

Abstract
A deletion variant of epidermal growth factor receptor (EGFRvIII) is a known driver mutation in a subset of primary and secondary glioblastoma multiforme. Adoptive transfer of genetically modified chimeric-antigen receptor (CAR) lymphocytes has demonstrated efficacy in hematologic malignancies but is still early in development for solid cancers. The surface expression of the truncated extracellular ligand domain created by EGFRvIII makes it an attractive target for a CAR-based cancer treatment. Patients with recurrent glioblastoma expressing EGFRvIII were enrolled in a dose escalation phase I trial, using a third-generation chimeric antigen receptor construct derived from a human antibody. Transduced cells were administered after lymphodepleting chemotherapy and supported post-transfer with intravenous interleukin-2. The dose escalation proceeded at half-log increments from 107 to >1010 cells. Primary endpoints were safety and progression-free survival. Eighteen patients were treated with final infusion products ranging from 6.3×106 to 2.6×1010 anti-EGFRvIII-CAR+ T cells. Median progression free survival was 1.3 months (interquartile range 1.1–1.9), with a single outlier of 12.5 months. Two patients experienced severe hypoxia, including one treatment related mortality after cell administration at the highest dose level. All patients developed expected transient hematologic toxicities from preparative chemotherapy. Median overall survival was 6.9 months (interquartile range 2.8–10). Two patients survived over one year, and a third patient was alive at 59 months. Persistence of CAR+ cells correlated with cell dose, but there were no objective responses. Administration of anti-EGFRvIII CAR-transduced T cells did not demonstrate clinically meaningful impact in patients with glioblastoma multiforme in this phase I pilot trial.
Keywords: glioblastoma, chimeric antigen receptor, immunotherapy, EGFRvIII
Introduction
Glioblastoma is the most aggressive subtype of primary brain cancer. Standard of care includes surgical resection or biopsy, followed by radiation and chemotherapy. Even with this strategy, median survival is 14.6 months.1 Molecular analysis of these tumors has demonstrated that increased signaling of the epidermal growth factor receptor (EGFR) is a frequent genetic alteration associated with glioblastoma.2,3 The most frequent variant, EGFR variant III (EGFRvIII), an in-frame deletion of exons 2–7, occurs in 25–64% of glioblastoma.4,5 When present, it truncates the extracellular ligand binding domain of the receptor and renders the protein constitutively active.6 This mutation drives a malignant phenotype by enhancing tumorgenicity7,8, promoting cellular motility9, and conferring resistance to radiation and chemotherapy10,11, but also creates a unique opportunity for immune-based therapy. Because EGFRvIII is not present in normal tissue4,5,12, the truncated extracellular portion of the protein is tumor-specific and could be an antigenic target.
Human vaccine trials with EGFRvIII peptides have demonstrated antigenicity with specific cellular and humoral responses, but no clinical efficacy has been seen. In the VICTORI trial, patients with EGFRvIII+ glioblastoma received intradermal injections of autologous dendritic cells pulsed with rindopepimut, a 13mer peptide spanning the fusion junction conjugated via a terminal cysteine with keyhole limpet hemocyanin. Ten of 12 patients demonstrated increased in vitro proliferation when post-vaccination blood was stimulated with EGFRvIII peptide. Comparison to historical controls did not demonstrate significant improvement in survival.13 In a series of trials (ACTIVATE, ACT II, ACT III) of intradermal peptide injections of rindopepimut admixed with GM-CSF, most patients demonstrated increased anti-EGFRvIII antibody titers over baseline. Delayed-type hypersensitivity skin tests converted to reactive in a minority of patients, but further T cell analysis was not performed.14,15 These studies suggested longer than expected survival, however a large, multinational phase III trial (ACT IV) randomizing patients to temozolomide ± rindopepimut was terminated when a planned interim analysis reached a futility boundary for overall survival.16
Chimeric antigen receptors (CAR) utilize antibody-derived specificity to signal T cell activation, and preclinical work from other groups has demonstrated that EGFRvIII-directed CAR T cells can inhibit the growth of human tumor cells in vitro17 and intracranial murine tumors18 expressing the variant. Human glioma cell lines, however, did not always retain the molecular characteristics of the primary tumor and required introduction of the EGFRvIII variant through transfection or transduction. To develop a more robust target to test the in vitro specificity of anti-EGFRvIII CAR T cells, glioblastoma stem cells derived from single cell suspensions and grown in neurospheres were shown to maintain mRNA expression of EGFRvIII.19
Multiple second-generation CAR constructs were created for initial testing, each combining a single chain antibody sequence (scFv) of one of four murine or three human anti-EGFRvIII antibodies with CD28 costimulation and CD3ζ signaling. Three constructs recognized cell lines engineered to express the variant without reactivity to wildtype, and the human scFv (139) was chosen for further development (139–28Z). A third-generation CAR construct incorporating additional 4–1BB costimulation (139–28BBZ, Figure 1A) demonstrated equivalent function to 139–28Z and the ability to recognize glioblastoma stem cells.19 Because animal models suggested that additional 4–1BB costimulatory signaling could increase persistence and tumor localization20–22, the third-generation construct was chosen for the clinical trial described herein.
Figure 1.
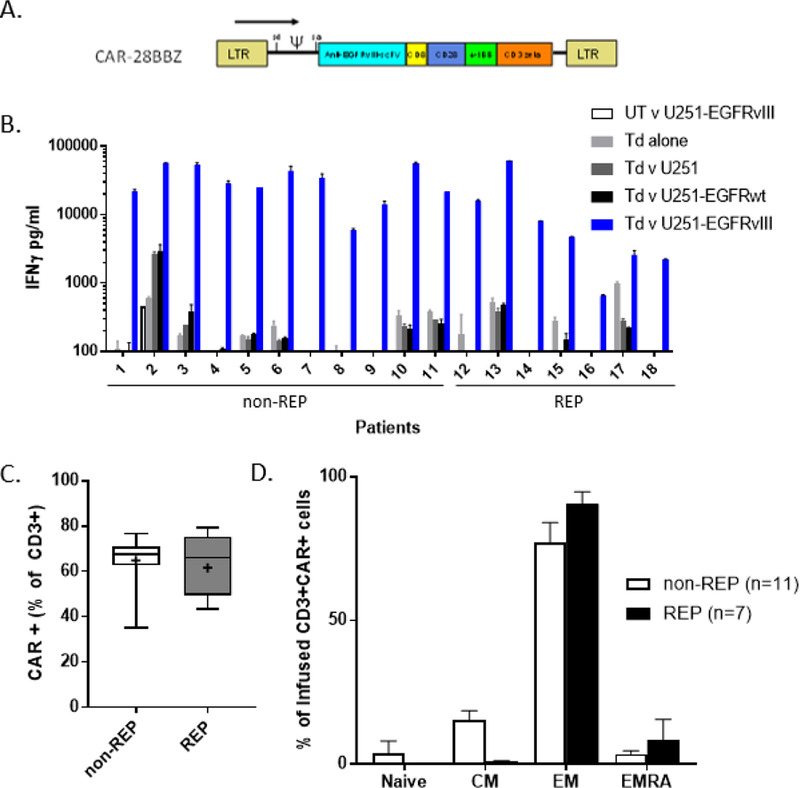
Characterization of clinical infusion product. A. Diagram of retroviral vector construct selected for clinical protocol detailing location of single chain variable fragment of human monoclonal Ab 139, CD8 linker domain, CD28 and 4–1BB costimulatory domains, and CD3ζ signaling domain. B. EGFRvIII-specific cytokine release of cell product 48–72 hours prior to infusion as measured by interferon-γ ELISA. UT: untransduced PBL, Td: PBL transduced with CAR-28BBZ, U251: glioblastoma cell line ± transduction to express EGFRwt or EGFRvIII, REP: rapid expansion protocol C. Measurement of CAR (+) cells in each infusion product by flow cytometry. Non-REP: median 67.5% (IQR 62.6–71), REP: median 66% (IQR 49.4–75.3). Gated on live, CD3+ cells. Whiskers indicate range; + denotes mean. D. Infused CAR+ cells were primarily of effector memory (EM) phenotype, as defined as CD45RA-, CCR7- T cells. There were significantly fewer central memory (CM) cells in REP products (p=0.0006).
During the accrual of this trial, other groups have explored EGFRvIII-directed CAR therapy, one using a second-generation lentiviral construct containing a 4–1BB costimulation domain. In that study of 10 patients, no objective responses nor persistent CAR+ cells were identified.23
Materials and Methods
Study Design
This clinical trial was designed to determine the maximum safe dose of autologous peripheral blood lymphocytes retrovirally transduced with an EGFRvIII–targeting chimeric antigen receptor (CAR) and whether this approach could impact progression free survival in patients with recurrent glioblastoma. Secondary endpoints included CAR persistence and radiologic response. The protocol was approved by the Institutional Review Board of the National Cancer Institute and registered with ClinicalTrials.Gov (NCT01454596). All patients gave informed consent.
Treatment began with nonmyeloablative preparative chemotherapy: two days of cyclophosphamide (60 mg/kg) followed by five days of fludarabine (25 mg/m2). On the next day, autologous CAR-transduced cells were infused over thirty minutes, with patient 18 receiving divided doses two hours apart. Low-dose intravenous interleukin-2 administration (72,000 IU/kg) began within 24 hours of cell transfer and continued every eight hours to tolerance. Tumor response was assessed by comparison to baseline dynamic contrast-enhanced magnetic resonance imaging (MRI) with perfusion using Neuro-oncology Working Group proposed guidelines starting one month after cell administration and proceeding at regular intervals thereafter24.
Patients
Adult patients with a pathologically confirmed diagnosis of EGFRvIII+ glioblastoma with radiologic recurrence after initial surgical resection or chemoradiotherapy were eligible for this clinical trial. Patients were required to have a Karnosfsky performance status of ≥60%. Concomitant steroids for symptom control did not preclude eligibility provided a stable dose was achieved at least five days prior to enrollment. Detection of EGFRvIII was confirmed by the Molecular Diagnostics Section, Laboratory of Pathology, NCI using a clinically validated RT-PCR assay modified from Yoshimoto et al25.
Anti-EGFRvIII CAR T Cell Production and Analysis
After determining eligibility, peripheral blood lymphocytes (PBL) were isolated from patients by leukapheresis and separated by centrifugation on a lymphocyte separation medium cushion. PBL were stimulated by OKT3 antibody (50 ng/mL) and transduced with a clinical grade γ-retroviral vector that encodes the EGFRvIII CAR, as previously described19. National Institutes of Health Guidelines for Research Involving Recombinant or Synthetic Nucleic Acid Molecules were followed. Final cell product was required to have a minimum of 10% CAR+ CD3+ cells and specific interferon-γ release to EGFRvIII+ cell lines. Cell dose was calculated by total number of CD3+ cells and proceeded at half-log increments from 107 to >1010 cells; higher doses (for patients 12–18) required additional lymphocyte rapid expansion protocols (REP), as previously described.26
Characterization of the infusion product was performed by flow cytometry. To assess the proportion of transduced T-cells, cryopreserved lymphocytes aliquoted from the final infusion product were thawed and cultured overnight in media without IL-2. Cells were first stained with biotinylated goat anti-human Fab (Jackson ImmunoResearch, West Grove, PA) for 45 minutes on ice. At the end of incubation, cells were washed and further stained with other antibodies including streptavidin-PE, anti-CD3, anti-CD4, anti-CD8, anti-CCR7 and anti-CD45RA from either BD Biosciences (Franklin Lakes, NJ) or BioLegend (San Diego, CA). Data acquisition was performed using a FACS Canto II (BD Biosciences), and data were analyzed using FlowJo software (FlowJo, Inc., Ashland, OR). T cell phenotypes were defined as: naive CCR7+CD45RA+, central memory CCR7+CD45RA-, effector memory CCR7-CD45RA-, and effector CCR7-CD45RA+.
CAR-engineered PBLs were tested for antigen-specific reactivity in cytokine release assays using wild type and EGFRvIII-modified U251 glioblastoma tumor lines. In these assays, effector cells (1×105) were cocultured with an equal number of target cells in AIM-V medium in a final volume of 0.2 mL in duplicate wells of a 96-well U-bottom microplate. Culture supernatants were harvested 18–24 hours after the initiation of coculture and assayed for IFN-γ by ELISA (Thermo Scientific, Rockford, IL).
To detect genetically modified T cells, genomic DNA was extracted from an aliquot of the final infusion product and PBL collected prior to treatment and at multiple time-points after treatment. Duplicate aliquots of 100 ng DNA from each sample were used for each real-time quantitative PCR reaction (TaqMan, Applied Biosystems Inc, Foster City, CA). The following primers/probe set was used to detect EGFRvIII-CAR: forward primer (5’-TGCTAGGGCTCTGGGTCATCT-3’), reverse primer (5’-TCGAGCATGGTTCTGCTGGTCA-3’) and the probe (5’-FAM-AGCCTGCTGCTGTGCGAACT-3’). A standard curve was established using a DNA sample prepared from a single patient CAR infusion product determined to contain 2.843×106 copies of CAR/µg DNA. TaqMan β-actin control reagents kit (Applied Biosystems Inc., Foster City, CA) was used to normalize reactions to input DNA amounts. CAR DNA in all samples prior to treatment was below the detectable limit in this assay.27
Statistics
A Mann-Whitney test was used to evaluate differences between REP and non-REP infusion products and patients ± concomitant steroid administration. Student’s t test was used to evaluate differences in phenotype and corrected using the Bonferroni-Dunn method. A nonparametric Spearman correlation was used to evaluate CAR+ cell persistence and cell dose. A log-rank test was applied to Kaplan-Meier survival analysis (Prism; GraphPad Software, La Jolla, CA). Data were reported as medians with interquartile ranges (IQR), where appropriate. Reported P values were two-tailed, and P < .05 was considered statistically significant.
Results
Patient and Treatment Characteristics
Initially, 33 patients with historical evidence of EGFRvIII+ GBM were clinically screened, three of whom were ineligible when the presence of EGFRvIII could not be confirmed by PCR. Of the remaining thirty patients with confirmed variant, three patients underwent biopsy of recurrent disease that did not demonstrate the presence of EGFRvIII. Nine patients pursued other treatment or developed progressive disease prior to enrollment. Eighteen patients were treated on this dose-escalating phase one study (Table 1). All had developed recurrent glioblastoma after surgery, radiation, and temozolomide chemotherapy, and 10 of 18 patients had received bevacizumab. Median interval from initial diagnosis of GBM to screening was 15.7 months (interquartile range 11.6–20.6). Four of the patients had tumors with an O6-methylguanine-DNA-methyltransferase (MGMT) methylated promoter. Fourteen patients entered the trial requiring medication for seizure control or prophylaxis. Repeat biopsy was not required for study entry, and the median interval between an EGFRvIII (+) biopsy and cell infusion was 11.1 months (interquartile range 3.1–17.0).
Table 1.
Summary of patients enrolled on trial
| TUMOR CHARACTERISTICS | SURVIVAL (MONTHS) | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| CONCURRENT STEROIDS (DAILY DOSE) | EGFRvIII qPCR |
||||||||||
| ID | AGE/SEX | PRIOR TREATMENTS | MGMT METHYLATION | TUMOR | % TUMOR | ∆Ct* | INTERVAL† (MONTHS) | RECIST RESPONSE | PROGRESSION FREE | OVERALL | |
| 1 | 45/M | Surgery, Radiation, temozolomide, bevacizumab, BCNU | Y (Dex 3mg) | N | Initial | >80 | −4.8 | 23.9 | NR | 1.1 | 2.2 |
| 2 | 43/M | Surgery, Radiation, temozolomide, bevacizumab | N | N | Recurrent | 50 | −1.2 | 10.9 | NR | 1.9 | 13.1 |
| 3 | 52/M | Surgery, Radiation, temozolomide, bevacizumab | Y (Dex 1mg) | N | Initial | 70 | 0.6 | 20.4 | NR | 1.1 | 4.5 |
| 4 | 46/M | Surgery, Radiation, temozolomide | Y‡ | N | Initial | >70 | −3.4 | 11.0 | NR | 2.0 | 11.1 |
| 5 | 55/M | Surgery, Radiation, temozolomide | Y‡ | N | Initial | 60 | −2.9 | 9.0 | NR | 1.5 | 9.0 |
| 6 | 57/M | Radiation, temozolomide, bevacizumab | Y (HCT 20mg) | N | Initial | 85 | −4.1 | 34.2 | NR | 1.3 | 6.9 |
| 7 | 53/M | Surgery, Radiation, temozolomide | N | N | Recurrent | 70–80 | 3.5 | 11.1 | NR | 1.2 | 9.7 |
| 8 | 56/M | Surgery, Radiation, temozolomide, AZD7451 | N | N | Recurrent | 60 | −2.6 | 2.7 | NR | 0.9 | 10.1 |
| 9 | 55/F | Surgery, Radiation, temozolomide, bevacizumab | N | N | Recurrent | 95 | −2.7 | 1.4 | NR | 1.1 | 2.0 |
| 10 | 55/M | Surgery, Radiation, temozolomide | N | N | Initial | 60 | −0.8 | 12.3 | NR | 1.3 | 4.4 |
| 11 | 61/F | Surgery, Radiation, temozolomide | Y (Dex 1mg) | Y | Initial | 80 | 0.4 | 16.2 | NR | 12.5 | 59+ |
| 12 | 66/M | Surgery, Radiation, temozolomide, veliparib, bevacizumab | Y (Dex 4mg) | Y | Initial | >60 | −1.3 | 18.5 | NR | 0.9 | 2.1 |
| 13 | 60/M | Surgery, Radiation, temozolomide, bevacizumab, EGFRvIII vaccine | Y (Dex 2mg) | N | Recurrent | 90 | −3.9 | 1.8 | NR | 2.7 | 4.5 |
| 14 | 64/F | Surgery, Radiation, temozolomide, IMA950 vaccine | N | Y | Recurrent | 60–80 | −4.8 | 1.9 | NR | 1.6 | 8.9 |
| 15 | 61/M | Surgery, Radiation, temozolomide, EGFRvIII vaccine vs. placebo trial, bevacizumab, trebananib | Y (Dex 4mg) | N | Initial | >90 | 0.1 | 12.5 | NR | 1.1 | 1.4 |
| 16 | 43/M | Surgery, Radiation, temozolomide, carotuximab, bevacizumab | Y (Dex 2mg) | N | Recurrent | 60 | −2.5 | 4.3 | NR | 1.1 | 6.9 |
| 17 | 47/M | Surgery, Radiation, temozolomide, EGFRvIII vaccine vs placebo trial, bevacizumab | Y (Dex 2mg) | N | Initial | 90 | −3.3 | 17.3 | NE | TRM | TRM |
| 18 | 57/M | Surgery, Radiation, temozolomide | N | Y | Recurrent | 95 | −1.5 | 1.9 | NR | 2.0 | 13.6 |
MGMT: O6-methylguanine-DNA-methyltransferase; Dex: dexamethasone; HCT: hydrocortisone; NR: no response; NE: not evaluable; TRM: treatment-related mortality.
∆Ct = EGFRvIII Ct – Mean Ct of housekeeping genes GUS and HPRT1. Values <8 indicate presence of variant.
Time from confirmed biopsy to protocol treatment.
indicate ongoing survival
The cell infusion products (Table 2) were predominantly CD3+ (97.9%, range 94.8–99.4), and while always CD8+ dominant, there was variability in the CD8:CD4 ratio (median 1.79, interquartile range 1.45–4.33). Three patients did not receive interleukin-2 (IL-2) after cell infusion: one developed transient mental status changes that did not resolve within 24 hours and two patients developed acute pulmonary symptoms. Infusion samples released on average >200-fold higher IFNγ in response to EGFRvIII+ targets than negative controls (Figure 1B). As planned, a rapid expansion protocol (REP) was used to achieve the desired cell number for patients treated at higher dose levels (≥3×109). The percentage of CAR+ cells was not statistically different (p=0.84) between the non-REP (median 67.5%, interquartile range 62.6–71) and REP (median 66%, interquartile range 49.4–75.3) infusion products (Figure 1C). As expected with additional stimulation, the REP infusion samples had fewer central memory T cells than non-REP (median 0.96% vs 15.2%, p=0.0006), however both were predominantly of an effector memory phenotype (Figure 1D).
Table 2.
Characteristics of Treatment
| PHENOTYPE (% OF INFUSED CD3+ CELLS) | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| ID | DOSE | % CAR (+) | EGFRvIII Transgene* | # CAR (+) | # IL-2 DOSES | CD4 | CD8 | TN CD45RA+ CCR7+ | TCM CD45RA− CCR7+ | TEM CD45RA− CCR7− | TEMRA CD45RA+ CCR7− |
| 1 | 1.00E+07 | 71.0% | 5.8E+04 | 7.10E+06 | 7 | 5 | 92 | 4 | 3 | 70 | 23 |
| 2 | 1.00E+07 | 62.6% | 1.1E+05 | 6.26E+06 | 10 | 40 | 58 | 1 | 3 | 88 | 8 |
| 3 | 3.00E+07 | 67.6% | 1.3E+05 | 2.03E+07 | 8 | 45 | 53 | 23 | 8 | 62 | 7 |
| 4 | 3.00E+07 | 66.5% | 1.1E+05 | 2.00E+07 | 4 | 46 | 52 | 10 | 15 | 67 | 8 |
| 5 | 1.00E+08 | 67.5% | 1.1E+05 | 6.75E+07 | 7 | 28 | 69 | 14 | 7 | 71 | 8 |
| 6 | 1.00E+08 | 62.9% | 4.0E+04 | 6.29E+07 | 6 | 18 | 80 | 4 | 5 | 89 | 3 |
| 7 | 3.00E+08 | 76.5% | 4.2E+04 | 2.30E+08 | 8 | 49 | 49 | 5 | 8 | 83 | 4 |
| 8 | 1.00E+09 | 62.0% | 4.3E+04 | 6.20E+08 | 6 | 40 | 57 | 19 | 11 | 61 | 10 |
| 9 | 1.00E+09 | 67.6% | 5.3E+04 | 6.76E+08 | 6 | 37 | 60 | 15 | 13 | 67 | 5 |
| 10 | 1.00E+09 | 73.3% | 4.7E+04 | 7.33E+08 | 5 | 12 | 85 | 2 | 2 | 94 | 3 |
| 11 | 2.48E+09 | 35.0% | 5.5E+04 | 8.68E+08 | 3 | 19 | 80 | 4 | 3 | 89 | 3 |
| 12 | 3.00E+09 | 67.9% | 2.8E+04 | 2.04E+09 | 5 | 16 | 83 | 1 | 8 | 90 | 2 |
| 13 | 3.00E+09 | 79.3% | 5.8E+04 | 2.38E+09 | 1 | 38 | 59 | 0 | 1 | 85 | 14 |
| 14 | 1.00E+10 | 49.4% | 4.2E+04 | 4.94E+09 | 5 | 44 | 51 | 1 | 0 | 12 | 87 |
| 15 | 1.00E+10 | 66.0% | 5.1E+04 | 6.60E+09 | 0 | 34 | 63 | 1 | 6 | 91 | 3 |
| 16 | 1.00E+10 | 75.3% | 4.2E+04 | 7.53E+09 | 1 | 6 | 90 | 0 | 6 | 93 | 1 |
| 17 | 6.00E+10 | 43.2% | n.d. | 2.59E+10 | 0 | 35 | 61 | 1 | 10 | 87 | 2 |
| 18 | 3.00E+10 | 49.9% | 3.8E+04 | 1.50E+10 | 0 | 30 | 65 | 0 | 5 | 91 | 4 |
CAR: chimeric antigen receptor; IL-2: interleukin-2. T cell subsets: TN – naïve; TCM – central memory; TEM – effector memory; TEMRA – effector memory RA.
Adverse Events
There were no dose-limiting toxicities associated with cell infusion until the highest dose level (≥1010) of CD3+ cells. Approximately one hour after administration of 6×1010 cells, one patient developed acute dyspnea and oxygen desaturation that was initially managed with bilevel positive airway pressure (BiPAP) but proceeded rapidly to intubation. Severe hypotension ensued, refractory to resuscitation efforts, and the patient expired four hours after completion of transfer, with significant pulmonary edema identified post-mortem. The next patient received two doses of 1.5×1010 cells separated by two hours for a total dose of 3×1010 cells and developed dyspnea approximately four hours later, just prior to planned IL-2 administration. The patient was managed successfully with continuous positive airway pressure (CPAP) during a brief stay in intensive care and discharged home without requiring oxygen support. Other than the treatment related mortality noted above, no patients required high-dose steroids to ameliorate symptoms of cytokine release syndrome. Worsening grade 2 neurologic symptoms or suspected seizure activity prompted brain imaging in ten patients; the results of which led to adjustments in steroids (n=3), anti-seizure medications (n=3), or both (n=1). Intravenous steroids were administered without imaging in two additional patients for symptoms that developed during preparative chemotherapy.
As a consequence of lymphodepleting chemotherapy, all patients experienced transient leukopenia and thrombocytopenia, while half (9/18) also developed anemia (Figure 2). During inpatient admission, patients were supported with transfusions of blood (median 2, n=11) for hemoglobin values approaching 8 g/dL and platelets (median 4, n=16) for values less than 30 K/uL.
Figure 2.
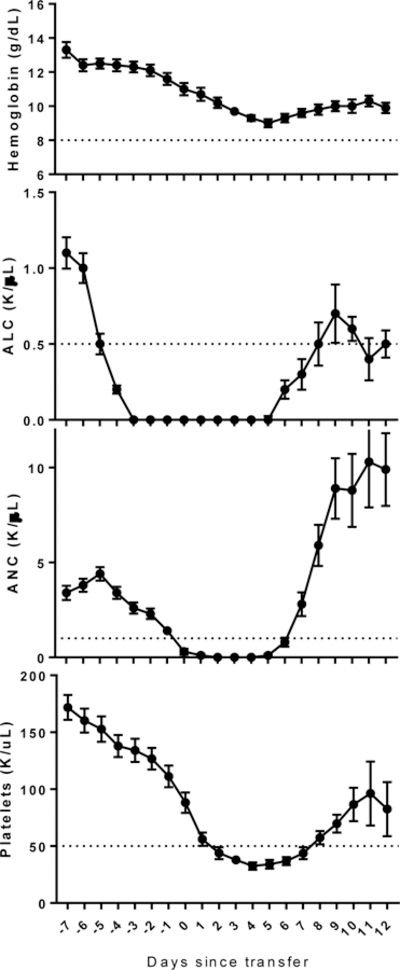
Most transient cytopenias after preparative chemotherapy resolved prior to hospital discharge. A. Hemoglobin. B. Absolute lymphocyte count. Delayed recovery was consistent with prior experience. C. Absolute neutrophil count. Filgrastim was used at the onset of neutropenia to promote neutrophil recovery. D. Platelet count. Values shown indicate mean ± SEM. Lines indicate threshold for Grade 3 toxicity (Common Terminology Criteria for Adverse Events version 3.0).
One patient developed a lower extremity deep venous thrombosis during preparative chemotherapy, and subsegmental pulmonary embolus was identified in a different patient one week after cell transfer. Both were treated with placement of an inferior vena cava filter rather than anti-coagulation given the bleeding risks associated with intracranial pathology. All patients underwent daily neutropenic surveillance blood cultures, and asymptomatic bacteremia was documented in eight patients. There were two additional patients with episodes of febrile neutropenia without bacteremia, but no patients exhibited overt signs of sepsis (Table 3). Median length of hospitalization (from enrollment to discharge) was 17 days (range 14 to 30) and was similar in patients whose pre-treatment symptoms did or did not require concomitant steroids (median 18 vs. 17, p=0.51)
Table 3.
Grade 3 and 4 Serious Adverse Events
| Description | # of patients |
|---|---|
| Cardiopulmonary | |
| Dyspnea/Hypoxia | 2* |
| Hypotension (not associated with sepsis) | 2 |
| Capillary leak syndrome |
1 |
| Infectious | |
| Febrile Neutropenia (without bacteremia) | 2 |
| Bacteremia (asymptomatic) | 8 |
| Eschericia coli | 4 |
| Enterobacter cloacae | 1 |
| Klebsiella pneumoniae | 2 |
| Staphylococcus epidermidis | 1 |
| Neurologic | |
| Motor weakness (transient) | 1 |
| Urinary incontinence (transient) | 1 |
| Coagulation | |
| Prolonged PTT | 1 |
| Deep Venous Thrombus | 1 |
| Pulmonary Embolism | 1 |
| Hematologic+ | |
| Anemia | 9 |
| Lymphopenia | 18 |
| Neutropenia | 18 |
| Thrombocytopenia | 18 |
| Metabolic | |
| Transaminitis | 2 |
includes 1 treatment-related mortality (Grade 5)
expected due to lymphodepleting chemotherapy
Response and Survival
Based on serial MRI imaging, there were no objective responses. Most patients demonstrated progressive disease at first follow-up with a median progression-free survival of 1.3 months (interquartile range 1.1–1.9). Sixteen of the 17 evaluable patients progressed less than three months after infusion, with no evidence of pseudoprogression. Increasing symptoms prompted immediate bevacizumab-based therapy (n=3), resection (n=1), or palliative care (n=7, median survival after progression 1.2 months, range 0.4–3.5). Five patients had confirmation of progression on further imaging. A single patient, with no post-CAR GBM treatment, is still alive at 59 months, and two additional patients survived greater than one year (13.1 and 13.6 months, median survival of all patients 6.9 months, interquartile range 2.8–10). There was no significant difference in overall survival when analyzed for MGMT promoter methylation status (p=0.08) or concomitant steroid administration (p=0.55).
Persistence
Persistence of CAR+ cells was measured indirectly by identification of the transgene product in DNA derived from the peripheral blood and an aliquot of the infusion product. For each sample, the quantitative PCR values at serial time points were normalized to 1×106 copies of β-actin. There was no correlation between % transduction and EGFRvIII transgene copies in the infusion products (Table 2, p=0.8). Patients were divided into dose level groups based on the number of CAR+ CD3+ cells in the infusion product and the use of REP (Figure 3A–D). Persistence at one month could be analyzed in 14 patients (median day 32), and while presence of EGFRvIII CAR correlated with cell dose, it did not correlate with survival (Figure 3E). There was no significant difference in transgene persistence (p=0.07) when the cohort was divided by concomitant steroid administration. Transcripts were still identified three months after infusion in five patients, four of whom had already demonstrated radiologic progression. An attempt was made to quantify the number of circulating CAR+ cells in the peripheral blood at one month, however a combination of low frequency events, non-specific binding of anti-human Fab’, and slow recovery of the lymphocyte compartment (median ALC 0.59 K/µL, range 0.17–2.08) yielded inconclusive results (Figure 3F).
Figure 3.
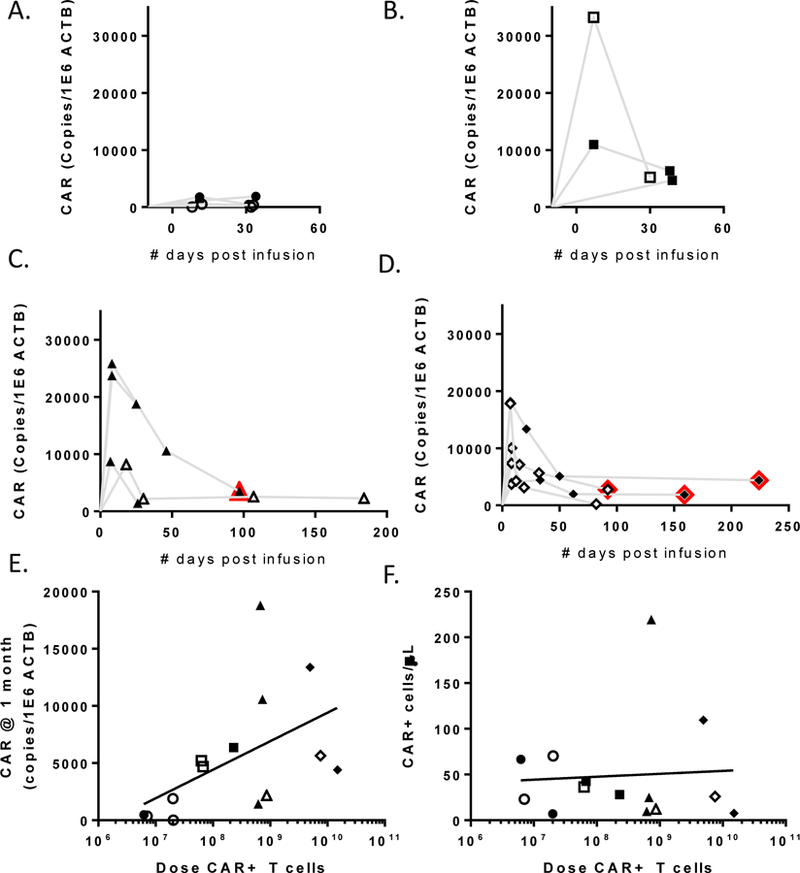
Persistence of infused CAR+ cells as measured by qPCR. A. Patients who received <3×107 CAR+ cells. B. Patients who received between 3×107 and 3×108 CAR+ cells. C. Patients who received >3×108 CAR+ cells without rapid expansion. D. Patients who received >3×108 CAR+ cells including rapid expansion. Open symbols indicate those patients receiving concurrent steroids. Red outlines indicate time points after radiologic progression. E. Persistence at one month (median day 32, n=14) was correlated with CAR+ cell dose (r=0.6, p=0.0261), but not survival (not shown). F. Persistence at one month was analyzed by FACS in conjunction with clinical lymphocyte counts, however there were too few events for reliable analysis.
Discussion
In this pilot clinical trial, adoptive transfer of autologous CAR T cells targeting EGFRvIII after lymphodepleting preparative chemotherapy was not capable of inducing objective tumor regression and did not appear to be either delaying progression or prolonging survival in patients with recurrent glioblastoma.
Adoptive transfer included a preparative chemotherapy to create an immune milieu that promotes lymphocyte expansion – cytokine-enhanced and depleted of myeloid-derived suppressor cells and regulatory T cells.28 The patients in this trial received a standard lymphodepleting regimen that induced the transient cytopenias associated with this strategy demonstrating that patients with recurrent glioblastoma can safely undergo the same preparative chemotherapy used successfully in large trials of patients without primary brain tumors.27,29,30 With transfusion support during periods of thrombocytopenia, there were no significant bleeding events. Underlying recurrent glioblastoma added complexity to the management of mild neurologic symptoms, and treatment was guided by liberal use of brain imaging.
Given our experience transferring activated lymphocytes and the associated risks of lymphodepletion, we balanced the first-in-human nature of this receptor with a low initial starting dose (107 CD3+ cells) and a rapid single patient escalation schema to achieve doses (>109 CD3+ cells) historically capable of conferring clinical benefit before beginning a traditional 3+3 escalation design. At the highest dose levels in this trial, patients began to develop respiratory symptoms within hours of cell infusion, likely demonstrating congestion of pulmonary vasculature from activated T cells in a dose-dependent fashion, unlike our previous experience with a trastuzumab-derived CAR, in which low levels of ERBB2 likely resulted in fatal on-target, off-tumor reactivity.31 The dose-limiting pulmonary toxicity was reached in this trial without any indication of clinical benefit. One promising case report of an interleukin-13 receptor alpha 2 CAR suggests that repeated intrathecal administration of anti-tumor CAR is feasible (2×106-1×107 cells).32 A potential future protocol with anti-EGFRvIII CAR could incorporate the use of an intrathecal reservoir (Ommaya) or intratumoral catheter to allow further dose-finding strategies to test efficacy without encountering limitations of the pulmonary vasculature.
In this study, we delivered cell doses capable of demonstrating initial engraftment and long-term persistence of EGFRvIII CAR in the peripheral blood. While qPCR measurements are indirect and cannot account for the number of copies of a transgene within an individual cell, there was only one patient among those analyzed in whom the anti-EGFRvIII-CAR sequence could not be amplified and detected one month after infusion. While the third-generation retroviral construct (139–28BBZ) was chosen based on preclinical evidence of improved persistence, the human data on the importance of persistence is mixed. In the use of adoptive transfer in patients with melanoma, objective responders had significantly greater persistence of autologous tumor infiltrating lymphocytes one month post-infusion than those patients without a response.33 In that trial, persistence was based on comparison of unique T cell receptor beta chain variable complementarity-determining region 3 (CDR3) sequences within the infusion product with those detected in post-treatment peripheral blood. The intratumoral population may reflect a baseline CDR3 distribution that is recapitulated with homeostatic reconstitution, and the persistent cells may or may not be tumor-reactive. However, in a pilot trial of lymphocytes with genetically modified T-cell receptors against the NY-ESO-1 cancer germline antigen, there was no difference in persistence between responding and non-responding patients.30 Another study using the same NY-ESO-1 TCR demonstrated a correlation between response and peak vector expression and suggested an association with long-term persistence.34 Specifically in a CAR setting, two independent studies of the same anti-CD19 construct demonstrated that clinical response correlated with early peak values but did not require long-term persistence.35,36
The frequent clinical use of steroids to manage edema-related symptoms in patients with intracranial pathology has the potential to confound immune-based strategies. While it is difficult to assert that concomitant steroid administration did not abrogate potential immune effects in a trial with no clinical responders, we have demonstrated that this patient cohort has similar one-month CAR persistence and overall survival to those patients enrolled without concomitant steroids, though the small sample size and broad range may obscure a potential difference. In another CAR trial targeting CD19 in B cell malignancies, glucocorticoids were administered to 27 of 101 patients as a management strategy for acute cytokine release syndrome, and response rates were not significantly different in patients who did (78%, 95% CI 58–91%) and did not (84%, 95% CI 73–91%) receive them.35 A delayed administration of steroids was occasionally necessary for patients treated with ipilimumab, an immune-based strategy based on checkpoint blockade inhibition, and the use of steroids did not affect the generation or duration of an objective response.37
While there has long been an assumption that the brain is a site of immune privilege, cell transfer immune therapies have demonstrated that it is possible to eradicate intracranial parenchymal brain metastases using autologous gene-engineered or tumor infiltrating lymphocytes in some patients with melanoma.29,38 In primary CNS cancer, specifically in histologic studies of glioblastoma, T cell infiltration could be identified using immunohistochemistry in a subset of tumors.39,40 The presence of T cells in brain parenchyma may imply that the trafficking signals necessary to penetrate intracranial pathology are intact or may reflect an increased permeability of tumor neovasculature. O’Rourke et al detected transferred lentiviral anti-EGFRvIII CAR cells in tumor parenchyma with the highest levels described in a subset of four patients that underwent surgical resection within two weeks of intravenous infusion.23 However, without corresponding normal tissue at a time post-infusion when peripheral blood levels remain high, it remains unclear if the presence of CAR T cells was target specific.
For successful CAR-directed therapy, the ideal target would be homogenously presented on the surface of tumor cells with no expression on normal tissue. Considerable intra- and intertumor heterogeneity of EGFRvIII expression has been reported. One study, published after the conception of and recruitment of this trial, examined paired samples of tumors resected before (primary) and after (recurrent) standard chemoradiotherapy. The variant was not expressed in nearly half of recurrent tumors from patients with EGFRvIII+ primary disease.41 While we did not see this level of discrepancy in our study population, we did screen three patients with confirmed EGFRvIII that was absent on subsequent biopsy of recurrent disease. Intertumoral heterogeneity has been better elucidated using single-cell sequencing techniques, and in one study, EGFR amplifications coexisted with other known EGFR variants (structural alterations and missense mutations) in 71% of analyzed samples.42 One model indicated that the EGFRvIII mutant actively augmented heterogeneity with paracrine signaling that drove wild type EGFR cells into accelerated proliferation.43 Another pre-clinical model indicated that tumors treated with tyrosine kinase inhibitors transiently downregulated the mutated protein, but extrachromosomal EGFRvIII DNA may have served as a repository for restoring the mutation phenotype and promoting heterogeneity.44
Although the presence or absence of EGFRvIII DNA or protein at any stage of tumor is likely reflective of this undulating heterogeneity and epigenetic regulation rather than treatment effect, the loss of this variant has been reported as evidence of clinical response. The phase II trial of rindopepimut peptide vaccine described a loss of EGFRvIII (as detected by immunohistochemistry) in nine of eleven tumors resected after vaccination.14 In the subsequent phase III trial comparing temozolomide ± rindopepimut, presence of the variant was assessed using RT-PCR. In the subset of patients with pre- and post-tumor samples available, the rate of EGFRvIII loss was the same with and without the administration of the EGFRvIII vaccine (57% vs 59%).16 In the O’Rourke EGFRvIII CAR trial, five of seven patients with post-treatment samples available for comparison had decreased EGFRvIII as measured by % of total EGFR reads on next generation sequencing of RNA.23
Despite demonstrating anti-EGFRvIII CAR persistence and reaching dose-limiting toxicity, only a single patient was free from progression at six months; that tumor was also MGMT-methylated, associated with longer survival.45
The paucity of safe normal self-proteins or tumor-specific mutated antigens to target on the surface of tumor cells is a severe limitation in the more widespread application of CAR T technology for solid cancers. This clinical trial of 18 patients is the only one to utilize high cell doses (≥ 1010), preceded by a lymphodepleting regimen and followed by IL-2 administration, the exact regimen associated with clinical responses in patients with melanoma, synovial sarcoma, and selected patients with colon, breast, cervical and bile duct cancer. Here, the inability to successfully treat patients with CAR T cells targeting one of these rare mutated surface tumor antigens in this pilot trial may be a harbinger of additional difficulties in translating the CAR T strategy to more common solid cancers.
Acknowledgments
Research was funded by the Center for Cancer Research, the intramural program of the National Cancer Institute.
Research Support: Center for Cancer Research at the National Cancer Institute of the US National Institutes of Health
Footnotes
The authors declare no potential conflicts of interest.
References
- 1.Stupp R, Mason WP, van den Bent MJ, et al. : Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med 352:987–96, 2005 [DOI] [PubMed] [Google Scholar]
- 2.Libermann TA, Nusbaum HR, Razon N, et al. : Amplification, enhanced expression and possible rearrangement of egf receptor gene in primary human brain tumours of glial origin. Nature 313:144–7, 1985 [DOI] [PubMed] [Google Scholar]
- 3.Yamazaki H, Fukui Y, Ueyama Y, et al. : Amplification of the structurally and functionally altered epidermal growth factor receptor gene (c-erbb) in human brain tumors. Mol Cell Biol 8:1816–20, 1988 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Saikali S, Avril T, Collet B, et al. : Expression of nine tumour antigens in a series of human glioblastoma multiforme: Interest of egfrviii, il-13ralpha2, gp100 and trp-2 for immunotherapy. J Neurooncol 81:139–48, 2007 [DOI] [PubMed] [Google Scholar]
- 5.Feldkamp MM, Lala P, Lau N, et al. : Expression of activated epidermal growth factor receptors, ras-guanosine triphosphate, and mitogen-activated protein kinase in human glioblastoma multiforme specimens. Neurosurgery 45:1442–53, 1999 [DOI] [PubMed] [Google Scholar]
- 6.Chu CT, Everiss KD, Wikstrand CJ, et al. : Receptor dimerization is not a factor in the signalling activity of a transforming variant epidermal growth factor receptor (egfrviii). Biochem J 324 ( Pt 3):855–61, 1997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Batra SK, Castelino-Prabhu S, Wikstrand CJ, et al. : Epidermal growth factor ligand-independent, unregulated, cell-transforming potential of a naturally occurring human mutant egfrviii gene. Cell Growth Differ 6:1251–9, 1995 [PubMed] [Google Scholar]
- 8.Lal A, Glazer CA, Martinson HM, et al. : Mutant epidermal growth factor receptor up-regulates molecular effectors of tumor invasion. Cancer Res 62:3335–9, 2002 [PubMed] [Google Scholar]
- 9.Boockvar JA, Kapitonov D, Kapoor G, et al. : Constitutive egfr signaling confers a motile phenotype to neural stem cells. Mol Cell Neurosci 24:1116–30, 2003 [DOI] [PubMed] [Google Scholar]
- 10.Lammering G, Valerie K, Lin PS, et al. : Radiation-induced activation of a common variant of egfr confers enhanced radioresistance. Radiother Oncol 72:267–73, 2004 [DOI] [PubMed] [Google Scholar]
- 11.Montgomery RB, Guzman J, O’Rourke DM, et al. : Expression of oncogenic epidermal growth factor receptor family kinases induces paclitaxel resistance and alters beta-tubulin isotype expression. J Biol Chem 275:17358–63, 2000 [DOI] [PubMed] [Google Scholar]
- 12.Humphrey PA, Wong AJ, Vogelstein B, et al. : Anti-synthetic peptide antibody reacting at the fusion junction of deletion-mutant epidermal growth factor receptors in human glioblastoma. Proc Natl Acad Sci U S A 87:4207–11, 1990 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sampson JH, Archer GE, Mitchell DA, et al. : An epidermal growth factor receptor variant iii-targeted vaccine is safe and immunogenic in patients with glioblastoma multiforme. Mol Cancer Ther 8:2773–9, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sampson JH, Heimberger AB, Archer GE, et al. : Immunologic escape after prolonged progression-free survival with epidermal growth factor receptor variant iii peptide vaccination in patients with newly diagnosed glioblastoma. J Clin Oncol 28:4722–9, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Schuster J, Lai RK, Recht LD, et al. : A phase ii, multicenter trial of rindopepimut (cdx-110) in newly diagnosed glioblastoma: The act iii study. Neuro Oncol 17:854–61, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Weller M, Butowski N, Tran DD, et al. : Rindopepimut with temozolomide for patients with newly diagnosed, egfrviii-expressing glioblastoma (act iv): A randomised, double-blind, international phase 3 trial. Lancet Oncol 18:1373–1385, 2017 [DOI] [PubMed] [Google Scholar]
- 17.Bullain SS, Sahin A, Szentirmai O, et al. : Genetically engineered t cells to target egfrviii expressing glioblastoma. J Neurooncol 94:373–82, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ohno M, Natsume A, Ichiro Iwami K, et al. : Retrovirally engineered t-cell-based immunotherapy targeting type iii variant epidermal growth factor receptor, a glioma-associated antigen. Cancer Sci 101:2518–24, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Morgan RA, Johnson LA, Davis JL, et al. : Recognition of glioma stem cells by genetically modified t cells targeting egfrviii and development of adoptive cell therapy for glioma. Hum Gene Ther 23:1043–53, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Song DG, Ye Q, Carpenito C, et al. : In vivo persistence, tumor localization, and antitumor activity of car-engineered t cells is enhanced by costimulatory signaling through cd137 (4–1bb). Cancer Res 71:4617–27, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhao Y, Wang QJ, Yang S, et al. : A herceptin-based chimeric antigen receptor with modified signaling domains leads to enhanced survival of transduced t lymphocytes and antitumor activity. J Immunol 183:5563–74, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhong XS, Matsushita M, Plotkin J, et al. : Chimeric antigen receptors combining 4–1bb and cd28 signaling domains augment pi3kinase/akt/bcl-xl activation and cd8+ t cell-mediated tumor eradication. Mol Ther 18:413–20, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.O’Rourke DM, Nasrallah MP, Desai A, et al. : A single dose of peripherally infused egfrviii-directed car t cells mediates antigen loss and induces adaptive resistance in patients with recurrent glioblastoma. Sci Transl Med 9, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wen PY, Macdonald DR, Reardon DA, et al. : Updated response assessment criteria for high-grade gliomas: Response assessment in neuro-oncology working group. Journal of Clinical Oncology 28:1963–1972, 2010 [DOI] [PubMed] [Google Scholar]
- 25.Yoshimoto K, Dang J, Zhu S, et al. : Development of a real-time rt-pcr assay for detecting egfrviii in glioblastoma samples. Clin Cancer Res 14:488–93, 2008 [DOI] [PubMed] [Google Scholar]
- 26.Dudley ME, Wunderlich JR, Shelton TE, et al. : Generation of tumor-infiltrating lymphocyte cultures for use in adoptive transfer therapy for melanoma patients. J Immunother 26:332–42, 2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Morgan RA, Dudley ME, Wunderlich JR, et al. : Cancer regression in patients after transfer of genetically engineered lymphocytes. Science 314:126–9, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dudley ME, Wunderlich JR, Yang JC, et al. : Adoptive cell transfer therapy following non-myeloablative but lymphodepleting chemotherapy for the treatment of patients with refractory metastatic melanoma. J Clin Oncol 23:2346–57, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Johnson LA, Morgan RA, Dudley ME, et al. : Gene therapy with human and mouse t-cell receptors mediates cancer regression and targets normal tissues expressing cognate antigen. Blood 114:535–46, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Robbins PF, Kassim SH, Tran TL, et al. : A pilot trial using lymphocytes genetically engineered with an ny-eso-1-reactive t-cell receptor: Long-term follow-up and correlates with response. Clin Cancer Res 21:1019–27, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Morgan RA, Yang JC, Kitano M, et al. : Case report of a serious adverse event following the administration of t cells transduced with a chimeric antigen receptor recognizing erbb2. Mol Ther 18:843–51, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Brown CE, Alizadeh D, Starr R, et al. : Regression of glioblastoma after chimeric antigen receptor t-cell therapy. N Engl J Med 375:2561–9, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rosenberg SA, Yang JC, Sherry RM, et al. : Durable complete responses in heavily pretreated patients with metastatic melanoma using t-cell transfer immunotherapy. Clin Cancer Res 17:4550–7, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.D’Angelo SP, Melchiori L, Merchant MS, et al. : Antitumor activity associated with prolonged persistence of adoptively transferred ny-eso-1 (c259)t cells in synovial sarcoma. Cancer Discov 8:944–957, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Neelapu SS, Locke FL, Bartlett NL, et al. : Axicabtagene ciloleucel car t-cell therapy in refractory large b-cell lymphoma. N Engl J Med 377:2531–2544, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kochenderfer JN, Somerville RPT, Lu T, et al. : Lymphoma remissions caused by anti-cd19 chimeric antigen receptor t cells are associated with high serum interleukin-15 levels. J Clin Oncol 35:1803–1813, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Downey SG, Klapper JA, Smith FO, et al. : Prognostic factors related to clinical response in patients with metastatic melanoma treated by ctl-associated antigen-4 blockade. Clin Cancer Res 13:6681–8, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hong JJ, Rosenberg SA, Dudley ME, et al. : Successful treatment of melanoma brain metastases with adoptive cell therapy. Clin Cancer Res 16:4892–8, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lee KS, Lee K, Yun S, et al. : Prognostic relevance of programmed cell death ligand 1 expression in glioblastoma. J Neurooncol, 2017 [DOI] [PubMed] [Google Scholar]
- 40.Samson A, Scott KJ, Taggart D, et al. : Intravenous delivery of oncolytic reovirus to brain tumor patients immunologically primes for subsequent checkpoint blockade. Sci Transl Med 10, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.van den Bent MJ, Gao Y, Kerkhof M, et al. : Changes in the egfr amplification and egfrviii expression between paired primary and recurrent glioblastomas. Neuro Oncol 17:935–41, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Furnari FB, Cloughesy TF, Cavenee WK, et al. : Heterogeneity of epidermal growth factor receptor signalling networks in glioblastoma. Nat Rev Cancer 15:302–10, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Inda MM, Bonavia R, Mukasa A, et al. : Tumor heterogeneity is an active process maintained by a mutant egfr-induced cytokine circuit in glioblastoma. Genes Dev 24:1731–45, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Nathanson DA, Gini B, Mottahedeh J, et al. : Targeted therapy resistance mediated by dynamic regulation of extrachromosomal mutant egfr DNA. Science 343:72–6, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Krex D, Klink B, Hartmann C, et al. : Long-term survival with glioblastoma multiforme. Brain 130:2596–606, 2007 [DOI] [PubMed] [Google Scholar]