

Abstract
Using a novel chemistry-based assay for identifying electrophilic natural products from unprocessed extracts, we identified the PI3-kinase/mTOR dual inhibitor neolymphostin A from Salinispora arenicola CNY-486. The method further showed that the vinylogous ester substituent on the neolymphostin core was the exact site for enzyme conjugation. Tandem MS/MS experiments on PI3Kα treated with the inhibitor revealed that neolymphostin covalently modified Lys802 with a shift in mass of +306 amu, corresponding to addition of the inhibitor and elimination of methanol. The binding pose of the inhibitor bound to PI3Kα was modelled, and hydrogen-deuterium exchange mass spectrometry experiments supported this model. Against a panel of kinases, neolymphostin showed good selectivity for PI3-kinase and mTOR. In addition, the natural product blocked AKT phosphorylation in live cells with an IC50 of ~3 nM. Taken together, neolymphostin is the first reported example of a covalent kinase inhibitor from the bacterial domain of life.
Graphic Abstract
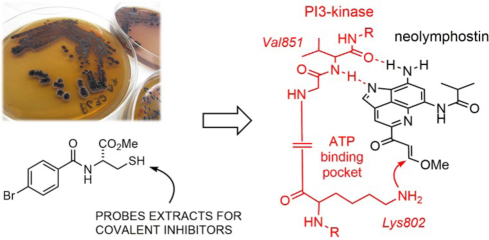
Introduction
Natural products have played a key role in our understanding of kinase inhibition and in the development of drugs.1 Natural product kinase inhibitors have been discovered from bacteria, fungi, plants, and marine sponges. Staurosporine, isolated in 1977 from Streptomyces staurosporeus, and its analogues are pan-kinase inhibitors that have been adapted to yield several clinical candidates.2 Recently, a semisynthetic derivative of staurosporine, midostaurin (N-benzoyl staurosporine), was approved by the FDA for the treatment of hematologic cancers in combination with standard chemotherapy.3 Other ATP-competitive natural product kinase inhibitors include flavonoids such as quercetin, genistein and myricetin, terpenoids like the nakijiquinones and celastrol, and nitrogenous heterocycles like olomucine and hymenialdisine. The allosteric mTOR inhibitor rapamycin from Streptomyces hygroscopicus is an immunosuppressive drug used to prevent organ transplant rejection.4
Spurred by the discovery of natural product kinase inhibitors, the development of novel potent and selective kinase inhibitors is a key priority of pharmaceutical companies, with 41 small molecule kinase inhibitors being clinically approved for a variety of diseases (39 protein kinase inhibitors and 2 lipid kinase inhibitors) as of April 2018.5 Extensive efforts have gone into the development of lipid kinase inhibitors, specifically for the phosphoinositide 3-kinase (PI3K) family of enzymes, due to the key role of PI3Ks in cancer, immunodeficiencies, and metabolic disorders.6 There are more than 80 clinical trials of PI3K inhibitors ongoing,7 with specific inhibitors of the p110δ isoform of PI3K approved by the FDA for the treatment of multiple blood cancers.8
Selective kinase inhibition at the ATP-binding site is difficult since this binding pocket is highly conserved.9 Leveraging the finding that several poorly conserved cysteine residues are present in the ATP binding pocket, a new strategy for designing selective ATP-competitive inhibitors was developed in the 1990s that involved appending an electrophilic warhead at a position on a small molecule that allows for conjugation to the cysteine residue.10–15 This approach has led to tĝhe FDA approval of selective protein tyrosine kinase inhibitors ibrutinib (2013), afatinib (2013), osimertinib (2015), neratinib (2017), and acalabrutinib (2017), which all contain an electrophilic acrylamide moiety for cysteine conjugation, for the treatment of breast cancer, non-small cell lung cancer, and several blood cancers.
Covalent kinase inhibition is a strategy that also evolved in fungi and plants.1 Wortmannin and related furanosteroids have been demonstrated to inhibit PI3K and the mammalian target of rapamycin (mTOR) in a covalent manner via conjugation to a key lysine residue.16–18 In addition, celastrol and parthenolide inhibit IκΒ kinase via cysteine conjugation,19,20 and hypothemycin and related resorcyclic acid lactones inhibit their kinase targets via cysteine conjugation.21 To date, there are no known bacterial natural products that function as covalent kinase inhibitors.
Results
In our ongoing efforts to identify naturally-occurring covalent inhibitors, we screened over 200 Salinispora extracts using thiol-based probe 1 (Fig. 1a).22 Probe 1 is highly conspicuous in an LC-MS chromatogram due to the UV absorbance of the benzamide ring and the isotopic pattern of the bromine atom, so adducts resulting from addition of 1 can be rapidly detected and isolated in complex extracts. To date, the probe has been demonstrated to selectively label a variety of validated electrophilic natural products in crude extracts, such as penicillin G (β-lactam), salinosporamide A (β-lactone), parthenolide (α,β-unsaturated enone), and wortmannin (α,β,γ,δ-unsaturated enone), regardless of the exact identity of the nucleophilic amino acid (Cys, Thr, Ser, Lys) in the cellular target that is responsible for covalent attachment. However, the method, and others like it,23–25 has yet to lead to the identification of a secondary metabolite with an unestablished covalent mechanism of action.
Figure 1.

Identification of neolymphostin as an electrophilic natural product. a) Cysteine-based probe 1 for the discovery of electrophilic natural products. b) Treatment of extract from Salinispora strain CNY-486 with 1 produced brominated adducts 2 and 3, both as a mixture of diastereomers, which are derived from neolymphostin A (4) and B (5).
Treatment of crude extract from Salinispora arenicola strain CNY-486 with 1 in triethylamine and N,N-dimethylformamide produced two pairs of diastereomeric adducts with a mass isotope pattern that indicated the presence of a single bromine atom (Fig. 1b and Supporting Information Figs. S1–S3). The formation of diastereomeric products, based on previous work,22 is indicative of an electrophilic natural product containing a Michael acceptor, since the reaction involves the formation of new stereocenters with little to no diastereoselectivity. A pair of diastereomers (2a, 2b) were purified by reversed phase C18 HPLC, and their structures were determined using a combination of NMR and MS techniques (Supporting Information Tables S1 and S2). Retrosynthetic analysis led to the identification of neolymphostin A (4), which revealed that reaction occurred at the vinylogous ester substituent (C-11) of the natural product.26–29 The other diasteromeric pair of adducts (3a, 3b) stemming from neolymphostin B (5) were also isolated and characterized (Fig. 1b, Supporting Information Tables S3 and S4).
As the lymphostins have been reported to inhibit lymphocyte-specific tyrosine kinase (Lck), phosphoinositide 3-kinase (PI3K), and the mammalian target of rapamycin (mTOR),26,28,29 we first set out to determine if our isolated material showed similar activity using an active-site dependent competition binding assay described in the Supporting Information.30 Indeed, neolymphostin (4) showed strong affinity for all four isoforms of PI3K [Kd = 0.88 (α), 5.7 (β), 3.5 (γ), and 4.9 (δ) nM] and mTOR (Kd = 10 nM) (Supporting Information Table S5). Binding to Lck, however, was less pronounced (Kd = 4.6 μM). For comparison, wortmannin, a fungal natural product and well-established covalent PI3K inhibitor, was also measured in the binding assay. This metabolite showed strong affinity for PI3K [Kd = 5.4 (α), 7.6 (β), 15 (γ), and 5.5 (δ) nM] but little affinity for mTOR (Kd = 9.2 μM) (Supporting Information Table S6).16–18
In order to quantify the electrophilicity of the vinylogous ester and to preserve precious natural product, the reactivity of model electrophile (E)-3-methoxy-1-(pyridin-2-yl)prop-2-en-1-one (6) toward glutathione was measured in 100 mM phosphate buffer at pH 7.4 according to the NMR-based method of Flanagan, et al. (Fig. 2).31 This reaction proceeded to 7 with a t1/2 = 5.4 min at 37 °C (t1/2 = 13 min at 25 °C) (Supporting Information Fig. S4). Highlighting the enhanced reactivity of the neolymphostin warhead over an FDA-approved acrylamide-containing kinase inhibitor, ibrutinib was reported to convert to its glutathione adduct with a t1/2 = 7.36 h under similar conditions.32
Figure 2.

Reactivity of neolymphostin model electrophile 6 with glutathione at pH 7.4.
To rapidly assess whether the interaction of neolymphostin with the PI3Ks was reversible or irreversible, we then measured the Kd for neolymphostin-treated PI3Kα and PI3Kγ with and without an intermediate dilution step according to the scanKINETIC platform (Fig. 3a and Supporting Information Table S7). In study arm A and C, the inhibitor and kinase were combined and equilibrated for 6 h and 1 h, respectively, before measuring the Kd. The fact that these dissociation constants are comparable suggests rapid association kinetics. In study arm B the inhibitor and kinase were combined and equilibrated for 1 h, diluted 30-fold, and re-equilibrated before measuring the Kd, while in study arm D the pair were immediately diluted and equilibrated before measuring the Kd. Importantly, dilution after inhibitor and kinase were first combined gave dissociation constants similar to study arm C, which is expected of a covalent inhibitor. Known covalent kinase inhibitors like wortmannin show the same pattern.
Figure 3.

The interaction of neolymphostin with the PI3Ks is covalent. a) Incubation studies with PI3Kα and PI3Kγ performed in duplicate (first: grey; second: blue) b) Tandem MS/MS spectrum of a neolymphostin-modified PI3Kα peptide containing Lys802. Different b and y fragments and their location are indicated, with both the observed and expected masses of the precursor annotated.
To verify that the inhibition identified was due to covalent attachment and to identify the site of modification, we carried out mass spectrometry tandem MS/MS experiments on the full-length complex of PI3Kα (composed of the proteins p110α and p85α). The tandem MS/MS data of pepsin-generated peptides was queried for modification of lysine side chains, since docking experiments suggested an appropriately positioned lysine residue was present in the binding pocket (see below). Searches were carried out for lysine modifications of either 306.11 or 338.14 amu, which correspond to the mass of neolymphostin with or without subsequent elimination of methanol. Covalent modification was detected for only a single amino acid in PI3Kα, corresponding to a +306.11 amu modification on Lys802 (Fig. 3b), and a timecourse experiment showed that modification of the kinase was complete within 2 min (Supporting Information Fig. S5). This is the same catalytic residue that is covalently modified upon addition of wortmannin, revealing that bacteria and fungi produce metabolites able to target this conserved residue in the PI3K family of enzymes.16
Docking neolymphostin into PI3Kα and mTOR indicated favorable binding at the hinge region for both enzymes with docking scores of −7.58 kcal/mol for PI3Kα and −7.45 kcal/mol for mTOR. In the best predicted binding pose for PI3Kα, hydrogen bonding occurs between the C-8 amino group of the inhibitor and the Val851 backbone carbonyl group of the kinase and between the N-1 endocyclic imine nitrogen atom of the inhibitor and the Val851 backbone amino group of the kinase (Fig. 4 and Supporting Information Fig. S6). Favorable pi-pi stacking interactions between the aromatic neolymphostin core and Tyr836 are also observed. In the mTOR model, comparable hydrogen bonding interactions occur between Val2240 and the inhibitor, as well as comparable pi-pi stacking and pi-cation interactions between Trp2239 and the neolymphostin core (Supporting Information Fig. S7). Homologous nucleophilic lysine residues in both enzymes, Lys802 in PI3Kα and Lys2187 in mTOR, lie close to the methoxyenone arm of neolymphostin. The distance between the nitrogen atom of the lysine side-chain and the electrophilic C-11 site of neolymphostin, which corresponds to the nascent bond, is 3.29 Å in PI3Kα and 6.08 Å in mTOR. Closer inspection of the neolymphostin binding pose in mTOR revealed that Lys2187 can position itself nearer to the reactive site via a slight C-C single bond rotation in its side chain. Taking active site flexibility into account is likely to generate a binding pose in mTOR similar to the case of PI3Kα. When we tried induced fit docking as an alternative approach, the binding poses for both cases were similar to the binding poses obtained by initial docking experiments except for some small active site rearrangements and slight differences in the ligand position and conformation (Supporting Information Figs. S8 and S9). Notably, the distance between the ε-amino nitrogen atom of Lys2187 in mTOR and C-11 of neolymphostin decreased to 3.98 Å. Finally, we computationally formed a chemical bond between ε-amino nitrogen atom of Lys802 and C-11 of neolymphostin, deleted the methoxy group that would be eliminated as a result of the chemical reaction, and energy-minimized the Lys802 adduct using Schrödinger software as depicted in Fig. 4 (Supporting Information Fig. S10).
Figure 4.

HDX-MS data mapped on the docked model of neolymphostin bound to PI3Kα (pdbID:5SW8, chain A) with the covalent modification site of Lys802 indicated. PI3Kα is shown in silver ribbons while amino acids that differ greater than 5% and 0.4 Da between apo- and neolymphostin-bound protein (amino acids 848–856) are highlighted in blue. Amino acids that were not covered in the HDX/MS experiment (amino acids 799–802) are highlighted in pink. The ligand and the amino acids that interact with it are shown in ball-and-stick form with N = blue, O = red, S = yellow, C(inhibitor) = orange, and C(ligand) = cyan. The black dashed lines indicate hydrogen bonds. H atoms are not shown for clarity.
We then used hydrogen deuterium exchange mass spectrometry (HDX-MS) to experimentally verify the putative docking pose from our docking experiments, This technique measures the exchange rate of amide hydrogen atoms with deuterated solvent, and as this exchange is dependent on secondary structure, it is a powerful tool to examine secondary structure dynamics.33 It has previously been used to examine the binding of certain small molecule inhibitors of PI3Kα.34 HDX-MS experiments were performed on the full length complex of p110α and p85α. Deuterium exchange was carried out for both 3 and 300 s in the presence and absence of neolymphostin, with deuterium incorporation being localized through digestion of the protein into peptide fragments and subsequent MS analysis. The full set of p110α peptides that were analyzed and their deuterium incorporation are shown in Supporting Information Table S8. Significant differences in H/D exchange rates (both >5% and >0.4 Da difference with a p value<0.01 from triplicate samples) was observed for only a single region, encompassing amino acids 848–856 in p110α (blue ribbon, Fig. 4). This region, corresponding to the hinge region at the interface of the N-lobe and C-lobe of the p110α kinase domain, contains the atoms that make hydrogen bonds with the inhibitor in the docked model, which is in agreement with the in silico model.
To gauge the electrophilicity of the vinylogous ester toward more relevant amine nucleophiles, the reactivities of model electrophile 6 toward Nα-acetyl lysine was measured. In 100 mM phosphate buffer at pH 7.4, no reaction was observed in the presence of a ten-fold excess of Nα-acetyl lysine at 25 °C. At pH 8.1, however, Nα-acetyl lysine did add to 6 with a t1/2 = 60 min at 25 °C to give adduct 8, while a competing hydrolysis reaction proceeded with a t1/2 ~ 24 h to produce 9 (Fig. 5, Supporting Information Fig. S11). Model electrophile 6 and a stoichiometric amount of Nα-acetyl lysine were then subjected to the same reaction conditions at higher concentration in order to unambiguously determine the structure of the covalent adduct by proton and 2D NMR analyses (Supporting Information Table S9). The pH requirement for reactivity between the two partners suggests that the pKa of Lys802 in PI3K (and Lys2187 in mTOR) is perturbed by the local environment.
Figure 5.

Competing reactivity of model electrophile 6 with Nα-acetyl lysine and water at pH 8.1.
Although we had at this point shown that neolymphostin strongly inhibits PI3K (and likely mTOR) in a covalent manner, we were interested in assessing the overall kinase selectivity of the inhibitor. So, the natural product was screened in vitro against a panel of 97 kinases including representatives from all seven major kinase families in addition to several atypical, lipid, pathogen, and mutant kinases (Fig. 6, Supporting Information Table S10 and Fig. S12).30,35 At a concentration of 1 μM neolymphostin showed strong affinity for the PI3Ks, as expected. The inhibitor also showed moderate affinity for Polo-like kinase 4 (PLK4), aurora kinases A and B (AURKA/B), and the c-Jun N-terminal kinases 1, 2, and 3 (JNK1/2/3) compared to control. The affinity of the inhibitor for AURKA/B may help counteract a recently established process in cancer cells for eluding the effects of PI3K inhibition.36 For the majority of kinases the alkaloid had very little affinity compared to control. A selectivity score, S-score(35), calculated by dividing the number of kinases with affinities to neolymphostin of less than 35% compared to control by the total number of kinases tested, was equal to 0.20 (Supporting Information Table S11). Neolymphostin’s S-score(35) is similar to that of FDA-approved drugs like erlotinib, sorafenib, and dasatinib.
Figure 6.

Kinase selectivity of neolymphostin and blockage of AKT phosphorylation in vivo. a) At a 1 μM concentration, neolymphostin showed strong affinity for PI3Kα (0% vs. control) and moderate affinity for Polo-like kinase 4 (0.95%), aurora kinases A (1.5%) and B (1.9%), and the c-Jun N-terminal kinases 1 (0.25%), 2 (0.15%), and 3 (0.2%). Larger red circles indicate greater affinities. b) Like wortmannin, neolymphostin blocked phosphorylation of AKT at S473 and T308 in EGF-stimulated cells and did not affect phosphorylation of EGFR or ERK1/2. Neolymphostinol did not significantly block AKT phosphorylation. c) Structure of neolymphostinol (10).
Next, we examined the ability of neolymphostin to inhibit PI3K activity in cells (Fig. 6b). Serum-starved HeLa cells were stimulated with the growth factor EGF to activate the EGFR, which leads to class I PI3K-dependent phosphorylation of AKT and PI3K-independent phosphorylation of ERK1/2. We first observed that EGF stimulation of serum-starved cells indeed led to robust phosphorylation of EGFR, AKT, and ERK1/2. As expected, pretreatment with the known PI3K inhibitor wortmannin blocked phosphorylation of AKT on Ser473 and Thr308 without affecting phosphorylation of EGFR or ERK1/2. Similarly, pretreatment with neolymphostin inhibited AKT phosphorylation, with an estimated IC50 of ~3nM, but had no apparent effect on EGFR or ERK1/2 phosphorylation. In contrast, AKT phosphorylation was largely unaffected by pretreatment with neolymphostinol (10), which lacks an electrophilic moiety (Fig 6c). Thus, we found that neolymphostin, but not the inactive neolymphostinol analogue, potently inhibits class I PI3K activity in cells.
Lastly, neolymphostin was tested in the National Cancer Institute 60-cell line anticancer screen (Supporting Information Figs. S13–S16). GI50 values, which indicate the concentration of compound that causes 50% inhibition of cell proliferation, ranged over two orders of magnitude from 6–840 nM. The natural product showed some selectivity toward leukemia, renal cancer, and breast cancer cell lines. At the highest concentration in the dose-response curves (50 μM) a uniform increase in percentage growth was observed when compared to the lower 5 μM concentration. The fact that neolymphostin appears to aggregate and “crash out” of solution at higher concentrations in buffer may explain this discrepancy. Finally, using GI50 endpoints for neolymphostin, the COMPARE algorithm showed a strong correlation to the mode of action of rapamycin (corr=0.45) and triciribine phosphate (corr=0.43), which likewise target the PI3K-AKT-mTOR pathway.37 Weaker correlations to fluorodopan (corr=0.38), caracemide (corr=0.36), asaley (corr=0.36), and pyrimidine-5-glycodialdehyde (corr=0.34) suggest that neolymphostin may alkylate DNA and other protein targets (Supporting Information Fig. S17).
Discussion and Conclusions
The structure and kinase inhibitory properties of lymphostin have been known for almost two decades but crucial information concerning the compound’s mechanism of action has been absent. Here, we show that neolymphostin, a closely-related derivative of lymphostin, is a covalent kinase inhibitor that competitively binds to the ATP-binding pocket of the PI3-kinases and mTOR. The alkaloid then reacts covalently with a conserved lysine residue within the pocket that is involved in the kinases’ key phosphor-transfer reaction, making the development of resistance through a point mutation highly unlikely.38 The vinylogous ester substituent of the lymphostins exhibits the precise electrophilicity needed to engage a pKa-perturbed amine functionality, and this conjugate addition reaction is then followed by an elimination reaction to produce a stable vinylogous amide. This study introduces a unique warhead that medicinal chemists may find useful in the development of synthetic lysine-targeting covalent inhibitors, an area which, compared to cysteine-targeting covalent inhibitors, is severely limited.39,40
As both are covalent PI3K inhibitors that target the same lysine residue, the lymphostins and wortmannin are strikingly similar in terms of function. However, there are key differences between the two metabolites. First, the lymphostins are produced by bacteria while wortmannin was discovered in fungi. The fact that the same mechanism of action evolved separately in the two kingdoms of life is remarkable. Second, according to binding experiments, lymphostin is a dual PI3K/mTOR inhibitor while wortmannin selectively targets the PI3Ks. Third, although they possess a similar electrophilic moiety, the two molecules have distinct biosynthetic origins; the lymphostins are tryptophan-derived alkaloids while wortmannin is an furanosteroid-type terpenoid. As such, the lymphostins are much more “drug-like,” possessing superior water solubility and bearing a combination of hydrogen bond donors and acceptors.
The pyrrolo[4,3,2-de]quinoline core of the lymphostins strongly resembles the purine heterocycle of ATP. In fact, the lymphostins appear to bind to the kinase ATP-binding pocket in the same manner as ATP, forming two hydrogen bonds to the kinase hinge region via the amine substituent (H bond donor) and the endocyclic imine nitrogen (H bond acceptor).41 The natural product is a “sticky” ATP mimic that first binds like ATP and then couples covalently to a conserved lysine residue that is essential for the enzyme’s function. Given that the lymphostins possess a relatively simple ATP-like structure devoid of any chirality, the level of selectivity that neolymphostin demonstrated in the kinase panel toward the PI3Ks and mTOR is somewhat surprising. Further studies are needed to understand the structural basis for the kinase selectivity of the inhibitors.
Thiol-reactive small molecules are considered pan-assay interference compounds (PAINS) and have historically been avoided in drug discovery efforts due to concerns that they possess significant off-target liabilities.42–44 Nevertheless, as many naturally-occurring electrophilic compounds appear to have a defined target or function in nature, the “reactivity-guided” isolation of thiol-reactive natural products may emerge as a powerful method to uncover medicinally-relevant covalent inhibitors hidden in extracts.22,45
Experimental Section
Chemistry
Reactions and compounds were analyzed with an analytical 1100 Series Agilent Technologies HPLC system coupled to an ELSD and UV/vis detector (210, 254, and 360 nm) using a Phenomenex Luna reversed-phase C18(2) column (100 × 4.6 mm, 5 μm, 100 Å) with a 10 min solvent gradient from 10% to 100% containing 0.1% formic acid and a flow rate of 1.0 mL min−1 or a 20 min solvent gradient from 10% to 100% containing 0.1% formic acid and a flow rate of 0.7 mL min−1. Using the same column and solvent gradients, liquid chromatography/high-resolution mass spectrometry was performed on an analytical Agilent 1260 Infinity Series LC system coupled to a 6530 Series Q-TOF mass spectrometer. Thiol 1 was synthesized as described previously.22 Neolymphostin A (4) was purified by normal-phase HPLC (4% CH3OH in DCM, silica(2) Phenomenex Luna, 100 × 10 mm, 5 μ, 3 mL min−1) and determined to have ≥95% purity by analytical HPLC. All other reagents and solvents were purchased commercially and were used without further purification. 1H NMR and 2D NMR spectra were recorded at 500 MHz in DMSO-d6 (residual solvent referenced to 2.50 ppm) on a Jeol 500 MHz NMR spectrometer. IR spectra were recorded on a Nicolet 100 FT-IR. Optical rotations were recorded on a Jasco P-2000 polarimeter.
Generation of extract
Salinispora arenicola strain CNY-486 was cultivated in three 2.8 L Fernbach flasks containing 1 L of a seawater-based A1 medium (10 g L−1 starch, 4 g L−1 yeast extract, 2 g L−1 peptone, 75% seawater, 25% deionized (DI) water) at 230 rpm and 27 °C under artificial light. After 6 days of cultivation, Amberlite XAD-16N and XAD-7HP resins (~10 g L−1 each) were added and left shaking for 1 day. The resin was filtered through cheesecloth, washed with deionized water, and extracted with ethyl acetate mixed with sodium sulfate. The ethyl acetate was removed under reduced pressure to give 246 mg of a dark red solid.
Labeling experiment
CNY-486 extract (124 mg) was treated with probe 1 (30.0 mg, 0.0943 mmol) and triethylamine (15.0 μL, 0.108 mmol) in dry and sparged DMF (2.0 mL) in a vial purged with N2. The reaction vessel was kept at room temperature and under an inert atmosphere using an N2 gas source and bubbler. Analysis of the reaction mixture after 3 h by LC-MS showed formation of two pairs of isomeric products with brominated pseudomolecular ions m/z (M+H)+ 642/644 (1:1) and m/z (M+H)+ 656/658 (1:1). The solvent was removed under vacuum, and the products were purified by reversed-phased HPLC [51% CH3CN in water, C18(2) Phenomenex Luna, 250 × 10 mm, 5 μ, 3 mL min−1] to yield 0.8 mg of 3a (tR = 22 min), 0.8 mg of 3b (tR = 24 min), 0.7 mg of 2a (tR = 30 min) and 0.7 mg of 2b (tR = 32 min) as red solids. 2a: UV/vis (CH3CN/water/0.1% formic acid) λmax 235, 330, 465 nm; 1H NMR see Tables S1; HRESI-Q-TOF-MS m/z (M+H)+ 656.1178 calcd for C29H31BrN5O6S, found 656.1166 (Δ1.8 ppm). 2b: UV/vis (CH3CN/water/0.1% formic acid) λmax 235, 330, 465 nm; 1H NMR and 2D NMR see Table S2; HRESI-Q-TOF-MS m/z (M+H)+ 656.1178 calcd for C29H31BrN5O6S, found 656.1158 (Δ3.0 ppm). 3a: UV/vis (CH3CN/water/0.1% formic acid) λmax 235, 330, 465 nm; 1H NMR see Table S3; HRESI-Q-TOF-MS m/z (M+H)+ 642.1022 calcd for C28H29BrN5O6S, found 642.1004 (Δ2.8 ppm). 3b: UV/vis (CH3CN/water/0.1% formic acid) λmax 235, 330, 465 nm; 1H NMR and 2D NMR see Table S4; HRESI-Q-TOF-MS m/z (M+H)+ 642.1022 calcd for C28H29BrN5O6S, found 642.1005 (Δ2.6 ppm).
Model electrophile kinetics experiments with glutathione
The method reported by Flanagan, et al, was slightly adapted.30 A 200 mM stock solution of 6 was prepared in DMSO-d6. A 2.0 mM aqueous electrophile solution was then prepared by diluting an aliquot of the stock solution 1:100 in phosphate-buffered D2O (100 mM, pH 7.4). A 20 mM glutathione solution in phosphate-buffered D2O was freshly prepared before the kinetics experiments. In a vial was added 100 μL of 20 mM glutathione solution, followed by 100 μL of 2.0 mM electrophile solution (final concentration 10 mM for glutathione and 1.0 mM for the electrophile). The reaction mixture was briefly swirled in the vial to mix and then quickly loaded into a 3 mm O.D. NMR tube, which was then capped. The tube was loaded into a Jeol 500 MHz NMR spectrometer at 25 °C or 37 °C. The sample was locked and shimmed, and then 1H NMR spectra were acquired every 5 min for 60 min. Each spectrum consisted of 40 transients with an acquisition time of 2.18383 s and a relaxation delay of 5 s. The resulting array of spectra were processed in MestReNova, and the residual DMSO solvent peak was referenced to 2.50 ppm. Non-overlapping pairs of peaks corresponding to the electrophile and adduct, or those pairs that overlapped perfectly, were identified and integrated. A plot was then constructed of ln([SM]) versus time for each appropriate signal, and a linear regression line was applied. For a given electrophile at a given temperature, the pseudo-first order half-life was calculated by averaging the linear regression slopes derived from each appropriate signal.
MS identification of covalent Lys802 modification
PI3Kα preincubated with 4 μM neolymphostin was brought to a final concentration of 0.8 μM in a total of 50 μL non-deuterated buffer (10 mM HEPES pH 7.5, 100 mM NaCl in water). After adding 20 μL of ice-cold quench buffer (2 M guanidine-HCl, 3% formic acid), the samples were immediately frozen in liquid nitrogen and stored at −80°C. Peptides were digested and separated in a similar manner to the HDX-MS method described below. Peptide identification was carried out in PEAKS 7. The search was performed with a mass error threshold of 5 ppm for precursors and 0.1 Da for fragments. Variable modification of Lys with a mass of 306.1117 was included. The false discovery rate for peptides was set at 0.1%.
Timecourse for covalent modification of PI3K
The timecourse of covalent modification was initiated by mixing 4 μM neolymphostin A with 2 μM PI3Kα (p110α/p85α) in a buffer containing 10 mM HEPES pH 7.5, 100 mM NaCl, and 5% DMSO. Different timepoints were taken by unfolding the protein in an ice-cold quench buffer (2 M guanidine HCl, 3% formic acid), with samples immediately frozen in liquid nitrogen and stored at −80°C. Samples were injected and analyzed on the mass spectrometer as described in the HDX-MS section.
Docking experiments
All steps for the docking experiments in this study were performed using the Schrödinger 2017–3 suite. PI3Kα (pdbID:5SW8, chain A) and mTOR (pdbID:4JT5, chain A) were first processed using Protein Preparation Wizard, which added missing hydrogen atoms, assigned protonation states of residues based on PropKA calculations, optimized H-bonds, and deleted all water molecules farther than 5 Å from protein atoms. The receptor grid was generated for a 30 Å × 30 Å × 30 Å cubic box centered at hinge residues Val850 and Val851 for PI3Kα and at hinge residues Trp2239 and Val2240 for mTOR. Neolymphostin was built in Maestro (Small-Molecule Drug Discovery Suite 2017–3: Maestro, Schrödinger, LLC, New York, NY, 2017) and docked into the active site of PI3Kα and mTOR using Glide (Small-Molecule Drug Discovery Suite 2017–3: Maestro, Schrödinger, LLC, New York, NY, 2017) with standard precision docking and default parameters. For induced fit docking experiments, search space was again centered at the hinge residues Val850 and Val851 for PI3Kα and at hinge residues Trp2239 and Val2240 for mTOR. The side chains of Lys802 of PI3Kα and Lys2187 of mTOR were trimmed before neolymphostin docking and restored and optimized together with all residue side chains within 5 Å of the ligand by Schrödinger’s Prime module after docking (Small-Molecule Drug Discovery Suite 2017–3: Prime, Schrödinger, LLC, New York, NY, 2017).
Hydrogen deuterium exchange mass spectrometry (HDX-MS) experiment
HDX experiments were performed as described in Vadas, et al.33 The experiments were conducted in 50 μL reaction mixtures with a final PI3Kα (p110α/p85α) concentration of 0.4 μM. Prior to the experiment, PI3Kα was incubated at 2 μM with neolymphostin (4 μM final concentration) or in DMSO for 30 minutes. The final DMSO concentration was 5%. Two conditions were tested: (i) PI3Kα alone and (ii) PI3Kα with neolymphostin. Exchange was carried out for two time-points (3 s on ice and 300 s at 20 °C) and terminated by the addition of 20 μL ice-cold quench buffer (2 M guanidine-HCl, 3% formic acid). Samples were immediately frozen in liquid nitrogen and stored at −80 °C. Protein samples were rapidly thawed and injected onto a UHPLC system at 2 °C. Protein was run over two immobilized pepsin columns (Applied Biosystems; porosyme, 2–3131-00) at 10 °C and 2 °C at 200 μL min−1 for 3 minutes, and peptides were collected onto a VanGuard precolumn trap (Waters). The trap was subsequently eluted in line with an Acquity 1.7 μm particle, 100 × 1 mm2 C18 UPLC column (Waters), using a gradient of 5–36% B (buffer A 0.1% formic acid, buffer B 100% acetonitrile) over 16 minutes. Mass spectrometry experiments were performed on an Impact II TOF-MS (Bruker) acquiring over a mass range from 150 to 2200 m/z using an electrospray ionization source operated at a temperature of 200 °C and a spray voltage of 4.5 kV. Peptides were identified using data-dependent acquisition methods following tandem MS/MS experiments (0.5 s precursor scan from 150–2000 m/z; twelve 0.25 s fragment scans from 150–2000 m/z). MS/MS datasets were analyzed using PEAKS 7 (PEAKS STUDIO), and a false discovery rate was set at 1% using a database of purified proteins and known contaminants.
HD-Examiner Software (Sierra Analytics) was used to automatically calculate the level of deuterium incorporation into each peptide. All peptides were manually inspected for correct charge state and presence of overlapping peptides. Deuteration levels were calculated using the centroid of the experimental isotope clusters. Results for these proteins are presented as relative levels of deuterium incorporation and the only control for back exchange was the level of deuterium present in the buffer (76.92%). Changes in any peptide at any time point greater than both 5% and 0.4 Da between conditions with a paired t-test value of p<0.01 was considered significant.
Model electrophile kinetics experiments with Nα-acetyl lysine
Kinetics experiments on model electrophile 6 with and without Nα-acetyl lysine were conducted in an identical manner to those with glutathione (see above), with the exception that phosphate-buffered D2O was prepared at pH 8.1 (instead of 7.4).
Synthesis and characterization of enamine 8
A 20 mM solution of 6 was prepared by diluting an aliquot of electrophile stock (200 mM, DMSO-d6) 1:10 in phosphate-buffered D2O (100 mM, pH 8.1). A 20 mM solution of Nα-acetyl lysine was freshly prepared in phosphate-buffered D2O (100 mM, pH 8.1). To a vial was added 100 μL of the Nα-acetyl lysine solution, followed by 100 μL of the electrophile solution. The vial was capped, swirled to mix, and allowed to stand overnight. The reaction mixture was then loaded into a 3 mm O.D. NMR tube and characterized by 1H, COSY, HSQC, and HMBC NMR experiments on a Jeol 500 MHz spectrometer.
Kinase selectivity panel
The affinity of neolymphostin for 97 kinases was assessed using the DiscoverX KINOMEscan profiling service. A detailed description of this assay can be found in Fabian, et al., and in the Supplementary Information.30
PI3K inhibition in live cells
After serum starvation overnight, HeLa cells were pretreated with the indicated concentration of wortmannin, neolymphostin A or neolymphostinol for 10 min followed by stimulation with 10ng mL−1 EGF for 5 min. Cells were lysed by the addition of boiling SDS sample buffer, collected into microfuge tubes, and quantified by Pierce 660 nm Protein Assay. Equal protein lysates were boiled at 100° for 5 min, resolved by SDS-PAGE, transferred to PVDF for Western blotting, and detected by enhanced chemiluminescence. AKT, pAKT (Ser473), pAKT (Thr308), ERK1/2, pERK1/2 (Thr202/Tyr204), EGFR, pEGFR (Tyr1068) and GAPDH antibodies were purchased from Cell Signaling Technologies.
NCI 60-cell line assay
5-Point dose-response curves were generated for leukemia, non-small cell lung cancer, colon cancer, CNS cancer, melanoma, ovarian cancer, renal cancer, prostate cancer, and breast cancer cell lines. 50% growth inhibition (GI50), total growth inhibition (TGI), and 50% lethal concentration (LC50) values were then calculated. A description of the NCI 60-cell line assay can be found in Shoemaker.46
Supplementary Material
Acknowledgments
This work was supported by seed funding from the Scripps Institution of Oceanography (E.W. Scripps Associates), a UCSD Academic Senate Research Award (RP42S-HUGHES), and an American Cancer Society Institutional Research Grant (ACS-IRG #15-172-45) to C.H. It was also funded in part by the Director’s New Innovator Award Program NIH DP2 OD007237 to R.A. Funding and support from the National Biomedical Computation Resource (NBCR) is provided through NIH P41 GM103426. G.C.F. is a Howard Hughes Medical Institute Gilliam Fellow. We also thank the NIH for an HRLC-MS instrument (S10 OD0106400).
Abbreviations
- PI3K
phosphoinositide 3-kinase
- Mtor
mammalian target of rapamycin
- AKT
protein kinase B
- Lck
lymphocyte-specific tyrosine kinase
- Kd
dissociation constant
- PIKK
phosphoinositide 3-kinase-related kinase
- HDX-MS
hydrogen deuterium exchange mass spectrometry
- PLK4
Polo-like kinase 4
- AURK
aurora kinase
- JNK
c-Jun N-terminal kinase
- ERK
extracellular signal-regulated kinase
- ELSD
evaporative light-scattering detector
- UV/vis
ultraviolet/visible light
- HRESI-Q-TOF-MS
high-resolution electrospray ionization quadrupole time-of-flight mass spectrometry
- PVDF
polyvinylidene fluoride
- GADPH
glyceraldehyde 3-phosphate dehydrogenase
Footnotes
Ancillary Information
Supporting Information The Supporting Information is available free of charge on the ACS Publications website at DOI: https://doi.org/10.1021/acs.jmedchem.8b00975.
Experimental procedures, compound characterization data (NMR, MS), curves for dissociation constant measurements, docking images, kinetics data, HDX-MS data, cytotoxicity data (PDF)
Molecular formula strings (CSV)
R.A. is a co-founder of, has equity interest in, and is on the scientific advisory board of Actavalon Inc.
References
- (1).Liu J; Hu Y; Waller DL; Wang J; Liu Q Natural products as kinase inhibitors. Nat. Prod. Rep 2012, 29, 392–403. [DOI] [PubMed] [Google Scholar]
- (2).Omura S; Iwai Y; Hirano A; Nakagawa A; Awaya J; Tsuchya H; Takahashi Y; Masuma R A new alkaloid AM-2282 of Streptomyces origin. taxonomy, fermentation, isolation and preliminary characterization. J. Antibiot. (Tokyo) 1977, 30, 275–282. [DOI] [PubMed] [Google Scholar]
- (3).Weisberg E; Boulton C; Kelly LM; Manley P; Fabbro D; Meyer T; Gilliland DG; Griffin JD Inhibition of mutant FLT3 receptors in leukemia cells by the small molecule tyrosine kinase inhibitor PKC412. Cancer Cell 2002, 1, 433–443. [DOI] [PubMed] [Google Scholar]
- (4).Vezina C; Kudelski A; Sehgal SN Rapamycin (AY-22,989), a new antifungal antibiotic. I. Taxonomy of the producing streptomycete and isolation of the active principle. J. Antibiot. (Tokyo) 1975, 28, 721–726. [DOI] [PubMed] [Google Scholar]
- (5).FDA-approved protein kinase inhibitors. http://www.brimr.org/PKI/PKIs.htm (accessed Aug 23, 2018).
- (6).Fruman DA; Chiu H; Hopkins BD; Bagrodia S; Cantley LC; Abraham RT The PI3K pathway in human disease. Cell 2017, 170, 605–635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Bergholz JS; Roberts TM; Zhao JJ Isoform-selective phosphatidylinositol 3-kinase inhibition in cancer. J. Clin. Oncol. 2018, 1–4. [DOI] [PubMed] [Google Scholar]
- (8).Furman RR; Sharman JP; Coutre SE; Cheson BD; Pagel JM; Hillmen P; Barrientos JC; Zelenetz AD; Kipps TJ; Flinn I; Ghia P; Eradat H; Ervin T; Lamanna N; Coiffier B; Pettitt AR; Ma S; Stilgenbauer S; Cramer P; Aiello M; Johnson DM; Miller LL; Li D; Jahn TM; Dansey RD; Hallek M; O’Brien SM Idelalisib and rituximab in relapsed chronic lymphocytic leukemia. N. Engl. J. Med. 2014, 370, 997–1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Zhang J; Yang PL; Gray NS Targeting cancer with small molecule kinase inhibitors. Nat. Rev. Cancer 2009, 9, 28–39. [DOI] [PubMed] [Google Scholar]
- (10).Fry DW; Bridges AJ; Denny WA; Doherty A; Greis KD; Hicks JL; Hook KE; Keller PR; Leopold WR; Loo JA; McNamara DJ; Nelson JM; Sherwood V; Smaill JB; Trumpp-Kallmeyer S; Dobrusin EM Specific, irreversible inactivation of the epidermal growth factor receptor and erbB2, by a new class of tyrosine kinase inhibitor. Proc. Natl. Acad. Sci. U. S. A. 1998, 95, 12022–12027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Cohen MS; Zhang C; Shokat KM; Taunton J Structural bioinformatics-based design of selective, irreversible kinase inhibitors. Science 2005, 308, 1318–1321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Pan Z; Scheerens H; Li SJ; Schultz BE; Sprengeler PA; Burrill LC; Mendonca RV; Sweeney MD; Scott KCK; Grothaus PG; Jeffery DA; Spoerke JM; Honigberg LA; Young PR; Dalrymple SA; Palmer JT Discovery of selective irreversible inhibitors for Bruton’s tyrosine kinase. ChemMedChem 2007, 2, 58–61. [DOI] [PubMed] [Google Scholar]
- (13).Liu Q; Sabnis Y; Zhao Z; Zhang T; Buhrlage SJ; Jones LH; Gray NS Developing irreversible inhibitors of the protein kinase cysteinome. Chem. Biol. 2013, 20, 146–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Kung A; Chen Y-C; Schimpl M; Ni F; Zhu J; Turner M; Molina H; Overman R; Zhang C Development of specific, irreversible inhibitors for a receptor tyrosine kinase EphB3. J. Am. Chem. Soc. 2016, 138, 10554–10560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Wu H; Wang W; Liu F; Weisberg EL; Tian B; Chen Y; Li B; Wang A; Wang B; Zhao Z; McMillin DW; Hu C; Li H; Wang J; Liang Y; Buhrlage SJ; Liang J; Liu J; Yang G; Brown JR; Treon SP; Mitsiades CS; Griffin JD; Liu Q; Gray NS Discovery of a potent, covalent BTK inhibitor for B-cell lymphoma. ACS Chem. Biol. 2014, 9, 1086–1091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Wymann MP; Bulgarelli-Leva G; Zvelebil MJ; Pirola L; Vanhaesebroeck B; Waterfield MD; Panayotou G Wortmannin inactivates phosphoinositide 3-kinase by covalent modification of Lys-802, a residue involved in the phosphate transfer reaction. Mol. Cell. Biol. 1996, 16, 1722–1733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Brunn GJ; Williams J; Sabers C; Wiederrecht G; Lawrence JC; Abraham RT Direct inhibition of the signaling functions of the mammalian target of rapamycin by the phosphoinositide 3-kinase inhibitors, wortmannin and LY294002. EMBO J. 1996, 15, 5256–5267. [PMC free article] [PubMed] [Google Scholar]
- (18).Arcaro A; Wymann MP Wortmannin is a potent phosphatidylinositol 3-kinase inhibitor: the role of phosphatidylinositol 3,4,5-trisphosphate in neutrophil responses. Biochem. J. 1993, 296, 297–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Lee JH; Koo TH; Yoon H; Jung HS; Jin HZ; Lee K; Hong YS; Lee JJ Inhibition of NF-κB activation through targeting IκB kinase by celastrol, a quinone methide triterpenoid. Biochem. Pharmacol. 2006, 72, 1311–1321. [DOI] [PubMed] [Google Scholar]
- (20).Kwok BHB; Koh B; Ndubuisi MI; Elofsson M; Crews CM The anti-inflammatory natural product parthenolide from the medicinal herb Feverfew directly binds to and inhibits IκB kinase. Chem. Biol. 2001, 8, 759–766. [DOI] [PubMed] [Google Scholar]
- (21).Schirmer A; Kennedy J; Murli S; Reid R; Santi DV Targeted covalent inactivation of protein kinases by resorcylic acid lactone polyketides. Proc. Natl. Acad. Sci. U. S. A. 2006, 103, 4234–4239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Castro-Falcon G; Hahn D; Reimer D; Hughes CC Thiol probes to detect electrophilic natural products based on their mechanism of action. ACS Chem. Biol. 2016, 11, 2328–2336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Miles CO; Sandvik M; Nonga HE; Rundberget T; Wilkins AL; Rise F; Ballot A Thiol derivatization for LC-MS identification of microcystins in complex matrices. Environmetal Sci. Technonlogy 2012, 46, 8937–8944. [DOI] [PubMed] [Google Scholar]
- (24).Cox CL; Tietz JI; Sokolowski K; Melby JO; Doroghazi JR; Mitchell DA Nucleophilic 1,4-additions for natural product discovery. ACS Chem. Biol. 2014, 4, 2014–2022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Rudolf GC; Koch MF; Mandl FAM; Sieber SA Subclass-specific labeling of protein-reactive natural products with customized nucleophilic probes. Chem. - A Eur. J. 2015, 21, 3701–3707. [DOI] [PubMed] [Google Scholar]
- (26).Aotani Y; Nagata H; Yoshida M Lymphostin (LK6-A), a novel immunosuppressant from Streptomyces sp. KY11783: Structural elucidation. J. Antibiot. (Tokyo) 1997, 50, 543–545. [DOI] [PubMed] [Google Scholar]
- (27).Nagata H; Ochiai K; Aotani Y; Ando K; Yoshida M; Takahashi I Taxonomy of the producing organism, fermentation, isolation. J. Antibiot. (Tokyo) 1997, 50, 537–542. [DOI] [PubMed] [Google Scholar]
- (28).Nagata H; Yano H; Sasaki K; Sato S; Nakanishi S; Takahashi I; Tamaoki T Inhibition of lymphocyte kinase Lck and phosphatidylinositol 3-kinase by a novel immunosuppressant, lymphostin. Biosci. Biotechnol. Biochem. 2002, 66, 501–507. [DOI] [PubMed] [Google Scholar]
- (29).Miyanaga A; Janso JE; McDonald L; He M; Liu H; Barbieri L; Eustáquio AS; Fielding EN; Carter GT; Jensen PR; Feng X; Leighton M; Koehn FE; Moore BS Discovery and assembly-line biosynthesis of the lymphostin pyrroloquinoline alkaloid family of mTOR inhibitors in Salinispora bacteria. J. Am. Chem. Soc. 2011, 133, 13311–13313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Fabian MA; Biggs WH; Treiber DK; Atteridge CE; Azimioara MD; Benedetti MG; Carter TA; Ciceri P; Edeen PT; Floyd M; Ford JM; Galvin M; Gerlach JL; Grotzfeld RM; Herrgard S; Insko DE; Insko MA; Lai AG; Lélias J-M; Mehta SA; Milanov Z V; Velasco, A. M.; Wodicka, L. M.; Patel, H. K.; Zarrinkar, P. P.; Lockhart, D. J. A small molecule-kinase interaction map for clinical kinase inhibitors. Nat. Biotechnol. 2005, 23, 329–336. [DOI] [PubMed] [Google Scholar]
- (31).Flanagan ME; Abramite JA; Anderson DP; Aulabaugh A; Dahal UP; Gilbert AM; Li C; Montgomery J; Oppenheimer SR; Ryder T; Schuff BP; Uccello DP; Walker GS; Wu Y; Brown MF; Chen JM; Hayward MM; Noe MC; Obach RS; Philippe L; Shanmugasundaram V; Shapiro MJ; Starr J; Stroh J; Che YJ Chemical and computational methods for the characterization of covalent reactive groups for the prospective design of irreversible inhibitors. Med. Chem 2014, 57, 10072–10079. [DOI] [PubMed] [Google Scholar]
- (32).Palkowitz MD; Tan B; Hu H; Roth K; Bauer RA Synthesis of diverse N-acryloyl azetidines and evaluation of their enhanced thiol reactivities. Org. Lett. 2017, 19, 2270–2273. [DOI] [PubMed] [Google Scholar]
- (33).Vadas O; Jenkins ML; Dornan GL; Burke JE Using Hydrogen–Deuterium Exchange Mass Spectrometry to Examine Protein–Membrane Interactions, 1st ed.; Elsevier Inc.: Cambridge, MA, 2017; pp. 143–172. [DOI] [PubMed] [Google Scholar]
- (34).Masson GR; Maslen SL; Williams RL Analysis of phosphoinositide 3-kinase inhibitors by bottom-up electron-transfer dissociation hydrogen/deuterium exchange mass spectrometry. Biochem. J. 2017, 474, 1867–1877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Karaman MW; Herrgard S; Treiber DK; Gallant P; Atteridge CE; Campbell BT; Chan KW; Ciceri P; Davis MI; Edeen PT; Faraoni R; Floyd M; Hunt JP; Lockhart DJ; Milanov ZV; Morrison MJ; Pallares G; Patel HK; Pritchard S; Wodicka LM; Zarrinkar PP A quantitative analysis of kinase inhibitor selectivity. Nat. Biotechnol. 2008, 26, 127–132. [DOI] [PubMed] [Google Scholar]
- (36).Donnella HJ; Webber JT; Levin RS; Camarda R; Momcilovic O; Bayani N; Shah KN; Korkola JE; Shokat KM; Goga A; Gordan JD; Bandyopadhyay S Kinome rewiring reveals AURKA limits PI3K-pathway inhibitor efficacy in breast cancer. Nat. Chem. Biol. 2018, 14, 768–777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Paull KD; Shoemaker RH; Hodes L; Monks A; Scudiero DA; Rubinstein L; Plowman J; Boyd MR Display and analysis of patterns of differential activity of drugs against human tumor cell lines: development of mean graph and COMPARE alogrithm. J. Natl. Cancer Inst. 1989, 81, 1088–1092. [DOI] [PubMed] [Google Scholar]
- (38).Carrera AC; Alexandrov K; Roberts TM The conserved lysine of the catalytic domain of protein kinases is actively involved in the phosphotransfer reaction and not required for anchoring ATP. Biochemistry 1993, 90, 442–446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Dalton SE; Dittus L; Thomas DA; Convery A; Nunes J; Bush JT; Evans JP; Werner T; Bantsche M; Murphy JA; Campos S Selectively targeting the kinome-conserved lysine of PI3Kδ as a general approach to covalent kinase inhibition. J. Am. Chem. Soc. 2018, 140, 932–939. [DOI] [PubMed] [Google Scholar]
- (40).Pettinger J; Jones K; Cheeseman MD Lysine-targeting covalent inhibitors Angew. Chemie - Int. Ed 2017, 56, 15200–15209. [DOI] [PubMed] [Google Scholar]
- (41).Jeffrey PD; Russo AA; Polyak K; Gibbs E; Hurwitz J; Massagué J; Pavletich NP Mechanism of CDK activation revealed by the structure of a cyclinA-CDK2 complex. Nature. 1995, pp 313–320. [DOI] [PubMed] [Google Scholar]
- (42).Huth JR; Mendoza R; Olejniczak ET; Johnson RW; Cothron DA; Liu Y; Lerner CG; Chen J; Hajduk PJ ALARM NMR: A rapid and robust experimental method to detect reactive false positives in biochemical screens. J. Am. Chem. Soc. 2005, 127, 217–224. [DOI] [PubMed] [Google Scholar]
- (43).Baell JB; Nissink JWM Seven year itch: Pan-assay interference compounds (PAINS) in 2017–utility and limitations. ACS Chem. Biol. 2018, 13, 36–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Dahlin JL; Nissink JWM; Strasser JM; Francis S; Higgins L; Zhou H; Zhang Z; Walters MA PAINS in the assay: Chemical mechanisms of assay interference and promiscuous enzymatic inhibition observed during a sulfhydryl-scavenging HTS. J. Med. Chem. 2015, 58, 2091–2113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Gersch M; Kreuzer J; Sieber SA Electrophilic natural products and their biological targets. Nat. Prod. Rep. 2012, 29, 659–682. [DOI] [PubMed] [Google Scholar]
- (46).Shoemaker RH; Shoemaker RH The NCI60 human tumour cell line anticancer drug screen. Nat. Rev. Cancer 2006, 6, 813–823. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.