

Abstract
Small molecules that target microtubules (MTs) represent promising therapeutics to treat certain types of cancer, including glioblastoma multiform (GBM). We synthesized modified carbazoles and evaluated their antitumor activity in GBM cells in culture. Modified carbazoles with an ethyl moiety linked to the nitrogen of the carbazole and a carbonyl moiety linked to distinct biaromatic rings exhibited remarkably different killing activities in human GBM cell lines and patient-derived GBM cells, with IC50 values from 67 to > 10,000 nM. Measures of the activity of modified carbazoles with tubulin and microtubules coupled to molecular docking studies show that these compounds bind to the colchicine site of tubulin in a unique low interaction space that inhibits tubulin assembly. The modified carbazoles reported here represent novel chemical tools to better understand how small molecules disrupt MT functions and kill devastating cancers such as GBM.
Keywords: Microtubules, colchicine, carbazole, gliomas
Graphical Abstract

1. Introduction
Many subtypes of cancers are successfully treated with microtubule-targeting agents (MTAs), including breast, lung and head and neck tumors, lymphoma and melanoma [1]. MTAs act by binding to tubulin or MTs and disrupting MT assembly, disassembly and dynamics, all of which impairs the precise orchestration of mitosis and triggers cell death by engaging the MT spindle check-point [2]. Starting in the 2000s, evidence suggested that GBM, the most common, devastating and incurable type of brain cancer, is particularly sensitive to MTAs [3–6]. The sensitivity of GBM cells likely results from the expression of mutated proteins that erroneously control MT dynamics and their interactions with kinetochores [7, 8]. Accordingly, several studies have shown that GBM cells treated with MTAs undergo death through MT spindle check-point arrest and ensuing apoptosis [4, 5]. Human clinical trials showed that MTA treatment reduces tumor burden in patients diagnosed with GBM [9], and the recent advent of tumor-treating fields for the treatment of patients diagnosed with GBM provide additional evidence that GBMs are sensitive to the disruption of MT function [10]. Specifically, the polarizing field established by tumor-treating fields affects tubulin assembly into MTs, disrupts mitosis and triggers the spindle check-point and ensuing cell death [11]. Together, these genetic, pharmacological and tumor-treating field findings show that GBM cells are particularly sensitive to alterations in MT function and highlights the need to develop novel MTAs for the treatment of this devastating disease.
There are several classes of MTAs that disrupt MT function through distinct mechanisms of action (MOA). Three of the most extensively studied classes of MTAs are vinca alkaloids, taxanes and colchicinoids, which bind to distinct sites on tubulin and differentially affect MT dynamics [1, 12]. Prototypical MTAs that bind to the colchicine sites include combretastatin A-4 (1), the carbazole-based analogues of combretastatin (2) that weakly inhibit tubulin polymerization and yet triggers pronounced apoptosis, and nocodazole (3), which rapidly and fully inhibits tubulin polymerization and triggers apoptosis in cancer cells (Figure 1) [13, 14].
Figure 1. Microtubule targeting agents that bind to the colchicine site and chemical strategy for the synthesis of modified carbazoles.

Combretastatin A-4 (1), carbazole-based analogues of combretastatin (2), nocodazole (3) and scaffold of modified carbazoles (4).
In a previous study, we identified a series of indole-based MTAs that bind to the colchicine site of tubulin, reduce MT polymerization in vitro, reduce MT dynamics in GBM cell lines in culture and kill GBM cells by apoptosis when tested in an orthotopic syngeneic mouse model [14]. Here, we used these results as the starting point for the development of a series of carbazole-based MTAs (4) and studied their antitumor activity in both GBM cell lines and patient-derived GBM cells in culture. We also studied how the antitumor activity of carbazole analogues relate to their ability to disrupt MT. Our studies emphasize the importance of substituting the carbonyl linker with either carbinol or thiocarbonyl functional groups, as well as the importance of substitutions at the nitrogen atom of the carbazole moiety (4). Finally, we provide a modeling study of the docking of the most potent modified carbazoles to the colchicine site and outline its unique binding mode and how it affects MT assembly.
2. Chemistry
The route for the synthesis of the carbazole derivatives is outlined in Scheme 1. Compounds 8, 10, 11, 12, 13, 14, 16, 17, 25 and 31 were prepared from commercially available carbazoles via consecutive substitution with the corresponding n-alkyl bromide under alkaline conditions, followed by a Friedel-Crafts reaction using the corresponding acyl chloride derivatives (Scheme 1). Carbazole 28 was obtained by alkylation of carbazole with 4-methylnaphthalene-1-carbonyl chloride under basic conditions. Bromine-lithium exchange of bromo-carbazole derivative (t-BuLi, −78 °C), followed by the addition of quinolone aldehyde derivatives, furnished the corresponding alcohols, 19 and 21, which were oxidized to form 20 and 22 (Scheme 2).
Scheme 1:

Reagents and conditions: a. Cs2CO3, alkylbromide, DMF, r.t.; b. acyl chloride, AlCl3, benzene 0 °C to r.t.; c. i. POCl3, DMF, μW, 1 h, 100 °C; ii. KMnO4, water/acetone, reflux; d. EDC, DMAP, DIPEA, amine, DMF.
8: R1 = H, R2 = ethyl, R3 = 4-methylnaphthalenyl;
9: R1 = H, R2 = ethyl, R3 = 4-methylpiperazinyl;
10: R1 = H, R2 = ethyl, R3 = 4-chlorophenyl;
11: R1 = H, R2 = ethyl, R3 = 4-fluorophenyl;
12: R1 = H, R2 = ethyl, R3 = p-tolyl;
13: R1 = H, R2 = ethyl, R3 = methylbenzene;
14: R1 = H, R2 = ethyl, R3 = naphthalenyl;
16: R1 = H, R2 = propyl, R3 = 4-methylnaphthalenyl;
17: R1 = H, R2 = 2,2,2-trifluoroethyl, R3 = 4-methylnaphthalenyl
31: R1 = MeO-, R2 = ethyl, R3 = 4-methylnaphthalenyl;
Scheme 2:

Reagents and conditions: a. Cs2CO3, ethylbromide, DMF; b. t-BuLi, THF, −78 to 0 °C; c. PDC, CH2Cl2, Molecular sieves r.t.
Quinoline derivatives 23 and 24 were conveniently prepared after reduction of compounds 20 and 22, respectively.
The thione and the carbinol derivatives (26 and 27) were obtained by heating amide 8 with Lawesson’s reagent under microwave conditions and by reducing its carbonyl with NaBH4, respectively (Scheme 3).
Scheme 3:

Reagents and conditions: a. Lawesson’s reagent, toluene; b. NaBH4, MeOH.
To validate the importance of the planar carbazole ring, we synthesized the carboline derivative 35 (Scheme 4) from phenylhydrazine and ethyl-4-oxocyclohexanecarboxylate.
Scheme 4:

Reagents and conditions: a. HCl, EtOH; b. NaH, alkylbromide, DMF; c. KOH, H2O, EtOH, reflux; d. EDC, DMAP, DIPEA, amine, DMF.
35: R1 = H, R2 = ethyl, R3 = 4-methylnaphthalenyl
3. Results and Discussion
3.1. Antitumor activity of reference MTAs and modified carbazoles in the human GBM cell line T98G.
The two prototypical, colchicine site targeting agents, combretastatin A-4 (1) and nocodazole (3), killed human GBM cells line, T98G, with the expected antitumor EC50 values (0.6 and 29 nM, respectively), but only killed T98G cells by a maximum of 30.4% ± 2.1 (at 10 nM) and 45.6% ± 1.7 (at 10 μM), respectively (Figure 2A). Supplementary Figure 1 shows that antitumor activity of 3 additional references MTAs also only partially killed T98G cells: EC50 and maximal killing values for colcemide were 113 nM and 35.9% ± 6.4 at 10 μM, for vinblastine were 184 nM and 36.0% ± 2.3 and for paclitaxel were 148 μM and 63.2% ± 2.5 at 30 μM. These results show that reference MTAs that both inhibit assembly and destabilize MTs exhibit moderate antitumor activities in T98G cells.
Figure 2: Antitumor activity of reference MTAs that target the colchicine site, as well as select modified-carbazoles, in T98G GBM cells in culture.

T98G cells in culture were treated with increasing concentrations of A) two MTAs acting through the colchicine site of tubulin, combretastatin A-4 (1) and nocodazole (3), B) compounds 8, 20 and 27, and C) compounds 23 and 25. Cell viability was measured 72 h following treatment using WST-1. Dotted line shows 100% vehicle control. Data are the mean ± SEM of at least three independent experiments performed in triplicate.
We then treated T98G cells with the 19 modified-carbazoles synthesized for this study and report the EC50 values and maximal killing activity values of their antitumor activities in Tables 1 and 2.
Table 1:
Antitumor activity of 18 modified carbazoles in T98G cells in culture. Cells were treated with increasing concentrations of the compounds and cell viability was measured 72 h following initiation of treatment using WST-1. EC50 values were calculated using Prism® software from curves shown in Figure 1 and Supplementary Figure 2. When the curves could not be extrapolated, the EC50 could not be calculated and values are indicated as > 10 μM. Unless otherwise noted, maximal killing activities were measured at 10 μM. Data are the mean of at least three independent experiments performed in triplicate.
 | ||||||
|---|---|---|---|---|---|---|
| Compound | R1 | R2 | R3 | Antitumor activity | ||
| EC50 | Maximal killing (% ± SEM) | |||||
| 8 | H | Ethyl |  |
87 nM | 45.3 ± 4.1 | |
| 9 | H | Ethyl |  |
> 10 μM | 13.2 ± 1.7 | |
| 10 | H | Ethyl |  |
1.5 μM | 71.1 ± 5.3 | |
| 11 | H | Ethyl | 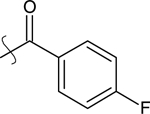 |
8 μM | 54.8 ± 4.0 | |
| 12 | H | Ethyl |  |
993 nM | 83.6 ± 2.2 (at 3 ^M) | |
| 13 | H | Ethyl |  |
> 10 μM | 14.0 ± 2.1 | |
| 14 | H | Ethyl |  |
1.7 μM | 72.0 ± 5.7 | |
| 16 | H | Propyl |  |
> 10 μM | 16.3 ± 2.2 | |
| 18 | H |  |
 |
5.6 μM | 35.6 ± 9.4 | |
| 20 | H | Ethyl |  |
184 nM | 74.4 ± 1.7 (at 1 μM) | |
| 22 | H | Ethyl |  |
8.8 μM | 60.0 ± 4.9 | |
| 23 | H | Ethyl |  |
> 10 μM | −17.0 ± 10.8 (at 3 μM) | |
| 24 | H | Ethyl |  |
> 10 μM | −18.0 ± 5.2 (at 100 nM) | |
| 25 | H | Ethyl |  |
757 nM | 89.0 ± 0.7 (at 1 μM) | |
| 26 | H | Ethyl | 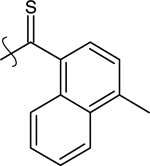 |
> 10 μM | −17.0 ± 10.8 (at 3 μM) | |
| 27 | H | Ethyl |  |
560 nM | 72.8 ± 7.6 (at 3 μM) | |
| 28 | H |  |
H | 957 nM | 89.1 ± 0.7 | |
| 31 | MeO- | Ethyl | 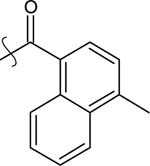 |
2.6 μM | 71.6 ± 6.1 | |
Table 2:
Antitumor activity of modified pyridoindole 35 in T98G cells in culture. Cells were treated with increasing concentrations of compound 35 and cell viability was measured 72 h following treatment initiation using WST-1. Since with 35 a curve of the concentration-dependent antitumor activity could not be extrapolated, the EC50 is indicated as > 10 μM. Data are the mean of at least 3 independent experiments performed in triplicate.
 | |||||
|---|---|---|---|---|---|
| Compound | Ri | R2 | R3 | Antitumor activity | |
| EC50 | Maximal killing (mean ± SEM) | ||||
| 35 | H | Ethyl |  |
> 10 μM | 45.1 ± 9.9 (at 3 μM) |
Figure 2B and 2C show the antitumor activities of five modified carbazoles (8, 20, 23, 25 and 27) that hold key SAR characteristics. The antitumor activities for the remaining compounds are in Supplementary Figure 2. Thus, a comparison of the chemical structures of the modified carbazoles and their antitumor activities expressed as EC50 values (potency) and maximal killing activities (efficacy) allows for multiple conclusions around the SAR of this response.
First, we found that fourteen modified carbazoles that all have an ethyl moiety linked to the nitrogen of the carbazole and a carbonyl moiety linked to distinct biaromatic rings exhibited remarkably different EC50s. Specifically, five such compounds killed T98G cells with EC50 values below 1 μM [8 (87 nM) < 20 (184 nM) < 27 (560 nM) < 25 (757 nM) < 12 (993 nM)], two compounds killed T98G cells with EC50s between 1 and 2 μM [10 (1.5 μM) and 14 (1.7 μM)] and seven compounds were only weakly active at concentrations above five micromolar or essentially inactive [9, 13, 16, 18, 23, 24 and 26]. Note that the replacement of the methylnaphthyl group present in the most potent compound 8 (87 nM) by either toluyl (as in 12, 993 nM) or a non-substituted naphthyl moiety (as in 14, 1.7 μM) or a benzyl moiety (as in 13, inactive), increased EC50s values by 11-fold, 20-fold and more than 115-fold, respectively.
Second, modified carbazoles and carboline exhibited remarkably different maximal killing activities. Specifically, nine compounds killed T98G cells by more than 50% (i.e. more than the maximal killing activities reached by reference MTAs). Thus, the rank order of these compounds is: 25 (89%) > 12 (84%) > 20 (74%) > 27 (73%) > 14 (72%) = 31 (72%) > 10 (71%) > 22 (60%) > 11 (55%). Three compounds killed T98G cells by 35–45% (8, 18 and 35) and six compounds did not significantly affect T98G cell viability (9, 13, 16, 23, 24 and 26). Note that the position of the nitrogen atom in the quinoline portion of modified carbazoles controls their maximal killing activities, as exemplified by the reduction in maximal killing activity when moving the nitrogen atom from position 1 (as in 25, 89% killing) to either position 5 (as in 20, 60% killing) or position 8 (as in 22, 60% killing).
Third, introduction of a nitrogen atom in position 6 of the carbazole scaffold increases the EC50 value by more than 115-fold as exemplified when comparing the response of 8 (87 nM) and the carboline analogue 35 (> 10 μM). A possible explanation for this change in EC50 value is that both the carbazole moiety and the aromatic moiety attached to the carbonyl will impact the positioning of the lipophilic moiety attached to the carbonyl. Thus, this result suggest a need for modified carbazoles to be planar for optimal activity [15]. Importantly, the nitrogen atom in position 6 of the carbazole scaffold only affect the EC50 values of 8 and 35, as the maximal killing reached by both compounds was 45%, indicating that a nitrogen atom in position 6 of the carbazole scaffold will control the EC50 value of the compound independently of the maximal killing activity.
Fourth, the carbonyl linker determined the compound’s EC50 values as evidenced by comparing 8 (87 nM) with both the carbinol 27 (560 nM) and the thiocarbonyl compound 26 (> 10 μM), which results in 6- and 115- fold increase in EC50 values, respectively. This result can be explained by a loss of a hydrogen bond interaction since thiocarbonyl derivatives are known to destabilize hydrogen bond formation through higher steric demands that are imposed by both the larger sulfur atom and the lower electronegativity compared with the oxygen atom [15]. Remarkably, changes in the carbonyl linker had an opposite effect on the maximal killing activity of these compound, as the carbinol 27 killed more T98G cells than the carbonyl 8 (72% versus 45%, respectively).
Fifth, the importance of the chain born from the endocyclic nitrogen is evident by comparing the EC50 values of the ethyl compound 8 (87 nM) with both the trifluoroethyl compound 18 (5.6 μM) and the propyl compound 16 (> 10 μM), corresponding to 64- and 115-fold increases in the EC50s values, respectively. Remarkably, the chain linked to the endocyclic nitrogen has less impact on the maximal killing activity: 8 (45%) and 18 (36%). This result suggests a critical interaction site between the chain borne by the endocyclic nitrogen of modified carbazoles and the target ((i.e. colchicine site of tubulin) that controls antitumor activity.
Sixth, a small methoxy R1 substituent in position 7 of the carbazole moiety affects the EC50 value by 30-fold (as exemplified by 8 (87 nM) and 31 (2.6 μM)), suggesting an essential interaction site between the R1 substituent in position 7 of the carbazole and the target. Remarkably, the R1 substituent in position 7 of the carbazole moiety had an opposite effect on their maximal killing activities: 31 (72%) killed more T98G cells compared with 8 (45%). Note that the positioning of the methylnaphthyl moiety has the inverse impact on the EC50 values (8 (87 nM) and 28 (957 nM)) and the maximal killing activities (8 (45%) and 28 (89%)). Together, these results show that the antitumor EC50 values (potency) and maximal killing activities (efficacy) of modified carbazoles can be mechanistically separated by specific chemical substitutions.
In conclusion, the SAR analysis of the antitumor activities of novel modified carbazoles measured in T98G cells identified chemical features that independently control their antitumor EC50s values and maximal killing activities, including a carbonyl moiety that links the carbazole to aromatic moieties, the ethyl moiety linked to the nitrogen atom of the carbazole and select chemical modifications in the second aromatic moiety.
3.2. Killing activity of select modified carbazoles in patient-derived GBM cells and the human liver cell line HepG2
To explore the therapeutic potential of the modified carbazoles, we measured their antitumor activity in human patient-derived GBM (PD-GBM) cells in culture. Specifically, these cells represent a recognized preclinical model system for testing for the efficacy of novel treatments of GBM as they are categorized in three subtypes based on their mutation profile (i.e., proneural, mesenchymal and classical) that differentially respond to treatment [16–20]. To test for potential liver toxicity, we treated the human liver cell line HepG2, a commonly used cell culture model system [21–24]. Therefore, we treated the PD-GBM cells in culture with the four most potent modified carbazoles (8, 20, 27 and 25) from our initial screen and found that they kill the PD-GBM cells with EC50 values and maximal efficacy values that both mirrored their activity on T98G cells and remained within similar values irrespective of the PD-GBM subtype (Table 3). Importantly, the four compounds exhibited lower cell-killing activity in HepG2 cells, reaching only 8–41% maximal killing activities as compared with 44–99% maximal killing activities reached in PD-GBM cells (Table 3).
Table 3:
Cell-killing activity of selected modified carbazoles in patient-derived GBM and liver HepG2 cells. PD-GBM and HepG2 cells in culture were treated with increasing concentrations of compound. Cell viability was measured using WST-1 72 h after treatment. EC50 values were calculated using Prism® software using the curves shown in Supplementary Figure 3. Maximal killing activity was measured at 5.6 μM. Data are the mean of at least three independent experiments performed in triplicate. Not converged (NC).
| Compounds | Cell Killing Activity | |||||||
|---|---|---|---|---|---|---|---|---|
| ECsg | Maximal Killing | ECsg | Maximal Killing | ECsg | Maximal Killing | ECsg | Maximal Killing | |
| (nM) | (mean ± sem) | (nM) | (mean ± sem) | (nM) | (mean ± sem) | (nM) | (mean ± sem) | |
| 8 | 542 | 53.3% ± 10.9 (at 5.6 μM) | 698 | 47.9% ± 6.5 (at 3 μM) | 268 | 64.3% ± 1.3 (at 1 μM) | 1689 | 25.0% ± 3.8 (at 5.6 μM) |
| 20 | 316 | 44.8% ± 10.5 (at 1 μM) | 407 | 53.9% ± 4.2 (at 1 μM) | 335 | 58.9% ± 1.4 (at 1 μM) | 66 | 28.6% ± 13.9 (at 0.6 μM) |
| 25 | 613 | 98.9% ± 0.1 (at 5.6 μM) | 846 | 98.6% ± 0.9 (at 5.6 μM) | 1823 | 97.2% ± 0.8 (at 10 μM) | NC | 8.3% ± 8.5 (at 5.6 μM) |
| 27 | 630 | 44.0% ± 14.2 (at 1 μM) | 633 | 56.0% ± 3.8 (at 3 μM) | 386 | 50.6% ± 6.2 (at 3 μM) | 101 | 30.8% ± 13.3 |
| (at 10 μM) | ||||||||
The preferential sensitivity of PD-GBM cells to the killing activity of modified carbazoles compared with HepG2 cells suggests a promising therapeutic index (i.e., the ratio of the amount of agent that causes the therapeutic effect to the amount that causes toxicity). One compound, 25, demonstrated a clear-cut result by killing 95–100% of the PD-GBM cells when applied at 1–10 μM while remaining inactive at these concentrations in HepG2 cells (Table 3 and Supplementary Figure 3). Another advantage of the modified carbazoles developed here is that they kill all PD-GBM subtypes independent of their genetic make-up. This addresses one of the most thorny and extensively highlighted issues concerning the selectivity of targeting GBM tumors, which are known to rapidly become heterogeneous in their genetic make-up, often encompassing all subtypes (proneural, mesenchymal and classical) [25, 26]. Therefore, the equally potent and efficacious killing activities of modified carbazole measured in PD-GBM subtypes shows their potential to kill the majority of GBM cells forming the heterogeneous tumor mass.
3.3. Cell-killing activities and MOA of modified carbazoles in the NCI-60 cancer cell line panel
We worked with The Developmental Therapeutics Program’s NCI 60 cell screen to study the MOA by which the three most potent modified carbazoles (8, 20 and 27) kill cancer cell lines and included the inactive analogue 23 as negative control. This screening platform measures the antitumor activities of compounds expressed as percent growth inhibition, which differentiates the compound’s activity at inhibiting cell proliferation and reducing cell number (i.e. killing cells) [27]. As expected, the inactive compound 23 did not significantly affect the viability and proliferation of any of the cell lines (growth inhibition: Mean 102.05; Delta 10.42, and Range 28.77) (Supplementary Figure 4). By contrast, the three active compounds 8, 20 and 27 killed 4 cell lines (13% to 60% loss in cell number), halted the proliferation of 11 cell lines (from 9% to −1% growth inhibition) and reduced the proliferation rate of 41 cell lines (from 67% to 10% percent growth inhibition) (Supplementary Figure 4). Of note, two GBM cells lines that are included in the NCI-60 panel, SF-295 and SF-539, were among the top ten cell lines that were the most susceptible to 8, 20 and 27 treatments (Figure 3 and Supplementary Table 1). The respective EC50s of the compounds measured in SF-295 and SF-530 cells mirrored their EC50s measured in T98G cells. Additional cell lines among the top ten cell lines that were sensitive to these three compounds were three cell lines of colon cancer origin: Colo205, HT-29 and HCC-2998 (Supplementary Figure 5).
Figure 3: Cell-killing activity of select modified-carbazoles in SF-539 and SF-295 GBM cells in culture.

A) SF-539 cells in culture and B) SF-295 cells in culture were treated with increasing concentrations of compounds 8, 20 and 27. Cell viability was measured 48 h following treatment using Alamar blue and is expressed as optical density (O.D.). Data are gathered at seeding and after 48 h, providing an index of the inhibition of cell proliferation (between both dotted lines) and triggering of cell death (below Seed dotted line). Parameters of cell-killing activity (GI50, Total Growth Inhibition [TGI] and LC50) are in Supplementary Table 1.
The COMPARE and CellMiner web applications allow for both the statistical analysis of the antitumor activities of drugs tested in the NCI-60 cell line panel and the identification of other compounds that similarly impact the proliferation and viability of NCI 60 cells [27, 28]. Using these applications, we found three MTAs (paclitaxel, thalicarpine and maytansine) among the top eight compounds that exhibit antitumor activities in the NCI-60 cell line panel that correlate with the antitumor activities of 8, 20 and 27 (Supplementary Table 2). While this result suggests that 8, 20 and 27 kill cancer cells by disrupting MTs, the respective correlation parameters were relatively low, pointing to a somewhat different MAO as compared to that of paclitaxel, thalicarpine and maytansine, and all other MTAs tested on NCI 60 cells. Based on these results, it was reasonable to expect that the modified carbazoles would interact with tubulin or MTs through a different MOA.
3.4. Activity of modified carbazoles with tubulin and microtubules
First, we determined whether modified carbazoles compete for [3H]colchicine binding to native purified tubulin as previously described [29]. In this assay, the reference MTAs 1 (5 μM) and 3 (5 μM) competed for binding by 98% and 74%, respectively (Table 4).
Table 4:
Activity of selected modified carbazole with tubulin and microtubules. Colchicine binding to tubulin and tubulin assembly (as assessed by turbidity development of tubulin solutions) were inhibited in a dose dependent manner by combretastatin A-4 (1) and nocodazole (3) (positive controls) and by the selected modified carbazoles. Results are the mean ± standard deviation (SD) of at least three independent experiments. MT and free tubulin partitioning was shifted toward free tubulin by all compounds indicating MT destabilization. EC50 values are the results from representative experiments that were repeated three times with comparable results. Not determined (ND).
| Compounds | Colchicine Binding | Tubulin Assembly | MT-Tubulin Partitioning | |
|---|---|---|---|---|
| % Inhibition | IC50 (μM) | EC50 (μM) | ||
| mean ± SD | mean ± SD | mean | ||
| at 5 μM | at 1 μM | |||
| 1 | 98 ± 0.1 | 89 ± 3 | 0.64 ± 0.01 | 0.6 |
| 3 | 74 ± 0.8 | 23 ± 0.8 | 0.48 ± 0.01 | 2.4 |
| 23 | 4 ± 1 | ND | > 20 | > 20 |
| 25 | 41 ± 4 | ND | 4.4 ± 0.4 | 5.3 |
| 8 | 53 ± 3 | ND | 1.4 ± 0.2 | 2.1 |
| 27 | 70 ± 1 | ND | 1.6 ± 0.01 | 6.4 |
| 20 | 91 ± 1 | 61 ± 4 | 0.89 ± 0.04 | 3.4 |
Under these experimental conditions, modified carbazoles competed for colchicine binding with increasing affinity (23 << 25 < 8 < 27 < 20) that mirrored their respective EC50s at killing GBM cells. We next tested their ability to inhibit the assembly of purified tubulin using an in vitro turbidity assay as previously described [29]. In this assay, where tubulin is present at 10 μM in 0.8 M glutamate with 0.4 mM GTP and the extent of assembly is measured after 20 min at 30 °C, 1 and 3 have IC50s of 0.6 μM and 0.5 μM, respectively (Table 4). Under these experimental conditions, 8, 20 and 27 exhibited IC50s of 1.4, 0.9 and 1.6 μM, respectively, and 23 was inactive at concentrations up to 20 μM (Table 4). To build on these results, we measured the ability of modified carbazoles to influence the partitioning of MT dimers and polymers, an index of MT assembly and disassembly as previously described [14]. In this assay, compounds are incubated with sheared MTs at 37°C and the resulting amounts of tubulin dimers (free tubulin) and polymer (MT) after 15 min is measured on polyacrylamide gels [30]. Under these experimental conditions, 1 and 3 have IC50s of 0.6 and 2.4 μM, respectively; 8, 20 and 27 have IC50s of 2.1, 3.4 and 6.6 μM, respectively; and 23 was inactive when tested up to 20 μM (Table 4). Thus, the IC50s values of these modified carbazoles to bind to the colchicine site on tubulin parallels their EC50s to kill GBM cells, whereas their IC50s to trigger MT disassembly does not correlate as well and remained in the micromolar range as measured in two in vitro assays.
The analysis of the SARs of modified carbazoles to bind to the colchicine site, disrupt MT assembly and favor disassembly provide initial mechanistic insights on how their binding to the colchicine site in tubulin might favor conformational changes in this protein that might affect MT dynamics. The potency of modified carbazole derivatives are in the micromolar range when affecting MT assembly and disassembly. A possible explanation is that the affinity of modified carbazoles for the colchicine site depends on specific interactions with a subset of amino acid within the binding pocket, whereas the conformational changes in tubulin that are stabilized by the modified carbazoles are only reached at high concentrations. Thus, the modified carbazoles that we developed here may represent new chemical tools to study how binding to the colchicine site might affect tubulin conformation and how this affects MT end dynamics.
3.5. Gas Phase Pharmacophore Study
To understand the structural basis of the MT-disrupting and antitumor activities of the modified carbazole series, we initiated a gas phase study to determine common pharmacophoric features with sterically-relevant colchicine site agent. The structure of 20, the most potent of the series to bind to the colchicine site, was used to compare complementary features and volumetric space filling models of ligand co-crystal structures that currently exist in the Protein Data Bank (PDB) for the colchicine site on tubulin. Two MTAs that target the colchicine site, colchicine and podophyllotoxin, were found to have the most steric, hydrophilic and hydrophobic spatial similarity with the carbazole analogues. Using methodology employed previously [31, 32], the X-ray based configurations and conformations of the two chiral molecules (colchicine and podophyllotoxin, taken from the X-ray co-crystals 402B [31], and 1SA1 [33], respectively) were employed as templates for the translational and conformational overlap modeling of 20. Figure 4 displays the results of the gas phase pharmacophore overlap study for colchicine, podophyllotoxin and 20.
Figure 4: Gas phase pharmacophore overlap model of colchicine, podophyllotoxin, and 20.

Panel A illustrates a three-dimensional stick rendering of the gas phase pharmacophoric overlap model for colchicine (carbons in magenta), podophyllotoxin (carbons in cyan), and 20 (carbons in yellow). For purposes of visual clarity, oxygen atoms are colored red in podophyllotoxin and deeper red for both colchicine and 20. In all three structures, nitrogen atoms are colored blue. The red circles correspond to the two 20 polar hydrogen bond acceptor features in common with colchicine or podophyllotoxin. Panels B, C and D are two-dimensional illustrations of the pharmacophore overlap model depicted in Panel A. Panel B illustrates colchicine. Panel C illustrates podophyllotoxin and Panel D illustrates 20. In Panels B-D, the bolded blue atoms correspond to the steric overlaps of the colchicine and podophyllotoxin atoms that are in common with 20; otherwise, atoms in Panels B-D are colored magenta for colchicine, cyan for podophyllotoxin and yellow for 20. As with Panel A, the red circles in Panels B-D correspond to polar hydrogen bond acceptor features of compound 20 that are in common with colchicine or podophyllotoxin.
The results from the gas-phase pharmacophore overlap study identified the common steric features between 20, podophyllotoxin and colchicine. The atoms of 20 are almost completely subsumed within the steric space of podophyllotoxin and approximately 80% of the steric space of colchicine. The common steric overlap between these three molecules is largely composed of hydrophobic atoms. However, the two key polar atoms of 20 are the basic quinoline N and its ketone O. The quinoline N atom of 20 shows remarkable spatial consistency with the central methoxy O’s of the trimethoxylated-aryl moieties of podophyllotoxin and colchicine (Figure 4), whereas the ketone O of 20 is isosteric with the lactone carbonyl of podophyllotoxin. Unique to the 20 scaffold is the relatively strong hydrogen bond acceptor quinoline N in spatial proximity to the comparatively much weaker hydrogen bond accepting ether O’s of podophyllotoxin and colchicine. Furthermore, the 10-membered aromatic quinoline ring system of 20 offers unique topology to satisfy the binding requirements of the subsite normally occupied by the trimethoxylated-aryl systems of podophyllotoxins and colchicine. An additional unique feature of the 20 scaffold is the ethyl moiety attached to the carbazole N (Figure 4). The consistent polar, hydrophobic and steric overlap identified by the gas phase pharmacophore study provides a template to conduct molecular docking studies to determine a detailed, all-atom rationalization of the modified carbazole series’ structure activity relationships in the colchicine site.
3.5. Molecular Docking Studies
Given the close steric congruency with podophyllotoxin, we employed the alpha and beta tubulin subunits of the recently solved 2.30 Å co-crystal of the podophyllotoxin analogue: 4β-(1,2,4-triazol-3-ylthio)-4- deoxypodophyllotoxin (PDB entry code =5JCB) [34] as the initial geometry to conduct the molecular docking studies of the modified carbazole series. Maestro [34] protein preparation tools (utilizing the OPLS force field) were utilized for X-ray co-crystal comparisons and to prepare the structure for molecular mechanics energy refinement simulations. The 5JCB coordinates were optimized in a stepwise fashion (hydrogens first, followed by side chain and backbone) resulting in an all-atom energy-refined structure used as the starting structure for the carbazole molecular docking studies. Superimposition of the 5JCB X-ray coordinates onto the resulting energy refined structure revealed only a 0.98 Å RMS deviation between the 13,646 heavy atoms. Of the 6880 backbone atoms compared, only a 0.74 Å deviation was observed. These data signify that very close geometries were obtained between the X-ray and energy refined structures that are well within the expected thermodynamic variation and resolution of the 2.3 Å 5JCB X-ray structure.
Carbazole structures were built and initially optimized with the MM2 force field and transferred into the cff91/cvff force fields for potential assignment [35]. For molecular docking studies, we used methodology employed previously with added refinements [36]. Briefly, the carbazoles were docked into the energy refined 5JCB X-ray co-crystal structure to examine all translation and rotational steric complementarity between the most potent ligand of the carbazole series, 20, its conformational isomers, and the side chains of the energy-refined podophyllotoxin binding site. The resulting all-atom model was derived from iterative constrained optimizations to refine a maximum complementarity between the atoms of 20 and its contacts with the amino acid side chains of the colchicine site. Figure 5 shows the optimally docked conformation of 20 and its overlap with the triazole-podophyllotoxin analogue in the model.
Figure 5: Comparison of modeling and X-ray poses.

Panel A displays the view of the optimized 20 (carbons in yellow) in the colchicine site from the molecular docking studies In Panel B, the pose of the podophyllotoxin-triazole analog (carbons in cyan) from the X-ray 5JCB is shown with 20 overlapped from molecular docking studies. In addition to the high degree of steric congruency with the podophyllotoxin-triazole analog, the ketone oxygens of both 20 and podophyllotoxin-triazole are isosteric. Unique to t 20, compared to podophyllotoxin and colchicine, is the relatively strong hydrogen bond acceptor quinoline N located in the trimethoxy-aryl subsite (near β-Cys-241). 20 can form a direct hydrogen bond (blue dashed lines in both panels) with the side chain S-H of β-Cys-241. The quinoline N of 20 can form a stronger hydrogen bond with a water bridge to polar backbone atoms compared to the ether O’s of the central methoxy groups of podophyllotoxin and colchicine (also see Supplementary Figure 6).
An overlap that includes colchicine can be found in Supplementary Figures 6 and 7. The optimally docked model of 20 served as a template for molecular docking studies with the rest of the carbazole series to rationalize the protein-atom ligand contacts’ SARs. Constrained optimizations were performed in an iterative fashion to eliminate bad contacts and biochemically unfeasible hydrophobic-polar interactions as identified by the HINT program [37]. When available, the in vitro binding data were prioritized to rank-order the carbazoles; otherwise the percent inhibition of the rank-ordered congeneric series served as the biological activity data for the SAR. A total of twenty structures (including the R and S enantiomers of 27) provided a structure-based explanation that rationalized the SAR of the carbazole series.
The modified carbazoles described herein exemplify a new chemotype for tubulin depolymerizers that interact at the colchicine site. Our modeling studies indicate that the 20 scaffold is almost entirely subsumed by the steric space of podophyllotoxin and approximately 80% within the steric space of colchicine. Potent carbazoles bind more efficiently (requiring fewer atoms) in the same steric space than podophyllotoxin because of the conformational restriction imposed by the 13-membered (carbazole) and 10-membered (quinoline/napthalene) rings systems when both are bridged by a methanone. The methanone carbonyl, which functions as a strong hydrogen bond acceptor, is important for potent activity in the carbazole series, as evidenced by a substantial loss of activity when the carbonyl is replaced by a thiocarbonyl (26). Thus, 26 forms a weaker hydrogen bond r and imposes the bulkier S atom in the sterically restricted space where the lactone carbonyl O of podophyllotoxins and carbazole Os overlap.
Although most of the atoms that are sterically congruent with podophyllotoxin and colchicine are hydrophobic, 20 introduces several unique structural elements to its colchicine site interactions. 20 poses a 10-membered quinoline aromatic ring system in the colchicine subsite normally occupied by the colchicine/podophyllotoxin trimethoxy-aryl system. With respect to enthalpic interactions, two hydrogen bond acceptors: the methanone O, and the quinoline N account for most of the potency of 20. Specifically, the methanone O can hydrogen bond with the backbone N-H of β-Asp-251, and the quinoline N located at the 5 position enables the formation of a strong can hydrogen bond with the S-H of β-Cys-241) (Supplementary Figure 6). Perhaps this occurs as the tubulin dimers polymerize and could therefore play a significant role in the prevention of MT assembly. By contrast, 22 possesses the quinoline N in the 8 position which greatly reduces activity due to its inability to form the hydrogen bonds characteristic of 20, podophyllotoxin, and colchicine (Supplementary Figure 7).
Our modeling studies indicate that the 8-quinoline N enables the formation a water-mediated intramolecular hydrogen bond that stabilizes a sterically-dissimilar conformation from both 20 and podophyllotoxin, which potentially also contributes to a loss of activity. Removal of the N atom altogether (as in in 8) to form a naphthalene, also reduces activity due to the loss of the hydrogen bonding N altogether. Our docking studies further indicate that the 4-methyl group of 8 forces the naphthalene deeper into the trimethoxy-aryl subsite to avoid unfavorable hydrophobic-polar contacts with backbone polar atoms. This then places the methanone O slightly further from the ketone O in 20’s optimal orientation. The net effect is that 8’s methanone O is less conformationally available to function as a strong hydrogen bond acceptor. This is also supported by 8’s close congener 14, which only differs structurally by the lack of the methyl at the napthalene 4 position, and 14 is markedly less active due to loss of a hydrogen bond acceptor. Conversely, adding a hydrophobic bulk to the carbazole ring system reduces activity, as exemplified by the 7-methoxy substituted carbazole of 31 where the methyl portion of its methoxy forms unfavorable hydrophobic-polar contacts with the backbone N-H atoms of b-Lys-352, b-Val-351–349 and b-Asn-350 (Supplementary Figure 8, Panel A).
A second unique structural feature of the most potent analogue 20 is the ethyl group attached to the carbazole. Both conformational analysis and docking studies indicate that this ethyl group helps stabilize the scaffold’s conformation through steric hindrance during binding. An additional attribute of the ethyl group is that it forms favorable hydrophobic contacts with the aliphatic component of the b-Lys-352 side chain (Supplementary Figure 8, Panel A). Accordingly, the less active compound 18, in which the hydrophobic distal carbon of the ethyl is replaced with an isosteric polar trifluoromethyl group, is closer to the hydrocarbon component of β-Lys-352 side chain (Supplementary Figure 8, Panel B). Moreover, replacement of this ethyl group with a bulkier hydrophobic propyl group (as with 16) reduces activity because of unfavorable hydrophobic-polar liabilities with the cationic NH3 of the β-Lys-352 side chain (Supplementary Figure 8, Panel B).
Compound 25 features a 10-membered system linked to the methanone by a N atom. Since it is a non-planar dihydroquinoline derivative, it also lacks ring aromaticity in this part of the molecule. There are several possible explanations for the reduced activity of 25. First, it could be due to the formation of a more rigid amide-like linkage that produces a less optimal orientation of the hydrogen bond accepting O due to the rigidity of the amide. Second, it could be due to the bulkier unsaturated ring that induces a twisted binding mode that potentially introduces an unfavorable hydrophobic-polar interaction liability near the side chain group of β-Ala-250. Third, it could be due to a loss of a hydrogen bond with β-Cys-241 (Supplementary Figure 9).
Several conclusions can be drawn from several less active compounds. For example, a feature to highlight is how removal of one the aromatic rings from the naphthalene/quinoline systems of the carbazoles reduces activity. Specifically, despite its p-tolyl methyl group, the single ring of 12 sterically occupies almost half of the trimethoxy-aryl subsite compared with colchicine, podophyllotoxins and 20 and yet it is less potent. An implication for this finding could be that binding to the colchicine site has a minimum steric requirement of occupation at the trimethoxy-aryl subsite to incur significant activity. However, replacement of the 10-membered ring with a 6-membered ring (as in 12 and 10) also increases the number of conformational isomers, which reduces conformational access compared to the more active carbazoles possessing double aromatic ring systems. Another example is 13, which also loses activity by reducing conformational access while increasing conformation isomerism as it extends the single ring linkage to the methanone by a methylene group. Thus, replacement of the p-tolyl methyl of 12 with a hydrophobic and isosteric Cl atom (as in 10) reduces activity by introducing similar disadvantages. The poor activity of 9 is due to the presence of a positive charge introduced by the likely protonated piperazinyl single ring system, reinforcing the finding that the trimethoxy-aryl subsite favors hydrophobic occupation with carefully positioned hydrogen bond acceptors. Accordingly, both 23 and 24 contain hydrogen bond donating N-Hs that are potentially charged.
Finally, our molecular docking studies indicate that the structural configuration of 28 requires a unique binding mode to form a biochemically-feasible complementarity with the colchicine site that accounts for its reduced biological activity compared to 20 (Supplementary Figure 10, Panel A). Specifically, the 4-tolyl-naphthalene methanone of 28 is attached directly to the carbazole N, which causes the carbazole moiety of 28 to bind less congruently with the dioxolane component of podophyllotoxin’s tetracycle. Instead, one of the carbazole’s aromatic rings finds steric overlap with the hydrophobic portion of the 5-membered lactone ring of the podophyllotoxin analog. Compared to the other carbazoles, 28 forms unique, favorable hydrophobic contacts with the side chain of β-Leu-248. Interestingly, the methanone O of 28 is still positioned close to both the other methanone O’s of the more potent modified carbazole and the lactone carbonyl O’s of podophyllotoxin (Supplemental Figure 10, Panel B). The significance of this novel binding mode is that 28 can be modified to include key features from the optimal carbazole 20, while retaining its novel interactions with the colchicine site.
Together, the structure-activity relationships of these carbazoles signify that there are key features for them to retain optimal activity in the colchicine site: 1) steric occupation of a moiety of similar size to a naphthyl, quinolinyl or trimethoxy-aryl system with preferably a hydrogen bond interacting with the β-Cys-241 sulfhydral, 2) lack of cationic ionizability for moieties that occupy the trimethoxy-aryl subsite, 3) reduction of conformational isomerism by either intramolecular steric hindrance or inherent rigidity, and 4) a strong hydrogen bond acceptor with an overlapping pose of the methanone O of 20 and the lactone carbonyl O of podophyllotoxins. Thus, the most potent 20 appears to share similar binding features with both podophyllotoxins and colchicine but provides better atom economy to achieve a comparable potency. In 20, the larger quinoline aromatic system fills the trimethoxy-aryl subsite in a sterically more compact manner than colchicine and podophyllotoxin. Moreover, the quinoline N serves as a strong hydrogen bond acceptor capable of forming diverse hydrogen bonding interactions either directly with the side chain of β-Cys-241 (Supplementary Figure 6) or via water bridges (models not shown). We have shown that the short N-ethyl substituent on the carbazole part of 20 is involved in key hydrophobic interactions with the side chain of β-Lys-352 (Supplementary Figure 8, Panel A). Our molecular docking studies provide a unified structure-based explanation for both positive and negative substitutions according to the SAR of the carbazole analogs.
4. Conclusion
We report here a novel series of modified carbazoles that destabilize MTs by binding to the colchicine site of tubulin in a similar mode to a podophyllotoxin analogue and appears to interact with a unique low interaction binding space. Several modified carbazoles trigger marked cell death in multiple GBM model systems while exhibiting a much less lower activity in the HepG2 liver cells, suggesting a promising therapeutic index. The use of the heterocyclic carbazole scaffold provides several advantages when considering the future optimization of small-molecule for therapeutic use. For example, this scaffold can be modified in a variety of ways using readily available and simple starting materials, and their chemical modification typically requires only as few steps of well-established chemistry, thereby providing a versatile scaffold for medicinal chemistry optimization. In summary, this work increases our understanding of how targeting the colchicine site by small molecules affects tubulin assembly and disrupts MT function, and provides a reference design approach to develop the next generation of MTAs for treating devastating cancers, such as GBM.
5. Experimental Section
General experimental methods.
Moisture sensitive reactions were performed in an inert, dry atmosphere of Ar in flame-dried glassware. Air sensitive liquids were transferred via syringe or cannula through rubber septa. Reagent grade solvents were used for extraction and flash chromatography. THF was distilled from Na/benzophenone under Ar. All commercially obtained reagents and solvents were used directly without further purification. The progress of reactions was checked by analytical thin-layer chromatography (Silica G TLC plates w/UV 254). Flash column chromatography was performed using prepacked Biotage SNAP/ZIP cartridges on a Biotage Isolera One instrument. The solvent compositions reported for all chromatographic separations are on a volume/volume (v/v) basis. 1H NMR spectra were recorded at 400 MHz and are reported in parts per million (ppm) on the δ scale relative to tetramethylsilane as an internal standard. 13C NMR spectra were acquired at 101 MHz operating with 1H decoupling and are reported in parts per million (ppm) on the δ scale relative to CDCl3. Melting points were determined on a Stuart melting point apparatus from Bibby Scientific Limited and are uncorrected. LC/MS and HRMS analyses were obtained on a Waters ACQUITY UPLC-series liquid chromatography system equipped with a diode array detector and coupled to a LCT PREMIER XE™ time of flight (TOF) mass spectrometer with electrospray ionization (ESI). The liquid chromatography conditions were as follows: a Phenomenex column (NX, 3μ, C18, 110A, 50.0×4.60 mm) was used, and bound compounds were eluted with the following gradient over 15 min at a rate of 0.4mL/min (water (0.1% formic acid)/acetonitrile): (90/10 to 2/98, 0–6.6 min), (2/98 isocratic, 6.6–13 min), (2/98 to 90/10, 13–15 min). Compound purity was assigned based on 254 nM detection data assessed by comparing relative peak areas of the signals. All final compounds were more than 95% pure.
9-ethyl-9H-carbazole (5).
Under Ar, a solution of carbazole (5.0 g, 29.9 mmol), bromoethane (4.45 mL, 35.9 mmol), and Cs2CO3 (14.6 g, 44.8 mmol) in DMF (20 mL) was stirred at 80°C for 16 h. The reaction mixture was cooled, diluted with EtOAc (50 mL), and filtered. The organic solvents were evaporated in vacuo. The resultant dark oil was purified by column chromatography on silica gel using heptanes/EtOAc in different proportions to afford the title compound as a light-yellow oil (5.075 g, 87%). 1H NMR (500 MHz, CDCl3) δ ppm 8.18 (d, J=7.79 Hz, 2H), 7.54 (t, J=7.66 Hz, 2H), 7.46 (d, J=8.06 Hz, 2H), 7.26 – 7.35 (m, 2H), 4.40 (q, J=7.25 Hz, 3H), 1.41 – 1.52 (m, 3H) 13C NMR (126 MHz, CDCl3) δ ppm 139.85, 125.54, 122.85, 120.37, 118.68, 108.37, 37.42, 13.75.
9-ethyl-9H-carbazole-3-carbaldehyde (6).
POC13 (1.4 mL, 10.2 mmol) was added, over a period of 10 min, to ice-cooled, stirred DMF (3.2 mL, 40.8 mmol) under Ar. The reddish solution was stirred at room temperature for 1 h. 9-ethyl-9H-carbazole 5 (1.0 g, 5.1 mmol) was added over 10 min, and the mixture was subjected to microwave irradiation at 100 °C for 1 h. The reaction mixture was cooled and then poured into crushed ice. After warming to room temperature, the resultant product was extracted with EtOAc. The organic phase was washed with water, and brine, dried (MgSO4), filtered, and evaporated in vacuo. The residue was purified by column chromatography on silica gel using heptanes/EtOAc in different proportions to afford the title compound as a white solid (1.054 g, 93%); 1H NMR (400 MHz, CDCl3) δ ppm 10.03 (s, 1H), 8.57 (s, 1H), 8.12 (d, J = 7.7 Hz, 1H), 7.94 (d, J = 8.5 Hz, 1H), 7.52 (t, J = 7.6 Hz, 1H), 7.41 (d, J = 8.6 Hz, 2H), 7.30 (t, J = 7.4 Hz, 1H), 4.26 (t, J=7.2 Hz, 2H), 1.45 (t, J = 7.2 Hz, 3H). 13C NMR (101 MHz, CDCl3) δ ppm 191.83, 144.18, 141.27, 128.56, 127.23, 126.82, 124.06, 123.14, 123.08, 120.84, 120.40, 109.53, 109.05, 42.76, 13.91.
9-ethyl-9H-carbazole-3-carboxylic acid (7).
To an ice-cold solution of 9-ethyl-3-carbaldehyde 6 (1.0 g, 4.5 mmol) in water/acetone (50 mL, 1:1) was added dropwise with stirring a solution of potassium permanganate (711 mg, 4.5 mmol) in acetone (25 mL). The mixture was heated 3 h at reflux and then allowed to cool to room temperature. After that, the reaction mixture was quenched with ethanol (20 mL) and then stirred for 30 min at reflux. After cooling to room temperature, the mixture was filtered through a pad of Celite© and concentrated in vacuo. The concentrated solution was diluted with water (100 mL), basified with NaOH to pH ca. 10, and extracted with heptane/ether (4:1, 50 mL × 3) to remove the unreacted starting material. The aqueous solution was cooled on an ice-water bath and then acidified with an ice-cold solution of 2 N HCl to pH ca. 2. The resultant precipitate was extracted with EtOAc (150 mL). The organic layer was washed with brine (30 mL), dried over MgSO4, filtered, and concentrated in vacuo. The precipitated product was collected by filtration, washed with heptanes (20 mL), and dried overnight to produce the title compound 3 (527.2 mg, 49%) as a yellow oil; 1H NMR (500 MHz, CDCl3) δ ppm 12.34 (s, 1H), 8.89 (d, J = 1.6 Hz, 1H), 8.31 (dd, J = 8.6, 1.6 Hz, 1H), 8.12 (d, J = 7.7 Hz, 1H), 7.51 (ddd, J = 8.3, 7.1, 1.3 Hz, 1H), 7.37 (d, J = 8.2 Hz, 1H), 7.34 (d, J = 8.7 Hz, 1H), 7.28 (ddd, 7.9, 6.9, 1 Hz, 1H), 4.32 (t, J = 7.2 Hz, 2H), 1.45 (t, J = 7.2 Hz, 3H). 13C NMR (126 MHz, CDCl3) δ ppm 173.30, 143.47, 141.17, 128.13, 126.62, 123.87, 123.14, 122.67, 120.88, 120.33, 119.82, 109.34, 108.45, 42.57, 14.01.
9-ethyl-3-[(4-methylnaphthalen-1-yl)carbonyl]-9H-carbazole (8).
Under Ar, AlCl3 (199 mg, 1.5 mmol) was added to a solution of 9-ethyl-9H-carbazole 7 (300 mg, 1.25 mmol) in anhydrous benzene (30 mL), and the solution was cooled by an ice bath for 20 min. 4-methyl-1-naphthoyl chloride (282 mL, 2.43 mmol) was added dropwise via a syringe to the solution, and the reaction mixture was stirred for 16 h while warming at room temperature. The reaction mixture was cooled on an ice-water bath, then poured onto a mixture of ice and a 4 M NaOH solution (50 mL) and extracted with ethyl acetate (150 mL). The obtained residue was purified by column chromatography on silica gel eluting with EtOAc/heptanes in different proportions to yield the target product (296.3 mg, 64%) as a light yellow solid: 1H NMR (400 MHz, CDCl3) δ ppm 8.63 (s, 1H), 8.00 – 8.18 (m, 4H), 7.37 −7.61 (m, 7H), 7.24 – 7.27 (m, 1H), 4.41 (q, J = 7.28 Hz, 2H), 2.81 (s, 3H), 1.47 (t, J = 7.28 Hz, 3H) 13C NMR (101 MHz, CDCl3) δ ppm 197.9, 142.9, 140.7, 137.3, 136.2, 132.9, 131.3, 129.8, 128.6, 126.9, 126.6, 126.6, 126.5, 126.2, 125.3, 124.4, 124.3, 123.3, 122.7, 120.9, 120.0, 109.0, 108.1, 37.9, 19.9, 13.9. HRMS calcd for C26H22NO (M+H)+: 364.1684, found HRMS: 364.1701.
9-ethyl-3-(4-methylpiperazine-1-carbonyl)-9H-carbazole (9).
9-ethyl-9H-carbazole-3-carboxylic acid 7 (200 mg, 0.84 mmol), 1-methylpiperazine (171 mg, 1.29 mmol), DIPEA (284 μL, 1.7 mmol), and DMAP (122 mg, 1 mmol) were added to DCM (26 mL) under N2. The obtained solution was cooled on an ice-water bath. EDC (275 mg, 1.44 mmol) was added to the solution, and the reaction mixture was then allowed to warm to room temperature and stirred for 16 h. The solvent was removed in vacuo, and the obtained residue was extracted into ethyl acetate (100 mL). The organic layer was washed consecutively with 5% citric acid solution (50 mL × 3), concentrated NaHCO3 (50 mL × 3), brine (50 mL), dried over MgSO4, filtered, and concentrated in vacuo. The residue was purified on silica gel using heptanes/ethyl acetate in different proportions to afford 199 mg (74%) of 9 as a yellow glass. 322.1952 1H NMR (400 MHz, CDCl3) δ ppm 8.24 (s, 1H), 8.10 (d, J=7.45 Hz, 1H), 7.50 – 7.59 (m, 2H), 7.41 – 7.48 (m, 2H), 7.29 (t, J=7.53 Hz, 1H), 4.40 (q, J=7.19 Hz, 2H), 3.72 (br s, 1H), 2.52 (br s, 1H), 2.44 (s, 3H), 2.17 (s, 1H), 1.40 – 1.49 (m, 4H) 13C NMR (101 MHz, CDCl3) δ ppm 167.78, 141.17, 140.49, 126.63, 125.27, 122.83, 122.51, 120.72, 120.66, 119.79, 108.93, 108.46, 53.47, 43.58, 37.79, 26.89, 13.78. HRMS calcd for C20H24 N3 O (M+H)+: 322.1913, found: 322.1952.
3-(4-chlorobenzoyl)-9-ethyl-9H-carbazole (10).
Under Ar, AlCl3 (1.13 g, 8.44 mmol) was added to a solution of 5 (1.5 g, 7.68 mmol) in anhydrous benzene (20 mL), and the solution was cooled by an ice bath for 20 min. 4-chlorobenzoic acid (1.12 mL, 8.83 mmol) was added dropwise via a syringe to the solution, which was tightly capped in a microwave vessel and subjected to microwave irradiation at 100 °C for 1 h and then cooled to room temperature. The reaction mixture was cooled on an ice-water bath, then poured onto a mixture of ice and a 4 M NaOH solution (50 mL) and extracted with ethyl acetate (150 mL). The organic phase was washed with saturated aqueous NaHCO3 and brine, dried (MgSO4), filtered and evaporated in vacuo. The residue was purified by column chromatography on silica gel eluting with EtOAc/heptanes in different proportions to yield the target product (1.374 g, 54%) as a white solid: 1H NMR (400 MHz, CDCl3) δ ppm 8.63 (d, J=1.30 Hz, 1H), 8.16 (d, J=7.77 Hz, 1H), 8.04 (dd, J=8.58, 1.67 Hz, 1H), 7.87 – 7.98 (m, 2H), 7.56 (td, J=7.64, 1.03 Hz, 1H), 7.45 – 7.53 (m, 2H), 7.29 – 7.37 (m, 1H), 7.17 – 7.28 (m, 2H), 4.43 (q, J=7.23 Hz, 2H), 1.51 (t, J=7.23 Hz, 3H) 13C NMR (101 MHz, CDCl3) δ ppm 195.06, 166.11, 163.60, 142.43, 140.57, 135.12, 135.09, 132.36, 132.28, 128.25, 128.22, 126.46, 123.76, 123.04, 122.51, 120.68, 119.92, 115.30, 115.08, 108.95, 107.99, 37.75, 13.74. ESI: m/z 334.1 (M + H)+. HRMS calcd for C21H17ClNO (M + H)+ 334.0999, found 334.0981.
9-ethyl-3-(4-fluorobenzoyl)-9H-carbazole (11).
Using 9-ethyl-9H-carbazole 5 (1.57 g, 8.05 mmol) and 4-fluorobenzoic acid (1.1 mL, 9.26 mmol), as starting compounds, the title compound was prepared by the procedures described in the preparation of compound 10 to yield 1.07 g (42%) of 11 as an orange oil. 1H NMR (400 MHz, CDCl3) δ ppm 8.56 (d, J = 1.7 Hz, 1H), 8.09 (d, J = 7.8 Hz, 1H), 7.97 (dd, J = 8.6, 1.7 Hz, 1H), 7.86 (dd, J = 8.6, 5.6 Hz, 2H), 7.50 (ddd, J = 8.3, 7.0, 1.2 Hz, 1H), 7.43 – 7.40 (m, 2H), 7.33 – 7.23 (m, 1H), 7.17 (t, J = 8.6 Hz, 2H), 4.36 (q, J = 7.2 Hz, 2H), 1.44 (t, J = 7.2 Hz, 3H). 13C NMR (101 MHz, CDCl3) δ ppm 195.28, 142.56, 140.62, 137.93, 137.30, 131.28, 128.60, 128.45, 128.26, 128.06, 126.53, 123.89, 123.08, 122.58, 120.75, 120.01, 109.00, 108.07, 37.83, 26.87, 13.79. HRMS calcd for C21H17FNO (M + H)+ 318.1294, found 318.0766.
9-ethyl-3-(4-methylbenzoyl)-9H-carbazole (12).
Using 9-ethyl-9H-carbazole 5 (325 mg, 1.67 mmol) and p-toluoyl chloride (246 mg, 2.00 mmol) as starting compounds, the title compound was prepared by the procedures described in the preparation of compound 10 to yield 241 mg (46%) of 12 as a yellow glass. 1H NMR (400 MHz, CDCl3) δ ppm 8.63 (d, J=1.30 Hz, 1H), 8.16 (d, J=7.77 Hz, 1H), 8.04 (dd, J=8.58, 1.67 Hz, 1H), 7.64 – 7.73 (m, 2H), 7.56 (td, J=7.64, 1.03 Hz, 1H), 7.29 – 7.37 (m, 1H), 7.17 – 7.28 (m, 4H), 4.43 (q, J=7.23 Hz, 2H), 2.31 (s, 3H) 1.51 (t, J=7.23 Hz, 3H) 13C NMR (101 MHz, CDCl3) δ ppm 197.02, 143.46, 141.52, 135.19, 135.07, 131.36, 131.28, 128.15, 128.12, 126.46, 123.76, 122.64, 122.22, 120.66, 119.72, 115.90, 115.68, 108.95, 107.54, 36.98, 21.43, 13.13. HRMS calcd for C22H20NO (M+H)+: 314.1539, found: 314.1545.
1-(9-ethyl-9H-carbazol-3-yl)-2-phenylethan-1-one (13).
Using 9-ethyl-9H-carbazole 5 (325 mg, 1.67 mmol) and phenylacetyl chloride (246 mg, 2.00 mmol) as starting compounds, the title compound was prepared by the procedures described in the preparation of compound 10 to yield 299 mg (57%) of 13 as a yellow glass. 1H NMR (400 MHz, CDCl3) δ ppm 8.81 (d, J=1.30 Hz, 1H), 8.24 (d, J=7.77 Hz, 1H), 8.09 (dd, J=8.58, 1.67 Hz, 1H), 7.59 – 7.69 (m, 2H), 7.29 – 7.37 (m, 2H), 7.23 – 7.36 (m, 5H), 4.43 (q, J=7.23 Hz, 2H), 4.12 (s, 2H), 2.31 (s, 3H), 1.51 (t, J=7.23 Hz, 3H) 13C NMR (101 MHz, CDCl3) δ ppm 195.38, 141.72, 135.66, 129.74, 129.13, 128.19, 128.42, 126.35, 124.32, 123.64, 122.22, 120.63, 119.79, 115.57, 115.35, 108.78, 107.54, 44.43, 36.98, 13.13. HRMS calcd for C22H20NO (M-H)− 314.1539, found: 314.1530.
9-ethyl-3-(naphthalene-1-carbonyl)-9H-carbazole (14).
Using 9-ethyl-9H-carbazole 4 (100 mg, 0.40 mmol) and 1-naphthoyl chloride (74 mL, 0.71 mmol) as starting compounds, the title compound was prepared following the procedures described in the preparation of compound 10. A yellowish viscous oil was obtained. Yield: 141.1 mg (41.5%). 1H NMR (400 MHz, CDCl3) δ 8.65 (s, 1H), 8.16(d, 1H), 8.04 (t, 2H), 7.96 (d, 1H), 7.51(d, 2H), 7.42(m, 2H), 7.33 (m, 4H), 7.18(t, 1H), 4.18 (q, 2H), 1.45 (t, 3H). 13C NMR (101 MHz, CDCl3) δ 197.79, 143.02, 140.73, 137.27, 133.77, 130.44, 128.64, 128.37, 126.56, 126.38, 125.97, 124.58, 124.33, 120.89, 120.12, 109.05, 108.13, 37.92, 13.87. HRMS calcd for C25 H19 NO (M+H)+: 350.1466, found: 350.1536.
9-propyl-9H-carbazole (15).
Using carbazole (550 mg, 2.52 mmol) and 1-bromopropane (369 mg, 3 mmol), as the starting compounds, the title compound was prepared by the procedures described in the preparation of compound 10 to yield 410 mg (74%) of 15 as a yellow oil. 1H NMR (400 MHz, CDCl3) δ 8.09 (d, J = 7.8 Hz, 2H), 7.44 (ddd, J = 8.2, 6.9, 1.2 Hz, 2H), 7.39 (d, J = 8.1 Hz, 2H), 7.21 (ddd, J = 8.0, 6.8, 1.1 Hz, 2H), 4.24 (t, J = 7.2 Hz, 2H), (m, 2H), 0.95 (t, J = 7.4 Hz, 3H). 13C NMR (101 MHz, CDCl3) δ 140.71, 125.75, 123.00, 120.53, 118.90, 108.89, 44.80, 22.51, 12.02.
9-propyl-3-[(4-methylnaphthalen-1-yl)carbonyl]-9H-carbazole (16).
Using 9-propyl-9H-carbazole (350 mg, 1.67 mmol) and 4-methyl-1-naphthoyl chloride (246 mg, 2.00 mmol) as starting compounds, the title compound was prepared by the procedures described in the preparation of compound 10 to yield 327 mg (52%) of 16 as a yellow glass. 1H NMR (400 MHz, CDCl3) δ ppm 8.62 (s, 1H), 8.14 (d, J=8.53 Hz, 1H), 8.04 (t, J=9.03 Hz, 2H), 7.96 (d, J=7.78 Hz, 1H), 7.47 – 7.55 (m, 2H), 7.38 – 7.47 (m, 2H), 7.27 – 7.38 (m, 3H), 7.18 (t, J=7.40 Hz, 1H), 4.18 (t, J=7.03 Hz, 2H), 2.73 (s, 3H), 1.78 – 1.90 (m, 2H), 0.97 (t, J=7.40 Hz, 3H) 13C NMR (101 MHz, CDCl3) δ ppm 197.63, 143.30, 141.06, 137.08, 132.71, 129.58, 128.35, 126.76, 126.41, 126.04, 125.42, 124.24, 122.97, 122.41, 120.53, 119.84, 118.56, 109.16, 108.57, 44.62, 60.38, 22.72, 19.67, 11.83. HRMS calcd for C27H24NO (M+H)+: 378.1852, found: 378.1851.
9-(2,2,2-trifluoroethyl)-9H-carbazole (17).
Using carbazole (300 mg, 1.80 mmol) and 2-chloro-1,1,1-trifluoroethane (0.25 mL, 1.28 mmol) as starting compounds, the title compound was prepared by the procedures described in the preparation of compound 10 to yield 372 mg (83%) of 17 as a yellow oil. 1H NMR (500 MHz, CDCl3) δ ppm 8.17 (d, J=7.86 Hz, 2H), 7.53 (t, J=7.63 Hz, 2H), 7.37 (d, J=7.86 Hz, 2H), 7.22 – 7.32 (m, 2H), 4.35 (s, 2H), 13C NMR (126 MHz, CDCl3) δ ppm 140.14, 127.44, 122.91, 120.53, 118.77, 111.23, 108.37, 72.14, 19.7.
3-(4-methylnaphthalene-1-carbonyl)-9-(2,2,2-trifluoroethyl)-9H-carbazole (18).
Using 17 (350 mg, 1.41 mmol) and 4-methyl-1-naphthoyl chloride (246 mg, 2.00 mmol) as starting compounds, the title compound was prepared by the procedures described in the preparation of compound 10 to yield 363 mg (62%) of 18 as a yellow glass.1H NMR (400 MHz, CDCl3) δ 8.64 (s, 1H), 8.00 – 8.19 (m, 4H), 7.36 – 7.59 (m, 7H), 7.24 – 7.27 (m, 1H), 4.37 (s, 2H), 2.79 (s, 3H), 13C NMR (101 MHz, CDCl3) δ ppm 197.6, 142.8, 139.7, 136.6, 136.1, 133.2, 131.2, 129.5, 128.8, 126.7, 126.9, 126.7, 126.5, 126.2, 125.2, 124.9, 124.6, 123.3, 122.4, 120.9, 120.0, 112.7 109.8, 108.6, 71.8, 19.7. HRMS calcd for C26H19F3NO (M+H)+: 418.1413, found: 418.1345.
(9-ethyl-9H-carbazol-3-yl)(quinolin-5-yl)methanol (19).
A solution of 1.5 M tert-buthyllithium in pentane (9 mL) was added dropwise in 20 min. at −78 °C to a solution of 3-bromo-9-ethyl-9H-carbazole (1.9 g; 0.007 mol) dissolved in dry THF (50 mL). The solution was stirred at −78 °C for 1 h. A brownish precipitate formed. TLC in cyclohexane/DCM 9/1: 100 % conversion. A solution of quinoline-5-carbaldehyde (1.1 g; 0.007 mol) dissolved in 50 mL of dry THF was added dropwise at −78 °C in 15 min. The resulting solution was stirred at −78 °C for 1.5 h. 220 mL of a saturated NH4Cl solution was added. The product was then extracted with EtOAc (200 mL and then 50 mL). The organic phases were combined and washed with water (2×100 mL) and dried over MgSO4. The solution was concentrated under reduced pressure. The residue was purified by column chromatography on silica gel eluting with EtOAc/cyclohexanes to afford the pure product as a pale yellow solid. Yield: 1.54 g (62.4%). 1H NMR (400 MHz, CDCl3) δ 8.78 (d, J = 4.0, 1H), 8.40 (d, J = 8.6, 1H), 8.12 (s, 1H), 8.06 (d, J = 8.4, 1H), 8.02 (d, J = 7.8, 1H), 7.82 (d, J = 7.1, 1H), 7.78 – 7.67 (m, 1H), 7.45 (t, J = 7.6, 1H), 7.39 (td, J = 4.7, 2.3, 2H), 7.31 (d, J = 8.5, 1H), 7.23 (d, J =4.2, 1H), 7.20 (t, J = 7.3, 1H), 6.65 (s, 1H), 4.32 (q, J = 7.2, 2H), 1.39 (t, J = 7.2, 3H). 13C NMR (101 MHz, CDCl3) δ 149.94, 139.74, 139.66, 133.53, 132.82, 129.69, 128.90, 125.94, 124.93, 124.89, 120.80, 120.53, 119.17, 119.01, 108.70, 108.61, 74.18, 37.65, 13.83.
9-ethyl-3-(quinoline-5-carbonyl)-9H-carbazole (20).
A suspension of 19 (1.4g; 3.97 mmol) dissolved in DCM (20 mL), PDC (2.95 g, 7.84 mmol) and molecular sieves 4 Å (2.95 g) was stirred for 2 h at r.t. The resulting solution was filtered over a pad of SiO2 and eluted with AcOEt. 350 mL of solvent (AcOEt and DCM) was collected, and the solutions were combined and concentrated under reduced pressure to afford the pure product as a pale yellow solid. Yield: 995 mg (78.7%). 1H NMR (400 MHz, CDCl3) δ 9.04 – 8.92 (m, 1H), 8.62 (s, 1H), 8.49 (d, J = 8.5, 1H), 8.31 (d, J = 8.3, 1H), 8.06 (d, J = 7.4, 2H), 7.87 – 7.77 (m, 1H), 7.75 (d, J = 6.9, 1H), 7.52 (t, J = 7.6, 1H), 7.49 – 7.43 (m, 2H), 7.43 – 7.39 (m, 1H), 7.31 – 7.25 (m, 2H), 4.42 (q, J = 7.2, 2H), 1.48 (t, J = 7.2, 3H). 13C NMR (101 MHz, CDCl3) δ 196.38, 150.91, 148.35, 143.07, 140.74, 137.72, 134.31, 132.13, 129.16, 128.54, 128.02, 127.78, 126.69, 126.63, 124.37, 123.19, 122.84, 121.95, 120.87, 120.24, 109.11, 108.24, 37.96, 13.86. HRMS calcd for C24H19N2O (M+H)+: 351.1491, found: 351.1487.
(9-ethyl-9H-fluoren-3-yl)(quinolin-7-yl)methanol (21).
To an oven-dried round-bottom flask flushed with N2 was added 3-bromo-9-ethyl-9H-carbazole (508 mg, 1.85 mmol) in 13 mL dry THF. The mixture was cooled to −78°C and tert-BuLi (2M in heptane) (1.85 mL, 3.7 mmol) was added dropwise. The mixture was stirred at −78°C for 1 hr, then 8-Quinolinecarboxaldehyde (291 mg, 1.85 mmol) was added. The resulting mixture was stirred at −78°C for 1.5 hr, then allowed to warm to 0°C. 50 mL sat. NH Cl was added, then organics were extracted with EtOAc (2×30 mL), washed with water and brine, and dried over MgSO4. Solvents were removed in vacuo, and the resulting oil was purified by column chromatography eluting with a gradient of 12–100% EtOAc in Heptane to yield the title compound as a dark purple oil (303 mg, 46.4% yield). 1H NMR (400 MHz, Chloroform-d) δ 8.90 (dd, J = 4.3, 1.8 Hz, 1H), 8.26 – 8.24 (m, 1H), 8.22 (dd, J = 8.4, 1.8 Hz, 1H), 8.06 (dt, J = 7.8, 1.0 Hz, 1H), 7.75 (dd, J = 8.0, 1.7 Hz, 1H), 7.60 (dd, J = 8.4, 1.7 Hz, 1H), 7.45 (dd, J = 6.1, 1.8 Hz, 2H), 7.39 (d, J = 5.0 Hz, 1H), 7.19 (ddd, J = 7.9, 6.9, 1.2 Hz, 1H), 6.97 (s, 1H), 6.67 (s, 1H), 4.36 (q, J = 7.2 Hz, 2H), 1.42 (t, J = 7.2 Hz, 3H).
9-ethyl-3-(quinoline-8-carbonyl)-9H-carbazole (22).
A suspension of 21 (300 mg, 0.85 mmol) dissolved in DCM (5 mL), PDC (641 mg, 1.70 mmol) and molecular sieves 4 Å (632 mg) was stirred for 3 h. at r.t. The resulting solution was filtered under a pad of SiO2, and eluted with AcOEt. 350 mL of solvent (AcOEt and DCM) were collected and the solutions were combined and then concentrated in vacuo. The resulting oil was purified using column chromatography eluting with a gradient of 7–50% EtOAc in Heptane to afford the pure product as a bright-orange solid (73 mg, 24.4% yield). 1H NMR (400 MHz, Chloroform-d) δ 8.87 (dd, J = 4.2, 1.8 Hz, 1H), 8.60 (d, J = 1.7 Hz, 1H), 8.25 (dd, J = 8.3, 1.8 Hz, 1H), 8.05 (dd, J = 8.7, 1.7 Hz, 1H), 8.03 – 7.97 (m, 2H), 7.81 (dd, J = 7.0, 1.5 Hz, 1H), 7.67 (dd, J = 8.2, 7.0 Hz, 1H), 7.48 (ddd, J = 8.3, 7.0, 1.2 Hz, 1H), 7.44 – 7.40 (m, 2H), 7.38 (d, J = 8.7 Hz, 1H), 7.23 (ddd, J = 8.0, 7.0, 1.1 Hz, 1H), 4.39 (q, J = 7.2 Hz, 2H), 1.45 (t, J = 7.2 Hz, 3H). 13C NMR (101 MHz, CDCl3) δ 197.39, 150.97, 146.36, 143.05, 140.69, 140.37, 136.04, 129.42, 129.26, 128.56, 128.37, 128.06, 126.37, 125.92, 124.20, 123.44, 122.79, 121.58, 120.83, 119.96, 108.97, 108.02, 77.36, 77.25, 77.05, 76.73, 37.86, 13.83. HRMS calcd for C24H18N2O (M+H)+: 351.1492, found: 351.1656.
(9-ethyl-9H-carbazol-3-yl)(1,2,3,4-tetrahydroquinolin-6-yl)methanone (23).
To a solution of 20 (106.3 mg, 0.30 mmol) in MeOH (1.5 mL) and THF (1 mL) was added sodium cyanoborohydride (95.46 mg, 1.52 mmol). Boron trifluoride diethyl etherate (190.9 L, 1.52 mmol) was added dropwise to the resulting solution, with evolution of gas. The solution was stirred and refluxed at 63 °C for 3.5 h under N2. 3 mL NH3 (25% in water) was added to the reaction mixture, then diluted with another 10 mL of water. The product was extracted with EtOAc (2 × 15 mL), and washed with brine (1 × 30 mL). The organic layers were dried over anhydrous MgSO, and the solvent was removed in vacuo. Raw product was purified with flash chromatography (16 – 40% EtOAc in heptane) to yield 23 as a sticky yellow solid (43.9 mg, 41.3% yield). 1H NMR (400 MHz, Chloroform-d) δ 8.68 – 8.52 (m, 1H), 8.11 (d, J = 7.7 Hz, 0H), 8.08 – 8.00 (m, 1H), 7.51 (ddd, J = 8.1, 6.3, 2.6 Hz, 1H), 7.42 (dd, J = 15.0, 8.5 Hz, 1H), 7.32 – 7.27 (m, 0H), 7.05 (t, J = 7.7 Hz, 1H), 6.75 – 6.65 (m, 1H), 6.61 (dt, J = 8.1, 2.2 Hz, 1H), 4.40 (q, J = 7.2 Hz, 1H), 4.10 (s, 1H), 3.32 (dd, J = 7.0, 3.9 Hz, 1H), 2.72 (t, J = 6.2 Hz, 1H), 1.88 (p, J = 6.0 Hz, 1H), 1.47 (t, J = 7.2 Hz, 2H). 13C NMR (101 MHz, CDCl3) δ 198.70, 145.01, 142.90, 140.68, 140.54, 129.03, 128.31, 126.44, 126.14, 124.07, 123.36, 122.73, 120.90, 120.03, 119.09, 116.93, 115.51, 108.97, 108.01, 41.73, 37.88, 24.76, 21.89, 13.85. HRMS calcd for C24H22N2O (M+H)+: 355.1804, found: 355.1810.
9-ethyl-3-(1,2,3,4-tetrahydroquinoline-8-carbonyl)-9H-carbazole (24).
To a solution of 22 (60 mg, 0.171 mmol) in MeOH (0.75 mL) and THF (0.5 mL) was added sodium cyanoborohydride (54.8 mg, 0.856 mmol). Boron trifluoride diethyl etherate (108 L, 0.856 mmol) was added dropwise to the resulting solution, with evolution of gas. The solution was stirred and refluxed at 60⁰C for 3.5 hours under nitrogen. 2 mL ammonia (25% in water) was added to the reaction mixture, which was then diluted with another 10 mL of water. The product was extracted with EtOAc (2 × 15 mL) and washed with brine (1 × 30 mL). The organic layers were dried over anhydrous MgSO4 and the solvent was removed in vacuo. Raw product was purified with flash chromatography twice eluting with a gradient of 15–55% EtOAc in Heptane and then 10–100% DCM in heptane to yield 24 as a yellow oil (12.6 mg, 20.8% Yield). 1H NMR (400 MHz, Chloroform-d) δ 8.43 (t, J = 2.5 Hz, 2H), 8.10 (d, J = 7.7 Hz, 1H), 7.84 (dd, J = 8.5, 1.7 Hz, 1H), 7.50 (ddd, J = 8.2, 6.9, 1.2 Hz, 1H), 7.47 – 7.40 (m, 3H), 7.27 (d, J = 8.1 Hz, 1H), 7.08 (dd, J = 7.1, 1.4 Hz, 1H), 6.47 – 6.39 (m, 1H), 4.41 (q, J = 7.2 Hz, 2H), 3.49 (td, J = 5.9, 2.7 Hz, 2H), 2.86 (t, J = 6.3 Hz, 2H), 2.02 – 1.94 (m, 2H), 1.47 (t, J = 7.2 Hz, 3H). 13C NMR (101 MHz, CDCl3) δ 199.08, 148.76, 141.63, 140.59, 133.38, 133.30, 131.50, 127.75, 126.19, 123.24, 122.92, 122.46, 122.28, 120.77, 119.61, 117.18, 112.80, 108.83, 107.77, 77.35, 77.23, 77.03, 76.71, 41.26, 39.00, 37.80, 35.45, 34.15, 31.91, 29.52, 29.45, 29.05, 27.93, 25.05, 22.71, 22.68, 20.81, 20.18, 19.19, 14.42, 14.12, 13.85, 11.41, 10.97.HRMS calcd C24H22N2 O (M+H)+: 355.1805, found 355.1815.
9-ethyl-3-[(1,2,3,4-tetrahydroquinolin-1-yl)carbonyl]-9H-carbazole (25).
Using 9-ethyl-9H-carbazole-3-carboxylic acid 7 (200 mg, 0.84 mmol) and 1,2,3,4-tetrahydroquinoline (171 mg, 1.29 mmol) as starting compounds, the title compound was prepared following the procedures described for the preparation of compound 9 to yield 241 mg (81%) of 25 as a yellow glass. 1H NMR (400 MHz, CDCl3) δ ppm 8.62 (s, 1H), 8.04 (t, J=9.03 Hz, 2H), 7.96 (d, J=7.78 Hz, 1H), 7.41 – 7.54 (m, 3H), 7.08 (t, J=7.40 Hz, 1H), 6.89 – 7.01 (m, 3H), 4.38 (t, J=7.03 Hz, 2H), 2.73–2.84 (m, 2H), 1.54 – 1.70 (m, 4H), 1.28 (t, J=7.40 Hz, 3H). 13C NMR (101 MHz, CDCl3) δ ppm 166.05, 141.72, 135.50, 131.12, 129.53, 128.00, 126.77, 125.17, 124.85, 124.46, 123.83, 123.56, 122.45, 121.39, 118.95, 118.55, 118.26, 107.58, 106.98, 51.10, 42.14, 28.87, 25.32, 20.09, 14.72. HRMS calcd for C24H23N2O (M+H)+: 355.1804, found: 355.1831.
9-ethyl-3-(4-methylnaphthalene-1-carbothioyl)-9H-carbazole (26).
Under Ar, a solution of carbazole 8 (62 mg, 0.17 mmol) and Lawesson’s reagent (49 mg, 0.12 mmol) in toluene (3 mL) was tightly capped in a 5 mL microwave vessel. The mixture was subjected to microwave irradiation at 140 °C for 4 h and then cooled to r.t. The organic solvent was evaporated in vacuo, and the residue was purified by column chromatography on silica gel using heptanes/ethyl acetate in different proportions to yield thioamide 26 as a yellow glass. Yield: 20 mg (31%). 1H NMR (400 MHz, CDCl3) δ ppm 8.63 (s, 1H), 8.10 (m, 4H), 7.49 (m, 7H), 7.26 (t, 1H), 4.41 (q, 2H), 2.82 (s, 3H), 1.46 (t, 3H). 13C NMR (101 MHz, CDCl3) δ ppm 218.89, 142.92, 140.70, 137.27, 129.82, 128.61, 126.94, 126.60, 126.49, 126.21, 125.32, 124.35, 120.88, 120.05, 109.01, 108.06, 37.91, 19.92, 13.87. HRMS calcd for C26H22NS (M+H)+: 380.1467, found: 380.1498.
(9-ethyl-9H-carbazol-3-yl)(4-methylnaphthalen-1-yl)methanol (27).
Carbazole 8 (2.59 g; 0.071 mol) was dissolved in 62 mL of dioxane, and 100 mL of MeOH was added. NaBH4 (400 mg; 0.01 mol) was added to the solution over 10 min. The solution was stirred for 14 h at room temperature. 110 mg of NaBH4 was added to complete the reduction, and the reaction mixture was stirred at r.t. for 14 h. 250 mL of water was added. The product was then extracted with EtOAc (250 mL) from water. The organic phases were combined and washed with water (2×100 mL) and dried over MgSO4. The solution was concentrated under reduced pressure. Flash-chromatography of the crude mixture using a DCM/Cyclohexane gradient afforded the pure product as a pale yellow solid. Yield: 1.735 g (66.6%). 1H NMR (400 MHz, CDCl3) δ 8.18 (s, 1H), 8.12 (d, J=8.2, 1H), 8.05 (dd, J=15.5, 8.8, 2H), 7.65 (d, J=7.3, 1H), 7.53 – 7.37 (m, 6H), 7.37 – 7.29 (m, 2H), 7.23 – 7.15 (m, 1H), 6.74 (d, J=3.8, 1H), 4.34 (q, J=7.2, 2H), 2.72 (s, 3H), 2.32 (d, J=3.9, 1H), 1.44 – 1.36 (m, 8H). 13C NMR (101 MHz, CDCl3) δ 140.31, 139.56, 137.57, 134.38, 134.12, 133.06, 130.84, 126.16, 125.72, 125.37, 125.11, 124.80, 124.69, 124.18, 122.99, 122.89, 120.56, 119.26, 118.84, 108.51, 74.17, 37.62, 26.95, 19.66, 13.84.
9-(4-methylnaphthalene-1-carbonyl)-9H-carbazole (28).
Using carbazole (300 mg, 1.80 mmol), and 4-methyl-1-naphthoyl chloride (0.25 mL, 1.28 mmol) as starting compounds, the title compound was prepared by the procedures described for the preparation of compound 5 to yield 96 mg (16%) of 28 as a yellow oil. (400 MHz, CDCl3) δ ppm 8.14 (d, J = 8.5 Hz, 1H), 8.05 – 7.93 (m, 3H), 7.60 (ddd, J = 8.4, 6.8, 1.3 Hz, 1H), 7.54 (d, J = 7.2 Hz, 1H), 7.48 (ddd, J = 8.5, 6.9, 1.4 Hz, 1H), 7.42 (t, J = 8.3 Hz, 2H), 7.36 – 7.30 (m, 2H), 7.25 – 7.18 (m, 1H), 2.84 (s, 3H), 13C NMR (101 MHz, CDCl3) δ ppm 167.81, 140.09, 139.36, 133.72, 132.29, 131.84, 129.61, 128.79, 126.76, 126.41, 126.04, 125.42, 120.53, 120.14, 119.49, 115.17, 19.64. HRMS calcd for C24H18 NO (M+H)+: 336.1383, found: 336.1737.
9-ethyl-2-methoxy-9H-carbazole (29).
Using 7-methoxycarbazole [15], (1.2 g, 4.7 mmol), bromoethane (0.697 mL, 9.4 mmol) and Cs2CO3 (3.46 mg, 10 mmol) in DMF (40 mL) as starting compounds, the title compound was prepared by the procedures described for the preparation of compound 5 to yield 1.03 g (77%) of 29 as a greenish solid. 1H NMR (400 MHz, CDCl3) δ 8.69 (dd, J = 1.6, 0.6 Hz, 1H), 8.08 (dd, J = 8.7, 1.7 Hz, 1H), 7.99 (d, J = 8.4 Hz, 1H), 7.31 (d, J = 8.6 Hz, 1H), 6.89 (dd, J = 8.5, 2.2 Hz, 1H), 6.85 (d, J = 2.1 Hz, 1H), 4.28 (q, J = 7.2 Hz, 2H), 3.96 (s, 3H), 3.93 (s, 3H), 1.41 (t, J = 7.2 Hz, 3H). 13C NMR (101 MHz, CDCl3) δ 168.02, 159.55, 142.81, 141.95, 130.20, 127.23, 126.14, 122.90, 121.91, 121.47, 120.78, 116.88, 108.06, 107.67, 93.43, 55.75, 51.91, 37.76, 13.68.
9-ethyl-7-methoxy-9H-carbazole-3-carboxylic acid (30):
Using 29 (1.413 g, 4.99 mmol), as the starting compound, the title compound was prepared by the procedures described for the preparation of compound 6 to yield 990 mg (52%) of 29 as an orange solid. 1H NMR (400 MHz, DMSO-d6) δ ppm 12.60 (br s, 1H), 8.68 (s, 1H), 8.12 (d, J=8.53 Hz, 1H), 7.99 (dd, J=8.53, 1.51 Hz, 1H), 7.58 (d, J=8.53 Hz, 1H), 7.18 (d, J=1.76 Hz, 1H), 6.86 (dd, J=8.53, 1.76 Hz, 1H), 4.42 (q, J=6.86 Hz, 2H), 3.89 (s, 3H), 1.24 – 1.37 (m, 4H) 13C NMR (101 MHz, DMSO-d6) δ ppm 168.11, 159.27, 142.18, 141.72, 125.76, 122.22, 121.53, 121.47, 121.16, 115.80, 108.56, 108.40, 93.58, 55.52, 37.13, 13.56.
9-ethyl-2-methoxy-6-(4-methylnaphthalene-1-carbonyl)-9H-carbazole (31).
Using 7-methoxy-9-pentyl-9H-carbazole-3-carboxylic acid (1.57 g, 8.05 mmol), and 1-methylnaphthalene (1.1 mL, 9.26 mmol) as starting compounds, the title compound was prepared by the procedures described for the preparation of compound 10 to yield 133 mg (42%) of 31 as an orange oil. 1H NMR (400 MHz, Chloroform-d) δ 8.53 (d, J = 1.7 Hz, 1H), 8.16 – 8.04 (m, 2H), 7.98 (dd, J = 8.6, 1.7 Hz, 1H), 7.91 (d, J = 8.4 Hz, 1H), 7.62 – 7.55 (m, 1H), 7.54 (d, J = 7.1 Hz, 1H), 7.48 (ddd, J = 8.2, 6.7, 1.3 Hz, 1H), 7.42 – 7.38 (m, 1H), 7.35 (d, J = 8.5 Hz, 1H), 6.86 (dd, J = 8.5, 2.2 Hz, 2H), 4.34 (q, J = 7.3 Hz, 2H), 3.94 (s, 3H), 2.80 (d, J = 1.0 Hz, 3H), 1.45 (t, J = 7.2 Hz, 3H). 13C NMR (101 MHz, DMSO-d6) δ ppm 168.10, 159.29, 142.19, 141.74, 125.77, 122.23, 121.55, 121.48, 121.16, 115.81, 108.58, 108.43, 93.60, 55.54, 37.15, 30.67, 13.58. HRMS calcd for C27H24NO2 (M + H)+: 394.1807, found 394.0875.
Ethyl 1H,2H,3H,4H,5H-pyrido[4,3-b]indole-2-carboxylate (32).
12 M HCl (1.2 mL) was added to a solution of phenylhydrazine (3.64 mL, 36.99 mmol) and ethyl-4-oxocyclohexanecarboxylate (0.98 mL, 5.73 mmol) in EtOH (10 mL). The solution was microwaved at 140 °C for 3 h. The reaction mixture was cooled on an ice-water bath, then poured onto a mixture of ice and a 4 M NaOH solution (50 mL) and extracted with ethyl acetate (150 mL). The organic phase was washed with saturated aqueous NaHCO3 and brine, dried (MgSO4), filtered and evaporated in vacuo. The residue was purified by column chromatography on silica gel eluting with EtOAc/heptanes in different proportions to yield 731 mg (52%) of 32 as an orange solid: 1H NMR (500 MHz, CDCl3) δ ppm 7.77 (br. s., 1H), 7.28 (t, J=7.80 Hz, 1H), 7.14 (t, J=7.80 Hz, 1H), 7.02 (t, J=8.08 Hz, 1H), 6.98 (d, J=8.08 Hz, 1H), 4.15 – 4.29 (m, 2H), 3.90 (s, 2H), 3.39 (dd, J=16.01, 5.15 Hz, 2H), 2.92 (m, 1H), 2.22 – 2.31 (m, 1H), 1.32 (t, J=7.10 Hz, 3H) 13C NMR (126 MHz, CDCl3) δ ppm 175.89, 137.20, 131.02, 127.33, 121.85, 117.32, 108.25, 103.93, 60.39, 45.13, 40.67, 25.86, 14.23.
Ethyl 5-ethyl-1H,2H,3H,4H,5H-pyrido[4,3-b]indole-2-carboxylate (33).
Under Ar, a solution of 320020(1.5 g, 6.15 mmol), bromoethane (0.20 mL, 26.8 mmol), and NaH (131 mg, 44.8 mmol) in DMF (30 mL) was stirred for 30 min at r.t. The reaction mixture was diluted with DCM (50 mL) and filtered through Celite©. The organic solvents were evaporated in vacuo. The concentrated residue was extracted with tBuOMe/EtOAc (5:1, v/v, 150 mL). The organic layer was washed with NaHCO3 (100 mL), dried over MgSO4, filtered, and concentrated in vacuo. The resultant oil was purified by column chromatography on silica gel using heptanes/EtOAc (4:1, v/v) to afford the title compound as an orange oil, 1455 mg (87%) 1H NMR (500 MHz, CDCl3) δ ppm 7.77 (br. s., 1H), 7.28 (t, J=7.80 Hz, 1H), 7.14 (t, J=7.80 Hz, 1H), 7.02 (t, J=8.08 Hz, 1H), 6.98 (d, J=8.08 Hz, 1H), 4.15 – 4.29 (m, 2H), 3.90 (s, 2H), 3.39 (dd, J=16.01, 5.15 Hz, 2H), 2.92 (m, 1H), 2.22 – 2.31 (m, 1H), 1.32 (t, J=7.10 Hz, 3H) 13C NMR (126 MHz, CDCl3) δ ppm 175.74, 136.24, 131.72, 127.12, 121.58, 117.21, 108.32, 103.83, 60.27, 45.42, 40.55, 37.22, 25.79, 15.25, 14.12.
5-ethyl-1H,2H,3H,4H,5H-pyrido[4,3-b]indole (34).
A solution of 33 (1400 mg, 5.15 mmol) and KOH (726 mg, 7.73 mmol) in a mixture of H2O (2 mL) and EtOH (9 mL) was refluxed for 48 h. The reaction mixture was cooled and diluted with EtOAc (50 mL), and the organic phase was washed with water and brine, dried (MgSO4), filtered, and evaporated in vacuo. The resultant oil was purified by column chromatography on silica gel using heptanes/EtOAc in different proportions to afford the title compound as a light yellow oil, 988 mg (96%). δ ppm 7.50–7.56 (m, 1H), 7.09 – 7.15 (m, 3H), 4.09 (q, J=7.06 Hz, 2H), 3.66 (br. s.,2 H), 3.26 – 3.34 (m, 2H), 2.87 – 2.96 (m, 2H), 1.16 (t, J=7.06 Hz, 3H) 13C NMR (101 MHz, DMSO-d6) δ ppm 135.11, 132.14, 129.12, 122.15, 119.24, 118.56, 108.33, 108.10, 44.99, 41.59, 37.18, 25.77, 15.15.
5-ethyl-2-[(4-methylnaphthalen-1-yl)carbonyl]-1H,2H,3H,4H,5H-pyrido[4,3-b]indole (35).
Using 34 (250 mg, 1.25 mmol), and 4-methyl-1-naphthoic acid (348 mg, 1.87 mmol) as starting compounds, the title compound was prepared by the procedures described for the preparation of compound 7 to yield 193 mg (42%) of 35 as an orange oil. 1H NMR (400 MHz, DMSO-d6) δ ppm 8.71 (d, J=8.20 Hz, 1H), 8.07 (d, J=8.42 Hz, 1H), 7.72 – 7.81 (m, 2H), 7.52 – 7.66 (m, 2H), 7.35 – 7.50 (m, 2H), 7.29 – 7.35 (m, 2H), 6.98 – 7.02 (m, 1H), 6.91 – 6.96 (m, 1H), 4.36 (br s, 2H), 4.02 (q, J=7.01 Hz, 2H), 3.41 – 3.53 (m, 2H), 3.04 – 3.14 (m, 2H), 2.64 (s, 3H), 1.18 (t, J=7.01 Hz, 3H) 13C NMR (101 MHz, DMSO-d6) δ ppm 168.86, 135.21, 135.14, 133.16, 132.09, 132.04, 129.15, 126.62, 126.32, 125.96, 125.91, 125.12, 124.65, 124.58, 123.15, 119.31, 118.70, 108.30, 108.00, 44.20, 42.78, 37.05, 25.47, 19.07, 15.15. HRMS calcd for C25H25N2O (M+H)+: 369.1961, found: 369.1936.
Cell culture.
Cells were cultured in DMEM supplemented with 10% FBS, 100 U/mL penicillin, and 100 μg/mL streptomycin at 37 °C in a 5% CO2 humidified atmosphere. T98G and HepG2 cells (ATCC, Manassas, VA) were authenticated by ATCC when purchased using human short tandem repeat analysis and maintained in culture for less than 6 months. Tumors were obtained from surgeries performed at the Swedish Medical Center (Seattle, WA) according to Institutional Review Board guidelines. Patient samples used in this study were diagnosed as WHO grade IV glioblastoma multiforme. Patient-derived GBM cells were established from the freshly resected tumor tissues and maintained in NeuroCult® NSA medium (Stem Cell Technologies) with B-27 serum-free supplement, 20 ng/mL epidermal growth factor and 20 ng/mL fibroblast growth factor 2 as previously described [18, 38].
Cell viability.
WST-1 (Roche, Pleasanton, CA) was used to evaluate cell viability as described [14] 72 h following drug treatment according to the manufacturer’s protocol. Maximal killing activity was identified as the maximal % reduction in cell viability measured at drug concentrations tested. EC50 values were calculated and reported only when a curve was reliably extrapolated by Prism software. The solvent for all modified carbazoles was DMSO (0.1% final). This solvent (negative reference, vehicle control) had no effect on cell viability.
[3H]Colchicine binding to purified tubulin and tubulin assembly.
[3H]Colchicine binding to purified tubulin and tubulin assembly (as assessed by turbidity development in purified bovine tubulin solutions) were both measured as previously described[29].
MT-tubulin partitioning.
Bovine brain tubulin was purified, polymerized into MTs and sheared as previously described[39]. Sheared MTs and drugs were incubated at 37 °C for 15 min and centrifuged for 10 min at 42,000 rpm at 37 °C prior to separation into supernatants containing free tubulin and pellets containing polymerized MTs as described[29]. Supernatants and pellets were run on a 4–12% polyacrylamide gel and stained with Coomassie G-250 for quantification. Peak intensities were quantified using ImageJ.
Supplementary Material
Highlights.
Synthesis of 19 new modified carbazoles that differentially bind to the colchicine site.
Docking of modified carbazoles in colchicine site shows unique interaction space.
Structure-activity relationship analysis identifies moieties that control activities.
Modified carbazoles kill human GBM cells (EC50 values from 67 to 10,000 nM).
ACKNOWLEDGEMENT
This work was supported by NIH grants: P30NS055022 (PD, RRP), R43CA165452 (EH). RO1DA014486 (CX,NS) and RO1GM069429 (MW and LW).
Abbreviations
- GBM
glioblastoma multiform
- MOA
mechanisms of action
- MTs
microtubules
- MTAs
microtubule-targeting agents
- NC
not converged
- ND
not determined
- PDB
protein data bank
- PD-GBM
patient-derived GBM
- OD
optical density
- TGI
total growth inhibition
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
DISCLAIMER
The content of this paper is solely the responsibility of the authors and does not necessarily reflect the official views of the National Institutes of Health.
References
- [1].Liu YM, Chen HL, Lee HY, Liou JP, Tubulin inhibitors: a patent review, Expert Opin Ther Pat, 24 (2014) 69–88. [DOI] [PubMed] [Google Scholar]
- [2].Matson DR, Stukenberg PT, Spindle poisons and cell fate: a tale of two pathways, Mol Interv, 11 (2011) 141–150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Yoshida D, Hoshino S, Shimura T, Takahashi H, Teramoto A, Drug-induced apoptosis by anti-microtubule agent, estramustine phosphate on human malignant glioma cell line, U87MG; in vitro study, Journal of neuro-oncology, 47 (2000) 133–140. [DOI] [PubMed] [Google Scholar]
- [4].Landen JW, Hau V, Wang M, Davis T, Ciliax B, Wainer BH, Van Meir EG, Glass JD, Joshi HC, Archer DR, Noscapine crosses the blood-brain barrier and inhibits glioblastoma growth, Clinical Cancer Research, 10 (2004) 5187–5201. [DOI] [PubMed] [Google Scholar]
- [5].Boumendjel A, McLeer-Florin A, Champelovier P, Allegro D, Muhammad D, Souard F, Derouazi M, Peyrot V, Toussaint B, Boutonnat J, A novel chalcone derivative which acts as a microtubule depolymerising agent and an inhibitor of P-gp and BCRP in in-vitro and in-vivo glioblastoma models, BMC cancer, 9 (2009) 242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Zhou R, Mazurchuk RV, Tamburlin JH, Harrold JM, Mager DE, Straubinger RM, Differential pharmacodynamic effects of paclitaxel formulations in an intracranial rat brain tumor model, Journal of Pharmacology and Experimental Therapeutics, 332 (2010) 479–488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Calinescu A-A, Castro MG, Microtubule targeting agents in glioma, Translational Cancer Research, 5 (2016) S54–S60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Herman JA, Toledo CM, Olson JM, DeLuca JG, Paddison PJ, Molecular pathways: regulation and targeting of kinetochore-microtubule attachment in cancer, Clin Cancer Res, 21 (2015) 233–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Oehler C, Frei K, Rushing EJ, McSheehy PM, Weber D, Allegrini PR, Weniger D, Lutolf UM, Knuth A, Yonekawa Y, Barath K, Broggini-Tenzer A, Pruschy M, Hofer S, Patupilone (epothilone B) for recurrent glioblastoma: clinical outcome and translational analysis of a single-institution phase I/II trial, Oncology, 83 (2012) 1–9. [DOI] [PubMed] [Google Scholar]
- [10].Stupp R, Taillibert S, Kanner AA, Kesari S, Steinberg DM, Toms SA, Taylor LP, Lieberman F, Silvani A, Fink KL, Barnett GH, Zhu JJ, Henson JW, Engelhard HH, Chen TC, Tran DD, Sroubek J, Tran ND, Hottinger AF, Landolfi J, Desai R, Caroli M, Kew Y, Honnorat J, Idbaih A, Kirson ED, Weinberg U, Palti Y, Hegi ME, Ram Z, Maintenance Therapy With Tumor-Treating Fields Plus Temozolomide vs Temozolomide Alone for Glioblastoma: A Randomized Clinical Trial, JAMA, 314 (2015) 2535–2543. [DOI] [PubMed] [Google Scholar]
- [11].Giladi M, Schneiderman RS, Voloshin T, Porat Y, Munster M, Blat R, Sherbo S, Bomzon Z, Urman N, Itzhaki A, Cahal S, Shteingauz A, Chaudhry A, Kirson ED, Weinberg U, Palti Y, Mitotic Spindle Disruption by Alternating Electric Fields Leads to Improper Chromosome Segregation and Mitotic Catastrophe in Cancer Cells, Sci Rep, 5 (2015) 18046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Jordan MA, Wilson L, Microtubules as a target for anticancer drugs, Nat Rev Cancer, 4 (2004) 253–265. [DOI] [PubMed] [Google Scholar]
- [13].Hu L, Li Z.-r., Li Y, Qu J, Ling Y-H, Jiang J.-d., Boykin DW, Synthesis and structure− acvity relaonships of carbazole sulfonamides as a novel class of antimitotic agents against solid tumors, Journal of medicinal chemistry, 49 (2006) 6273–6282. [DOI] [PubMed] [Google Scholar]
- [14].Cherry AE, Haas BR, Naydenov AV, Fung S, Xu C, Swinney K, Wagenbach M, Freeling J, Canton DA, Coy J, Horne EA, Rickman B, Vicente JJ, Scott JD, Ho RJ, Liggitt D, Wordeman L, Stella N, ST-11: a new brainpenetrant microtubule-destabilizing agent with therapeutic potential for glioblastoma multiforme, Mol Cancer Ther, 15 (2016) 2018–2029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Petrov RR, Knight L, Chen S-R, Wager-Miller J, McDaniel SW, Diaz F, Barth F, Pan H-L, Mackie K, Cavasotto CN, Mastering tricyclic ring systems for desirable functional cannabinoid activity, European journal of medicinal chemistry, 69 (2013) 881–907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Verhaak RG, Hoadley KA, Purdom E, Wang V, Qi Y, Wilkerson MD, Miller CR, Ding L, Golub T, Mesirov JP, Alexe G, Lawrence M, O’Kelly M, Tamayo P, Weir BA, Gabriel S, Winckler W, Gupta S, Jakkula L, Feiler HS, Hodgson JG, James CD, Sarkaria JN, Brennan C, Kahn A, Spellman PT, Wilson RK, Speed TP, Gray JW, Meyerson M, Getz G, Perou CM, Hayes DN, Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1, Cancer Cell, 17 (2010) 98–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Lee J, Kotliarova S, Kotliarov Y, Li A, Su Q, Donin NM, Pastorino S, Purow BW, Christopher N, Zhang W, Tumor stem cells derived from glioblastomas cultured in bFGF and EGF more closely mirror the phenotype and genotype of primary tumors than do serum-cultured cell lines, Cancer cell, 9 (2006) 391–403. [DOI] [PubMed] [Google Scholar]
- [18].Pollard SM, Yoshikawa K, Clarke ID, Danovi D, Stricker S, Russell R, Bayani J, Head R, Lee M, Bernstein M, Glioma stem cell lines expanded in adherent culture have tumor-specific phenotypes and are suitable for chemical and genetic screens, Cell stem cell, 4 (2009) 568–580. [DOI] [PubMed] [Google Scholar]
- [19].Garcia C, Dubois LG, Xavier AL, Geraldo LH, da Fonseca AC, Correia AH, Meirelles F, Ventura G, Romao L, Canedo NH, de Souza JM, de Menezes JR, Moura-Neto V, Tovar-Moll F, Lima FR, The orthotopic xenotransplant of human glioblastoma successfully recapitulates glioblastoma-microenvironment interactions in a non-immunosuppressed mouse model, BMC Cancer, 14 (2014) 923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].D’Alessandris QG, Biffoni M, Martini M, Runci D, Buccarelli M, Cenci T, Signore M, Stancato L, Olivi A, De Maria R, Larocca LM, Ricci-Vitiani L, Pallini R, The clinical value of patient-derived glioblastoma tumorspheres in predicting treatment response, Neuro Oncol, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Nguyen DG, Funk J, Robbins JB, Crogan-Grundy C, Presnell SC, Singer T, Roth AB, Bioprinted 3D primary liver tissues allow assessment of organ-level response to clinical drug induced toxicity in vitro, PloS one, 11 (2016) e0158674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Almela MJ, Lozano S, Lelièvre J, Colmenarejo G, Coterón JM, Rodrigues J, Gonzalez C, Herreros E, A new set of chemical starting points with Plasmodium falciparum transmission-blocking potential for antimalarial drug discovery, PLoS One, 10 (2015) e0135139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Filipuzzi I, Cotesta S, Perruccio F, Knapp B, Fu Y, Studer C, Pries V, Riedl R, Helliwell SB, Petrovic KT, High-Resolution Genetics Identifies the Lipid Transfer Protein Sec14p as Target for Antifungal Ergolines, PLoS genetics, 12 (2016) e1006374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Sadhukhan P, Saha S, Sinha K, Brahmachari G, Sil PC, Selective pro-apoptotic activity of novel 3, 3′-(aryl/alkyl-methylene) bis (2-hydroxynaphthalene-1, 4-dione) derivatives on human cancer cells via the induction reactive oxygen species, PloS one, 11 (2016) e0158694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Vartanian A, Singh SK, Agnihotri S, Jalali S, Burrell K, Aldape KD, Zadeh G, GBM’s multifaceted landscape: highlighting regional and microenvironmental heterogeneity, Neuro-oncology, 16 (2014) 1167–1175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Patel AP, Tirosh I, Trombetta JJ, Shalek AK, Gillespie SM, Wakimoto H, Cahill DP, Nahed BV, Curry WT, Martuza RL, Single-cell RNA-seq highlights intratumoral heterogeneity in primary glioblastoma, Science, 344 (2014) 1396–1401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Reinhold WC, Sunshine M, Liu H, Varma S, Kohn KW, Morris J, Doroshow J, Pommier Y, CellMiner: a web-based suite of genomic and pharmacologic tools to explore transcript and drug patterns in the NCI-60 cell line set, Cancer research, 72 (2012) 3499–3511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Paull KD, Lin CM, Malspeis L, Hamel E, Identification of novel antimitotic agents acting at the tubulin level by computer-assisted evaluation of differential cytotoxicity data, Cancer research, 52 (1992) 3892–3900. [PubMed] [Google Scholar]
- [29].Fung S, Xu C, Hamel E, Wager-Miller JB, Woodruff G, Miller A, Sanford C, Mackie K, Stella N, Novel indole-based compounds that differentiate alkylindole-sensitive receptors from cannabinoid receptors and microtubules: Characterization of their activity on glioma cell migration, Pharmacol Res, (2016). [DOI] [PubMed] [Google Scholar]
- [30].Wagenbach M, Domnitz S, Wordeman L, Cooper J, A kinesin-13 mutant catalytically depolymerizes microtubules in ADP, The Journal of cell biology, 183 (2008) 617–623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Nguyen TL, McGrath C, Hermone AR, Burnett JC, Zaharevitz DW, Day BW, Wipf P, Hamel E, Gussio R, A common pharmacophore for a diverse set of colchicine site inhibitors using a structure-based approach, J Med Chem, 48 (2005) 6107–6116. [DOI] [PubMed] [Google Scholar]
- [32].Nguyen TL, Cera MR, Pinto A, Presti LL, Hamel E, Conti P, Gussio R, De Wulf P, Evading Pgp activity in drug-resistant cancer cells: a structural and functional study of antitubulin furan metotica compounds, Molecular cancer therapeutics, 11 (2012) 1103–1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Ravelli RB, Gigant B, Curmi PA, Jourdain I, Lachkar S, Sobel A, Knossow M, Insight into tubulin regulation from a complex with colchicine and a stathmin-like domain, Nature, 428 (2004) 198–202. [DOI] [PubMed] [Google Scholar]
- [34].Zhao W, Zhou C, Guan Z-Y, Yin P, Chen F, Tang Y-J, Structural Insights into the Inhibition of Tubulin by the Antitumor Agent 4β-(1, 2, 4-triazol-3-ylthio)-4-deoxypodophyllotoxin, ACS chemical biology, 12 (2017) 746–752. [DOI] [PubMed] [Google Scholar]
- [35].Ewig CS, Berry R, Dinur U, Hill JR, Hwang MJ, Li H, Liang C, Maple J, Peng Z, Stockfisch TP, Derivation of class II force fields. VIII. Derivation of a general quantum mechanical force field for organic compounds, Journal of computational chemistry, 22 (2001) 1782–1800. [DOI] [PubMed] [Google Scholar]
- [36].Burnett JC, Lim C, Peyser BD, Samankumara LP, Kovaliov M, Colombo R, Bulfer SL, LaPorte MG, Hermone AR, McGrath CF, A threonine turnstile defines a dynamic amphiphilic binding motif in the AAA ATPase p97 allosteric binding site, Organic & Biomolecular Chemistry, 15 (2017) 4096–4114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Kellogg GE, Semus SF, Abraham DJ, HINT: a new method of empirical hydrophobic field calculation for CoMFA, Journal of computer-aided molecular design, 5 (1991) 545–552. [DOI] [PubMed] [Google Scholar]
- [38].Hothi P, Martins TJ, Chen L, Deleyrolle L, Yoon J-G, Reynolds B, Foltz G, High-throughput chemical screens identify disulfiram as an inhibitor of human glioblastoma stem cells, Oncotarget, 3 (2012) 1124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Hyman A, Drechsel D, Kellogg D, Salser S, Sawin K, Steffen P, Wordeman L, Mitchison T, Preparation of modified tubulins, Methods Enzymol, 196 (1991) 478–485. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.