

Abstract

Mycobacterium abscessus (Mab) is a rapidly growing species of multidrug-resistant nontuberculous mycobacteria that has emerged as a growing threat to individuals with cystic fibrosis and other pre-existing chronic lung diseases. Mab pulmonary infections are difficult, or sometimes impossible, to treat and result in accelerated lung function decline and premature death. There is therefore an urgent need to develop novel antibiotics with improved efficacy. tRNA (m1G37) methyltransferase (TrmD) is a promising target for novel antibiotics. It is essential in Mab and other mycobacteria, improving reading frame maintenance on the ribosome to prevent frameshift errors. In this work, a fragment-based approach was employed with the merging of two fragments bound to the active site, followed by structure-guided elaboration to design potent nanomolar inhibitors against Mab TrmD. Several of these compounds exhibit promising activity against mycobacterial species, including Mycobacterium tuberculosis and Mycobacterium leprae in addition to Mab, supporting the use of TrmD as a target for the development of antimycobacterial compounds.
Introduction
Mycobacterium abscessus (Mab) is an opportunistic human pathogen responsible for a wide range of lung, skin, and soft tissue infections. Although acquisition was thought to occur through environmental exposure (from soil and/or water), it has become apparent that indirect person-to-person transmission may be an important route of infection in cystic fibrosis (CF) patients (via fomite or long-lived infectious aerosol spread).1,2 In CF patients, Mab infection results in accelerated inflammatory lung damage and impaired quality and quantity of life.3−5Mab is challenging to treat because of its unique combination of drug-modifying enzymes, high number of efflux pumps, and genetic polymorphism of target genes, in addition to its complex and impermeable multilayered cell envelope. As a result, Mab possesses both intrinsic and acquired resistance to currently available antibiotics6 and is therefore currently difficult or sometimes impossible to treat.5 Consequently, there is an acute need for novel antibiotics with improved efficacy against Mab infection.
tRNA (m1G37) methyltransferase (TrmD), a member of the SpoU-TrmD (SPOUT) RNA methyltransferase family, catalyzes the transfer of a methyl group from S-adenosyl methionine (SAM) to the N1 position of guanosine 37 in bacterial tRNA when preceded by another guanosine in the sequence.7 The addition of this marker immediately adjacent to the anticodon acts to improve reading frame maintenance on the ribosome, preventing frameshift errors that would result in truncated and inactive peptides.8 TrmD has been shown to be essential for growth in a range of bacterial species from Staphylococcus aureus and Pseudomonas aeruginosa to mycobacteria, including Mycobacterium tuberculosis (Mtb) and Mab.9−12
The X-ray crystal structure of Mab TrmD shows a fold consistent with that of previously described TrmD enzymes,13−15 with a homodimeric structure exhibiting deep trefoil knots for SAM binding in two symmetry-related sites. These sites are formed by contributions from the N- and C-terminal domains of alternate subunits, which are separated by catalytically relevant interdomain linkers that show organization upon tRNA binding.16 The analogue of TrmD in archaea and eukaryotes, Trm5, is structurally distinct with a differing SAM binding mode.17 This reduces the chance of an inhibitor of Mab TrmD binding off-site in the human host, making this essential enzyme an attractive target for drug development.
The attractiveness of TrmD as a target is reflected in the recent application of a high-throughput screen against TrmD from P. aeruginosa to identify low-micromolar inhibitors.18 However, there are currently on-going efforts to validate the in vivo mechanism of these compounds before they are further developed as antibiotics. Structure-driven fragment-based methods provide an alternative methodology for the efficient design of potent inhibitors from low-molecular weight starting points and are now firmly established in both academia and industry.19 A fragment-based approach was used in a prior study against Haemophilus influenzae TrmD to develop selective inhibitors that ordered the interdomain linker in a similar manner to tRNA.20 Disappointingly, in general, these compounds only displayed weak activity when screened against a range of Gram-positive and Gram-negative pathogens, including efflux mutant strains of Escherichia coli and H. influenzae. In this work, a fragment-based approach was employed to design potent inhibitors against Mab TrmD that show growth inhibition across a range of pathogenic mycobacteria.
Results and Discussion
A fragment library of 960 fragments was screened against TrmD using differential scanning fluorimetry (DSF) as a primary screen. This resulted in 53 hits with a thermal shift cut-off value of 3 standard deviations from the negative control. These hits were then carried forward for soaking experiments using X-ray crystallography. Of these hits, density was observed for 27 fragments, all of which were shown to bind at the SAM binding pocket of Mab TrmD. The remaining 26 fragments did not show any electron density.12 This work herein describes the use of fragment-growing and merging strategies on fragment hits to develop novel compounds to inhibit Mab TrmD. Using the fragment-merging strategy, compounds have been developed that afford up to a 4-order of magnitude improvement in affinity against Mab TrmD, combined with inhibition of Mab growth in vitro and in a human macrophage infection model. A number of these key compounds display potent inhibition of Mtb growth in vitro, while one of the lead molecules also exhibits growth inhibition of intracellular Mycobacterium leprae.12
Fragment-Growing Strategy
The SAM co-factor has been shown by X-ray crystallography to bind to Mab TrmD simultaneously in the two symmetry-related active sites, with the adenine ring “anchoring” the molecule in place through hydrogen bonds to the backbone amides of residues Ile133, Gly134, Tyr136, and Leu138 (Figure 1a).12 This is formed from the loop of Val131 to Leu138, bordered behind and above by Pro83 and Pro85, and below by Ile133, Tyr136, Leu138, and Ala144, encompassing the “adenine binding pocket”. Fragment hit 1 (Kd 89 μM, LE 0.55, Table 1) was observed by X-ray crystallography to occupy the adenine binding pocket, with the pyrazole ring hydrogen bonding to the backbone amides of Gly134 and Ile133, and the carbonyl oxygen engaging the backbone amide of Leu138 (Figure 1b). This highly ligand-efficient fragment was chosen as a starting point for a fragment-growing strategy (Table 1). DSF screening was used as a first line screen with the elaborated compounds as a guide to prioritize compounds for isothermal titration calorimetry (ITC). The ester group of 1 was shown to be resistant to replacement, with the amide analogue 2 giving a thermal shift below +0.5 °C at 5 mM (+4.0 °C for 1) by DSF. From the X-ray crystal structure, the ethyl chain of 1 extends into the region occupied by the ribose portion of SAM, the “ribose binding pocket” (Figure 1a,b). This was used as a vector for a fragment-growing strategy. The phenyl ring introduced in 3 (Kd 33 μM, LE 0.41) was shown by X-ray crystallography to occupy the ribose-binding pocket (Figure S1a). This induced a movement in Tyr111 (2.5–5.1 Å at C-α depending on the active site), similar to that shown in the SAM-bound Mab TrmD X-ray crystal structure. From the synthesized analogues of 3 with varying substituents, 7 (Kd 14 μM, LE 0.39) demonstrated a ligand-efficient improvement in binding affinity. The X-ray crystal structures of 7- and 3-bound Mab TrmD both showed evidence of a second ligand molecule in the active site bound to the backbone amide nitrogen of Glu180 through its carbonyl oxygen (Figures 1c and S1a).
Figure 1.

(a) X-ray crystal structures of Mab TrmD in complex with SAM (PDB code 6NW6),12 illustrating both the whole dimer (individual protomers in blue/gray) with the positions of the active sites highlighted and one of the active sites in detail, and Mab TrmD in complex with (b) 1 (PDB code 6QOS)12 and (c) 7 (PDB code 6QRE), illustrating one of the active sites.
Table 1. Change in the Melting Temperatures (ΔTm), Affinities (Kd), and Ligand Efficiencies (LE) of Fragment Hit 1 and Compounds 2–7.

| compound | ΔTma (°C) | R | Kd (μM) | LEb |
|---|---|---|---|---|
| 1 | +0.4c | 89 ± 4 | 0.55 | |
| 2 | NDd | ND | ||
| 3 | +3.1 | H | 33 ± 2 | 0.41 |
| 4 | +0.4 | 4-NO2 | ND | |
| 5 | +0.9 | 4-CO2Me | ND | |
| 6 | +2.8 | 3-CN | 30 ± 1 | 0.36 |
| 7 | +3.5 | 3-OMe | 14 ± 1 | 0.39 |
100 μM ligand and 10 μM Mab TrmD.
kcal mol–1 per heavy atom.
Determined to be +4.0 °C with 5 mM ligand and 10 μM Mab TrmD.12
Determined to be +0.4 °C with 5 mM ligand and 10 μM Mab TrmD.
Compound 7 was used as the basis for further derivatives (Table 2), initially from the use of the 3-methoxy group as a vector for growth (see Figure S1). The use of aromatic rings in 9 (ΔTm +2.5 °C) and 11 (ΔTm +3.0 °C), an ester group in 8 (Kd 10 μM, LE 0.32) or chain extension in 10 (Kd 11 μM, LE 0.38) did not result in either a significant improvement in binding affinity or, when this was not determinable by ITC, an increase in thermal shift. The observation of a second ligand molecule for 7 in the active site was exploited for a fragment linking approach, with the attachment of a molecule of 1 to the scaffold of 7 through a propynyl linker in 12 (Kd 4.6 μM, LE 0.24); however, while an improved binding affinity was achieved, the ligand efficiency was adversely affected. The 5-position of the phenyl ring of 7 was also utilized as a vector for growth for the insertion of ring systems into the volume bordered by residues Glu112, Val137, Arg154, and Glu180. However, while the added pyrrolidinyl and pyridyl groups were tolerated in 13 (Kd 10 μM, LE 0.31) and 14 (Kd 6.7 μM, LE 0.31), a significant improvement in affinity was again not achieved, including with the addition of a methylene linker in 15 (Kd 12 μM, LE 0.28). Protein X-ray crystallography showed that the added groups in 8 and 14 did reach the desired regions of the active site in Mab TrmD (Figure S1b,c); however, attempts to improve the affinity using the stated strategies proved challenging. The lack of increase in affinity and the liability of the ester functional group in these compounds pointed to the need to develop an alternative strategy to target Mab TrmD.
Table 2. Change in the Melting Temperatures (ΔTm), Affinities (Kd), and Ligand Efficiencies (LE) of Compounds 8–15.


100 μM ligand and 10 μM Mab TrmD.
kcal mol–1 per heavy atom.
Not determined due to solubility.
Fragment-Merging Strategy
In a similar way to compound 1 (Kd 89 μM, LE 0.55, Table 2), the aminopyrazole ring system of fragment hit 16 (Kd 170 μM, LE 0.37) occupied the adenine binding pocket, with the pyrazole ring hydrogen bonding to the backbone amides of Tyr136 and Leu138 and the amino group engaging Gly134 and the alcohol side chain of Ser132 (Figure 2a). In contrast, the 4-methoxyphenyl ring of 16 extended into the ribose binding pocket. The methoxy group showed no direct interactions with the surrounding residues; however, the corresponding phenyl (17) and 4-tolyl (18) analogues lacking the methoxy group afforded relatively low ΔTm values (1.4–1.5 °C). The methoxy group of 16 was also shown to possess a preference for the 4-position of the phenyl ring, with the 3-methoxyphenyl analogue 19 providing a negative thermal shift (Table 3). The ribose binding pocket was shown to be occupied by the indole ring of fragment hit 20 (Kd 260 μM, LE 0.41), where the indole nitrogen indirectly interacts with the backbone carbonyl of Leu138 through a water molecule. The 6-boronic acid group of 20 partially extends into the adenine binding pocket, engaging the backbone amides of residues Tyr136 and Leu138 through hydrogen bonds, as well as two water molecules that occupy the remaining space in the pocket, interacting with the backbone amides of Val131, Ile133, and Gly134 in addition to the side chain of Ser132 (Figure 2b). The isosteric replacement of the boronic acid group with a carboxylic acid, as shown in fragment 21, was shown to not be tolerated, with no ΔTm observed. Further, examination of the structure with 5-boronic acid isomer 22 bound using X-ray crystallography showed the indole ring “flipping” to maintain the hydrogen bonding interactions of the boronic acid, with the indole nitrogen interacting with an extensive hydrogen-bonded water network in the rear of the active site (Figure S2a). Even though the boronic acid of 22 was shown to form the same hydrogen-bonding water network in the adenine binding pocket as 20, it gave a lower ΔTm (Table 3). The overlap of the respective 4-methoxyphenyl and indole ring systems of 16 and 20 (Figure 2c), and their spanning of the adenine and ribose binding pockets, offers the possibility of a fragment-merging strategy. This was explored successfully with 23 (Kd 110 μM, LE 0.36), providing a new aminopyrazole-indole scaffold with both improved affinity and prospects for further elaboration relative to the parent fragments (Figure 2d). Compound 23 was shown by X-ray crystallography to maintain the hydrogen-bonding interactions of the original fragment 16, which remained unaffected throughout all subsequent additions to the scaffold, with the indole nitrogen providing a vector for elaboration that the fragment hit 16 lacked.
Figure 2.

X-ray crystal structures of Mab TrmD in complex with (a) 16 (PDB code 6QOT), (b) 20 (PDB code 6QOU), (c) 16 and 20 (overlay of PDB code 6QOT and PDB code 6QOU), and (d) 23 (PDB code 6QQS), illustrating one of the active sites.12
Table 3. Change in the Melting Temperatures (ΔTm), Affinities (Kd), and Ligand Efficiencies (LE) of Fragment Hits 16 and 20, Their Structural Analogues 17–19 and 21–22, and Compound 23.

Elaboration of Compound 23
Elaboration of 23 was initially carried out from the indole nitrogen with the introduction of a 4-methoxybenzyl group in 24a (Kd 59 μM, LE 0.24) (Table 4). While the gain in affinity was less than desired, the added group reached the portion of the active site occupied by the methionine moiety of SAM defined by Pro85, Glu112, Val137, Arg154, and Glu180 (Figure 3a). The synthesis of the 3-methoxybenzyl 24b (Kd 13 μM, LE 0.28) and benzyl 24c (Kd 19 μM, LE 0.29) derivatives demonstrated improved affinities (GE 0.14 and 0.15, respectively). While the 2-benzamide 24e gave a lower ΔTm of +1.4 °C, further improvements in affinity were realized relative to 24c with the synthesis of the 2-cyanobenzyl 24d (Kd 12 μM, LE 0.28) and 2-picolyl 24f (Kd 12 μM, LE 0.30) analogues. Attention was also focused on elaboration from the 4-position of the pyrazole ring of 23. This vector was shown by X-ray crystallography to face a narrow and elongated pocket formed by Pro83, Thr84, Val131, Ser132, Ile133, and Ala144 (Figure 2d). The addition of a nitrile group in 25 (Kd 5.0 μM, LE 0.43) afforded a jump in affinity from 23 (Kd 110 μM, LE 0.36), with the added functionality highly group efficient (GE 0.91). An X-ray crystal structure of 25 bound to TrmD showed that the nitrile extended into the pocket, engaging the backbone amides of Thr84 and Ile133, with no major changes in the ligand binding orientation or conformation of neighboring residues (Figure 3e). The nitrile was subsequently incorporated into 24f to afford 26f (Kd 0.50 μM, LE 0.36), with the added nitrile functional group affording a similar jump in affinity as in 25 (GE 0.95 and 0.91 respectively).
Table 4. Change in the Melting Temperatures (ΔTm), Affinities (Kd), and Efficiency Metrics of Compounds 24a–f, 25, 26c, and 26f.
![]()

100 μM ligand and 10 μM Mab TrmD.
kcal mol–1 per heavy atom.
Kd and LE previously reported.12
Not determined due to solubility.
Figure 3.

X-ray crystal structures of Mab TrmD in complex with (a) 24a (PDB code 6QQT), (b) 24b (PDB code 6QRC), (c) 24c (PDB code 6QQW), (d) 24f (PDB code 6QQX),12 (e) 25 (PDB code 6QQU), and (f) 26f (PDB code 6QQY),12 illustrating one of the active sites.
Following the development of 26f, the corresponding 3-pyridyl isomer 26g (Kd 0.18 μM, LE 0.38, Table 5) afforded a greater than 2-fold improvement in binding affinity without additional interactions being observed in the X-ray crystal structure of ligand-bound Mab TrmD (Figures 3f and 4a). The late stage functionalization of 26g through addition of a chlorine atom on the 3-position of its indole ring in 27g (Kd 8.5 μM, LE 0.28) was not tolerated (Table 5). While this modification of 26g did not result in a significant shift in the indole ring, one of the ordered water molecules in the rear of the active site bound to Pro83 was displaced (Figure 4b). 2-Hydroxypyridyl/pyridone derivatives of 26g, 26j (Kd 3.2 μM, LE 0.30), and 26k (Kd 1.3 μM, LE 0.32) were synthesized to engage neighboring residues Arg154 and Glu180, respectively, through the added oxygen atoms. While the X-ray crystal structure of Mab TrmD with 26j bound did show evidence of a hydrogen bond between the added oxygen atom of the ligand and Arg154 (Figure 4c), neither derivative possessed an improved binding affinity. In contrast, the exploration of the alternative 4-pyridyl isomer 26l (Kd 0.12 μM, LE 0.39) resulted in a 4-fold decrease of Kd value relative to 26f, with the nitrogen atom of its pyridyl ring lying 2.9 Å from the carboxylate group of Glu180 (Figure 4d).
Table 5. Change in the Melting Temperatures (ΔTm), Affinities (Kd), and Efficiency Metrics of Compounds 26g, 26j–o, and 27g.
![]()

100 μM ligand and 10 μM Mab TrmD.
kcal mol–1 per heavy atom.
Not determined due to solubility.
Figure 4.

X-ray crystal structures of Mab TrmD in complex with (a) 26g (PDB code 6QQZ), (b) 27g (PDB code 6QR1), (c) 26j (PDB code 6QR2), and (d) 26l (PDB code 6QR0), illustrating one of the active sites, and Mab TrmD in complex with 26o (PDB code 6QR3), illustrating active sites 1 (e) and 2 (f).
Modification of the pyridyl ring of 26l to the corresponding 4-quinolyl ring system in 26m afforded a significant thermal shift (ΔTm +10.3 °C at 100 μM), suggesting that the bicyclic ring was tolerated (Table 5). However, this modification adversely affected solubility, precluding the determination of binding affinity by ITC at the desired concentrations. The replacement of the pyridyl ring with more soluble saturated alternatives resulted in the development of racemic compounds N-methyl piperidin-2-yl 26n (Kd 0.59 μM, LE 0.34) and N-methyl piperidin-3-yl 26o (Kd 0.36 μM, LE 0.35), whose ITC isotherms could be fitted by a one-site binding model (Figures S7d and S8a). However, racemic mixtures can afford deceptively simple isotherms under certain circumstances, such as when the enthalpies of binding of the individual enantiomers are equivalent, with the curve mimicking the Kd of the weaker component.21 Hence, a reverse ITC titration was also performed for 26o. This afforded a simple curve that could be fitted by similar parameters to the forward titration (Figure S8b), implying that the enantiomers of 26o are equipotent. The X-ray crystal structure of 26n showed that the nitrogen atom of its piperidinyl ring interacts with the backbone carbonyl of Tyr111 (Figure S2c), while that of 26o formed electrostatic interactions with either of Glu112 or Glu180 depending on the active site (Figure 4e,f).
The targeting of Glu112 and Glu180 through electrostatic interactions with saturated heterocyclic ring systems was continued, with the pyridyl ring of 26f providing a good starting point for the presentation of amines at the top of the active site (Table 6). The implementation of a similar strategy against H. influenzae TrmD with engagement of the catalytic residue Asp169 resulted in improved inhibition and the ordering of the interdomain linker.20 The addition of a pyrrolidinyl ring through a methylene linker to 26f in 28 (Kd 92 nM, LE 0.32) showed that the strategy was applicable to this lead series. Modification of the scaffold through replacement of the pyridyl ring of 28 with a phenyl ring in 29a (Kd 27 nM, LE 0.34) was beneficial to the binding affinity, in contrast to the difference in affinities between the phenyl and pyridyl derivatives 24c (Kd 19 μM, LE 0.29) and 24f (Kd 12 μM, LE 0.30). The X-ray crystal structure of Mab TrmD in complex with 29a showed the pyrrolidinyl ring occupying the binding sites in two conformations, thereby interacting with the carboxylates of either Glu112 or Glu180, while Asp169 and the rest of the interdomain linker remained unstructured (Figure 5a,b). Replacement of the nitrile group on the 4-position of the pyrazole ring of 29a was also explored through the methyl-substituted alternative 30a (Kd 2.0 μM, LE 0.27), which showed a higher binding affinity than the unsubstituted derivative 31a (Kd 0.49 μM, LE 0.31). Derivatives of 29a with alternative substituents in place of its pyrrolidinyl ring were trialed, including 29b (Kd 70 nM, LE 0.31) whose piperidinyl ring was shown to interact with Glu112 in preference to Glu180 (Figure 5c). The use of a morpholinyl ring resulted in a significant attenuation of binding affinity in 29c (Kd 0.19 μM, LE 0.30), possibly because of the impact of reduced basicity of the morpholinyl nitrogen on its interactions with Glu112 and Glu180. Protein X-ray crystallography showed that compound 29c adopted a similar binding pose as 29b, but with the morpholinyl ring rotated and orienting its oxygen atom toward Pro57 (Figure 5d). In contrast to 29c, the N-methyl piperazinyl ring of 29d (Kd 73 nM, LE 0.30) was oriented away from Glu112 with its methyl group extending further into the pocket defined by the loop from Ala176 to Glu180 (Figure 5e). The methyl substituent on the piperazinyl ring of 29d was expanded to an isopropyl group in 29e (Kd 0.10 μM, LE 0.28), whose electron density maps suggest two alternative binding poses with the isopropyl group oriented toward either Glu112 or the loop from Ala176 to Glu180 at the top of the active site (Figure 5f).
Table 6. Change in the Melting Temperatures (ΔTm), Affinities (Kd), and Efficiency Metrics of Compounds 28, 29a–e, 30a, and 31a–b.
![]()

Figure 5.

X-ray crystal structures of Mab TrmD in complex with 29a (PDB code 6QR6),12 illustrating active sites 1 (a) and 2 (b), and Mab TrmD in complex with (c) 29b (PDB code 6QR7), (d) 29c (PDB code 6QR9), (e) 29d (PDB code 6QR8),12 and (f) 29e (PDB code 6QRD), illustrating one of the active sites.
Native Mass Spectrometry
Native mass spectrometry (native MS) was used to investigate the structure and interactions of Mab TrmD. The protein alone presented as the dimer with an observed mass of 53 187 Da, centered around the 15+ charge state (Figure 6). The observed mass is slightly higher than the theoretical mass of 52 520 Da because of the presence of solvent and buffer molecules that remain weakly bound to the protein under the soft ionization conditions employed.22 In the presence of 5% dimethylsulfoxide (DMSO), the native mass spectrum of Mab TrmD shifted to a lower charge state distribution (Figure 6). This is consistent with previous studies on the effect of DMSO on protein charging in electrospray ionization (ESI).23,24
Figure 6.

Native mass spectrometry of Mab TrmD with compounds 24c, 26c, and 29a. Experiments with ligands contain 5% DMSO.
Native MS was used to study the interaction between the small molecules and Mab TrmD. A titration experiment performed with 24c revealed increasing occupancy of the protein as the concentration of the ligand was increased (Figure 6). At 20 μM of Mab TrmD and 12.5 μM of 24c (with 5% DMSO), a single molecule of the ligand could be observed to bind to the 13+ charge state of the protein, although the majority of that charge state remained unbound. At 25 μM of 24c, both singly and doubly bound Mab TrmD could be detected at the 13+ charge state. The proportion of doubly bound Mab TrmD increased at 50 μM of 24c, while at 100 μM, the 13+ charge state of the protein was entirely doubly bound. A similar trend could be observed for the 14+ charge state, except that the degree of occupancy of the protein was lower at each concentration of 24c due to the greater Coulombic repulsion experienced by the protein-ligand complexes at 14+ compared to at 13+. The lack of triply bound Mab TrmD observed even at excess concentrations of 24c suggests that little, if any, of the small molecule bound non-specifically to the protein dimer.
Compounds 26c and 29a were also screened against Mab TrmD using native MS at a ligand concentration of 100 μM (Figure 6). Interestingly, despite its tight binding affinity for Mab TrmD as measured by ITC, complexes formed with 29a showed low gas-phase stabilities in native MS. Dissociation of 29a from Mab TrmD led to a shift in the charge state distribution of the protein to lower charge states, which was confirmed for individual precursor charge states by MS/MS (data not shown). This could be rationalized by the dissociation of positively charged 29a molecules from Mab TrmD, leading to a reduction of the overall positive charge of the protein. In contrast, 24c and 26c, being potentially less cationic than 29a, did not exhibit this charge reduction behavior. Overall, the native MS data confirm the direct binding of the ligands to Mab TrmD and suggest that the interaction is specific, with each Mab TrmD dimer binding to two molecules of the ligand.
Synthetic Chemistry
Synthesis of compound 2 (Scheme 1) began with addition of a benzyl protecting group to fragment hit 1 through heating at reflux with benzyl bromide in acetone (93% yield), followed by alkaline hydrolysis of the ester group of 32 to a carboxylic acid (94% yield). Acid 33 was converted by the action of 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide (EDC) to the amide 34 (84% yield) and then debenzylated to 2 through transfer hydrogenation with palladium on carbon, formic acid, and tert-butanol (64% yield). The production of esters 3–7 (Scheme 1) started with hydrolysis of 1 to the corresponding carboxylic acid 35 (96% yield), whose pyrazole was protected with a tetrahydropyran ring through treatment with 3,4-dihydro-2H-pyran under acidic conditions to give 36 (43% yield). Carboxylic acid 36 was used to synthesize the final ester compounds 3–7 by two alternative two step protocols, involving either initial treatment with a benzyl halide and base in dimethylformamide (DMF) or EDC-mediated ester coupling with a benzyl alcohol. Both protocols ended with removal of the tetrahydropyran protecting group by trifluoroacetic acid (TFA; 5–57% yield overall).
Scheme 1. Synthesis of Compounds 2–7.
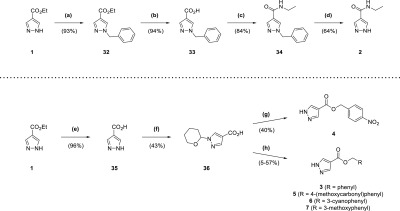
Reagents and conditions: (a) BnBr, K2CO3, acetone, reflux, 7 h; (b) NaOH, THF, H2O, 70 °C, 150 min; (c) EtNH2·HCl, DIPEA, DMAP, EDC·HCl, DCM, 15 h; (d) Pd/C (10 wt % loading), formic acid, tBuOH, 50 °C, 2 h; (e) NaOH, H2O, reflux, 2 h; (f) 3,4-dihydro-2H-pyran, TsOH·H2O, EtOAc, DMF, overnight; (g) (i) 4-nitrobenzyl alcohol, DIPEA, DMAP, EDC·HCl, DCM, overnight (ii) TFA, 5 min; (h) (i) RCH2Br, DIPEA, DMF, 90 min to 2 h (ii) TFA, 10–12 min.
The routes for synthesis of compounds 8–11 (Scheme 2) involved protection of phenol 37 with a triisopropylsilyl protecting group (81% yield), followed by reduction of the ester group of 38 to the corresponding alcohol 39 using lithium aluminium hydride (83% yield). Alcohol 39 was put through an EDC-mediated ester coupling reaction with 36, with the silyl protecting group removed using tetra-n-butylammonium fluoride (TBAF; 28% yield overall). Phenol 40 was treated with an alkyl bromide and base in DMF, and then the tetrahydropyran-protecting group was removed with TFA to afford 8 and 9 (50–53% yield overall). In contrast, 10 and 11 were synthesized from 40 through initial coupling with an alcohol under Mitsunobu conditions with triphenylphosphine and diisopropyl azodicarboxylate, followed by treatment with TFA (24–46% yield overall). Compounds 12–15 all utilized bromoarene 44 in their respective syntheses (Scheme 2), itself synthesized by a sequence involving methyl iodide alkylation of 41 (97% yield), followed by lithium aluminium hydride reduction of ester 42 to alcohol 43 (71% yield) that was coupled with 36 using EDC (63% yield). The Sonogashira cross-coupling of 44 with alkyne 45, itself produced by treatment of fragment hit 1 with propargyl bromide in a suspension of potassium carbonate and DMF (81% yield), afforded 46 (43% yield). TFA-mediated deprotection of 46 provided 12 (63% yield). 13 was synthesized by Buchwald–Hartwig amination of 44 with pyrrolidine, followed by TFA deprotection (7% yield overall). In contrast, 14 was produced through a Suzuki–Miyaura cross-coupling reaction of 44 with 3-(bromomethyl)pyridine, followed by TFA deprotection (44% yield overall). For 15, 44 was converted to the corresponding pinacol boronic ester 47 by a Miyaura-borylation with bis(pinacolato)diboron (44% yield), which was then cross-coupled with 3-(bromomethyl)pyridine in a Suzuki–Miyaura reaction and deprotected with TFA (18% yield overall).
Scheme 2. Synthesis of Compounds 8–15.

Reagents and conditions: (a) TIPSCl, imidazole, DMF, 2 h; (b) LiAlH4 (2.4 M in THF), Et2O, 0 °C to rt, 3 h; (c) (i) 36, DIPEA, DMAP, EDC·HCl, DCM, overnight (ii) TBAF (1 M in THF), 10 min; (d) (i) Cs2CO3, RCH2Br (·HBr), DMF, 2 h (ii) TFA, 15 min; (e) (i) PPh3, RCH2OH, DIAD, THF, 1 h to overnight (ii) TFA, 15 min; (f) MeI, K2CO3, DMF, 3 d; (g) LiAlH4 (2.4 M in THF), Et2O, 0 °C to rt, overnight; (h) 36, DIPEA, DMAP, EDC·HCl, DCM, overnight; (i) propargyl bromide (80 wt % solution in toluene), K2CO3, DMF, 3 d; (j) 44, Pd(dppf)Cl2, CuI, NEt3, DMF, 100 °C, 4 h; (k) TFA, 15 min; (l) (i) pyrrolidine, Cs2CO3, RuPhos, RuPhos Pd G1 methyl tert-butyl ether adduct, tBuOH, 85 °C, 15 h (ii) TFA, 30 min; (m) (i) 3-pyridinylboronic acid, Pd(dppf)Cl2, K2CO3, dioxane, H2O, 100 °C μW, 30 min (ii) TFA, 15 min; (n) B2pin2, Pd(dppf)Cl2, KOAc, dioxane, 80 °C, 3 h; (o) (i) 3-(bromomethyl)pyridine HBr, Pd(PPh3)4, K2CO3, DME, H2O, 95 °C, 10 h (ii) TFA, 30 min.
The synthetic route for the 5-aminopyrazole analogue 19 (Scheme 3) began with nucleophilic attack of ester 48 by a nitrile anion, generated from n-butyllithium and acetonitrile. This resulted in the corresponding β-ketonitrile 49 (99% yield), which was heated at reflux with hydrazine in ethanol to form 19 (73% yield).25 The synthesis of 23 (Scheme 3) was similar to 19, using indole 50 as the starting point, which was protected with a tert-butyldimethylsilyl group to form 51 (59% yield). After synthesis of the corresponding β-ketonitrile, deprotection was accomplished by TBAF without prior purification affording 52 (99% yield overall), which was heated at reflux with hydrazine in ethanol (59% yield). The deprotected β-ketonitrile 52 was also utilized in the synthesis of 25 (Scheme 3), with its activated methylene group treated with trichloroacetonitrile and sodium acetate.26 The resultant functionalized compound 53 was purified (87% yield) before heating at reflux with hydrazine in ethanol to afford 25 (24% yield).
Scheme 3. Synthesis of Compounds 19, 23, 24a–f, 25, 26c, and 26f.
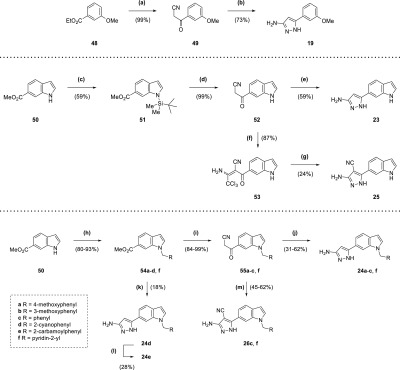
Reagents and conditions: (a) n-butyllithium (1.6 M in hexanes), acetonitrile, THF, −78 °C, 30 min; (b) N2H4·H2O, EtOH, reflux, 6 h; (c) NaH, TBDMSCl, THF, 0 °C to rt, 10 h; (d) (i) n-butyllithium (1.6 M in hexanes), acetonitrile, toluene, −78 °C, 1 h (ii) TBAF (1 M in THF), THF, 20 min; (e) N2H4·H2O, EtOH, reflux, 12 h; (f) CCl3CN, NaOAc, EtOH, 90 min; (g) N2H4·H2O, EtOH, reflux, 22 h; (h) RCH2X (·HX), Cs2CO3, acetonitrile, reflux, 1–15 h; (i) n-butyllithium (1.6 M in hexanes), acetonitrile, toluene/THF, −78 °C, 15 min to 1 h; (j) N2H4·H2O, EtOH, reflux, 5–13 h; (k) (i) n-butyllithium (1.6 M in hexanes), acetonitrile, toluene, −78 °C, 1 h (ii) N2H4·H2O, EtOH, reflux, 5 h; (l) NaOH, H2O, 100 °C, 7 h; (m) (i) CCl3CN, NaOAc, EtOH, 90 min to 9 h (ii) N2H4·H2O, EtOH, reflux, 5–15 h.
Synthesis of compounds 24a–c and 24f (Scheme 3) utilized formation of β-ketonitriles 55a–c and 55f from the corresponding methyl esters 54a–c and 54f (84–99% yield) and subsequent reaction with hydrazine (31–62% yield). Unlike 23, the indole nitrogen was protected by the benzyl or picolyl groups. In the cases of 54a–d and 54f, these were added to indole 50 by heating at reflux with caesium carbonate and benzyl or picolyl halides in acetonitrile (80–93% yield). Because of the production of significant amounts of side products in the synthesis of the corresponding β-ketonitrile from 54d, the resultant product was taken forward crude in the synthesis of 24d (18% yield overall) (Scheme 3). The nitrile group of 24d was also hydrolyzed to an amide, affording 24e (28% yield) (Scheme 3). Compounds 26c and 26f were synthesized in a similar manner to 25, using the previously generated β-ketonitriles 55c and 55f from the production of 24c and 24f (Scheme 3). However, the intermediates from the reaction with trichloroacetonitrile were taken forward crude, after aqueous work up, to the subsequent reaction with hydrazine (45–62% yield overall).
The production of compounds 26g–m and 27g (Scheme 4) involved the synthesis of methyl esters 54g–i and 54l–m from alkylation of indole 50 using a mixture of the corresponding electrophile with sodium hydride in DMF. Following the consumption of starting material, the reaction was diluted with methanol and sulfuric acid and heated at reflux to recover the carboxylic acid side-product. While the electrophiles for the synthesis of methyl esters 54g and 54l were commercially available, 54h–i and 54m required the synthesis of precursors. Alkyl halide 58 for 54i was generated from alcohol 57 by treatment with tosyl chloride and DMAP (17% yield), itself produced by the reduction of ester 56 using lithium aluminium hydride (91% yield). Alcohol 60, itself a product of the reduction of ester 59 using sodium borohydride (67% yield), was treated with tosyl chloride in a similar manner to 57; however, the product was taken forward crude for reaction with indole 50 to afford 54h (31% yield overall). With 54m, aldehyde 61 was reduced to the corresponding alcohol 62 by sodium borohydride (79% yield),27 which was heated at reflux in aqueous HBr to afford a crude material that was taken forward for reaction with 50 (65% yield overall). The methyl esters 54g–i and 54l–m were converted to the corresponding β-ketonitriles 55g–i and 55l–m (26–82% yield), which were used to produce the 4-cyanopyrazole compounds in the same manner as 26c and 26f (41–61% yield overall). 2-Hydroxypyridyl/pyridone compounds 26j and 26k were synthesized from methoxypyridyl precursors 26h and 26i, respectively, using nucleophilic demethylation with lithium chloride and p-toluenesulfonic acid in DMF (47–51% yield).28 Meanwhile, 3-chloroindole derivative 27g was synthesized directly from 26g through treatment with N-chlorosuccinimide in DMF (63% yield). Synthesis of compounds 26n and 26o (Scheme 4) began with conversion of racemic alcohols 63 and 65, respectively, to electrophilic alkylating agents. Alkyl chloride 64 was produced from 63 using thionyl chloride (58% yield), while treatment of 65 with tosyl chloride, triethylamine, and DMAP generated the alkyl tosylate 66 (36% yield). Synthesis of the methyl esters 54n–o from indole 50 and the agents 64 and 66 with sodium hydride in DMF involved heating at 60 °C (27–45% yield), with NaI also added as a catalyst for 54o. These esters were converted to the corresponding β-ketonitriles 55n–o (32–67% yield), which were used to produce the 4-cyanopyrazole compounds in the same manner as 26c and 26f (38–57% yield overall).
Scheme 4. Synthesis of Compounds 26g–o and 27g.

Reagents and conditions: (a) LiAlH4, THF, 0 °C, 1 h; (b) TsCl, DMAP, NEt3, DCM, 18 h; (c) (i) RCH2X·HX (58 for 54i), NaH, DMF, 0 °C to rt, 45 min to 1 h (ii) MeOH, H2SO4, reflux, 1–16 h; (d) NaBH4, EtOH, 0 °C to rt, 1 d; (e) (i) TsCl, DMAP, NEt3, DCM, 2 h (ii) 50, NaH, DMF, 0 °C to rt, 30 min (iii) MeOH, H2SO4, reflux, 90 min; (f) NaBH4, MeOH, 0 °C, 90 min; (g) (i) aqueous HBr (48%), reflux, 90 min (ii) 50, NaH, DMF, 0 °C to rt, 15 min (iii) MeOH, H2SO4, reflux, 2 h; (h) n-butyllithium (1.6 M in hexanes), acetonitrile, toluene/THF, −78 °C, 30 min to 1 h; (i) (i) CCl3CN, NaOAc, EtOH, 13–36 h (ii) N2H4·H2O, EtOH, reflux, 6–21 h; (j) NCS, DMF, 7 h 30 min; (k) LiCl, TsOH·H2O, DMF, 120 °C, 25 min; (l) SOCl2, DCM, reflux, 7 h; (m) 50, NaH, DMF, 0–60 °C, 1 h (ii) MeOH, H2SO4, reflux, 1 h; (n) TsCl, DMAP, NEt3, DCM, 4 h; (o) (i) 50, NaH, NaI, DMF, 0–60 °C, 1 h; (p) n-butyllithium (1.6 M in hexanes), acetonitrile, THF, −78 °C, 30 min to 1 h; (q) (i) CCl3CN, NaOAc, EtOH, 3 h 30 min to 18 h (ii) N2H4·H2O, EtOH, reflux, 15 h to 1 d.
Synthesis of 28 (Scheme 5) began with regioselective reduction of diester 67 using a combination of sodium borohydride and CaCl2 (84% yield),29 with protection of the resultant alcohol 68 as a tetrahydropyranyl ether in 69 (77% yield). The ester of 69 was reduced with lithium aluminium hydride to the corresponding alcohol 70 (64% yield), which was converted to a mesylate then reacted with pyrrolidine and Cs2CO3 in DMF to afford 71 (66% yield overall). The tetrahydropyranyl ether 71 was deprotected with p-toluenesulfonic acid in ethanol (86% yield), and the resultant alcohol 72 converted to a mesylate that was reacted with indole 50 using sodium hydride and NaI in DMF at elevated temperatures (7% yield overall). The methyl ester 73 was converted to the corresponding β-ketonitrile 74 (71% yield), which was used to produce 28 in the same manner as 26c and 26f (6% yield overall). Compounds 29a–e (Scheme 5) were all produced from methyl esters 79a–e. Esters 79a–d were synthesized by the microwave-assisted condensation of aldehydes 78a–d and indoline 75 (53–67% yield),30 itself produced by the reduction of indole 50 using sodium cyanoborohydride in acetic acid (66% yield).31 Aldehydes 78a–d were generated in two steps by reductive amination of 4-cyanobenzaldehyde 76 with various secondary amines to form 77a–d (70–85% yield), followed by reduction using diisobutylaluminium hydride (46–75% yield). In comparison, ester 79e required the protection of one of the alcohols of 80 with a tert-butyldiphenylsilyl group (38% yield), followed by conversion of the remaining alcohol of 81 to a crude mixture of the mesylate and chloride that was used to alkylate 50 with sodium hydride and DMF, with subsequent deprotection of the silyl protecting group with TBAF (72% yield overall). The benzyl alcohol of 82 was treated with mesyl chloride and triethylamine, then functionalized with 1-isopropylpiperazine to afford 79e (43% yield overall). The methyl esters 79a–e were converted to the corresponding β-ketonitriles 83a–e (77–87% yield), which were used to produce 29a–e in the same manner as 26c and 26f (32–75% yield overall) or 31a–b by heating at reflux with hydrazine in ethanol (22–52% yield). Methyl ester 79a was also used to produce 30a (Scheme 5) through initial treatment with n-butyllithium and propionitrile to afford β-ketonitrile 84a (35% yield), which was then heated at reflux with hydrazine in ethanol (21% yield).
Scheme 5. Synthesis of Compounds 28, 29a–e, 30a, and 31a–b.

Reagents and conditions: (a) NaBH4, CaCl2, MeOH, THF, 0 °C, 90 min; (b) MsOH, 3,4-dihydro-2H-pyran, DCM, 150 min; (c) LiAlH4, THF, 0 °C, 45 min; (d) (i) MsCl, NEt3, DCM, 0 °C to rt, 1 h (ii) pyrrolidine, Cs2CO3, DMF, 14 h; (e) TsOH·H2O, ethanol, 50 °C, 30 min; (f) (i) MsCl, NEt3, DCM, 0 °C to rt, 150 min; (ii) 50, NaH, NaI, DMF, 0 to 60 °C, 75 min (iii) MeOH, H2SO4, reflux, 14 h; (g) n-butyllithium (1.6 M in hexanes), acetonitrile, THF, −78 °C, 1 h; (h) (i) CCl3CN, NaOAc, EtOH, 36 h (ii) N2H4·H2O, EtOH, reflux, 1 d; (i) NaCNBH3, AcOH, 0 °C to rt, 7 h; (j) Na(OAc)3BH, RH, AcOH, DCM, 80 min to 15 h; (k) DIBAL-H, THF, 0 °C to rt, 90 min to 1 h; (l) PhCO2H, toluene, 200 °C μW, 20–30 min; (m) TBDPSCl, imidazole, DMF, 21 h; (n) (i) MsCl, NEt3, DCM, 0 °C to rt, 1 h (ii) 50, NaH, DMF, 0 °C to rt, 45 min (iii) TBAF (1 M in THF), 30 min; (o) (i) MsCl, NEt3, DCM, 0 °C to rt, 90 min (ii) RH, Cs2CO3, DMF, 12 h; (p) n-butyllithium (1.6 M in hexanes), acetonitrile, THF, −78 °C, 20 min to 1 h; (q) (i) CCl3CN, NaOAc, EtOH, 4 h to 2 d (ii) N2H4·H2O, EtOH, reflux, 7 h to 1 d; (r) N2H4·H2O, EtOH, reflux, 12–21 h; (s) n-butyllithium (1.6 M in hexanes), propionitrile, toluene, −78 to 0 °C, 2 h; (t) N2H4·H2O, EtOH, reflux, 1 d.
Screening of Compounds against Mycobacteria
The effect of compounds on Mab and Mtb growth in liquid culture was determined and revealed promising minimal inhibitory concentrations (MIC) for many compounds (Table 7). A lack of correlation between target binding affinity and activity against bacteria was observed, indicating likely effects of differential permeability, retention, and metabolism of compounds on their in vitro activity. While compounds performed similarly against Mab, 29e afforded the best results against Mtb across different media types and carbon sources, with MIC values of 1.6 and 2.3 μM achieved with 7H9 supplemented by dipalmitoyl phosphatidylcholine (DPPC) and glucose respectively. Both glucose and DPPC are carbon sources predicted to be relevant during the in vivo pathogenesis of Mtb,32 and the activity of the compounds during catabolism of both of these supports the notion that TrmD inhibition will inhibit bacterial growth under any metabolic condition. The poor activity in 7H9/ADC was likely due to high protein binding because this medium contains 0.4% bovine serum albumin. Hence, overcoming protein binding will be an important component of future drug design strategies.
Table 7. Minimum Inhibitory Concentrations (MIC) against Mab and Mtb of Compounds 26l, 28, 29a–e, and 31a–b.
| H37Rv Mtb MIC (μM) |
||||||
|---|---|---|---|---|---|---|
| compound | Mab TrmD Kd (μM) | Mab MIC (μM) | GAST-Fe | 7H9/ADC | 7H9/glucose | 7H9/DPPC |
| 26l | 0.12 ± 0.02 | 50 | 50 | >100 | ND | ND |
| 28 | 0.092 ± 0.018 | 50 | ND | ND | ND | ND |
| 29aa | 0.027 ± 0.004 | 50 | 50 | >100 | 25 | 12.5 |
| 29b | 0.070 ± 0.029 | 50 | 50 | 100 | 25 | 12.5 |
| 29c | 0.19 ± 0.02 | 50 | 50 | >100 | 100 | 50 |
| 29da | 0.073 ± 0.030 | 50 | 25 | 50 | 12.5 | 6.3 |
| 29e | 0.10 ± 0.02 | 50 | 6.3 | 12.5 | 2.3 | 1.6 |
| 31aa | 0.49 ± 0.21 | 50 | 25 | 100 | 12.5 | 6.3 |
| 31b | 0.71 ± 0.09 | 50 | 50 | >100 | 25 | 12.5 |
Mab MIC and H37Rv Mtb MIC (7H9/DPPC) previously reported.12
In addition to the results against Mab and Mtb in liquid culture, activity was observed by compounds from the lead series in macrophage infection models against Mab and M. leprae, demonstrating the utility of the series against mycobacteria in vivo.12
Conclusions
With the application of a fragment-growing approach to a highly ligand-efficient fragment hit of Mab TrmD failing to afford a significant improvement in binding affinity, a fragment-merging approach was explored. The combination of structural features of two fragments, 16 (Kd 170 μM, LE 0.37) and 20 (Kd 0.26 mM, LE 0.41), proved promising with the resultant aminopyrazole-indole compound 23 (Kd 110 μM, LE 0.36) possessing improved affinity. Controlled elaboration with 23, guided by extensive structural information and biophysical data, resulted in the development of a ligand-efficient low-nanomolar affinity compound 29a (Kd 27 nM, LE 0.34) against Mab TrmD. Compounds from this fragment-merging series were subsequently screened against both Mab and Mtb growth in liquid culture, with many affording promising MIC values. These results are encouraging and support the use of TrmD as a target for the development of compounds with antimycobacterial activity. As a result, the work described in this study is currently being developed in the pursuit of further compounds with potent activity against Mab.
Experimental Section
General Chemistry
All reactions were carried out in oven-dried glassware under a positive pressure of dry nitrogen atmosphere. Temperatures of 0 and −78 °C were obtained by submerging the reaction vessel in a bath containing either ice or a mixture of solid CO2 pellets and acetone respectively. The solvents dichloromethane (DCM), ethyl acetate, acetonitrile, methanol, petroleum ether, and toluene were distilled over calcium hydride under a dry nitrogen atmosphere prior to use, with tetrahydrofuran (THF) distilled over a mixture of lithium aluminium hydride, calcium hydride, and triphenylphosphine. DMF was purchased as anhydrous from commercial suppliers, with ethanol and acetic acid obtained in the absolute and glacial forms, respectively. All purchased chemicals were used as received. Solutions of Na2CO3, NaHCO3, NaCl (brine), and NH4Cl were aqueous and saturated. Solutions of LiCl were aqueous and 5% w/v.
Flash column chromatography was performed using automated Biotage Isolera Spektra purification systems with appropriately sized Biotage SNAP cartridges, containing either KP 50 μm silica in “normal phase” purification or HP-sphere 25 μm C18 silica in “reverse phase” purification. Microwave heating was performed using a Biotage Initiator+ system with sealed Biotage microwave reaction vials. Analytical thin layer chromatography (TLC) was performed using Merck glass-backed silica plates, with visualization by 254 or 365 nm ultraviolet light.
Liquid chromatography mass spectrometry (LCMS) was carried out using a Waters Acquity UPLC H-Class system, with samples run on a solvent gradient from 0 to 95% acetonitrile in water (+0.1% formic acid) over 4 min. Peaks corresponding to the desired product are described, including the retention time (rt) and % purity by integration. High resolution mass spectrometry (HRMS) was mainly performed using ThermoFinnigan Orbitrap Classic, Waters LCT Premier or Waters Vion IMS QTof systems. A PerkinElmer Spectrum One FT-IR spectrometer fitted with a universal attenuated total reflectance accessory was used to record infrared spectra, with wavelengths of maximum absorbance (νmax) quoted in wavenumbers (cm–1) for signals outside of the fingerprint region (br = broad). Only peaks corresponding to key functional groups were characterized. Nuclear magnetic resonance (NMR) spectra were recorded in the indicated deuterated solvents with Avance III HD (400 MHz), QNP Cryoprobe (400 MHz), or DCH Cryoprobe (500 MHz) Bruker spectrometers. 1H NMR data are presented in the following order: chemical shift (in ppm on a δ scale relative to the residual solvent resonance peak), integration, multiplicity (s = singlet, d = doublet, t = triplet, q = quartet, quin = quintet, sep = septet, m = multiplet), and coupling constant (J, in Hz). 13C NMR spectra were proton-decoupled, with chemical shifts recorded, and further description are provided for certain peaks (br = broad).
A combination of TLC and LCMS analysis was used to monitor reactions. All tested compounds possessed a purity of at least 95% as determined by LCMS analysis (except for compound 2, for which LCMS analysis was not possible).
The synthesis of 23, 24f, 26f, 28, 29a, 29d, and 31a, and associated intermediates including 51, 52, 54f, 55f, 68–75, 77a, 77d, 78a, 78d, 79a, 79d, 83a, and 83d, have previously been reported.12
N-Ethyl-1H-pyrazole-4-carboxamide (2)
Palladium on carbon (10 wt % loading, 0.150 g) was added to a suspension of 1-benzyl-N-ethyl-1H-pyrazole-4-carboxamide 34 (75 mg, 0.33 mmol) in tert-butanol (15 mL) and formic acid (1.5 mL). The reaction mixture was heated to 50 °C over 2 h, then filtered, and concentrated in vacuo. Purification by flash column chromatography (0–15% methanol in DCM) afforded 2 (29 mg, 64% yield). 1H NMR: (400 MHz, CD3OD) 8.03 (2H, s), 3.36 (2H, q, J = 7.3 Hz), 1.20 (3H, t, J = 7.3 Hz); 13C NMR: (100 MHz, CD3OD) 165.3, 119.1, 35.2, 15.0 (1 peak missing); νmax/cm–1: 3302 (N–H), 3110, 2974, 2876, 1632 (C=O), 1584, 1541, 1515; HRMS (ESI)+: m/z calcd for [C6H9N3O + H]+, 140.0818; observed, 140.0818.
Benzyl 1H-Pyrazole-4-carboxylate (3)
Benzyl bromide (0.11 mL, 0.92 mmol) and N,N-diisopropylethylamine (0.20 mL, 1.1 mmol) were added to a solution of 1-(tetrahydro-2H-pyran-2-yl)-1H-pyrazole-4-carboxylic acid 36 (0.150 g, 0.765 mmol) in DMF (2.5 mL). The reaction mixture was stirred over 2 h, then diluted with ethyl acetate (200 mL), washed with LiCl solution (2 × 100 mL) and brine (2 × 200 mL), dried (MgSO4), and concentrated in vacuo to afford the crude reaction intermediate. TFA (1 mL) was added, and the reaction mixture was stirred over 12 min, then diluted with NaHCO3 solution (20 mL) at 0 °C, and extracted into DCM (3 × 50 mL). The combined organic extracts were washed with NaHCO3 solution (50 mL) and brine (50 mL), dried (MgSO4), and concentrated in vacuo. Purification by flash column chromatography (0–85% ethyl acetate in petroleum ether) afforded 3 (76 mg, 50% yield). LCMS (ESI+): m/z 203.2 [M + H]+, (ESI–): m/z 201.1 [M – H]−, rt 1.61 min, >99%; 1H NMR: (500 MHz, CDCl3) 10.83 (1H, br s), 8.09 (2H, s), 7.45–7.32 (5H, m), 5.31 (2H, s); 13C NMR: (125 MHz, CDCl3) 163.0, 136.8 (br), 136.2, 128.7, 128.41, 128.36, 115.1, 66.2; νmax/cm–1: 3168, 3124, 3043, 2947, 2835, 1714, 1702 (C=O), 1578, 1521; HRMS (ESI)+: m/z calcd for [C11H10N2O2 + H]+, 203.0815; observed, 203.0823.
4-Nitrobenzyl 1H-Pyrazole-4-carboxylate (4)
EDC·HCl (0.293 g, 1.53 mmol) was added to a solution of 1-(tetrahydro-2H-pyran-2-yl)-1H-pyrazole-4-carboxylic acid 36 (0.200 g, 1.02 mmol), 4-nitrobenzyl alcohol (0.188 g, 1.22 mmol), N,N-diisopropylethylamine (0.267 mL, 1.53 mmol), and DMAP (19 mg, 0.15 mmol) in DCM (5 mL). The reaction mixture was stirred overnight, then diluted with water (30 mL), and extracted into DCM (3 × 20 mL). The combined organic extracts were washed (brine), dried (MgSO4), and concentrated in vacuo. Purification by flash column chromatography (30–80% ethyl acetate in petroleum ether) afforded the reaction intermediate. TFA (2 mL) was added to a solution of the reaction intermediate in DCM (4 mL). The reaction mixture was stirred over 5 min, then diluted with NaHCO3 solution (50 mL) at 0 °C, and extracted into DCM (3 × 50 mL). The combined organic extracts were washed (brine), dried (MgSO4), and concentrated in vacuo. Purification by flash column chromatography (0–7% methanol in DCM) afforded 4 (98 mg, 40% yield). LCMS (ESI–): m/z 246.1 [M – H]−, rt 1.62 min, >99%; 1H NMR: (400 MHz, CDCl3) 8.27–8.22 (2H, m), 8.11 (2H, s), 7.58 (2H, d, J = 8.9 Hz), 5.40 (2H, s); 13C NMR: (100 MHz, CDCl3) 162.6, 147.9, 143.5, 136.9, 128.5, 124.0, 114.4, 64.7; νmax/cm–1: 3120, 2930, 2855, 1715 (C=O), 1606, 1518 (N–O), 1342 (N–O); HRMS (ESI)−: m/z calcd for [C11H9N3O4 – H]−, 246.0520; observed, 246.0512.
4-(Methoxycarbonyl)benzyl 1H-Pyrazole-4-carboxylate (5)
Methyl-4-(bromomethyl) benzoate (0.248 g, 1.08 mmol) and N,N-diisopropylethylamine (0.24 mL, 1.5 mmol) were added to a solution of 1-(tetrahydro-2H-pyran-2-yl)-1H-pyrazole-4-carboxylic acid 36 (0.177 g, 0.903 mmol) in DMF (3 mL). The reaction mixture was stirred over 2 h, then diluted with ethyl acetate (200 mL), washed with LiCl solution (2 × 100 mL) and brine (2 × 200 mL), dried (MgSO4), and concentrated in vacuo to afford the crude reaction intermediate. TFA (2 mL) was added, and the reaction mixture was stirred over 10 min, then diluted with NaHCO3 solution (50 mL) at 0 °C, and extracted into DCM (3 × 50 mL). The combined organic extracts were washed with NaHCO3 solution (50 mL) and brine (50 mL), dried (MgSO4), and concentrated in vacuo. Purification by flash column chromatography (0–70% ethyl acetate in petroleum ether) afforded 5 (0.115 g, 49% yield). LCMS (ESI–): m/z 259.1 [M – H]−, rt 1.62 min, >99%; 1H NMR: (400 MHz, CDCl3) 8.10 (2H, s), 8.08–8.02 (2H, m), 7.48 (2H, d, J = 8.2 Hz), 5.36 (2H, s), 3.92 (3H, s); 13C NMR: (100 MHz, CDCl3) 166.9, 162.8, 141.2, 136.9, 130.1, 130.0, 127.8, 114.7, 65.4, 52.4; νmax/cm–1: 3114, 2950, 1712 (C=O), 1617, 1578, 1523; HRMS (ESI)+: m/z calcd for [C13H12N2O4 + H]+, 261.0870; observed, 261.0860.
3-Cyanobenzyl 1H-Pyrazole-4-carboxylate (6)
3-(Bromomethyl)benzonitrile (0.360 g, 1.84 mmol) and N,N-diisopropylethylamine (0.53 mL, 3.1 mmol) were added to a solution of 1-(tetrahydro-2H-pyran-2-yl)-1H-pyrazole-4-carboxylic acid 36 (0.300 g, 1.53 mmol) in DMF (3.5 mL). The reaction mixture was stirred over 90 min, then diluted with ethyl acetate (200 mL), washed with LiCl solution (2 × 100 mL) and brine (2 × 200 mL), dried (MgSO4), and concentrated in vacuo to afford the crude reaction intermediate. TFA (2 mL) was added, and the reaction mixture was stirred over 10 min, then diluted with NaHCO3 solution (50 mL) at 0 °C, and extracted into DCM (3 × 50 mL). The combined organic extracts were washed with NaHCO3 solution (50 mL) and brine (50 mL), dried (MgSO4), and concentrated in vacuo. Purification by flash column chromatography (0–10% methanol in DCM) afforded 6 (0.197 g, 57% yield). LCMS (ESI–): m/z 226.1 [M – H]−, rt 1.54 min, 96%; 1H NMR: (500 MHz, CDCl3) 8.10 (2H, s), 7.74–7.70 (1H, m), 7.67–7.61 (2H, m), 7.50 (1H, t, J = 7.8 Hz), 5.32 (2H, s); 13C NMR: (125 MHz, CDCl3) 162.6, 137.8, 136.9, 132.5, 132.0, 131.6, 129.6, 118.6, 114.4, 113.0, 64.8; νmax/cm–1: 3179, 3125, 2951, 2227 (C≡N), 1721 (C=O), 1576, 1522; HRMS (ESI)+: m/z calcd for [C12H9N3O2 + H]+, 228.0768; observed, 228.0782.
3-Methoxybenzyl 1H-Pyrazole-4-carboxylate (7)
3-Methoxybenzyl bromide (0.13 mL, 0.92 mmol) and N,N-diisopropylethylamine (0.20 mL, 1.1 mmol) were added to a solution of 1-(tetrahydro-2H-pyran-2-yl)-1H-pyrazole-4-carboxylic acid 36 (0.150 g, 0.765 mmol) in DMF (2.5 mL). The reaction mixture was stirred over 2 h, then diluted with ethyl acetate (200 mL), washed with LiCl solution (2 × 100 mL) and brine (2 × 200 mL), dried (MgSO4), and concentrated in vacuo. Purification by flash column chromatography (0–85% ethyl acetate in petroleum ether) afforded the reaction intermediate. TFA (1 mL) was added, and the reaction mixture was stirred over 10 min, then diluted with NaHCO3 solution (20 mL) at 0 °C, and extracted into DCM (3 × 15 mL). The combined organic extracts were washed with NaHCO3 solution (50 mL) and brine (50 mL), dried (MgSO4), and concentrated in vacuo. Purification by flash column chromatography (30–60% ethyl acetate in petroleum ether) afforded 7 (8.2 mg, 5% yield). LCMS (ESI–): m/z 231.2 [M – H]−, rt 1.64 min, 98%; 1H NMR: (400 MHz, CDCl3) 8.09 (2H, s), 7.30 (1H, t, J = 7.9 Hz), 7.00 (1H, d, J = 7.7 Hz), 6.97–6.93 (1H, m), 6.88 (1H, dd, J = 8.1, 2.5 Hz), 5.28 (2H, s), 3.82 (3H, s); 13C NMR: (100 MHz, CDCl3) 162.9, 159.9, 137.7, 136.9, 129.8, 120.5, 115.0, 113.9, 113.8, 66.1, 55.4; νmax/cm–1: 3276, 3113, 2942, 1718, 1678 (C=O), 1598, 1558; HRMS (ESI)+: m/z calcd for [C12H12N2O3 + H]+, 233.0921; observed, 233.0927.
3-(2-Methoxy-2-oxoethoxy)benzyl 1H-Pyrazole-4-carboxylate (8)
Caesium carbonate (0.336 g, 1.03 mmol) and methyl bromoacetate (54 μL, 0.57 mmol) were added to a solution of 3-hydroxybenzyl 1-(tetrahydro-2H-pyran-2-yl)-1H-pyrazole-4-carboxylate 40 (0.156 mg, 0.516 mmol) in DMF (3 mL). The reaction mixture was stirred over 2 h, then diluted with ethyl acetate (200 mL), washed with LiCl solution (2 × 100 mL) and brine (2 × 200 mL), dried (MgSO4), and concentrated in vacuo to afford the crude reaction intermediate. TFA (2 mL) was added, and the reaction mixture was stirred over 15 min, then diluted with NaHCO3 solution (50 mL) at 0 °C, and extracted into DCM (3 × 50 mL). The combined organic extracts were washed with NaHCO3 solution (50 mL) and brine (50 mL), dried (MgSO4), and concentrated in vacuo. Purification by flash column chromatography (0–100% ethyl acetate in petroleum ether) afforded 8 (75 mg, 50% yield). LCMS (ESI+): m/z 291.2 [M + H]+, (ESI–): m/z 289.1 [M – H]−, rt 1.61 min, >99%; 1H NMR: (400 MHz, CDCl3) 10.75 (1H, br s), 8.08 (2H, s), 7.30 (1H, t, J = 7.9 Hz), 7.05 (1H, d, J = 7.5 Hz), 6.98 (1H, s), 6.86 (1H, dd, J = 8.2, 2.2 Hz), 5.27 (2H, s), 4.65 (2H, s) 3.80 (3H, s); 13C NMR: (100 MHz, CDCl3) 169.5, 162.9, 158.1, 138.0, 137.0 (br), 130.0, 121.6, 114.9, 114.6, 114.3, 65.8, 65.5, 52.5; νmax/cm–1: 3303, 1745 (C=O), 1714 (C=O), 1587, 1561; HRMS (ESI)+: m/z calcd for [C14H14N2O5 + Na]+, 313.0795; observed, 313.0792.
3-(Pyridin-2-ylmethoxy)benzyl 1H-Pyrazole-4-carboxylate (9)
Caesium carbonate (0.970 g, 2.98 mmol) and 2-bromomethylpyridine hydrobromide (0.282 g, 1.09 mmol) were added to a solution of 3-hydroxybenzyl 1-(tetrahydro-2H-pyran-2-yl)-1H-pyrazole-4-carboxylate 40 (0.300 g, 0.992 mmol) in DMF (3.5 mL). The reaction mixture was stirred over 2 h, then diluted with ethyl acetate (80 mL), washed with LiCl solution (2 × 100 mL) and brine (2 × 150 mL), dried (MgSO4), and concentrated in vacuo to afford the crude reaction intermediate. TFA (2 mL) was added, and the reaction mixture was stirred over 15 min, then diluted with NaHCO3 solution (50 mL) at 0 °C, and extracted into DCM (3 × 50 mL). The combined organic extracts were washed with NaHCO3 solution (50 mL) and brine (50 mL), dried (MgSO4), and concentrated in vacuo. Purification by flash column chromatography (20–100% ethyl acetate in petroleum ether) afforded 9 (0.163 g, 53% yield). LCMS (ESI+): m/z 310.2 [M + H]+, (ESI–): m/z 308.1 [M – H]−, rt 1.55 min, 97%; 1H NMR: (400 MHz, CDCl3) 11.01 (1H, br s), 8.59 (1H, d, J = 4.8 Hz), 8.08 (2H, s), 7.72 (1H, td, J = 7.7, 1.7 Hz), 7.52 (1H, d, J = 7.8 Hz), 7.29 (1H, t, J = 7.9 Hz), 7.23 (1H, dd, J = 7.1, 5.1 Hz), 7.08–7.04 (1H, m), 7.01 (1H, d, J = 7.7 Hz), 6.95 (1H, dd, J = 8.2, 2.2 Hz), 5.27 (2H, s), 5.22 (2H, s); 13C NMR: (100 MHz, CDCl3) 162.9, 158.7, 157.2, 149.3, 137.9, 137.1, 129.9, 122.9, 121.5, 120.9, 114.9, 114.7, 114.6, 70.7, 65.9 (1 peak missing); νmax/cm–1: 3118, 2947, 1707 (C=O), 1598, 1570, 1516; HRMS (ESI)−: m/z calcd for [C17H15N3O3 – H]−, 308.1041; observed, 308.1030.
3-Ethoxybenzyl 1H-Pyrazole-4-carboxylate (10)
Triphenylphosphine (0.217 g, 0.831 mmol), ethanol (49 μL, 0.83 mmol), and diisopropyl azodicarboxylate (0.16 mL, 0.83 mmol) were added to a solution of 3-hydroxybenzyl 1-(tetrahydro-2H-pyran-2-yl)-1H-pyrazole-4-carboxylate 40 (0.250 g, 0.831 mmol) in THF (5 mL). The reaction mixture was stirred over 1 h, then diluted with NaHCO3 solution (35 mL), and extracted into diethyl ether (3 × 30 mL). The combined organic extracts were washed with NaHCO3 solution (40 mL) and brine (40 mL), dried (Na2SO4), and concentrated in vacuo. Purification by flash column chromatography (20–100% ethyl acetate in petroleum ether) afforded the reaction intermediate. TFA (2 mL) was added, and the reaction mixture was stirred over 15 min, then diluted with NaHCO3 solution (50 mL) at 0 °C, and extracted into DCM (3 × 50 mL). The combined organic extracts were washed (brine), dried (MgSO4), and concentrated in vacuo. Purification by flash column chromatography (0–90% ethyl acetate in petroleum ether) afforded 10 (94 mg, 46% yield). LCMS (ESI+): m/z 247.2 [M + H]+, (ESI–): m/z 245.1 [M – H]−, rt 1.79 min, >99%; 1H NMR: (500 MHz, CDCl3) 8.09 (2H, s), 7.28 (1H, t, J = 7.9 Hz), 6.98 (1H, d, J = 7.5 Hz), 6.96–6.94 (1H, m), 6.86 (1H, dd, J = 8.2, 2.4 Hz), 5.27 (2H, s), 4.04 (2H, q, J = 7.0 Hz), 1.41 (3H, t, J = 7.0 Hz); 13C NMR: (125 MHz, CDCl3) 163.0, 159.3, 137.6, 136.9, 129.8, 120.4, 115.0, 114.4, 114.3, 66.1, 63.6, 15.0; νmax/cm–1: 3256, 3118, 2979, 2942, 2888, 1686 (C=O), 1601, 1559; HRMS (ESI)+: m/z calcd for [C13H14N2O3 + H]+, 247.1077; observed, 247.1084.
3-Phenethoxybenzyl 1H-Pyrazole-4-carboxylate (11)
Triphenylphosphine (0.261 g, 0.997 mmol), phenethyl alcohol (0.12 mL, 1.0 mmol), and diisopropyl azodicarboxylate (0.20 mL, 1.0 mmol) were added to a solution of 3-hydroxybenzyl 1-(tetrahydro-2H-pyran-2-yl)-1H-pyrazole-4-carboxylate 40 (0.300 g, 0.997 mmol) in THF (5 mL). The reaction mixture was stirred overnight, then diluted with NaHCO3 solution (30 mL), and extracted into diethyl ether (3 × 30 mL). The combined organic extracts were washed with NaHCO3 solution (40 mL) and brine (40 mL), dried (Na2SO4), and concentrated in vacuo. Purification by flash column chromatography (0–100% ethyl acetate in petroleum ether) afforded the reaction intermediate. TFA (2 mL) was added, and the reaction mixture was stirred over 15 min, then diluted with NaHCO3 solution (50 mL) at 0 °C, and extracted into DCM (3 × 50 mL). The combined organic extracts were washed with NaHCO3 solution (50 mL) and brine (50 mL), dried (MgSO4), and concentrated in vacuo. Purification by flash column chromatography (0–100% ethyl acetate in petroleum ether) afforded 11 (77 mg, 24% yield). LCMS (ESI+): m/z 323.2 [M + H]+, (ESI–): m/z 321.1 [M – H]−, rt 2.08 min, >99%; 1H NMR: (500 MHz, CDCl3) 10.69 (1H, br s), 8.08 (2H, s), 7.35–7.21 (6H, m), 7.01–6.97 (1H, m), 6.95 (1H, t, J = 1.9 Hz), 6.87 (1H, ddd, J = 8.3, 2.5, 0.7 Hz), 5.26 (2H, s), 4.19 (2H, t, J = 7.1 Hz), 3.10 (2H, t, J = 7.1 Hz); 13C NMR: (125 MHz, CDCl3) 162.9, 159.1, 138.3, 137.7, 136.9, 129.8, 129.1, 128.7, 126.7, 120.6, 115.0, 114.6, 114.4, 68.8, 66.1, 35.9; νmax/cm–1: 1714 (C=O), 1610, 1588, 1559; HRMS (ESI)+: m/z calcd for [C19H18N2O3 + Na]+, 345.1210; observed, 345.1195.
Ethyl 1-(3-(3-(((1H-Pyrazole-4-carbonyl)oxy)methyl)-5-methoxyphenyl)prop-2-yn-1-yl)-1H-pyrazole-4-carboxylate (12)
TFA (1 mL) was added to 3-(3-(4-(ethoxycarbonyl)-1H-pyrazol-1-yl)prop-1-yn-1-yl)-5-methoxybenzyl 1-(tetrahydro-2H-pyran-2-yl)-1H-pyrazole-4-carboxylate 46 (75 mg, 0.15 mmol). The reaction mixture was stirred over 15 min, then diluted with NaHCO3 solution (20 mL) at 0 °C, and extracted into DCM (3 × 20 mL). The combined organic extracts were washed (brine), dried (MgSO4), and concentrated in vacuo. Purification by flash column chromatography (0–100% ethyl acetate in petroleum ether) afforded 12 (42 mg, 63% yield). LCMS (ESI+): m/z 409.3 [M + H]+, (ESI–): m/z 407.2 [M – H]−, rt 1.89 min, >99%; 1H NMR: (500 MHz, CDCl3) 8.19 (1H, s), 8.10 (2H, s), 7.95 (1H, d, J = 0.5 Hz), 7.15–7.11 (1H, m), 6.98–6.94 (2H, m), 5.24 (2H, s), 5.17 (2H, s), 4.31 (2H, q, J = 7.2 Hz), 3.81 (3H, s), 1.35 (3H, t, J = 7.2 Hz); 13C NMR: (125 MHz, CDCl3) 163.1, 162.8, 159.7, 141.7, 138.2, 137.0 (br), 132.4, 124.0, 123.0, 116.5, 115.8, 115.4, 114.8, 86.9, 81.0, 65.3, 60.5, 55.6, 43.0, 14.5; νmax/cm–1: 3127, 2977, 1706 (C=O), 1592, 1555; HRMS (ESI)+: m/z calcd for [C21H20N4O5 + H]+, 409.1506; observed, 409.1519.
3-Methoxy-5-(pyrrolidin-1-yl)benzyl 1H-Pyrazole-4-carboxylate (13)
Pyrrolidine (0.110 mL, 1.32 mmol) and tert-butanol (2 mL) were added to a mixture of 3-bromo-5-methoxybenzyl 1-(tetrahydro-2H-pyran-2-yl)-1H-pyrazole-4-carboxylate 44 (0.500 g, 1.10 mmol), caesium carbonate (0.430 g, 1.32 mmol), RuPhos (26 mg, 0.055 mmol), and RuPhos Pd G1 methyl tert-butyl ether adduct (45 mg, 0.055 mmol). The reaction mixture was stirred at 85 °C over 15 h, then diluted with ethyl acetate (25 mL), washed with water (15 mL) and brine (15 mL), dried (MgSO4), and concentrated in vacuo. TFA (4 mL) was added to the crude residue, and the reaction mixture was stirred over 30 min. The reaction mixture was diluted with ethyl acetate (100 mL), washed with NaHCO3 solution (3 × 100 mL) and brine (100 mL), dried (MgSO4), and concentrated in vacuo. Purification by reverse phase chromatography (0–45% acetonitrile in water (+0.1% NH3)), followed by flash column chromatography (0–4% methanol in DCM), afforded 13 (23 mg, 7% yield). LCMS (ESI+): m/z 302.2 [M + H]+, (ESI–): m/z 300.1 [M – H]−, rt 1.91 min, >99%; 1H NMR: (500 MHz, CDCl3) 8.09 (2H, s), 6.31–6.28 (1H, m), 6.25–6.22 (1H, m), 6.07 (1H, t, J = 2.2 Hz), 5.22 (2H, s), 3.80 (3H, s), 3.31–3.25 (4H, m), 2.02–1.96 (4H, m); 13C NMR: (125 MHz, CDCl3) 163.1, 161.0, 149.4, 138.0, 136.8, 115.1, 104.8, 100.7, 97.6, 66.9, 55.3, 47.8, 25.6; νmax/cm–1: 3200 (br, N–H), 2962, 2840, 1712 (C=O), 1586; HRMS (ESI)+: m/z calcd for [C16H19N3O3 + H]+, 302.1499; observed, 302.1500.
3-Methoxy-5-(pyridin-3-yl)benzyl 1H-Pyrazole-4-carboxylate (14)
Dioxane (2 mL) and water (0.5 mL) were added to a mixture of 3-bromo-5-methoxybenzyl 1-(tetrahydro-2H-pyran-2-yl)-1H-pyrazole-4-carboxylate 44 (0.200 g, 0.435 mmol), 3-pyridinylboronic acid (59 mg, 0.479 mmol), Pd(dppf)Cl2 (16 mg, 0.022 mmol), and potassium carbonate (0.241 g, 1.74 mmol). The reaction mixture was heated to 100 °C by microwave for 30 min, then diluted with DCM (25 mL), washed with NaHCO3 solution (3 × 25 mL), dried (MgSO4), and concentrated in vacuo. TFA (2 mL) was added to the crude residue, and the reaction mixture was stirred over 15 min. The reaction mixture was diluted with DCM (100 mL), washed with NaHCO3 solution (3 × 100 mL) and brine (100 mL), dried (MgSO4), and concentrated in vacuo. Purification by flash column chromatography (75% ethyl acetate in petroleum ether) afforded 14 (59 mg, 44% yield). LCMS (ESI+): m/z 310.2 [M + H]+, (ESI–): m/z 308.1 [M – H]−, rt 1.45 min, >99%; 1H NMR: (500 MHz, CD3OD) 8.79 (1H, dd, J = 2.3, 0.7 Hz), 8.52 (1H, dd, J = 5.0, 1.5 Hz), 8.32–7.90 (2H, br s), 8.09 (1H, ddd, J = 8.0, 2.3, 1.6 Hz), 7.52 (1H, ddd, J = 8.0, 4.9, 0.8 Hz), 7.31–7.27 (1H, m), 7.16 (1H, t, J = 2.0 Hz), 7.10–7.04 (1H, m), 5.34 (2H, s), 3.87 (3H, s); 13C NMR: (125 MHz, CD3OD) 164.6, 162.1, 148.9, 148.3, 140.4, 140.3, 138.3, 136.8, 133.6 (br), 125.5, 120.1, 115.3, 114.6, 113.5, 66.7, 56.0; νmax/cm–1: 3100 (br, N–H), 2940, 1708 (C=O), 1598, 1562; HRMS (ESI)+: m/z calcd for [C17H15N3O3 + H]+, 310.1186; observed, 310.1186.
3-Methoxy-5-(pyridin-3-ylmethyl)benzyl-1H-pyrazole-4-carboxylate (15)
1,2-Dimethoxyethane (4 mL) and water (1 mL) were added to a mixture of 3-methoxy-5-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)benzyl 1-(tetrahydro-2H-pyran-2-yl)-1H-pyrazole-4-carboxylate 47 (0.192 g, 0.347 mmol), 3-(bromomethyl)pyridine hydrobromide (0.105 g, 0.417 mmol), tetrakis(triphenylphosphine)palladium(0) (40 mg, 0.035 mmol), and potassium carbonate (0.240 g, 1.74 mmol). The reaction mixture was heated to 95 °C over 10 h, then diluted with water (15 mL), and extracted into DCM (3 × 25 mL). The combined organic extracts were washed (brine), dried (MgSO4), and concentrated in vacuo. Purification by flash column chromatography (0–100% ethyl acetate in petroleum ether) was attempted, and TFA (0.5 mL) was added to the resultant residue. The reaction mixture was stirred over 30 min, then diluted with NaHCO3 solution (15 mL), and extracted into DCM (3 × 25 mL). The combined organic extracts were washed with NaHCO3 solution (25 mL) and brine (25 mL), dried (MgSO4), and concentrated in vacuo. Purification by flash column chromatography (0–100% ethyl acetate in petroleum ether, 0–10% methanol in DCM) afforded 15 (20 mg, 18% yield).
LCMS (ESI+): m/z 324.2 [M + H]+, (ESI–): m/z 322.1 [M – H]−, rt 1.30 min, 98%; 1H NMR: (500 MHz, CDCl3) 8.51 (1H, s), 8.47 (1H, d, J = 4.9 Hz), 8.06 (2H, s), 7.52–7.47 (1H, m), 7.22 (1H, dd, J = 7.8, 4.9 Hz), 6.84–6.79 (2H, m), 6.70–6.66 (1H, m), 5.23 (2H, s), 3.96 (2H, s), 3.78 (3H, s); 13C NMR: (125 MHz, CDCl3) 162.9, 160.2, 150.2, 147.8, 141.8, 138.1, 136.9, 136.7, 136.3, 123.7, 120.9, 114.9, 114.6, 111.7, 65.9, 55.5, 39.1; νmax/cm–1: 3150 (br, N–H), 2936, 1709 (C=O), 1597, 1562; HRMS (ESI)+: m/z calcd for [C18H17N3O3 + H]+, 324.1343; observed, 324.1330.
5-(3-Methoxyphenyl)-1H-pyrazol-3-amine (19)25
Hydrazine monohydrate (1.1 mL, 23 mmol) was added to a suspension of 3-(3-methoxyphenyl)-3-oxopropanenitrile 49 (0.400 g, 2.28 mmol) in ethanol (10 mL). The reaction mixture was heated under reflux for 6 h, then quenched with excess acetone at room temperature, and concentrated in vacuo. Purification by flash column chromatography (0–50% ethyl acetate in petroleum ether, 0–6% methanol in DCM) afforded 19 (0.314 g, 73% yield). LCMS (ESI+): m/z 190.2 [M + H]+, rt 1.21 min, >99%; 1H NMR: (500 MHz, CDCl3) 7.28 (1H, t, J = 8.0 Hz), 7.13 (1H, dt, J = 7.7, 1.2 Hz), 7.09 (1H, t, J = 2.0 Hz), 6.86 (1H, ddd, J = 8.2, 2.6, 0.8 Hz), 5.89 (1H, s), 3.80 (3H, s); 13C NMR: (125 MHz, CDCl3) 160.1, 154.5, 145.7, 131.8, 130.1, 118.0, 114.0, 111.2, 90.7, 55.4; νmax/cm–1: 3200 (br, N–H), 2837, 1612, 1589, 1567, 1506; HRMS (ESI)+: m/z calcd for [C10H11N3O + H]+, 190.0975; observed, 190.0972.
5-(1-(4-Methoxybenzyl)-1H-indol-6-yl)-1H-pyrazol-3-amine (24a)
Hydrazine monohydrate (42 μL, 0.67 mmol) was added to a solution of 3-(1-(4-methoxybenzyl)-1H-indol-6-yl)-3-oxopropanenitrile 55a (0.200 g, 0.611 mmol) in ethanol (5 mL). The reaction mixture was heated under reflux for 30 min, and then further hydrazine monohydrate (0.114 mL, 1.83 mmol) was added at room temperature. The reaction mixture was heated under reflux for 4 h, then quenched with excess acetone at room temperature, and concentrated in vacuo. Purification by flash column chromatography (0–10% methanol in DCM) afforded 24a (0.120 g, 62% yield). LCMS (ESI+): m/z 319.3 [M + H]+, rt 1.65 min, >99%; 1H NMR: (500 MHz, CDCl3) 7.63 (1H, d, J = 8.3 Hz), 7.46 (1H, s), 7.30–7.25 (1H, m), 7.12 (1H, d, J = 3.1 Hz), 7.06–7.02 (2H, m), 6.83–6.78 (2H, m), 6.52 (1H, d, J = 3.0 Hz), 5.88 (1H, s), 5.22 (2H, s), 3.74 (3H, s); 13C NMR: (125 MHz, CDCl3) 159.3, 155.0, 146.7, 136.5, 129.5, 129.3, 129.1, 128.3, 123.8, 121.6, 117.7, 114.3, 106.8, 101.9, 90.5, 55.4, 49.7; νmax/cm–1: 3150 (br, N–H), 2927, 1610, 1585, 1510; HRMS (ESI+): m/z calcd for [C19H18N4O + H]+, 319.1553; observed, 319.1557.
5-(1-(3-Methoxybenzyl)-1H-indol-6-yl)-1H-pyrazol-3-amine (24b)
Hydrazine monohydrate (0.104 mL, 2.14 mmol) was added to a solution of 3-(1-(3-methoxybenzyl)-1H-indol-6-yl)-3-oxopropanenitrile 55b (65 mg, 0.21 mmol) in ethanol (4 mL). The reaction mixture was heated under reflux for 5 h and then concentrated in vacuo. Purification by flash column chromatography (0–100% ethyl acetate in petroleum ether, 0–10% methanol in DCM) was carried out, followed by reverse phase chromatography [0–40% acetonitrile in water (+0.1% formic acid)] with addition of NaHCO3 solution (10 mL) to the combined fractions and extraction into DCM (3 × 25 mL). The combined organic extracts were washed (brine), dried (MgSO4), and concentrated in vacuo to afford 24b (27 mg, 40% yield). LCMS (ESI+): m/z 319.2 [M + H]+, rt 1.65 min, >99%; 1H NMR: (500 MHz, CDCl3) 7.66 (1H, d, J = 8.3 Hz), 7.42 (1H, s), 7.29–7.25 (1H, m), 7.21 (1H, t, J = 7.9 Hz), 7.17 (1H, d, J = 3.2 Hz), 6.80 (1H, dd, J = 8.2, 2.4 Hz), 6.69 (1H, d, J = 7.6 Hz), 6.66–6.63 (1H, m), 6.56 (1H, dd, J = 3.2, 0.7 Hz), 5.89 (1H, s), 5.30 (2H, s), 3.72 (3H, s); 13C NMR: (125 MHz, CDCl3) 160.2, 155.2, 146.6, 138.9, 136.6, 130.1, 129.8, 129.2, 123.8, 121.7, 119.1, 117.7, 113.0, 112.8, 106.8, 102.1, 90.7, 55.3, 50.2; νmax/cm–1: 3150 (br, N–H), 1585, 1505; HRMS (ESI+): m/z calcd for [C19H18N4O + H]+, 319.1553; observed, 319.1542.
5-(1-Benzyl-1H-indol-6-yl)-1H-pyrazol-3-amine (24c)
Hydrazine monohydrate (0.271 mL, 5.58 mmol) was added to a solution of 3-(1-benzyl-1H-indol-6-yl)-3-oxopropanenitrile 55c (0.170 g, 0.558 mmol) in ethanol (10 mL). The reaction mixture was heated under reflux for 8 h, then quenched with excess acetone at room temperature, and concentrated in vacuo. Purification by flash column chromatography (0–10% methanol in DCM) afforded 24c (50 mg, 31% yield). LCMS (ESI+): m/z 289.3 [M + H]+, rt 1.65 min, >99%; 1H NMR: (400 MHz, CD3OD) 7.63 (1H, s), 7.58 (1H, d, J = 8.3 Hz), 7.34 (1H, dd, J = 8.4, 1.5 Hz), 7.31–7.20 (4H, m), 7.16 (2H, d, J = 7.2 Hz), 6.50 (1H, d, J = 3.1 Hz), 5.90 (1H, s), 5.42 (2H, s); 13C NMR: (125 MHz, CD3OD) 155.6, 148.7, 139.5, 137.9, 130.7, 130.4, 129.7, 128.5, 127.9, 125.5, 122.0, 118.5, 107.7, 102.6, 90.2, 50.7; νmax/cm–1: 2920, 2849, 1583, 1503; HRMS (ESI+): m/z calcd for [C18H16N4 + H]+, 289.1448; observed, 289.1446.
2-((6-(3-Amino-1H-pyrazol-5-yl)-1H-indol-1-yl)methyl)benzonitrile (24d)
n-Butyllithium (1.6 M in hexanes, 4.09 mL, 6.54 mmol) was added dropwise at −78 °C to a mixture of acetonitrile (0.68 mL, 13 mmol) and toluene (5 mL). The reaction mixture was stirred at −78 °C over 30 min. A solution of methyl 1-(2-cyanobenzyl)-1H-indole-6-carboxylate 54d (0.380 g, 1.31 mmol) in toluene (5 mL) was added dropwise at −78 °C over 30 min. The reaction mixture was stirred at −78 °C over 1 h, and then aqueous HCl (1 M, 15 mL) was added dropwise at 0 °C. The product was extracted into ethyl acetate (3 × 25 mL). The combined organic extracts were washed (brine), dried (MgSO4), and concentrated in vacuo. Purification by flash column chromatography (0–80% ethyl acetate in petroleum ether) was attempted. Ethanol (20 mL) was added to the crude residue, followed by hydrazine monohydrate (0.50 mL, 10 mmol). The reaction mixture was heated under reflux for 5 h, then quenched with excess acetone at room temperature, and concentrated in vacuo. Purification by flash column chromatography (0–10% methanol in DCM) afforded 24d (73 mg, 18% yield), with 14 mg subjected to further purification by flash column chromatography (0–100% ethyl acetate in petroleum ether, 0–5% methanol in DCM). LCMS (ESI+): m/z 314.3 [M + H]+, rt 1.58 min, 96%; 1H NMR: (400 MHz, CD3OD) 7.77 (1H, dd, J = 7.6, 1.1 Hz), 7.63 (1H, s), 7.61 (1H, d, J = 8.4 Hz), 7.49 (1H, td, J = 7.7, 1.2 Hz), 7.44–7.35 (2H, m), 7.34 (1H, d, J = 3.2 Hz), 6.90 (1H, d, J = 7.9 Hz), 6.56 (1H, d, J = 3.2 Hz), 5.92 (1H, s), 5.64 (2H, s); 13C NMR: (125 MHz, CD3OD) 155.5, 148.5, 142.9, 137.9, 134.6, 134.2, 130.8, 130.4, 129.4, 128.8, 126.0, 122.2, 118.8, 118.3, 112.0, 107.5, 103.3, 90.2 (1 peak missing); νmax/cm–1: 3200 (br, N–H), 2225 (C≡N), 1585, 1505; HRMS (ESI+): m/z calcd for [C19H15N5 + H]+, 314.1400; observed, 314.1402.
2-((6-(3-Amino-1H-pyrazol-5-yl)-1H-indol-1-yl)methyl)benzamide (24e)
A suspension of 2-((6-(3-amino-1H-pyrazol-5-yl)-1H-indol-1-yl)methyl)benzonitrile 24d (50 mg, 0.16 mmol) in aqueous NaOH (10 M, 4 mL) was heated under reflux for 7 h. The reaction mixture was adjusted to pH 3 and extracted into DCM/methanol (10:1, 3 × 75 mL). The combined organic extracts were washed (brine), dried (MgSO4), and concentrated in vacuo. Purification by reverse phase column chromatography (0–35% acetonitrile in water) afforded 24e (15 mg, 28% yield). LCMS (ESI+): m/z 332.3 [M + H]+, (ESI–): m/z 330.1 [M – H]−, rt 1.47 min, >99%; 1H NMR: (500 MHz, CD3OD) 7.62–7.52 (3H, m), 7.37–7.24 (4H, m), 6.82–6.75 (1H, m), 6.51 (1H, d, J = 3.0 Hz), 5.90 (1H, s), 5.64 (2H, s); 13C NMR: (125 MHz, CD3OD) 174.6, 138.0, 137.7, 135.9, 131.6, 131.2, 130.3, 128.7, 128.5, 125.3 (br), 122.0, 118.5, 107.7, 102.7, 90.5 (br), 48.3 (3 peaks missing); νmax/cm–1: 3200 (br), 1635 (C=O), 1505; HRMS (ESI)+: m/z calcd for [C19H17N5O + H]+, 332.1506; observed, 332.1504.
3-Amino-5-(1H-indol-6-yl)-1H-pyrazole-4-carbonitrile (25)
Hydrazine monohydrate (0.163 mL, 3.36 mmol) was added to a suspension of 3-amino-4,4,4-trichloro-2-(1H-indole-6-carbonyl)but-2-enenitrile 53 (0.690 g, 2.10 mmol) in ethanol (10 mL). The reaction mixture was heated under reflux for 22 h and then quenched with excess acetone at room temperature and concentrated in vacuo. Purification by flash column chromatography (0–100% ethyl acetate in petroleum ether) afforded 25 (0.113 g, 24% yield). LCMS (ESI+): m/z 224.2 [M + H]+, (ESI–): m/z 222.1 [M – H]−, rt 1.50 min, >99%; 1H NMR: (400 MHz, CD3OD) 7.89 (1H, s), 7.63 (1H, d, J = 8.0 Hz), 7.45 (1H, d, J = 7.0 Hz), 7.33 (1H, d, J = 2.1 Hz), 6.50 (1H, d, J = 2.7 Hz); 13C NMR: (125 MHz, CD3OD) 137.5, 130.6, 127.8, 121.7, 118.5, 116.9, 110.6, 102.6 (4 peaks missing); νmax/cm–1: 3411 (N–H), 3200 (br, N–H), 2221 (C≡N), 1634, 1584, 1527; HRMS (ESI+): m/z calcd for [C12H9N5 + H]+, 224.0931; observed, 224.0935.
3-Amino-5-(1-Benzyl-1H-indol-6-yl)-1H-pyrazole-4-carbonitrile (26c)
Trichloroacetonitrile (0.168 mL, 1.67 mmol) was added to a suspension of 3-(1-benzyl-1H-indol-6-yl)-3-oxopropanenitrile 55c (0.170 g, 0.558 mmol) and sodium acetate (0.229 g, 2.79 mmol) in ethanol (5 mL). The reaction mixture was stirred over 9 h, then concentrated in vacuo, diluted with water (20 mL), and extracted into DCM (3 × 25 mL). The combined organic extracts were washed (brine), dried (MgSO4), and concentrated in vacuo. The crude residue was dissolved in ethanol (10 mL), and hydrazine monohydrate (0.271 mL, 5.58 mmol) was added. The reaction mixture was heated under reflux for 5 h, then quenched with excess acetone at room temperature, and concentrated in vacuo. Purification by flash column chromatography (0–100% ethyl acetate in petroleum ether) afforded 26c (78 mg, 45% yield). LCMS (ESI+): m/z 314.2 [M + H]+, (ESI–): m/z 312.1 [M – H]−, rt 1.84 min, 100%; 1H NMR: (500 MHz, CDCl3) 7.72 (1H, s), 7.67 (1H, d, J = 8.3 Hz), 7.41 (1H, dd, J = 8.2, 1.5 Hz), 7.29–7.19 (4H, m), 7.13–7.08 (2H, m), 6.57 (1H, dd, J = 3.1, 0.8 Hz), 5.26 (2H, s), 4.12 (2H, br s); 13C NMR: (125 MHz, CDCl3) 157.0, 150.1, 136.9, 136.2, 130.8, 130.4, 129.0, 128.0, 127.1, 122.0, 120.8, 117.6, 115.3, 108.2, 102.3, 76.1, 50.4; νmax/cm–1: 3200 (br, N–H), 2211 (C≡N), 1621, 1582, 1524, 1501; HRMS (ESI+): m/z calcd for [C19H15N5 + H]+, 314.1400; observed, 314.1404.
3-Amino-5-(1-(pyridin-3-ylmethyl)-1H-indol-6-yl)-1H-pyrazole-4-carbonitrile (26g)
Trichloroacetonitrile (0.647 mL, 6.45 mmol) was added to a suspension of 3-oxo-3-(1-(pyridin-3-ylmethyl)-1H-indol-6-yl)propanenitrile 55g (0.592 g, 2.15 mmol) and sodium acetate (0.882 g, 10.8 mmol) in ethanol (20 mL). The reaction mixture was stirred over 13 h, then concentrated in vacuo, diluted with water (20 mL), and extracted into DCM (3 × 25 mL). The combined organic extracts were washed (brine), dried (MgSO4), and concentrated in vacuo. The crude residue was dissolved in ethanol (20 mL), and hydrazine monohydrate (1.1 mL, 22 mmol) was added. The reaction mixture was heated under reflux for 21 h, then quenched with excess acetone at room temperature, and concentrated in vacuo. Purification by flash column chromatography (0–100% ethyl acetate in DCM, 5% methanol in DCM) afforded 26g (0.367 g, 54% yield). LCMS (ESI+): m/z 315.2 [M + H]+, (ESI–): m/z 313.1 [M – H]−, rt 1.38 min, >99%; 1H NMR: (400 MHz, (CD3)2SO) 12.09 (1H, br s), 8.56 (1H, s), 8.46 (1H, dd, J = 4.7, 1.3 Hz), 7.90 (1H, s), 7.74–7.56 (3H, m), 7.50 (1H, d, J = 8.2 Hz), 7.32 (1H, dd, J = 7.7, 4.8 Hz), 6.55 (1H, d, J = 2.8 Hz), 6.26 (2H, br s), 5.49 (2H, s); 13C NMR: (100 MHz, (CD3)2SO) 148.8, 148.5, 135.4, 135.0, 133.5, 130.6, 128.9, 123.7, 120.9, 117.5, 116.5, 107.4, 101.6, 46.8 (4 peaks missing); νmax/cm–1: 3345 (N–H), 3100 (br, N–H), 2215 (C≡N), 1662, 1600, 1502; HRMS (ESI–): m/z calcd for [C18H14N6 – H]−, 313.1207; observed, 313.1195.
3-Amino-5-(1-((2-hydroxypyridin-3-yl)methyl)-1H-indol-6-yl)-1H-pyrazole-4-carbonitrile (26j)
Lithium chloride (12 mg, 0.29 mmol) and p-toluenesulfonic acid monohydrate (55 mg, 0.29 mmol) were added to a solution of 3-amino-5-(1-((2-methoxypyridin-3-yl)methyl)-1H-indol-6-yl)-1H-pyrazole-4-carbonitrile 26h (20 mg, 0.058 mmol) in DMF (1 mL). The reaction mixture was heated to 120 °C over 25 min, then diluted with ethyl acetate (100 mL), washed with water (3 × 100 mL) and brine (100 mL), dried (MgSO4), and concentrated in vacuo. Purification by flash column chromatography (50–100% ethyl acetate in petroleum ether, 0–10% methanol in DCM) afforded 26j (10 mg, 51% yield). LCMS (ESI+): m/z 331.2 [M + H]+, (ESI–): m/z 329.1 [M – H]−, rt 1.55 min, 97%; 1H NMR: (500 MHz, CD3OD) 7.82 (1H, s), 7.66 (1H, d, J = 8.1 Hz), 7.51 (1H, d, J = 7.5 Hz), 7.45 (1H, s), 7.32 (1H, dd, J = 6.5, 2.0 Hz), 7.08 (1H, d, J = 4.8 Hz), 6.55 (1H, d, J = 2.8 Hz), 6.25 (1H, t, J = 6.7 Hz), 5.28 (2H, s); 13C NMR: (125 MHz, CD3OD) 164.1, 140.1, 137.4, 135.0, 132.1, 131.4, 129.6, 122.3, 119.0, 116.9, 109.0, 108.0, 102.7, 46.2 (4 peaks missing); νmax/cm–1: 3150 (br, N–H/O–H), 2925, 2207 (C≡N), 1647, 1611, 1564, 1529, 1501; HRMS (ESI+): m/z calcd for [C18H14N6O + Na]+, 353.1121; observed, 353.1105.
3-Amino-5-(1-((6-hydroxypyridin-3-yl)methyl)-1H-indol-6-yl)-1H-pyrazole-4-carbonitrile (26k)
Lithium chloride (26 mg, 0.61 mmol) and p-toluenesulfonic acid monohydrate (0.116 g, 0.610 mmol) were added to a solution of 3-amino-5-(1-((6-methoxypyridin-3-yl)methyl)-1H-indol-6-yl)-1H-pyrazole-4-carbonitrile 26i (42 mg, 0.12 mmol) in DMF (1 mL). The reaction mixture was heated to 120 °C over 25 min, then diluted with ethyl acetate (100 mL), washed with water (3 × 100 mL) and brine (100 mL), dried (MgSO4), and concentrated in vacuo. Purification by flash column chromatography (50–100% ethyl acetate in petroleum ether, 0–12% methanol in DCM) afforded 26k (19 mg, 47% yield). LCMS (ESI+): m/z 331.2 [M + H]+, (ESI–): m/z 329.1 [M – H]−, rt 1.47 min, >99%; 1H NMR: (500 MHz, (CD3)2SO) 12.01 (1H, br s), 11.54 (1H, br s), 7.94 (1H, s), 7.73–7.29 (5H, m), 6.51 (1H, d, J = 2.4 Hz), 6.39 (1H, br s), 6.26 (1H, d, J = 9.3 Hz), 5.15 (2H, s); 13C NMR: (125 MHz, (CD3)2SO) 161.9, 141.0, 135.3, 133.9, 130.3, 128.8, 125.6, 120.8, 120.3, 117.4, 116.6, 114.6, 107.5, 101.4, 45.7 (3 peaks missing); νmax/cm–1: 3436, 3340, 3231, 2941, 2211 (C≡N), 1661, 1614, 1533; HRMS (ESI)+: m/z calcd for [C18H14N6O + H]+, 331.1302; observed, 331.1289.
3-Amino-5-(1-(pyridin-4-ylmethyl)-1H-indol-6-yl)-1H-pyrazole-4-carbonitrile (26l)
Trichloroacetonitrile (0.309 mL, 3.08 mmol) was added to a suspension of 3-oxo-3-(1-(pyridin-4-ylmethyl)-1H-indol-6-yl)propanenitrile 55l (0.283 g, 1.03 mmol) and sodium acetate (0.422 g, 5.14 mmol) in ethanol (20 mL). The reaction mixture was stirred over 13 h, then concentrated in vacuo, diluted with water (20 mL), and extracted into DCM (4 × 25 mL). The combined organic extracts were washed (brine), dried (MgSO4), and concentrated in vacuo. The crude residue was dissolved in ethanol (20 mL), and hydrazine monohydrate (0.500 mL, 10.3 mmol) was added. The reaction mixture was heated under reflux for 21 h, then quenched with excess acetone at room temperature, and concentrated in vacuo. Purification by flash column chromatography (0–100% ethyl acetate in DCM, 8% methanol in DCM) afforded 26l (0.183 g, 57% yield). LCMS (ESI+): m/z 315.2 [M + H]+, (ESI–): m/z 313.1 [M – H]−, rt 1.35 min, >99%; 1H NMR: (500 MHz, CD3OD) 8.40–8.36 (2H, m), 7.73 (1H, s), 7.66 (1H, d, J = 8.3 Hz), 7.53 (1H, dd, J = 8.2, 1.1 Hz), 7.37 (1H, d, J = 3.2 Hz), 7.12–7.08 (2H, m), 6.58 (1H, dd, J = 3.2, 0.8 Hz), 5.43 (2H, s); 13C NMR: (125 MHz, CD3OD) 157.8, 152.5, 150.3, 149.7, 137.4, 131.7, 131.3, 124.6, 123.3, 122.4, 119.2, 116.9, 108.8, 103.3, 73.9, 49.7; νmax/cm–1: 2204 (C≡N), 1605, 1554, 1500; HRMS (ESI+): m/z calcd for [C18H14N6 + H]+, 315.1353; observed, 315.1350.
3-Amino-5-(1-(quinolin-4-ylmethyl)-1H-indol-6-yl)-1H-pyrazole-4-carbonitrile (26m)
Trichloroacetonitrile (92 μL, 0.91 mmol) was added to a suspension of 3-oxo-3-(1-(quinolin-4-ylmethyl)-1H-indol-6-yl)propanenitrile 55m (99 mg, 0.30 mmol) and sodium acetate (0.125 g, 1.52 mmol) in ethanol (10 mL). The reaction mixture was stirred over 36 h, then diluted with water (50 mL), and extracted into DCM (3 × 50 mL). The combined organic extracts were washed (brine), dried (MgSO4), and concentrated in vacuo. The crude residue was dissolved in ethanol (10 mL), and hydrazine monohydrate (0.148 mL, 3.04 mmol) was added. The reaction mixture was heated under reflux for 6 h and then concentrated in vacuo. Purification by flash column chromatography (50–100% ethyl acetate in petroleum ether) afforded 26m (45 mg, 41% yield). LCMS (ESI+): m/z 365.3 [M + H]+, (ESI–): m/z 363.2 [M – H]−, rt 1.55 min, >99%; 1H NMR: (400 MHz, CD3OD) 8.61 (1H, d, J = 4.6 Hz), 8.23 (1H, d, J = 8.3 Hz), 8.07 (1H, d, J = 8.6 Hz), 7.81 (1H, t, J = 7.9 Hz), 7.76–7.65 (3H, m), 7.57 (1H, d, J = 7.9 Hz), 7.41 (1H, s), 6.65 (1H, d, J = 2.9 Hz), 6.62 (1H, d, J = 4.6 Hz), 5.99 (2H, s); 13C NMR: (125 MHz, CD3OD) 151.2, 148.5, 146.2, 137.8, 131.6, 131.2, 129.9, 128.6, 127.3, 124.3, 122.4, 119.6, 119.5, 117.0, 108.8, 103.6, 47.7 (5 peaks missing); νmax/cm–1: 3150 (br, N–H), 2210 (C≡N), 1621, 1595, 1504; HRMS (ESI+): m/z calcd for [C22H16N6 + Na]+, 387.1329; observed, 387.1328.
3-Amino-5-(1-((1-methylpiperidin-2-yl)methyl)-1H-indol-6-yl)-1H-pyrazole-4-carbonitrile (26n)
Trichloroacetonitrile (42 μL, 0.42 mmol) was added to a suspension of 3-(1-((1-methylpiperidin-2-yl)methyl)-1H-indol-6-yl)-3-oxopropanenitrile 55n (43 mg, 0.14 mmol) and sodium acetate (58 mg, 0.71 mmol) in ethanol (5 mL). The reaction mixture was stirred over 18 h, then diluted with NaHCO3 solution (12.5 mL) and water (12.5 mL), and extracted into DCM (3 × 25 mL). The combined organic extracts were washed (brine), dried (MgSO4), and concentrated in vacuo. The crude residue was dissolved in ethanol (5 mL), and hydrazine monohydrate (69 μL, 1.4 mmol) was added. The reaction mixture was heated under reflux for 24 h and then concentrated in vacuo. Purification by flash column chromatography (50–100% ethyl acetate in petroleum ether, 0–16% methanol in DCM) afforded 26n (27 mg, 57% yield). LCMS (ESI+): m/z 335.3 [M + H]+, (ESI–): m/z 333.2 [M – H]−, rt 1.22 min, >99%; 1H NMR: (500 MHz, CD3OD) 7.90 (1H, s), 7.64 (1H, d, J = 8.4 Hz), 7.50 (1H, dd, J = 8.4, 0.9 Hz), 7.31 (1H, d, J = 3.2 Hz), 6.52 (1H, dd, J = 3.2, 0.6 Hz), 4.74 (1H, dd, J = 14.3, 4.7 Hz), 4.07 (1H, dd, J = 14.4, 9.5 Hz), 3.11–3.03 (1H, m), 2.97–2.87 (1H, m), 2.66 (3H, s), 2.51 (1H, td, J = 11.3, 3.4 Hz), 1.74–1.58 (3H, m), 1.41–1.18 (3H, m); 13C NMR: (125 MHz, CD3OD) 157.9 (br), 152.8 (br), 137.5, 131.8, 131.3, 124.4 (br), 122.4, 119.0, 117.2, 109.1, 102.9, 74.3 (br), 64.2, 57.7, 42.8, 29.5, 25.5, 23.7 (1 peak missing); νmax/cm–1: 3150 (br, N–H), 2927, 2210 (C≡N), 1623, 1589, 1525, 1503; HRMS (ESI+): m/z calcd for [C19H22N6 + H]+, 335.1979; observed, 335.1971.
3-Amino-5-(1-((1-methylpiperidin-3-yl)methyl)-1H-indol-6-yl)-1H-pyrazole-4-carbonitrile (26o)
Trichloroacetonitrile (0.143 mL, 1.42 mmol) was added to a suspension of 3-(1-((1-methylpiperidin-3-yl)methyl)-1H-indol-6-yl)-3-oxopropanenitrile 55o (0.140 g, 0.474 mmol) and sodium acetate (0.194 g, 2.37 mmol) in ethanol (3 mL). The reaction mixture was stirred over 3 h 30 min, then diluted with NaHCO3 solution (10 mL), and extracted into DCM (3 × 25 mL). The combined organic extracts were washed (brine), dried (MgSO4), and concentrated in vacuo. The crude residue was dissolved in ethanol (5 mL), and hydrazine monohydrate (0.231 mL, 4.74 mmol) was added. The reaction mixture was heated under reflux for 15 h and then concentrated in vacuo. Purification by flash column chromatography (50–100% ethyl acetate in petroleum ether, 0–20% methanol in DCM) was carried out, followed by reverse phase chromatography (0–8% acetonitrile in water (+0.1% formic acid)) with addition of NaHCO3 solution (40 mL) to the combined fractions and extraction into DCM/methanol (9:1, 4 × 50 mL). The combined organic extracts were washed (brine), dried (MgSO4), and concentrated in vacuo to afford 26o (61 mg, 38% yield). LCMS (ESI+): m/z 335.3 [M + H]+, (ESI–): m/z 333.2 [M – H]−, rt 1.23 min, >99%; 1H NMR: (500 MHz, CDCl3) 7.78 (1H, s), 7.63 (1H, d, J = 8.3 Hz), 7.41 (1H, dd, J = 8.2, 1.4 Hz), 7.11 (1H, d, J = 3.2 Hz), 6.49 (1H, dd, J = 3.0, 0.6 Hz), 4.44 (2H, br s), 4.05–3.91 (2H, m), 2.68 (1H, d, J = 9.6 Hz), 2.61 (1H, d, J = 10.4 Hz), 2.32–2.21 (1H, m), 2.17 (3H, s), 2.05–1.88 (1H, m), 1.77 (1H, t J = 10.2 Hz), 1.69–1.45 (3H, m), 1.06–0.92 (1H, m); 13C NMR: (125 MHz, CDCl3) 157.1, 150.2, 136.2, 130.8, 130.1, 121.8, 121.1, 117.6, 115.7, 108.2, 101.7, 76.0, 59.4, 56.1, 50.4, 46.4, 37.0, 28.0, 24.5; νmax/cm–1: 3200 (br, N–H), 2932, 2788, 2209 (C≡N), 1622, 1590, 1524, 1501; HRMS (ESI+): m/z calcd for [C19H22N6 + H]+, 335.1979; observed, 335.1983.
3-Amino-5-(3-chloro-1-(pyridin-3-ylmethyl)-1H-indol-6-yl)-1H-pyrazole-4-carbonitrile (27g)
N-Chlorosuccinimide (21 mg, 0.16 mmol) was added to a solution of 3-amino-5-(1-(pyridin-3-ylmethyl)-1H-indol-6-yl)-1H-pyrazole-4-carbonitrile 26g (50 mg, 0.16 mmol) in DMF (1 mL). The reaction mixture was stirred over 6 h, and then further N-chlorosuccinimide (7 mg, 0.05 mmol) was added. The reaction mixture was stirred over 90 min, then diluted with ethyl acetate (100 mL), washed with NaHCO3 solution (2 × 100 mL) and brine (100 mL), dried (MgSO4), and concentrated in vacuo. Purification by flash column chromatography (0–100% ethyl acetate in petroleum ether, 0–7% methanol in DCM), followed by reverse phase column chromatography (0–50% acetonitrile in water), afforded 27g (35 mg, 63% yield). LCMS (ESI+): m/z 349.2 [M + H]+, (ESI–): m/z 347.1 [M – H]−, rt 1.57 min, >99%; 1H NMR: (500 MHz, (CD3)2SO) 8.60 (1H, d, J = 1.8 Hz), 8.47 (1H, dd, J = 4.8, 1.6 Hz), 7.97 (1H, s), 7.88 (1H, s), 7.67 (1H, dt, J = 7.9, 1.9 Hz), 7.61 (1H, dd, J = 8.4, 1.3 Hz), 7.58 (1H, d, J = 8.4 Hz), 7.33 (1H, ddd, J = 7.9, 4.7, 0.8 Hz), 6.22 (2H, br s), 5.48 (2H, s); 13C NMR: (125 MHz, (CD3)2SO) 155.7, 149.6, 149.0, 148.6, 135.1, 134.6, 133.0, 127.2, 126.0, 125.4, 123.8, 118.4, 118.0, 116.5, 108.0, 103.5, 70.8, 47.0; νmax/cm–1: 3344 (N–H), 2218 (C≡N), 1623, 1586, 1525; HRMS (ESI)−: m/z calcd for [C18H13ClN6 – H]−, 347.0817; observed, 347.0804.
3-Amino-5-(1-(4-(piperidin-1-ylmethyl)benzyl)-1H-indol-6-yl)-1H-pyrazole-4-carbonitrile (29b)
Trichloroacetonitrile (0.138 mL, 1.37 mmol) was added to a suspension of 3-oxo-3-(1-(4-(piperidin-1-ylmethyl)benzyl)-1H-indol-6-yl)propanenitrile 83b (0.170 g, 0.458 mmol) and sodium acetate (0.188 g, 2.29 mmol) in ethanol (5 mL). The reaction mixture was stirred over 44 h, then diluted with NaHCO3 solution (10 mL) and water (10 mL), and extracted into DCM (3 × 20 mL). The combined organic extracts were washed (brine), dried (MgSO4), and concentrated in vacuo. The crude residue was dissolved in ethanol (10 mL), and hydrazine monohydrate (0.223 mL, 4.58 mmol) was added. The reaction mixture was heated under reflux for 24 h, then concentrated in vacuo. Purification by flash column chromatography (0–100% ethyl acetate in petroleum ether, 0–15% methanol in DCM) was carried out, followed by reverse phase chromatography (0–35% acetonitrile in water (+0.1% formic acid)) with adjustment of the combined fractions to pH 8 and extraction into DCM/methanol (10:1, 3 × 50 mL). The combined organic extracts were washed (brine), dried (MgSO4), and concentrated in vacuo to afford 29b (60 mg, 32% yield). LCMS (ESI+): m/z 411.3 [M + H]+, rt 1.44 min, >99%; 1H NMR: (500 MHz, CDCl3) 7.70 (1H, s), 7.63 (1H, d, J = 8.2 Hz), 7.40 (1H, dd, J = 8.2, 1.4 Hz), 7.21 (2H, d, J = 8.1 Hz), 7.19 (1H, d, J = 3.1 Hz), 7.04 (2H, d, J = 7.9 Hz), 6.54 (1H, dd, J = 3.2, 0.6 Hz), 5.20 (2H, s), 4.33 (2H, br s), 3.42 (2H, s), 2.36 (4H, br s), 1.50 (4H, quin, J = 5.6 Hz), 1.42–1.33 (2H, m); 13C NMR: (125 MHz, CDCl3) 156.8, 150.2, 137.4, 136.2, 136.0, 130.6, 130.21, 130.17, 127.1, 121.9, 121.4, 117.7, 115.7, 108.1, 102.2, 75.7, 63.3, 54.5, 50.1, 25.6, 24.2; νmax/cm–1: 3200 (br, N–H), 2931, 2211 (C≡N), 1621, 1584, 1502; HRMS (ESI+): m/z calcd for [C25H26N6 + H]+, 411.2292; observed, 411.2287.
3-Amino-5-(1-(4-(morpholinomethyl)benzyl)-1H-indol-6-yl)-1H-pyrazole-4-carbonitrile (29c)
Trichloroacetonitrile (0.142 mL, 1.42 mmol) was added to a suspension of 3-(1-(4-(morpholinomethyl)benzyl)-1H-indol-6-yl)-3-oxopropanenitrile 83c (0.190 g, 0.473 mmol) and sodium acetate (0.194 g, 2.37 mmol) in ethanol (5 mL). The reaction mixture was stirred over 14 h, then diluted with NaHCO3 solution (15 mL), and extracted into DCM (3 × 25 mL). The combined organic extracts were washed (brine), dried (MgSO4), and concentrated in vacuo. The crude residue was dissolved in ethanol (5 mL), and hydrazine monohydrate (0.230 mL, 4.73 mmol) was added. The reaction mixture was heated under reflux for 22 h and then concentrated in vacuo. Purification by flash column chromatography (0–100% ethyl acetate in petroleum ether, 0–10% methanol in DCM) was carried out, followed by reverse phase chromatography (100% water (+0.1% formic acid)) with adjustment of the combined fractions to pH 8 and extraction into DCM/methanol (10:1, 3 × 50 mL). The combined organic extracts were washed (brine), dried (MgSO4), and concentrated in vacuo to afford 29c (81 mg, 42% yield). LCMS (ESI+): m/z 413.3 [M + H]+, rt 1.36 min, >99%; 1H NMR: (500 MHz, CDCl3) 7.73 (1H, s), 7.66 (1H, d, J = 8.2 Hz), 7.39 (1H, dd, J = 8.3, 1.3 Hz), 7.25–7.20 (3H, m), 7.08 (2H, d, J = 8.1 Hz), 6.56 (1H, dd, J = 3.2, 0.6 Hz), 5.26 (2H, s), 4.20 (2H, s), 3.64 (4H, t, J = 4.6 Hz), 3.41 (2H, s), 2.47–2.31 (4H, m); 13C NMR: (125 MHz, CDCl3) 157.0, 150.1, 137.6, 136.2, 135.9, 130.7, 130.4, 129.9, 127.1, 122.0, 121.0, 117.6, 115.4, 108.2, 102.3, 76.1, 67.0, 63.1, 53.7, 50.2; νmax/cm–1: 3200 (br, N–H), 2922, 2212 (C≡N), 1740, 1624, 1584, 1502; HRMS (ESI+): m/z calcd for [C24H24N6O + H]+, 413.2084; observed, 413.2079.
3-Amino-5-(1-(4-((4-isopropylpiperazin-1-yl)methyl)benzyl)-1H-indol-6-yl)-1H-pyrazole-4-carbonitrile (29e)
Trichloroacetonitrile (0.129 mL, 1.29 mmol) was added to a suspension of 3-(1-(4-((4-isopropylpiperazin-1-yl)methyl)benzyl)-1H-indol-6-yl)-3-oxopropanenitrile 83e (0.178 g, 0.429 mmol) and sodium acetate (0.176 g, 2.15 mmol) in ethanol (5 mL). The reaction mixture was stirred over 10 h, then diluted with NaHCO3 solution (7.5 mL) and water (7.5 mL), and extracted into DCM (3 × 25 mL). The combined organic extracts were washed (brine), dried (MgSO4), and concentrated in vacuo. The crude residue was dissolved in ethanol (3 mL), and hydrazine monohydrate (0.209 mL, 4.29 mmol) was added. The reaction mixture was heated under reflux for 7 h, then quenched with excess acetone at room temperature, and concentrated in vacuo. Purification by flash column chromatography (50–100% ethyl acetate in petroleum ether, 0–20% methanol in DCM (+0.1% NH3)) afforded 29e (0.147 g, 75% yield). LCMS (ESI+): m/z 454.3 [M + H]+, (ESI–): m/z 452.3 [M – H]−, rt 1.40 min, >99%; 1H NMR: (500 MHz, CD3OD) 7.81 (1H, s), 7.63 (1H, d, J = 8.3 Hz), 7.48 (1H, d, J = 8.1 Hz), 7.39 (1H, d, J = 2.6 Hz), 7.25 (2H, d, J = 8.1 Hz), 7.21 (2H, d, J = 8.2 Hz), 6.53 (1H, d, J = 3.2 Hz), 5.36 (2H, s), 3.55 (2H, s), 3.36–3.29 (1H, m), 3.10 (4H, br s), 2.69 (4H, br s), 1.27 (6H, d, J = 6.9 Hz); 13C NMR: (125 MHz, CD3OD) 138.6, 137.3, 137.1, 131.6, 131.4, 130.8, 128.5, 122.2, 118.7, 117.0, 109.2, 102.7, 62.3, 59.0, 51.0, 50.8, 49.4, 17.3 (4 peaks missing); νmax/cm–1: 2210 (C≡N), 1623, 1587, 1501; HRMS (ESI)+: m/z calcd for [C27H31N7 + H]+, 454.2714; observed, 454.2715.
4-Methyl-5-(1-(4-(pyrrolidin-1-ylmethyl)benzyl)-1H-indol-6-yl)-1H-pyrazol-3-amine (30a)
Hydrazine monohydrate (0.147 mL, 3.01 mmol) was added to a solution of 2-methyl-3-oxo-3-(1-(4-(pyrrolidin-1-ylmethyl)benzyl)-1H-indol-6-yl)propanenitrile 84a (0.140 g, 0.301 mmol) in ethanol (5 mL). The reaction mixture was heated under reflux for 28 h, then quenched with excess acetone at room temperature, and concentrated in vacuo. Purification by flash column chromatography [50–100% ethyl acetate in petroleum ether, 0–20% methanol (+0.1% NH3) in DCM] afforded 30a (24 mg, 21% yield). LCMS (ESI+): m/z 386.3 [M + H]+, rt 1.25 min, >99%; 1H NMR: (500 MHz, CDCl3) 7.69 (1H, d, J = 8.2 Hz), 7.30 (1H, s), 7.26 (2H, d, J = 8.1 Hz), 7.20 (1H, d, J = 3.2 Hz), 7.18 (1H, dd, J = 8.4, 1.5 Hz), 7.08 (2H, d, J = 8.1 Hz), 6.57 (1H, dd, J = 3.1, 0.7 Hz), 5.31 (2H, s), 3.58 (2H, s), 2.53–2.43 (4H, m), 1.98 (3H, s), 1.81–1.71 (4H, m); 13C NMR: (125 MHz, CDCl3) 154.4, 142.7, 139.1, 136.4, 135.8, 129.7, 129.6, 128.8, 126.9, 124.3, 121.5, 119.0, 108.8, 101.9, 99.3, 60.3, 54.2, 50.3, 23.5, 7.9; νmax/cm–1: 3200 (br, N–H), 2916, 2788, 1606, 1502; HRMS (ESI)+: m/z calcd for [C24H27N5 + H]+, 386.2339; observed, 386.2354.
5-(1-(4-(Piperidin-1-ylmethyl)benzyl)-1H-indol-6-yl)-1H-pyrazol-3-amine (31b)
Hydrazine monohydrate (0.136 mL, 2.80 mmol) was added to a solution of 3-oxo-3-(1-(4-(piperidin-1-ylmethyl)benzyl)-1H-indol-6-yl)propanenitrile 83b (0.104 g, 0.280 mmol) in ethanol (5 mL). The reaction mixture was heated under reflux for 21 h and then concentrated in vacuo. Purification by flash column chromatography (0–100% ethyl acetate in petroleum ether, 0–15% methanol in DCM) was carried out, followed by reverse phase chromatography [0–20% acetonitrile in water (+0.1% formic acid)] with adjustment of the combined fractions to pH 14 and extraction into DCM (3 × 25 mL). The combined organic extracts were washed (brine), dried (MgSO4), and concentrated in vacuo to afford 31b (56 mg, 52%). LCMS (ESI+): m/z 386.3 [M + H]+, rt 1.24 min, >99%; 1H NMR: (500 MHz, CDCl3) 7.64 (1H, d, J = 8.3 Hz), 7.44 (1H, s), 7.30–7.25 (1H, m), 7.23 (2H, d, J = 8.1 Hz), 7.14 (1H, d, J = 3.2 Hz), 7.03 (2H, d, J = 8.2 Hz), 6.54 (1H, dd, J = 3.1, 0.7 Hz), 5.88 (1H, s), 5.27 (2H, s), 3.68 (2H, br s), 3.41 (2H, s), 2.33 (4H, br s), 1.58–1.50 (4H, m), 1.45–1.36 (2H, m); 13C NMR: (125 MHz, CDCl3) 155.1 (br), 146.6 (br), 138.4, 136.6, 135.8, 129.8, 129.7, 129.1, 126.7, 123.9, 121.6, 117.7, 106.8, 102.0, 90.6, 63.5, 54.6, 50.0, 26.0, 24.4; νmax/cm–1: 2926, 2848, 2789, 2757, 1586, 1505; HRMS (ESI+): m/z calcd for [C24H27N5 + H]+, 386.2339; observed, 386.2336.
Expression and Purification of Full Length Mab TrmD
Expression and purification was carried out as previously described.12 AVA0421 plasmid with an N-His-3C Protease site-TrmD full-length insert (kindly provided by the Seattle Structural Genomics Consortium) was used for expression. E. coli BL21 (DE3) strain containing the above plasmid was grown overnight at 37 °C in 2xYT media, supplemented with ampicillin (100 μg/mL), until optical density (A600nm) reached 0.6. The expression of recombinant construct was induced by the addition of isopropyl β-d-1-thiogalactopyranoside (IPTG) to a final concentration of 0.5 mM and further allowed to grow at 18 °C for 16 h. Cells were harvested by centrifugation at 4 °C for 20 min at 5000g and the pellet re-suspended in buffer A [25 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES) pH 7.5, 500 mM NaCl, 5% glycerol, 10 mM MgCl2, 1 mM tris(2-carboxyethyl)phosphine (TCEP), 20 mM imidazole]. 0.1% Triton (Sigma), 10 μg/mL DNaseI, 5 mM MgCl2, and 3 protease inhibitor cocktail tablets (New England Biolabs) were added to the cell suspension. The clarified cell lysate was filtered using a 0.45 μm syringe filter and passed through a pre-equilibrated (with buffer A), 10 mL pre-packed nickel-sepharose column (HiTrap IMAC FF, GE Healthcare). The column was washed with buffer A and the bound protein eluted using buffer B (25 mM HEPES pH 7.5, 500 mM NaCl, 5% glycerol, 1 mM TCEP, 500 mM imidazole). 3C Protease was added to the pooled elutes and subjected to dialysis, against 2 L of buffer C (25 mM HEPES pH 7.5, 500 mM NaCl, 5% glycerol, 1 mM TCEP) overnight at 4 °C, to cleave the His-tag. The dialyzed protein was then passed through a pre-equilibrated (buffer A) 5 mL HiTrap IMAC FF Nickel column (GE Healthcare). The flow through from the column was collected and concentrated to 3 mL using a 10 kDa centrifugal concentrator (Sartorius Stedim) and loaded onto a pre-equilibrated (with buffer D: 25 mM HEPES pH 7.5, 500 mM NaCl, 5% glycerol) 120 mL Superdex200 16/600 column (GE Healthcare), and 2 mL fractions were collected and analyzed on a 15% SDS-PAGE gel and by MALDI fingerprinting.
Soaking and Co-crystallization of Mab TrmD with Fragments and Compounds
Experiments for SAM, 1, 16, 20, 23, 24f, 26f, 29a, 29d, and 31a have been described.12 Soaking experiments with the remaining compounds were performed as previously described.12Mab TrmD apo crystals were grown at 19 °C in 48-well sitting drop plates (Swiss CDI) using 0.08 mM sodium cacodylate pH 6.5–7.0, 1–2 M ammonium sulfate and 20 mg/mL of the protein in storage buffer (25 mM HEPES pH 7.5, 500 mM NaCl, 5% glycerol) at drop ratio 1 μL/1 μL (protein: reservoir respectively) and equilibrated against 250 μL reservoir solution. The apo crystals formed were allowed to soak in a 4 μL drop containing reservoir solution and 10 mM compound (in DMSO) and equilibrated against 700 μL of the corresponding reservoir solution overnight at 19 °C in 24-well hanging drop vapor diffusion plates.
X-ray Data Collection and Processing
The Mab TrmD crystals were cryo-cooled in mother liquor containing 27.5% ethylene glycol. Crystallographic data collection and processing was performed as previously described.12 X-ray data sets were collected on I04, I03, I02, I04-1, or I24 beamlines at the Diamond Light Source (UK), using the rotation method at a wavelength of 0.979 Å, omega start: 0°, omega oscillation: 0.1°–0.2°, total images: 2100–2400, exposure time: 0.05–0.08 s. The diffraction images were processed using AutoPROC, utilizing XDS for indexing, integration, followed by POINTLESS, AIMLESS and TRUNCATE programs from CCP4 Suite for data reduction, scaling, and the calculation of structure factor amplitudes and intensity statistics.33−38
Mab TrmD ligand-bound structures were solved by molecular replacement using PHASER with the atomic coordinates of the solved Mab TrmD apo structure (PDB code 6NVR) as a search model.39 Structure refinement was carried out using REFMAC and PHENIX.40,41 The models obtained were manually re-built using the COOT interactive graphics program and electron density maps calculated with 2|Fo| – |Fc| and |Fo| – |Fc| coefficients.42,43 Positions of ligands and water molecules were located in difference electron density maps, and OMIT difference maps |mFo – DFc| were calculated and analyzed to further verify positions of ligands.44
Differential Scanning Fluorimetry
Experiments for fragments 1, 16–18, and 20–21 at 5 mM concentration have been described.12 DSF was performed using a BioRad CFX Connect system, from 25 to 95 °C in 0.5 °C increments of 30 s duration. Samples were run in 96-well plates, with each well containing a final volume of 25 μL. Screening was conducted with 50 mM HEPES pH 7.5, 500 mM NaCl, 5x SyproOrange, 10 μM Mab TrmD, and either 5% DMSO or 5% ligand stock solution in DMSO.
Isothermal Titration Calorimetry
Experiments for 16, 20, 23, 24f, 26f, 28, 29a, 29d, and 31a have been described.12 ITC experiments with the remaining compounds to quantify binding to Mab TrmD were performed as previously described,12 using Malvern MicroCal iTC200 or Auto-iTC200 systems at 25 °C. Titrations consisted of an initial injection (0.2 μL), discarded during data processing, followed by either 19 (2 μL) or 39 (1 μL) injections separated by intervals of 60–150 s duration. Protein was dialyzed overnight at 4 °C in storage buffer (50 mM HEPES pH 7.5, 500 mM NaCl, 5% glycerol). Sample cell and syringe solutions were prepared using the same storage buffer, with a final DMSO concentration of 2–10% according to ligand solubility in the buffer. TrmD concentrations of either 33 or 100 μM were used, with ligand to protein concentration ratios ranging from 10 to 20:1. Control titrations without protein were also performed and subtracted from ligand to protein titrations. Titrations were fitted with Origin software (OriginLab, Northampton, MA, USA), using a one-site binding model with N fixed to 1 only for weakly binding ligands. Titrations were typically performed once (n = 1), with multiple isotherms obtained (n > 1) for key compounds of interest. Kd values are reported to 2 significant figures. Error provided by Origin software due to model fit is reported when n = 1, whereas standard deviation is reported when n > 1.
For the reverse ITC titration with 26o (Figure S8b), protein solution (420 μM TrmD) was injected into the sample cell (35 μM ligand). Other aspects of the experiment setup and analysis were identical to the forward titrations.
Native Mass Spectrometry
Native nanoelectrospray ionization mass spectrometry spectra were recorded on a Synapt HDMS mass spectrometer (Waters, Manchester, UK) modified for studying high masses. Mab TrmD was exchanged into 200 mM ammonium acetate (pH 7.0) solution using Micro Bio-Spin 6 chromatography columns (Bio-Rad, UK). Protein samples were equilibrated at room temperature in the absence or presence of the indicated concentrations of ligands for at least 15 min before analysis. Protein solution (2.5 μL) was injected into a gold-coated borosilicate emitter (Thermo Scientific, UK) for sampling. Typical conditions were capillary voltage 1.5 kV, cone voltage 50 V, trap collision voltage 20 V, transfer collision voltage 20 V, source temperature 20 °C, backing pressure 3–4 mbar, trap pressure 3–4 × 10–2 mbar, IM (N2) pressure 5–6 × 10–1 mbar and TOF pressure 7–8 × 10–7 mbar. All mass spectra were calibrated externally with caesium iodide (10 mg/mL). Data acquisition and processing were performed using MassLynx 4.1 (Waters).
Screening against Mab
Mab subspecies abscessus (ATCC 19977) was transformed with pmv310 plasmid expressing Lux ABDCE operon, grown in Middlebrook 7H9 broth supplemented with ADC (Sigma, UK). Minimum Inhibitory Concentrations (MIC) were determined according to the Clinical and Laboratory Standards Institute (CLSI) method M07-A9. Mycobacteria were grown to an OD600 of 0.2–0.3 in liquid culture, and 1 × 105 bacteria were added to each well of 96-well plates containing serial dilutions of compound (400, 200, 100, 50, 25, 12.5, 6.3, 3.1, 1.6, 0.8, 0.4, 0 μM), in triplicate wells per condition, and incubated at 37 °C until growth was seen in the control wells. The MIC value was determined as the last well which showed no bacterial growth.
Screening against H37Rv Mtb
Antimicrobial susceptibility testing against Mtb H37Rv was performed in various media including: GAST-Fe, 7H9/ADC, 7H9/DPPC (4.7 g/L 7H9 base, 14 mg/L DPPC, 0.81 g/L NaCl, 0.3 g/L Casitone, and 0.05% Tyloxapol), and 7H9/Glucose (4.7 g/L 7H9 base, 4 g/L glucose, 0.81 g/L NaCl, 0.3 g/L Casitone, and 0.05% Tyloxapol). H37Rv was grown in the corresponding media to OD 0.2–0.4 prior to use. A two-fold serial dilution series (50 μL) was placed in each well of a sterile 96-well round bottom plate, and then 50 μL of H37Rv diluted to OD 0.0002 was added. Plates were incubated at 37 °C for 2 weeks prior to determining MIC. The MIC is defined here as the drug concentration that completely inhibits growth of cells.
Acknowledgments
The authors would like to thank Dr Glyn Williams for his advice on ITC and Dr Sitthivut Charoensutthivarakul for the collection of HRMS data. The authors would like to thank the Seattle Structural Genomics Consortium for kindly providing the Mab trmD AVA0421 plasmid, Diamond Light Source for beam-time (proposals mx9537, mx14043, mx18548) and the staff of beamlines I03, I02, I04, I04-1 and I24 for assistance with data collection.
Glossary
Abbreviations
- ΔTm
change in melting temperature
- CF
cystic fibrosis
- DPPC
dipalmitoyl phosphatidylcholine
- DSF
differential scanning fluorimetry
- ESI
electrospray ionization
- GE
group efficiency
- ITC
isothermal titration calorimetry
- Kd
binding affinity
- LE
ligand efficiency
- Mab
Mycobacterium abscessus
- Mtb
Mycobacterium tuberculosis
- MIC
minimum inhibitory concentration
- native MS
native mass spectrometry
- SAM
S-adenosyl methionine
- TrmD
tRNA (m1G37) methyltransferase
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.jmedchem.9b00809.
Synthesis of intermediates, X-ray crystal structures of Mab TrmD in complex with compounds 3, 8, and 14, X-ray crystal structures of Mab TrmD in complex with compounds 22, 24d, 26n, and 31b, X-ray crystallographic data collection and refinement statistics, and ITC traces (PDF)
Molecular formula strings (CSV)
Accession Codes
Atomic coordinates for the X-ray structures of compounds 1 (PDB code 6NW6), 3 (PDB code 6QRF), 7 (PDB code 6QRE), 8 (PDB code 6QRG), 14 (PDB code 6QQQ), 16 (PDB Code 6QOT), 20 (PDB code 6QOU), 22 (PDB code 6QQR), 23 (PDB code 6QQS), 24a (PDB code 6QQT), 24b (PDB code 6QRC), 24c (PDB code 6QQW), 24d (PDB code 6QQV), 24f (PDB code 6QQX), 25 (PDB code 6QQU), 26f (PDB code 6QQY), 26g (PDB code 6QQZ), 26n (PDB code 6QR4), 27g (PDB code 6QR1), 26j (PDB code 6QR2), 26l (PDB code 6QR0), 26o (PDB code 6QR3), 29a (PDB code 6QR6), 29b (PDB code 6QR7), 29c (PDB code 6QR9), 29d (PDB code 6QR8), 29e (PDB code 6QRD), 31b (PBD code 6QRA) are available from the RCPB Protein Data Bank (www.rcpb.org). Authors will release the atomic coordinates and experimental data upon article publication.
Author Contributions
A.J.W. and S.E.T. contributed equally. The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
A.J.W. is funded by the EPSRC; SET is funded by the Cystic Fibrosis Trust and Foundation Botnar; V.M. is funded by the Bill and Melinda Gates Foundation SHORTEN-TB (OPP1158806); T.L.B. is funded by the Wellcome Trust (Wellcome Trust Investigator Award 200814_Z_16_Z); R.A.F. is funded by the Cystic Fibrosis Trust Strategic Research Centre (SRC010) and Innovation Hub (IH01) awards, Wellcome Trust (107032AIA), and Royal Papworth Hospital Research Award. This work was funded in part by the Intramural Research Program of NIH, NIAID (AI000693-25).
The authors declare no competing financial interest.
Supplementary Material
References
- Bryant J. M.; Grogono D. M.; Rodriguez-Rincon D.; Everall I.; Brown K. P.; Moreno P.; Verma D.; Hill E.; Drijkoningen J.; Gilligan P.; Esther C. R.; Noone P. G.; Giddings O.; Bell S. C.; Thomson R.; Wainwright C. E.; Coulter C.; Pandey S.; Wood M. E.; Stockwell R. E.; Ramsay K. A.; Sherrard L. J.; Kidd T. J.; Jabbour N.; Johnson G. R.; Knibbs L. D.; Morawska L.; Sly P. D.; Jones A.; Bilton D.; Laurenson I.; Ruddy M.; Bourke S.; Bowler I. C. J. W.; Chapman S. J.; Clayton A.; Cullen M.; Dempsey O.; Denton M.; Desai M.; Drew R. J.; Edenborough F.; Evans J.; Folb J.; Daniels T.; Humphrey H.; Isalska B.; Jensen-Fangel S.; Jönsson B.; Jones A. M.; Katzenstein T. L.; Lillebaek T.; MacGregor G.; Mayell S.; Millar M.; Modha D.; Nash E. F.; O’Brien C.; O’Brien D.; Ohri C.; Pao C. S.; Peckham D.; Perrin F.; Perry A.; Pressler T.; Prtak L.; Qvist T.; Robb A.; Rodgers H.; Schaffer K.; Shafi N.; van Ingen J.; Walshaw M.; Watson D.; West N.; Whitehouse J.; Haworth C. S.; Harris S. R.; Ordway D.; Parkhill J.; Floto R. A. Emergence and spread of a human-transmissible multidrug-resistant nontuberculous mycobacterium. Science 2016, 354, 751–757. 10.1126/science.aaf8156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haworth C. S.; Banks J.; Capstick T.; Fisher A. J.; Gorsuch T.; Laurenson I. F.; Leitch A.; Loebinger M. R.; Milburn H. J.; Nightingale M.; Ormerod P.; Shingadia D.; Smith D.; Whitehead N.; Wilson R.; Floto R. A. British Thoracic Society guidelines for the management of non-tuberculous mycobacterial pulmonary disease (NTM-PD). Thorax 2017, 72, ii1–ii64. 10.1136/thoraxjnl-2017-210927. [DOI] [PubMed] [Google Scholar]
- Esther C. R. Jr; Esserman D. A.; Gilligan P.; Kerr A.; Noone P. G. Chronic Mycobacterium abscessus infection and lung function decline in cystic fibrosis. J. Cystic Fibrosis 2010, 9, 117–123. 10.1016/j.jcf.2009.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qvist T.; Taylor-Robinson D.; Waldmann E.; Olesen H. V.; Hansen C. R.; Mathiesen I. H.; Høiby N.; Katzenstein T. L.; Smyth R. L.; Diggle P. J.; Pressler T. Comparing the harmful effects of nontuberculous mycobacteria and gram negative bacteria on lung function in patients with cystic fibrosis. J. Cystic Fibrosis 2016, 15, 380–385. 10.1016/j.jcf.2015.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Floto R. A.; Olivier K. N.; Saiman L.; Daley C. L.; Herrmann J.-L.; Nick J. A.; Noone P. G.; Bilton D.; Corris P.; Gibson R. L.; Hempstead S. E.; Koetz K.; Sabadosa K. A.; Sermet-Gaudelus I.; Smyth A. R.; van Ingen J.; Wallace R. J.; Winthrop K. L.; Marshall B. C.; Haworth C. S. US Cystic Fibrosis Foundation and European Cystic Fibrosis Society consensus recommendations for the management of non-tuberculous mycobacteria in individuals with cystic fibrosis. Thorax 2016, 71, i1–i22. 10.1136/thoraxjnl-2015-207360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nessar R.; Cambau E.; Reyrat J. M.; Murray A.; Gicquel B. Mycobacterium abscessus: a new antibiotic nightmare. J. Antimicrob. Chemother. 2012, 67, 810–818. 10.1093/jac/dkr578. [DOI] [PubMed] [Google Scholar]
- Goto-Ito S.; Ito T.; Yokoyama S. Trm5 and TrmD: Two Enzymes from Distinct Origins Catalyze the Identical tRNA Modification, m1G37. Biomolecules 2017, 7, 32. 10.3390/biom7010032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gamper H. B.; Masuda I.; Frenkel-Morgenstern M.; Hou Y.-M. Maintenance of protein synthesis reading frame by EF-P and m1G37-tRNA. Nat. Commun. 2015, 6, 7226. 10.1038/ncomms8226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forsyth R. A.; Haselbeck R. J.; Ohlsen K. L.; Yamamoto R. T.; Xu H.; Trawick J. D.; Wall D.; Wang L.; Brown-Driver V.; Froelich J. M.; King P.; McCarthy M.; Malone C.; Misiner B.; Robbins D.; Tan Z.; Zhu Z.-y.; Carr G.; Mosca D. A.; Zamudio C.; Foulkes J. G.; Zyskind J. W. A genome-wide strategy for the identification of essential genes in Staphylococcus aureus. Mol. Microbiol. 2002, 43, 1387–1400. 10.1046/j.1365-2958.2002.02832.x. [DOI] [PubMed] [Google Scholar]
- Turner K. H.; Wessel A. K.; Palmer G. C.; Murray J. L.; Whiteley M. Essential genome of Pseudomonas aeruginosa in cystic fibrosis sputum. Proc. Natl. Acad. Sci. U.S.A. 2015, 112, 4110–4115. 10.1073/pnas.1419677112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sassetti C. M.; Boyd D. H.; Rubin E. J. Genes required for mycobacterial growth defined by high density mutagenesis. Mol. Microbiol. 2003, 48, 77–84. 10.1046/j.1365-2958.2003.03425.x. [DOI] [PubMed] [Google Scholar]
- Thomas S. E.; Whitehouse A. J.; Brown K.; Belardinelli J. M.; Lahiri R.; Libardo M. D. J.; Gupta P.; Malhotra S.; Boshoff H. I. M.; Jackson M.; Abell C.; Coyne A. G.; Blundell T. L.; Floto R. A.; Mendes V.. Fragment-based Discovery of a New Class of Inhibitors Targeting Mycobacterial tRNA Modification. 2019, arXiv: 564013, DOI: https://doi.org/10.1101/564013.bioRxiv, https://www.biorxiv.org/content/10.1101/564013v1 (accessed May 09, 2019). [DOI] [PMC free article] [PubMed]
- Baugh L.; Phan I.; Begley D. W.; Clifton M. C.; Armour B.; Dranow D. M.; Taylor B. M.; Muruthi M. M.; Abendroth J.; Fairman J. W.; Fox D. III; Dieterich S. H.; Staker B. L.; Gardberg A. S.; Choi R.; Hewitt S. N.; Napuli A. J.; Myers J.; Barrett L. K.; Zhang Y.; Ferrell M.; Mundt E.; Thompkins K.; Tran N.; Lyons-Abbott S.; Abramov A.; Sekar A.; Serbzhinskiy D.; Lorimer D.; Buchko G. W.; Stacy R.; Stewart L. J.; Edwards T. E.; Van Voorhis W. C.; Myler P. J. Increasing the structural coverage of tuberculosis drug targets. Tuberculosis 2015, 95, 142–148. 10.1016/j.tube.2014.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elkins P. A.; Watts J. M.; Zalacain M.; van Thiel A.; Vitazka P. R.; Redlak M.; Andraos-Selim C.; Rastinejad F.; Holmes W. M. Insights into catalysis by a knotted TrmD tRNA methyltransferase. J. Mol. Biol. 2003, 333, 931–949. 10.1016/j.jmb.2003.09.011. [DOI] [PubMed] [Google Scholar]
- Ahn H. J.; Kim H.-W.; Yoon H.-J.; Lee B. I.; Suh S. W.; Yang J. K. Crystal structure of tRNA(m1G37)methyltransferase: Insights into tRNA recognition. EMBO J. 2003, 22, 2593–2603. 10.1093/emboj/cdg269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito T.; Masuda I.; Yoshida K.-i.; Goto-Ito S.; Sekine S.-i.; Suh S. W.; Hou Y.-M.; Yokoyama S. Structural basis for methyl-donor-dependent and sequence-specific binding to tRNA substrates by knotted methyltransferase TrmD. Proc. Natl. Acad. Sci. U.S.A. 2015, 112, E4197–E4205. 10.1073/pnas.1422981112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goto-Ito S.; Ito T.; Kuratani M.; Bessho Y.; Yokoyama S. Tertiary structure checkpoint at anticodon loop modification in tRNA functional maturation. Nat. Struct. Mol. Biol. 2009, 16, 1109–1115. 10.1038/nsmb.1653. [DOI] [PubMed] [Google Scholar]
- Zhong W.; Koay A.; Ngo A.; Li Y.; Nah Q.; Wong Y. H.; Chionh Y. H.; Ng H. Q.; Koh-Stenta X.; Poulsen A.; Foo K.; McBee M.; Choong M. L.; El Sahili A.; Kang C.; Matter A.; Lescar J.; Hill J.; Dedon P. Targeting the bacterial epitranscriptome for antibiotic development: Discovery of novel tRNA-(N1G37) methyltransferase (TrmD) inhibitors. ACS Infect. Dis. 2019, 5, 326–335. 10.1021/acsinfecdis.8b00275. [DOI] [PubMed] [Google Scholar]
- Scott D. E.; Coyne A. G.; Hudson S. A.; Abell C. Fragment-based approaches in drug discovery and chemical biology. Biochemistry 2012, 51, 4990–5003. 10.1021/bi3005126. [DOI] [PubMed] [Google Scholar]
- Hill P. J.; Abibi A.; Albert R.; Andrews B.; Gagnon M. M.; Gao N.; Grebe T.; Hajec L. I.; Huang J.; Livchak S.; Lahiri S. D.; McKinney D. C.; Thresher J.; Wang H.; Olivier N.; Buurman E. T. Selective Inhibitors of Bacterial t-RNA-(N1G37) Methyltransferase (TrmD) That Demonstrate Novel Ordering of the Lid Domain. J. Med. Chem. 2013, 56, 7278–7288. 10.1021/jm400718n. [DOI] [PubMed] [Google Scholar]
- Lundbäck T.; O’Brien R.; Williams G.. The Mix: Simultaneous Affinity Determination for Isomers and Enantiomers; MicroCal: Northampton, MA, USA, 2006. [Google Scholar]
- Hernández H.; Robinson C. V. Determining the stoichiometry and interactions of macromolecular assemblies from mass spectrometry. Nat. Protoc. 2007, 2, 715–726. 10.1038/nprot.2007.73. [DOI] [PubMed] [Google Scholar]
- Chan D. S.-H.; Matak-Vinković D.; Coyne A. G.; Abell C. Insight into protein conformation and subcharging by DMSO from native ion mobility mass spectrometry. ChemistrySelect 2016, 1, 5686–5690. 10.1002/slct.201601402. [DOI] [Google Scholar]
- Sterling H. J.; Prell J. S.; Cassou C. A.; Williams E. R. Protein conformation and supercharging with DMSO from aqueous solution. J. Am. Soc. Mass Spectrom. 2011, 22, 1178–1186. 10.1007/s13361-011-0116-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zanaletti R.; Bettinetti L.; Castaldo C.; Cocconcelli G.; Comery T.; Dunlop J.; Gaviraghi G.; Ghiron C.; Haydar S. N.; Jow F.; Maccari L.; Micco I.; Nencini A.; Scali C.; Turlizzi E.; Valacchi M. Discovery of a Novel Alpha-7 Nicotinic Acetylcholine Receptor Agonist Series and Characterization of the Potent, Selective, and Orally Efficacious Agonist 5-(4-Acetyl[1,4]diazepan-1-yl)pentanoic Acid [5-(4-Methoxyphenyl)-1H-pyrazol-3-yl] Amide (SEN15924, WAY-361789). J. Med. Chem. 2012, 55, 4806–4823. 10.1021/jm300247y. [DOI] [PubMed] [Google Scholar]
- Hassaneen H. M. E. New Approach to 4- and 5-Aminopyrazole Derivatives. Synth. Commun. 2007, 37, 3579–3588. 10.1080/00397910701557564. [DOI] [Google Scholar]
- Wender P. A.; Beckham S.; O’Leary J. G. A second generation photochemically activatable dynemicin analog: A concise synthesis and DNA cleavage studies. Synthesis 1994, 1994, 1278–1282. 10.1055/s-1994-25681. [DOI] [Google Scholar]
- Soni A.; Dutt A.; Sattigeri V.; Cliffe I. A. Efficient and selective demethylation of heteroaryl methyl ethers in the presence of aryl methyl ethers. Synth. Commun. 2011, 41, 1852–1857. 10.1080/00397911.2010.493262. [DOI] [Google Scholar]
- Soler M.; Figueras E.; Serrano-Plana J.; González-Bártulos M.; Massaguer A.; Company A.; Martínez M. Á.; Malina J.; Brabec V.; Feliu L.; Planas M.; Ribas X.; Costas M. Design, preparation, and characterization of Zn and Cu metallopeptides based on tetradentate aminopyridine ligands showing enhanced DNA cleavage activity. Inorg. Chem. 2015, 54, 10542–10558. 10.1021/acs.inorgchem.5b01680. [DOI] [PubMed] [Google Scholar]
- Deb I.; Das D.; Seidel D. Redox Isomerization via Azomethine Ylide Intermediates:N-Alkyl Indoles from Indolines and Aldehydes. Org. Lett. 2011, 13, 812–815. 10.1021/ol1031359. [DOI] [PubMed] [Google Scholar]
- Doebelin C.; Patouret R.; Garcia-Ordonez R. D.; Chang M. R.; Dharmarajan V.; Kuruvilla D. S.; Novick S. J.; Lin L.; Cameron M. D.; Griffin P. R.; Kamenecka T. M. N-Arylsulfonyl Indolines as Retinoic Acid Receptor-Related Orphan Receptor γ (RORγ) Agonists. ChemMedChem 2016, 11, 2607–2620. 10.1002/cmdc.201600491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehrt S.; Rhee K. Mycobacterium tuberculosis metabolism and host interaction: mysteries and paradoxes. Curr. Top Microbiol. Immunol. 2013, 374, 163–188. 10.1007/82_2012_299. [DOI] [PubMed] [Google Scholar]
- Vonrhein C.; Flensburg C.; Keller P.; Sharff A.; Smart O.; Paciorek W.; Womack T.; Bricogne G. Data processing and analysis with theautoPROCtoolbox. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2011, 67, 293–302. 10.1107/s0907444911007773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabsch W. XDS. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2010, 66, 125–132. 10.1107/s0907444909047337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans P. R. An introduction to data reduction: Space-group determination, scaling and intensity statistics. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2011, 67, 282–292. 10.1107/s090744491003982x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans P. R.; Murshudov G. N. How good are my data and what is the resolution?. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2013, 69, 1204–1214. 10.1107/s0907444913000061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- French S.; Wilson K. On the treatment of negative intensity observations. Acta Crystallogr., Sect. A: Cryst. Phys., Diffr., Theor. Gen. Crystallogr. 1978, 34, 517–525. 10.1107/s0567739478001114. [DOI] [Google Scholar]
- Winn M. D.; Ballard C. C.; Cowtan K. D.; Dodson E. J.; Emsley P.; Evans P. R.; Keegan R. M.; Krissinel E. B.; Leslie A. G. W.; McCoy A.; McNicholas S. J.; Murshudov G. N.; Pannu N. S.; Potterton E. A.; Powell H. R.; Read R. J.; Vagin A.; Wilson K. S. Overview of theCCP4 suite and current developments. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2011, 67, 235–242. 10.1107/s0907444910045749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCoy A. J.; Grosse-Kunstleve R. W.; Adams P. D.; Winn M. D.; Storoni L. C.; Read R. J. Phasercrystallographic software. J. Appl. Crystallogr. 2007, 40, 658–674. 10.1107/s0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murshudov G. N.; Skubák P.; Lebedev A. A.; Pannu N. S.; Steiner R. A.; Nicholls R. A.; Winn M. D.; Long F.; Vagin A. A. REFMAC5 for the refinement of macromolecular crystal structures. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2011, 67, 355–367. 10.1107/s0907444911001314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adams P. D.; Afonine P. V.; Bunkóczi G.; Chen V. B.; Davis I. W.; Echols N.; Headd J. J.; Hung L.-W.; Kapral G. J.; Grosse-Kunstleve R. W.; McCoy A. J.; Moriarty N. W.; Oeffner R.; Read R. J.; Richardson D. C.; Richardson J. S.; Terwilliger T. C.; Zwart P. H. PHENIX: A comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2010, 66, 213–221. 10.1107/s0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emsley P.; Cowtan K. Coot: Model-building tools for molecular graphics. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2004, 60, 2126–2132. 10.1107/s0907444904019158. [DOI] [PubMed] [Google Scholar]
- Emsley P.; Lohkamp B.; Scott W. G.; Cowtan K. Features and development ofCoot. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2010, 66, 486–501. 10.1107/s0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodel A.; Kim S. H.; Brünger A. T. Model bias in macromolecular crystal structures. Acta Crystallogr., Sect. A: Found. Crystallogr. 1992, 48, 851–858. 10.1107/s0108767392006044. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.