

Abstract
Dysregulated redox signaling in pulmonary vasculature is central to the development of pulmonary arterial hypertension (PAH) and lung injury. Modulators of reactive oxygen species (ROS) production and downstream signaling targets are critical for mediating the physiological or pathological effects of ROS. Understanding the complex interactions between the modulators and signaling targets of ROS is essential for developing novel strategies to prevent or attenuate lung pathologies. In this review, we discuss recent studies on the modulators and targets of ROS in pulmonary endothelial and smooth muscle cells, their cellular effects, and the disease conditions associated with dysregulated redox signaling.
Keywords: Redox signaling, pulmonary hypertension, lung injury, endothelial cell, smooth muscle cell
Graphical Abstract
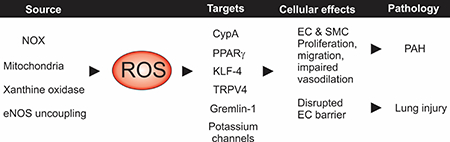
New redox signaling mechanisms in pulmonary vascular diseases. Reactive oxygen species (ROS) in pulmonary circulation are generated mainly by the electron transport chain in the mitochondria, NADPH oxidase (NOX) and xanthine oxidase enzymes, and endothelial nitric oxide synthase (eNOS) uncoupling. ROS can act on a diverse set of downstream signaling targets to alter cellular function. The cellular effects of excessive ROS production include increased proliferation and migration, contributing to the development of pulmonary arterial hypertension (PAH), and disruption of endothelial cell barrier, which results in lung injury.
1. Introduction
Reactive oxygen species (ROS) comprise a set of reactive chemical intermediates of oxygen that have one or more unpaired electrons in their outer orbits. The main ROS in pulmonary circulation include superoxide radicals, hydrogen peroxide, and hydroxyl radicals. Additionally, superoxide can react with nitric oxide (NO) to form peroxynitrite, a reactive nitrogen species (RNS) that inhibits endogenous antioxidant systems via posttranslational modifications [1, 2]. Due to their high reactivity, ROS or RNS can oxidize proteins, DNA, lipids, or cellular membranes and alter their functions [3]. The most common sites for redox modifications of proteins are oxidant-sensitive amino acids (cysteine, selenocysteine, and methionine residues) and redox-active transition metal ion centers (heme groups, iron-sulfur centers, zinc-thiolate centers). Thus, ROS signaling plays important roles in maintaining cellular homeostasis, however, dysregulated redox signaling results in oxidative stress, which can have damaging effects on cellular functions [4].
Mitochondria and the NADPH oxidase (NOX) family of enzymes are widely regarded as the major sources of cellular ROS (Figure 1, Figure 2). Aerobic metabolism within the mitochondrial electron transport chain produces ROS, while NOX enzymes generate ROS (superoxide or H2O2) as their primary product. Four different NOX isoforms, NOX1–4, have been detected in the pulmonary vasculature [5–8]. ROS can also be generated by uncoupled nitric oxide synthase (NOS), xanthine oxidase, lipid peroxidases, and cytochrome P450 enzymes [9]. Notably, lungs express endogenous antioxidants that prevent excessive formation of ROS or limit the ROS-induced cellular damage. These include enzymes that metabolize ROS (superoxide dismutase, catalase, glutathione peroxidases, peroxiredoxins) [10], thiol reductases that reverse cysteine oxidation (thioredoxin, glutaredoxin), phase II metabolic enzymes (glutathione-S-transferases), small molecular weight antioxidants (glutathione and vitamins), and metal binding proteins (transferrin, lactoferrin). Several of these antioxidant systems are altered or impaired under disease conditions.
Figure 1. Endothelial redox signaling mechanisms in pulmonary arterial hypertension (PAH) and acute lung injury (ALI).

(A) The major sources of reactive oxygen species (ROS) generation in pulmonary vascular endothelial cells include enzymes NOX1–3 and xanthine oxidase, mitochondria, and endothelial nitric oxide synthase (eNOS) uncoupling. In PAH, excessive ROS generation promotes endothelial cell proliferation and attenuates endothelium-dependent vasodilation. On the other hand, NOX4 is thought to be protective. It releases H2O2, which exerts protective effects on endothelium via activation of antioxidant enzyme HO-1 and formation of NO. (B) Excessive ROS formation can lead to endothelial cell apoptosis and disruption of endothelial barrier, thus contributing to ALI. Hsp70-TLR4 signaling, KATP channels, and HO-1 have protective effects on ROS-induced inflammation and apoptosis in ALI.
Figure 2. Smooth muscle cell redox signaling mechanisms in PAH.

In pulmonary artery smooth muscle cells, hypoxia induces mitochondrial ROS generation, which inhibits voltage-gated K+ (KV) channels to cause membrane depolarization and vasoconstriction. ROS generation by NOX enzyme activity is mainly responsible for smooth muscle cell proliferation and vascular remodeling in PAH. ROS generation by NOX1–3 enzymes can be activated by serotonin, 16α-hydroxyestrone (16α-OHE1), and galectin-3 (Gal-3).
Increased NOX activity or mitochondrial dysfunction or altered endogenous antioxidant systems [10] is usually associated with higher ROS levels. In the pulmonary vasculature, an imbalance between oxidant production and antioxidant protective mechanisms is associated with deleterious effects on both smooth muscle cells (SMCs) and endothelial cells (ECs) [2, 11, 12]. Oxidative stress has been associated with excessive pulmonary vasoconstriction, inflammation, and vascular remodeling, all of which contribute to the development of pulmonary arterial hypertension (PAH, Graphical Abstract). Abnormal redox signaling has also been implicated in the disruption of pulmonary endothelial barrier function and pathogenesis of acute lung injury (ALI, Graphical Abstract). Evidence for the connection between ROS and pulmonary abnormalities have been diligently summarized in previous review papers [9, 13–16]. Here, we discuss the most recent discoveries on the physiological and pathological roles of ROS in the pulmonary vasculature.
2. Modulators of ROS production in ECs
NOX family of enzymes
Among the NOX isoforms, NOX1–3, which primarily release superoxide, are thought to contribute to pulmonary vascular disease pathogenesis via ROS production (Figure 1). NOX4 primarily releases H2O2 [17] and has been suggested to have a protective effect on endothelial function [18, 19]. In NOX4−/− mice, pulmonary endothelial tube formation and angiogenesis were attenuated [19]. Lowering the concentrations of H2O2 also reversed EC tube formation and angiogenesis in wild-type mice, supporting the role of NOX4-H2O2 signaling in EC tube formation and angiogenesis in lungs (Figure 1) [19]. Similarly, in an angiotensin II (Ang II) - induced model of oxidative stress, NOX4-deficient mice displayed endothelial dysfunction [19]. One possibility is that NOX4-H2O2 signaling exerts protective effects on endothelial function via increases in NO production and expression of heme oxygenase-1 (HO-1) [19]. While NO is a vasodilator and anti-inflammatory signaling molecule, HO-1 is an endogenous antioxidant enzyme [19]. Interestingly, NOX4 can localize to the endoplasmic reticulum (ER) [20, 21], and this ER localization may be important for establishing the target specificity of NOX4-dependent signaling [21, 22]. Additionally, endothelial NOX4-H2O2 signaling has also been linked with maintenance of vascular endothelial growth factor (VEGF) expression [18]. Despite numerous studies supporting an essential role for NOX4 in pulmonary vascular cells, the precise downstream mechanisms that mediate functional effects of NOX4 remain unclear.
Another enzyme of the NOX family, NOX3, was previously thought to be an insignificant player in pulmonary vasculature. However, recent studies have identified a key role for NOX3 in pulmonary vascular diseases [23, 24]. For example, Yin et al. targeted 143 single nucleotide polymorphisms (SNPs) for 14 candidate genes and showed that the rs6557421 variant in NOX3 may be important for individual susceptibility to pulmonary hypertension (PH) [25]. Among the SNP candidates were NOX4, NOX5, and the structural protein of caveolae, caveolin-1. However, only NOX3 SNP was associated with overall PH susceptibility. In another study, NOX3-induced ROS production led to pulmonary EC death and lung injury in mice [23]. Additionally, activation of heat shock protein 70 (Hsp70)-toll like receptor 4 (TLR-4)-Trif signaling was shown to inhibit NOX3 activity in lung ECs, attenuating ROS generation and oxidant-induced ALI (Table 1, Figure 1) [23, 26]. Moreover, TLR-4-dependent activation of stanniocalcin-1 (STC-1), an endogenous antioxidant in the lungs, had a beneficial effect in ALI (Table 1) [26]. Thus, inhibition of NOX3 via a TLR-4/STC1-mediated mechanism could be a potential strategy for the prevention or treatment of ALI. In this regard, Hsp70 upregulation suppressed mitochondrial ROS production and disruption of pulmonary endothelial barrier in response to bacterial toxin pneumolysin [27], suggesting that Hsp70 upregulation may also be a therapeutic approach against pneumonia-associated lung injury (Table 1).
Table 1.
Novel modulators of ROS production in pulmonary vasculature
| Modulator | Target cell | Effect | References |
|---|---|---|---|
| Hsp70-TLR4-Trif signaling | EC | Attenuates NOX3 activity, ROS production, and ALI | [23, 26] |
| TLR4-STC1 signaling | EC | Endogenous antioxidant STC1 lowers oxidant-induced lung injury | [26, 27] |
| Pneumolysin | EC | Elevates ROS, damages endothelial barrier | [27] |
| KATP channel | EC | Inhibits of NF-κB and MAPK signaling, lower ROS production | [33] |
| CypA/acetylated CypA | EC | Increases ROS levels, inflammation and apoptosis, PAH phenotype | [32] |
| Serotonin (5-HT) | SMC | Increases NOX1 signaling and ROS production | [47] |
| Gal-3 | SMC | Increases NOX expression, PA remodeling and fibrosis in PAH | [48, 49] |
| 16α-hydroxyestrone | SMC | Increased NOX1-mediated ROS production, cell proliferation | [46] |
Another prime candidate for ROS generation, NOX1, was elevated in cultured human pulmonary artery ECs exposed to hypoxia. This was accompanied by increased ROS levels and gremlin 1 expression, which in turn stimulated EC proliferation and migration [28]. Consistent with these findings, increased levels of NOX subunit p22phox were detected in the lungs from a mouse model of hypoxia-induced PH [29]. Importantly, mice harboring a loss of function mutation in p22phox were protected against hypoxia-induced PH, revealing an essential role of p22phox in the development of PH [29]. p22phox was also found to promote ROS generation, vascular proliferation, migration, and angiogenesis. Thus, NOX1-ROS-gremlin 1 signaling and p22phox subunit could be targeted for potential therapy against vascular remodeling in PAH (Figure 1).
Cyclophilin A (CypA)
CypA is both a target and a modulator of cellular ROS (Figure 1). CypA is a ubiquitously expressed protein that has also been identified as a secreted oxidative stress-induced factor [30, 31]. EC-derived CypA promotes inflammation through paracrine and autocrine mechanisms [32]. Importantly, oxidative stress-induced release of CypA and acetylated CypA from mouse and human MVECs, which further promoted EC oxidative stress (Table 1) [32]. A study utilizing transgenic mice that express high levels of CypA revealed a PAH phenotype in these mice by three months of age. In the same study, extracellular CypA and acetylated CypA enhanced inflammatory signals and apoptosis in MVECs [32]. This series of findings suggests that CypA or acetylated CypA release from ECs may be a potential therapeutic target for treating PAH.
KATP channels
Activation of ATP-sensitive K+ (KATP) ion channels was recently shown to curtail ROS generation (Table 1, Figure 1) [33], thus revealing KATP channels as a novel regulator of ROS generation and a potential therapeutic target. Stimulation of endothelial KATP channels reduced lipopolysaccharide (LPS)-induced ALI by lowering ROS levels, preventing apoptosis, and suppressing endothelial inflammation. These effects were mainly attributed to the inhibition of NF-κB and MAPK signaling pathways by KATP channels [33].
3. Targets of ROS in ECs
Heme oxygenase (HO-1)
HO-1 is an endogenous antioxidant and anti-inflammatory enzyme in the lungs [34]. Searching for a potential treatment for LPS-induced acute lung injury (ALI), Lin et al. recently investigated the role of HO-1 in ECs and other pulmonary cells [35]. Under healthy conditions, HO-1 lowered the levels of pro-inflammatory cytokines (TNF-α, IL-6 and IL-1β) and stimulated SOD activity, thereby reducing ROS levels in lung cells [35]. This observation was further supported by subsequent findings that HO-1 inhibited pro-inflammatory PI3K/Akt/NF-κB signaling and upregulated anti-inflammatory AMPK activity; effects that would be beneficial in LPS-induced ALI [34, 36]. Further research is needed to determine whether the protective effect of HO-1 is derived from pulmonary ECs or other cell types.
TRPV4 channels
Recent studies on Ca2+ signaling mechanisms in pulmonary ECs have focused on TRP (transient receptor potential) cation channels at the EC membrane. It appears that Ca2+ influx through endothelial TRPV4 (TRP vanilloid 4) channels controls physiological function of pulmonary microcirculation [37] but has also been implicated in a ROS-activated mechanism for the pathogenesis of PAH (Figure 1) [37]. Marziano et al. recently demonstrated that a spatially restricted TRPV4-eNOS signaling mechanism in the endothelium promotes vasodilation of small, resistance-sized pulmonary arteries [37]. A subsequent study by Suresh et al. showed that cultured lung microvascular ECs (MVECs) from a rat model of PAH (SU5416 injection + chronic hypoxia) exhibited higher basal levels of intracellular Ca2+ and elevated migratory and proliferative capacity [38] when compared to MVECs from normoxic rat. Interestingly, ROS levels were elevated in MVECs from PAH rats, and mitochondrial antioxidants lowered TRPV4 channel-dependent increases in intracellular Ca2+. Similarly, TRPV4 channel inhibition also reduced intracellular Ca2+ levels and reversed MVEC migration and proliferation [38], implying that abnormal migration and proliferation of MVECs in PAH may be modulated by mitochondrial ROS-induced Ca2+ influx via TRPV4 channels (Figure 1). The TRPV4 channel has a high Ca2+ conductance [39]. Therefore, it is plausible that the extent of TRPV4 channel activation determines whether the ion channel plays a beneficial or a harmful role in pulmonary vasculature.
Transcription factors
Recent studies have focused mainly on two transcription factors that can be targeted by ROS (Figure 1): peroxisome proliferator-activated receptor gamma (PPARγ) and Kruppel-like factor 4 (KLF4). The expression of PPARγ, a nuclear hormone transcription factor, was found to be reduced in ECs and SMCs during hypoxia-induced PH. Moreover, this decrease in PPARγ expression was dependent on NOX4-mediated H2O2 release [40–43]. In this regard, Bijli et al. showed that hypoxia triggers the activation of proline-rich tyrosine kinase 2 (Pyk2), which stimulates extracellular signal–regulated kinase (ERK) 1/2, promoting NF-κB-mediated NOX4 expression. The resulting H2O2 generation downregulates PPARγ [40]. It is not known whether PPARγ downregulation in ECs and SMCs is essential for the development of PH.
Another transcription factor, KLF4, belongs to the Kruppel-like transcription factor family and is known to be important for normal endothelial function and vascular homeostasis [44]. KLF4 is constitutively expressed in pulmonary arteries and is associated with modulation of inflammation, coagulation and phenotypic switching [44]. In PAH, S-nitrosation of endothelial KFL4 was shown to contribute to endothelial dysfunction. Thus, ROS-mediated post-translational modifications of endothelial KLF4 may underlie pulmonary vascular dysfunction and development of PAH.
4. Modulators and targets of ROS in SMCs
Pulmonary vasoconstriction and vascular remodeling are key causative factors for increased pulmonary arterial pressures in PAH. A recent study by Jernigan et al. investigated the contribution of ROS in increasing pulmonary vasoconstrictor activity in a rat model of PAH (SU5416 injection + chronic hypoxia) [12]. Hypoxia/SU5416 rats showed greater vasoconstriction of pulmonary arteries in response to endothelin-1 and this effect was dependent on ROS generation. Although ROS production increased pulmonary vasoconstriction, it did not seem to play a role in promoting vascular remodeling in this model [12]. As discussed below, several other studies support an important role for ROS production in pulmonary vascular remodeling.
NOX and transcription factors
Among the NOX family, NOX1 and NOX4 have been consistently found to be expressed in SMCs, where they play crucial roles in cell growth [17, 45]. In pulmonary artery SMCs, estrogen metabolite 16α-hydroxyestrone (16α-OHE1, Table 1, Figure 2) induced NOX1-mediated ROS production, expression of nuclear factor erythroid-related factor 2 (Nrf-2), and cell proliferation [46]. Moreover, genetic ablation of NOX1 protected the animals against development of PAH, supporting an important role for SMC NOX1 in ROS production and PAH pathogenesis.
Serotonin
Hood et al. recently provided evidence that serotonin (5-HT) signaling via 5-HT1B receptors can stimulate NOX1-dependent ROS production in pulmonary SMCs (Table 1, Figure 2) [47]. In human pulmonary artery SMCs, 5-HT stimulated 5-HT1B receptor-Src-related kinase-NOX1 signaling and ROS production. 5-HT-mediated ROS production also lowered the expression of transcription factor Nrf-2 and catalase activity [47]. The fact that 5-HT, more popular for its neurotransmitter properties in mood disorders, appears to regulate redox-sensitive signaling pathways in pulmonary SMCs opens a wide range of possibilities for further exploration of neurotransmitters’ role in pulmonary vascular pathologies.
Galectin-3 (Gal-3)
Gal-3 is a chimeric lectin that may regulate NOX expression and redox signaling, and is a potent driver of fibrosis [48]. In rat models of PAH, Gal-3 expression in pulmonary artery SMCs was increased, which correlated with a higher SMC migration and fibrosis. Genetic deletion of Gal-3 lowered SMC migration and fibrosis, supporting an important role for galectin-3 in pulmonary artery remodeling and fibrosis during PAH (Table 1, Figure 2) [49, 50].
5. Concluding remarks
The physiological and pathological importance of ROS in pulmonary vasculature is firmly established. Recent studies have made significant strides into understanding novel regulators and targets of ROS (Table 1), although the specific mechanisms that can be therapeutically targeted remain elusive. Therefore, further research on targetable ROS-dependent mechanisms is needed for therapeutic benefit in pulmonary vascular diseases. It should also be noted that a significant amount of evidence for ROS regulators and their cellular effects has been derived from studies in cultured ECs or SMCs. Because of the vastly different environment in the intact blood vessels, it is plausible that native vascular cells employ different ROS signaling pathways and regulators. In this regard, studies in cell-specific knockout mice models and intact pulmonary blood vessels may provide useful insights. Identifying the source of ROS production and targeting specific ROS involved in the pathological mechanisms will also result in better therapeutic strategies.
Highlights.
Abnormal redox signaling has deleterious effects in pulmonary endothelial and smooth muscle cells.
Oxidative stress contributes to vasoconstriction and vascular remodeling in pulmonary hypertension.
Oxidative stress underlies the disruption of endothelial barrier function in acute lung injury.
Understanding the sources and targets of reactive oxygen species is important for identifying targetable mechanisms.
Studies in native pulmonary vascular cells and cell-specific knockout models are more likely to identify physiological and pathological redox signaling mechanisms.
Acknowledgement
This work was supported by the grants from the NIH to SKS (HL121484, HL138496, and HL146914) and VEL (HL133293 and HL130053).
List of Abbreviations
- 5-HT
Serotonin
- 16α-OHE1
16α-hydroxyestrone
- AKT
Protein kinase B
- AMPK
5’ AMP-activated protein kinase
- ALI
Acute lung injury
- Ang II
Angiotensin II
- ATP
Adenosine triphosphate
- CypA
Cyclophilin A
- DNA
Deoxyribonucleic acid
- EC
Endothelial cells
- eNOS
Endothelial nitric oxide synthase
- ER
Endoplasmic reticulum
- ERK1/2
Extracellular signal-regulated protein kinases 1 and 2
- HO-1
Heme oxygenase-1
- H2O2
Hydrogen peroxide
- Hsp70
Heat shock protein 70
- IL-1β
Interleukin 1 beta
- IL-6
Interleukin 6
- KATP
ATP-sensitive potassium channels
- KLF4
Kruppel-like factor 4
- LPS
Lipopolysaccharide
- MVEC
Microvascular endothelial cells
- NADPH
Nicotinemide adenine dinucleotide phosphate
- NF-κB
Nuclear factor kappa-light-chain-enhancer of activated B cells
- NO
Nitric oxide
- NOS
Nitric oxide synthase
- NOX
NADPH oxidase
- Nrf-2
Nuclear factor erythroid 2–related factor 2
- PAH
Pulmonary arterial hypertension
- PASMC
Pulmonary artery smooth muscle cells
- PH
Pulmonary hypertension
- PI3K
Phosphoinositide 3-kinases
- PPARγ
Peroxisome proliferator-activated receptor gamma
- Pyk2
Proline-rich tyrosine kinase 2
- ROS
Reactive oxygen species
- SMC
Smooth muscle cells
- SNP
Single nucleotide polymorphisms
- SOD2
Superoxide dismutase
- STC-1
Stanniocalcin-1
- TLR-4
Toll-like receptor 4
- TNF-α
Tumor necrosis factor alpha
- TRPV4
Transient Receptor Potential vanilloid 4
- VEGF
Vascular endothelial growth factor
Footnotes
Conflict of Interest Statement.
The authors have no conflicts of interest to declare.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
Annotated References.
** Of special note
* Of note
- [1].Rafikova O, Rafikov R, Kumar S, Sharma S, Aggarwal S, Schneider F, Jonigk D, Black SM, Tofovic SP, Bosentan inhibits oxidative and nitrosative stress and rescues occlusive pulmonary hypertension, Free Radic Biol Med 56 (2013) 28–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Fulton DJR, Li X, Bordan Z, Haigh S, Bentley A, Chen F, Barman SA, Reactive Oxygen and Nitrogen Species in the Development of Pulmonary Hypertension, Antioxidants (Basel) 6(3) (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Montezano AC, Touyz RM, Oxidative stress, Noxs, and hypertension: experimental evidence and clinical controversies, Ann Med 44 Suppl 1 (2012) S2–16. [DOI] [PubMed] [Google Scholar]
- [4].Elko EA, Cunniff B, Seward D, Chia SB, Aboushousha R, van de Wetering C, van der Velden J, Manuel A, Shukla A, Heintz NHP, Anathy V, van der Vliet A, Janssen-Heininger YMW, Peroxiredoxins and beyond; redox systems regulating lung physiology and disease, Antioxid Redox Signal (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Yu B, Meng F, Yang Y, Liu D, Shi K, NOX2 Antisense Attenuates Hypoxia-Induced Oxidative Stress and Apoptosis in Cardiomyocyte, Int J Med Sci 13(8) (2016) 646–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Chen F, Haigh S, Barman S, Fulton DJ, From form to function: the role of Nox4 in the cardiovascular system, Front Physiol 3 (2012) 412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Veit F, Pak O, Egemnazarov B, Roth M, Kosanovic D, Seimetz M, Sommer N, Ghofrani HA, Seeger W, Grimminger F, Brandes RP, Schermuly RT, Weissmann N, Function of NADPH oxidase 1 in pulmonary arterial smooth muscle cells after monocrotaline-induced pulmonary vascular remodeling, Antioxid Redox Signal 19(18) (2013) 2213–31. [DOI] [PubMed] [Google Scholar]
- [8].Barman SA, Chen F, Su Y, Dimitropoulou C, Wang Y, Catravas JD, Han W, Orfi L, Szantai-Kis C, Keri G, Szabadkai I, Barabutis N, Rafikova O, Rafikov R, Black SM, Jonigk D, Giannis A, Asmis R, Stepp DW, Ramesh G, Fulton DJ, NADPH oxidase 4 is expressed in pulmonary artery adventitia and contributes to hypertensive vascular remodeling, Arterioscler Thromb Vasc Biol 34(8) (2014) 1704–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Huetsch JC, Suresh K, Shimoda LA, Regulation of Smooth Muscle Cell Proliferation by NADPH Oxidases in Pulmonary Hypertension, Antioxidants (Basel) 8(3) (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Comhair SA, Erzurum SC, Antioxidant responses to oxidant-mediated lung diseases, Am J Physiol Lung Cell Mol Physiol 283(2) (2002) L246–55. [DOI] [PubMed] [Google Scholar]
- [11].Suresh K, Shimoda LA, Endothelial Cell Reactive Oxygen Species and Ca(2+) Signaling in Pulmonary Hypertension, Adv Exp Med Biol 967 (2017) 299–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Jernigan NL, Naik JS, Weise-Cross L, Detweiler ND, Herbert LM, Yellowhair TR, Resta TC, Contribution of reactive oxygen species to the pathogenesis of pulmonary arterial hypertension, PLoS One 12(6) (2017) e0180455.* This study showed that pulmonary vasoconstriction is increased in a rat model of PAH, and is normalized by antioxidant therapy. Interestingly, antioxidant therapy was unable to rescue vascular remodeling in this model of PAH.
- [13].Siques P, Brito J, Pena E, Reactive Oxygen Species and Pulmonary Vasculature During Hypobaric Hypoxia, Front Physiol 9 (2018) 865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Rosanna DP, Salvatore C, Reactive oxygen species, inflammation, and lung diseases, Curr Pharm Des 18(26) (2012) 3889–900. [DOI] [PubMed] [Google Scholar]
- [15].Incalza MA, D’Oria R, Natalicchio A, Perrini S, Laviola L, Giorgino F, Oxidative stress and reactive oxygen species in endothelial dysfunction associated with cardiovascular and metabolic diseases, Vascul Pharmacol 100 (2018) 1–19. [DOI] [PubMed] [Google Scholar]
- [16].Yang M, Silverstein RL, CD36 signaling in vascular redox stress, Free Radic Biol Med (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Dikalov SI, Dikalova AE, Bikineyeva AT, Schmidt HH, Harrison DG, Griendling KK, Distinct roles of Nox1 and Nox4 in basal and angiotensin II-stimulated superoxide and hydrogen peroxide production, Free Radic Biol Med 45(9) (2008) 1340–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Craige SM, Chen K, Pei Y, Li C, Huang X, Chen C, Shibata R, Sato K, Walsh K, Keaney JF Jr., NADPH oxidase 4 promotes endothelial angiogenesis through endothelial nitric oxide synthase activation, Circulation 124(6) (2011) 731–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Schroder K, Zhang M, Benkhoff S, Mieth A, Pliquett R, Kosowski J, Kruse C, Luedike P, Michaelis UR, Weissmann N, Dimmeler S, Shah AM, Brandes RP, Nox4 is a protective reactive oxygen species generating vascular NADPH oxidase, Circ Res 110(9) (2012) 1217–25. [DOI] [PubMed] [Google Scholar]
- [20].Sedeek M, Hebert RL, Kennedy CR, Burns KD, Touyz RM, Molecular mechanisms of hypertension: role of Nox family NADPH oxidases, Curr Opin Nephrol Hypertens 18(2) (2009) 122–7. [DOI] [PubMed] [Google Scholar]
- [21].Chen K, Kirber MT, Xiao H, Yang Y, Keaney JF Jr., Regulation of ROS signal transduction by NADPH oxidase 4 localization, J Cell Biol 181(7) (2008) 1129–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Yang Q, Wu FR, Wang JN, Gao L, Jiang L, Li HD, Ma Q, Liu XQ, Wei B, Zhou L, Wen J, Ma TT, Li J, Meng XM, Nox4 in renal diseases: An update, Free Radic Biol Med 124 (2018) 466–472. [DOI] [PubMed] [Google Scholar]
- [23].Zhang Y, Shan P, Srivastava A, Jiang G, Zhang X, Lee PJ, An Endothelial Hsp70-TLR4 Axis Limits Nox3 Expression and Protects Against Oxidant Injury in Lungs, Antioxid Redox Signal 24(17) (2016) 991–1012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Dorfmuller P, Chaumais MC, Giannakouli M, Durand-Gasselin I, Raymond N, Fadel E, Mercier O, Charlotte F, Montani D, Simonneau G, Humbert M, Perros F, Increased oxidative stress and severe arterial remodeling induced by permanent high-flow challenge in experimental pulmonary hypertension, Respir Res 12 (2011) 119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Yin C, Li K, Yu Y, Huang H, Wang Z, Yan J, Pu Y, Li Z, Li D, Chen P, Chen F, Genome-wide association study identifies loci and candidate genes for non-idiopathic pulmonary hypertension in Eastern Chinese Han population, BMC Pulm Med 18(1) (2018) 158.* Yin et al. targeted 143 tag SNPs of 14 candidate genes in healthy controls and patients diagnosed with non-idiopathic PH. The results revealed a SNP in NOX3 gene that was associated with PH, suggesting that a genetic alteration of NOX3 may be responsible for individual susceptibility to PH.
- [26].Zhang Y, Shan P, Srivastava A, Li Z, Lee PJ, Endothelial Stanniocalcin 1 Maintains Mitochondrial Bioenergetics and Prevents Oxidant-Induced Lung Injury via Toll-Like Receptor 4, Antioxid Redox Signal (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Li X, Yu Y, Gorshkov B, Haigh S, Bordan Z, Weintraub D, Rudic RD, Chakraborty T, Barman SA, Verin AD, Su Y, Lucas R, Stepp DW, Chen F, Fulton DJR, Hsp70 Suppresses Mitochondrial Reactive Oxygen Species and Preserves Pulmonary Microvascular Barrier Integrity Following Exposure to Bacterial Toxins, Front Immunol 9 (2018) 1309.** Li et al. showed that Hsp70 upregulation suppresses mitochondrial ROS production and disruption of pulmonary endothelial barrier in response to bacterial toxin pneumolysin. These results provide the first evidence that Hsp70 upregulation can be used therapeutically to treat pneumonia-associated lung injury.
- [28].de Jesus DS, DeVallance E, Li Y, Falabella M, Guimaraes D, Shiva S, Kaufman BA, Gladwin MT, Pagano PJ, Nox1/Ref-1-mediated activation of CREB promotes Gremlin1-driven endothelial cell proliferation and migration, Redox Biol 22 (2019) 101138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Zhang Z, Trautz B, Kracun D, Vogel F, Weitnauer M, Hochkogler K, Petry A, Gorlach A, Stabilization of p22phox by Hypoxia Promotes Pulmonary Hypertension, Antioxid Redox Signal 30(1) (2019) 56–73.** This study investigated the relationship between the NADPH oxidase subunit p22phox and development of hypoxia-induced PH. The p22phox subunit was elevated in PH and genetic deletion of p22phox protected against hypoxia-induced PH. The study points to a direct link between p22phox-dependent NADPH oxidases and PH pathogenesis.
- [30].Marks AR, Cellular functions of immunophilins, Physiol Rev 76(3) (1996) 631–49. [DOI] [PubMed] [Google Scholar]
- [31].Jin ZG, Melaragno MG, Liao DF, Yan C, Haendeler J, Suh YA, Lambeth JD, Berk BC, Cyclophilin A is a secreted growth factor induced by oxidative stress, Circ Res 87(9) (2000) 789–96. [DOI] [PubMed] [Google Scholar]
- [32].Xue C, Sowden M, Berk BC, Extracellular Cyclophilin A, Especially Acetylated, Causes Pulmonary Hypertension by Stimulating Endothelial Apoptosis, Redox Stress, and Inflammation, Arterioscler Thromb Vasc Biol 37(6) (2017) 1138–1146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].mengyu h., wen s., min y., xiang l., jian x., jiali z., linling j., weiping x., hui k., Nicorandil attenuates LPS-induced acute lung injury by pulmonary endothelial cell protection via NF-κB and MAPK pathways, Oxidative Medicine and Cellular Longevity, 2019, p. 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Lee JW, Chun W, Kwon OK, Park HA, Lim Y, Lee JH, Kim DY, Kim JH, Lee HK, Ryu HW, Oh SR, Ahn KS, 3,4,5-Trihydroxycinnamic acid attenuates lipopolysaccharide (LPS)-induced acute lung injury via downregulating inflammatory molecules and upregulating HO-1/AMPK activation, Int Immunopharmacol 64 (2018) 123–130. [DOI] [PubMed] [Google Scholar]
- [35].Lin Y, Qiu D, Huang L, Zhang S, Song C, Wang B, Wu J, Chen C, A novel chalcone derivative, L2H17, ameliorates lipopolysaccharide-induced acute lung injury via upregulating HO-1 activity, Int Immunopharmacol 71 (2019) 100–108. [DOI] [PubMed] [Google Scholar]
- [36].Jiang K, Guo S, Yang C, Yang J, Chen Y, Shaukat A, Zhao G, Wu H, Deng G, Barbaloin protects against lipopolysaccharide (LPS)-induced acute lung injury by inhibiting the ROS-mediated PI3K/AKT/NF-kappaB pathway, Int Immunopharmacol 64 (2018) 140–150. [DOI] [PubMed] [Google Scholar]
- [37].Marziano C, Hong K, Cope EL, Kotlikoff MI, Isakson BE, Sonkusare SK, Nitric Oxide-Dependent Feedback Loop Regulates Transient Receptor Potential Vanilloid 4 (TRPV4) Channel Cooperativity and Endothelial Function in Small Pulmonary Arteries, J Am Heart Assoc 6(12) (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Suresh K, Servinsky L, Jiang H, Bigham Z, Yun X, Kliment C, Huetsch J, Damarla M, Shimoda LA, Reactive oxygen species induced Ca(2+) influx via TRPV4 and microvascular endothelial dysfunction in the SU5416/hypoxia model of pulmonary arterial hypertension, Am J Physiol Lung Cell Mol Physiol 314(5) (2018) L893–l907.* Suresh et al. showed that in a rat model of PAH, mitochondrial ROS generation activated Ca2+ influx through TRPV4 channels in lung microvascular ECs. The ROS-TRPV4 Ca2+ signaling resulted in increased EC proliferation and migration.
- [39].Mercado J, Baylie R, Navedo MF, Yuan C, Scott JD, Nelson MT, Brayden JE, Santana LF, Local control of TRPV4 channels by AKAP150-targeted PKC in arterial smooth muscle, J Gen Physiol 143(5) (2014) 559–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Bijli KM, Kang BY, Sutliff RL, Hart CM, Proline-rich tyrosine kinase 2 downregulates peroxisome proliferator-activated receptor gamma to promote hypoxia-induced pulmonary artery smooth muscle cell proliferation, Pulm Circ 6(2) (2016) 202–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Nisbet RE, Bland JM, Kleinhenz DJ, Mitchell PO, Walp ER, Sutliff RL, Hart CM, Rosiglitazone attenuates chronic hypoxia-induced pulmonary hypertension in a mouse model, Am J Respir Cell Mol Biol 42(4) (2010) 482–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Lu X, Bijli KM, Ramirez A, Murphy TC, Kleinhenz J, Hart CM, Hypoxia downregulates PPARgamma via an ERK1/2-NF-kappaB-Nox4-dependent mechanism in human pulmonary artery smooth muscle cells, Free Radic Biol Med 63 (2013) 151–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Blanquicett C, Kang BY, Ritzenthaler JD, Jones DP, Hart CM, Oxidative stress modulates PPAR gamma in vascular endothelial cells, Free Radic Biol Med 48(12) (2010) 1618–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Ban Y, Liu Y, Li Y, Zhang Y, Xiao L, Gu Y, Chen S, Zhao B, Chen C, Wang N, S-nitrosation impairs KLF4 activity and instigates endothelial dysfunction in pulmonary arterial hypertension, Redox Biol 21 (2019) 101099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Suh YA, Arnold RS, Lassegue B, Shi J, Xu X, Sorescu D, Chung AB, Griendling KK, Lambeth JD, Cell transformation by the superoxide-generating oxidase Mox1, Nature 401(6748) (1999) 79–82. [DOI] [PubMed] [Google Scholar]
- [46].Hood KY, Montezano AC, Harvey AP, Nilsen M, MacLean MR, Touyz RM, Nicotinamide Adenine Dinucleotide Phosphate Oxidase-Mediated Redox Signaling and Vascular Remodeling by 16alpha-Hydroxyestrone in Human Pulmonary Artery Cells: Implications in Pulmonary Arterial Hypertension, Hypertension 68(3) (2016) 796–808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Hood KY, Mair KM, Harvey AP, Montezano AC, Touyz RM, MacLean MR, Serotonin Signaling Through the 5-HT1B Receptor and NADPH Oxidase 1 in Pulmonary Arterial Hypertension, Arterioscler Thromb Vasc Biol 37(7) (2017) 1361–1370.** Hood et al. showed that serotonin (5-HT) can increase NOX1-mediated ROS production in pulmonary SMCs in a 5-HT1B receptor-dependent manner. This is the first study to indicate a role for a neurotransmitter in the regulation of pulmonary vascular redox signaling.
- [48].Fulton D, Li X, Bordan Z, Wang Y, Mahboubi K, Rudic RD, Haigh S, Chen F, Barman SA, Galectin-3: A Harbinger of Reactive Oxygen Species, Fibrosis and Inflammation in Pulmonary Arterial Hypertension, Antioxid Redox Signal (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Barman SA, Li X, Haigh S, Kondrikov D, Mahboubi K, Bordan Z, Stepp DW, Zhou J, Wang Y, Weintraub DS, Traber P, Snider W, Jonigk D, Sullivan JC, Crislip GR, Butcher JT, Thompson J, Su Y, Chen F, Fulton DJR, Galectin-3 is Expressed in Vascular Smooth Muscle Cells and Promotes Pulmonary Hypertension through changes in Proliferation, Apoptosis and Fibrosis, Am J Physiol Lung Cell Mol Physiol (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Barman SA, Chen F, Li X, Haigh S, Stepp DW, Kondrikov D, Mahboubi K, Bordan Z, Traber P, Su Y, Fulton DJR, Galectin-3 Promotes Vascular Remodeling and Contributes to Pulmonary Hypertension, Am J Respir Crit Care Med 197(11) (2018) 1488–1492. [DOI] [PMC free article] [PubMed] [Google Scholar]