

ABSTRACT
The use of assisted reproductive technologies (ART) can induce a congenital overgrowth condition in humans and ruminants, namely Beckwith-Wiedemann syndrome (BWS) and large offspring syndrome (LOS), respectively. Shared phenotypes and epigenotypes have been found between BWS and LOS. We have observed global misregulation of transcripts in bovine foetuses with LOS. microRNAs (miRNAs) are important post-transcriptional gene expression regulators. We hypothesize that there is miRNA misregulation in LOS and that this misregulation is shared with BWS. In this study, small RNA sequencing was conducted to investigate miRNA expression profiles in bovine and human samples. We detected 407 abundant known miRNAs and predicted 196 putative miRNAs from the bovine sequencing results and identified 505 abundant miRNAs in human tongue. Differentially expressed miRNAs (DE-miRNAs) were identified between control and LOS groups in all tissues analysed as well as between BWS and control human samples. DE-miRNAs were detected from several miRNA clusters including DLK1-DIO3 genomic imprinted cluster in LOS and BWS. DNA hypermethylation was associated with downregulation of miRNAs in the DLK1-DIO3. mRNA targets of the DE-miRNAs were predicted and signalling pathways associated with control of organ size (including the Hippo signalling pathway), cell proliferation, apoptosis, cell survival, cell cycle, and cell adhesion were found to be enriched with these genes. Yes associated protein 1 (YAP1) is the core effector of the Hippo signalling pathway, and increased level of active (non-phosphorylated) YAP1 protein was detected in skeletal muscle of LOS foetuses. Overall, our data provide evidence of miRNA misregulation in LOS and BWS.
KEYWORDS: microRNA, large offspring syndrome, Beckwith-Wiedemann syndrome, assisted reproductive technologies, bovine foetus, DNA methylation, DLK1-DIO3, imprinted miRNA cluster, Hippo signalling pathway, YAP1
Introduction
Assisted reproductive technologies (ART) refer to a series of procedures commonly used in cattle [1] and humans [2,3] to conceive offspring. These procedures include collection of oocytes from ovaries, in vitro maturation of oocytes, in vitro fertilization, intracytoplasmic sperm injection, embryo culture, and embryo transfer. In cattle, ART is used to shorten the length of time required to improve the genetics of a herd. Approximately 500,000 bovine ART embryos are currently transferred worldwide [1]. In humans, ART is used primarily to circumvent infertility and to preserve fertility [2,3]. ART conceived babies account for as many as 6.1% of children born in developed countries (2012 figure) [2]. In the United States that figure was ~1.7% in 2016 (76,930 ART babies; http://www.cdc.gov/art/artdata/index.html). In these species, offspring conceived by ART have an increased incidence of altered foetal development [4–10] when compared to the naturally conceived population.
Congenital overgrowth syndrome is one disorder that is shared by a subpopulation of ART-conceived humans and ruminants. In ruminants, the syndrome is known as large offspring syndrome (LOS [11]; also referred to as abnormal offspring syndrome [12]) while in humans it is referred to as Beckwith-Wiedemann Syndrome (BWS [13], OMIM #130650). Shared phenotypes of these syndromes include macrosomia (increased body size), macroglossia (enlarged tongue), omphalocele (umbilical hernia), abnormal organ growth and abnormal placental development [9,14,15]. LOS can affect the dam and cause death of the afflicted offspring bringing financial loss to producers. Children diagnosed with BWS may require corrective surgery for their malformations [16] and may need to undergo frequent blood draws and ultrasounds until they are seven years of age [17] as they have a 4–7.5% increased incidence of tumorigenesis [18]. The most commonly observed molecular alteration in BWS and LOS is a loss of DNA methylation at the imprinting centre 2 (i.e. KvDMR1; IC2), biallelic expression of KCNQ1 overlapping transcript 1 (KCNQ1OT1) and decreased expression of cyclin dependent kinase inhibitor 1C (CDKN1C) [9,19–24]. In addition, we have also reported misregulation of 12 other imprinted genes associated with BWS in LOS [25].
Additional molecular characterization of the overgrowth phenomenon in cattle identified global misregulation of genes as well as global misregulation of the DNA methylome [26,27]. However, most of the misregulation of messenger RNA (mRNA) could not be explained by the identified altered DNA methylation states [27].
microRNAs (miRNAs) are small non-coding RNAs (~23 nucleotides) with important roles in post-transcriptional gene regulation [28]. A function of miRNAs is to target mRNA for degradation [29–32]; therefore, our goal is to identify if the difference in transcript levels observed between LOS and controls [26] may be partly explained by differences in type and abundance of miRNAs.
In this study, we have analysed global miRNA expression profiles in tissues (liver, kidney, skeletal muscle, and tongue) of control, ART-conceived normal and ART-induced LOS bovine foetuses as well as human control and BWS tongue. We detected 407 abundant known miRNAs and predicted 196 putative miRNAs from the bovine sequencing results and identified 505 abundant miRNAs in human tongue. Differentially expressed miRNAs (DE-miRNAs) were identified between control and LOS groups in all tissues analysed as well as between BWS and control human samples. DE-miRNAs were detected from several miRNA clusters including DLK1-DIO3 genomic imprinted cluster in LOS and BWS. DNA hypermethylation was associated with downregulation of miRNAs in the DLK1-DIO3. mRNA targets of the DE-miRNAs were predicted and signalling pathways associated with control of organ size (including the Hippo signalling pathway), cell proliferation, apoptosis, cell survival, cell cycle, and cell adhesion were found to be enriched with these genes. Yes associated protein 1 (YAP1) is the core effector of the Hippo signalling pathway, and increased level of active (non-phosphorylated) YAP1 protein was detected in skeletal muscle of LOS foetuses. Overall, our data provides evidence of miRNA misregulation in LOS and BWS.
Results
Small RNA sequencing
To investigate the differences of miRNA profiles between day ~105 control, LOS, and ART bovine foetuses, we performed small RNA-seq for skeletal muscle (referred to as muscle), kidney, liver, and tongue. Reads were trimmed of sequencing adapters and low quality bases were removed, resulting in ~93%, ~94%, ~85%, and ~96% of reads retained for muscle, kidney, liver, and tongue, respectively (Supplementary Table S1A). Trimmed reads were aligned to the UMD3.1.1 bovine genome assembly by miRDeep2 [33]. On average, ~60%, ~76%, ~44%, and ~60% of the trimmed reads were aligned to miRNA genes for muscle, kidney, liver, and tongue, respectively (Supplemental Table S1A). Samples within a tissue show similar patterns of trimmed read length distribution (Supplementary Figure S1A).
Detection of miRNAs
In total, we detected the expression of 884 known bovine miRNAs (Supplementary Table S2A), of which 407 were further analysed. The remaining 477 detected miRNAs did not meet our cut-off for abundance and were not included in our analysis. In addition to the known miRNAs we identified 196 previously unreported bovine miRNAs which met our cut-off for abundance and were included in further analyses (henceforth referred to as putative miRNA; Supplementary Table S2A). They were subdivided into three categories: (1) unknown 3p or 5p counterparts of known bovine miRNAs (n = 133; Table 1A and Supplementary Table S3A); (2) putative bovine miRNAs conserved in at least one vertebrate species of all (n = 74) miRBase represented species (n = 20; Table 1B and Supplementary Table S3B); and (3) putative miRNAs without conservation in other species (n = 43; Table 1C and Supplementary Table S3C).
Table 1.
An example of putative miRNAs predicted by miRDeep2.
| A. Unknown 3p or 5p counterparts of known bovine miRNAs. | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| miRNA | Pre-miRNA | Type | Blast results | Mismatch | Chr. | Strand | Start | End | miRNA sequence (5ʹ-3ʹ) | Pre-miRNA sequence (5ʹ-3ʹ) |
| bta-let-7d-3p | bta-let-7d | known | hsa-let-7d-3p | 0 | 8 | + | 86,887,437 | 86,887,513 | CUAUACGACCUGCUGCCUUUCU | CCUAGGAAGAGGUAGUAGGUUGCAUAGUUUUCGGGCAGGGAUUUUGCCCACAAGGAGGUAACUAUACGACCUGCUGCCUUUCUUAGG |
| bta-miR-101–1-5p | bta-mir-101–1 | known | hsa-miR-101-5p | 1 | 3 | + | 80,666,430 | 80,666,486 | CAGUUAUCACAGUGCUGAUGC | AGGCUGCCCUGGCUCAGUUAUCACAGUGCUGAUGCUGUCCAUUCUAAAGGUACAGUACUGUGAUAACUGAAGGAUGGCAGCCA |
| bta-miR-24–2-5p | bta-mir-24–2 | known | dno-miR-24b-5p | 0 | 7 | - | 12,981,641 | 12,981,702 | GUGCCUACUGAGCUGAAACACAGU | CUCUGCCUCCCGUGCCUACUGAGCUGAAACACAGUUGAUUUGUGCACACUGGCUCAGUUCAGCAGGAACAGG |
| bta-miR-339b-3-3p | bta-mir-339b-3 | putative | ssc-miR-339-3p | 1 | 25 | + | 42,294,010 | 42,294,067 | CGCUCCUCGAGGCCAGAGCCC | UCCCUGUCCUCCAGGAGCUCACUUGGUCCGGCCGUGCGCUCCUCGAGGCCAGAGCCC |
| bta-miR-432-3p | bta-mir-432 | known | hsa-miR-432-3p | 0 | 21 | + | 67,431,249 | 67,431,318 | CUGGAUGGCUCCUCCAUGUCU | GCAUGACUCCUCCAAGUCUUGGAGUAGGUCAUUGGGUGGAUCCUUUAUUUCCCUAUGUGGGCCACUGGAUGGCUCCUCCAUGUCUUGGCG |
| bta-miR-500-3p | bta-mir-500 | known | pha-miR-502b | 0 | X | + | 92,899,709 | 92,899,771 | AUGCACCUGGGCAAGGAUUCUGA | GCUCCCCCUCUCUAAUCCUUGCUACCUGGGUGAGAGUGCUUUCUGAAUGCAAUGCACCUGGGCAAGGAUUCUGAGAGAGGGAGC |
| B. Putative bovine miRNAs with conservation in vertebrate species represented by miRBase. | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| miRNA | Blast results | Mismatch | Align to tRNA | Chr. | Strand | Start | End | miRNA sequence (5ʹ-3ʹ) | Pre-miRNA sequence (5ʹ-3ʹ) |
| bta-miR-temp11-5p | hsa-miR-4531 | 2 | yes | 5 | - | 39,604,147 | 39,604,189 | AUGGAUAAGGCGUCUGA | AUGGAUAAGGCGUCUGAUUCUGGAUCAGAAGAUUGAAGGUUC |
| bta-miR-temp14-3p | mdo-miR-1388-3p | 3 | - | 13 | - | 54,375,941 | 54,376,002 | UUCUCAGGUUGGACAGUCCUGA | CGGGCUGACAAACCUGAGAUUGUCCCUGGAUGCUCACCAUUCUCAGGUUGGACAGUCCUGA |
| bta-miR-temp15-5p | mmu-miR-802-5p | 1 | - | 1 | + | 149,720,314 | 149,720,371 | UCAGUAACAAAGAUUCAUCCUUG | UCAGUAACAAAGAUUCAUCCUUGUGUCCAUCAAGCAACAAGGAGAAUCUUUGUCACU |
| bta-miR-temp5-3p | bta-miR-2285f | 3 | - | 14 | + | 62,420,781 | 62,420,842 | AAAGCUGAAUGAACUUUUUGGC | CAAAAAGUUUGUUCAGGUUGUUAGUAAAACCGUACAGCUAAAGCUGAAUGAACUUUUUGGC |
| bta-miR-temp5-3p | bta-miR-2285f | 3 | - | X | - | 118,460,421 | 118,460,481 | AAAGCUGAAUGAACUUUUUGGC | ACAAGUUCAUUUGGGGUUUUCUGUACCAGCUUCUGGUUAAAGCUGAAUGAACUUUUUGGC |
| bta-miR-temp91-5p | mml-miR-7193-5p | 5 | - | 7 | + | 41,231,662 | 41,231,740 | GAAUACUGGGUGCUGUAGGCUU | GAAUACUGGGUGCUGUAGGCUUAAAAAUAAAGAAAGAAAAGAAAAUAUUAACACUUACAGUGGUUCAGUUGGCUUCUC |
| C. Putative bovine miRNAs without conservation in other species. | |||||||
|---|---|---|---|---|---|---|---|
| miRNA | Align to tRNA | Chr. | Strand | Start | End | miRNA sequence (5ʹ-3ʹ) | Pre-miRNA sequence (5ʹ-3ʹ) |
| bta-miR-temp103-5p | yes | 3 | + | 116,209,371 | 116,209,419 | GGGGGUGUAGCUCAGUGGUAGAGU | GGGGGUGUAGCUCAGUGGUAGAGUGCAUACUUAGCAUCCAUGAGGCCC |
| bta-miR-temp12-3p | - | 11 | - | 72,573,707 | 72,573,763 | AACCGCGAGUGGCUGAACCUUG | UCGGUUUUUGCCCCAUGCGUUGGAAGACUUCCCCAACCGCGAGUGGCUGAACCUUG |
| bta-miR-temp1-5p | - | 4 | + | 95,112,096 | 95,112,156 | AACUUUUGCCCCUAGUAACGGACU | AACUUUUGCCCCUAGUAACGGACUGAAGGCAUGAGGAAGUCCGUGCAGGAGGCAAAGCAA |
| bta-miR-temp26-5p | yes | 25 | - | 36,566,883 | 36,566,976 | GUGGACUUCCCUGGUAGCUC | GUGGACUUCCCUGGUAGCUCAGUGGUAAAGAAUCCACCUGCCAAUACAGGAGACAUGGGUUUGAUCCCUGGGUCGGGACGAUCCACUGGAGAA |
| bta-miR-temp45-3p | yes | 22 | + | 52,110,608 | 52,110,675 | GUGGUUAUCACAUUCGCCUC | GGGAAAAAAAGUAUAUUCACAGCAGUGUAUCAAGUUUCCAUACUGUUGUGGUUAUCACAUUCGCCUC |
| bta-miR-temp58-5p | - | 24 | + | 32,811,237 | 32,811,285 | CCCACCCAGGGACGCCC | CCCACCCAGGGACGCCCUUCCCUGCUAUUUGGGCUUCCCUGGUGGGUC |
miRNA = unofficial names of putative miRNAs used in this paper. Pre-miRNA = official/unofficial precursor names of the known bovine miRNAs depending on pre-miRNA type (known = official; putative = unofficial). Type = pre-miRNA types. Blast results = known miRNAs of vertebrates with sequence similarity to the predicted miRNAs. Mismatch = number of not matched bases between the putative miRNA and the blast results. Chr. = chromosome. Start/end = starting/ending positions of the pre-miRNA on the chromosome. miRNA sequence = RNA sequences of putative miRNAs in 5' to 3' direction. Pre-miRNA sequence = RNA sequences of known/putative precursors of known bovine miRNAs in 5ʹ to 3ʹ direction. Align to tRNA = whether the putative miRNAs can be aligned to bovine tRNA sequences. Complete table refers to Supplementary Table S3A-C.
Data visualization by principal component analysis (PCA) and hierarchical clustering
PCA and hierarchical cluster analyses were conducted for abundant miRNAs using normalized read counts, and the results can be found in Figure 1 and Supplementary Figure S1B. For skeletal muscle, the four control libraries clustered in the PCA plot while the four LOS libraries were different from controls and did not cluster as a group (Figure 1(a)). Hierarchical cluster analyses showed similar results (Figure 1(b)). Similar patterns for PCA and hierarchical cluster analyses were found for kidney, liver, and tongue (Supplementary Figure S1B). Only female samples were analysed for liver, kidney, and muscle; however, both female and male samples were analysed for tongue. It should be noted that the PCA plot for tongue showed sex-specific variation (Supplementary Figure S1B.E), thus only sex-specific analyses were conducted.
Figure 1.
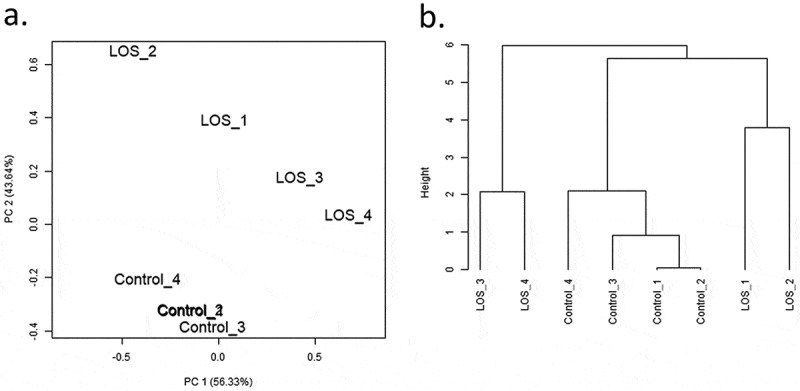
Principal component analyses (PCA) and hierarchical clustering of miRNAs detected in bovine muscle samples of control and LOS groups. (a) The PCA plot illustrates the variance of miRNA expression within or between groups through two principal components – the first (PC1, x-axis) and second (PC2, y-axis) principal component. PC1 explains ~56% of the variance, and PC2 explains ~44% of the variance. (b) The dendrogram shows the hierarchical clusters of bovine control and LOS libraries. Height represents the closeness between individual samples or clusters by measuring the correlations. Results of kidney, liver, and tongue refer to Supplementary Figure S1B.
Identification of de-miRNAs
We compared miRNA abundance using two R packages, namely edgeR and DESeq2. In addition, two strategies of grouping LOS libraries were applied during statistical analyses. In method one, all LOS were compared to all controls for each tissue (Figure 2(c), Supplementary Figure S2A, S2B.C, S2C.C, and S2D.C – rightmost columns). For example, this comparison for muscle resulted in 20 DE-miRNAs detected by edgeR and 21 DE-miRNAs detected by DESeq2 (Supplementary Figure S2A). As LOS is a syndrome, method two employed a combination of LOS made for each tissue and compared with the average of the controls. This type of analysis was done in order to address the variation in expression between LOS foetuses [25,27]. An example of DE-miRNAs detected in skeletal muscle by both strategies is shown in Figure 2(c), and the entire list can be found in Supplementary Figure S2A. The number of shared DE-miRNAs between two-LOS combinations in muscle are shown in Figure 2(a) (EdgeR) and Figure 2(b) (DESeq2). One special comparison between LOS #3 and the control group was performed for tongue since LOS #3 was the only foetus with macroglossia (tongue weight = 4.6g vs. controls ranging from 3.5 to 3.9g; Supplementary Figure S2D.C and Supplementary Table S4K).
Figure 2.
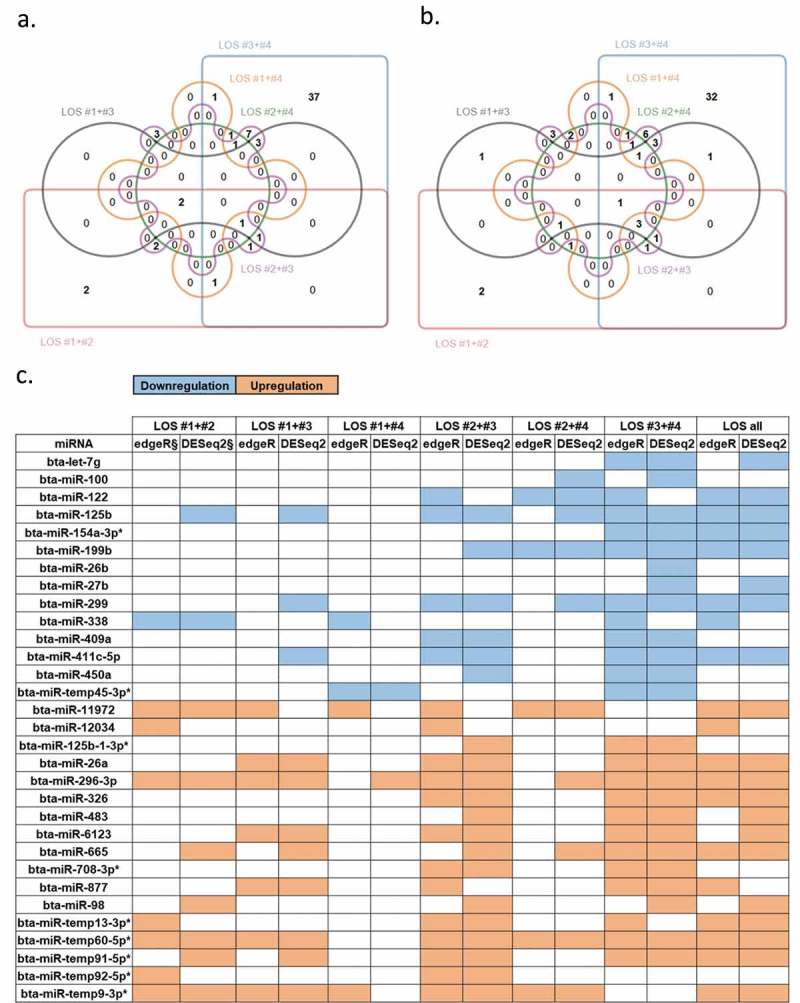
Differentially expressed miRNAs (DE-miRNAs) in muscle of LOS foetuses when compared with controls. (a) The Edwards’ Venn diagram shows the number of DE-miRNAs detected by edgeR package. Different colours/shapes represent different two-LOS combinations. Numbers within unique or overlapped regions indicate numbers of DE-miRNA shared by one or multiple combinations. (b) Same as (a) but shows the results of DESeq2 package. (c) An example of DE-miRNAs detected by various combinations and methods (complete summary table of muscle refers to Supplementary Figure S2A). Leftmost column shows the name of the DE-miRNAs. Space filled with colour indicate that miRNA was detected as significant in the corresponding combination and method. § = FDR < 0.05 for edgeR or padj < 0.05 for DESeq2. Blue = downregulation. Orange = upregulation. * = putative miRNAs. Kidney, liver, and tongue results refer to Supplementary Figure S2B, S2C, and S2D, respectively.
In total, 78 (muscle), 33 (kidney), 63 (liver), and 41 (tongue) DE-miRNAs were detected between control and LOS (Supplementary Figure S2A, S2B.C, S2C.C, and S2D.C). Out of the 78 DE-miRNAs detected by at least one method between controls and LOS in muscle, 38 were found to be unique in the LOS #3-#4 combination. This higher number of DE-miRNAs is associated with the heavier body weights of LOS #3 and #4 foetuses, which were 620g and 714g, respectively, when compared to the other two LOS foetuses (mean ~516g) and controls (mean ~405g). As in muscle, the LOS #3-#4 combination also had the largest number of DE-miRNAs in kidney and liver (Supplementary Figure S2B.C and S2C.C). The raw data for the statistical analyses can be found in Supplementary Table S4A–J. It should be noted that only data generated with the suggested small sample size k value parameter (RUVseq), namely k = 1 are presented in the text (for analysis with k = 2 and k = 3 please refer to Supplementary Table S4A-E). miRNAs identified as misregulated by DESeq2 and edgeR were combined for further analysis.
ART-specific miRNAs
To determine how much of the variation in miRNA expression between LOS and control can be explained by the method of conception, namely ART, we compared the levels of miRNA between control, ART and LOS in liver. On the PCA and hierarchical cluster plots, the five control libraries and the four ART libraries clustered together, but not the four LOS libraries (Supplementary Figure S1B.C and S1B.D). Statistical comparisons of miRNA expression were made between (1) control group and ART group; (2) ART group and LOS group; (3) control group and the combined ART and LOS groups. For the first comparison (control vs. ART) we detected four upregulated putative miRNAs (i.e. bta-miR-temp32-5p, bta-miR-temp34-3p, bta-miR-temp43-3p, bta-miR-temp82-3p; Table 2). When comparing ART and LOS, we found 29 DE-miRNAs (Table 2). Only two putative miRNAs, namely bta-miR-485-3p, bta-miR-temp43-3p (Table 2) were differentially expressed when comparing the control vs. combined ART/LOS group. The raw data for the statistical analyses can be found in Supplementary Table S5A–C.
Table 2.
DE-miRNAs associated with ART in liver.
| Comparison | Direction | DE-miRNAs |
|---|---|---|
| ART vs. Control | upregulation | bta-miR-temp32-5p*, bta-miR-temp34-3p*, bta-miR-temp43-3p*, bta-miR-temp82-3p* |
| LOS vs. ART | downregulation | bta-miR-187, bta-miR-192, bta-miR-2408, bta-miR-2904, bta-miR-423-3p, bta-miR-485-3p*, bta-miR-temp13-3p*, bta-miR-temp32-5p*, bta-miR-temp34-3p*, bta-miR-temp36-3p*, bta-miR-temp77-5p*, bta-miR-temp81-5p*, bta-miR-temp82-3p*, bta-miR-temp91-5p*, bta-miR-temp94-3p*, bta-miR-temp97-5p* |
| upregulation | bta-let-7a-5p, bta-let-7c, bta-let-7d, bta-let-7f, bta-let-7f-1-3p*, bta-let-7g, bta-let-7i, bta-miR-2285ca, bta-miR-33a-3p*, bta-miR-3431, bta-miR-98, bta-miR-temp1-5p*, bta-miR-temp43-3p* | |
| ART+LOS vs. Control | downregulation | bta-miR-485-3p* |
| upregulation | bta-miR-temp43-3p* |
Comparison = statistical comparison between two group. DE-miRNA = miRNAs with FDR (edgeR) or Padj (DESeq2) less than 0.05, ‘*’ indicates putative miRNAs. The raw data for the statistical analyses can be found in Supplementary Table S5A-C.
Genomic location of de-miRNAs
DE-miRNAs were detected from several miRNA clusters including DLK1-DIO3 genomic imprinted cluster and miR-143/145 cluster. Figure 3 depicts a ~ 610 kilobase (kb) region on bovine chromosome 21, which contains the DLK1-DIO3 imprinted cluster. DE-miRNAs detected in muscle reside in this region and six of them belong to that cluster (Figure 3). miRNA misregulation at this imprinted locus was also evident in the tongue of LOS #3 (foetus with macroglossia; Supplementary Table S6D). In addition, when analysing misregulation of miRNA based on genomic position of the pre-miRNA, some of the DE-miRNAs were found to be in close proximity (within 50 kb) and had a tendency to be similarly misregulated. Of the 78 (muscle), 33 (kidney), 63 (liver), and 41(tongue) DE-miRNAs between control and LOS, 36, 6, 27, and 14, respectively were found within 50kb of another DE-miRNA (Supplementary Table S6A–D).
Figure 3.
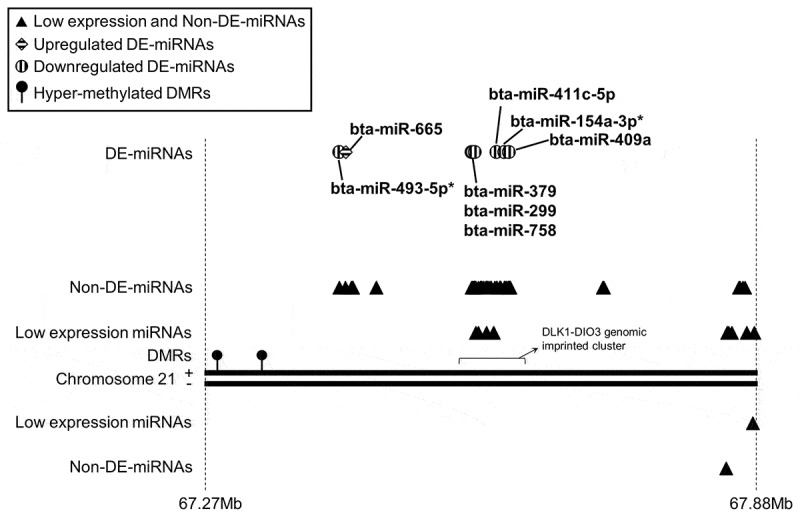
An example of differentially expressed miRNAs (DE-miRNAs) detected in bovine muscle with close proximity (<50 kb) on chromosome 21. In muscle of LOS foetuses, eight DE-miRNAs were found from this ~610-kb region and six of them belong to the DLK1-DIO3 genomic imprinted cluster. Two hypermethylated differentially methylated regions (DMRs) were detected at ~230-kb and ~279-kb upstream regions of DLK1-DIO3 cluster. Triangles/circles/rhombuses = locations of miRNAs on the chromosome (not drawn to scale). Filled lollipops = hypermethylated DMRs. Solid lines = chromosome 21. Dashed lines = start and end positions of this chromosome region with million bases (Mb) as unit. ‘+’ = sense strand of chromosome 21. ‘-’ = antisense strand of chromosome 21. Low expression miRNAs = miRNAs that were not detected or failed to pass the low expression cut-off in muscle. Non-DE-miRNAs = miRNAs that passed the low expression cut-off but were not detected as DE-miRNAs by any methods and grouping strategies in muscle. DE-miRNAs = miRNAs detected as DE-miRNAs in muscle by any of the methods and grouping strategies. For entire lists of genome locations of DE-miRNAs in muscle, kidney, liver, and tongue please refer to Supplementary Table S6A, S6B, S6C, and S6D, respectively.
Associations between de-miRNAs and differentially methylated regions (DMRs)
The associations between DE-miRNAs and DMRs were investigated using the day ~105 bovine foetal muscle whole genome bisulfite sequencing data from our previous publication [27]. In this study, we used two human databases [34,35] to predict transcription start sites (TSS) and upstream regulatory regions of conserved miRNAs in bovine since this information is not yet available for bovine. DMRs were detected within 10-kb upstream regions of the predicted TSS regions for bta-miR-132, bta-miR-196a, bta-miR-26a, and bta-miR-296-3p (Table 3). bta-miR-196a may be transcribed from two loci (Table 3) and both putative TSS were hypomethylated. Figure 4 illustrates one of the cases in which upregulated bta-miR-132 was associated with a hypomethylated DMR in LOS.
Table 3.
Muscle DE-miRNAs with DMRs in upstream region of predicted TSS.
|
Bovine miRNA TSS prediction based on human database mirTrans | ||||||||||
|
Bovine |
Human |
|||||||||
|
Pre-miRNA |
DE-miRNA |
Chr. |
Strand |
Predicted TSS regions |
DMRs (10 kb upstream) |
Pre-miRNA |
Chr. |
Strand |
TSS regions |
TSS numbers |
| bta-mir-132 | bta-miR-132 | 19 | - | 23651124-23652605 | 23658550-23659389 hypo-methylation | hsa-mir-132 | 17 | - | 2049750-2051271 | 6 |
| 23654400-23655941 | 2053392-2055221 | 5 | ||||||||
| 23655550-23656356 | 2054780-2055780 | 1 | ||||||||
| |
|
|
|
23659251-23660616 |
|
|
|
|
2058698-2059698 |
1 |
| bta-mir-196a-2 | bta-miR-196a | 5 | - | 26205331-26205996 | 26213420-26215619 hypo-methylation | hsa-mir-196a-2 | 12 | + | 53985345-53986345 | 1 |
| |
|
|
|
26204650-26205506 |
|
|
|
|
53985933-53986933 |
1 |
|
Bovine miRNA TSS prediction based on human database miRStart | ||||||||||
|
Bovine |
Human |
|||||||||
|
Pre-miRNA |
DE-miRNA |
Chr. |
Strand |
Predicted TSS regions |
DMRs (10 kb upstream) |
Pre-miRNA |
Chr. |
Strand |
TSS regions |
TSS numbers |
| bta-mir-132 | bta-miR-132 | 19 | - | 23651124-23652234 | 23658550-23659389 hypo-methylation | hsa-mir-132 | 17 | - | 1952818-1954251 | 2 |
| 23652410-23653246 | 1954267-1955267 | 1 | ||||||||
| 23656460-23658263 | 1959006-1961027 | 5 | ||||||||
| 23657985-23658829 | 1960569-1961569 | 1 | ||||||||
| |
|
|
|
23658965-23660332 |
|
|
|
|
1961706-1962706 |
1 |
| bta-mir-196a-1 |
bta-miR-196a |
19 |
+ |
38495732-38496157 |
38493580-38494349 hypo-methylation |
hsa-mir-196a-1 |
17 |
- |
46710434-46711434 |
1 |
| bta-mir-196a-2 | bta-miR-196a | 5 | - | 26224532-26225761 | 26222020-26225019 hypo-methylation; 26226320-26228319 hypo-methylation | hsa-mir-196a-2 | 12 | + | 54359273-54360486 | 2 |
| |
|
|
|
26216530-26217547 |
|
|
|
|
54367586-54368646 |
2 |
| bta-mir-196a-2 | bta-miR-196a | 5 | - | 26210597-26211395 | 26213420-26215619 hypo-methylation | hsa-mir-196a-2 | 12 | + | 54373662-54374662 | 1 |
| |
|
|
|
26204238-26205212 |
|
|
|
|
54379926-54381208 |
2 |
| bta-mir-26a-1 |
bta-miR-26a |
22 |
+ |
11436811-11437037 |
11430423-11431382 hyper-methylation |
hsa-mir-26a-1 |
3 |
+ |
37984525-37985525 |
1 |
| bta-mir-296 | bta-miR-296-3p | 13 | + | 58054240-58054654 | 58046118-58046817 hyper-methylation; 58048708-58049757 hyper-methylation | hsa-mir-296 | 20 | - | 57408489-57409674 | 3 |
| 58053302-58053794 | 57409262-57410262 | 1 | ||||||||
Chr. = chromosome. Predicted TSS regions: conserved region in bovine of human TSS region of the conserved miRNAs. TSS regions: region of adjacent TSS (within 500bp) + 500bp in both up- and downstream.
Figure 4.
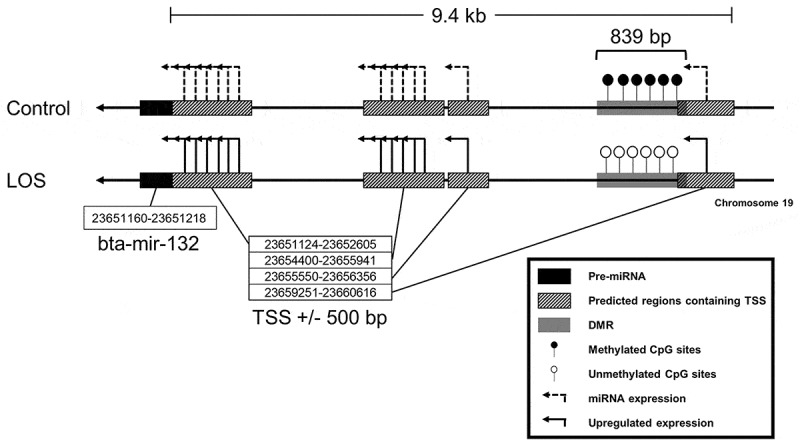
An example of differentially expressed miRNAs (DE-miRNAs) associated with differentially methylated regions (DMRs) within 10-kb upstream regions of their transcription start sites (TSS). A hypomethylated DMR was found within 10-kb upstream regions of predicted TSS regions of upregulated bta-miR-132 in LOS muscle. The TSS regions were predicted based on two human databases, namely mirTrans and miRStart. Line arrows = chromosome 19 antisense strand. Dashed elbow arrows = predicted TSS and miRNA expression. Solid elbow arrows = predicted TSS and upregulated miRNA expression. Solid filled black rectangles = pre-miRNAs. Striped rectangles = predicted TSS regions. Grey rectangles = DMRs. Open lollipops = unmethylated CpG sites. Filled lollipops = methylated CpG sites. For all the association between DE-miRNAs and DMRs refer to Table 3.
In addition, based on mouse studies [36] there is a DMR ~200-kb upstream of the DLK1-DIO3 imprinted cluster. We identified two hypermethylated DMRs at ~230-kb and ~279-kb upstream regions of the DLK1-DIO3 cluster, although they were not identified in the analysis results based on the TSS databases (Figure 3).
Validation of de-miRNAs by qRT-PCR and different data analysis pipeline
To validate the small RNA-seq analyses results, qRT-PCR was performed on DE-miRNAs in bovine muscle. Four abundant miRNAs with constant expression in all muscle libraries, namely bta-miR-181a, bta-miR-20a, bta-miR-30b-5p, and bta-miR-378, were selected as endogenous controls, and the geometric means of their Ct value were used for normalization [37,38]. Three upregulated miRNAs (bta-miR-145, bta-miR-143, bta-miR-296-3p) and three downregulated miRNAs (bta-miR-409a, bta-miR-299, bta-miR-450b) were chosen as targets for validation. As per the sequencing results, bta-miR-145, bta-miR-143, and bta-miR-296-3p were found to be upregulated by qRT-PCR (Figure 5 and Supplementary Figure S3A); however, none of the sequencing-identified downregulated miRNAs were validated (Figure 5 and Supplementary Figure S3A). Since the chosen downregulated miRNAs were not confirmed by qRT-PCR, we suspected that there were issues with our miRNA sequencing analysis pipeline. In order to corroborate our results, the small RNA-seq data was blindly re-analysed by a collaborator using a different pipeline (i.e. ‘Corroboration of DE-miRNA with a different pipeline’ in the methods section). Results were similar for both pipelines utilized (Supplementary Table S7).
Figure 5.
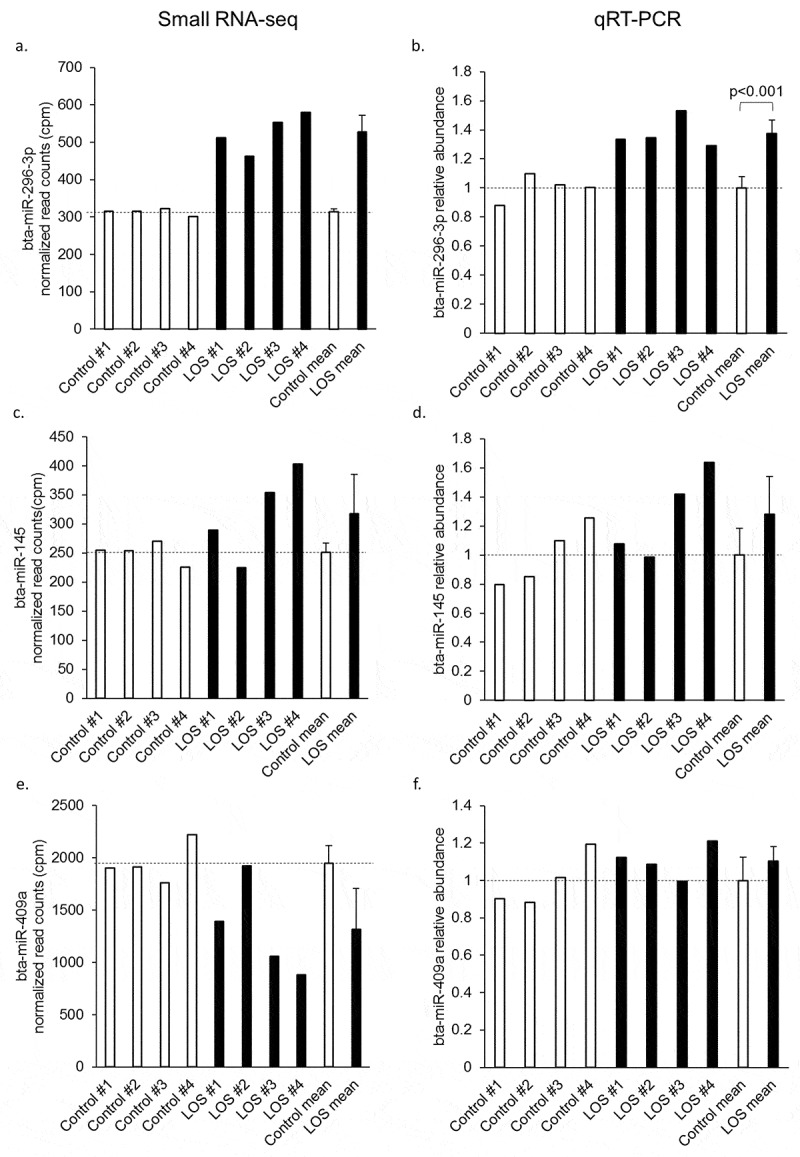
qRT-PCR validation of differentially expressed miRNAs (DE-miRNAs) in bovine muscle. Normalized read counts from small RNA-seq results for each DE-miRNA tested are shown in panels a, c, and e and the corresponding qRT-PCR results are shown in b, d, and f. The normalized read counts were acquired from all control vs. all LOS comparisons after all the normalization steps were done. For qRT-PCR results, the level of each DE-miRNAs was normalized to the geometric mean of four selected endogenous control miRNAs. Student’s t tests were performed for qRT-PCR results and significant difference between control and LOS groups was indicated by p value. Dashed lines = control means. Unfilled bars = control muscle samples. Filled bars = LOS muscle samples. The mean and standard deviation of the groups are shown at the right of each panel. Sequencing results of bta-miR-409a were not confirmed by qRT-PCR. Results for the other three DE-miRNAs may be found in Supplementary Figure S3A.
Levels of bta-miR-145 and bta-miR-143 were also measured in bovine tongue RNA samples by qRT-PCR with the same set of endogenous controls miRNAs for normalization (not differentially expressed in tongue). We only compared control group with the LOS group, and observed higher levels of these miRNAs in LOS #3, which was the only LOS foetus with a large protruding tongue (Supplementary Figure S3B).
Prediction of mRNA targets and enriched signalling pathways of de-miRNAs in LOS
The 3ʹUTR of all bovine mRNAs found in Ensembl and RefSeq were used to predict putative targets of the DE-miRNAs using miRanda. DAVID was used to determine functionally related signalling pathways enriched in genes predicted as targets of DE-miRNAs in muscle, kidney, liver, and tongue (Supplementary Table S8A–D). In addition, DE-miRNAs conserved in human (Supplementary Table S9) were selected to corroborate the muscle, kidney, and liver bovine target and pathway predictions using TarBase (Supplementary Table S8E–G). Pathways associated with control of organ size, cell proliferation, apoptosis, cell survival, cell cycle, and cell adhesion were enriched in both bovine and human prediction results. An example of pathways predicted for bovine muscle is illustrated in Figure 6.
Figure 6.
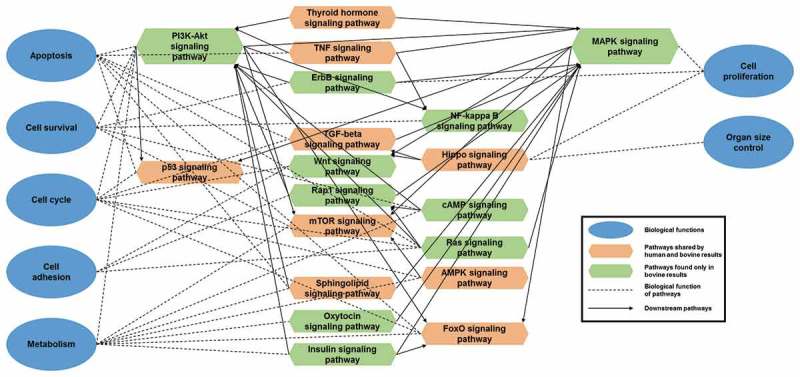
Bovine muscle differentially expressed miRNAs (DE-miRNAs) enriched KEGG signalling pathways and its corroboration with human data. KEGG pathways were enriched by DAVID with predicted mRNA targets of DE-miRNAs in LOS muscle. Pathways associated with several important biological functions are illustrated. mirPath and human library of TarBase was used to corroborate bovine prediction results. Green hexagons = pathways detected significantly (corrected p value < 0.05) in bovine but not shown in human results. Orange hexagons = significant bovine pathways that were also confirmed by human results. Blue circle = important biological functions associated with development and growth. Dashed lines = link pathways to their biological functions. Solid arrows = link pathways to their downstream pathways. The upper and lower corners of the hexagons on both sides connect the upstream pathways, and the middle corners connect the downstream pathways or biological functions.
Detection of YAP1 protein abundance
The Hippo signalling pathway, a pathway involved in control of organ size, was detected in both bovine and human enriched signalling pathways across all the tissues analysed (Supplementary Table S8A–F). We queried our previous dataset [27] and identified that the transcript of YAP1, the core effector of the Hippo signalling pathway, increased in muscle of LOS #3 and #4 (Figure 7(a)). We performed western blot analyses to further investigate YAP1 protein abundance and phosphorylation state at serine 127 (i.e. pYAP1 = inactive state) in muscle. Protein levels of glyceraldehyde-3-phosphate dehydrogenase (GAPDH) were used for data normalization. No changes in pYAP1 were detected in LOS #1, #3, and #4 although a slight decrease was evident for LOS #2 (Figure 7(b)). However, levels of active (non-phosphorylated) YAP1 protein were higher in LOS #3 and #4 when compared to controls (Figure 7(c)).
Figure 7.
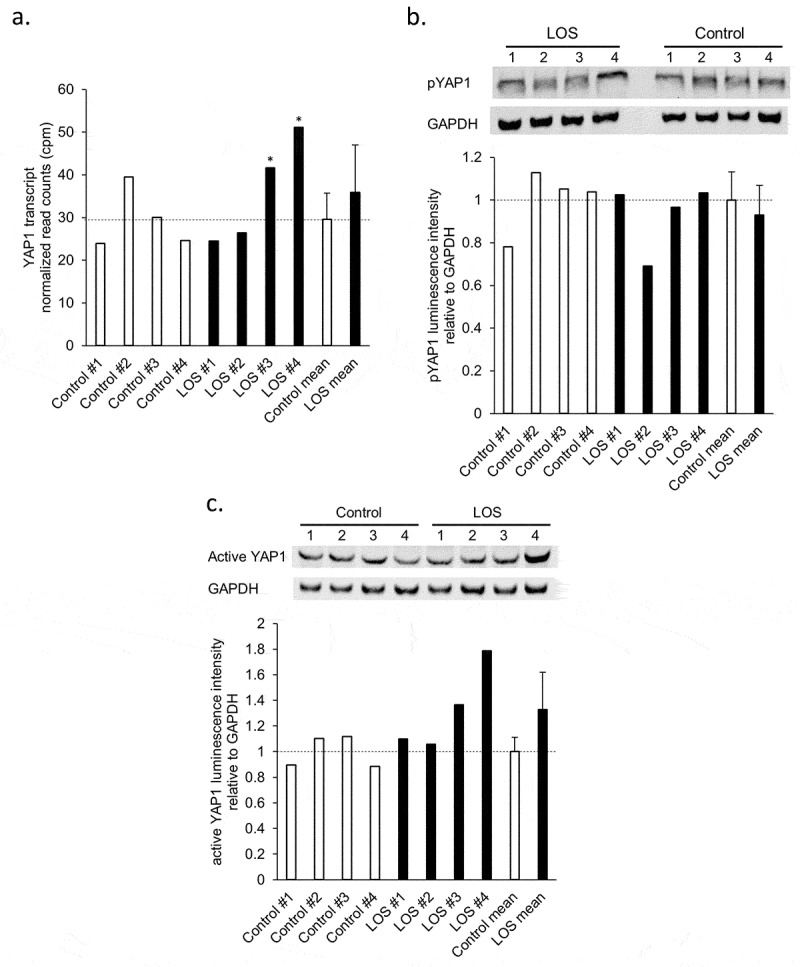
YAP1 transcript and protein abundance in bovine muscle samples of control and LOS foetuses. (a) Normalized read counts of YAP1 transcript were acquired from previous studies (* = FDR < 0.05 [27];). (b) and (c) Upper panel: Western blotting results of phosphorylated (inactive) YAP1 protein (b) or active YAP1 protein (c) with GAPDH as endogenous control. pYAP1 = phosphorylated YAP1. Lower panel: Relative luminescent signal intensity of pYAP1 or active YAP1 to GAPDH measured from the pictures of the membrane. Dashed lines = control means. Unfilled bars = control muscle samples. Filled bars = LOS muscle samples. The mean and standard deviation of the groups are shown at the right of each panel.
YAP1 mRNAs were predicted by miRanda to be target of several DE-miRNAs (Supplementary Table S10). Noticeably one of the predicted bovine YAP1 transcripts had a much longer (3,414 bp; Figure 8) 3ʹ UTR sequence than the others and most of the target prediction results came from this transcript, namely XM_015474584.1. To further investigate the target potential between YAP1 and DE miRNAs, primers were designed to test the expression of XM_015474584.1 in control foetal muscle (Figure 8). Additionally, intron-spanning primers for exons 6 and 7 of XM_015474589.1 were used as positive control for YAP1 cDNA since these two exons were shared by all the predicted YAP1 transcripts (Figure 8(a,c,d)). As shown in Figure 8(b), products were amplified by PCR when genomic DNA was used as a template but not when muscle cDNA was used, which suggests that YAP1 transcript XM_015474584.1 may not be expressed in muscle at this gestation stage.
Figure 8.
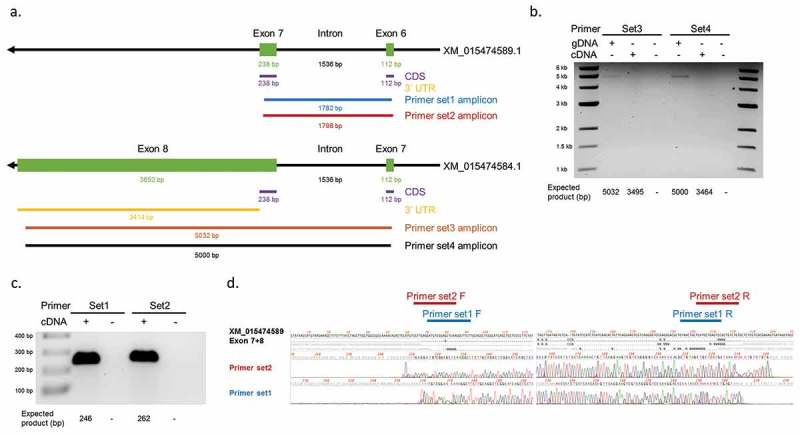
Detection of 3ʹUTR sequences of YAP1 transcripts in bovine muscle. (a) Design of primer sets for YAP1 transcript XM_015474584.1 and XM_015474589.1. Black arrows = YAP1 gene. Green rectangles = exons. Purple lines = coding sequences. Yellow lines = 3ʹUTR sequences. Blue/red/brown/black lines = amplicon sequences of different primer sets. Numbers indicate expected length of each sequence. (b) PCR products of primer set3 and set4 with genomic DNA (gDNA), cDNA, and water (-/-) as templates on 0.5% agarose gel. (c) PCR products of primer set1 and set2 with cDNA and water (-) as templates on 1% agarose gel. (d) Sanger sequencing results of the two bands detected in (c). Locations of the primers are indicated on the top. F = forward primer. R = reverse primer.
Identification of DE-miRNAs in BWS
RNA from tongue samples of adult human control (hControl) and BWS children were sequenced and data was processed following the same procedure as for bovine. On average, ~84% of reads were retained after adapter and quality trimming, and ~36% of the trimmed reads were aligned to miRNA genes (Supplementary Table S1B). BWS #1 was eliminated from further analyses since its mapping ratio was below 5%. Samples show similar patterns of trimmed read length distribution (Supplementary Figure S1A.E). We detected the expression of 1919 human miRNAs, of which 505 passed the cut-off for abundance and were kept for further analyses (Supplementary Table S2B). PCA and hierarchical cluster analyses results showed that the two hControl libraries clustered, while the 12 BWS libraries were different from hControls and were loosely clustered (Supplementary Figure S1B.G and S1B.H). In total, 206 DE-miRNAs were detected between hControl and BWS group (Table 4). The raw data for the statistical analyses can be found in Supplementary Table S11.
Table 4.
DE-miRNAs of human tongue.
| Comparison | Direction | DE-miRNAs |
|---|---|---|
| BWS vs. hControl | downregulation | hsa-let-7b-3p, hsa-let-7d-3p, hsa-miR-100-5p, hsa-miR-10a-5p, hsa-miR-12,136, hsa-miR-1247-3p, hsa-miR-1247-5p, hsa-miR-125a-5p, hsa-miR-125b-5p, hsa-miR-1271-5p, hsa-miR-1296-5p, hsa-miR-1304-3p, hsa-miR-1306-5p, hsa-miR-132-3p, hsa-miR-143-5p, hsa-miR-145-5p, hsa-miR-146b-3p, hsa-miR-146b-5p, hsa-miR-150-5p, hsa-miR-18a-3p, hsa-miR-191-5p, hsa-miR-193b-3p, hsa-miR-193b-5p, hsa-miR-197-3p, hsa-miR-210-5p, hsa-miR-211-5p, hsa-miR-2110, hsa-miR-2116-3p, hsa-miR-212-5p, hsa-miR-215-5p, hsa-miR-221-5p, hsa-miR-222-3p, hsa-miR-223-3p, hsa-miR-224-5p, hsa-miR-2277-5p, hsa-miR-28-3p, hsa-miR-29a-3p, hsa-miR-29b-2-5p, hsa-miR-29b-3p, hsa-miR-29c-3p, hsa-miR-29c-5p, hsa-miR-30d-5p, hsa-miR-31-3p, hsa-miR-3195, hsa-miR-320a-3p, hsa-miR-320b, hsa-miR-328-3p, hsa-miR-330-3p, hsa-miR-339-5p, hsa-miR-342-3p, hsa-miR-345-5p, hsa-miR-361-3p, hsa-miR-361-5p, hsa-miR-365a-3p, hsa-miR-365b-3p, hsa-miR-423-3p, hsa-miR-4488, hsa-miR-484, hsa-miR-499a-5p, hsa-miR-5010-3p, hsa-miR-504-5p, hsa-miR-505-3p, hsa-miR-550a-3p, hsa-miR-5690, hsa-miR-574-3p, hsa-miR-576-5p, hsa-miR-616-5p, hsa-miR-625-3p, hsa-miR-628-3p, hsa-miR-6511a-3p, hsa-miR-652-3p, hsa-miR-664a-3p, hsa-miR-671-3p, hsa-miR-744-3p, hsa-miR-766-3p, hsa-miR-7706, hsa-miR-7977, hsa-miR-885-5p, hsa-miR-891a-5p, hsa-miR-92a-3p, hsa-miR-92b-3p, hsa-miR-93-3p, hsa-miR-941, hsa-miR-942-5p, hsa-miR-96-5p, hsa-miR-99a-5p, hsa-miR-99b-5p |
| upregulation | hsa-let-7c-5p, hsa-let-7e-5p, hsa-let-7f-2-3p, hsa-let-7i-5p, hsa-miR-100-3p, hsa-miR-101-3p, hsa-miR-1185–1-3p, hsa-miR-1185–2-3p, hsa-miR-1185-5p, hsa-miR-1197, hsa-miR-12,135, hsa-miR-127-3p, hsa-miR-127-5p, hsa-miR-1307-5p, hsa-miR-134-3p, hsa-miR-134-5p, hsa-miR-136-3p, hsa-miR-136-5p, hsa-miR-144-3p, hsa-miR-152-5p, hsa-miR-153-3p, hsa-miR-154-5p, hsa-miR-181a-5p, hsa-miR-181b-5p, hsa-miR-181d-5p, hsa-miR-188-5p, hsa-miR-190a-5p, hsa-miR-199b-5p, hsa-miR-206, hsa-miR-20b-5p, hsa-miR-27b-3p, hsa-miR-299-3p, hsa-miR-299-5p, hsa-miR-301a-3p, hsa-miR-301b-3p, hsa-miR-30d-3p, hsa-miR-30e-5p, hsa-miR-3200-3p, hsa-miR-323a-3p, hsa-miR-323b-3p, hsa-miR-329-3p, hsa-miR-335-5p, hsa-miR-337-3p, hsa-miR-337-5p, hsa-miR-33a-5p, hsa-miR-340-5p, hsa-miR-362-3p, hsa-miR-369-3p, hsa-miR-369-5p, hsa-miR-370-3p, hsa-miR-370-5p, hsa-miR-376a-2-5p, hsa-miR-376a-3p, hsa-miR-376a-5p, hsa-miR-376b-3p, hsa-miR-376b-5p, hsa-miR-376c-3p, hsa-miR-376c-5p, hsa-miR-377-3p, hsa-miR-377-5p, hsa-miR-378e, hsa-miR-379-3p, hsa-miR-379-5p, hsa-miR-380-3p, hsa-miR-381-3p, hsa-miR-381-5p, hsa-miR-382-3p, hsa-miR-382-5p, hsa-miR-409-3p, hsa-miR-409-5p, hsa-miR-410-3p, hsa-miR-411-3p, hsa-miR-411-5p, hsa-miR-412-5p, hsa-miR-424-5p, hsa-miR-431-3p, hsa-miR-431-5p, hsa-miR-432-3p, hsa-miR-432-5p, hsa-miR-433-3p, hsa-miR-433-5p, hsa-miR-450b-5p, hsa-miR-4677-3p, hsa-miR-483-5p, hsa-miR-485-3p, hsa-miR-485-5p, hsa-miR-487a-3p, hsa-miR-487a-5p, hsa-miR-487b-3p, hsa-miR-487b-5p, hsa-miR-493-3p, hsa-miR-493-5p, hsa-miR-494-3p, hsa-miR-495-3p, hsa-miR-496, hsa-miR-502-5p, hsa-miR-539-3p, hsa-miR-539-5p, hsa-miR-541-3p, hsa-miR-541-5p, hsa-miR-542-3p, hsa-miR-542-5p, hsa-miR-543, hsa-miR-598-3p, hsa-miR-6505-5p, hsa-miR-653-5p, hsa-miR-654-3p, hsa-miR-654-5p, hsa-miR-655-3p, hsa-miR-655-5p, hsa-miR-656-3p, hsa-miR-660-5p, hsa-miR-668-3p, hsa-miR-675-5p, hsa-miR-758-3p, hsa-miR-770-5p, hsa-miR-889-3p, hsa-miR-98-5p, hsa-miR-99a-3p |
Comparison = statistical comparison between two group. DE-miRNA = miRNAs with FDR (edgeR) or Padj (DESeq2) less than 0.05. The raw data for the statistical analyses can be found in Supplementary Table S11.
Similar to bovine, DE-miRNAs were detected from human miRNA clusters including DLK1-DIO3 cluster, miR-143/145 cluster, miR-181a/181b cluster, miR-125b/let7a/100 cluster, and miR-99b/let7e/125a cluster (Supplementary Table S6E). In total, there are 121 DE-miRNAs found within 50kb of another DE-miRNA in BWS (Supplementary Table S6E).
Enriched signalling pathways of DE-miRNAs were predicted using TarBase, and pathways associated with control of organ size, cell proliferation, apoptosis, cell survival, cell cycle, and cell adhesion were detected, as for LOS (Supplementary Table S8H).
To compare the miRNA expression in LOS and BWS, DE-miRNAs detected in tongue samples of both LOS and BWS individuals were investigated. More than one third (n = 17) of the DE-miRNAs found in LOS tongue were detected in BWS tongue (Figure 9(a)). Eight of the 17 shared DE-miRNAs are located within the DLK1-DIO3 cluster, namely hsa-miR-369-5p; hsa-miR-379-3p; hsa-miR-379-5p; hsa-miR-411–3p; hsa-miR-411-5p; hsa-miR-485-5p; hsa-miR-487b-3p; hsa-miR-654-3p. Interestingly, almost all the DE-miRNAs from DLK1-DIO3 cluster are deregulated in the opposite direction in LOS (downregulation) and BWS (upregulation) including the eight mentioned above (Supplementary Table S6A–E). Among the other nine shared DE-miRNAs, six are deregulated in the same direction in LOS and BWS, and three are deregulated in the opposite direction (Figure 9(a); Supplementary Table S6A–E). Not all DE-miRNAs identified were conserved between human and bovine tongue. For example, of the 24 bovine DE-miRNAs not shared with BWS 10 are not conserved and of the 189 human DE-miRNAs not shared with LOS 46 are not conserved.
Figure 9.
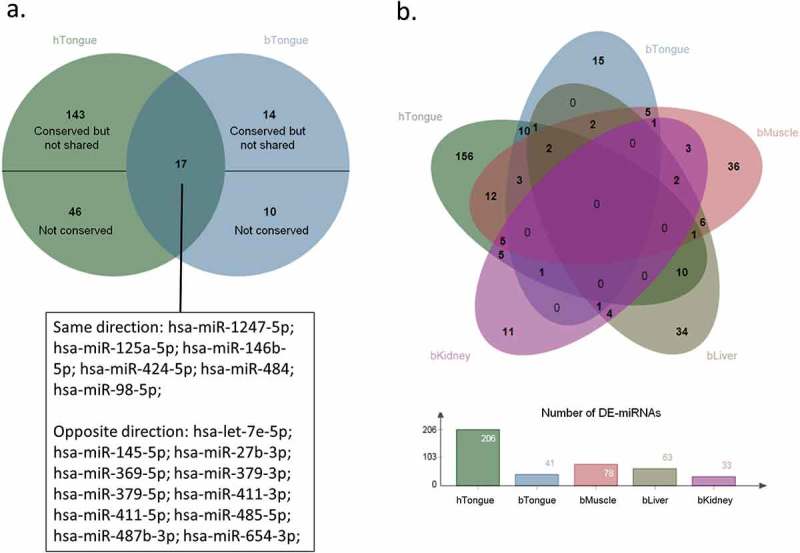
Differentially expressed miRNAs (DE-miRNAs) shared by LOS and BWS. (a) A classic Venn diagram of shared DE-miRNAs between LOS tongue and BWS tongue. Overlapping region indicates shared DE-miRNAs. The human nomenclature of the shared DE-miRNAs is shown below and separated into two lists indicating the direction of deregulation in LOS and BWS. Not shared DE-miRNAs are subdivided into two group: (1) conserved in the other species but not shared; and (2) not conserved in the other species. hTongue = human tongue. bTongue = bovine tongue. (b) Top: A classic Venn diagram of shared DE-miRNAs between LOS muscle, kidney, liver, and tongue, and BWS tongue. Overlapping regions indicate shared DE-miRNAs. hTongue = human tongue. bMuscle = bovine muscle. bKidney = bovine kidney. bLiver = bovine liver. bTongue = bovine tongue. Bottom: bar graph showing the number of DE-miRNA in each tissue.
In order to determine if global misregulation of specific miRNAs exist, all tissues of LOS together with the human tongue samples were compared. About 24% (n = 50) of DE-miRNAs found in BWS tongue samples were also detected in at least one tissue of LOS as DE-miRNAs (Figure 9(b)). Among these 50 DE-miRNAs, 25 are deregulated in the same direction in LOS and BWS, and the remaining 25, including 15 from DLK1-DIO3 cluster, show deregulation in the opposite direction. Five DE-miRNAs (hsa-let-7e-5p, hsa-miR-145-5p, hsa-miR-27b-3p, hsa-miR-379-5p, hsa-miR-98-5p) are shared by LOS muscle, LOS tongue, and BWS tongue, and only hsa-miR-98-5p is deregulated in the same direction in LOS and BWS.
DNA methylation status in BWS
DNA methylation status at known regulatory DMRs (i.e. maternally expressed 3 [MEG3, within the DLK1-DIO3 cluster], growth factor receptor bound protein 10 [GRB10], and small nuclear ribonucleoprotein N [SNPRN]) was determined by pyrosequencing. No difference in DNA methylation was detected between the hControl and the BWS samples for any of the loci analysed (data not shown).
Expression of insulin like growth factor 2 (IGF2) in BWS tongue
Relative expression of the DE-miRNA (miR-483) host gene, IGF2, was determined by qRT-PCR using GAPDH, beta actin (ACTB), and ATP synthase peripheral stalk-membrane subunit b (ATP5PB) as endogenous control genes. Transcript amount of IGF2 were increased (P < 0.03) in BWS tongue when compared to hControls (Figure 10).
Figure 10.
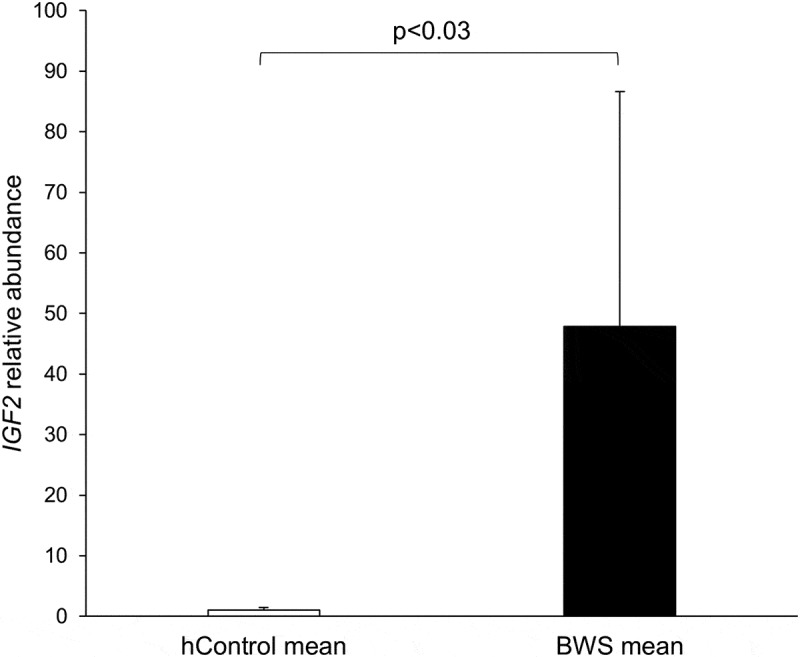
qRT-PCR determination of IGF2 expression in human tongue. The level of IGF2 was normalized to the mean of three endogenous control genes (GAPDH, ACTB, and ATP5PB). Mann-Whitney U Test was performed for qRT-PCR results and significant difference between hControl and BWS groups was indicated by p value. Unfilled bars = mean and standard deviation of hControl tongue samples. Filled bars = mean and standard deviation of BWS tongue samples.
Discussion
In this study, we performed small RNA-seq to investigate the miRNA expression profiles in various tissues of a congenital overgrowth syndrome observed in bovine and human, namely large offspring syndrome and Beckwith-Wiedemann syndrome, respectively. For the bovine LOS study, we tested skeletal muscle, kidney, liver, and tongue. Skeletal muscle was selected since in cattle and human muscle accounts for ~35% of the body weight [39,40] which could be the top contributor to the overgrowth phenotype. Wilms’ tumour of the kidney and hepatoblastoma are two of the most commonly observed tumours in BWS [41], thus kidney and liver were also included in the bovine LOS study. We also included tongue since macroglossia is defined as a cardinal clinical feature of BWS [14] and enlarged tongue was also observed in a LOS foetus. For the human BWS study, we only tested tongue as other tissues are not readily available from BWS patients.
Cases of multi-locus imprinting disturbance with IC2 hypomethylation have been reported, these also include hypomethylation of other maternal ICs controlling expression of genes such as GRB10 and SNPRN [14,42–44]. In this study, only tongue samples from patients with IC2 hypomethylation were used. These samples did not show altered DNA methylation at GRB10, SNPRN, and MEG3 regulatory DMRs, thus suggesting that these BWS patients do not have multi-locus methylation defects.
In the bovine sequencing data, over 86% of the annotated miRNAs (n = 1,025) from the most updated version of miRBase [45] were detected in at least one of the tissues analysed, and ~40% were abundantly expressed. Human (n = 2,654) and mouse (n = 1,978) are the two species contributing the largest number of annotated miRNAs. When considering that these species have a similar number of protein coding genes (22,686 for human, 22,396 for mouse, and 19,994 for bovine according to Ensembl Release 94) and about 80% of the genes are conserved among them, the number of bovine annotated miRNAs should be higher than the current number. Indeed, we detected 196 bovine putative miRNAs in our dataset, and ~74% of them (n = 146) are conserved in at least one of the vertebrate species annotated in miRBase. Experimental validation will be required to confirm the functionalities of these putative miRNAs which may increase the bovine miRNA database by ~20%. For human sequencing data, ~72% of the annotated human miRNAs were detected, and ~19% were abundantly expressed.
DE-miRNAs in bovine were found between the LOS group and the control group in each tissue analysed. For liver, we also analysed a group of ART-produced foetuses that were not LOS (based on weight) to determine what contribution of the DE-miRNAs observed could be ascribed to the method of conception. We found that only five putative miRNAs were differentially expressed when compared to controls. The function of these five putative miRNAs have not been studied, although bta-miR-temp34-3p and bta-miR-temp82-3p have sequence similarity to the uncharacterized platypus and human miRNAs oan-miR-1386 and hsa-miR-6765-3p, respectively. When considering the greater number (n = 29) of DE-miRNAs detected between the LOS group and the ART group in bovine liver, we hypothesize that there are other factors that together with the ART procedure trigger the development of LOS.
Bovine miRNA bta-miR-21-5p was found to be upregulated in liver of LOS #3 and #4. The conserved human miRNA hsa-miR-21-5p targets CDKN1C transcript in the coding region, and decreases both the mRNA and protein amount in prostate cancer cells [46]. The bovine CDKN1C transcript amount decreased in LOS #1 and #4 of liver although LOS #3 is not different [9]. Biallelic expression of KCNQ1OT1 was also observed in LOS #1 and #4 of liver which may also explain the downregulation of this transcript [9]. The complementary sequence of the seed region of bta-miR-21-5p was also found in the coding region of bovine CDKN1C transcript, but this targeting relationship is not yet confirmed for bovine. In addition, bta-miR-199a-5p was also upregulated in liver of LOS #3 and #4. The conserved human miRNA hsa-miR-199a-5p targets CDKN1C transcript in the 3ʹUTR region, and inhibition of hsa-miR-199a-5p causes reduced cell proliferation rate [47]. We found one predicted binding site for bta-miR-199a-5p in the 3ʹUTR region of the bovine CDKN1C transcript. Future work will determine the accuracy of this targeting prediction in bovine.
We found bta-miR-483 to be upregulated in muscle and downregulated in kidney of LOS foetuses. Bta-miR-483 is conserved with human hsa-miR-483-3p. Human miR-483 (3p and 5p) have been reported as oncomiRs (miRNAs associated with cancer) and is located within the second intron of the insulin like growth factor 2 (IGF2) gene, a location conserved in many mammals including mouse, rat, cow, dog, and gorilla according to our analysis of the various genome assemblies. Both hsa-miR-483-5p and hsa-miR-483-3p are functional mature miRNAs and the expression of hsa-miR-483-3p and IGF2 have been shown to be positively correlated in human [48]. Increased hsa-miR-483-3p is the main type of misregulation found in Wilm’s tumours, although there are a few Wilm’s tumour cases showing significant decrease of this miRNA [48]. Inhibition of hsa-miR-483-3p but not IGF2 contributes to in vivo suppression of tumorigenesis. hsa-miR-483-3p inhibits apoptosis through directly targeting BCL2 binding component 3 (BBC3/PUMA) which functions in p53 modulated apoptosis [48]. Consistent with the increased bta-miR-483 in LOS muscle, an increase of IGF2 transcript was also found in muscle from LOS foetuses [27]. Similar results were observed in BWS. Upregulation of hsa-miR-483-5p is correlated with decreased survival rate of patients with adrenocortical carcinomas [49], a cancer observed in BWS [50].
miRNAs within 50-kb regions show higher correlation of expression patterns, which indicates the length of 50 kb is a range for miRNA clusters [51]. In this study, we used 50-kb windows and identified many DE-miRNAs that are clustered in the bovine and human genomes. DE-miRNAs were detected from several miRNA clusters including miR-99a/let7c/125b, miR-17/92, and DLK1-DIO3 in both human and bovine.
The miR-99a/let7c/125b cluster is located on chromosome 1 in bovine and 21 in human. miRNAs from this cluster had tissue specific deregulation in liver, kidney, and muscle in LOS and were also misregulated in BWS tongue. In human, this cluster is co-transcribed with the host gene MIR99AHG/LINC00478 and is directly repressed by binding of androgen receptor to the host gene [52]. Human insulin like growth factor 1 receptor (IGF1R) transcript is a target of this cluster and androgen-induced cell proliferation requires the completeness of this pathway. IGF1R transcript was also found to be downregulated in kidney of LOS #3 and #4 [27].
DE-miRNAs were also detected from the miR-17/92 cluster. This cluster has two copies in human and bovine. In bovine this cluster is located on chromosomes 12 and X and in human is located on chromosomes 13 and X. From this cluster, bta-miR-19b and bta-miR-92a were misregulated in LOS liver, and hsa-miR-18a-3p, hsa-miR-92a-3p, and hsa-miR-20b-5p were misregulated in BWS tongue. This cluster is known for its oncogenic properties and has been reported in various tumour types including Wilm’s tumour and hepatoblastoma [53–57]. Expression of miR-17/92 cluster is activated by MYC/c-Myc and E2F transcription factors (E2F1, E2F2, and E2F3) are targets of miRNAs from this cluster [58,59]. In addition, a negative feedback loop allows E2F transcription factors to activate the expression of miR-17/92 cluster [59].
DLK1-DIO3 cluster is a conserved imprinted cluster of miRNAs in mammalian species [36]. According to the newest miRBase version, this cluster contains ~42 miRNA genes in humans (chromosome 14; each may have a 5p and a 3p arm) and ~50 miRNA genes in bovine (chromosome 21). In total, 17 DE-miRNAs were detected from DLK1-DIO3 cluster across all the tissues analysed in LOS, most of them downregulated. For BWS tongue, 45 DE-miRNAs were detected from this cluster and all of them are upregulated. miRNAs in this cluster are extensively expressed during embryo development but are confined to the brain in adult mice [36]. They have wide roles in various biological processes and their aberrant expression is often observed in various tumours [60,61]. Interestingly, the DE-miRNAs from this cluster are different between LOS muscle, liver, and tongue, which suggests tissue specific or primary germ layer specific mechanisms for miRNA misregulation.
The deregulation of miRNAs in LOS could be partially caused by aberrant DNA methylation of regulatory CpG sites. Although our ability to answer this question is limited by the lack of miRNA TSS data available for bovine, we could find five examples of potential regulation by DNA methylation based on conservation with human for the TSS’ surrounding sequence. For instance, the expression of miR-132 is silenced by hypermethylation of a CpG island located within 2 kb of miR-132 promoter in human pancreatic tumour cells [62]. Differential methylation at other CpG islands have also been reported to regulate the expression of this miRNA in human prostate cancer [63] and oral cancer [64]. In our study, we observed hypomethylation of a DMR located 9.4 kb upstream of bta-mir-132 gene (Figure 4), which may help explain the observed upregulation of bta-miR-132 detected in muscle of LOS foetuses.
In mouse, miRNAs in the DLK1-DIO3 cluster are maternally expressed and regulated by a differentially methylated region (DMR) ~200-kb upstream of the cluster [36]. In muscle of LOS foetuses, two hypermethylated DMRs were detected at ~230-kb and ~279-kb upstream regions of DLK1-DIO3 cluster (Figure 3). All the DE-miRNAs found in LOS muscle from this cluster are downregulated, which coincides with the hypermethylated state of the DMRs, suggesting a possible regulatory function of this locus on gene expression in bovine. Further, although miRNAs in this cluster are regulated by the same DMR in mice, most of them are not transcribed in a single pri-miRNA [36], which may help explain why two miRNAs are upregulated and 15 are downregulated in LOS.
Target prediction and enriched signalling pathway analyses identified the Hippo signalling pathway as misregulated in LOS and BWS of all the tissues analysed. Hippo pathway functions in control of organ size, and the main components in humans are kinases macrophage stimulating 1/2 (MST1/2) and large tumour suppressor kinase 1/2 (LATS1/2), cofactors Salvador family WW domain containing protein 1 (SAV1) and MOB kinase activator 1A (MOB1A/MOB1), and co-transcription factors tafazzin (TAZ) or YAP1 [65]. When Hippo pathway is not activated, YAP1/TAZ proteins remain in an unphosphorylated state and are transferred into nucleus. In the nucleus, YAP1/TAZ interact with SMAD family member 1/2/3/4 (SMAD1/2/3/4; signal transducers of the TGF-beta signalling pathway), or hepatocyte nuclear factor 4 alpha (HNF4A/TCF) and TEADs (transcriptional enhancer factors of the Wnt signalling pathway), to promote cell proliferation and inhibit apoptosis [65]. Extracellular signals such as high cell density activates the Hippo pathway [66], and a phosphorylation cascade occurs in which LATS1/2 is phosphorylated and activated by MST1/2 and phosphorylate YAP1/TAZ on 5 serine motifs. YAP1/TAZ will be retained in cytoplasm when serine 127 is phosphorylated and results in apoptosis and control of organ size. Since the 3'UTR region of YAP1 transcript XM_015474584.1 cannot be amplified by PCR in bovine muscle, most of the predicted targeting relationships between DE-miRNAs and YAP1 may not exist at this gestation stage. The higher levels of YAP1 transcript and active (non-phosphorylated) YAP1 protein in LOS #3 and #4 (the heaviest LOS foetuses) when compared to controls may not be directly regulated by miRNA misregulation, but still contribute to enhanced cell proliferation activities and loosely controlled organ size of muscle in LOS #3 and #4.
The upregulation of bta-miR-296-3p was detected in LOS muscle, kidney, and liver, and this upregulation was confirmed by qRT-PCR in muscle. Neurofibromin 2 (NF2) has been found to be a negative regulator of the human conserved miRNA hsa-miR-296-3p and through the Hippo signalling pathway suppress the invasiveness of brain tumour cells [67]. Unphosphorylated YAP1 promotes the expression of hsa-miR-296-3p, and NF2 induces phosphorylation on YAP1. Both YAP1 and hsa-miR-296-3p positively regulate the expression of cysteine rich angiogenic inducer 61 (CYR61/CCN1) which promotes cell proliferation and tumour growth [68,69]. Signal transducer and activator of transcription 5A (STAT5A) mRNA is a target of hsa-miR-296-3p [67]. Suppressed expression of STAT5A reduces suppressor of cytokine signalling 2 (SOCS2) and thereby increases phosphorylation level of STAT3 proteins and increases invasiveness of tumour cells. Decreased SOCS2 transcript amount was also found in muscle of LOS foetuses, but no change was observed for CYR61 and STAT5A on transcript level.
In summary, through profiling the miRNA expression in bovine tissues, the differences between LOS and controls were clearly illustrated. Similarly, the miRNA expression patterns were different between BWS and controls in human tongue. Shared DE-miRNAs were found between LOS and BWS studies, which indicates mechanistic similarities in these overgrowth conditions. The clustering tendency of detected DE-miRNAs suggests genome location-based mechanisms such as DNA methylation that affect the miRNAs expression in LOS and BWS. In addition, altered Hippo pathway signalling could be a contributor to the overgrowth phenotype. In conclusion, our data support our hypothesis that changes in miRNA expression profiles play important roles in development of LOS and BWS.
Materials and methods
Animals and tissue samples
Day ~105 Bos taurus indicus (B. t. indicus; Nelore breed) × Bos taurus taurus (B. t. taurus; Holstein breed) F1 hybrid control, ART-produced normal (similar weight as controls; referred to as ART) and LOS (body weight greater than 97th centile of controls) conceptuses were previously generated by our laboratory [9]. One B. t. indicus bull (ABS CSS MR N OB 425/1,677,344 29NE0001 97,155) sired all the foetuses. In brief, for the control group, B. t. taurus dam underwent estrus synchronization and were artificially inseminated. For the ART group, B. t. taurus cumulus-oocyte complexes were matured and fertilized in vitro, and the embryos were cultured in vitro. Estrus cow serum (10%) was used to supplement the culture medium on day 5 after insemination. On day 7, blastocysts were transferred into synchronized B. t. taurus recipients. On day ~105, conceptuses were collected by caesarean section to maintain nucleic acid integrity. Tissues were well diced, mixed, and snap frozen in liquid nitrogen immediately and stored at −80°C. The sex, weight, and identifier of the experimental samples were as follows: (1) female control foetuses: 392g (#1), 404g (#2), 408g (#3), 416g (#4), and 360g (#5), (2) male control foetuses: 380g (#6), 400g (#7), 410g (#8), and 496g (#9), (3) female LOS foetuses: 514g (LOS#1), 518g (LOS#2), 620g (LOS#3), and 714g (LOS#4), (4) male LOS foetuses: 478g (LOS#5), 482g (LOS#6), and 584g (LOS#7), and (5) female ART foetuses: 360g (ART#1), 376g (ART#2), 390g (ART#3), and 466g (ART#4).
De-identified human control (referred to as hControl) tongue samples were purchased from the Eurofin Pre-Transplant Testing Laboratory in Centennial Colorado. De-identified human BWS (referred to as BWS) tongue samples were from the Beckwith–Wiedemann Syndrome Registry and Biorepository at the Children’s Hospital of Philadelphia collected under IRB 13–010658. Tongue samples of two hControl adult cadavers (female: hControl #2 and male: hControl #3) and 12 BWS children with IC2 hypomethylation (female: BWS #3, #4, #6, #11 and male: BWS #1, #2, #5, #7, #8, #9, #10, #12) were used in this study.
RNA isolation
Total RNA from bovine skeletal muscle (referred to as muscle), kidney, liver, and tongue was isolated using TRIzol™ Reagent (Invitrogen, 15596026) following the manufacturer’s instructions. RNA samples were stored at −80°C. The concentration and integrity of RNA samples were measured by using a NanoDrop® ND-1000 Spectrophotometer (Thermo Fisher Scientific) and a Fragment Analyzer™ (Advanced Analytical Technologies, Inc.), respectively. Bovine RNA samples with RNA Quality Number greater than 7.0 were used for small RNA-sequencing.
Total RNA from human samples was isolated by homogenization in TRIzol™ Reagent based on manufacturer’s protocol and using a VWR 25D Bench Top Homogenizer.
Library preparation and small RNA-seq
Small RNA library preparation and Illumina Sequencing were conducted at the University of Missouri DNA Core Facility. TruSeq® Small RNA Library Prep Kit (Illumina, Inc.) was used for library preparation following the manufacturer’s instructions. Briefly, 3ʹ and 5ʹ RNA adapters were ligated to RNAs by T4 RNA ligase for each RNA sample separately. cDNA constructs were created by reverse transcription from the adapter ligated RNAs, and the libraries were amplified by polymerase chain reactions (PCR) to make sufficient templates for sequencing. Unique indexes were used in each library during PCR amplification. The PCR amplified libraries were pooled in equal volumes and size selection of 140–160 bp (enriched in 15–35 bp small RNA sequences) was performed by polyacrylamide gel electrophoresis. The libraries were purified and concentrated by ethanol precipitation. Finally, the quantity and quality of the libraries was confirmed by Fragment Analyzer.
The pooled libraries were sequenced using Illumina NextSeq 500 platform (Illumina, Inc.). Bovine muscle and kidney libraries of control #1, #2, #3, #4 and LOS #1, #2, #3, #4 were sequenced initially with 32 samples per lane. Bovine liver libraries of control #1, #2, #3, #4, #5, LOS #1, #2, #3, #4 and ART #1, #2, #3, #4 were sequenced in a second round with 23 samples per lane. Bovine and human tongue libraries were sequenced in a third round with 16 samples per lane each. Approximately 400 million single-end reads of 75 bases were obtained from each lane.
Processing of small RNA-seq data
Adapter sequence (TGGAATTCTCGGGTGCCAAGG) was removed from the raw sequencing reads by using cutadapt v1.18 [70]. Reads with low quality bases (Phred score < 20) were trimmed using the dynamictrim function of SolexaQA++ v3.1.7.1 [71]. The Burrows-Wheeler Aligner trimming algorithm was applied instead of the default algorithm of dynamictrim. Reads with lengths shorter than 17 bases were discarded after adapter and quality trimming, and the remaining reads were aligned to the bovine genome (UMD3.1.1) for bovine libraries and to the human genome (GRCh38) for human libraries using miRDeep2 v0.0.8 (Bowtie algorithm [33]). A configuration file was used to allow all the sequencing files to be processed together. Reads aligned to known bovine or human miRNAs from miRBase (release 22 [45]) were quantified using the default settings of miRDeep2 in which only one mismatch was allowed within the read. In addition, only reads that fit the specified default setting region, namely the length of the miRNA (≥17-≤25 nt) and 7 nucleotide flanking region (i.e. ≤2 nt upstream and ≤5 nt downstream of the miRNA) were counted. Reads mapped to miRNAs with multiple precursors were only counted once for each read per miRNA.
Putative bovine miRNAs were predicted using the default settings of miRDeep2 and using human and mouse known miRNAs (miRBase release 22) as reference sequences. miRDeep2 score and randfold p value (significance indicating high tendency to form a secondary stem-loop structure due to low folding free energy change) were the two parameters given by miRDeep2 to evaluate the likelihood of a miRNA to be true. Since not all the known miRNAs have high miRDeep2 score or significant randfold p value, no cut-off on miRDeep2 score or randfold p-value was set for putative miRNA prediction. For example bta-miR-532 which has a low miRDeep2 score and does not have a significant randfold p value is abundant in three of the tissues analysed, is conserved in humans, and it has been shown to be a functional miRNA [72]. Three types of putative miRNAs were predicted: (1) unknown 3p or 5p counterparts of known bovine miRNAs (the two strands of the miRNA duplex are now known as ‘5p’ or ‘3p’ to differentiate the specific arms they locate on), (2) putative bovine miRNAs which are conserved in at least one vertebrate species of all miRBase represented species (n = 74), and (3) putative miRNAs without conservation in other species. Only one name was kept for putative mature miRNAs that had multiple pre-miRNAs. Temporary names were given by us to these putative miRNAs and official names will be given by miRBase if they are accepted. Reads mapped to putative miRNAs were quantified with the same rules as for known miRNAs.
Differentially expressed miRNAs (DE-miRNAs) analyses
For bovine, known and putative miRNAs with greater than five counts per million (cpm) in all the control or all the ART or at least two LOS libraries for each tissue were considered as abundant miRNAs and were kept for differential expression analyses. For human tongue, known miRNAs with greater than five cpm in all the hControl or at least two BWS libraries were considered as abundant miRNAs. DE-miRNAs were determined using R environment [73]. Raw read counts of abundant miRNAs were normalized by using RUVseq v1.14.0 [74]. Briefly, upper-quartile normalization and RUVs function (with user-specified k value) were used to estimate the factors of unwanted variation. A small k value (1 or 2) is recommended for small sample sizes, and in this study, we used k = 1-3 for comparisons. Two packages, namely edgeR v3.22.5 [75] and DESeq2 v1.20.0 [76], were used for statistical analyses. We adopted both packages since the order of their performance may change on different datasets [77,78]. The trimmed mean of M values method was also used with the edgeR method. Specifically, false discovery rate (FDR) was controlled at 0.05 for both edgeR (labelled as FDR) and DESeq2 (labelled as Padj) by the Benjamini-Hochberg method.
For bovine samples, two strategies of grouping were applied for statistical analyses to compare the miRNA expression of muscle, kidney, liver, and tongue between the control group and LOS group. For the first method (i.e. group comparison), miRNA abundance was compared within tissues between LOS and the control group while for method two, a combination of LOS group was made and each of the two-LOS combinations was compared with the entire control group for each tissue. Furthermore, in order to determine how much miRNA misregulation is due to method of conception three comparisons were done, (1) control vs. ART, (2) ART vs. LOS, and (3) control vs. combined ART+LOS. We used liver for these comparisons. It should be noted that all the statistical comparisons were performed within the same sequencing round, since we observed variations caused by batch effects between the two sequencing rounds (data not shown).
For human samples, only group comparisons (BWS vs hControl) were made.
Principal component analyses (PCA) and hierarchical cluster analyses of the normalized read counts were performed using the plotPCA function of DESeq2 and hclust function of R stats package, respectively. Classic and Edwards’ Venn diagrams were generated using jvenn [79] to show the numbers of DE-miRNAs that are shared by the six two-LOS combinations or are unique to one combination for control vs. LOS studies in bovine.
Corroboration of DE-miRNA with a different pipeline
For a complete explanation of the method used, please refer to Supplementary methods. Briefly, FastQC v0.11.0 [80] was used to perform primary quality control of the small RNA-Seq data. Raw data was trimmed for quality and adapter sequences using Trimmomatic v0.35 [81]. Small RNA annotation was performed using the Unitas pipeline [82] to remove non-miRNA sequences. mirPRo [83] and miRDeep2 [33] were used to quantify known miRNAs and predict putative miRNAs. Reads were collapsed into a set of unique reads and were aligned to Ensembl UMD3.1 bovine genome [84] and miRNAs sequences (miRBase, release 22) using both methods, individually. Raw read counts of known miRNAs were used for differential expression analysis. RUVg module (k = 2) of RUVSeq [74] along with DESeq2 R package [76] were used to identify differentially expressed miRNAs between control and LOS samples in muscle. By this approach, two lists of differentially expressed miRNAs (Padj < 0.05) were obtained for mirPRo and miRDeep2 methods.
Genomic locations of miRNAs
All the genome locations of miRNAs presented in this study were acquired from bovine reference genome UMD3.1.1 for bovine miRNAs and from human genome GRCh38 for human miRNAs.
Statistical method for detecting differentially methylated regions (DMRs)
LOS – The muscle whole genome bisulphite sequencing data from our previous publication [27] was re-analysed using a newly developed method namely hmmDMR [85]. More specifically, the developed hidden Markov model assumes three hidden states at each CpG site: (1) the normal state where the LOS group and the control group have the same level of methylation rate; (2) the hyper-state where the LOS group has an elevated methylation rate compared to the control group; and (3) the hypo-state where the LOS group has a lower methylation rate compared to the control group. The hmmDMR method is applied to one chromosome at a time to allow flexibility between chromosomes. Then the hmmDMR method finds the best possible sequence of methylation states across an entire chromosome by maximizing the likelihood of all observations. Upon model fitting, we report DMRs with a minimum length of 500 bps that contain at least 10 CpGs with no two adjacent CpGs being more than 300 bps apart. The hmmDMR software is freely available for download on GitHub (https://github.com/Tieming/hmmDMR-package).
BWS – DNA methylation pyrosequencing results were analysed by Mann-Whitney U Test using Prism software.
Association of DE-miRNAs with differentially methylated regions
The regions containing transcription start sites (TSS) of bovine miRNAs that are conserved in human were predicted based on two human miRNA databases, namely mirTrans [35] and miRStart [34]. One or more TSS regions for every human pre-miRNAs were defined as adjacent TSS (within 500 bp) plus 500 bp in both up- and downstream directions. For each muscle DE-miRNA that is conserved in human, the sequence of the human TSS regions was acquired from the corresponding genome (mirTrans using GRCh38 and miRStart using GRCh37) and aligned to the bovine genome UMD3.1.1 using BLAT [86] with parameters replicating web-based results (-stepSize = 5 -repMatch = 2253 -minScore = 0 -minIdentity = 0). The top hit for each human TSS region was kept as predicted bovine pre-miRNA TSS only if (1) the sequence was on the same chromosome as the bovine pre-miRNA; (2) the distance between bovine pre-miRNA and the sequence was similar as in human; (3) the percentage of mapped length is greater than 20% (at least 200 bp); and (4) the mapped region should contain the TSS. Then DMRs were searched within 10 kb upstream of each predicted bovine TSS region of DE-miRNAs.
Prediction of mRNA targets for DE-miRNAs and enriched signalling pathway analyses
For bovine control vs. LOS studies, mRNA targets of DE-miRNAs were predicted and enriched signalling pathways were analysed for each tissue. The sequences of 3' untranslated regions (3'UTRs) of all the bovine protein-coding genes were acquired based on annotations from NCBI Bos taurus Annotation Release 105 [87] and Ensembl Release 92 [84] for the reference genome UMD3.1.1. miRanda v3.3a [88] was used to predict mRNA targets of DE-miRNAs using the 3'UTR as reference sequences with score cut-off = 150 and default free energy change settings under strict mode (perfect complementarity within the seed region). DAVID v6.8 [89] was used to determine functionally related signalling pathways enriched in genes predicted as targets of DE-miRNAs. Predicted pathways with corrected p-value ≤ 0.05 (Benjamini-Hochberg method used by DAVID) were considered significant.
Published human data was used to corroborate target and pathway predictions in bovine muscle, kidney, and liver, since experimentally-examined miRNA-mRNA targeting relationships are much more abundant in human than in bovine, and bovine miRNAs are largely conserved in human. Human miRNAs which have conserved sequences among bovine DE-miRNAs (referred to as human conserved DE-miRNAs) were acquired from miRBase. mirPath v.3 [90] was used to analyse enriched signalling pathways using TarBase algorithm based on human conserved DE-miRNAs and TarBase v.8 (human library) [91]. Predicted pathways with corrected p value ≤ 0.05 (Benjamini-Hochberg method used by mirPath) were considered significant.
For human BWS vs. hControl tongue DE-miRNA analysis, enriched signalling pathways were predicted using mirPath v.3 with the same setting mentioned above.
Quantitative reverse transcription PCR (qRT-PCR) validation of miRNA expressions
To validate the miRNA expressions detected by small RNA-seq, qRT-PCR assays were conducted on a QuantStudio™ 3 platform (Thermo Fisher Scientific). cDNAs were synthesized from equal amounts of total RNA of bovine muscle and tongue by using TaqMan® Advanced miRNA cDNA Synthesis Kit (Applied Biosystems, A28007) following the manufacturer’s instructions and stored at −20°C until use. TaqMan® Advanced miRNA Assays (Applied Biosystems, A25576) were used and qRT-PCR was performed in a 20 ul reaction. The programme used for qRT-PCR starts with 20 s enzyme activation at 95°C, followed by 40 cycles of 1 s denaturing at 95°C and 20 s annealing/extension at 60°C according to the manufacturer’s instructions. Four miRNAs (i.e. bta-miR-181a, bta-miR-20a, bta-miR-30b-5p, and bta-miR-378), which were identified as being highly expressed and not different between treatment (FDR/padj ≥ 0.7 and log2(cpm) ≥ 5 and log2(fold change) ≤ 0.1), were employed as endogenous controls for qRT-PCR assays. Assays used were: bta-miR-181a (hsa-miR-181a-5p, 477857_mir), bta-miR-20a (hsa-miR-20a-5p, 478586_mir), bta-miR-30b-5p (hsa-miR-30b-5p, 478007_mir), bta-miR-378 (hsa-miR-378a-3p, 478349_mir), bta-miR-299 (hsa-miR-299-5p, 478793_mir), bta-miR-409a (hsa-miR-409-5p, 478872_mir), bta-miR-143 (hsa-miR-143-3p, 477912_mir), bta-miR-145 (hsa-miR-145-5p, 477916_mir), bta-miR-450b (hsa-miR-450b-5p, 478914_mir), and bta-miR-296-3p (custom design, btamiR296p_1_498).
Determination of gene expression in human samples by qRT-PCR
qRT-PCR assays were conducted on a 7900HT Fast Real-Time PCR System (Applied Biosystems) with Power SYBR™ Green PCR Master Mix (Applied Biosystems, 4367659) following the manufacturer’s instructions. The programme used for qRT-PCR starts with 2 min incubation at 50°C, 10 min enzyme activation at 95°C, followed by 40 cycles of 15 s denaturing at 95°C and 1 min annealing/extension at 60°C, and final stages for dissociation curve of 15 s incubation at 95°C, 15 s incubation at 60°C, and 15 s incubation at 95°C. Primer sequences are: GAPDH (forward 5ʹ-TGTCATCAATGGAAATCCCATCACC-3ʹ and reverse 5ʹ-CATGAGTCCTTCCACGATACCAAA-3ʹ [92]), ACTB (forward 5ʹ-CTAAGGCCAACCGTGAAAAG-3ʹ and reverse 5ʹACCAGAGGCATACAGGGACA-3ʹ [93]), ATP5PB (forward 5ʹ-TCGAGTCGCGTCCACC-3ʹ and reverse 5ʹ-GGGAGCATCGTCGCCC-3ʹ [94]), and IGF2 (forward 5ʹ- CCGTGCTTCCGGACAACTT-3ʹ and reverse 5ʹ-CTGCTTCCAGGTGTCATATTGG-3ʹ, designed in BLAST). Results were analysed by Mann-Whitney U Test using Prism software.
Protein extraction and Western blot
Proteins were extracted using NP-40 buffer (0.15 M NaCl, 1% (v/v) Triton X-100, and 0.05 M Tris). Alternatively, PhosphoSafe™ Extraction Reagent (Novagen, 712964) was used for extracting phosphorylated proteins following the manufacturer’s instructions. Protein samples were stored at −20°C after extraction. Protein concentrations were measured using the Pierce™ BCA Protein Assay Kit (Thermo Scientific, 23225) following the manufacturer’s instructions.
Proteins were resolved by molecular weight and charge using sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE; running buffer = 0.025 M Tris, 0.192 M Glycine, and 0.1% (w/v) SDS). After electrophoresis, the proteins were transferred to NitroPure™ Supported Nitrocellulose membranes (0.22 microns; GVS Life Sciences, WP2HY00010) with transfer buffer (0.025 M Tris, 0.192 M Glycine, and 20% (v/v) methanol). Then, membranes were blocked for four hours at room temperature with blocking buffer. The blocking buffer for the portion of the membrane containing the samples was TBST (0.02 M Tris, 0.137 M NaCl, and 0.1% (v/v) Tween 20) with the addition of 5% (v/v) donkey serum (SIGMA, D9663) and 10% (w/v) Amicase (SIGMA, A2427). The blocking buffer for the portion of the membrane containing the ladder was TBST with the addition of 10% (w/v) Amicase. The membranes were then incubated with primary antibodies in blocking buffers at 4°C overnight with rocking. On day two, the membranes were incubated with secondary antibodies in blocking buffers at room temperature for one hour with rocking. SuperSignal™ West Pico PLUS Chemiluminescent Substrate (Thermo Scientific, #34577) was used for luminescent signal development following the manufacturer’s instructions. ChemiDoc™ Touch Imaging System (Bio-Rad) was used for exposure of the membranes. Antibodies used were: GAPDH (rabbit anti-human; Cell Signaling #2118; as endogenous control), active YAP1 (rabbit anti-human; Abcam ab205270), Phospho-YAP1 (rabbit anti-human; Thermo Fisher PA5-17481), and secondary antibody (donkey anti-rabbit IgG; Jackson ImmunoResearch #76763). The protein sequence of epitopes of active YAP1 and Phospho-YAP1 antibodies were confirmed to be conserved in bovine by BLAST.
RNA-seq data from our previous studies (NCBI Gene Expression Omnibus (GEO) accession numbers GSE63509 [26]) were used to query transcript levels.
Genomic DNA isolation
Tissues were lysed in lysis buffer (0.05 M Tris-HCl (pH 8.0), 0.1 M EDTA, and 0.5% (w/v) SDS) with proteinase K (Fisher BioReagents, BP1700) at 55°C overnight. On day two, Phenol:Chloroform:Isoamyl Alcohol (SIGMA, P3803) was used to isolate genomic DNA following the manufacturer’s instructions. Genomic DNA samples were stored at −20°C. The DNA concentration was measured by using a NanoDrop® ND-1000 Spectrophotometer (Thermo Fisher Scientific) and DNA integrity was confirmed by electrophoresis on a 0.7% agarose gel.
Reverse transcription of mRNAs
Total RNA samples (isolated by Trizol) were treated with RQ1 RNase-Free DNase (Promega, M6101) following the manufacturer’s instructions to ensure no DNA contamination of the RNA. cDNA was synthesized from total RNA using SuperScript® IV Reverse Transcriptase (Invitrogen, 18090010) with random hexamers (Promega, C1181) following the manufacturer’s instructions. cDNA samples were stored at −20°C.
Polymerase chain reaction (PCR) verification of YAP1 transcripts
GoTaq® Flexi DNA Polymerase (Promega, M8295) was used for PCR following the manufacturer’s instructions.
Two sets of intron-spanning primers (set1 and set2) were designed for exons 6 to 7 of YAP1 transcript XM_015474589.1. The sequences of set1 are 5ʹ-GATGTCGGAGCTCAAGGCTTCT-3ʹ (forward) and 5ʹ-CGGACTCAGCATGAGCAGCTA-3ʹ (reverse). The sequences of set2 are 5ʹ-CGTTGAGGATGTCGGAGCTCAA-3ʹ (forward) and 5ʹ-CACAGACAGCGGACTCAGCAT-3ʹ (reverse). Touchdown PCR was applied and the thermal conditions for cDNA template are: initial denaturation at 95°C for two minutes; denaturation for each cycle at 95°C for 30 s; annealing starts at 70°C, and decreases to 60°C by 2°C per cycle, and maintains 30 cycles at 60°C for 30 s; extension for each cycle at 72°C for 20 s; final extension at 72°C for five minutes. The PCR products were visualized on a 1% agarose gel with 100 bp DNA Ladder (New England Biolabs, N3231S). Bands between 200 bp and 300 bp were cut and DNA was retrieved from the gel using Wizard® SV Gel and PCR Clean-Up System (Promega, A9282). Sanger sequencing for the retrieved DNA was performed at the University of Missouri DNA Core Facility.
Two additional sets of intron-spanning primers (set3 and set4) were designed for exons 7–8 of YAP1 transcript XM_015474584.1. The sequences of set3 are 5ʹ-ACTGTTAAGATAGGCACACCCACAA-3ʹ (forward) and 5ʹ-CGGACTCAGCATGAGCAGCTA-3ʹ (reverse). The sequences of set4 are 5ʹ-AACTGATCCGAGCATTCACTTCTCA-3ʹ (forward) and 5ʹ-TCTCGCGACGAGAGCACA-3ʹ (reverse). The thermal conditions for genomic DNA and cDNA template are: initial denaturation at 95°C for two minutes; denaturation for each cycle at 95°C for one minute; annealing starts at 70°C, and decreases to 60°C by 2°C per cycle, and maintains 30 cycles at 60°C for one minute; extension for each cycle at 72°C for five minutes and 20 s; final extension at 72°C for five minutes. The PCR products were visualized on a 0.5% agarose gel with Quick-Load® 1 kb Extend DNA Ladder (New England Biolabs, N3239S).
Pyrosequencing
Human control and BWS DNA samples were bisulfite converted using the EpiTect® Fast DNA Bisulfite Kit (QIAGEN, 59824). PCR and sequencing primers were designed or verified using the PyroMark Q48 Autoprep software (QIAGEN, sequences listed in Supplementary Table S12). Bisulfite-converted DNA was amplified by PCR using the PyroMark PCR kit (QIAGEN, 978705) and pyrosequencing was performed on the PyroMark Q48 Autoprep (QIAGEN) using PyroMark Q48 Advanced Reagents (QIAGEN, 974022) according to the manufacturer’s instructions. Data were analysed using the Pyro Q-CpG software (QIAGEN).
Small RNA sequencing data availability statement
The small RNA-seq reads from bovine libraries are available in the GEO database with accession numbers (GSE117015). The small RNA-seq reads from human libraries are available in the dbGaP database with accession numbers (phs001794.v1.p1). The sequences of the bovine putative miRNAs identified in this study have been submitted to miRBase and official names for these putative miRNAs will be assigned by miRBase.
Ethics approval
Human control tongue samples were purchased from the Eurofin Pre-Transplant Testing Laboratory in Centennial Colorado. Human BWS tongue samples were from the Beckwith–Wiedemann Syndrome Registry and Biorepository at the Children’s Hospital of Philadelphia collected under IRB 13–010658.
Funding Statement
This work was supported by the National Institutes of Health [5R21HD062920; K08 CA193915], the National Science Foundation [1615789], Agriculture and Food Research Initiative Competitive Grant No. [2017-08953] from the USDA National Institute of Food and Agriculture, University of Missouri Research Board [RB 16-07 HAGEN], the Food for the 21st Century Program at the University of Missouri – Animal Reproduction [RMR], St Baldrick's Scholar Award [JMK], Alex's Lemonade Stand Foundation [JMK], Margaret Q. Landenberger Foundation [JMK], University of Pennsylvania Orphan Disease Center Beckwith-Wiedemann Syndrome Program of Excellence Pilot Grant [JMK].
Acknowledgments
The authors would like to thank Zhiyuan Chen, Linda Rowland, Olivia Styron, and Bhaumik Patel for their technical help and Anna Goldkamp for critically reviewing the manuscript. The authors would also thank the University of Missouri DNA Core Facility, especially Dr Nathan Bivens, Mingyi Zhou, and Karen Bromet for library preparation and their work on small RNA-sequencing.
Disclosure statement
No potential conflict of interest was reported by the authors.
Supplementary Material
Supplementary data for this article can be accessed here.
References
- [1].Perry G. 2016 statistics of embryo collection and transfer in domestic farm animals. Unpublished. 2016. DOI: 10.13140/RG.2.2.24793.42087 [Google Scholar]
- [2].Calhaz-Jorge C, de Geyter C, Kupka M, et al. Assisted reproductive technology in Europe, 2012: results generated from European registers by ESHRE. Hum Reprod. 2016;31(8):1638–1652. [DOI] [PubMed] [Google Scholar]
- [3].Centers for Disease Control and Prevention 2015 assisted reproductive technology national summary report. Atlanta (GA): US Dept of Health and Human Services; 2017. [Google Scholar]
- [4].Lazzari G, Wrenzycki C, Herrmann D, et al. Cellular and molecular deviations in bovine in vitro-produced embryos are related to the large offspring syndrome. Biol Reprod. 2002. September;67(3):767–775. PubMed PMID: 12193383; Eng. [DOI] [PubMed] [Google Scholar]
- [5].Behboodi E, Anderson GB, BonDurant RH, et al. Birth of large calves that developed from in vitro-derived bovine embryos. Theriogenology. 1995. July 15;44(2):227–232. PubMed PMID: 16727722; Eng. [DOI] [PubMed] [Google Scholar]
- [6].Hasler J, Henderson W, Hurtgen P, et al. Production, freezing and transfer of bovine IVF embryos and subsequent calving results. Theriogenology. 1995;43(1):141–152. [Google Scholar]
- [7].van Wagtendonk-de Leeuw AM, Mullaart E, de Roos AP, et al. Effects of different reproduction techniques: AI MOET or IVP, on health and welfare of bovine offspring. Theriogenology. 2000. January 15;53(2):575–597. PubMed PMID: 10735051; Eng. [DOI] [PubMed] [Google Scholar]
- [8].Farin PW, Farin CE.. Transfer of bovine embryos produced in vivo or in vitro: survival and fetal development. Biol Reprod. 1995;52(3):676–682. [DOI] [PubMed] [Google Scholar]
- [9].Chen Z, Robbins KM, Wells KD, et al. Large offspring syndrome: a bovine model for the human loss-of-imprinting overgrowth syndrome Beckwith-Wiedemann. Epigenetics. 2013. June;8(6):591–601. PubMed PMID: 23751783; PubMed Central PMCID: PMCPMC3857339. Eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Hansen M, Kurinczuk JJ, Milne E, et al. Assisted reproductive technology and birth defects: a systematic review and meta-analysis. Hum Reprod Update. 2013;19(4):330–353. [DOI] [PubMed] [Google Scholar]
- [11].Young LE, Sinclair KD, Wilmut I.. Large offspring syndrome in cattle and sheep. Rev Reprod. 1998. September;3(3):155–163. PubMed PMID: 9829550. [DOI] [PubMed] [Google Scholar]
- [12].Farin PW, Piedrahita JA, Farin CE. Errors in development of fetuses and placentas from in vitro-produced bovine embryos. Theriogenology. 2006. January 7;65(1):178–191. PubMed PMID: 16266745; Eng. [DOI] [PubMed] [Google Scholar]
- [13].Filippi G, Mckusick VA. The Beckwith-Wiedemann syndrome: the Exomphalos-Macroglossia-Gigantism syndrome: report of two cases and review of the literature. Medicine (Baltimore). 1970;49(4):279–298. [DOI] [PubMed] [Google Scholar]
- [14].Brioude F, Kalish JM, Mussa A, et al. Expert consensus document: clinical and molecular diagnosis, screening and management of Beckwith–Wiedemann syndrome: an international consensus statement. Nat Rev Endocrinol. 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Farin P, Crosier A, Farin C. Influence of in vitro systems on embryo survival and fetal development in cattle. Theriogenology. 2001;55(1):151–170. [DOI] [PubMed] [Google Scholar]
- [16].Style CC, Cruz SM, Lau PE, et al. Surgical outcomes of patients with Beckwith-Wiedemann syndrome. J Pediatr Surg. 2018;53:1042–1045. [DOI] [PubMed] [Google Scholar]
- [17].Clericuzio CL. Recognition and management of childhood cancer syndromes: a systems approach. Am J Med Genet Part A. 1999;89(2):81–90. [DOI] [PubMed] [Google Scholar]
- [18].DeBaun MR, Siegel MJ, Choyke PL. Nephromegaly in infancy and early childhood: a risk factor for Wilms tumor in Beckwith-Wiedemann syndrome. J Pediatr. 1998. March;132(3 Pt 1):401–404. PubMed PMID: 9544890; Eng. [DOI] [PubMed] [Google Scholar]
- [19].Lee MP, DeBaun MR, Mitsuya K, et al. Loss of imprinting of a paternally expressed transcript, with antisense orientation to KVLQT1, occurs frequently in Beckwith–Wiedemann syndrome and is independent of insulin-like growth factor II imprinting. Proc Nat Acad Sci. 1999;96(9):5203–5208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Cooper WN, Luharia A, Evans GA, et al. Molecular subtypes and phenotypic expression of Beckwith–wiedemann syndrome. Eur J Hum Genet. 2005;13(9):1025. [DOI] [PubMed] [Google Scholar]
- [21].Mussa A, Peruzzi L, Chiesa N, et al. Nephrological findings and genotype–phenotype correlation in Beckwith–Wiedemann syndrome. Pediatr Nephrol. 2012;27(3):397–406. [DOI] [PubMed] [Google Scholar]
- [22].Brioude F, Lacoste A, Netchine I, et al. Beckwith-Wiedemann syndrome: growth pattern and tumor risk according to molecular mechanism, and guidelines for tumor surveillance. Hormone Res Paediatrics. 2013;80(6):457–465. [DOI] [PubMed] [Google Scholar]
- [23].Ibrahim A, Kirby G, Hardy C, et al. Methylation analysis and diagnostics of Beckwith-Wiedemann syndrome in 1,000 subjects. Clin Epigenetics. 2014;6(1):11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Mussa A, Russo S, De Crescenzo A, et al. (Epi) genotype–phenotype correlations in Beckwith–wiedemann syndrome. Eur J Hum Genet. 2016;24(2):183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Chen Z, Hagen DE, Elsik CG, et al. Characterization of global loss of imprinting in fetal overgrowth syndrome induced by assisted reproduction. Proc Natl Acad Sci U S A. 2015. April 14;112(15):4618–4623. PubMed PMID: 25825726; PubMed Central PMCID: PMCPMC4403167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Chen Z, Hagen DE, Wang J, et al. Global assessment of imprinted gene expression in the bovine conceptus by next generation sequencing. Epigenetics. 2016. July 2;11(7):501–516. PubMed PMID: 27245094; PubMed Central PMCID: PMCPMC4939914. Eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Chen Z, Hagen DE, Ji T, et al. Global misregulation of genes largely uncoupled to DNA methylome epimutations characterizes a congenital overgrowth syndrome. Sci Rep. 2017;7(1):12667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Bartel DP. MicroRNAs: target recognition and regulatory functions. Cell. 2009. January 23;136(2):215–233. PubMed PMID: 19167326; PubMed Central PMCID: PMCPMC3794896. Eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Hendrickson DG, Hogan DJ, McCullough HL, et al. Concordant regulation of translation and mRNA abundance for hundreds of targets of a human microRNA. PLoS Biol. 2009;7(11):e1000238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Hutvagner G, Zamore PD. A microRNA in a multiple-turnover RNAi enzyme complex. Science (New York, NY). 2002. September 20;297(5589):2056–2060. PubMed PMID: 12154197; Eng. [DOI] [PubMed] [Google Scholar]
- [31].Behm-Ansmant I, Rehwinkel J, Izaurralde E, editors.. MicroRNAs silence gene expression by repressing protein expression and/or by promoting mRNA decay. Cold Spring Harbor symposia on quantitative biology. Long Island (NY): Cold Spring Harbor Laboratory Press; 2006. [DOI] [PubMed] [Google Scholar]
- [32].Liu J, Valencia-Sanchez MA, Hannon GJ, et al. MicroRNA-dependent localization of targeted mRNAs to mammalian P-bodies. Nat Cell Biol. 2005;7(7):719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Friedlander MR, Mackowiak SD, Li N, et al. miRDeep2 accurately identifies known and hundreds of novel microRNA genes in seven animal clades. Nucleic Acids Res. 2012. January;40(1):37–52. PubMed PMID: 21911355; PubMed Central PMCID: PMCPMC3245920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Chien C-H, Sun Y-M, Chang W-C, et al. Identifying transcriptional start sites of human microRNAs based on high-throughput sequencing data. Nucleic Acids Res. 2011;39(21):9345–9356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Hua X, Tang R, Xu X, et al. mirTrans: a resource of transcriptional regulation on microRNAs for human cell lines. Nucleic Acids Res. 2017;46(D1):D168–D174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Seitz H, Royo H, Bortolin M-L, et al. A large imprinted microRNA gene cluster at the mouse Dlk1-Gtl2 domain. Genome Res. 2004;14(9):1741–1748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Peltier HJ, Latham GJ. Normalization of microRNA expression levels in quantitative RT-PCR assays: identification of suitable reference RNA targets in normal and cancerous human solid tissues. Rna. 2008;14(5):844–852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Vandesompele J, De Preter K, Pattyn F, et al. Accurate normalization of real-time quantitative RT-PCR data by geometric averaging of multiple internal control genes. Genome Biol. 2002;3(7):research0034.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Green RE, Taggart MA, Das D, et al. Collapse of Asian vulture populations: risk of mortality from residues of the veterinary drug diclofenac in carcasses of treated cattle. J Appl Ecol. 2006;43(5):949–956. [Google Scholar]
- [40].Janssen I, Heymsfield SB, Wang Z, et al. Skeletal muscle mass and distribution in 468 men and women aged 18–88 yr. J Appl Physiol. 2000;89(1):81–88. [DOI] [PubMed] [Google Scholar]
- [41].DeBaun MR, Tucker MA. Risk of cancer during the first four years of life in children from The Beckwith-Wiedemann Syndrome Registry. J Pediatr. 1998. March;132(3 Pt 1):398–400. PubMed PMID: 9544889; Eng. [DOI] [PubMed] [Google Scholar]
- [42].Bliek J, Verde G, Callaway J, et al. Hypomethylation at multiple maternally methylated imprinted regions including PLAGL1 and GNAS loci in Beckwith–Wiedemann syndrome. Eur J Hum Genet. 2009;17(5):611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Rossignol S, Steunou V, Chalas C, et al. The epigenetic imprinting defect of patients with Beckwith—wiedemann syndrome born after assisted reproductive technology is not restricted to the 11p15 region. J Med Genet. 2006;43(12):902–907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Arima T, Kamikihara T, Hayashida T, et al. ZAC, LIT1 (KCNQ1OT1) and p57 KIP2 (CDKN1C) are in an imprinted gene network that may play a role in Beckwith–Wiedemann syndrome. Nucleic Acids Res. 2005;33(8):2650–2660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Kozomara A, Griffiths-Jones S. miRBase: annotating high confidence microRNAs using deep sequencing data. Nucleic Acids Res. 2013;42(D1):D68–D73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Mishra S, Lin C-L, Huang TH, et al. MicroRNA-21 inhibits p57 Kip2 expression in prostate cancer. Mol Cancer. 2014;13(1):212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Sun L, Zhu J, Wu M, et al. Inhibition of MiR-199a-5p reduced cell proliferation in autosomal dominant polycystic kidney disease through targeting CDKN1C. Med Sci Monit. 2015;21:195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Veronese A, Lupini L, Consiglio J, et al. Oncogenic role of miR-483-3p at the IGF2/483 locus. Cancer Res. 2010;70(8):3140–3149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Soon PSH, Tacon LJ, Gill AJ, et al. miR-195 and miR-483-5p identified as predictors of poor prognosis in adrenocortical cancer. Clin Cancer Res. 2009;15(24):7684–7692. [DOI] [PubMed] [Google Scholar]
- [50].Else T. Association of adrenocortical carcinoma with familial cancer susceptibility syndromes. Mol Cell Endocrinol. 2012;351(1):66–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Baskerville S, Bartel DP. Microarray profiling of microRNAs reveals frequent coexpression with neighboring miRNAs and host genes. Rna. 2005;11(3):241–247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Sun D, Lee YS, Malhotra A, et al. miR-99 family of microRNAs suppresses the expression of prostate specific antigen and prostate cancer cell proliferation. Cancer Res. 2011;71(4):1313–1324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Connolly E, Melegari M, Landgraf P, et al. Elevated expression of the miR-17–92 polycistron and miR-21 in hepadnavirus-associated hepatocellular carcinoma contributes to the malignant phenotype. Am J Pathol. 2008;173(3):856–864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Tsuchida A, Ohno S, Wu W, et al. miR-92 is a key oncogenic component of the miR‐17–92 cluster in colon cancer. Cancer Sci. 2011;102(12):2264–2271. [DOI] [PubMed] [Google Scholar]
- [55].He L, Thomson JM, Hemann MT, et al. A microRNA polycistron as a potential human oncogene. Nature. 2005;435(7043):828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Calvano Filho CMC, Calvano-Mendes DC, Carvalho KC, et al. Triple-negative and luminal A breast tumors: differential expression of miR-18a-5p, miR-17-5p, and miR-20a-5p. Tumor Biol. 2014;35(8):7733–7741. [DOI] [PubMed] [Google Scholar]
- [57].Kort EJ, Farber L, Tretiakova M, et al. The E2F3-Oncomir-1 axis is activated in Wilms’ tumor. Cancer Res. 2008;68(11):4034–4038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].O’donnell KA, Wentzel EA, Zeller KI, et al. c-Myc-regulated microRNAs modulate E2F1 expression. Nature. 2005;435(7043):839. [DOI] [PubMed] [Google Scholar]
- [59].Sylvestre Y, De Guire V, Querido E, et al. An E2F/miR-20a autoregulatory feedback loop. J Biol Chem. 2007;282(4):2135–2143. [DOI] [PubMed] [Google Scholar]
- [60].Luk JM, Burchard J, Zhang C, et al. DLK1-DIO3 genomic imprinted microRNA cluster at 14q32. 2 defines a stemlike subtype of hepatocellular carcinoma associated with poor survival. J Biol Chem. 2011;286(35):30706–30713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Benetatos L, Hatzimichael E, Londin E, et al. The microRNAs within the DLK1-DIO3 genomic region: involvement in disease pathogenesis. Cell Mol Life Sci. 2013;70(5):795–814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Zhang S, Hao J, Xie F, et al. Downregulation of miR-132 by promoter methylation contributes to pancreatic cancer development. Carcinogenesis. 2011;32(8):1183–1189. [DOI] [PubMed] [Google Scholar]
- [63].Formosa A, Lena A, Markert E, et al. DNA methylation silences miR-132 in prostate cancer. Oncogene. 2013;32(1):127. [DOI] [PubMed] [Google Scholar]
- [64].Kozaki K-I, Imoto I, Mogi S, et al. Exploration of tumor-suppressive microRNAs silenced by DNA hypermethylation in oral cancer. Cancer Res. 2008;68(7):2094–2105. [DOI] [PubMed] [Google Scholar]
- [65].Zhao B, Li L, Lei Q, et al. The Hippo–YAP pathway in organ size control and tumorigenesis: an updated version. Genes Dev. 2010;24(9):862–874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Harvey KF, Zhang X, Thomas DM. The Hippo pathway and human cancer. Nat Rev Cancer. 2013;13(4):nrc3458. [DOI] [PubMed] [Google Scholar]
- [67].Lee H, Hwang SJ, Kim HR, et al. Neurofibromatosis 2 (NF2) controls the invasiveness of glioblastoma through YAP-dependent expression of CYR61/CCN1 and miR-296-3p. Biochimica Et Biophysica Acta (Bba)-Gene Regulatory Mechanisms. 2016;1859(4):599–611. [DOI] [PubMed] [Google Scholar]
- [68].Kireeva ML, Mo F-E, Yang GP, et al. Cyr61, a product of a growth factor-inducible immediate-early gene, promotes cell proliferation, migration, and adhesion. Mol Cell Biol. 1996;16(4):1326–1334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Babic AM, Kireeva ML, Kolesnikova TV, et al. CYR61, a product of a growth factor-inducible immediate early gene, promotes angiogenesis and tumor growth. Proc Nat Acad Sci. 1998;95(11):6355–6360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Martin M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J. 2011;17(1):10–12. [Google Scholar]
- [71].Cox MP, Peterson DA, Biggs PJ. SolexaQA: at-a-glance quality assessment of Illumina second-generation sequencing data. BMC Bioinformatics. 2010. September 27;11:485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Song X, Wang Z, Jin Y, et al. Loss of miR-532-5p in vitro promotes cell proliferation and metastasis by influencing CXCL2 expression in HCC. Am J Transl Res. 2015;7(11):2254. [PMC free article] [PubMed] [Google Scholar]
- [73].R Core Team R: a language and environment for statistical computing. Vienna (Austria): R Foundation for Statistical Computing; 2018. Available from: https://www.R-project.org [Google Scholar]
- [74].Risso D, Ngai J, Speed TP, et al. Normalization of RNA-seq data using factor analysis of control genes or samples. Nat Biotechnol. 2014;32(9):896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Robinson MD, McCarthy DJ, Smyth GK. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2010;26(1):139–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15(12):550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Ching T, Huang S, Garmire LX. Power analysis and sample size estimation for RNA-Seq differential expression. Rna. 2014;20(11):1684–1696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Rajkumar AP, Qvist P, Lazarus R, et al. Experimental validation of methods for differential gene expression analysis and sample pooling in RNA-seq. BMC Genomics. 2015;16(1):548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Bardou P, Mariette J, Escudié F, et al. jvenn: an interactive Venn diagram viewer. BMC Bioinformatics. 2014;15(1):293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Andrews S. FastQC a quality control tool for high throughput sequence data. 2014. Available from: http://www.bioinformatics.babraham.ac.uk/projects/fastqc/ [Google Scholar]
- [81].Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 2014;30(15):2114–2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Gebert D, Hewel C, Rosenkranz D. unitas: the universal tool for annotation of small RNAs. BMC Genomics. 2017;18(1):644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Shi J, Dong M, Li L, et al. mirPRo–a novel standalone program for differential expression and variation analysis of miRNAs. Sci Rep. 2015;5:14617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Zerbino DR, Achuthan P, Akanni W, et al. Ensembl 2018. Nucleic Acids Res. 2017;46(D1):D754–D761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Ji T. A Bayesian hidden Markov model for detecting differentially methylated regions. Biometrics 2018. Epub ahead of print. DOI:10.1111/biom.13000 [DOI] [PubMed] [Google Scholar]
- [86].Kent WJ. BLAT—the BLAST-like alignment tool. Genome Res. 2002;12(4):656–664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Pruitt KD, Brown GR, Hiatt SM, et al. RefSeq: an update on mammalian reference sequences. Nucleic Acids Res. 2013;42(D1):D756–D763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].Enright AJ, John B, Gaul U, et al. MicroRNA targets in Drosophila. Genome Biol. 2003;5(1):R1 PubMed PMID: 14709173; PubMed Central PMCID: PMCPMC395733. Eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [89].Huang DW, Sherman BT, Lempicki RA. Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 2008;37(1):1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].Vlachos IS, Zagganas K, Paraskevopoulou MD, et al. DIANA-miRPath v3. 0: deciphering microRNA function with experimental support. Nucleic Acids Res. 2015;43(W1):W460–W466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [91].Vlachos IS, Paraskevopoulou MD, Karagkouni D, et al. DIANA-TarBase v7. 0: indexing more than half a million experimentally supported miRNA: mRNA interactions. Nucleic Acids Res. 2014;43(D1):D153–D159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].Bendall SC, Stewart MH, Menendez P, et al. IGF and FGF cooperatively establish the regulatory stem cell niche of pluripotent human cells in vitro. Nature. 2007;448(7157):1015. [DOI] [PubMed] [Google Scholar]
- [93].Carbognin E, Betto RM, Soriano ME, et al. Stat3 promotes mitochondrial transcription and oxidative respiration during maintenance and induction of naive pluripotency. Embo J. 2016;35(6):618–634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [94].Panina Y, Germond A, Masui S, et al. Validation of common housekeeping genes as reference for qpcr gene expression analysis during iPS reprogramming process. Sci Rep. 2018;8(1):8716. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.