L, Dillingham K, & Pitts CD (2001). Paroxetine in the treatment of chronic posttraumatic stress disorder: results of a placebo-controlled, flexible-dosage trial. J Clin Psychiatry, 62(11), 860–868. [DOI] [PubMed] [Google Scholar]
- van der Kolk B, Greenberg M, Boyd H, & Krystal J (1985). Inescapable shock, neurotransmitters, and addiction to trauma: toward a psychobiology of post traumatic stress. Biol Psychiatry, 20(3), 314–325. [DOI] [PubMed] [Google Scholar]
- Yeh MS, Mari JJ, Costa MC, Andreoli SB, Bressan RA, & Mello MF (2011). A double-blind randomized controlled trial to study the efficacy of topiramate in a civilian sample of PTSD. CNS Neurosci Ther, 17(5), 305–310. doi: 10.1111/j.1755-5949.2010.00188.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young MB, Andero R, Ressler KJ, & Howell LL (2015). 3,4-Methylenedioxymethamphetamine facilitates fear extinction learning. Transl Psychiatry, 5, e634. doi: 10.1038/tp.2015.138 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.