

Summary
Voltage-gated sodium (NaV) channels initiate action potentials in nerve, muscle, and other electrically excitable cells. The structural basis of voltage gating is uncertain because the resting state exists only at deeply negative membrane potentials. To stabilize the resting conformation, we inserted voltage-shifting mutations and introduced a disulfide crosslink in the VS of the ancestral bacterial sodium channel NaVAb. Here we present a cryo-EM structure of the resting state and a complete voltage-dependent gating mechanism. The S4 segment of the VS is drawn intracellularly with three gating charges passing through the transmembrane electric field. This movement forms an elbow connecting S4 to the S4-S5 linker, tightens the collar around the S6 activation gate, and prevents its opening. Our structure supports the classical ‘sliding-helix’ mechanism of voltage-sensing and provides a complete gating mechanism for voltage-sensor function, pore-opening, and activation-gate closure based on high-resolution structures of a single sodium channel protein.
Keywords: Cryo-EM, disulfide crosslinking, electrophysiology, ion channel, membrane protein, NaV, X-ray crystallography, voltage-gated sodium channel
Graphical Abstract
eTOC Blurb
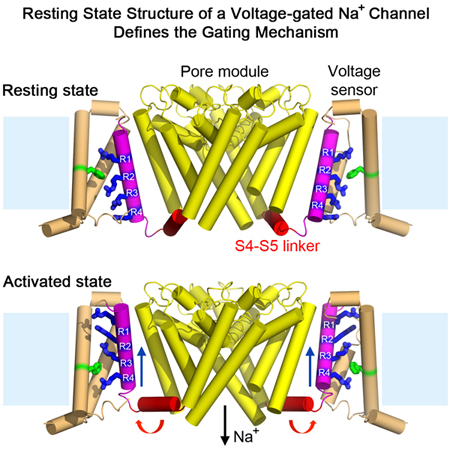
An engineered bacterial NaV channel adopts a resting conformation in the absence of a membrane potential, enabling structural analysis and elucidation of the full voltage-dependent gating mechanism.
Introduction
Voltage-gated sodium (NaV) channels are transmembrane proteins that initiate and propagate action potentials in response to membrane depolarization in nerve, muscle, and other electrically excitable cells (Hille, 2001). Their steep voltage dependence of gating is essential for electrical excitability from bacteria to man. Eukaryotic NaV channels are composed of a single pore-forming polypeptide of approximately 2,000 amino acid residues containing four homologous, but nonidentical domains connected by intracellular linkers (Catterall, 1984; Noda et al., 1984). Each homologous domain is composed of six transmembrane helices that form a voltage-sensing module (VS) containing S1 to S4 and a pore module (PM) containing the S5, P, and S6 segments (Ahern et al., 2016; Catterall et al., 2017). The VS and PM are connected to each other by the S4-S5 helical linker located on the cytoplasmic edge of the protein. In contrast, bacterial sodium channels like NaVAb are composed of a tetramer of identical subunits containing a VS, S4-S5 linker, and PM, and they are the likely evolutionary ancestors of both NaV and CaV channels (Catterall et al., 2017; Ren et al., 2001; Yu and Catterall, 2004). High-resolution structures of bacterial and eukaryotic sodium channels have been determined in multiple states with activated VS (Catterall et al., 2017; Lenaeus et al., 2017; McCusker et al., 2012; Pan et al., 2018; Payandeh et al., 2012; Payandeh et al., 2011; Shaya et al., 2014; Shen et al., 2017), but structures of sodium channels in resting states have not been resolved.
The S4 helices of sodium channels contain three or more positive gating charges (usually Arg) at three-residue intervals, which are required for voltage-dependent activation (Ahern et al., 2016; Catterall et al., 2017). They are thought to move across the membrane electric field in response to depolarization and repolarization and initiate conformational changes to open and close the pore (Ahern et al., 2016; Catterall et al., 2017). The outward movement of the gating charges upon depolarization has been measured electrically as a capacitive gating current (Armstrong and Bezanilla, 1973) and documented directly by chemical labeling and disulfide crosslinking (DeCaen et al., 2008; Yang et al., 1996; Yang and Horn, 1995; Yarov-Yarovoy et al., 2012). The steeply voltage-dependent gating of sodium channels implies the movement of 2–3 gating charges per VS during activation. Three general molecular models have been proposed for coupling membrane potential changes to pore opening and closing: the ‘sliding-helix’ model in which S4 moves outward and rotates along its axis as it translocates the gating charges through the VS (Catterall, 1986; Guy and Seetharamulu, 1986; Yarov-Yarovoy et al., 2012), the ‘transporter’ model in which the gating charges do not make major movements themselves but the VS rearranges to alternately expose them to the extracellular or intracellular milieu (Bezanilla, 2008), and the ‘paddle’ model in which the S3-S4 helical hairpin pivots through the lipid bilayer independent of the VS (Jiang et al., 2003). This electromechanical coupling mechanism is the key to electrical signaling from bacteria to man, but its structural basis has been elusive owing to lack of information on the structure of the resting state. Our experiments revealing the structure of the resting state of a voltage-gated sodium channel allow this fundamental mechanistic question to be addressed at the structural level for the first time.
Results
Voltage Shifting and Disulfide Locking the Resting State
The resting state of sodium channels is present at the resting membrane potential of approximately −90 mV in nerve and muscle or <−160 mV in bacteria. This huge electrical force must be overcome to determine the structure of the sodium channel in the resting state at 0 mV. Because of the domain-swapped organization of the subunits of bacterial sodium channels (Payandeh et al., 2011), the VS of one subunit of NaVAb interacts most closely with the PM of its neighbor (Figure 1A). As a first step, we identified pairs of substituted Cys residues that can form disulfide bonds, crosslink the VS of one subunit to the PM of its neighbor, and thereby stabilize the resting state of the VS in E. coli at the resting membrane potential of −160 mV. As NaVAb is Cys-free, only substituted Cys residues can form disulfide bonds. We used an unbiased search method to identify pairs of substituted Cys residues that would covalently link S5 of the PM to S4 (Figures 1B and S1A; STAR Methods). After an extensive search of a matrix of paired-Cys substitutions, we found that V100C on S4 formed disulfide bonds with Q150C on S5, as detected by a change in subunit migration on SDS-PAGE (Figures 1B and S1B). Unfortunately, only neighboring residues of V100C crosslinked strongly to Q150C, indicating that S4 is mainly in an outward conformation consistent with the structure of the activated state of the VS (Figures 1A, 1B, and S1A, green). To further favor disulfide-crosslinking of the resting state, we introduced the N49K mutation that shifts the voltage dependence of activation of NaVAb from V1/2=−98.7 mV to V1/2=−21.9 mV and abolishes late use-dependent inactivation (Gamal El-Din et al., 2013). With this voltage-shifting mutation, additional residues in S4 on the extracellular side of V100 formed disulfide crosslinks with Q150C, including G94 (Figures 1B; S1A and S1C, red and yellow), indicating that S4 had moved inward with respect to the PM and the resting state of NaVAb had been captured by the combination of voltage-shifting and disulfide-locking at the strongly negative bacterial membrane potential.
Figure 1. NaVAb Double Cysteine Substitutions Reveal State-dependent Disulfide Crosslinking.

(A) Design of double-cysteine substitution experiment for disulfide crosslinking. The crystal structure of NaVAb in the activated state (Payandeh et al., 2011) was used as a starting template. The domain-swapped architecture of the NaVAb homotetramer permits intermolecular disulfide crosslinking between one subunit (purple) and the neighboring subunit (blue) that results in a formation of a covalently linked tetramer. Cysteine substitutions that lead to structures are shown as spheres at Cα for G94 (red) and V100 (green) and Q150 (blue).
(B) Close-up view for intermolecular disulfide crosslinking design in the double-cysteine substitution experiment. Twelve residues on S3-S4 loop and S4 were screened against 5 residues on S5 that yielded a total of 60 variants. Key S4 residues that showed disulfide-crosslinked tetramer with S5 residue (Q150, blue) are highlighted in red (G94) for resting state, and green (V100) for activated states.
(C) Effects of KAV mutations on state-dependent disulfide crosslinking under hyperpolarization/oxidation condition.
(D) Effects of KAV mutations on state-dependent disulfide crosslinking under control condition. Percent crosslink denotes the extent of crosslinking quantified from the intensity of the tetramer band divided by the total stain intensity. The bar graph (left axis) shows % crosslink of double-cysteine substitutions for NaVAb (yellow) and NaVAb KAV (red). The line graph shows differences (Δ) in % crosslink between the same double-cysteine substitutions for NaVAb KAV vs. NaVAb (black circles). Error bars represent SEM with n = 5.
See also Figures S1 and S2.
To further shift the voltage dependence of activation of NaVAb, we screened 49 combinations of 18 mutations previously shown to shift voltage dependence of activation of NaV or KV channels positively (Figure 2; McCord et al., in preparation). The mutations N49K, L109A, and M116V (Gamal El-Din et al., 2013; Lopez et al., 1991) each caused large positive shifts of the voltage dependence of activation for NaVAb constructs expressed in insect cells, and the combination of these three mutations (termed KAV) shifted voltage-dependent activation from V1/2=−98mV for WT (Gamal El-Din et al., 2013) to V1/2=+59mV (Figures 2A and 2B; gray). Importantly, NaVAb/KAV is fully in the resting state at 0 mV (Figure 2A).
Figure 2. Electrophysiological Recordings of NaVAb/WT, NaVAb/N49K, NaVAb/KAV and NaVAb/KAV Cysteine Mutants.

(A) Normalized conductance-voltage (G/V) relationships and Boltzmann fits for WT and each mutant. Sf9 cells expressing NaVAb KAV mutants (all C-terminal Δ28 truncation constructs) were stimulated with 50 ms pulses from 0–150 mV in 10 mV increments from a holding potential of −150 mV. Dashed: WT from (Gamal El-Din et al., 2013); Green, NaVAb/N49K, V1/2=−21.8±1.8 mV, k=7.9±1.0 mV, n=4; Gray: NaVAb/N49K/L109A/M116V (KAV) Δ28, V1/2=59.1±0.8 mV, k=7.7±0.8 mV, n=3; Blue: NaVAb/KAV/Q150C Δ28, V1/2=76.8±3.1 mV, k=10.0±0.9 mV, n=3; Red: NaVAb/KAV/G94C Δ28, V1/2=88.4±1.1 mV, k=10.6±0.4 mV, n=4; Purple: NaVAb/KAV/G94C/Q150C Δ28 in 50 mM DTT, V1/2=105.7±0.8 mV, k=10.2±0.2 mV, n=3; Black: NaVAb/KAV/G94C/Q150C Δ28 with no DTT, V1/2=n/a, k=n/a, n=5. Markers and error bars represent average G/Gmax ± standard error of the mean (SEM). Half activation (V1/2) and slope (k) values are averages of individual fits ± SEM. Curves = 1/(1+ê(( V1/2−Vm)/k)). Data points within 15 mV of Vrev were omitted to reduce noise.
(B) Representative current families of NaVAb/KAV Δ28 (gray), NaVAb/KAV/Q150C Δ28 (blue), NaVAb/KAV/Q94C Δ28 (red), and NaVAb/KAV/G94C/Q150C Δ28 in 50 mM DTT (purple). Transiently transfected Sf9 cells were held at −150 mV and stimulated for 50 ms to depolarized voltages in 10 mV increments. Scale bars represent 10 msec x 1 nAmp.
See also Figure S3.
We next combined voltage-shifting mutations with Cys substitutions and measured disulfide crosslinking in Trichoplusia ni insect cells (Hi5), the standard cell-type for expression of NaVAb for structural biology (Payandeh et al., 2011). In disulfide-crosslinking experiments, Hi5 cells expressing each double-Cys mutant were treated with a hyperpolarization/oxidation solution containing gramicidin/Cu-1,10-phenathroline and subjected to detergent extraction, SDS-PAGE, and immunoblotting. The percent formation of disulfide-crosslinked NaVAb tetramers for G94C, E96C, and V100C increased steadily to 20%, 40%, and 80%, respectively (Figure 1C, yellow bars; Figures S2A and S2B). When we tested NaVAb double-Cys mutants containing the KAV triple mutation, there was a significant change in the disulfide-crosslinking profile, such that G94C and E96C were both ~80% crosslinked in tetramers (Figure 1C, red bars; Figures 2C and 2D). The effect of the KAV mutation was greatest for crosslinking of Q150C to G94C, indicating that this residue pair approaches closely in the resting state (Figure 1C, black circles). In the absence of hyperpolarization/oxidation, a lower level of crosslinking was observed, but the effect of the KAV mutation was again greatest for crosslinking of the G94C and E96C (Figures 1D, S2C and S2D). These crosslinking profiles (Figures 1C and 1D ) indicated that NaVAb was stabilized in the resting state by the KAV mutations that cause S4 to move inward, allowing Cys on the extracellular side of G94C to crosslink with Q150C. These crosslinking results correlate well with our electrophysiological results for NavAb/KAV (Figure 2).
We further tested the functional effects of Cys substitutions in NaVAb/KAV by electrophysiology (Figures 2 and S3). Mutations G94C and Q150C shifted the voltage dependence of activation of NaVAb/KAV positively to V1/2=+88mV and +77mV, respectively (Figure 2A, red, blue). Moreover, in the presence of dithiothreitol (DTT) to reduce disulfide bonds, the double-Cys mutant NaVAb/KAV/G94C/Q150C showed an additive shift to V1/2=+106 mV, greater than 200 mV more positive than WT (Figure 2A, purple). Importantly, no current was observed for NaVAb/KAV/G94C/Q150C without DTT (Figure 2A, black), indicating that the substituted Cys residues disulfide-locked the VS in a resting state, which was reversed following disulfide bond reduction.
Conformations of the Reduced and Disulfide-Locked VS in the Activated State
To assure that G94C and Q150C did not cause structural changes themselves, independent of state-dependent disulfide-locking, we determined the structure of NaVAb/G94C/Q150C + DTT by X-ray crystallography (Figures 3A, 3B and S4A). No significant structural changes from NaVAb/WT were observed (Cα r.m.s.d. ~0.9 Å; Figure 3B). We also determined the structure of NaVAb/V100C/Q150C, which is expected to disulfide-lock in the activated state (Figures 3C, 3D, S4B, and S4C). The structure was nearly identical to NaVAb/WT (Cα r.m.s.d. ~0.9 Å) (Figure S5B) and the side chains of V100C and Q150C clearly formed a disulfide bond, as judged by strong FO−FC peaks from sulfur atoms with S-S bond length of ~2.1 Å (Figure 3D). These structures confirm that disulfide crosslinking does not distort the VS. Together, these electrophysiological and structural results show that NaVAb/KAV/G94C/Q150C is suitable for determination of the structure of the NaVAb resting state.
Figure 3. Structures of NaVAb G94C/Q150C and Disulfide-crosslinked NaVAb V100C/Q150C in the Activated State.

(A) Overall structure of NaVAb/G94C/Q150C Δ28 activated state. The VS S0 to S3 segments are colored in orange and S4 in magenta. The S4-S5 linker is shown in blue and the pore module S5 to S6 in yellow. Ordered DMPC phospholipid molecules are highlighted in green for acyl chains and orange/red for phosphate head groups.
(B) Superposition of NaVAb/G94C/Q150C Δ28 structure with NaVAb/WT structure (PDB 4EKW). Cα backbone structures are shown with NaVAb/WT colored in gray with Cα r.m.s.d. of ~0.9 Å.
(C) Overall structure of NaVAb/V100C/Q150C Δ28 disulfide-crosslinked activated state. The structure is rendered as in (A). Difference in the C-terminal tail is due to crystallization pH condition (pH 4.8 vs. pH 5.6–6.0 for NaVAb/G94C/Q150C Δ28).
(D) Electron density map for V100C-Q150C disulfide bond. The FO−FC difference map is shown for V100C and Q150C side chains at 3.5σ (green) and 7σ (red) contour levels. Sulfur atoms on cysteine side chains are shown in gold.
Structure of Disulfide-Locked NaVAb/KAV/G94C/Q150C in the Resting State
Extensive efforts to crystallize NaVAb/KAV/G94C/Q150C were unsuccessful, suggesting that its structure differs significantly from NaVAb/WT. Because NaVAb is too small for accurate structure determination by cryo-EM, we formed a fusion protein by linking the C-terminal alpha helix of maltose-binding protein (MBP) to the N-terminal alpha helix of NaVAb with a rigid Asn-Ala linker (Waugh, 2016), which increased the size of tetrameric NaVAb to 290 kDa (Figures S5A and S5B). Purified MBP-NaVAb/KAV/G94C/Q150C showed nearly complete disulfide crosslinking in SDS-PAGE (Figure S5B). We imaged MBP-NaVAb/KAV/G94C/Q150C in digitonin detergent by cryo-EM (Figures S5C and S5D) and reconstructed a 3D map to 4.0 Å overall resolution (Figures 4A and S6), as assessed by the gold standard FSC criterion (Henderson et al., 2012) (Figure S6B). The density map shows high-resolution features of side chains that allow atomic model building (Figures 4A and S7). The map resolution is 3.0–3.9Å in the PM (Figures 4A and S7B), revealing most side chains in positions consistent with NaVAb in the pre-open state (r.m.s.d.~1.4 Å)(Payandeh et al., 2011). Although the map resolution in the VS from S0 to S2 is limited to 4–5 Å, the density for S3, S4, and the S4-S5 linker is better than 3.9 Å and gives clear side-chain images, enabling us to assign most residues with confidence during model building and refinement (Figures 4A and S7). Close inspection of the VS showed three striking changes highlighted in color in Figure 4B: a new fold in the S3-S4 linker and the extracellular end of the S4 segment (Figure 4B, red), striking inward movement of the S4 segment (Figure 4B, magenta), and a new sharply angled bend in the S4-S5 linker (Figure 4B, blue).
Figure 4. Cryo-EM Structure of NaVAb Resting State at 4.0 Å Resolution.

(A) Cryo-EM density map of MBP-NaVAb/KAV/G94C/Q150C disulfide-crosslinked resting state in digitonin detergent. Side view (left) and top (extracellular) view (right) are shown. The density map is colored by local resolution according to the MonoRes color code (side bar). Density map for MBP regions is blurry and not displayed for clarity but can be viewed in Figure S5D.
(B) Overall structure of NaVAb/KAV/G94C/Q150C disulfide-crosslinked resting state. Side view (left) and top view (right) are shown in the same orientation as in (A). S0 to S3 segments of the VS are colored in orange, S3-S4 loop in red, and S4 in magenta. The S4-S5 linker is highlighted in blue and the pore module in yellow. Major structural changes are observed in the S3-S4 loop, S4 segment, and the S4-S5 linker.
See also Figures S5–S7 and Table S2.
Resting State Conformation of the VS
To examine these structural changes in detail, we compared the disulfide-locked resting state crosslinked through G94C-Q150C with the disulfide-locked activated state crosslinked through V100C-Q150C. The VS in the resting state has an extracellular aqueous cleft formed between the S1-S2 and S3-S4 helical hairpins (Figure 5A). While S1 and S2 remain the same in resting and activated states, the S3 helix tilts slightly toward S4 and the S3-S4 loop is shorter as the C-terminal part of the loop changed conformation to become the N-terminal segment of S4 (Figure 5A, red). This conformational change narrows the extracellular aqueous cleft and allows a major inward translocation of the S4 gating charges.
Figure 5. Comparison of NaVAb Disulfide-crosslinked Structures in the Resting State and the Activated State.

(A) Structures of the VS are shown as backbone cartoon superimposed with the solvent accessibility surface. S0 to S3 are shown in gray, S3-S4 loop in red, and S4 in magenta. A wider and shallower aqueous cleft between S1-S2 and S3-S4 helix-loop-helix is present in the resting state compared to a deeper cleft in the activated state.
(B) Gating charge movement. Four Arg gating charges R1-R4 (blue), extracellular negative charge (ENC) cluster of E32 and N49(K) and intracellular negative charge (INC) cluster of E59, E80 (red), Phe in the hydrophobic constriction site (HCS) (green), conserved W76 (gray) and E96 (yellow) are shown in sticks. S4 (magenta) moves outward by 11.5 Å, passing two gating charges through the HCS on S2. Part of S3 is omitted for clarity.
(C) Side view of the structures focusing on S4 (magenta) and the S4-S5 linker (blue), with the S0 to S3 segments shown in gray and the pore module in yellow. The S4 segment moves outward across the membrane from the resting to the activated states while the S1 to S3 segments remain relatively unchanged with respect to the membrane. The S4-S5 linker acts as an elbow that connects the S4 movement to modulate the pore.
(D) Bottom (intracellular) view of the structures in (C) with S0 to S3 omitted for clarity. The S4-S5 linker (blue) undergoes a large conformational change that tightens the collar around the S5 and S6 segments (yellow) of the pore in the resting state and loosens the collar in the activated state.
In this resting state, the S4 helix adopts a 310 conformation from G94C to R108 (R4), producing five complete helical turns with E96, R99 (R1), R102 (R2), R105 (R3), and R108 (R4) located on the same face of the helix with side chains pointing toward S2 (Figure 5B). The conserved HCS residue F56 wedges between R99 (R1) and R102 (R2), as S4 is shifted inward almost vertically toward the cytoplasm by two complete 310 helical turns. R102 (R2) makes a new charge-pair interaction with E59 in the INC on S3. R105 (R3) makes a new charge-pair interaction with E80 in the INC. The final helical turn of S4 after R108 (R4) adopts a loose helical conformation and protrudes into the cytoplasm. R3 and R4 are located near conserved W76 on the cytoplasmic side of S3 (Figure 5B), which may be important for resting-state stabilization. Overall, these major conformational changes in S4 extend the length of 310 helix, exchange ion pair partners between gating charges and counterions in the ENC and INC, and translocate three gating charges fully through the HCS. Remarkably, the S4 segment moves 11.5 Å inward in this process.
Coupling to the Activation Gate
Because S4 extends into the cytoplasm, the S4-S5 linker connecting the intracellular ends of S4 and S5 adopts an angled conformation that projects into the cytoplasm from S4 and rises back into the membrane like an ‘elbow’ to connect with S5 (Figure 5C, blue). These rearrangements of the four S4-S5 linkers tighten the collar around the S5 and S6 segments of the PM, which prevents opening the S6 activation gate (Figure 5D, blue vs. yellow). Therefore, the conformation of the S4-S5 linker is critical for stabilizing the closed pore and for triggering pore opening. The coupling between the VS and PM during pore opening must involve loosening the collar formed by the S4-S5 linkers as S4 moves outward and pulls the elbow of the S4-S5 segment up to the inner surface of the membrane. This direct connection between S4 movement and pore opening may be crucial for rapid pore opening of the sodium channel, which is complete in much less than 1 msec (Hille, 2001).
Comparison between structures of NaVAb in closed and open states (Lenaeus et al., 2017) further reveals the coupling mechanism between S4 movement and pore opening (Figure 6A). The open state of the pore is 10.5 Å in diameter at the activation gate and fills all of the space available within the collar of S4-S5 linkers (Lenaeus et al., 2017). When S4 is in the resting state, the collar of S4-S5 linkers is tightened by interactions of their hydrophobic residues (I119, L123, and V126) with S6 segments (at conserved N211 and I216) and prevents the pore from opening by keeping the activation gate tightly closed (at residue I217) (Figures 6B and 6C). When S4 moves outward to reach the activated state, the collar of S4-S5 linkers is loosened by exchanging interactions with N211 and allows the full opening of the S6 activation gate to 10.5 Å diameter (Figures 6B and 6C), as required for effective conductance of hydrated Na+ (Lenaeus et al., 2017). This mode of S4-S5 linker-mediated activation is likely to be a shared mechanism among many VS-containing ion channels.
Figure 6. Gating Mechanism for Voltage-gated Sodium Channels.

(A) Superposition of structures of NaVAb in the resting/closed state and the activated/open state (PDB 5VB8) viewed from the intracellular side. Major structural changes are observed in the S4-S5 linker, and the S6 activation gate (red for the resting/closed state vs. green for the activated/open state).
(B) Interactions between the S4-S5 linker and S6 near the activation gate. The elbow movement of the S4-S5 linker from the resting/closed state (left) to the activated/open state (right) causes an exchange in the interactions between the S4-S5 linker residues (blue) and the S6 residues (red for the resting/closed state vs. green for the activated/open state) surrounding the activation gate of the pore.
(C) Solvent accessibility surface of NaVAb in the resting closed state (left) and the activated/open state (right). The pore is tightly closed at the activation gate in the resting state (red) but wide open in the activated open state (green).
(D) Structural transition between resting and activated states. Images of structures in the resting (left), intermediate (middle), and activated/open states (right) are captured from Video S1 and rendered as in Figure 5. Part of S3 is omitted for clarity.
See also Video S2.
Implications for Sodium Channel Pharmacology
The structure of the resting state gives important insights into the mechanism of state-dependent modulation of sodium channels by drugs and toxins. Local anesthetics and antiarrhythmics reach their blocking site in the pore rapidly through the open activation gate (Figures 6B and 6C). In contrast, these drugs reach their receptor site in the resting state by moving more slowly through fenestrations in the sides of the pore (Gamal El-Din et al., 2018b; Payandeh et al., 2011), which our structures show are indeed open for drug entry (Figure 7A). Drug discovery research is aimed at finding new generations of NaV blockers. Unexpectedly, the activated state structure of NaVAb/G94C/Q150C shows an array of phospholipid molecules interacting with the S4-S5 linker (Figure 7B). This location presents a great opportunity for structure-based design of next-generation drugs targeting NaV channels. Finally, the resting state structure also reveals the basis for voltage-dependent binding of gating-modifier toxins and drugs. Peptide toxins from scorpions and spiders (Bosmans et al., 2008; Cestèle et al., 1998; Cestele et al., 2006; Clairfeuille et al., 2019; Rogers et al., 1996; Schmalhofer et al., 2008; Shen et al., 2018; Shen et al., 2019; Sokolov et al., 2008; Wang et al., 2011; Xiao et al., 2008; Xu et al., 2019; Zhang et al., 2011, 2012) and an experimental analgesic (Ahuja et al., 2015) bind in a state-dependent manner to a receptor formed by the S3-S4 helical hairpin and the aqueous cleft in the VS. They impede gating charge movement, shift the voltage dependence of activation, and slow its coupling to inactivation. Changes in the size and organization of the extracellular aqueous cleft in the resting state (Figures 5A and 7C) are likely to be responsible for state-dependent binding of these gating-modifier drugs and toxins to the VS.
Figure 7. Implications of the Resting State Structure for Sodium Channel Pharmacology.

(A) Drug access to the pore of NaVAb via the fenestrations in the resting state. Top view cutaway section below the selectivity filter of PM shows fenestration and hydrophobic access to central cavity of the pore.
(B) Phospholipid binding to a new drug target site in NaVAb G94C/Q150C. Electron density map for phospholipids near the S4-S5 linker. The 2FO−FC electron density is contoured at 1σ level (black mesh). Residues W76 on S2, and S121 and P128 on the S4-S5 linker interact with phosphate head groups, and V120, I124 and I127 interact with acyl chains of the lipids (not rendered).
(C) Conformational change between resting and activated states at the receptor site for gating-modifier toxins and drugs in the VS.
The Gating Mechanism of NaVAb
The series of conformational changes that connect resting and activated states of the VS is illustrated in detail in Videos S1 and S2 and in abbreviated form in Figure 6D by freeze-frame images from those videos. As shown by our resting state structure, transition from the previously characterized activated state to the resting state involves major coupled conformational changes (Videos S1 and S2). The extracellular S3-S4 loop and the extracellular end of the S4 segment adapt to allow inward movement of the S4 gating charges. The inward movement of the S4 segment translocates R1 to the HCS and passes R2 to R4 fully through the HCS. This movement bends the junction of the S4 segment with the S4-S5 linker to give an inwardly projecting elbow. Formation of this elbow tightens the collar of S4-S5 linkers around the activation gate and stabilizes the closed state. This resting state structure resembles a cocked gun with the inwardly drawn S4 segment poised to shoot outward upon depolarization, relieving the inward force on the elbow, loosening the collar of S4-S5 linkers, and rapidly opening the activation gate.
Discussion
Voltage Shifting and Disulfide Locking Capture the Resting State
The structure of NaV channels in the resting state has remained elusive because this state exists only at deeply negative resting membrane potential. To overcome this huge energetic barrier, we inserted voltage-shifting mutations to stabilize the resting state for structural study at 0 mV and introduced a disulfide crosslink to lock the conformation of the VS in a homogeneous configuration. The disulfide-locked VS was inactive, but reduction of disulfide bonds released the VS and allowed activation of the sodium channel, confirming that the functional resting state was indeed captured in these constructs.
The Resting State Conformation is Unique Among Sodium Channel Structures
The resting state structure of NaVAb is unique among sodium channel structures to date. Neither the previous structures of native eukaryotic sodium channels (Pan et al., 2018; Shen et al., 2018; Shen et al., 2019; Shen et al., 2017; Yan et al., 2017) nor the recent structure of the hybrid VS in a chimera of NaVAb and NaV1.7 trapped in an intermediate ‘deactivated state’ by toxin binding (Xu et al., 2019) show the characteristic conformational changes in the S3-S4 linker, S4 segment, elbow, or S4-S5 linker, or the symmetrically closed activation gate that the resting state structure of NaVAb reveals. Detailed comparison with the postulated ‘deactivated state’ induced by toxin binding in the nonfunctional NaVAb/NaV1.7 chimera (Xu et al., 2019) reveals that the S4 gating charges are only partially drawn inward toward the cytosol and the characteristic sharply bent elbow is not formed. Unlike in the resting state, the activation gate formed by the intracellular ends of the S6 segments in the toxin-bound structure of Xu et al. adopts a partially collapsed, two-fold symmetric conformation characteristic of the known structure of the inactivated state of NaVAb (Payandeh et al., 2012). Therefore, the comparison of the functionally defined resting and activated states of NaVAb presented here provides unprecedented insight into the details of the gating mechanism.
The Structure of the Resting State Supports a Sliding Helix Mechanism of Gating
Our structural evidence reveals major transmembrane movements of S4 and its gating charges. S4 moves vertically ~11.5Å and changes its conformation from 310 helix (from G94C to R4) in the resting state to loop + alpha helix (from E96 to R1) followed by 310 helix (from R1 to R4) in the activated state. Not only does S4 move almost vertically across the membrane, it also rotates significantly, and the gating charges exchange ion pair partners between the INC and ENC as S4 translocates through the HCS. These features are consistent with the classical sliding-helix mechanism of voltage-dependent gating (Catterall, 1986; Guy and Seetharamulu, 1986; Yarov-Yarovoy et al., 2012). On the other hand, our structural studies do not provide evidence for the transporter (Bezanilla, 2008) or paddle (Jiang et al., 2003) mechanisms of gating. Therefore, we consider it likely that the sliding-helix model correctly captures the fundamental mechanism of electromechanical coupling in voltage-gated sodium channels.
The Sliding Helix Model is Consistent with Studies of Other Voltage Sensitive Proteins
Structural studies of distantly related voltage-sensitive proteins with VS modules, including a voltage-sensitive phosphatase (Li et al., 2014) and calcium-and voltage-gated tandem-pore channels (Guo et al., 2016; Kintzer and Stroud, 2016; She et al., 2018), suggest conformational changes that are consistent with the sliding-helix mechanism of gating, but do not reveal the structure of the resting state. Similarly, several studies of KV channels using optical probes, site-directed mutagenesis, and disulfide-locking methods suggest a combination of outward and twisting movement of the S4 gating charges through the VS, consistent with the sliding helix mechanism of gating charge movement (Broomand and Elinder, 2008; Broomand et al., 2003; Gandhi et al., 2003; Glauner et al., 1999; Henrion et al., 2012; Larsson et al., 1996; Lin et al., 2011; Mannuzzu et al., 1996; Schow et al., 2010; Tao et al., 2010; Vargas et al., 2012). Together with our work on NaV channels, which are the most steeply voltage-sensitive and most rapidly activated among voltage-sensitive proteins, these complementary results on a wide range of other voltage-sensitive proteins support the conclusion that all of electrical signaling depends on a sliding-helix mechanism of voltage-dependent gating.
The Resting State of NaVAb Provides a Template for Eukaryotic NaV and CaV Channels
All NaV and CaV channels have a resting state that is stable at large negative resting membrane potentials (Hille, 2001). Because a homolog of NaVAb is likely to be the ancestor of eukaryotic NaV and CaV channels (Ren et al., 2001; Yu and Catterall, 2004), our resting state structure provides a template for understanding these more complex ion channel proteins. Thus, based on the structure of the NaVAb resting state, we expect that the resting states of eukaryotic NaV and CaV channels will have their homologous S4 segments in all four domains drawn intracellularly by the electrostatic force of the large negative membrane potential on their gating charges, such that only a single conserved gating charge remains within, or on the extracellular side of, the HCS. This movement would induce increased bending of the elbow at the junction of the S4 helix and S4-S5 linkers in each domain and tighten the collar of S4-S5 linkers around the activation gate in a pseudosymmetric fourfold bundle, ensuring its tight closure and poising it for re-opening. As we have modeled for NaVAb (Videos S1 and S2), activation from the resting state would be initiated by outward sliding-helix movement of the four S4 segments through the HCS, likely in specific sequence (Chanda and Bezanilla, 2002), thereby reducing the bend in the elbow to an angle of ~90°, widening the collar of S4-S5 segments, and opening the activation gate by similar concerted bending and rotation of the four homologous S6 segments as for NaVAb. These conformational movements are likely to be the conserved trigger for electrical signaling in both prokaryotes and eukaryotes.
STAR METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, William A. Catterall (wcatt@uw.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Microbe strains
E. coli GC10 was cultured at 37°C in LB medium supplemented with 100μg/ml of ampicillin for plasmid DNA extraction. E. coli BL21(DE3) was cultured in MagicMedia E. coli expression medium at 37°C for screening of disulfide-crosslinking mutants. E. coli DH10Bac was cultured at 37°C in LB medium supplemented with 50 µg/mL kanamycin sulfate, 7 µg/mL gentamicin and 10 µg/mL tetracycline for bacmid production.
Cell lines
Sf9 (Spodoptera frugiperda) insect cells were maintained in Grace’s Insect Medium and supplemented with 10% FBS and penicillin/streptomycin at 27°C and passaged at 80–95% confluence for electrophysiology. Sf9 and Hi5 (Trichoplusia ni) insect cells were maintained and infected in Grace’s Insect Medium supplemented with 7.5% FBS and glutamine/penicillin/streptomycin at 27°C for baculovirus production and protein expression, respectively.
METHOD DETAILS
Screening of disulfide-crosslinking mutants in E. coli
We screened for favorable Cys pairs that could disulfide-lock the resting state of NaVAb in E. coli because of its ease of expression of large numbers of constructs and its very negative membrane potential (~ −160 mV at early stationary phase) (Bot and Prodan, 2010) that would stabilize the resting state. Double-Cys mutations were designed for disulfide-crosslinking experiments based on a hypothesis that the S4 helix moves vertically across the membrane as a function of membrane potential and exchanges interactions between S4 residues from one subunit with S5 residues from a neighboring subunit (Shimomura et al., 2011). Therefore, favorable interactions could result in inter-subunit disulfide crosslinking that can be visualized by SDS-PAGE and immunoblotting as a measure of formation of higher order oligomers. The gene encoding N-terminal FLAG-tagged NaVAb was PCR-amplified from pFastBacDual FLAG-NaVAb vector (Payandeh et al., 2012) and cloned into pET21b vector (Life Technologies). Oligonucleotide primers containing a Cys mutation on S5 helix at positions 143, 146, 147, 150, or 151 were used to generate a set of vectors containing five different S5 Cys mutants by QuickChange Lightening site-directed mutagenesis (Agilent). Oligonucleotide primers containing a Cys mutation on S3-S4 loop to S4 helix at positions S93, G94, F95, E96, I97, L98, R99, V100, R102, L103, R105, or L106 were used to generate a set of double-Cys mutations using the set of vectors containing S5 cysteine mutants as templates. All clones were verified by DNA sequencing to contain correct mutations. The double-Cys vectors (60 in total) were transformed into the BL21(DE3) strain of E. coli, and the cultures were grown in 1 ml of MagicMedia E. coli auto expression medium (Life Technology) for 3 days at 37°C (Shimomura et al., 2011). Cells were harvested by centrifugation at 3000xg for 20 min. Cell pellets were resuspended in a buffer containing 20 mM Tris-HCl and 140 mM NaCl. Ten μl of cell suspension were mixed with 40 μl of 2X LDS sample buffer (Life Technologies) and assayed by SDS-PAGE and immunoblotting. For SDS-PAGE, 10 μl of the LDS samples were loaded on 4–12% Bis-Tris polyacrylamide gel and electrophoresed with 1X MOPS-SDS running buffer (Life Technologies). For immunoblots, protein bands from SDS-PAGE gels were transferred onto nitrocellulose membranes; blocked with 5% non-fat dried milk in Tris-saline buffer with 0.05% Tween 20; probed by mouse anti-FLAG M2 IgG primary antibody (MilliporeSigma) followed by goat anti-mouse IgG-HRP conjugated secondary antibody (Jackson ImmonuResearch Laboratories); detected with AmerSham ECL Prime Western Blotting Detection Reagent (GE Healthcare Life Sciences); and imaged using Bio-Rad ChemiDoc XRS+ Imaging System (Bio-Rad).
Disulfide-crosslinking assays in Hi5 (T. ni) insect cells
Our previous studies showed that the S4 segment of NaVAb expressed in cultured cells can be disulfide-locked in resting, intermediate, and activated conformations, as assessed by voltage clamp analysis, by insertion of specific pairs of Cys residues and catalysis of disulfide bond formation with Cu-1,10-phenanthroline (DeCaen et al., 2011; DeCaen et al., 2009; DeCaen et al., 2008). Plasmids containing double-Cys mutations were made using the same procedure as above but pFastBacDual FLAG-NaVAb was used as a template. Additional mutations including N49K, L109A, M116V, and 28-residue C-terminal truncation with stop codon (Δ28) were introduced by mutagenesis using the same procedure. Baculoviruses containing double-Cys mutants were prepared using the Bac-to-Bac protocol according to the manufacturer with Sf9 insect cells (Life Technologies). Third passage (P3) baculoviruses were used to infect Hi5 cells in single layer-culture dishes, and the cells were incubated at 27°C for 48–72 h. Cells were harvested by centrifugation. For the Control group, cell pellets from half of one dish were resuspended in 10 ml of 20 mM Tris-HCl, pH 7.5 and 140 mM NaCl and incubated at room temperature for 1 h. For the Hyperpolarization-Oxidation group, cell pellets from the other half of a dish were resuspended in 10 ml of 20 mM Tris-HCl (sodium free), pH 7.5, 140 mM choline chloride, and 10 μg/ml Gramicidin (MilliporeSigma) to hyperpolarize cell membrane potential, followed by an addition of 100 μM of copper (II) phenanthroline -- a mild oxidizing agent made freshly by mixing CuSO4 and 1,10-phenanthroline solutions at 1:3 molar ratio and incubation at room temperature for 1 h. To quench the oxidation reaction, 1 mM EDTA was added to the cell suspension. Cells from both groups were centrifuged and cell pellets were resuspended in 1 mL of 20 mM Tris-HCl and 140 mM NaCl. Five μl of cell suspension were mixed with 45 μl of 2X LDS sample buffer plus 25 mM iodoacetamide (prepared freshly within the hour) and protease inhibitor cocktails (MilliporeSigma). The samples were subsequently assayed by SDS-PAGE and immunoblotting as described above; imaged using Bio-Rad ChemiDoc XRS+ Imaging System; and quantified using Bio-Rad Image Lab Software according to manufacturer (Bio-Rad).
Channel expression for electrophysiology
Sf9 cells below passage 25 were transiently transfected in 24-well plates using a 2:4 ratio of µg DNA: µl PolyJet transfection reagent (SignaGen) in unsupplemented Grace’s insect media with a plasmid encoding GFP-P2A-NaVAb gene under an OpIE2 promoter modified from pIZT vector (Life Technologies) and purified using an endonuclease-free miniprep kit (Omega). To construct the modified vector, the eGFP was moved from the Zeocin resistance gene to upstream of the FLAG-channel sequence, and a GSG-P2A site (porcine teschovirus-1) was inserted between the two using standard site directed mutagenesis, restriction digestion, and ligation methods. This was done to better identify transfected cells with optimal expression (0.5–3 nA peak current) by utilizing the 1:1 stoichiometric ratio of GFP to the channel conferred by the P2A self-cleaving peptide. Cells were replated onto 10 mm glass coverslips 24 hours post-transfection and used for recording for the following 5–7 days.
Electrophysiological Data Collection
1.5–3 MΩ glass pipettes were used to patch-clamp fluorescent cells, and currents were recorded via whole-cell voltage clamp (EPC10, Pulse, HEKA), sampled at 250 kHz, and filtered at 3 kH. Membrane capacitance ranged from 5–25 pF. Rs was below 10 MΩ, with a 2 µs lag and at least 70% compensation. A P/4 leak subtraction protocol was used. In order to analyze NaVAb mutants with positively shifted voltage dependence, solutions were designed to give a reversal potential of 0 mV for Na+. Intracellular solution (in mM): NaCl (35) CsF (105) EGTA (10) HEPES (10). Extracellular solution contained: NaCl (35) NMDG-Cl (105) MgCl2 (2) CaCl2 (2) HEPES (10). The pH for both solutions was 7.4, adjusted with CsOH and NMDG+, respectively. Osmolarity was adjusted with sucrose to match that of the cells’ media each day. The measured liquid junction potential was −7 mV for all recordings, and the reported voltages were not adjusted. NaVAb /N49K, which activates at negative voltages, was studied as previously described (Gamal El-Din et al., 2013). Briefly, Hi5 cells were infected with baculovirus and recordings were made 18–24 hours later. The intracellular solution composition was as above, and the extracellular solution was identical except NaCl concentration was 140 mM, no NMDG was used, and pH was adjusted with NaOH. Current families were generated by holding cells at −150 mV (−120 for NaVAb/N49K) and stimulating with a series of 50-ms pulses at 10 mV intervals. To determine reversal potential of outward-only current, peak current was elicited by pulsing to an appropriate potential for each construct and decreasing the voltage in 10 mV steps until substantial inward current was seen in the tail currents. The time between the start of the initial voltage step and the subsequent steps was determined for each construct as roughly the time to peak (data not shown). An agar bridge consisting of extracellular solution with 3% Agar was placed between the reference electrode and the bath in experiments utilizing perfusion of extracellular solution containing reducing agents.
Expression and protein purification of NaVAb activated state disulfide-crosslinked mutant
Hi5 cells were infected with P3 virus for NaVAb V100C/Q150C double-Cys mutant (Δ28 construct (Gamal El-Din et al., 2018a)) and incubated for 48–72 h at 27°C. Cells were harvested by centrifugation. Cell pellets were subjected to oxidation treatment in Buffer A (50 mM Tris-HCl pH 7.5 and 200 mM NaCl, 20 ml per cells from 1 dish) instead of hyperpolarization buffer using the procedure as described above. Cells were harvested by centrifugation, resuspended in Buffer B (Buffer A supplemented with 1 mM PMSF, 2X SigmaFast protease inhibitor cocktails, benzamidine HCl, and DNAse I), lysed by sonication, and the membranes solubilized as described above. After centrifugation, the supernatant was incubated with FLAG-M2 affinity gel (MilliporeSigma) for 1 hour at 4°C with gentle mixing. The resins were washed with Buffer C (Buffer A supplemented with 0.12% digitonin) and bound protein was eluted with Buffer C supplemented with 200 μM FLAG peptide. Eluted protein was concentrated to 1 ml using Vivaspin20 100 kDa MWCO and further purified with Superdex S200 size-exclusion chromatography with using 10 mM Tris-HCl pH 7.5, 100 mM NaCl, and 0.12% digitonin as a running buffer. Elution fractions were evaluated using SDS-PAGE and peak fractions were combined and concentrated using Vivaspin6 100 kDa MWCO to final concentration ~20 mg/ml. NaVAb/G94C/Q150C Δ28 mutant was expressed and purified as described above, but without the oxidation treatment. FLAG-eluted protein was treated with 50mM DTT and subsequent purification step contains 5 mM DTT in the buffers.
Crystallization of NaVAb activated state disulfide-crosslinked mutant
NaVAb-bicelle complexes were prepared by mixing NaVAb double Cys mutant with 10% bicelle (7.5% DMPC and 2.5% CHAPSO) at 1:5 volume ratio and screened for crystallization conditions as described previously. Best crystals appeared under 1.8–2.0 M ammonium sulfate and 0.1 M sodium citrate, pH 4.8–5.0 for NaVAb/V100C/Q150C Δ28 mutant and pH 5.6 for NaVAb/G94C/Q150C Δ28 mutant. Crystals were cryo-protected by step-wise transfers to a series of cryo-protectant solutions containing 6–30% glucose with the same concentration of ammonium sulfate and sodium citrate pH 4.8 for NaVAb/V100C/Q150C Δ28 and pH 6.0 for NaVAb/G94C/Q150C Δ28.
X-ray data collection and structural determination
Crystals were tested for diffraction and data were collected at Advanced Light Source (ALS) beamline 8.2.1 and 8.2.2 (HHMI). Diffraction data were processed using HKL2000 software (Otwinowski and Minor, 1997) and structures were determined by molecular replacement using PHASER (McCoy et al., 2007) and refined using REFMAC (Murshudov et al., 2011) in CCP4 program suite (Winn et al., 2011). Manual model building and local real space refinement was carried out in COOT (Emsley et al., 2010), followed by structure refinement in REFMAC (Table S1).
Expression and purification of NaVAb/KAV/G94C/Q150C in the resting state
We developed a fusion protein strategy to generate MBP-NavAb fusion proteins for structural study by cryo-EM. Several rigid linkers between MBP and NaVAb were designed and tested by protein purification and cryo-EM imaging using a Tecnai G2 F20 microscope (FEI). The best linker that produced the most homogenous population with good particle image contrast was chosen for the disulfide-locked resting state NaVAb. The gene encoding MBP from E. coli was N-terminally fused to NaVAb/KAV/G94C/Q150C with an Asn-Ala linker sequence by PCR-driven overlap extension method (Heckman and Pease, 2007), and the fusion gene was cloned into pFastBacDual vector (Life Technologies). Baculovirus harboring the fusion gene was prepared using the Bac-to-Bac protocol. Hi5 cells were infected with P3 virus and incubated for 72–96 hours at 27°C for maximum expression. Cells were harvested by centrifugation. Cell pellets were subjected to hyperpolarization-oxidation treatment by re-suspending in hyperpolarization buffer (20 mM Tris-HCl (sodium free) pH 7.5, 140 mM choline chloride, 20 ml per cells from 1 dish) plus 100 μM copper (II) phenanthroline prepared as described above and incubated at room temperature for 1 h with gently shaking. The reaction was quenched with 1 mM EDTA, and the cells were pelleted by centrifugation and washed with buffer A followed by centrifugation. Cell pellets were resuspended in Buffer B, lysed by sonication and membranes were solubilized with 1% high purity digitonin (Calbiochem) for 1 h at 4°C with gentle mixing. The mixture was centrifuged at 15,000xg for 30 min at 4°C, and the supernatant was incubated with amylose resin (NEB) for 1 h at 4°C with gentle mixing. The resins were washed with Buffer C and bound protein was eluted with Buffer C supplemented with 10 mM maltose). Eluted protein was concentrated to 1 ml using Vivaspin20 100kDa MWCO and further purified with Superose 6 Increase size-exclusion chromatography using 10 mM Tris HCl pH 7.5, 100 mM NaCl, and 0.12% digitonin as a column running buffer. Elution fractions were evaluated using SDS-PAGE and peak fractions were combined and concentrated using Vivaspin6 100 kDa MWCO to final concentration ~5–8 mg/ml.
Cryo-EM sample preparation and data acquisition
Cryo-EM grids were prepared by applying freshly purified MBP-disulfide locked resting state NaVAb in digitonin detergent to glow-discharged holey carbon grids (C-Flat Cu R1.2/1.3, EMS). Grids were manually blotted for 4 to 5 s and immediately plunge-frozen in liquid ethane cooled by liquid nitrogen. Cryo-EM data were recorded on a Titan Krios (FEI) operated at 300 kV, equipped with a GIF-quantum energy filter (Gatan) at 20 eV slit width and a K2 Summit direct detector (Gatan). LEGINON (Suloway et al., 2005) was used for automated data collection. Movies were collected at a nominal magnification of 130,000x in super-resolution mode resulting in a pixel size of 0.528 Å, with a small defocus range of –0.5 to –2.5 μm. The dose rate on the camera was set to be ~8 counts per physical pixel per second. The total exposure time was 8.6 s (0.2 s/frame), leading to a total accumulate dose of 60 electrons per Å2 on the specimen (Table S2).
Cryo-EM Image processing
Movie frames were aligned and 2x binned to a pixel size off 1.056 Å using MotionCor2 (Zheng et al., 2017) and the contrast transfer function parameters for each motion-corrected image were estimated using Gctf (Zhang, 2016). Subsequent image processing steps were performed using RELION (Scheres, 2012) on an NVIDIA GPU-accelerated workstation. Particles were auto-picked using templates derived from 2D class averages that were initially generated from manually picked particles from ~50 images. A total of ~476,000 particles from ~5,000 good images were extracted into 304 × 304-pixel boxes and subjected to several rounds of 2D classification to remove bad particles. 3D classification and refinement were carried out in RELION using the crystal structure of a NaVAb pore-only model that had been low-pass filtered to 60 Å as an initial template. The resulting 3D reconstructions were routinely better than 5 Å resolution. Subsequently, 333,899 particles were subjected to cycles of global angular search in cisTEM (Grant et al., 2018) with an earlier reconstruction from RELION as a starting reference and a 3D mask that included only the transmembrane region and low-pass filtered the region outside to 50 Å. After global angular search, the particles were further subjected to cycles of local angular search with the resolution limit set to 8 Å for particle alignment and the 3D mask. The resolution was estimated using the gold-standard Fourier shell correlation (FSC) 0.143 criterion (Henderson et al., 2012) to be 4.0 Å (Table S2). Local resolution was evaluated using MonoRes (Vilas et al., 2018).
Model building and refinement of the resting state structure
The NaVAb crystal structure (PDB 3RVY) was initially docked into cryo-EM density map using Chimera (Pettersen et al., 2004). The atomic model was then rebuilt using Coot (Emsley et al., 2010) and subsequently refined using the real space refinement feature in Phenix (Adams et al., 2010) (Table S2).
QUANTIFICATION AND STATISTICAL ANALYSIS
Analysis of electrophysiological data
Pulse data was analyzed using IGOR Pro (6.37). Peak current at each voltage of the current family was plotted as a function of the stimulus voltage to visualize the current vs voltage (I/V) relationship. As the I/V curves contained only outward current, the reversal potential (Vrev) was determined from the tail currents at peak activation. The instantaneous current of the tail current family was plotted as a function of voltage, fit with a line near 0 mV, and the X intercept was used as Vrev to generate the conductance vs voltage (G/V) curve from the I/V curve by calculating G=I/(Vm−Vrev). The calculated Vrev was near 7 mV for all recordings. Normalized G/V curves were fit with a simple two-state Boltzmann equation 1/(1+exp(V1/2−Vm)/k) in which Vm is the stimulus potential, V1/2 is the half-activation voltage, and k is a slope factor. I/V plots of inward currents for NaVAb/N49K were fit with (Vm−Vrev)*(Imin/(1+exp((Vm−V1/2)/k)) to generate the above Boltzmann curve fit to normalized G/V.
Quantification and analysis of immunoblot data
Quantification for bands from immunoblots in Figures S2A, and S2C were performed using Bio-Rad Image Lab Software version 5.1 according to manufacturer. Details of experiments can be found in the figure legends. Data are represented as mean ± standard error of the mean (SEM) from 5 independent experiments (n=5). Significance was determined by Student’s t-test.
Estimation
Protein concentrations were estimated using an A280 extinction coefficient of 33,920 M−1 cm−1 on a spectrophotometer.
Global resolution estimation of cryo-EM density map is based on the 0.143 Fourier Shell Correlation criterion.
DATA AND SOFTWARE AVAILABILITY
The coordinates and structure factors for the reported crystal structures have been deposited in the Protein Data Bank under accession codes 6P6X (NaVAb G94C/Q150C Δ28) and 6P6Y (NaVAb V100C/Q150C Δ28). Cryo-EM map has been deposited in the Electron Microscopy Data Bank (EMDB) under accession code EMD-20265 (MBP-NaVAb KAV/G94C/Q150C). The coordinates of the NaVAb KAV/G94C/Q150C atomic model have been deposited in the PDB under accession code 6P6W.
Supplementary Material
(A) Matrix of cysteine substitutions used in this study. Twelve residues on S4 were screened against 5 residues on S5 that yielded a total of 60 double-cysteine variants. Key residues are highlighted in red for resting, yellow for intermediate, and green for activated state crosslinking.
(B) Immunoblot result for double-cysteine substitutions in E. coli for NaVAb/Q150C and S4 cysteine mutants. R99C and V100C (green) show large crosslinking yields while E96C (yellow) shows a small yield with Q150C.
(C) Immunoblot result for double-cysteine substitution in E. coli for NaVAb/N49K/Q150C and S4 cysteine mutants. R99C and V100C (green) shows large crosslinking yields while E96C and I97C (yellow) and G94C (red) shows moderate yield with Q150C.
Morph movie between the resting/closed and activated/open states were calculated using Pymol. The VS from S0 to S3 segments are shown in orange, S4 in magenta, the S4-S5 linker in blue and the PM in yellow. Only one VS, and two S4-S5 linkers and PM from two opposing subunits are shown for clarity. Side chains of key residues are rendered as sticks and colored as in Figures 5B and 6D.
Morph movie between the resting/closed and activated/open states were calculated using Pymol. The VS from S0 to S3 segments are shown in orange, S4 in magenta, the S4-S5 linker in blue and the PM in yellow. Only one VS is shown for clarity. Side chains of key residues are rendered as sticks and the structure is colored as in Video S1. I217 at the S6 activation gate is shown in yellow as sticks overlaid with transparent atomic spheres.
(A) Immunoblot result from disulfide-crosslinking assay for NaVAb double-cysteine substitutions in Hi5 insect cells. Lanes with (−) denotes cells from control experiments; (+) from hyperpolarization/oxidation experiments with gramicidin/Cu-1,10-phenathroline.
(B) Quantification of percent crosslink in disulfide-crosslinking assays for NaVAb double-cysteine substitutions with Q150C. Percent crosslink denotes the extent of crosslinking quantified from intensity of a tetramer band divided by the total intensity from immunoblots. Control (dark red) vs. hyperpolarization/oxidation (gray).
(C) Immunoblot result from disulfide-crosslinking assays for NaVAb KAV double cysteine substitutions in Hi5 insect cells. Lanes with (−) denotes cells from control experiments; (+) from hyperpolarization/oxidation experiments with gramicidin/Cu-1,10-phenathroline.
(D) Quantification of percent crosslink in the disulfide-crosslinking assays for NaVAb double-cysteine substitutions with Q150C. Error bars represent SEM with n = 5.
(A) Normalized conductance-voltage (G/V) relationships and Boltzmann fits for full-length (FL) compared to C-terminal truncated (Δ28) channels. Due to decreased current density in cells expressing channels containing G94C, the C-terminal truncation mutation Δ28 was added to all constructs for electrophysiological recordings to increase total signal, but the truncation did not change the voltage-dependent properties of the channels. The G/V relationship was not altered for NaVAb/KAV (black, circles; V1/2=61.3 ± 2.1 mV, k= 7.2 ± 0.4 mV, n = 3) compared to NaVAb/KAV Δ28 (gray, triangles; V1/2=59.1 ± 0.8 mV, k=7.7 ± 0.8 mV, n = 3) nor was it changed in NaVAb/KAV/Q150C (green, circles; V1/2=77.8 ±0.8 mV, k=9.9 ± 0.3 mV, n = 3) compared to NaVAb/KAV/Q150C Δ28 (blue, triangles; V1/2=76.8 ± 3.1 mV, k=10.0 ± 0.9 mV, n = 3). Dashed (FL) and solid (Δ28) curves = 1/(1+ê(( V1/2−Vm)/k)). Half activation (V1/2) and slope (k) values are averages of individual fits. Data points within 15 mV of Vrev were omitted to reduce noise. Markers and error bars represent average G/Gmax ± SEM.
(B) Representative current families of NaVAb/KAV (black) and NaVAb/KAV/Q150C (green) full length. Transiently transfected Sf9 cells were held at −150 mV and stimulated for 50 ms to depolarized voltages in 10 mV increments. Scale bars represent 10 msec x 1 nAmp.
(A) Size-exclusion chromatography profile of protein purification on a Superdex 200 column for NavAb/G94C/Q150C Δ28 in the presence of DTT. The protein was expressed and purified as reported previously (Payandeh et al., 2011) except for pre-treatment of FLAG-eluted protein with 50 mM DTT and for inclusion of 5 mM DTT in the running buffer for size-exclusion chromatography. The peak fractions used for protein crystallization are highlighted in light blue. (Inset) SDS-PAGE of the peak fractions of NavAb/G94C/Q150C Δ28 in the presence of DTT. The NaVAb bands migrated at the molecular weight of a monomer indicating complete reduction of disulfide crosslink.
(B) Size-exclusion chromatography profile of protein purification on a Superdex 200 column for disulfide-crosslinked NaVAb/V100C/Q150C Δ28. Hi5 cells were treated with Cu-1,10-phenathroline prior to cell lysis and protein purification. The peak fractions used for protein crystallization are highlighted in light blue. (inset) SDS-PAGE of the peak fractions of disulfide-crosslinked NavAb/V100C/Q150C Δ28. The NaVAb bands migrated at the molecular weight of a tetramer indicating complete disulfide crosslinking.
(C) Superposition of NaVAb/V100CC/Q150C Δ28 structure with NaVAb/WT structure (PDB 4EKW). Cα backbone structures are shown with NaVAb/WT colored in gray with Cα r.m.s.d. of ~0.9 Å.
(A) Size-exclusion chromatography profile of protein purification on a Superose 6 column for disulfide-crosslinked MBP-NaVAb/KAV/G94C/Q150C. Hi5 cells were resuspended in a sodium-free choline chloride buffer and treated with Cu-1,10-phenathroline prior to cell lysis and protein purification. The peak fractions used for cryo-EM are highlighted in light blue.
(B) SDS-PAGE of purified disulfide crosslinked MBP-NaVAb/KAV/G94C/Q150C. The protein bands migrated as a molecular weight of a tetramer indicating a complete disulfide crosslink.
(C) Representative electron micrograph (left) and contrast transfer function (CTF) estimation (right).
(D) Reference-free 2D classification of particle images. The box size is 321 × 321 Å (304 × 304 pixels). The particle largest dimension is ~150 Å. The 2D averages indicate particles of c4 symmetry and contain features of intracellular MBP, NaVAb transmembrane helices and its intracellular C-terminal tail.
(A) Angular distribution of particle orientations in 3D reconstruction.
(B) Resolution assessment for 3D reconstruction of the cryo-EM map using Gold Standard Fourier Shell Correlation (FSC) plot from two half maps. Refinement of particle alignment was carried out using resolution limit of 8 Å. The density map has an overall resolution of 4.0 Å using the FSC criterion of 0.143.
(A) FSC plot of the refined model vs. summed map. The model vs. map correlation is 4.1 Å and 4.5 Å using FSC criterion of 0.143 and 0.5, respectively.
(B) Cryo-EM map and model for VS S0 and S1, S2, S3 and S4 segments, S4-S5 linker, and PM S5, P-loop, and S6 segments. NaVAb stick model is shown superimposed with cryo-EM density in black mesh. High-resolution features for side chains are apparent for the VS S4 segment, the S4-S5 linker, and the PM S5 to S6 segments.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse Monoclonal Anti-FLAG M2 Antibody | Sigma-Aldrich | Cat# F1804 |
| Peroxidase AffiniPure Goat Anti-Mouse IgG (H+L) | Jackson ImmonuResearch Laboratories | Cat# 115–035-003 |
| Bacterial and Virus Strains | ||
| E. coli BL21(DE3) | NEB | Cat# C2527I |
| E. coli DH10Bac | ThermoFisher Scientific | Cat# 10361–012 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Gramicidin | Sigma-Aldrich | Cat# G5002 |
| Digitonin, High Purity | Millipore Sigma | Cat# 300410 |
| 1,2-Dimyristoyl-sn-Glycero-3-Phosphocholine (DMPC) | Anatrace | Cat# D514 |
| Anti-FLAG M2 Affinity Gel | Millipore Sigma | Cat# A2220 |
| FLAG peptide DYKDDDDK | Bio Basic | N/A |
| Deposited Data | ||
| Coordinates and Structure Factors of NaVAb G94C/Q150C ∆28 | This study | PDB: 6P6X |
| Coordinates and Structure Factors of NaVAb V100C/Q150C ∆28 | This study | PDB: 6P6Y |
| Coordinates of NaVAb KAV/G94C/Q150C | This study | PDB: 6P6W |
| Cryo-EM Map of NaVAb KAV/G94C/Q150C | This study | EMDB: EMD-20265 |
| Experimental Models: Cell Lines | ||
| Spodoptera frugiperda (Sf9) | Life Technologies | B825–01 |
| Trichoplusia ni (High Five) | Life Technologies | B855–02 |
| Recombinant DNA | ||
| pET-21b FLAG-NaVAb | This study | N/A |
| pFastBacDual FLAG-NaVAb | Payandeh et al., 2012 | N/A |
| pFastBacDual FLAG-NaVAb G94C/Q150C ∆28 | This study | N/A |
| pFastBacDual FLAG-NaVAb V100C/Q150C ∆28 | This study | N/A |
| pFastBacDual MBP-NaVAb KAV/G94C/Q150C | This study | N/A |
| pIZT GFP-P2A-NaVAb | This study | N/A |
| Software and Algorithms | ||
| HKL2000 | HKL Research | http://www.hkl-xray.com |
| CCP4i 7.0.068 | CCP4 | http://www.ccp4.ac.uk |
| Coot 0.8.9.1 | MRC | https://www2.mrc-lmb.cam.ac.uk/personal/pemsley/coot |
| Leginon 3.3 | NRAMM | http://leginon.org |
| MotionCor2 | UCSF | https://msg.ucsf.edu/software |
| Gctf | MRC | https://www.mrc-lmb.cam.ac.uk/kzhang |
| Relion 3.0 beta | MRC | https://www2.mrc-lmb.cam.ac.uk/relion |
| CisTEM 1.0.0-beta | https://cistem.org | |
| MonoRes | Scipion | http://scipion.cnb.csic.es/m/myresmap |
| Chimera 1.11.2 | UCSF | https://www.cgl.ucsf.edu/chimera |
| Phenix 1.14–3260 | https://www.phenix-online.org | |
| IGOR Pro 6.37 | WaveMetrics | https://www.wavemetrics.com |
| Pymol 1.7.2 | Schrödinger | https://pymol.org |
Highlights.
Voltage shifting and disulfide locking capture a resting state structure of NaVAb
Three gating charges translocate intracellularly through transmembrane electric field
Voltage sensor couples to pore opening by an elbow connecting S4 to the S4-S5 linker
Resting state structure supports a sliding helix mechanism of gating
Acknowledgements
We thank the beamline staff at the Advanced Light Source (BL8.2.1 and BL8.2.2) for assistance during X-ray data collection, J.D. Quispe (University of Washington Cryo-EM Facility) for technical assistance on cryo-EM, Dr. H. Shi (Department of Pharmacology and Howard Hughes Medical Institute, University of Washington) for computational support, and Dr. Jin Li (Department of Pharmacology, University of Washington) for technical and editorial support. This research was supported by National Institutes of Health research grants R01 NS015751 (W.A.C.) and R01 HL112808 (W.A.C. and N.Z.), research training grant T32 GM007750 (E.M.), and by the Howard Hughes Medical Institute (N.Z.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of interests
The authors declare no competing interests.
REFERENCES
- Adams PD, Afonine PV, Bunkoczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, et al. (2010). PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D Biol Crystallogr 66, 213–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahern CA, Payandeh J, Bosmans F, and Chanda B (2016). The hitchhiker’s guide to the voltage-gated sodium channel galaxy. J Gen Physiol 147, 1–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahuja S, Mukund S, Deng L, Khakh K, Chang E, Ho H, Shriver S, Young C, Lin S, Johnson JP Jr., et al. (2015). Structural basis of Nav1.7 inhibition by an isoform-selective small-molecule antagonist. Science 350, aac5464. [DOI] [PubMed] [Google Scholar]
- Armstrong CM, and Bezanilla F (1973). Currents related to movement of the gating particles of the sodium channels. Nature 242, 459–461. [DOI] [PubMed] [Google Scholar]
- Bezanilla F (2008). How membrane proteins sense voltage. Nature reviews Molecular cell biology 9, 323–332. [DOI] [PubMed] [Google Scholar]
- Bosmans F, Martin-Eauclaire MF, and Swartz KJ (2008). Deconstructing voltage sensor function and pharmacology in sodium channels. Nature 456, 202–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bot CT, and Prodan C (2010). Quantifying the membrane potential during E. coli growth stages. Biophys Chem 146, 133–137. [DOI] [PubMed] [Google Scholar]
- Broomand A, and Elinder F (2008). Large-scale movement within the voltage-sensor paddle of a potassium channel-support for a helical-screw motion. Neuron 59, 770–777. [DOI] [PubMed] [Google Scholar]
- Broomand A, Mannikko R, Larsson HP, and Elinder F (2003). Molecular movement of the voltage sensor in a K channel. The Journal of general physiology 122, 741–748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Catterall WA (1984). The molecular basis of neuronal excitability. Science 223, 653–661. [DOI] [PubMed] [Google Scholar]
- Catterall WA (1986). Molecular properties of voltage-sensitive sodium channels. AnnuRevBiochem 55, 953–985. [DOI] [PubMed] [Google Scholar]
- Catterall WA, Wisedchaisri G, and Zheng N (2017). The chemical basis for electrical signaling. Nature chemical biology 13, 455–463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cestèle S, Qu Y, Rogers JC, Rochat H, Scheuer T, and Catterall WA (1998). Voltage sensor-trapping: Enhanced activation of sodium channels by beta-scorpion toxin bound to the S3-S4 loop in domain II. Neuron 21, 919–931. [DOI] [PubMed] [Google Scholar]
- Cestele S, Yarov-Yarovoy V, Qu Y, Sampieri F, Scheuer T, and Catterall WA (2006). Structure and function of the voltage sensor of sodium channels probed by a beta-scorpion toxin. The Journal of biological chemistry 281, 21332–21344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chanda B, and Bezanilla F (2002). Tracking voltage-dependent conformational changes in skeletal muscle sodium channel during activation. J Gen Physiol 120, 629–645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clairfeuille T, Cloake A, Infield DT, Llongueras JP, Arthur CP, Li ZR, Jian Y, Martin-Eauclaire MF, Bougis PE, Ciferri C, et al. (2019). Structural basis of alpha-scorpion toxin action on Nav channels. Science [DOI] [PubMed]
- DeCaen PG, Yarov-Yarovoy V, Scheuer T, and Catterall WA (2011). Gating charge interactions with the S1 segment during activation of a Na+ channel voltage sensor. Proc Natl Acad Sci U S A 108, 18825–18830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeCaen PG, Yarov-Yarovoy V, Sharp EM, Scheuer T, and Catterall WA (2009). Sequential formation of ion pairs during activation of a sodium channel voltage sensor. Proc Natl Acad Sci U S A 106, 22498–22503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeCaen PG, Yarov-Yarovoy V, Zhao Y, Scheuer T, and Catterall WA (2008). Disulfide locking a sodium channel voltage sensor reveals ion pair formation during activation. Proc Natl Acad Sci U S A 105, 15142–15147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emsley P, Lohkamp B, Scott WG, and Cowtan K (2010). Features and development of Coot. Acta Crystallogr D Biol Crystallogr 66, 486–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gamal El-Din TM, Lenaeus MJ, Ramanadane K, Zheng N, and Catterall WA (2018a). Molecular dissection of multiphase inactivation of the bacterial sodium channel NaVAb. J Gen Physiol 151, 174–185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gamal El-Din TM, Lenaeus MJ, Zheng N, and Catterall WA (2018b). Fenestrations control resting-state block of a voltage-gated sodium channel. Proc Natl Acad Sci U S A [DOI] [PMC free article] [PubMed]
- Gamal El-Din TM, Martinez GQ, Payandeh J, Scheuer T, and Catterall WA (2013). A gating charge interaction required for late slow inactivation of the bacterial sodium channel NavAb. J Gen Physiol 142, 181–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gandhi CS, Clark E, Loots E, Pralle A, and Isacoff EY (2003). The orientation and molecular movement of a k(+) channel voltage-sensing domain. Neuron 40, 515–525. [DOI] [PubMed] [Google Scholar]
- Glauner KS, Mannuzzu LM, Gandhi CS, and Isacoff EY (1999). Spectroscopic mapping of voltage sensor movement in the Shaker potassium channel. Nature 402, 813–817. [DOI] [PubMed] [Google Scholar]
- Grant T, Rohou A, and Grigorieff N (2018). cisTEM, user-friendly software for single-particle image processing. eLife 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo J, Zeng W, Chen Q, Lee C, Chen L, Yang Y, Cang C, Ren D, and Jiang Y (2016). Structure of the voltage-gated two-pore channel TPC1 from Arabidopsis thaliana. Nature 531, 196–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guy HR, and Seetharamulu P (1986). Molecular model of the action potential sodium channel. ProcNatlAcadSciUSA 508, 508–512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heckman KL, and Pease LR (2007). Gene splicing and mutagenesis by PCR-driven overlap extension. Nat Protoc 2, 924–932. [DOI] [PubMed] [Google Scholar]
- Henderson R, Sali A, Baker ML, Carragher B, Devkota B, Downing KH, Egelman EH, Feng Z, Frank J, Grigorieff N, et al. (2012). Outcome of the first electron microscopy validation task force meeting. Structure 20, 205–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henrion U, Renhorn J, Borjesson SI, Nelson EM, Schwaiger CS, Bjelkmar P, Wallner B, Lindahl E, and Elinder F (2012). Tracking a complete voltage-sensor cycle with metal-ion bridges. Proc Natl Acad Sci U S A 109, 8552–8557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hille B (2001). Ionic Channels of Excitable Membranes, 3rd Ed (Sunderland, MA: Sinauer Associates Inc.). [Google Scholar]
- Jiang Y, Ruta V, Chen J, Lee A, and MacKinnon R (2003). The principle of gating charge movement in a voltage-dependent potassium channel. Nature 423, 42–48. [DOI] [PubMed] [Google Scholar]
- Kintzer AF, and Stroud RM (2016). Structure, inhibition and regulation of two-pore channel TPC1 from Arabidopsis thaliana. Nature 531, 258–262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larsson HP, Baker OS, Dhillon DS, and Isacoff EY (1996). Transmembrane movement of the Shaker potassium channel S4. Neuron 16, 387–397. [DOI] [PubMed] [Google Scholar]
- Lenaeus MJ, Gamal El-Din TM, Ing C, Ramanadane K, Pomes R, Zheng N, and Catterall WA (2017). Structures of closed and open states of a voltage-gated sodium channel. Proc Natl Acad Sci U S A 114, E3051–E3060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Q, Wanderling S, Paduch M, Medovoy D, Singharoy A, McGreevy R, Villalba-Galea CA, Hulse RE, Roux B, Schulten K, et al. (2014). Structural mechanism of voltage-dependent gating in an isolated voltage-sensing domain. Nat Struct Mol Biol 21, 244–252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin MC, Hsieh JY, Mock AF, and Papazian DM (2011). R1 in the Shaker S4 occupies the gating charge transfer center in the resting state. The Journal of general physiology 138, 155–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez GA, Jan YN, and Jan LY (1991). Hydrophobic substitution mutations in the S4 sequence alter voltage-dependent gating in Shaker K+ channels. Neuron 7, 327–336. [DOI] [PubMed] [Google Scholar]
- Mannuzzu LM, Moronne MM, and Isacoff EY (1996). Direct physical measure of conformational rearrangement underlying potassium channel gating. Science 271, 213–216. [DOI] [PubMed] [Google Scholar]
- McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, and Read RJ (2007). Phaser crystallographic software. J Appl Crystallogr 40, 658–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCusker EC, Bagneris C, Naylor CE, Cole AR, D’Avanzo N, Nichols CG, and Wallace BA (2012). Structure of a bacterial voltage-gated sodium channel pore reveals mechanisms of opening and closing. Nature communications 3, 1102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murshudov GN, Skubak P, Lebedev AA, Pannu NS, Steiner RA, Nicholls RA, Winn MD, Long F, and Vagin AA (2011). REFMAC5 for the refinement of macromolecular crystal structures. Acta Crystallogr D Biol Crystallogr 67, 355–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noda M, Shimizu S, Tanabe T, Takai T, Kayano T, Ikeda T, Takahashi H, Nakayama H, Kanaoka Y, Minamino N, et al. (1984). Primary structure of Electrophorus electricus sodium channel deduced from cDNA sequence. Nature 312, 121–127. [DOI] [PubMed] [Google Scholar]
- Otwinowski Z, and Minor W (1997). Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol 276, 307–326. [DOI] [PubMed] [Google Scholar]
- Pan X, Li Z, Zhou Q, Shen H, Wu K, Huang X, Chen J, Zhang J, Zhu X, Lei J, et al. (2018). Structure of the human voltage-gated sodium channel Nav1.4 in complex with beta1. Science 362. [DOI] [PubMed] [Google Scholar]
- Payandeh J, Gamal El-Din TM, Scheuer T, Zheng N, and Catterall WA (2012). Crystal structure of a voltage-gated sodium channel in two potentially inactivated states. Nature 486 135–139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Payandeh J, Scheuer T, Zheng N, and Catterall WA (2011). The crystal structure of a voltage-gated sodium channel. Nature 475, 353–358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, and Ferrin TE (2004). UCSF Chimera--a visualization system for exploratory research and analysis. J Comput Chem 25, 1605–1612. [DOI] [PubMed] [Google Scholar]
- Ren D, Navarro B, Xu H, Yue L, Shi Q, and Clapham DE (2001). A prokaryotic voltage-gated sodium channel. Science 294, 2372–2375. [DOI] [PubMed] [Google Scholar]
- Rogers JC, Qu Y, Tanada TN, Scheuer T, and Catterall WA (1996). Molecular determinants of high affinity binding of alpha-scorpion toxin and sea anemone toxin in the S3-S4 extracellular loop in domain IV of the Na+ channel alpha subunit. JBiolChem 271, 15950–15962. [DOI] [PubMed] [Google Scholar]
- Scheres SH (2012). RELION: implementation of a Bayesian approach to cryo-EM structure determination. J Struct Biol 180, 519–530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmalhofer WA, Calhoun J, Burrows R, Bailey T, Kohler MG, Weinglass AB, Kaczorowski GJ, Garcia ML, Koltzenburg M, and Priest BT (2008). ProTx-II, a selective inhibitor of NaV1.7 sodium channels, blocks action potential propagation in nociceptors. Molecular pharmacology 74, 1476–1484. [DOI] [PubMed] [Google Scholar]
- Schow EV, Freites JA, Gogna K, White SH, and Tobias DJ (2010). Down-state model of the voltage-sensing domain of a potassium channel. Biophys J 98, 2857–2866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaya D, Findeisen F, Abderemane-Ali F, Arrigoni C, Wong S, Nurva SR, Loussouarn G, and Minor DL Jr. (2014). Structure of a prokaryotic sodium channel pore reveals essential gating elements and an outer ion binding site common to eukaryotic channels. J Mol Biol 426, 467–483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- She J, Guo J, Chen Q, Zeng W, Jiang Y, and Bai XC (2018). Structural insights into the voltage and phospholipid activation of the mammalian TPC1 channel. Nature 556, 130–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen H, Li Z, Jiang Y, Pan X, Wu J, Cristofori-Armstrong B, Smith JJ, Chin YKY, Lei J, Zhou Q, et al. (2018). Structural basis for the modulation of voltage-gated sodium channels by animal toxins. Science 362. [DOI] [PubMed] [Google Scholar]
- Shen H, Liu D, Wu K, Lei J, and Yan N (2019). Structures of human Nav1.7 channel in complex with auxiliary subunits and animal toxins. Science [DOI] [PubMed]
- Shen H, Zhou Q, Pan X, Li Z, Wu J, and Yan N (2017). Structure of a eukaryotic voltage-gated sodium channel at near-atomic resolution. Science [DOI] [PubMed]
- Shimomura T, Irie K, Nagura H, Imai T, and Fujiyoshi Y (2011). Arrangement and mobility of the voltage sensor domain in prokaryotic voltage-gated sodium channels. The Journal of biological chemistry 286, 7409–7417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sokolov S, Kraus RL, Scheuer T, and Catterall WA (2008). Inhibition of sodium channel gating by trapping the domain II voltage sensor with protoxin II. Molecular pharmacology 73, 1020–1028. [DOI] [PubMed] [Google Scholar]
- Suloway C, Pulokas J, Fellmann D, Cheng A, Guerra F, Quispe J, Stagg S, Potter CS, and Carragher B (2005). Automated molecular microscopy: the new Leginon system. J Struct Biol 151, 41–60. [DOI] [PubMed] [Google Scholar]
- Tao X, Lee A, Limapichat W, Dougherty DA, and MacKinnon R (2010). A gating charge transfer center in voltage sensors. Science 328, 67–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vargas E, Yarov-Yarovoy V, Khalili-Araghi F, Catterall WA, Klein ML, Tarek M, Lindahl E, Schulten K, Perozo E, Bezanilla F, et al. (2012). An emerging consensus on voltage-dependent gating from computational modeling and molecular dynamics simulations. The Journal of general physiology 140, 587–594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vilas JL, Gomez-Blanco J, Conesa P, Melero R, Miguel de la Rosa-Trevin J, Oton J, Cuenca J, Marabini R, Carazo JM, Vargas J, et al. (2018). MonoRes: Automatic and Accurate Estimation of Local Resolution for Electron Microscopy Maps. Structure 26, 337–344 e334. [DOI] [PubMed] [Google Scholar]
- Wang J, Yarov-Yarovoy V, Kahn R, Gordon D, Gurevitz M, Scheuer T, and Catterall WA (2011). Mapping the receptor site for alpha-scorpion toxins on a Na+ channel voltage sensor. Proc Natl Acad Sci U S A 108, 15426–15431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waugh DS (2016). Crystal structures of MBP fusion proteins. Protein Sci 25, 559–571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winn MD, Ballard CC, Cowtan KD, Dodson EJ, Emsley P, Evans PR, Keegan RM, Krissinel EB, Leslie AG, McCoy A, et al. (2011). Overview of the CCP4 suite and current developments. Acta Crystallogr D Biol Crystallogr 67, 235–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao Y, Bingham JP, Zhu W, Moczydlowski E, Liang S, and Cummins TR (2008). Tarantula huwentoxin-IV inhibits neuronal sodium channels by binding to receptor site 4 and trapping the domain ii voltage sensor in the closed configuration. The Journal of biological chemistry 283, 27300–27313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu H, Li T, Rohou A, Arthur CP, Tzakoniati F, Wong E, Estevez A, Kugel C, Franke Y, Chen J, et al. (2019). Structural basis of Nav1.7 inhibition by a gating-modifier spider toxin. Cell [DOI] [PubMed]
- Yan Z, Zhou Q, Wang L, Wu J, Zhao Y, Huang G, Peng W, Shen H, Lei J, and Yan N (2017). Structure of the Nav1.4-beta1 complex from electric eel. Cell 170, 470–482 e411. [DOI] [PubMed] [Google Scholar]
- Yang N, George AL Jr., and Horn R (1996). Molecular basis of charge movement in voltage-gated sodium channels. Neuron 16, 113–122. [DOI] [PubMed] [Google Scholar]
- Yang N, and Horn R (1995). Evidence for voltage-dependent S4 movement in sodium channels. Neuron 15, 213–218. [DOI] [PubMed] [Google Scholar]
- Yarov-Yarovoy V, Decaen PG, Westenbroek RE, Pan CY, Scheuer T, Baker D, and Catterall WA (2012). Structural basis for gating charge movement in the voltage sensor of a sodium channel. Proc Natl Acad Sci USA 109 E93–E102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu FH, and Catterall WA (2004). The VGL-chanome: a protein superfamily specialized for electrical signaling and ionic homeostasis. Sci STKE 2004, re15. [DOI] [PubMed] [Google Scholar]
- Zhang JZ, Yarov-Yarovoy V, Scheuer T, Karbat I, Cohen L, Gordon D, Gurevitz M, and Catterall WA (2011). Structure-function map of the receptor site for beta-scorpion toxins in domain II of voltage-gated sodium channels. The Journal of biological chemistry 286, 33641–33651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang JZ, Yarov-Yarovoy V, Scheuer T, Karbat I, Cohen L, Gordon D, Gurevitz M, and Catterall WA (2012). Mapping the interaction site for a beta-scorpion toxin in the pore module of domain III of voltage-gated Na+ channels. The Journal of biological chemistry 287, 30719–30728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang K (2016). Gctf: Real-time CTF determination and correction. J Struct Biol 193, 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng SQ, Palovcak E, Armache JP, Verba KA, Cheng Y, and Agard DA (2017). MotionCor2: anisotropic correction of beam-induced motion for improved cryo-electron microscopy. Nat Methods 14, 331–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(A) Matrix of cysteine substitutions used in this study. Twelve residues on S4 were screened against 5 residues on S5 that yielded a total of 60 double-cysteine variants. Key residues are highlighted in red for resting, yellow for intermediate, and green for activated state crosslinking.
(B) Immunoblot result for double-cysteine substitutions in E. coli for NaVAb/Q150C and S4 cysteine mutants. R99C and V100C (green) show large crosslinking yields while E96C (yellow) shows a small yield with Q150C.
(C) Immunoblot result for double-cysteine substitution in E. coli for NaVAb/N49K/Q150C and S4 cysteine mutants. R99C and V100C (green) shows large crosslinking yields while E96C and I97C (yellow) and G94C (red) shows moderate yield with Q150C.
Morph movie between the resting/closed and activated/open states were calculated using Pymol. The VS from S0 to S3 segments are shown in orange, S4 in magenta, the S4-S5 linker in blue and the PM in yellow. Only one VS, and two S4-S5 linkers and PM from two opposing subunits are shown for clarity. Side chains of key residues are rendered as sticks and colored as in Figures 5B and 6D.
Morph movie between the resting/closed and activated/open states were calculated using Pymol. The VS from S0 to S3 segments are shown in orange, S4 in magenta, the S4-S5 linker in blue and the PM in yellow. Only one VS is shown for clarity. Side chains of key residues are rendered as sticks and the structure is colored as in Video S1. I217 at the S6 activation gate is shown in yellow as sticks overlaid with transparent atomic spheres.
(A) Immunoblot result from disulfide-crosslinking assay for NaVAb double-cysteine substitutions in Hi5 insect cells. Lanes with (−) denotes cells from control experiments; (+) from hyperpolarization/oxidation experiments with gramicidin/Cu-1,10-phenathroline.
(B) Quantification of percent crosslink in disulfide-crosslinking assays for NaVAb double-cysteine substitutions with Q150C. Percent crosslink denotes the extent of crosslinking quantified from intensity of a tetramer band divided by the total intensity from immunoblots. Control (dark red) vs. hyperpolarization/oxidation (gray).
(C) Immunoblot result from disulfide-crosslinking assays for NaVAb KAV double cysteine substitutions in Hi5 insect cells. Lanes with (−) denotes cells from control experiments; (+) from hyperpolarization/oxidation experiments with gramicidin/Cu-1,10-phenathroline.
(D) Quantification of percent crosslink in the disulfide-crosslinking assays for NaVAb double-cysteine substitutions with Q150C. Error bars represent SEM with n = 5.
(A) Normalized conductance-voltage (G/V) relationships and Boltzmann fits for full-length (FL) compared to C-terminal truncated (Δ28) channels. Due to decreased current density in cells expressing channels containing G94C, the C-terminal truncation mutation Δ28 was added to all constructs for electrophysiological recordings to increase total signal, but the truncation did not change the voltage-dependent properties of the channels. The G/V relationship was not altered for NaVAb/KAV (black, circles; V1/2=61.3 ± 2.1 mV, k= 7.2 ± 0.4 mV, n = 3) compared to NaVAb/KAV Δ28 (gray, triangles; V1/2=59.1 ± 0.8 mV, k=7.7 ± 0.8 mV, n = 3) nor was it changed in NaVAb/KAV/Q150C (green, circles; V1/2=77.8 ±0.8 mV, k=9.9 ± 0.3 mV, n = 3) compared to NaVAb/KAV/Q150C Δ28 (blue, triangles; V1/2=76.8 ± 3.1 mV, k=10.0 ± 0.9 mV, n = 3). Dashed (FL) and solid (Δ28) curves = 1/(1+ê(( V1/2−Vm)/k)). Half activation (V1/2) and slope (k) values are averages of individual fits. Data points within 15 mV of Vrev were omitted to reduce noise. Markers and error bars represent average G/Gmax ± SEM.
(B) Representative current families of NaVAb/KAV (black) and NaVAb/KAV/Q150C (green) full length. Transiently transfected Sf9 cells were held at −150 mV and stimulated for 50 ms to depolarized voltages in 10 mV increments. Scale bars represent 10 msec x 1 nAmp.
(A) Size-exclusion chromatography profile of protein purification on a Superdex 200 column for NavAb/G94C/Q150C Δ28 in the presence of DTT. The protein was expressed and purified as reported previously (Payandeh et al., 2011) except for pre-treatment of FLAG-eluted protein with 50 mM DTT and for inclusion of 5 mM DTT in the running buffer for size-exclusion chromatography. The peak fractions used for protein crystallization are highlighted in light blue. (Inset) SDS-PAGE of the peak fractions of NavAb/G94C/Q150C Δ28 in the presence of DTT. The NaVAb bands migrated at the molecular weight of a monomer indicating complete reduction of disulfide crosslink.
(B) Size-exclusion chromatography profile of protein purification on a Superdex 200 column for disulfide-crosslinked NaVAb/V100C/Q150C Δ28. Hi5 cells were treated with Cu-1,10-phenathroline prior to cell lysis and protein purification. The peak fractions used for protein crystallization are highlighted in light blue. (inset) SDS-PAGE of the peak fractions of disulfide-crosslinked NavAb/V100C/Q150C Δ28. The NaVAb bands migrated at the molecular weight of a tetramer indicating complete disulfide crosslinking.
(C) Superposition of NaVAb/V100CC/Q150C Δ28 structure with NaVAb/WT structure (PDB 4EKW). Cα backbone structures are shown with NaVAb/WT colored in gray with Cα r.m.s.d. of ~0.9 Å.
(A) Size-exclusion chromatography profile of protein purification on a Superose 6 column for disulfide-crosslinked MBP-NaVAb/KAV/G94C/Q150C. Hi5 cells were resuspended in a sodium-free choline chloride buffer and treated with Cu-1,10-phenathroline prior to cell lysis and protein purification. The peak fractions used for cryo-EM are highlighted in light blue.
(B) SDS-PAGE of purified disulfide crosslinked MBP-NaVAb/KAV/G94C/Q150C. The protein bands migrated as a molecular weight of a tetramer indicating a complete disulfide crosslink.
(C) Representative electron micrograph (left) and contrast transfer function (CTF) estimation (right).
(D) Reference-free 2D classification of particle images. The box size is 321 × 321 Å (304 × 304 pixels). The particle largest dimension is ~150 Å. The 2D averages indicate particles of c4 symmetry and contain features of intracellular MBP, NaVAb transmembrane helices and its intracellular C-terminal tail.
(A) Angular distribution of particle orientations in 3D reconstruction.
(B) Resolution assessment for 3D reconstruction of the cryo-EM map using Gold Standard Fourier Shell Correlation (FSC) plot from two half maps. Refinement of particle alignment was carried out using resolution limit of 8 Å. The density map has an overall resolution of 4.0 Å using the FSC criterion of 0.143.
(A) FSC plot of the refined model vs. summed map. The model vs. map correlation is 4.1 Å and 4.5 Å using FSC criterion of 0.143 and 0.5, respectively.
(B) Cryo-EM map and model for VS S0 and S1, S2, S3 and S4 segments, S4-S5 linker, and PM S5, P-loop, and S6 segments. NaVAb stick model is shown superimposed with cryo-EM density in black mesh. High-resolution features for side chains are apparent for the VS S4 segment, the S4-S5 linker, and the PM S5 to S6 segments.
Data Availability Statement
The coordinates and structure factors for the reported crystal structures have been deposited in the Protein Data Bank under accession codes 6P6X (NaVAb G94C/Q150C Δ28) and 6P6Y (NaVAb V100C/Q150C Δ28). Cryo-EM map has been deposited in the Electron Microscopy Data Bank (EMDB) under accession code EMD-20265 (MBP-NaVAb KAV/G94C/Q150C). The coordinates of the NaVAb KAV/G94C/Q150C atomic model have been deposited in the PDB under accession code 6P6W.