

Nanoparticle-assisted protein NMR opens window to observation of novel functional dynamics in the nano- to microsecond range.
Abstract
Protein function depends critically on intrinsic internal dynamics, which is manifested in distinct ways, such as loop motions that regulate protein recognition and catalysis. Under physiological conditions, dynamic processes occur on a wide range of time scales from subpicoseconds to seconds. Commonly used NMR spin relaxation in solution provides valuable information on very fast and slow motions but is insensitive to the intermediate nanosecond to microsecond range that exceeds the protein tumbling correlation time. Presently, very little is known about the nature and functional role of these motions. It is demonstrated here how transverse spin relaxation becomes exquisitely sensitive to these motions at atomic resolution when studying proteins in the presence of nanoparticles. Application of this novel cross-disciplinary approach reveals large-scale dynamics of loops involved in functionally critical protein-protein interactions and protein-calcium ion recognition that were previously unobservable.
INTRODUCTION
Nuclear magnetic resonance (NMR) spin relaxation measurements of proteins offer a wealth of information at atomic resolution about internal motional amplitudes and time scales under physiological conditions (1–4). This dynamics information provides critical mechanistic and thermodynamic insights into protein function involving loop motions, interdomain dynamics, recognition dynamics with small ligands, nucleic acids or other proteins, and partial unfolding/refolding events (5–13).
Every type of NMR parameters—such as scalar J-couplings, residual dipolar couplings, and average chemical shifts—depends in a unique way on both molecular structure and dynamics (1). In the case of spin relaxation parameters, the observable range of intramolecular protein motions covers fast time scales in the picosecond to low nanosecond range via longitudinal spin relaxation R1, transverse spin relaxation R2, and the heteronuclear Overhauser enhancement (NOE) experiments (1). By contrast, the chemical exchange and conformational exchange regime on tens of microseconds to seconds is covered by rotating frame relaxation experiments [R1ρ, Carr-Purcell-Meiboom-Gill (CPMG), and chemical exchange saturation transfer] (14–17). R1, R2, and NOE relaxation data represent the convolution of overall rotational tumbling and intramolecular dynamics, which renders motions unobservable if they take place on time scales slower than the overall tumbling correlation time τP, which is typically around 10 ns. Thus, the intermediate time scale regime between low nano- and microsecond motions represents a critical gap in our ability to directly observe protein dynamics. The mere existence and atomic-detail character of these motions and their role in protein function are therefore largely uncharted territory.
As described in the paper, this situation can be addressed by studying protein dynamics in the presence of aqueous colloidal dispersions of synthetic nanoparticles (NPs) (Fig. 1). With their much larger size, NPs have tumbling correlation times τNP >> τP into the hundreds of nanosecond to microsecond range (18). For proteins that transiently interact with the NP surface and are in rapid exchange between a free and an NP-bound state, their spin relaxation reflects dynamics on the much broader picosecond to τNP range, thereby offering an unobstructed view of pico- to microsecond motions.
Fig. 1. Protein dynamics into the hundreds of nanosecond to microsecond range accessible to NP-assisted solution NMR.
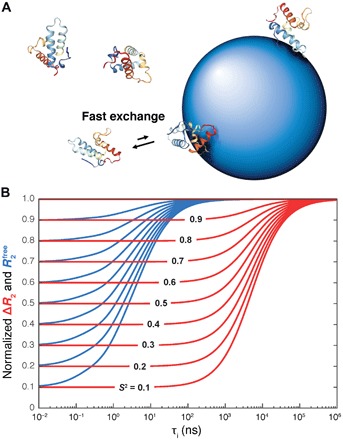
(A) Protein molecules are in fast exchange between their rapidly tumbling free state and a slowly tumbling NP-bound state giving rise to effective transverse spin relaxation rates versus in the absence of NPs. (B) Simulated dependence of in the absence of NPs (blue) and in the presence and absence of NPs (red) on the internal correlation time τi and motional restriction (S2 order parameter), which demonstrates the wide range of time scales sensitively probed by ΔR2. The blue and red curves were normalized by setting their maximal values to 1.0.
RESULTS
Transverse R2 spin relaxation experiments (1) are particularly sensitive to the presence of NPs. Consider a protein with an overall tumbling correlation time τP, which is intermittently bound to an NP with correlation time τNP with an exchange rate that is fast on the NMR chemical shift time scale (>103 s−1) but slow on the molecular tumbling time scale (<107 to 108 s−1). The effective transverse relaxation rate of a protein 15N spin is then
| (1) |
where p and 1 − p are the bound and free protein populations with transverse relaxation rates and , respectively. R2 is dominated by the spectral density of motion at zero frequency R2 = cJ(0), where c is a constant (see the Supplementary Materials). When internal dynamics is represented for each 15N site in a model-free way with an S2 order parameter and an internal correlation time τi (19), and , where and (used below). S2 is a general measure of the motional restriction of a 15N-1H bond vector varying between 0 (highly mobile) and 1 (static). The difference of R2 in the presence and absence of NPs, ΔR2, can then be expressed as
| (2) |
If ∣τNP − τP∣ ≫ ∣τi,NP − τi,P∣, which applies when τi < τNP/10 and S2 is nonzero, then Eq. 2 reduces to
| (3) |
It follows that the site-specific S2 order parameters can be directly extracted from experimental ΔR2 values whereby the global scaling factor (c pτNP)−1 is identical for all residues. Since ΔR2-derived S2 reflects the cumulative effect of all internal motions with correlation times τi < τNP/10, it exceeds the time scale range τi < τP of standard model-free S2 values determined in the absence of NPs by several orders of magnitude (Fig. 1B). The validity range of Eq. 3 is depicted in fig. S2.
NP-assisted NMR relaxation is demonstrated for the two globular proteins colicin E7 immunity protein (Im7) and calcium-binding domain 1 of the canine sodium-calcium exchanger NCX1 (CBD1). Im7 forms a four-helix bundle and binds to the deoxyribonuclease (DNase) domain of bacteriocin colicin E7 (ColE7), thereby inhibiting its strong toxic effect (20). Because Im7 exhibits an on-pathway folding intermediate, it has served as a model system for studying protein folding (21, 22). CBD1 is a globular domain with a β-sandwich fold and is part of the large cytosolic loop of the sodium-calcium exchanger (NCX) (23). CBD1, which is studied here in the absence of Ca2+, can bind up to four Ca2+ ions that produce an allosteric response that allows the exchange of intracellular Ca2+ with extracellular Na+ ions across the transmembrane domain of NCX (24).
Backbone 15N R2 relaxation parameters were measured in the presence and absence of submicromolar to low micromolar anionic silica NPs (SNPs) of 20 nm diameter (Figs. 2A and 3A) with minimal effect on solvent viscosity (25). Because of the transient interactions of the protein molecules with SNPs, their average overall tumbling is slowed down, which explains why R2 in the presence of SNPs exceeds that of the free state. Several residues display enhanced R2 relaxation, which is caused by chemical exchange Rex on the millisecond time scale and remains largely unaffected by the presence of SNPs. When focusing on ΔR2 profiles (Fig. 2, B and C), Rex effects cancel out, allowing an interpretation of ΔR2 solely in terms of S2 order parameters (Eq. 3). Conformationally rigid regions with large ΔR2 belong to regular secondary structures and certain loops. At the same time, both proteins show large amounts of dynamics in the N- and/or C-terminal tails and in selected loop regions, manifested in a substantial decrease of their ΔR2 values. To obtain a quantitative measure of the dynamics, ΔR2 values were converted to S2(ΔR2) order parameters (Eq. 3) by global scaling so that rigid secondary structures have average S2(ΔR2) values of 0.85 (see the Supplementary Materials). This allows a direct comparison with traditional model-free S2(MF) values (blue circles in Figs. 2C and 3C) (22, 26) derived from standard 15N R1, R2, and NOE data of free protein reporting on picosecond to low nanosecond motions only.
Fig. 2. Dynamics of Im7 protein by backbone 15N-NMR spin relaxation and molecular dynamics (MD) simulations.
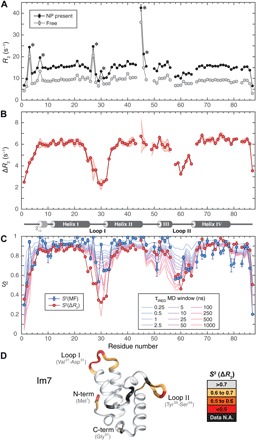
(A) 15N-R2 relaxation rates measured in the absence (gray) and presence (black) of NPs. Data points with asterisks (*) indicate substantial chemical exchange Rex effects. (B) R2 differences (ΔR2) of (A) with secondary structure of Im7 indicated at the bottom (4 α helices and 310 helix at N terminus). Experimental uncertainty (1 SD) is depicted by the shaded red area based on five independently measured ΔR2 profiles (see fig. S6). (C) Comparison of ΔR2-derived S2 (red circles) with standard model-free S2 order parameters (blue circles) and S2 values determined from 1-μs MD trajectory with variable averaging time window (from 250 ps to 1 μs). (D) S2(ΔR2) values mapped on three-dimensional (3D) crystal structure [Protein Data Bank (PDB) code 1AYI] show loops and tails that undergo substantial dynamics on pico- to microsecond time scales. N.A., not available.
Fig. 3. Dynamics of CBD1 protein domain from backbone 15N-NMR spin relaxation and MD simulations.
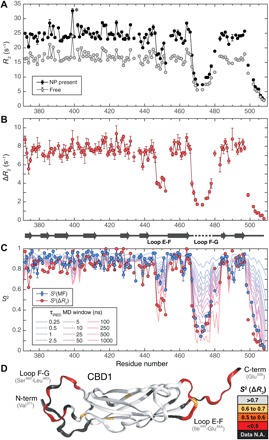
(A) 15N-R2 relaxation rates measured in the absence (gray) and presence (black) of NPs. Gly399 (*) shows substantial chemical exchange Rex. (B) R2 differences (ΔR2) of (A) with secondary structure of CBD1 indicated at the bottom (nine β strands). (C) Comparison of ΔR2-derived S2 (red circles) with standard model-free S2 order parameters (blue circles) and S2 values determined from 1-μs MD trajectory with variable averaging time window (from 250 ps to 1 μs). (D) S2(ΔR2) values mapped on the 3D crystal structure of CBD1 (PDB code 2DPK) show loops and tails that undergo substantial dynamics on pico- to microsecond time scales.
For Loop II of Im7, the S2(ΔR2) and S2(MF) profiles reflect a very similar degree of mobility with minima around 0.5, which indicates that relevant loop conformations are mostly explored on the fast picosecond to low nanosecond time scale. By contrast, Loop I has S2(ΔR2) values that are substantially lower than S2(MF), revealing the presence of fast dynamics with S2(MF) > 0.53 and additional slower motions on the nano- to microsecond range with S2(ΔR2) between 0.32 and 0.53. The S2(ΔR2) profile for Loop I is wider than the S2(MF) profile and reaches into the C terminus of Helix I (residues Lys24-Val27). This fraying of Helix I is consistent with submicrosecond folding and unfolding of the last helical turn observed in the molecular dynamics (MD) simulation (Fig. 2C). These nano- to microsecond motions were missed in previous studies based on spin relaxation data in the absence of SNPs. Further fraying and partial unfolding occur in a lowly populated folding intermediate, where the stabilizing Glu21-Lys24 salt bridge is absent and Glu25 exhibits random-coil behavior (27).
CBD1 has a total of eight loops connecting the nine β strands (strands A to G). Loop F-G, which is missing in the x-ray crystal structure [Protein Data Bank (PDB) code 2DPK], is most flexible with S2(ΔR2) values <0.2 that are in excellent agreement with the corresponding S2(MF) values. It suggests that this loop is highly dynamic with dominant correlation times on the pico- to nanosecond time scale that are fully reflected by spin relaxation data both in the presence and absence of SNPs. Loop E-F, which is located at the other end of the protein (Fig. 3D), has a distinctly different behavior. According to standard 15N-relaxation analysis, it is only moderately flexible with S2(MF) > 0.60. However, S2(ΔR2) values dip as low as 0.35, reflecting the presence of substantial amounts of additional dynamics into the hundreds of nanosecond range. Loop E-F therefore probes a much broader ensemble of conformations than suggested by traditional 15N-relaxation data alone. Similarly, the C-terminal residues of CBD1 also exhibit S2(ΔR2) < S2(MF) caused by dynamics on both the pico- to nanosecond and nano- to microsecond time scales.
Independent corroborating evidence of the presence and location of fast and slow time scale dynamics can be gleaned from extended MD computer simulations. MD trajectories in explicit water solvent were computed and analyzed for both protein systems. 1H-15N S2(MD) values, computed from MD trajectories using 12 different isotropic reorientational eigenmode dynamics (iRED) (28) time-averaging windows τiRED ranging from 250 ps to 1 μs, are plotted in Figs. 2C and 3C. Loops I and II in Im7 and Loops E-F and F-G in CBD1 all show a steady drop of S2(MD) when averaging over slower time scales (longer τiRED windows), whereas secondary structures and other loops remain notably rigid. Adequate sampling of Loop I conformations is only achieved when considering the full-length trajectory, whereas Loop II samples most of the relevant conformations already within τiRED ~25 ns, which corresponds to the time scale window accessible by 15N spin relaxation of the free protein (28). Similarly, for Loop E-F and the C terminus, the entire 1-μs trajectory length is required to reach good agreement with experimental S2(ΔR2), supporting the experimental finding that this loop displays large amplitude motions on time scales inaccessible by standard NMR relaxation methods.
The fast and slow dynamics of Im7 Loop I and CBD1 Loop E-F were further analyzed by principal component analysis (PCA) in backbone dihedral angle space (29). The score plots of Fig. 4 (A and C) show that both loops transition between multiple conformational clusters with representative cluster snapshots depicted in Fig. 4 (B and D). The diverse nature of the conformational loop ensembles reveals multiple possible interaction modes with partner molecules.
Fig. 4. Visualization of complex loop motions by 1-μs MD trajectories.
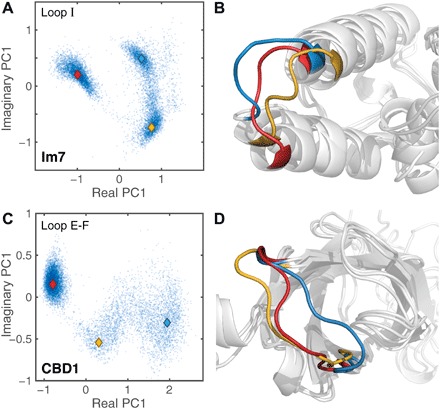
(A and C) Backbone dihedral angle–based PCA score plots of (A) Loop I of Im7 and (C) Loop E-F of CBD1 display multiple distinct conformational loop clusters. (B and D) 3D visualization of three cluster centers for each protein indicated by colored diamond (♦) symbols in score plots.
DISCUSSION
Despite their potential significance for biological function, observation of internal protein motions on the nano- to microsecond range has been a major challenge in the past. To make this motional regime accessible, rotational tumbling is sometimes slowed down by increasing solvent viscosity through the addition of ethylene glycol or glycerol. However, this also tends to stiffen or slow down internal motions, keeping slower motions mostly out of range of NMR relaxation experiments (30, 31). Alternatively, increasing the size of the molecule, as was shown for RNA, can slow down tumbling and open up observation of slower nanosecond motions (13). A recent NMR relaxometry approach, measuring R1 over a wide range of magnetic field strengths (0.33 to 22.3 T), could access internal protein motions into the low nanosecond range (32).
As demonstrated here, the use of slowly tumbling NPs to which a biomolecule can bind in fast exchange offers a general solution to this long-standing challenge. Rapid exchange has been observed in NMR-based NP binding studies of globular (33) and intrinsically disordered proteins (34). The binding equilibrium between NPs and proteins can be shifted by adjusting the NP concentration in the submicromolar to low micromolar range to optimize NMR line broadening (ΔR2 < 10 s−1 with p ≈ 0.01), allowing ΔR2 measurements with high accuracy.
For both CBD1 and Im7, NP-assisted spin relaxation reveals previously unknown slow time scale dynamics of loops displaying low S2(ΔR2) values that are directly involved in protein function through electrostatic interactions with their binding partners. Im7 has pico- to femtomolar affinity to its target colicin protein partner (35), whereby acidic residues Asp31 and Asp35 of Im7 Loop I form strong salt bridges with basic colicin residues Arg520, Lys525, and Lys528 (fig. S7). For CBD1, Ca2+ ions bind to negatively charged side chains of Asp446 and Asp447 of Loop E-F, which allosterically triggers the exchange of Ca2+ versus Na+ ions through the transmembrane domain (24). The central role of electrostatic interactions in biological complex formation is well established (36). The dynamic nature and high plasticity of the interacting loops identified here are likely to help fine-tune these interactions and optimize the affinity and specificity with interacting partner proteins and metal ions.
Low S2(ΔR2) values are reporters of previously unknown contributions to the conformational entropy, analogous to S2(MF) (12, 37), allowing a more quantitative and complete understanding of the thermodynamics of protein-protein and protein-ligand interactions. As demonstrated here (Figs. 2C and 3C), S2(ΔR2) provide powerful benchmarks for the testing (and potential improvement) of computational models, in particular MD simulation protocols and their underlying force fields. As MD trajectories now routinely extend into the hundreds of nano- to microsecond range, there is a pressing need for experimental benchmarks (38) that permit evaluation of these trajectories for which S2(ΔR2) profiles are highly suitable.
Because of their spherical shape, SNPs tumble isotropically along with the proteins bound to them (Fig. 1A), which further simplifies the model-free interpretation of ΔR2. It assumes that protein-NP interactions do not significantly affect protein structure and dynamics, similar to alignment media used for NMR residual dipolar coupling measurements (39, 40). This should hold when a globular protein interacts with NPs mostly in a nonspecific manner, for example, by making contacts with many different surface sections. Specific binding, for example, to a mobile loop, may systematically alter its mobility in the presence of NPs. By comparing ΔR2, profiles measured for different types of NPs could help identify these situations. For Im7, few residues (Asn79, Gly80, Gly83, Gln86, and Gly87) display S2(ΔR2) > S2(MF). This modest S2 inversion may reflect chemical exchange contributions with the SNPs, although no line broadening effects were observed in these regions. Because these residues are neither cationic nor hydrophobic, it is unlikely that these residues are limited in their mobility because of direct interactions with the SNPs.
We used anionic SNPs, but other types of NPs should work similarly well for the proteins studied here and other biomolecules. Anionic SNPs have partially dissociated silanol groups at their surface, which give rise to attractive and repulsive electrostatic interactions with charged protein residues. SNPs can also display hydrophobic interactions that are presumably mediated by siloxane groups at the NP surface, but the details of this interaction mechanism are not fully understood (41, 42).
The cross-disciplinary NP-assisted relaxation method introduced here uncovers previously undetected motions on submicrosecond time scales of protein regions that play key roles in the function of these systems by mediating receptor and ligand interactions. It seems likely that protein motions on these time scales are widespread. NP-assisted spin relaxation enables their comprehensive characterization at atomic resolution and shed new light on biomolecular function.
MATERIALS AND METHODS
The full description of Materials and Methods can be found in the Supplementary Materials. A brief summary is provided here.
Sample preparation
Uniformly 15N-labeled and/or 13C-labeled proteins Im7 and CBD1 were overexpressed in Escherichia coli with final NMR concentrations of 500 and 400 μM, respectively. For SNP-containing samples, Bindzil 2040 colloidal SNPs (AkzoNobel) with a 20 nm diameter were dialyzed in pH 7.0 buffers and mixed directly with proteins. All samples were stable over the entire course of the NMR data acquisition.
NMR spectroscopy
NMR experiments were performed on Bruker AVANCE III HD spectrometers operating at 850-MHz 1H frequency (19.97 T) for resonance assignments and relaxation measurements at 298 K (for Im7) and 306 K (for CBD1). 15N spin relaxation rates (R1 and R1ρ) for protein samples both in the absence and presence of SNPs, as well as a {1H}-15N steady-state NOE experiment in the absence of SNPs, were measured and analyzed using standard two-dimensional NMR 15N relaxation methods (43, 44).
MD simulations
MD simulations were performed by standard methods using the GROMACS 5.1.2 package (45) with initial structures of Im7 and CBD1 built on the basis of crystal structures (PDB codes, 1CEI and 2DPK). AMBER ff99SBnmr1 protein force field (46) together with the TIP3P explicit water model (47) was used. The integration time step was set to 2 fs, and Na+ ions were added to neutralize the total charge of the system. Particle mesh Ewald summation with a grid spacing of 1.2 Å was used to calculate long-range electrostatic interactions. After equilibration, the production run was performed in the NPT ensemble at 300 K and 1 atm for 1 μs. Amide order parameters S2(MD) were back-calculated from MD trajectories using the iRED method with varying lengths of the time-averaging window (48). PCA was performed on the backbone dihedral angles of loop residues.
Supplementary Material
Acknowledgments
We thank L. E. Kay for providing the plasmid of Im7 and AkzoNobel for the SNPs. Funding: This work was supported by the U.S. NSF (grant MCB-1715505 to R.B.). NMR experiments were performed at the CCIC NMR facility at The Ohio State University, and MD simulations were performed at the Ohio Supercomputer Center. Author contributions: M.X. prepared NP samples, performed NMR relaxation experiments, and analyzed data. L.Y. performed and analyzed MD simulations and compared them with experimental data. L.B.-L. subcloned Im7, expressed and isotopically labeled Im7 and CBD1, and prepared samples with NPs and for assignments. X.X. performed resonance assignments of Im7. A.L.H. performed NMR relaxation and assignment experiments, confirmed CBD1 assignments, and analyzed data. R.B. conceived the project, developed the theory, interpreted data, and wrote the manuscript with input from M.X., L.Y., and A.L.H. Competing interests: The corresponding author filed an invention disclosure related to this work. The authors declare no other competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/5/8/eaax5560/DC1
Supplementary Materials and Methods
Fig. S1. Simulated dependence of and ΔR2 on internal correlation time τi and S2 order parameter.
Fig. S2. Range of validity of Eq. 3 for the extraction of S2 from ΔR2.
Fig. S3. Experimental 15N spin relaxation parameters of Im7 in the absence of NPs.
Fig. S4. Experimental 15N spin relaxation parameters of CBD1 in the absence of NPs.
Fig. S5. Comparison between NMR S2(ΔR2) and x-ray B-factors of backbone nitrogen atoms in crystal structures.
Fig. S6. Dependence of ΔR2 values on SNP concentration.
Fig. S7. Mapping of experimental S2(ΔR2) onto the structural model of Im7 when bound to the DNase domain of colicin E7.
REFERENCES AND NOTES
- 1.J. Cavanagh, W. J. Fairbrother, A. G. Palmer III, M. Rance, N. J. Skelton, Protein NMR Spectroscopy: Principles and Practice (Academic Press, ed. 2, 2007). [Google Scholar]
- 2.Ishima R., Torchia D. A., Protein dynamics from NMR. Nat. Struct. Mol. Biol. 7, 740–743 (2000). [DOI] [PubMed] [Google Scholar]
- 3.Mittermaier A., Kay L. E., New tools provide new insights in NMR studies of protein dynamics. Science 312, 224–228 (2006). [DOI] [PubMed] [Google Scholar]
- 4.Brüschweiler R., New approaches to the dynamic interpretation and prediction of NMR relaxation data from proteins. Curr. Opin. Struct. Biol. 13, 175–183 (2003). [DOI] [PubMed] [Google Scholar]
- 5.Redfield C., Boyd J., Smith L. J., Smith R. A. G., Dobson C. M., Loop mobility in a four-helix-bundle protein: Nitrogen-15 NMR relaxation measurements on human interleukin-4. Biochemistry 31, 10431–10437 (1992). [DOI] [PubMed] [Google Scholar]
- 6.Brüschweiler R., Liao X., Wright P. E., Long-range motional restrictions in a multidomain zinc-finger protein from anisotropic tumbling. Science 268, 886–889 (1995). [DOI] [PubMed] [Google Scholar]
- 7.Eisenmesser E. Z., Millet O., Labeikovsky W., Korzhnev D. M., Wolf-Watz M., Bosco D. A., Skalicky J. J., Kay L. E., Kern D., Intrinsic dynamics of an enzyme underlies catalysis. Nature 438, 117–121 (2005). [DOI] [PubMed] [Google Scholar]
- 8.Boehr D. D., McElheny D., Dyson H. J., Wright P. E., The dynamic energy landscape of dihydrofolate reductase catalysis. Science 313, 1638–1642 (2006). [DOI] [PubMed] [Google Scholar]
- 9.Whittier S. K., Hengge A. C., Loria J. P., Conformational motions regulate phosphoryl transfer in related protein tyrosine phosphatases. Science 341, 899–903 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tollinger M., Skrynnikov N. R., Mulder F. A. A., Forman-Kay J. D., Kay L. E., Slow dynamics in folded and unfolded states of an SH3 domain. J. Am. Chem. Soc. 123, 11341–11352 (2001). [DOI] [PubMed] [Google Scholar]
- 11.Schneider R., Maurin D., Communie G., Kragelj J., Hansen D. F., Ruigrok R. W. H., Jensen M. R., Blackledge M., Visualizing the molecular recognition trajectory of an intrinsically disordered protein using multinuclear relaxation dispersion NMR. J. Am. Chem. Soc. 137, 1220–1229 (2015). [DOI] [PubMed] [Google Scholar]
- 12.Lee A. L., Kinnear S. A., Wand A. J., Redistribution and loss of side chain entropy upon formation of a calmodulin–peptide complex. Nat. Struct. Biol. 7, 72–77 (2000). [DOI] [PubMed] [Google Scholar]
- 13.Zhang Q., Sun X., Watt E. D., Al-Hashimi H. M., Resolving the motional modes that code for RNA adaptation. Science 311, 653–656 (2006). [DOI] [PubMed] [Google Scholar]
- 14.Palmer A. G., III, Chemical exchange in biomacromolecules: Past, present, and future. J. Magn. Reson. 241, 3–17 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Reddy J. G., Pratihar S., Ban D., Frischkorn S., Becker S., Griesinger C., Lee D., Simultaneous determination of fast and slow dynamics in molecules using extreme CPMG relaxation dispersion experiments. J. Biomol. NMR 70, 1–9 (2018). [DOI] [PubMed] [Google Scholar]
- 16.Vallurupalli P., Bouvignies G., Kay L. E., Studying “invisible” excited protein states in slow exchange with a major state conformation. J. Am. Chem. Soc. 134, 8148–8161 (2012). [DOI] [PubMed] [Google Scholar]
- 17.Anthis N. J., Clore G. M., Visualizing transient dark states by NMR spectroscopy. Q. Rev. Biophys. 48, 35–116 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Heyes D. M., Nuevo M. J., Morales J. J., Branka A. C., Translational and rotational diffusion of model nanocolloidal dispersions studied by molecular dynamics simulations. J. Phys. Condens. Matter 10, 10159–10178 (1998). [Google Scholar]
- 19.Lipari G., Szabo A., Model-free approach to the interpretation of nuclear magnetic resonance relaxation in macromolecules. 1. Theory and range of validity. J. Am. Chem. Soc. 104, 4546–4559 (1982). [Google Scholar]
- 20.Papadakos G., Wojdyla J. A., Kleanthous C., Nuclease colicins and their immunity proteins. Q. Rev. Biophys. 45, 57–103 (2012). [DOI] [PubMed] [Google Scholar]
- 21.Capaldi A. P., Shastry M. C. R., Kleanthous C., Roder H., Radford S. E., Ultrarapid mixing experiments reveal that Im7 folds via an on-pathway intermediate. Nat. Struct. Biol. 8, 68–72 (2001). [DOI] [PubMed] [Google Scholar]
- 22.Whittaker S. B.-M., Clayden N. J., Moore G. R., NMR characterisation of the relationship between frustration and the excited state of Im7. J. Mol. Biol. 414, 511–529 (2011). [DOI] [PubMed] [Google Scholar]
- 23.Weber C. R., Ginsburg K. S., Philipson K. D., Shannon T. R., Bers D. M., Allosteric regulation of Na/Ca exchange current by cytosolic Ca in intact cardiac myocytes. J. Gen. Physiol. 117, 119–131 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nicoll D. A., Sawaya M. R., Kwon S., Cascio D., Philipson K. D., Abramson J., The crystal structure of the primary Ca2+ sensor of the Na+/Ca2+ exchanger reveals a novel Ca2+ binding motif. J. Biol. Chem. 281, 21577–21581 (2006). [DOI] [PubMed] [Google Scholar]
- 25.Murshed S. M. S., Estellé P., A state of the art review on viscosity of nanofluids. Renewable Sustainable Energy Rev. 76, 1134–1152 (2017). [Google Scholar]
- 26.Johnson E., Bruschweiler-Li L., Showalter S. A., Vuister G. W., Zhang F., Brüschweiler R., Structure and dynamics of Ca2+-binding domain 1 of the Na+/Ca2+ exchanger in the presence and in the absence of Ca2+. J. Mol. Biol. 377, 945–955 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hansen A. L., Kay L. E., Quantifying millisecond time-scale exchange in proteins by CPMG relaxation dispersion NMR spectroscopy of side-chain carbonyl groups. J. Biomol. NMR 50, 347–355 (2011). [DOI] [PubMed] [Google Scholar]
- 28.Gu Y., Li D.-W., Brüschweiler R., NMR order parameter determination from long molecular dynamics trajectories for objective comparison with experiment. J. Chem. Theory Comput. 10, 2599–2607 (2014). [DOI] [PubMed] [Google Scholar]
- 29.Wang J., Brüschweiler R., 2D entropy of discrete molecular ensembles. J. Chem. Theory Comput. 2, 18–24 (2006). [DOI] [PubMed] [Google Scholar]
- 30.Zeeb M., Jacob M. H., Schindler T., Balbach J., 15N relaxation study of the cold shock protein CspB at various solvent viscosities. J. Biomol. NMR 27, 221–234 (2003). [DOI] [PubMed] [Google Scholar]
- 31.Xu J., Xue Y., Skrynnikov N. R., Detection of nanosecond time scale side-chain jumps in a protein dissolved in water/glycerol solvent. J. Biomol. NMR 45, 57–72 (2009). [DOI] [PubMed] [Google Scholar]
- 32.Cousin S. F., Kadeřávek P., Bolik-Coulon N., Gu Y., Charlier C., Carlier L., Bruschweiler-Li L., Marquardsen T., Tyburn J.-M., Brüschweiler R., Ferrage F., Time-resolved protein side-chain motions unraveled by high-resolution relaxometry and molecular dynamics simulations. J. Am. Chem. Soc. 140, 13456–13465 (2018). [DOI] [PubMed] [Google Scholar]
- 33.Assfalg M., Ragona L., Pagano K., D'Onofrio M., Zanzoni S., Tomaselli S., Molinari H., The study of transient protein–nanoparticle interactions by solution NMR spectroscopy. Biochim. Biophys. Acta Proteins Proteom. 1864, 102–114 (2016). [DOI] [PubMed] [Google Scholar]
- 34.Xie M., Li D.-W., Yuan J., Hansen A. L., Brüschweiler R., Quantitative binding behavior of intrinsically disordered proteins to nanoparticle surfaces at individual residue level. Chem. Eur. J. 24, 16997–17001 (2018). [DOI] [PubMed] [Google Scholar]
- 35.N. G. Housden, C. Kleanthous, Thermodynamic dissection of colicin interactions, in Methods in Enzymology, M. L. Johnson, J. M. Holt, G. K. Ackers, Eds. (Academic Press, 2011), vol. 488, chap. 6, pp. 123–145. [DOI] [PubMed] [Google Scholar]
- 36.Schreiber G., Haran G., Zhou H.-X., Fundamental aspects of protein–protein association kinetics. Chem. Rev. 109, 839–860 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Akke M., Brüschweiler R., Palmer A. G. III, NMR order parameters and free energy: An analytical approach and its application to cooperative Ca2+ binding by calbindin D9k. J. Am. Chem. Soc. 115, 9832–9833 (1993). [Google Scholar]
- 38.Beauchamp K. A., Lin Y.-S., Das R., Pande V. S., Are protein force fields getting better? A systematic benchmark on 524 diverse NMR measurements. J. Chem. Theory Comput. 8, 1409–1414 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bax A., Grishaev A., Weak alignment NMR: A hawk-eyed view of biomolecular structure. Curr. Opin. Struct. Biol. 15, 563–570 (2005). [DOI] [PubMed] [Google Scholar]
- 40.Lange O. F., Lakomek N.-A., Farès C., Schröder G. F., Walter K. F. A., Becker S., Meiler J., Grubmüller H., Griesinger C., de Groot B. L., Recognition dynamics up to microseconds revealed from an RDC-derived ubiquitin ensemble in solution. Science 320, 1471–1475 (2008). [DOI] [PubMed] [Google Scholar]
- 41.Shi B., Shin Y. K., Hassanali A. A., Singer S. J., DNA binding to the silica surface. J. Phys. Chem. B 119, 11030–11040 (2015). [DOI] [PubMed] [Google Scholar]
- 42.Xie M., Hansen A. L., Yuan J., Brüschweiler R., Residue-specific interactions of an intrinsically disordered protein with silica nanoparticles and their quantitative prediction. J. Phys. Chem. C 120, 24463–24468 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lakomek N.-A., Ying J., Bax A., Measurement of 15N relaxation rates in perdeuterated proteins by TROSY-based methods. J. Biomol. NMR 53, 209–221 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gairí M., Dyachenko A., González M. T., Feliz M., Pons M., Giralt E., An optimized method for 15N R1 relaxation rate measurements in non-deuterated proteins. J. Biomol. NMR 62, 209–220 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Abraham M. J., Murtola T., Schulz R., Páll S., Smith J. C., Hess B., Lindahl E., GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 1-2, 19–25 (2015). [Google Scholar]
- 46.Li D.-W., Brüschweiler R., NMR-based protein potentials. Angew. Chem. Int. Ed. Engl. 49, 6778–6780 (2010). [DOI] [PubMed] [Google Scholar]
- 47.Jorgensen W. L., Chandrasekhar J., Madura J. D., Impey R. W., Klein M. L., Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 79, 926–935 (1983). [Google Scholar]
- 48.Prompers J. J., Brüschweiler R., General framework for studying the dynamics of folded and nonfolded proteins by NMR relaxation spectroscopy and MD simulation. J. Am. Chem. Soc. 124, 4522–4534 (2002). [DOI] [PubMed] [Google Scholar]
- 49.Showalter S. A., Bruschweiler-Li L., Johnson E., Zhang F., Brüschweiler R., Quantitative lid dynamics of MDM2 reveals differential ligand binding modes of the p53-binding cleft. J. Am. Chem. Soc. 130, 6472–6478 (2008). [DOI] [PubMed] [Google Scholar]
- 50.Zhang B., Xie M., Bruschweiler-Li L., Bingol K., Brüschweiler R., Use of charged nanoparticles in NMR-based metabolomics for spectral simplification and improved metabolite identification. Anal. Chem. 87, 7211–7217 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Mulder F. A. A., de Graaf R. A., Kaptein R., Boelens R., An off-resonance rotating frame relaxation experiment for the investigation of macromolecular dynamics using adiabatic rotations. J. Magn. Reson. 131, 351–357 (1998). [DOI] [PubMed] [Google Scholar]
- 52.A. G. Palmer III, C. D. Kroenke, J. P. Loria, Nuclear magnetic resonance methods for quantifying microsecond-to-millisecond motions in biological macromolecules, in Methods in Enzymology, T. L. James, V. Dötsch, U. Schmitz, Eds. (Academic Press, 2001), vol. 339, chap. 10, pp. 204–238. [DOI] [PubMed] [Google Scholar]
- 53.Delaglio F., Grzesiek S., Vuister G. W., Zhu G., Pfeifer J., Bax A., NMRPipe: A multidimensional spectral processing system based on UNIX pipes. J. Biomol. NMR 6, 277–293 (1995). [DOI] [PubMed] [Google Scholar]
- 54.Lee W., Tonelli M., Markley J. L., NMRFAM-SPARKY: Enhanced software for biomolecular NMR spectroscopy. Bioinformatics 31, 1325–1327 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wangsness R. K., Bloch F., The dynamical theory of nuclear induction. Phys. Rev. 89, 728–739 (1953). [Google Scholar]
- 56.Bloch F., Dynamical theory of nuclear induction. II. Phys. Rev. 102, 104–135 (1956). [Google Scholar]
- 57.Redfield A. G., On the theory of relaxation processes. IBM J. Res. Dev. 1, 19–31 (1957). [Google Scholar]
- 58.A. Abragam, The Principles of Nuclear Magnetism (Clarendon Press, 1961). [Google Scholar]
- 59.Lipari G., Szabo A., Model-free approach to the interpretation of nuclear magnetic resonance relaxation in macromolecules. 2. Analysis of experimental results. J. Am. Chem. Soc. 104, 4559–4570 (1982). [Google Scholar]
- 60.Clore G. M., Szabo A., Bax A., Kay L. E., Driscoll P. C., Gronenborn A. M., Deviations from the simple two-parameter model-free approach to the interpretation of nitrogen-15 nuclear magnetic relaxation of proteins. J. Am. Chem. Soc. 112, 4989–4991 (1990). [Google Scholar]
- 61.Šali A., Blundell T. L., Comparative protein modelling by satisfaction of spatial restraints. J. Mol. Biol. 234, 779–815 (1993). [DOI] [PubMed] [Google Scholar]
- 62.Altis A., Nguyen P. H., Hegger R., Stock G., Dihedral angle principal component analysis of molecular dynamics simulations. J. Chem. Phys. 126, 244111 (2007). [DOI] [PubMed] [Google Scholar]
- 63.Ko T.-P., Liao C.-C., Ku W.-Y., Chak K.-F., Yuan H. S., The crystal structure of the DNase domain of colicin E7 in complex with its inhibitor Im7 protein. Structure 7, 91–102 (1999). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/5/8/eaax5560/DC1
Supplementary Materials and Methods
Fig. S1. Simulated dependence of and ΔR2 on internal correlation time τi and S2 order parameter.
Fig. S2. Range of validity of Eq. 3 for the extraction of S2 from ΔR2.
Fig. S3. Experimental 15N spin relaxation parameters of Im7 in the absence of NPs.
Fig. S4. Experimental 15N spin relaxation parameters of CBD1 in the absence of NPs.
Fig. S5. Comparison between NMR S2(ΔR2) and x-ray B-factors of backbone nitrogen atoms in crystal structures.
Fig. S6. Dependence of ΔR2 values on SNP concentration.
Fig. S7. Mapping of experimental S2(ΔR2) onto the structural model of Im7 when bound to the DNase domain of colicin E7.