

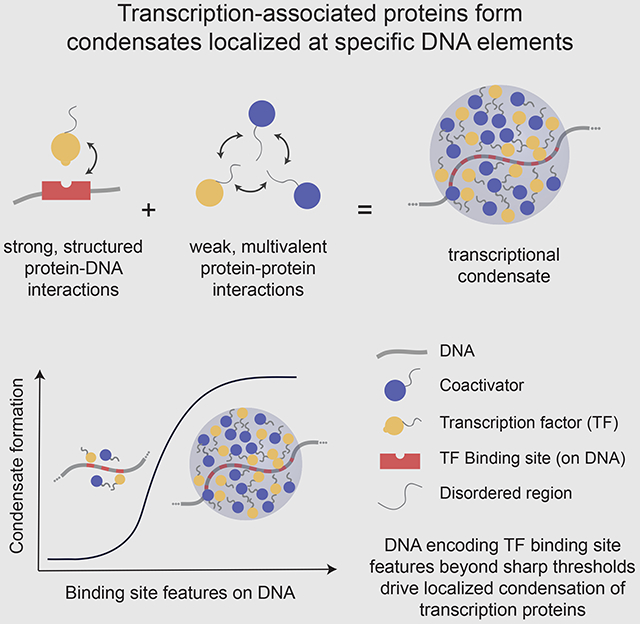
Summary
Enhancers are DNA elements that are bound by transcription factors (TFs), which recruit coactivators and the transcriptional machinery to genes. Phase-separated condensates of TFs and coactivators have been implicated in assembling the transcription machinery at particular enhancers, yet the role of DNA sequence in this process has not been explored. We show that DNA sequences encoding TF binding site number, density, and affinity above sharply defined thresholds drive condensation of TFs and coactivators. A combination of specific structured (TF-DNA) and weak multivalent (TF-coactivator) interactions allows for condensates to form at particular genomic loci determined by the DNA sequence and the complement of expressed TFs. DNA features found to drive condensation promote enhancer activity and transcription in cells. Our study provides a framework to understand how the genome can scaffold transcriptional condensates at specific loci, and how the universal phenonomenon of phase separation might regulate this process.
Graphical Abstract
eTOC Blurb
Shrinivas et al. demonstrate that specific types of motif compositions encoded in DNA drive localized formation of transcriptional condensates. These findings explain how phase separation can occur at specific genomic locations and shed light on why only some genomic loci become highly active enhancers.
Introduction
The precise regulation of gene transcription during development and in response to signals is established by the action of enhancer elements, which act as platforms for the recruitment of the gene control machinery at specific genomic loci (Levo and Segal, 2014; Long et al., 2016; Maniatis et al., 1998; Ptashne and Gann, 1997; Shlyueva et al., 2014; Spitz and Furlong, 2012). Imprecision in this process can cause disease, including cancer (Lee and Young, 2013; Smith and Shilatifard, 2014) Enhancer sequences contain short DNA motifs recognized by DNA-binding transcription factors (TFs), which recruit various coactivators that act together to engage RNA Polymerase II (RNAPII) resulting in transcriptional activity (Ptashne and Gann, 1997; Stampfel et al., 2015). Eukaryotic TFs typically recognize short DNA motifs of the order of 6-12 base pairs (Weirauch et al., 2014). There are many such similar affinity motifs in the genome (Lambert et al., 2018; Wunderlich and Mirny, 2009). As a result, active enhancer regions represent only a small fraction of putative binding sites for any given TF (Levo and Segal, 2014; Slattery et al., 2014; Spitz and Furlong, 2012; Wunderlich and Mirny, 2009). Determining whether a DNA motif participates in formation of an active enhancer element is thought to require defining a specific set of molecules and the mechanisms by which they act cooperatively to assemble the transcriptional machinery. Because this choice is made from a large set of possibilities, predicting enhancer elements is a significant challenge that has been referred to as the “futility theorem” (Wasserman and Sandelin, 2004).
Previous studies into the rules that govern enhancer formation have focused on cooperativity between TFs, mediated through direct protein-protein interactions or indirectly through changes in chromatin accessibility, nucleosome occupancy, local changes in DNA shape upon binding, and motif organization (Jolma et al., 2015; Lambert et al., 2018; Levo and Segal, 2014; Long et al., 2016; Maniatis et al., 1998; Morgunova and Taipale, 2017; Spitz and Furlong, 2012). The presence of clusters of TF binding sites at a genomic locus has been found to be predictive of enhancer elements (Berman et al., 2002; Markstein et al., 2002; Rajewsky et al., 2002). Clusters of TF binding sites can also occur without producing enhancer activity, and enhancer function can be realized upon small insertions (Mansour et al., 2014). The mechanisms by which TF binding site clusters enable the recruitment and stabilization of the appropriate transcriptional machinery at such loci are not well understood.
Recent studies suggest that the cooperative process of phase separation involving an ensemble of multivalent interactions among TFs, coactivators, and RNA Polymerase II can assemble these factors at specific enhancer elements as dynamic clusters, or condensates (Boija et al., 2018; Cho et al., 2018; Chong et al., 2018; Fukaya et al., 2016; Hnisz et al., 2017; Sabari et al., 2018; Tsai et al., 2017). While transcriptional condensates have been observed at specific genomic loci and features of proteins with intrinsically disordered regions (IDRs) have been implicated in their formation (Boija et al., 2018; Cho et al., 2018; Sabari et al., 2018), the features encoded in the DNA elements that facilitate this process have not been explored. We reasoned that if transcriptional condensate formation contributes to assembling certain active enhancers, investigating how features encoded in the DNA element regulate this process should shed light on the cooperative mechanisms that enable the recruitment of the transcriptional machinery, and provide insights into how enhancer regions in the genome are defined.
Using a combination of computational modeling and in vitro reconstitutions, we first demonstrate that DNA elements with specific types of TF binding site valence, density, and specificity drive condensation of TFs and coactivators. We show that modulating the affinities, number, or density of TF-DNA interactions and strength of IDR-IDR interactions impacts condensate formation. Because of the cooperative nature of phase separation, condensates form above sharply defined values of these quantities. We then show that the DNA sequence features that promote condensation in vitro also promote enhancer activity in cell-based reporter assays. Genome-wide bioinformatic analyses show that these features also characterize known enhancer regions. Importantly, we show that condensation localized to a specific genetic locus requires a combination of both weak multivalent IDR-mediated interactions and structured TF-DNA interactions. Our results also suggest that transcriptional condensate formation may contribute to long-range genomic interactions and organization, potentially promoting compartmentalization of actively transcribed regions.
Together, these results suggest that specific features encoded in DNA elements and the universal cooperative mechanism of phase separation contribute to localization of the transcriptional machinery at enhancers (especially, super-enhancers), and subsequent enhancer activity. Our studies provide a framework to understand how the genome can scaffold condensates at specific loci and how these condensates might be regulated.
Results
Development of a computational model
To explore how the complex interactions among regulatory DNA elements, TFs, and coactivators impact formation of transcriptional condensates, we first developed a simplified computational model (Figure 1A, Figure S1A). Since enhancers are typically short regions of DNA that are bound by multiple TFs (Levo and Segal, 2014; Spitz and Furlong, 2012), we modeled regulatory DNA elements as a polymer with varying numbers of TF binding sites. Each TF binding site mimics a short (6-12bp) DNA sequence. Specific recognition of DNA motifs by TFs (Weirauch et al., 2014) is mediated by typical TF-DNA binding strengths corresponding to nanomolar dissociation equilibrium constants (Jung et al., 2018), which is the range of TF-DNA interaction energies that we have studied in our simulations (Methods). TFs and coactivators contain large IDRs that interact with each other (Boija et al., 2018; Sabari et al., 2018). Thus, we modeled IDRs of TFs and coactivators as flexible chains attached to their respective structured domains. The IDRs interact with each other via multiple low-affinity interactions. The range of IDR-IDR interaction energies that we have studied in our simulations (Methods) corresponds to those that have been determined by in vitro studies of such systems (Brady et al., 2017; Nott et al., 2015; Wei et al., 2017). Our computational studies were focused on obtaining qualitative mechanistic insights that could then be tested by focused experiments.
Figure 1: Interactions between TFs and multivalent DNA drives phase separation of TFs and coactivators at low concentrations.

A. Schematic depiction of the stochastic computational model and key interactions between molecules. The model consists of a DNA polymer with variable number of TF binding sites, TFs, and coactivators. TFs bind TF binding sites with strong monovalent interactions, and TFs and coactivators interact via weak multivalent interactions between their flexible chains, which mimic the disordered regions of these proteins.
B. Scaled size is calculated from simulation trajectories, defined as the size of the largest cluster normalized by the number of DNA binding sites. This value is used as a proxy to differentiate stoichiometric binding of TFs to DNA (scaled size ≈ 1, top illustration) from phase-separated super-stoichiometric assemblies (scaled size >1, bottom illustration). For all reported simulation results, reported quantities are averaged over 10 replicate trajectories.
C. Simulations predict that multivalent DNA-TF interactions result in phase separation of TF and coactivator at dilute concentrations, as shown by scaled size >1 upon addition of DNA.
D. Schematic depiction of experimental workflow and image analysis for in vitro droplet assay. DNA, OCT4, and varying concentrations of MED1-IDR are incubated together in the presence of 10% PEG-8000 as a molecular crowder (illustrated with test tubes, see methods for detail). Fluorescence microscopy of these mixtures is used to detect droplet formation (illustrated by black square with or without white droplets). Multiple images per condition are then analyzed to calculate condensed fraction (c.f.) as intensity of fluorescence signal within droplets divided by total intensity in the image.
E. Representative images of MED1-IDR droplets in the presence of OCT4 and ODNA_20 (top row) or with only OCT4 (bottom row) at indicated MED1-IDR concentrations. See Table S2 for sequence of DNAs used in droplet assays.
F. Condensed fraction of MED1-IDR (in units of percentage) with DNA (purple) or without DNA (green) across a range of MED1-IDR concentrations (log scale). The respective inferred Csat values are shown in dashed lines and p-values are estimated from a two-sided Welch’s t-test. Higher condensed fraction implies higher fraction of total signal in droplet phase. Solid lines represent mean and error bars represent boundaries of mean±std from replicates. See methods for details on calculation of condensed fraction and Csat.
We simulated this model using standard Langevin molecular dynamics methods to calculate spatiotemporal trajectories of the participating species (see methods, (Anderson et al., 2008)). To distinguish stoichiometrically bound complexes from larger assemblies of transcriptional molecules, we computed the size of the largest molecular cluster scaled by the number of TF binding sites present on DNA. Values of this scaled size greater than 1 represent super-stoichiometric assemblies, while values close to 1 correspond to stoichiometrically bound TFs (Figure 1B). The scaled size is a direct measure of recruitment of transcription machinery and captures finite-size effects, an important factor in characterizing transcriptional condensates, which have been shown to contain ~ 100s-1000s of molecules (Cho et al., 2018). To study whether the super-stoichiometric assemblies are phase separated condensates, we calculated fluctuations in the scaled size spectra when appropriate (see Methods). A characteristic signature of a phase transition is that the fluctuation spectrum exhibits a peak across the threshold value of the titrated parameter. Using the scaled size and its fluctuation spectrum as measures of transcriptional condensate formation, we studied how particular motif compositions on DNA, as well as TF-DNA interactions and interactions between TF and coactivator IDRs regulate transcriptional condensate formation at DNA loci.
Interactions between TFs and multivalent DNA drive formation of condensates of TFs and coactivators
TFs and coactivators form condensates in vitro at supra-physiological concentrations (Boija et al., 2018; Lu et al., 2018; Sabari et al., 2018). Our simulation results (Figure 1C) predict that a dilute solution of TFs and coactivators that does not phase separate by itself forms condensates (scaled size greater than 1) upon adding multivalent DNA (DNA with 30 TF binding sites). To test this prediction, we developed an in vitro phase separation droplet assay containing the three components present in our simulations: TF, coactivator, and DNA (Figure 1D). For TF and coactivator, we employed purified OCT4, a master transcription factor in murine embryonic stem cells (mESCs), and MED1-IDR, the intrinsically disordered region of the largest subunit of the Mediator coactivator complex. We have previously shown that these proteins phase separate together in vitro and in vivo (Boija et al., 2018; Sabari et al., 2018). For DNA, we used various synthetic DNA sequences containing varying numbers of OCT4 binding sites (see methods and Table S2). Each of the three components was fluorescently labeled either by fluorescent protein fusion, mEGFP-OCT4 and mCherry-MED1-IDR, or a fluorescent dye, Cy5-DNA.
Formation of phase-separated droplets was monitored over a range of MED1-IDR concentrations by fluorescence microscopy with a fixed concentration of OCT4 in the presence or absence of multivalent DNA (DNA with 20 OCT4 binding sites, 8bp motif with 8bp spacers, ODNA_20, see methods and Table S2). The fluorescence microscopy results were quantified by calculating the condensed fraction as a function of MED1-IDR concentration (Figure 1D, also see methods). From the condensed fraction, a saturation concentration (Csat) is inferred (see methods under Image analysis and Statistical Analyses) to estimate the phase separation threshold under the specified experimental condition. Experimental variables with lower values of the inferred Csat promote phase separation at lower MED1-IDR concentrations than ones with higher Csat.
Consistent with model predictions, addition of DNA promoted phase separation at low MED1-IDR concentrations (Figure 1E). Addition of DNA lowered the inferred Csat by ~68 fold from ~2100 nM to 30 nM (Figure 1F). These results demonstrate that multivalent DNA promotes the phase separation of TFs and coactivators at low protein concentrations, comparable to concentrations observed in vivo (Figure S1B).
To further study how DNA influences condensate stability, we performed simulations where TF and coactivator condensates were allowed to form in the presence of DNA, followed by a simulated disruption of TF-DNA interactions (grey box in Figure 2A). At dilute protein concentrations, disrupting TF-DNA interactions resulted in dissolution of condensates (Figure 2A green line, Movie S1), demonstrating that, under these conditions, DNA is required for both formation and stability of condensates. Computing the radial density function around DNA (see methods) confirmed that TFs and coactivators form a largely uniform dense phase dependent on TF-DNA interactions (Figure S1C). While addition of DNA at high protein concentrations increased the rate of condensate assembly (Figure S1D; Movie S2), by reducing the nucleation barrier, disruption of TF-DNA interactions at these high concentrations did not lead to condensate dissolution (Figure 2A; grey line). We observed a drop in scaled size upon TF-DNA interaction disruption in this case, but this was primarily due to the condensate being broken into smaller droplets as the DNA was ejected from the condensate (as depicted in Figure 2A; grey box, Movie S2). Together, these results predict that, at dilute protein concentrations, specific TF-DNA interactions are required for both formation and stability of condensates at particular genomic loci.
Figure 2: Transcriptional condensate stability is governed by a combination of TF-DNA and IDR-IDR interactions between TFs and coactivators.

A. Simulation results for dynamics of condensate assembly/disassembly at two different protein concentrations is represented by average scaled size on the ordinate, and time (in simulation steps after initialization) on the abscissa. TF-DNA interactions are disrupted after stable condensate assembly (shown by a dark gray background). Schematic depiction of phase behavior is shown enclosed in boxes whose colors match the respective lines. See Movies S1 and S1.
B. Scatter plot depiction of experimentally determined MED1-IDR partition ratio (see methods) between condensate and background, at high (2500 nM, gray) and low concentrations (39nM, green) of MED1-IDR in the presence of OCT4 and ODNA_20, in the absence (−) or in the presence (+) of DNase I. The partition ratio is normalized to the (−) condition, and lower partition ratios imply lesser enrichment of MED1 in the droplet phase. Individual data points are presented with mean ± std, p-values represent Student’s t-test.
C. Energetic attractions, arising from a combination of TF-DNA (brown) and IDR (black) interactions, compensate for entropy loss (grey) of forming a condensate.
To mimic disruption of TF-DNA interactions in vitro, we added DNase I to droplets formed at high or low concentrations of MED1-IDR in presence of OCT4 and ODNA_20 (see methods). As expected, DNA was significantly degraded in both conditions (Figure S1E). Consistent with our model predictions, droplets formed at the lower concentrations were more sensitive to the degradation of DNA than those formed at higher concentrations (Figure 2B). While enzymatic degradation of DNA did not completely dissolve droplets in our in vitro experiments, MED1-IDR enrichment within droplets was significantly diminished only at the lower protein concentration (Figure 2B). Together, the in silico and in vitro results indicate that DNA can nucleate and scaffold phase-separated condensates of TFs and coactivators at low protein concentrations.
Physical mechanisms that underlie localized formation of transcriptional condensates
To understand the mechanisms driving DNA-mediated condensate formation, we cast our results in terms of the competing thermodynamic forces that govern phase separation. For computational efficiency, further characterization of our model was carried out with a simplified implicit-IDR model (Figure S2A), which recapitulated all features (Figure S2B–C) of the explicit-IDR model. Typically, condensate formation results in entropy loss because the molecules in the droplet are more confined than if they were in free solution. A condensate is stable only if this entropy loss is compensated by the energetic gain from enhanced attractive interaction energies between molecules confined in the condensate. We computed the energetic gain by summing up all pairwise molecular interactions in the condensate. Entropy loss due to confinement was calculated by adding a factor of for each molecule in the condensate. This loss in free volume is the principal source of entropy loss in our coarse-grained model. Other sources of entropy loss like solvent/ion effects are effectively incorporated in our affinity parameters. Our simulations show that energetic gains arising from a sum of specific TF-DNA interactions and weak IDR interactions (TFs-coactivator interactions) are necessary to compensate for the entropy loss of forming condensates at low concentrations (Figure 2C). IDR interactions alone are insufficient to compensate for the entropy loss of condensate formation, thus disruption of TF-DNA interactions results in condensate dissolution (Figures 2 and S2B–S2D, dark grey background). Likewise, TF-DNA interactions alone are insufficient to compensate for the entropy loss of condensate formation and disruption of IDR-IDR interactions results in condensate dissolution (see next section). The same features are observed in explicit-IDR simulations (Figures 2A; orange line, and S2E), though our simplified calculation of the entropy loss in this case (see above) is an underestimate, as contributions from the change in configurational entropy of IDR chains is not accounted. These results provide a mechanistic framework to understand how the combination of TF-DNA interactions and weak IDR interactions determine assembly and stability of transcription condensates at low concentrations.
Specific TF-DNA interactions and weak multivalent IDR interactions regulate formation of transcriptional condensates
Given that TF-DNA interactions are necessary for condensate formation, we next investigated the effect of modulating TF-DNA affinity at dilute protein concentrations. Simulations predict that condensates form above a sharply defined affinity threshold (Figure 3A) and that high affinity TF-DNA interactions result in condensate formation at low coactivator concentration thresholds (Figure 3B). The normalized fluctuation spectrum of the scaled size (see methods for details) showed a peak across the threshold affinity value, characteristic of phase separation (Figure S3A–B). Using the in vitro droplet assay, we probed the effect of TF-DNA interactions by comparing phase separation of MED1-IDR over a range of concentrations, with fixed concentrations of both OCT4 and either ODNA_20 or a scrambled ODNA_20 which does not contain any consensus binding sites for OCT4 (ODNA_20sc, Table S2). High-affinity OCT4-ODNA_20 interactions promoted phase separation at lower MED1-IDR concentrations when compared to OCT4-ODNA_20sc (Figure 3C). Quantifying the MED1-IDR condensed fraction further corroborated our finding, showing a ~2-fold decrease in inferred saturation concentrations in presence of higher affinity OCT4-ODNA_20 interactions (Figure 3D). Similar results were obtained by quantifying the condensed fraction of OCT4 or DNA (Figure S4A–B). These results demonstrate that higher TF-DNA affinities promote phase separation above sharply defined thresholds. Therefore, TFs, which exhibit higher affinity for specific DNA binding sites compared to random DNA, can drive transcriptional condensate formation at specific DNA loci.
Figure 3: Phase separation is regulated through strong specific TF-DNA interactions and weak multivalent interactions between TF and coactivator IDRs.

A. Simulations predict a shift in scaled size from stoichiometric binding (≈1) to phase separation (>1) with increasing normalized affinity (darker arrow in schematic); affinity normalized to threshold affinity of E=12kT.
B. Scaled size predictions for high (normalized affinity ≈50, purple) and low (normalized affinity ≈5e-2, green) TF-DNA affinities as a function of coactivator concentration. Coactivator concentrations are normalized to value of NcoA = 150.
C. Representative images of MED1-IDR droplets with OCT4 and ODNA_20 (top row) or ODNA_20scramble (sc) (bottom row) at indicated MED1-IDR concentrations. See Table S2 for sequence of DNAs used in droplet assays.
D. Condensed fraction of MED1-IDR (in units of percentage) for ODNA_20 (purple) and ODNA_20sc (green) across a range of MED1-IDR concentrations (log scale). The respective inferred Csat values are shown in dashed lines and p-values are estimated from a two-sided Welch’s t-test. Higher condensed fraction implies higher fraction of total signal in droplet phase.
E. Simulations predict a shift in scaled size from phase separation (>1) to stoichiometric binding (≈1) upon decreasing IDR interaction (from right to left, lighter arrow in schematic).
F. Scaled size predictions for high (IDR = 1.5kT, purple) and low (IDR=1.0 KT, green) IDR interaction as a function of coactivator concentration (normalized as in 3B).
G. Representative images of MED1-IDR droplets with ODNA_20 and OCT4 (top row) or an OCT4-mutant with reduced affinity for MED1-IDR (bottom row) at indicated MED1-IDR concentrations.
H. Condensed fraction of MED1-IDR (in units of percentage) for OCT4 (purple) and OCT4-mutant (green) across a range of MED1-IDR concentrations (log scale). The respective inferred Csat values are shown in dashed lines and p-values are estimated from a two-sided Welch’s t-test.
In all condensed fraction plots, solid lines represent mean and error bars represents boundaries of mean±std from replicates. For simulation plots, the solid lines represent mean and the shaded background represents mean±std from 10 replicate trajectories. See methods for details on calculation of condensed fraction and Csat.
We next investigated the effect of modulating the affinities of multivalent IDR interactions, whose effective affinity can be regulated in vivo through post-translational modifications (Banani et al., 2017; Shin and Brangwynne, 2017). Reducing the strength of IDR interactions between TFs and coactivators in our simulations predicts that condensates dissolve below a sharply defined interaction threshold (Figure 3E), and strong IDR interactions result in condensate formation at lower coactivator concentration thresholds (Figure 3F). To test this prediction, we monitored MED1-IDR phase separation over a range of MED1-IDR concentrations with fixed concentrations of both ODNA_20 and either OCT4 or a previously characterized OCT4 activation-domain mutant (acidic to alanine mutant) with reduced interaction with MED1-IDR (Boija et al., 2018). Consistent with simulation predictions, the OCT4 mutant was much less effective at promoting phase separation at low concentrations, with a nearly 8-fold higher inferred Csat as compared to OCT4 (Figures 3G–H). These results further highlight the importance of weak multivalent interactions between coactivators and TFs in the formation of transcriptional condensates.
Our results thus far suggest the following model. Specific TF-DNA interactions localize TFs to particular genomic loci. Transcriptional condensate formation is a cooperative process, which occurs at these loci when the weak multivalent interactions between TFs and coactivators exceed a threshold. While other processes may also be involved (e.g. DNA bending, removal and modification of nucleosomes, and interactions with RNA), this cooperative phenomenon of condensate formation by TF and coactivator phase separation contributes to assembling the transcriptional machinery at enhancers.
Specific motif compositions encoded in DNA facilitate localized transcriptional condensate formation
To begin defining the specific DNA sequence features that result in condensate formation, we explored the effects of modulating the valence and density of TF binding sites with the same TF-DNA affinities. We reasoned that the same energetic compensation for entropy loss we observed by increasing TF-DNA affinities (Figures 2C and 3A–D) could be obtained instead through increasing the number of DNA binding sites (i.e. valence). Our simulations predict that, for the same TF-DNA binding energies, condensates form above a sharply defined valence threshold (Figure 4A, Figures S3C–D), and higher valence results in condensate formation at lower coactivator concentrations (Figure 4B). Consistent with this prediction, in vitro assays revealed that ODNA_20 promoted phase separation of MED1-IDR and OCT4 at lower concentrations, with an inferred Csat ~2-fold lower than the threshold for DNA with fewer binding sites (ODNA_5) (Table S2) (Figures 4C–4D).
Figure 4: Motif valence encoded in DNA drives phase separation.

A. Simulations predict a shift in scaled size from stoichiometric binding (≈1) to phase separation (>1) with increasing number of TF binding sites on DNA (schematic depicts increasing number of binding sites).
B. Scaled size predictions for many (N =30, purple) and few (N=10, green) binding sites as a function of coactivator concentration (normalized as in 3B).
C. Representative images of MED1-IDR droplets with OCT4 and ODNA_20 (top row) or ODNA_5 (bottom row) at indicated MED1-IDR concentrations. See Table S2 for sequence of DNAs used in droplet assays
D. Condensed fraction of MED1-IDR (in units of percentage) for ODNA_20 (purple) and ODNA_5 (green) across a range of MED1-IDR concentrations (log scale). The respective inferred Csat values are shown in dashed lines and p-values are estimated from a two-sided Welch’s t-test.
E. Enhancer activity increases over a sharply defined TF binding site threshold. The left panel shows a schematic depiction of the luciferase reporter construct and the synthetic DNA sequences tested. The right panel shows the luciferase signal from constructs with the indicated number of binding sites transfected into murine embryonic stem cells (see methods). Inset presents data for 0-4 binding sites graphed on a different scale for the ordinate. Luciferase signal is normalized to the construct with 0 motifs. Data is graphed as average of three biological replicates ± std. **** = Student’s t-test p-value < 0.0001. See Table S3 for sequence of DNAs used in luciferase reporter assays
In all plots of condensed fraction, solid lines represent mean and error bars represent boundaries of mean±std from replicates. For simulation plots, the solid lines represent mean and the shaded background represents mean±std from 10 replicate trajectories. See methods for details on calculation of condensed fraction and Csat.
To test how motif valence impacts enhancer activity in cells, we cloned synthetic DNA sequences with varying number of OCT4 binding sites into previously characterized luciferase reporter constructs (Whyte et al., 2013) that were subsequently transfected into mESCs (see methods and figure 4E schematic). In these reporter assays, expression of the luciferase gene, read out as luminescence, is a measure of the strength of enhancer activity. Our computational studies and in vitro results show (Fig 4D) that for any concentration of MED1 less than Csat of ODNA_5, but higher than Csat of ODNA_20, only DNA with valence greater than a threshold can drive condensate formation. Since cellular protein levels are tightly regulated, these results predict that condensate assembly, and thus enhancer function, will be a digital function above a threshold valence of binding sites. Using a series of DNAs with 0 to 8 binding sites (8bp motif with 24bp spacers, see Table S3) we found that enhancer activity increased above a sharply defined valence threshold (Figure 4E), in striking qualitative agreement with expectations from our computational and in vitro studies.
To distinguish whether this behavior stemmed from motif valence alone or local motif density, we carried out simulations of DNA chains with a fixed number of binding sites, but different distributions along the chain (Figure 5A). We found that high local density, as compared to the same number of binding sites at lower density, promoted condensate formation at low protein concentration (Figure 5A). In vitro experiments were carried out with DNA containing the same binding site number (5 binding sites), but different densities (DNA_5M with higher density than DNA_5, see methods and Table S2). Quantifying the microscopy data validated simulation predictions, evidenced by a ~30% increase in inferred Csat for DNA_5 over DNA_5M (Figure 5C–D). To test the effect of binding site density on enhancer activity in cells, we compared the enhancer activity of 5 binding sites with different densities (see Table S3) in luciferase assays in mESCs (Figure 5B). In agreement with the model predictions, reducing density of binding sites led to reduced enhancer activity.
Figure 5: High DNA motif density, not overall number, drives phase separation.

A. Scaled size versus simulation time steps comparing two different distribution of binding site densities (shown in the schematic below), but same overall number of binding sites. TF-DNA interactions are disrupted after stable condensate assembly (dark gray background).
B. DNA sequences with the same number of binding sites, but higher density, shows increase in transcription activity. Left half shows a schematic depiction of the luciferase reporter construct and the synthetic DNA sequences tested. The right half shows the luciferase signal from constructs with indicated binding site density transfected into mouse embryonic stem cells. Data graphed as in E. **** = Student’s t-test p-value < 0.0001.
C. Representative images of MED1-IDR droplets with OCT4 and high motif density (ODNA_5M) (top row) or low motif density (ODNA_5) (bottom row) at indicated MED1-IDR concentrations. See Table S2 for sequence of DNAs used in droplet assays.
D. Condensed fraction of MED1-IDR (in units of percentage) for ODNA_5M (purple) and ODNA_5 (green) across a range of MED1-IDR concentrations. The respective inferred Csat values are shown in dashed lines and p-values are estimated from a two-sided Welch’s t-test. Solid lines represent mean and error bars represent boundaries of mean±std from replicates. See methods for details on calculation of condensed fraction and inferred Csat.
The results in Figures 4 and 5 show that dense clusters of a particular TF’s binding sites, with the valence of binding sites exceeding a sharply defined threshold, drive localized formation of transcriptional condensates, and that these same features influence enhancer activity in cells. The condensates form by the universal cooperative mechanism of phase separation which, in turn, requires weak cooperative interactions between the IDRs of TFs and coactivators (Figure 3). IDR-IDR interactions are relatively non-specific, and the same coactivator IDRs can assemble the transcriptional machinery in stable condensates at different enhancers upon cognate TF binding.
Transcriptional condensate formation may facilitate long-range interactions and higher-order genome organization
Given that regulatory elements often communicate over long linear distances, we next investigated whether two dense clusters of TF binding sites in DNA separated by a linker could assemble a single condensate. Our simulations show that this is indeed the case (Figure 6A; green line). Contact frequency maps, computed from the simulation data (see methods) show long-range interactions between the dense clusters of binding sites, which are absent (Figure 6B) at conditions with a low density of TF binding sites distributed uniformly (Figure 6A; black line). Further, removing a single cluster strongly diminished the ability of DNA to assemble a condensate (Figure S5), suggesting that both clusters of binding sites worked cooperatively over intervening linker DNA to assemble a condensate. These results suggest that condensate formation could explain recent observations of CTCF/cohesin-independent long-range interactions between active regions of the genome (Rowley et al., 2017; Schwarzer et al., 2017). More generally, our results suggest that localized transcriptional condensate formation can facilitate higher-order organization of the 3D-genome and contribute to long-range communication between enhancer-promoter pairs.
Figure 6: Transcriptional condensate formation facilitates long-range interactions.

A. Scaled size versus simulation time steps comparing two different distribution of binding site densities (as shown in the schematic legend). TF-DNA interactions are disrupted after stable condensate assembly (as shown in dark gray background).
B. Contact frequency maps (see methods) show long-range interactions (right panel, checkerboard-like patterns) for high local motif density (computed for green line in Figure 5A), and not for low motif density (left panel, computed for black line in Figure 5A). Illustrations depicting the organization of model components are provided for each condition below their respective contact map.
Mammalian genomes encode specific motif features in enhancers to assemble high densities of transcription apparatus
We next investigated whether enhancer features that our results suggest promote transcriptional condensate formation are present in mammalian genomes. Given that our results show that a linear increase in TF binding site valence can result in an exponential increase in coactivator recruitment by condensate formation (Figure 4), we investigated the relationship between TF binding site valence (i.e. occurrence of TF motifs) and coactivator recruitment in mESCs. We gathered genome-wide distribution of TF motif occurrence for highly expressed mESC master TFs – OCT4, SOX2, KLF4, ESRRB (OSKE). Super-enhancers, genomic regions with unusually high densities of transcriptional molecules (Whyte et al., 2013), where transcriptional condensates have recently been observed (Boija et al., 2018; Cho et al., 2018; Sabari et al., 2018), have higher OSKE motif densities when compared to typical enhancers or random loci (Figures 7A–B, methods). Consistent with our results, we found a highly non-linear (roughly exponential) correlation between OSKE motif density and ChIP-Seq data for MED1, RNA Pol II (Figure 7C), and BRD4 (Figure S6A) across genetic regions including SEs, TEs, and random loci. This correlation was minimal when input control data was analyzed (Figure S6B). These results suggest that enhancer elements that encode specific DNA sequence features we have described can recruit unusually high densities of transcriptional apparatus by transcriptional condensate formation, consistent with our results. The same features enable recruitment of varied cofactors – BRD4, MED1, and Pol II, thus suggesting that phase separation contributes to stabilization of transcription machinery at specific genomic loci.
Figure 7: Mammalian genomes leverage high motif density to assemble high density of transcriptional apparatus at key regulatory elements.

A. Box-plot depiction of motif density (per kb) of master mESC TFs − OCT4 + SOX2 + KLF4 + ESRRB (OSKE), over 20kb regions centered on super-enhancers (SEs, orange), typical enhancers (TEs, black), and random loci (light gray).
B. OSKE motif density over a 100kb window centered at SEs (orange), TEs (black), and random loci (gray).
C. MED1 (left) and RNA Pol II (right) ChIP-Seq counts (ordinate, reads-per-million, log scale) against total OSKE motifs over 20kb regions centered on SEs (orange), TEs (black), and random loci (gray). The black line is a fit inferred between the logarithmic ChIP signal and the linearly graphed motif count across all regions, and so the fit represents a highly non-linear (exponential) correlation. The grey shaded regions represent 95% confidence intervals in the value of the inferred slope. The exponential fit describes a sizable fraction of the observed variance i.e. R2 ≈ 0.25, ρ ≈ 0.5 for both inferred lines.
Discussion
Enhancers are DNA elements that control gene expression by promoting assembly of transcriptional machinery at specific genomic loci. Recent studies have suggested that phase-separated condensates of molecules involved in transcription form at enhancers (Boija et al., 2018; Cho et al., 2018; Chong et al., 2018; Fukaya et al., 2016; Hnisz et al., 2017; Sabari et al., 2018; Tsai et al., 2017), providing a potential mechanism for concentrating transcriptional machinery at specific loci. Here we investigated how features encoded in DNA elements can regulate the formation of transcriptional condensates. Our results identify features of DNA sequences that can enable assembly of the transcriptional machinery at specific genomic loci by the general cooperative mechanism of phase separation.
We first demonstrated that interactions between TFs, co-activators, and multivalent DNA elements can form condensates at protein concentrations that are too low for such a phase transition in the absence of the DNA. We suggest that these results help explain why condensates of coactivators and TFs form at enhancers in cells wherein protein concentrations are much lower than that required for phase separation without DNA in vitro. We also found that at low protein concentrations, DNA elements with multiple TF binding sites serve as scaffolds for the phase separated transcriptional condensates. However, at high protein concentrations, the DNA elements act only as a nucleation seed, and are not necessary for condensate stability. These results suggest an explanation for why co-activator overexpression is often linked to pathological gene expression programs (Bouras et al., 2001; Zhu et al., 1999).
By considering the competing thermodynamic forces of entropy loss and energy gain that control phase separation, we described how a combination of specific TF-DNA interactions and weak cooperative interactions between IDRs of TFs and coactivators are required for transcriptional condensate formation. These parameters must be above sharply defined thresholds for phase separation to occur. The necessary sharp threshold for TF-DNA interactions results in formation of transcriptional condensates at specific genomic loci containing cognate TF binding sites. That there is a threshold affinity and valence between IDRs of the interacting species for condensate formation implies that molecules with IDRs with complementary characteristics, such as those contained in TFs and coactivators, will be incorporated in transcriptional condensates. Therefore, different TFs with IDRs that are statistically matched with co-activator IDRs can mediate transcriptional condensate formation at different genomic loci via similar weak cooperative interactions. This may be the reason underlying recent observations that TFs with different disordered activation domains can co-localize with MED-1 condensates (Boija et al., 2018). Biomolecular condensates can exhibit diverse material properties and phase behavior as a function of their specific IDR sequences (Banani et al., 2017; Dignon et al., 2018; Shin and Brangwynne, 2017). For example, recent studies focused on electrostatic interactions in IDRs have shown that particular statistical patterns of charged residues dictate overall phase behavior (Das and Pappu, 2013; Huihui et al., 2018; Lin et al., 2017) and enable specific protein interactions (Borgia et al., 2018; Sherry et al., 2017) . Similarly, the “spacer-sticker” framework (Harmon et al., 2017; Wang et al., 2018), which builds on previous mean-field models (Semenov and Rubinstein, 1998), has been successfully used to elucidate the interplay of gelation and phase separation in prion-like proteins (Wang et al., 2018). Leveraging these techniques to characterize IDRs of transcription-associated proteins will provide insights on the molecular grammar underlying their interactions and enable better understanding of the biophysical properties of transcription condensates.
Importantly, we find that DNA elements with dense clusters of TF binding sites that exceed a sharply defined valence threshold promote transcriptional condensate formation, and the same findings are mirrored for enhancer activity in cells. Our results also provide insights on specific combinations of DNA features that facilitate transcription condensate formation. For example, low-affinity TF binding sites can contribute to scaffolding a transcriptional condensate, if present in sufficiently high valence and density, as the total energy gain comes from a combination of these parameters. This may explain recent intriguing descriptions of enhancer regulation through clusters of weak TF binding sites (Crocker et al., 2015; Tsai et al., 2017). In contrast, DNA sequences with many high affinity TF binding sites (high valence) distributed at low local density are not enhancer regions because they will not enable formation of transcriptional condensates. This may explain why many high-affinity sites that are not enhancers remain largely unbound and contribute to a deeper understanding of the futility theorem (Wasserman and Sandelin, 2004). Thus, our framework elucidates the key parameters, or specific combinations of these parameters, that must be above sharply defined thresholds for phase separated transcriptional condensates to form at specific genomic loci that function as enhancers.
Bioinformatic analyses reveal that the DNA sequence features that we have described as important for transcriptional condensate formation also characterize enhancer regions in mammalian genomes, and increases in the recruitment of transcriptional molecules at different loci are correlated in a highly non-linear way with motif density.
Taken together our results suggest the following model for a general cooperative mechanism that contributes to assembling the transcriptional machinery at enhancers, perhaps especially at super-enhancers. Dense clusters of a particular TF’s binding sites, with the number of binding sites exceeding a sharply defined threshold, drive localized formation of transcriptional condensates at a specific genomic locus. The condensate, which recruits and stabilizes various transcriptional molecules, forms by the universal cooperative mechanism of phase separation. Thus, a threshold number of cooperative binding events have to occur at a particular genomic locus, before phase separation occurs to robustly assemble the transcription machinery. Although included only implicitly in our model, past data suggests that TF binding to DNA can be cooperative and sequential (for example, due to DNA bending)(Levo and Segal, 2014; Spitz and Furlong, 2012). Thus, a series of sequential steps occurs when TFs bind to a sufficiently large number of binding sites that serve as enhancers. This is analogous to kinetic proofreading in cell signaling processes(Hopfield, 1974; Ninio, 1975), such as T cell receptor signaling that discriminates between self and cognate ligands to mediate pathogen-specific immune responses. In the latter situation, a sequence of biochemical steps need to occur before productive downstream signaling can lead to activation; only the cognate ligands can complete these steps with high probability. In T cell signaling, once the kinetic proofreading steps are completed, a positive feedback loop amplifies signal levels to result in robust downstream signaling leading to activation(Das et al., 2009). At enhancers, after TFs have bound to a sufficiently large number of cognate binding sites on DNA, amplification of the recruitment of transcriptional machinery occurs by condensate formation. Intriguingly, the mathematical description of a first order phase transition and a positive feedback loop’s effect on signaling are isomorphic, suggesting that perhaps biological processes have evolved similar strategies in diverse contexts.
Condensate formation requires weak cooperative interactions between the IDRs of TFs and coactivators (Figure 3). Although different molecular grammars may describe different types of IDR-IDR interactions, these interactions are relatively non-specific, and the same coactivator IDRs can assemble within condensates at different enhancers. This model is consistent with the observation that clusters of TF binding can often correctly predict active enhancers because this feature of the DNA sequence drives formation of transcriptional condensates by a common mechanism (Berman et al., 2002; Markstein et al., 2002; Rajewsky et al., 2002).
Our model can also describe situations where insertion of a relatively small DNA element that binds to a master TF that regulates cell type specific gene expression programs can stabilize TFs that bind weakly to adjacent binding sites, and recruit the transcriptional machinery in condensates. We carried out simulations with a DNA sequence comprised of two types of binding sites – those that bind strongly to a TF and others that bind weakly. As Figures S3E–F show, a transcriptional condensate forms at such a locus beyond a threshold fraction of high-affinity (master) TF binding sites. This is because the cooperative process of condensate formation recruits and stabilizes the transcriptional machinery once the number of strong TF binding sites exceeds a certain value. This result may explain why a relatively small insertion of a TF binding site into a region that contained an inactive cluster of binding sites for other TFs, resulted in the formation of a super-enhancer in T-ALL cells (Mansour et al., 2014).
While our model explicitly incorporates enhancer DNA, TFs, and coactivators, the underlying mechanistic framework can be extended to understand diverse condensates that form at specific genomic loci. Examples may include condensates in heterochromatin-organization (Larson et al., 2017; Strom et al., 2017), histone locus body assembly (Nizami et al., 2010), lncRNA-mediated paraspeckle formation (Fox et al., 2018; Yamazaki et al., 2018), nucleolar formation (Feric et al., 2016; Pederson, 2011) and in polycomb-mediated transcriptional silencing (Tatavosian et al., 2018). Recent advances in microscopy at the nano-scales (Li et al., 2019) can potentially shed light into whether transcription-associated condensates form higher-order substructures, like the nucleolus (Feric et al., 2016).
Our study provides a framework towards understanding how the genome can scaffold condensates at specific loci and implicates particular TF binding site compositions. In addition to TF binding sites, processes that dynamically modulate valence and specificity of interacting species at specific genetic loci, such as local RNA synthesis or chromatin modifications, are likely to play a role in the formation of transcriptional condensates.
STAR METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Arup K. Chakraborty (arupc@mit.edu)
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cells
V6.5 murine embryonic stem cells were a gift from R. Jaenisch of the Whitehead Institute. V6.5 are male cells derived from a C57BL/6(F) x 129/sv(M) cross.
Cell culture conditions
V6.5 murine embryonic stem cells were grown in 2i + LIF conditions on 0.2% gelatinized (Sigma, G1890) tissue culture plates. 2i + LIF media contains the following: 967.5 mL DMEM/F12 (GIBCO 11320), 5 mL N2 supplement (GIBCO 17502048), 10 mL B27 supplement (GIBCO 17504044), 0.5mML-glutaminae (GIBCO 25030), 0.5X non-essential amino acids (GIBCO 11140), 100 U/mL Penicillin-Streptomycin (GIBCO 15140), 0.1 mM b-mercaptoethanol (Sigma), 1 uM PD0325901 (Stemgent 04-0006), 3 uM CHIR99021 (Stemgent 04-0004), and 1000 U/mL recombinant LIF (ESGRO ESG1107). Cells were negative for mycoplasma.
METHOD DETAILS
Developing coarse-grained simulations of DNA, TFs, and coactivators
We set up a coarse-grained molecular-dynamics simulation to model 3 different components – TFs, DNA, and coactivators, employing the HOOMD simulation framework (Anderson et al., 2008; Glaser et al., 2015). Briefly, the DNA chain was modeled as beads on a string, with two types of monomers. “Active” DNA units were modeled by tessellating a sphere (diameter = 1/3 unit), using the rigid-body feature(Nguyen et al., 2011), to form a roughly cubical monomer of unit side length (Fig 1A). Binding patches were modeled as rigid particles along the cubic face centers, with as many patches added as number of binding sites per monomer. Tessellation of active DNA monomers enabled 1:1 binding interactions, facilitated by excluded volume interactions from other tessellated spheres. “Inactive” DNA monomers were modeled as spherical monomers of unit diameter without any binding patches. TFs and coactivators were modeled employing two different methods – explicit-IDR (Fig 1A) and implicit-IDR models (Fig S3A). In the explicit-IDR framework, TFs and coactivators were designed in a modular fashion (Fig S1A). The “structured” domain was modeled as a spherical monomer of diameter d = 0.75 units.s. IDRs were constructed by tethering a polymeric tail to the spherical domain, with TFs having shorter chains (4 monomers of d = 1/3 unit) than coactivators (9 monomers of d = 1/3 unit), to mimic the differential size of disordered regions. In the implicit-IDR model, TFs and coactivators were modeled as spherical monomers of unit diameter. All monomers had the same density. The sizes of the modeled monomers of DNA and proteins mimics the relative similarity in sizes between TFs/coactivators and nucleosomes. In both methods, DNA binding patches on proteins were modeled as rigid particles buried in the “structured” domains.
Non-bonding interactions between any two particles (including binding patches) were modeled using a truncated, shifted, and size-normalized LJ potential (U) with hard-core repulsion (particles don’t overlap), derived in the following form:
Bonding interactions between neighboring monomers on a chain were modeled using a harmonic potential with hard-cores, with a spring constant k = 1e4. All energy units are scaled to kT units, with kT=1.
The strength of various interactions was set based on the rationale stated in main text. Typical TF-DNA binding affinities are strong and in the range of nanomolar (Jung et al., 2018) disassociation constants i.e. KD ≈ 10−9M. The Gibbs free enthalpy change of binding can be approximately calculated as ΔG ≈ −kTln(KD) ≈ 20kT. Thus, specific monovalent DNA interactions were set to high affinities - for e.g. ϵDNA–TF = 20kT in fig 1B,2A, ϵDNA-TF = 16 kT in fig S3A–B. IDR interactions were much weaker and individual interactions are often of the order of thermal fluctuations (Brady et al., 2017; Nott et al., 2015; Wei et al., 2017) i.e. order kT, though their energetic contributions can effectively multiply through multivalence. Thus, we set ϵIDR ~ kT between monomers on the IDR chain. For the implicit-IDR model reported in Fig S2, multivalent interactions were approximated by a weak LJ potential between particles, for e.g., ϵTF-coA = 1.5kT, ϵcoA–coA = 1.5kT, ϵTF–TF = 1.0 kT. The key qualitative results i.e multivalent DNA acts as scaffold for phase separation at low protein concentrations and seed at higher protein levels, has been reproduced for different choices of interaction parameters guided by the rationale above.
Particles are randomly initialized in the periodic simulation box, and randomly re-seeded for each replicate trajectory, with the Langevin thermostat. Friction coefficients were γ = 1 for proteins and γ = 100 for DNA, to mimic chromosomal motion damping. Initial velocities were drawn from the Boltzmann distribution. First, simulations were run with small time steps (dt = 5 × 10−6) to prevent randomly generated “high-energy” configurations from blowing up and to relax the system to the thermostat temperature. These “warm-up” period (t ~ 0.1 units) is much smaller than the time to reach steady-state tss ~ (1000 units), so these warm-up data points are not used in any analysis. All simulations are run with a single DNA chain.
Explicit-IDR simulations are run for at least 45e6 steps to accurately recapitulate dynamics and reach steady-state, whilst implicit-IDR simulations are run for 5e6 steps. The slowing down of explicit-IDR simulations (due to slower explicit-IDR dynamics), combined with additional pair-wise interaction computations (explicit pairwise calls for all monomers, which are an order of magnitude more particles for explicit-IDR simulations, scale as ~ N2 for N monomers), cause computation times for single trajectories to be ~50-100 times longer than the implicit-IDR version. Trajectory states were logged in the highly compressed, binarized GSD format every 50000 steps, while observables were logged every 20000 steps.
To probe the role of DNA in our simulations, after steady-state is reached, interactions with the DNA binding sites are switched off. Interactions are switched off by replacing all binding patches with “ghost” patches, with no energetic benefits. Simulations are typically continued for the same amount of steps before disrupting TF-DNA interactions to accurately sample steady-state. A brief overview of key parameters used in main/supplementary figures is found below in Table S1. The MD code for running analysis will be made freely available upon publication.
Analysis of simulation data
Broadly, analyses of simulation data were split into on-the-fly calculations employing the Freud package (https://freud.readthedocs.io/en/stable/installation.html), as well as post-simulation calculations that leverage a combination of various libraries which interface with python – including numpy, scipy, freud, matplotlib, and fresnel. On-the-fly calculations include:
In-built functions for logging potential energy, kinetic energy, and temperature.
Number of monomers in largest cluster and radius of largest cluster: A call-back routine was implemented that used Freud to estimate the size of the largest connected cluster with r = 1.4dmax (dmax diameter of largest monomer) to identify largest cluster. This largest cluster size is relatively insensitive to studied choices of parameter r = 1.25,1.35,1.45 dmax. Every reported plot with scaled size at steady state, which is the number of molecules in the largest cluster divided by number of binding sites (Fig 1B), reports the mean in the dark line, and one standard deviation in the shaded background.
For post-simulation calculations, data was read from GSD formats using the gsd module. Explicit-IDR simulation trajectory data was parsed to convert from number of molecules to number of chains, while following the other steps as mentioned above.
The entropy was calculated in Fig 2 and Fig S2 by identifying the number of molecules in the largest cluster (in the case of the explicit-IDR simulations, each polymer was counted as one molecule), and adding a value of for each molecule in the condensed phase. Vfree was computed as the total volume minus the excluded volume occupied by all molecules.
For the fluctuation analysis in Fig S3, the variance in largest cluster size of individual stochastic trajectories was computed and averaged at steady state. This value was normalized by the scaled cluster size, to compute the scaling of fluctuations beyond the usual finite-size effects.
Contact frequency analysis of simulation data
For contact frequency maps, which are similar to Hi-C maps, represented in Fig 6B and S5, the following analysis protocol was employed. After individual trajectories reached steady-state, the position of each DNA monomer along the chain was logged at every time step. Monomers closer than (r = 3.0 units) a distance at a time t are “cross-linked” i.e. they count as an interacting pair. The qualitative interaction maps reported in Fig 6B are robust to other tested values of crosslinking radius in the regime of 2.5 < r < 4 units. The pairwise contact frequency matrix is then constructed by averaging over interactions over a time window at steady state per trajectory, as well as averaging over 10 replicate trajectories per simulation condition. The contact matrix is visualized using the seaborn and matplotlib packages in python3.
Computing radial density profiles from simulation data
Simulations were analyzed at steady-state to estimate the radial density of TFs and coactivators around the DNA chain (g(r) from DNA). The freud rdf analysis package was used to compute the rdf around reference positions of DNA for both distributions of TFs and coactivator molecules. In case of explicit-IDR simulation, the structured domains of the respective molecules were used to probe their locations. The final g(r) from DNA is obtained by averaging over 50 distinct simulation frames (typically logged once every 50,000 steps) per trajectory, and over 10 trajectories. The g(r) is visualized for both explicit-IDR (Fig S1C) and implicit-IDR (Fig S2C) at low concentrations, before and after disruption of TF-DNA interactions, using matplotlib in python3.
Visualization of simulation data:
All simulation data-sets were analyzed in python3, with the aid of matplotlib, to generate publication-ready figures. Simulation movies were generated by stitching together down-sampled frames (once every 100000 steps) of individual stochastic trajectories, using Fresnel to render scenes with the same color palette used in Fig 1A, and PIL to store image arrays as gifs. After storing the gifs, these files were converted to .mp4 movies externally and subs are added at the frame at which TF-DNA interactions are turned off.
Quantitative immunoblot
Determination of number of MED1 molecules per cell and concentration by linear regression analysis. Quantitative Western Blotting was carried out as described in (Lin et al., 2012). Cell number was determined using a Countless II FL Automated Cell Counter (Thermo Fisher Scientific). Cells were lysed with Cell Lytic M (Sigma) with protease inhibitors at various concentrations and denatured in DTT and XT Sample Buffer (Biorad) at 90°C for 5 minutes. Purified rec ombinant MED1-IDR was used as a standard and loaded in the amounts depicted in the figure in the same gel as the cell lysates. Lysates and standards were run on a 3%–8% Tris-acetate gel at 80 V for ~2 hrs, followed by 120 V until dye front reached the end of the gel. Protein was then wet transferred to a 0.45 μm PVDF membrane (Millipore, IPVH00010) in ice-cold transfer buffer (25 mM Tris, 192 mM glycine, 10% methanol) at 300 mA for 2 hours at 4°C. After transfer the membrane was blocked with 5% non-fat milk in TBS for 1 hour at room temperature, shaking. Membrane was then incubated with 1:1,000 anti-MED1 (Assay Biotech B0556) diluted in 5% non-fat milk in TBST and incubated overnight at 4°C, with shaking. The membrane was then washed three times with TBST for 5 minutes at room temperature shaking for each wash. Membrane was incubated with 1:10,000 secondary antibody conjugated to HRP for 1 hr at RT and washed three times in TBST for 5 minutes. Membranes were developed with ECL substrate (Thermo Scientific, 34080) and imaged using a CCD camera.(BioRad ChemiDoc). Band intensities were determined using ImageJ. Number of molecules per cell was determined by linear regression analysis through the origin using Prism 7. The concentration of MED1 was calculated using nuclear volumes obtained by analysis of Hoechst (Life Technologies)-stained mouse embryonic stem cells in ImageJ and assuming all MED1 molecules reside in the nucleus.
Protein purification
Proteins were purified as in (Boija et al., 2018; Sabari et al., 2018). cDNA encoding the genes of interest or their IDRs were cloned into a modified version of a T7 Pet expression vector. The base vector was engineered to include a 5’ 6xHIS followed by either mEGFP or mCherry and a 14 amino acid linker sequence “GAPGSAGSAAGGSG.” NEBuilder® HiFi DNA Assembly Master Mix (NEB E2621S) was used to insert these sequences (generated by PCR) in-frame with the linker amino acids. Mutant sequences were synthesized as gBlocks (IDT) and inserted into the same base vector as described above. All expression constructs were sequenced to ensure sequence identity. For protein expression, plasmids were transformed into LOBSTR cells (gift of Chessman Lab) and grown as follows. A fresh bacterial colony was inoculated into LB media containing kanamycin and chloramphenicol and grown overnight at 37°C. Cells containing the MED1-IDR co nstructs were diluted 1:30 in 500ml room temperature LB with freshly added kanamycin and chloramphenicol and grown 1.5 hours at 16°C. IPTG was added to 1mM and growth continued for 18 hours. Cells were collected and stored frozen at −80°C. Cells containing all other constructs were treated in a similar manner except they were grown for 5 hours at 37°C after IPTG induction. 500ml cell pellets were resuspended in 15ml of Buffer A (50mM Tris pH7.5, 500 mM NaCl) containing 10mM imidazole and complete protease inhibitors, sonicated, lysates cleared by centrifugation at 12,000g for 30 minutes at 4°C, added to 1ml of preequilibrated Ni-NTA agarose, and rotated at 4°C for 1.5 hours. The slurry was poured into a column, washed with 15 volumes of Buffer A containing 10mM imidazole and protein was eluted 2 X with Buffer A containing 50mM imidazole, 2 X with Buffer A containing 100mM imidazole, and 3 X with Buffer A containing 250mM imidazole.
Production of fluorescent DNA
Gene fragments were synthesized by either GeneWiz or IDT and cloned into a pUC19 vector using HiFi Assembly (NEB) so that the sequence was immediately flanked by M13(−21) and M13 reverse primer sequences. 5’-fluorescently labeled (Cy5) M13(−21) (/5Cy5/ TGTAAAACGACGGCCAGT) and M13 reverse (/5Cy5/ CAGGAAACAGCTATGAC) primers (IDT) were used to PCR amplify the synthetic DNA sequence, yielding a fluorescently labeled PCR product. Fluorescent PCR products were gel-purified (Qiagen) and eluted products were further purified using NEB Monarch PCR purification to remove any residual contaminants. The octamer motif sequence “ATTTGCAT” from the immunoglobulin kappa promoter was used as the TF binding site. All PCR products used are 377 bp. The sequences of PCR products are provided in Table S2.
In vitro droplet assay
Recombinant GFP or mCherry fusion proteins were concentrated and desalted to an appropriate protein concentration and 125mM NaCl using Amicon Ultra centrifugal filters (30K MWCO, Millipore) in Buffer D(125) (50mM Tris-HCl pH 7.5, 10% glycerol, 1mM DTT). Fluorescent PCR products were concentration normalized in Buffer D(0) (50mM Tris-HCl pH 7.5, 10% glycerol, 1mM DTT). For all droplet assays, DNA was included at 50nM, mEGFP-OCT4 at 1250nM, and mCherry-MED1-IDR at the indicated concentration. Recombinant proteins and DNA were mixed with 10% PEG-8000 as a crowding agent. The final buffer conditions were 50mM Tris-HCl pH 7.5, 100mM NaCl, 10% glycerol, 1mM DTT. The solution was immediately loaded onto a homemade chamber comprising a glass slide with a coverslip attached by two parallel strips of double-sided tape. Slides were then imaged with an Andor spinning disk confocal microscope with a 150x objective. Unless indicated, images presented are of droplets settled on the glass coverslip.
For DNase I experiment, MED1-IDR droplets were formed at indicated concentration in the presence of OCT4 (1250nM) and ODNA_20 (50nM). The solution containing droplets was split into two equal volumes, to one volume DNase I (Turbo DNase, Invitrogen, 3U) was added with manufacturer provided reaction buffer and to the second volume enzyme storage buffer and reaction buffer were added. These were loaded onto slides, incubated at 37° C for 2 hours and subsequently imaged as described above.
Image analysis for reconstructing experimental phase curves
A custom analysis pipeline was developed in MATLAB™, building on code described in (Boija et al., 2018). Briefly, droplets were identified by employing a two-step thresholding procedure. First, the image was segmented in the MED1-IDR channel with an intensity threshold (Ipixel > μ* + 3σ, where μ* is the most probable intensity, representative of background, and σ is the width of the distribution) to identify bright pixels. Subsequently, the identified bright pixels were labeled as “condensed” droplet phase after enforcing a minimum droplet size of 9 pixels i.e. atleast 9 clustered pixels had to simultaneously pass the intensity threshold to belong to the condensed phase. In the absence of phase separation, no pixels are identified as belonging to the condensed phase.
For each image, the total intensity in the condensed droplet phase was summed in each channel (Ichannel,droplet), as well as the total background intensity outside droplets (Ichannel,bulk). The condensed fraction in each channel was defined as :
The condensed fraction was averaged over replicate images (≥10 per condition). At very low concentrations or in the absence of observable phase separation, c.f. is close to 0. We repeated the c.f. analysis with different intensity thresholds (I > μ* + 2.5σ, I > μ* + 3.5σ, I > μ* + 4σ) and size thresholds (9,16,25 pixels). The qualitative results reported in main and supplementary figures did not change under these tested conditions.
In all plots of the c.f., solid lines represent the mean condensed fraction and error bars refer to values one standard deviation above and below the mean, computed from replicates (n≥10). Plots of the condensed fraction were generated by using the matplotlib library in python3. In all plots in the main figures (Figs 1F, 3D, 3H, 4D, 5B), the condensed fraction in the MED1-IDR channel is reported.
For inferring saturation concentrations from the condensed fraction curves, a linear interpolation was fit using the linear-least squares approach to the data from the replicates across the data points above and below the threshold (0.4% - for all data reported). The apparent saturation concentration (Csat) was estimated as the concentration at which the condensed fraction reached the threshold value. The standard deviation in inferred values were computed from the standard error of the regression.
The difference between the inferred values of saturation concentrations across any set of conditions (as measured by their ratio) was insensitive to other tested values of the threshold in the range 0.3-0.6 %. Lower values of the threshold (<0.3%) led to unreliable estimates, confounded by noise from replicates, as well as specking from background, and were thus not employed. A T-test (with unequal variances, Welch’s test, refer - scipy.stats.ttest_ind_from_stats) was performed to test for significance between inferred saturation concentrations.
DNase I image analysis
Building on the above-described analysis, for each condition, the partition ratio for each replicate image is calculated as in various channels for each image-set. The key difference is that a background intensity subtraction (of 80 pixel units) is performed to aid in droplet identification and partition calculation at low concentrations. The partition ratio is a proxy for the relative enrichment of molecules in the condensed phase over the bulk phase. For any given experimental condition, the sample of partition ratios are obtained over replicate images (n≥ 10).
Subsequently, the partition ratios for control (without DNAsel) and DNAsel experiments were normalized to the mean partition ratio for the control at same concentration of MED1-IDR. Scatter plots with mean +/− std were generated using the normalized partition ratios in the 561(MED1-IDR) channel for Fig. 2B, and in the 640 channel (DNA) for Fig S1D, using PRISM.
Luciferase reporter assays
For enhancer activity reporter assays, synthetic enhancer DNA sequences with varying valences or densities of OCT4 binding sites (see Table S3) were cloned into a previously characterized pGL3-basic construct containing a minimal OCT4 promoter (pGL3-pOCT4)(Whyte et al., 2013). The synthetic enhancer sequences were cloned into the SalI site of the pGL3-pOct4 vector by HiFi DNA Assembly (NEB E2621) with a SalI digested vector and PCR-amplified insert. All cloned constructs were sequenced to ensure sequence identity. 0.4μg of the pGL3-based enhancer plasmids were used to transfect 1x105 murine ESCs in 24-well plates using Lipofectamine 3000 (Thermo Fisher L3000015) according to the manufacturer’s instructions. 0.1 μg of the pRL-SV40 plasmid was co-transfected in each condition as a luminescence control. Transfected cells were harvested after 24 hours, and luciferase activity was measured using the Dual-Glo Luciferase Assay System (Promega E2920). Luciferase signal was normalized to the signal measured in cells transfected with a construct containing zero OCT4 motifs. Experiments were performed in triplicates.
Bioinformatic analysis
Position-weight matrices (PWMS) for Mus musculus stem cell master TFs −SOX2 (MA0143), OCT4+SOX2 (MA0142), KLF4 (MA0039), and ESRRB (MA0141), were obtained from the JASPAR database (Khan et al., 2018). 100kb DNA sequences centered on super-enhancers (SEs, N=231), as annotated in (Whyte et al., 2013) were gathered. The same number and length of sequences were randomly subsampled from enhancers (typical enhancers, TEs) annotated in (Whyte et al., 2013), as well as from random genetic loci (Random) on the mm9 reference genome. FIMO was used to predict individual motif instances in all sequences, against a background uniform random distribution, at a p-value threshold of 1e-4.
For the boxplots in Fig 6A, the average motif density is calculated as total number of motifs divided by length of sequence over a 20kb sequence region centered on SEs, TEs, and random loci, normalized in units of motifs/kb. For the line plots in Fig 6B, the whole distribution of motif density is represented along the 100 KB sequence, in bins of 2kb with similar units.
Published ChIP-Seq data-sets are gathered from (Sabari et al., 2018) for MED1, BRD4, RNA Pol II, and input control from GEO: GSE112808. Reads-per-million (rpm) are summed in previously defined regions for SEs, TEs, and random using BedTools. For Fig 6C, and supplementary figure S5, the summed rpm values are plotted on a log scale. On the x-axis, the total number of motifs calculated in a 20kb window centered on SEs, TEs, and random loci is plotted. Finally, a linear model is inferred between log(ChIP) signal and motif values using ordinary least squares regression. The inferred line is plotted in black and 95% confidence intervals are plotted as a shaded gray background. The data is visualized using the matplotlib library in python3.
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical analysis for simulation data:
Steady-state analysis of simulation data-sets in Fig 3 & 4 are reported with solid lines represented by the mean (μ) and averaged fluctuations at steady state (across trajectories) in the shaded background, whose boundaries are characterized by one standard deviation away from the mean on either side (μ ± σ). In all figures, the mean represents an average over 10 trajectories. In Fig 1B, the steady state value is reported for 2 specific conditions (+/− DNA at low protein concentrations), with mean and 1 standard deviance (n=10 trajectories). For dynamical plots reported in Figs 2,4,5, the mean trajectory (n=10) is reported.
Statistical analysis for bioinformatics:
The inferred linear lines in Fig 6C and S5 are generated between the logarithm of the ChIP signal and the motif density, and the R2 reported in the respective captions. The 95% confidence interval in the inferred slope of the linear fit is reported in the grey background, calculated from statsmodels.api in python.
Statistical analysis for in vitro condensate assays:
Condensed fraction reported at any given concentration in all figures are averaged over > 10 image-sets, with error bars representing one standard deviation from the mean condensed fraction. Saturation concentrations are inferred (mean and std error) from the above data (n>10 data sets, ref methods above for details). The T-test (with unequal variances, Welch’s test, refer - scipy.stats.ttest_ind_from_stats) was performed to test for significance. Pairwise student’s t-test for DNase experiment (Figure 2B) and luciferase experiments (Figure 4C,5B) were performed using PRISM 7 (GraphPad).
DATA AND SOFTWARE AVAILABILITY
All software and code generated in this project are publicly available at https://github.com/krishna-shrinivas/2019_Shrinivas_Sabari_enhancer_features. The raw experimental data can be found at https://dx.doi.org/10.17632/c36nyy79y4.1 .
Supplementary Material
Highlights.
Transcription machinery forms condensates localized to specific DNA elements
Combination of structured and dynamic interactions enables localized condensation
DNA encoding binding site features above threshold values drives condensation
DNA features that drive condensation promote enhancer activity in cells
Acknowledgments:
We acknowledge members of Chakraborty and Young labs for helpful discussions. We acknowledge the support of Wendy Salmon and the W.M Keck Microscopy Facility at the Whitehead Institute. This work was supported by grants from the NSF (PHY-1743900) (A.K.C., R.A.Y., and P.A.S), the NIH (GM123511) (R.A.Y.), (P01-CA042063) (P.A.S.), and, in part, by a Koch Institute Support (core) grant (P30-CA14051) from the National Cancer Institute (P.A.S). Additional support was provided by: Damon Runyon Cancer Research Foundation Fellowship (2309-17) (B.R.S.), NSF Graduate Research Fellowship (A.V.Z.), NWO Rubicon Fellowship (J.S.), Swedish Research Council Postdoctoral Fellowship (VR 2017-00372) (A.B.), and an NIH training grant (T32CA009172) (I.A.K.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of interests: R.A.Y. is a founder and shareholder of Syros Pharmaceuticals, Camp4 Therapeutics, Omega Therapeutics and Dewpoint Therapeutics. I.A.K. is a consultant to InfiniteMD, Best Doctors, and Foundation Medicine, and is a shareholder of InfiniteMD. P.A.S. is a member of the Board and shareholder in Syros and a member of the Scientific Advisory Board of Dewpoint. A.K.C is a member of the Scientific Advisory Boards of Dewpoint Therapeutics and Omega Therapeutics. All other authors declare no competing interests.
References:
- Anderson JA, Lorenz CD, and Travesset A (2008). General purpose molecular dynamics simulations fully implemented on graphics processing units. J. Comput. Phys 227, 5342–5359. [Google Scholar]
- Banani SF, Lee HO, Hyman AA, and Rosen MK (2017). Biomolecular condensates: organizers of cellular biochemistry. Nat. Rev. Mol. Cell Biol. 18, 285–298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berman BP, Nibu Y, Pfeiffer BD, Tomancak P, Celniker SE, Levine M, Rubin GM, and Eisen MB (2002). Exploiting transcription factor binding site clustering to identify cis-regulatory modules involved in pattern formation in the Drosophila genome. Proc. Natl. Acad. Sci. U. S. A 99, 757–762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boija A, Klein IA, Sabari BR, Dall’Agnese A, Coffey EL, Zamudio AV, Li CH, Shrinivas K, Manteiga JC, Hannett NM, et al. (2018). Transcription Factors Activate Genes through the Phase-Separation Capacity of Their Activation Domains. Cell 175, 1842–1855.e16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borgia A, Borgia MB, Bugge K, Kissling VM, Heidarsson PO, Fernandes CB, Sottini A, Soranno A, Buholzer KJ, Nettels D, et al. (2018). Extreme disorder in an ultrahigh-affinity protein complex. Nature. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouras T, Southey MC, and Venter DJ (2001). Overexpression of the steroid receptor coactivator AIB1 in breast cancer correlates with the absence of estrogen and progesterone receptors and positivity for p53 and HER2/neu. Cancer Res. 61, 903–907. [PubMed] [Google Scholar]
- Brady JP, Farber PJ, Sekhar A, Lin Y-H, Huang R, Bah A, Nott TJ, Chan HS, Baldwin AJ, Forman-Kay JD, et al. (2017). Structural and hydrodynamic properties of an intrinsically disordered region of a germ cell-specific protein on phase separation. Proc. Natl. Acad. Sci. U. S. A 114, E8194–E8203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho W-K, Spille J-H, Hecht M, Lee C, Li C, Grube V, and Cisse II (2018). Mediator and RNA polymerase II clusters associate in transcription-dependent condensates. Science 361, 412–415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chong S, Dugast-Darzacq C, Liu Z, Dong P, Dailey GM, Cattoglio C, Heckert A, Banala S, Lavis L, Darzacq X, et al. (2018). Imaging dynamic and selective low-complexity domain interactions that control gene transcription. Science 361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crocker J, Abe N, Rinaldi L, McGregor AP, Frankel N, Wang S, Alsawadi A, Valenti P, Plaza S, Payre F, et al. (2015). Low Affinity Binding Site Clusters Confer Hox Specificity and Regulatory Robustness. Cell 160, 191–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das RK, and Pappu RV (2013). Conformations of intrinsically disordered proteins are influenced by linear sequence distributions of oppositely charged residues. Proc. Natl. Acad. Sci. U. S. A 110, 13392–13397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das J, Ho M, Zikherman J, Govern C, Yang M, Weiss A, Chakraborty AK, and Roose JP (2009). Digital Signaling and Hysteresis Characterize Ras Activation in Lymphoid Cells. Cell 136, 337–351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dignon GL, Zheng W, Best RB, Kim YC, and Mittal J (2018). Relation between single-molecule properties and phase behavior of intrinsically disordered proteins. Proc. Natl. Acad. Sci. U. S. A 115, 9929–9934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feric M, Vaidya N, Harmon TS, Mitrea DM, Zhu L, Richardson TM, Kriwacki RW, Pappu RV, and Brangwynne CP (2016). Coexisting Liquid Phases Underlie Nucleolar Subcompartments. Cell 165, 1686–1697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fox AH, Nakagawa S, Hirose T, and Bond CS (2018). Paraspeckles: Where Long Noncoding RNA Meets Phase Separation. Trends Biochem. Sci. 43, 124–135. [DOI] [PubMed] [Google Scholar]
- Fukaya T, Lim B, and Levine M (2016). Enhancer Control of Transcriptional Bursting. Cell 166, 358–368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glaser J, Nguyen TD, Anderson JA, Lui P, Spiga F, Millan JA, Morse DC, and Glotzer SC (2015). Strong scaling of general-purpose molecular dynamics simulations on GPUs. Comput. Phys. Commun 192, 97–107. [Google Scholar]
- Harmon TS, Holehouse AS, Rosen MK, and Pappu RV (2017). Intrinsically disordered linkers determine the interplay between phase separation and gelation in multivalent proteins. Elife 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hnisz D, Shrinivas K, Young RA, Chakraborty AK, and Sharp PA (2017). A Phase Separation Model for Transcriptional Control. Cell 169, 13–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hopfield JJ (1974). Kinetic proofreading: a new mechanism for reducing errors in biosynthetic processes requiring high specificity. Proc. Natl. Acad. Sci. U. S. A 71, 4135–4139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huihui J, Firman T, and Ghosh K (2018). Modulating charge patterning and ionic strength as a strategy to induce conformational changes in intrinsically disordered proteins. J. Chem. Phys 149, 85101. [DOI] [PubMed] [Google Scholar]
- Jolma A, Yin Y, Nitta KR, Dave K, Popov A, Taipale M, Enge M, Kivioja T, Morgunova E, and Taipale J (2015). DNA-dependent formation of transcription factor pairs alters their binding specificity. Nature 527, 384–388. [DOI] [PubMed] [Google Scholar]
- Jung C, Bandilla P, von Reutern M, Schnepf M, Rieder S, Unnerstall U, and Gaul U (2018). True equilibrium measurement of transcription factor-DNA binding affinities using automated polarization microscopy. Nat. Commun 9, 1605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan A, Fornes O, Stigliani A, Gheorghe M, Castro-Mondragon JA, van der Lee R, Bessy A, Cheneby J, Kulkarni SR, Tan G, et al. (2018). JASPAR 2018: update of the open-access database of transcription factor binding profiles and its web framework. Nucleic Acids Res. 46, D260–D266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambert SA, Jolma A, Campitelli LF, Das PK, Yin Y, Albu M, Chen X, Taipale J, Hughes TR, and Weirauch MT (2018). The Human Transcription Factors. Cell 172, 650–665. [DOI] [PubMed] [Google Scholar]
- Larson AG, Elnatan D, Keenen MM, Trnka MJ, Johnston JB, Burlingame AL, Agard DA, Redding S, and Narlikar GJ (2017). Liquid droplet formation by HP1a suggests a role for phase separation in heterochromatin. Nature 236–240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee TI, and Young RA (2013). Transcriptional Regulation and Its Misregulation in Disease. Cell 152, 1237–1251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levo M, and Segal E (2014). In pursuit of design principles of regulatory sequences. Nat. Rev. Genet 15, 453–468. [DOI] [PubMed] [Google Scholar]
- Li J, Dong A, Saydaminova K, Chang H, Wang G, Ochiai H, Yamamoto T, and Pertsinidis A (2019). Single-Molecule Nanoscopy Elucidates RNA Polymerase II Transcription at Single Genes in Live Cells. Cell 0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin CY, Loven J, Rahl PB, Paranal RM, Burge CB, Bradner JE, Lee TI, and Young RA (2012). Transcriptional amplification in tumor cells with elevated c-Myc. Cell 151, 56–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin Y-H, Brady JP, Forman-Kay JD, and Chan HS (2017). Charge pattern matching as a “fuzzy” mode of molecular recognition for the functional phase separations of intrinsically disordered proteins. New J. Phys. 19, 115003. [Google Scholar]
- Long HK, Prescott SL, and Wysocka J (2016). Ever-Changing Landscapes: Transcriptional Enhancers in Development and Evolution. Cell 167, 1170–1187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu H, Yu D, Hansen AS, Ganguly S, Liu R, Heckert A, Darzacq X, and Zhou Q (2018). Phase-separation mechanism for C-terminal hyperphosphorylation of RNA polymerase II. Nature 558, 318–323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maniatis T, Falvo JV, Kim TH, Kim TK, Lin CH, Parekh BS, and Wathelet MG (1998). Structure and function of the interferon-beta enhanceosome. Cold Spring Harb. Symp. Quant. Biol. 68, 609–620. [DOI] [PubMed] [Google Scholar]
- Markstein M, Markstein P, Markstein V, and Levine MS (2002). Genome-wide analysis of clustered Dorsal binding sites identifies putative target genes in the Drosophila embryo. Proc. Natl. Acad. Sci 99, 763–768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgunova E, and Taipale J (2017). Structural perspective of cooperative transcription factor binding. Curr. Opin. Struct. Biol 47, 1–8. [DOI] [PubMed] [Google Scholar]
- Nguyen TD, Phillips CL, Anderson JA, and Glotzer SC (2011). Rigid body constraints realized in massively-parallel molecular dynamics on graphics processing units. Comput. Phys. Commun 182, 2307–2313. [Google Scholar]
- Ninio J (1975). Kinetic amplification of enzyme discrimination. Biochimie 57, 587–595. [DOI] [PubMed] [Google Scholar]
- Nizami Z, Deryusheva S, and Gall JG (2010). The Cajal Body and Histone Locus Body. Cold Spring Harb. Perspect. Biol. 2, a000653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nott TJ, Petsalaki E, Farber P, Jervis D, Fussner E, Plochowietz A, Craggs TD, Bazett-Jones DP, Pawson T, Forman-Kay JD, et al. (2015). Phase Transition of a Disordered Nuage Protein Generates Environmentally Responsive Membraneless Organelles. Mol. Cell 57, 936–947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pederson T (2011). The nucleolus. Cold Spring Harb. Perspect. Biol. 3, a000638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ptashne M, and Gann A (1997). Transcriptional activation by recruitment. Nature 386, 569–577. [DOI] [PubMed] [Google Scholar]
- Rajewsky N, Vergassola M, Gaul U, and Siggia ED (2002). Computational detection of genomic cis-regulatory modules applied to body patterning in the early Drosophila embryo. BMC Bioinformatics 3, 30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rowley MJ, Nichols MH, Lyu X, Ando-Kuri M, Rivera ISM, Hermetz K, Wang P, Ruan Y, and Corces VG (2017). Evolutionarily Conserved Principles Predict 3D Chromatin Organization. Mol. Cell 67, 837–852.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabari BR, Dall’Agnese A, Boija A, Klein IA, Coffey EL, Shrinivas K, Abraham BJ, Hannett NM, Zamudio AV, Manteiga JC, et al. (2018). Coactivator condensation at super-enhancers links phase separation and gene control. Science 361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwarzer W, Abdennur N, Goloborodko A, Pekowska A, Fudenberg G, Loe-Mie Y, Fonseca NA, Huber W, Haering C, Mirny L, et al. (2017). Two independent modes of chromatin organization revealed by cohesin removal. Nature 551, 51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Semenov AN, and Rubinstein M (1998). Thermoreversible Gelation in Solutions of Associative Polymers. 1. Statics. Macromolecules 31, 1373–1385. [Google Scholar]
- Sherry KP, Das RK, Pappu RV, and Barrick D (2017). Control of transcriptional activity by design of charge patterning in the intrinsically disordered RAM region of the Notch receptor. Proc. Natl. Acad. Sci. U. S. A 114, E9243–E9252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin Y, and Brangwynne CP (2017). Liquid phase condensation in cell physiology and disease. Science 357. [DOI] [PubMed] [Google Scholar]
- Shlyueva D, Stampfel G, and Stark A (2014). Transcriptional enhancers: from properties to genome-wide predictions. Nat. Rev. Genet 15, 272–286. [DOI] [PubMed] [Google Scholar]
- Slattery M, Zhou T, Yang L, Dantas Machado AC, Gordan R, and Rohs R (2014). Absence of a simple code: how transcription factors read the genome. Trends Biochem. Sci. 39, 381–399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith E, and Shilatifard A (2014). Enhancer biology and enhanceropathies. Nat. Struct. Mol. Biol 21, 210–219. [DOI] [PubMed] [Google Scholar]
- Spitz F, and Furlong EEM (2012). Transcription factors: from enhancer binding to developmental control. Nat. Rev. Genet 13, 613–626. [DOI] [PubMed] [Google Scholar]
- Strom AR, Emelyanov AV, Mir M, Fyodorov DV, Darzacq X, and Karpen GH (2017). Phase separation drives heterochromatin domain formation. Nature 547, 241–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tatavosian R, Kent S, Brown K, Yao T, Duc HN, Huynh TN, Zhen CY, Ma B, Wang H, and Ren X (2018). Nuclear condensates of the Polycomb protein chromobox 2 (CBX2) assemble through phase separation. J. Biol. Chem jbc.RA118.006620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai A, Muthusamy AK, Alves MR, Lavis LD, Singer RH, Stern DL, and Crocker J (2017). Nuclear microenvironments modulate transcription from low-affinity enhancers. Elife 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Choi J-M, Holehouse AS, Lee HO, Zhang X, Jahnel M, Maharana S, Lemaitre R, Pozniakovsky A, Drechsel D, et al. (2018). A Molecular Grammar Governing the Driving Forces for Phase Separation of Prion-like RNA Binding Proteins. Cell 174, 688–699.e16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wasserman WW, and Sandelin A (2004). Applied bioinformatics for the identification of regulatory elements. Nat. Rev. Genet 5, 276–287. [DOI] [PubMed] [Google Scholar]
- Wei M-T, Elbaum-Garfinkle S, Holehouse AS, Chen CC-H, Feric M, Arnold CB, Priestley RD, Pappu RV, Brangwynne CP, Chih C, et al. (2017). Phase behaviour of disordered proteins underlying low density and high permeability of liquid organelles. Nat. Chem 9, 1118–1125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weirauch MT, Yang A, Albu M, Cote AG, Montenegro-Montero A, Drewe P, Najafabadi HS, Lambert SA, Mann I, Cook K, et al. (2014). Determination and inference of eukaryotic transcription factor sequence specificity. Cell 158, 1431–1443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whyte WA, Orlando DA, Hnisz D, Abraham BJ, Lin CY, Kagey MH, Rahl PB, Lee TI, and Young RA (2013). Master Transcription Factors and Mediator Establish Super-Enhancers at Key Cell Identity Genes. Cell 153, 307–319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wunderlich Z, and Mirny LA (2009). Different gene regulation strategies revealed by analysis of binding motifs. Trends Genet. 25, 434–440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamazaki T, Souquere S, Chujo T, Kobelke S, Chong YS, Fox AH, Bond CS, Nakagawa S, Pierron G, and Hirose T (2018). Functional Domains of NEAT1 Architectural IncRNA Induce Paraspeckle Assembly through Phase Separation. Mol. Cell 70, 1038–1053.e7. [DOI] [PubMed] [Google Scholar]
- Zhu Y, Qi C, Jain S, Le Beau MM, Espinosa R, Atkins GB, Lazar MA, Yeldandi AV, Rao MS, and Reddy JK (1999). Amplification and overexpression of peroxisome proliferator-activated receptor binding protein (PBP/PPARBP) gene in breast cancer. Proc. Natl. Acad. Sci. U. S. A 96, 10848–l0853. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All software and code generated in this project are publicly available at https://github.com/krishna-shrinivas/2019_Shrinivas_Sabari_enhancer_features. The raw experimental data can be found at https://dx.doi.org/10.17632/c36nyy79y4.1 .