

We describe a safe and natural carbon-based system to actively deliver drugs to mucosal surfaces
Abstract
Current drug-delivery systems are designed primarily for parenteral applications and are either lipid or polymer drug conjugates. In our quest to inhibit herpes simplex virus infection via the compounds found in commonly used cosmetic products, we found that activated carbon particles inhibit infection and, in addition, substantially improve topical delivery and, hence, the efficacy of a common antiviral drug, acyclovir (ACV). Our in vitro studies demonstrate that highly porous carbon structures trapped virions, blocked infection and substantially improved efficacy when ACV was loaded onto them. Also, using murine models of corneal and genital herpes infections, we show that the topical use of drug-encapsulated carbon (DECON) reduced dosing frequency, shortened treatment duration, and exhibited higher therapeutic efficacy than currently approved topical or systemic antivirals alone. DECON is a nontoxic, cost-effective and nonimmunogenic alternative to current topical drug-delivery systems that is uniquely triggered for drug release by virus trapping.
INTRODUCTION
The development of a novel therapeutic or prophylactic against one of the world’s most common pathogens, herpes simplex virus (HSV), has been under extensive investigation in recent years with hopes of ultimately finding a more effective way to treat the infection (1). Herpesviridae is a family of double-stranded DNA–enveloped viruses that encompasses some of the most widespread viruses among the human population (2). Among these is HSV, which has two serotypes: HSV-1 and HSV-2. HSV-1 is the leading cause of infectious blindness, and HSV-2 is associated with genital lesions and warts (3, 4). Notably, it is estimated that more than a third of the world population currently suffers from HSV infection (2–4). In addition, HSV infection has been implicated with increased risk of contracting other sexually transmitted diseases (STDs), such as HIV and human papilloma virus, and, as a result, has garnered much attention in the recent years (1, 5–8).
The most common mode of treatment for HSV infection includes daily dosing of orally administered acyclovir (ACV) (9). While this treatment is effective in most cases, various reports have shown that long-term systemic use of ACV results in resistance against the drug and may cause renal injury (9–11). Sustained delivery of pharmacologically active ACV is also an issue, and drug efficacy is often compromised because of this problem. While topical medications in the form of ganciclovir (ACV analog) gels and trifluridine (TFT) have been used for HSV infection, they have shown low retention time on corneal and vaginal surfaces and require repeated dosing (6 to 10 times daily) (12). To provide a better alternative, experimental treatments such as antibodies (13), peptides (14, 15), small-molecule inhibitors (16, 17), aptamers (18), and nanoparticles (19–22) have been tested. While most demonstrated effective antiviral activity against HSV both in vitro and in vivo, they all also suffer from low retention time and delivery issues on mucosal surfaces (23–27). A simple yet effective way of resolving this problem is to develop a topical treatment that protects the site of infection while providing antiviral relief for an extended period.
In this study, we used highly porous activated carbon (HPAC) particles as a model for restricting HSV-1 and HSV-2 from entering target cells. Charcoal black is a common ingredient used in the cosmetic industry, especially for the eye (8, 28). Traditionally, black soot obtained from burning clarified butter was used as an eyeliner and is still used today in various cultures across the world (28, 29). We hypothesized that charcoal in its surface-active form could trap the virions and provide antiviral effects. In line with our hypothesis, our studies demonstrate the antiviral activity of HPAC. In addition, synergy studies with ACV revealed that ACV may be adsorbed and/or is encapsulated in HPAC, resulting in a sustained release of ACV that generates a stronger therapeutic effect. We termed this antiviral drug delivery platform as drug-encapsulated carbon (DECON) and demonstrate that DECON can provide better therapeutic efficacy via sustained release of ACV, and given its nontoxic nature and kidney function–related benefits, it can also circumvent renal toxicity issues (30, 31) associated with ACV (32). Our results raise a strong possibility that DECON can be a novel delivery platform for many drugs for human/nonhuman diseases.
RESULTS
HPAC inhibits HSV from entering cells
To understand whether HPAC was able to trap viruses in its pores, we conducted a reporter-based virus entry assay using a prophylactic treatment model. Various cell types were prophylactically treated for 90 min with HPAC, followed by HSV-1 or HSV-2 infection at a multiplicity of infection (MOI) of 10 for 6 hours. A 40 to 60% reduction in HSV-1 and HSV-2 entry was shown using concentrations of HPAC as low as 1 mg/ml (Fig. 1, A and D). The half maximal inhibitory concentration (IC50) value of HPAC during HSV-1 and HSV-2 infection in prophylactic treatment was found to be 0.8 and 1 mg/ml, respectively. These values are substantially below the clinically accepted half maximal toxicity concentration (TC50) value of HPAC (>50 mg/ml), making them a viable material for inhibiting virus entry. We calculated the selectivity index (SI = TC50/IC50) for HPAC in the prophylactic model for HSV-1 and HSV-2 as 62.5 and 50, respectively.
Fig. 1. Prophylactic, neutralization, and therapeutic efficacy of HPAC.
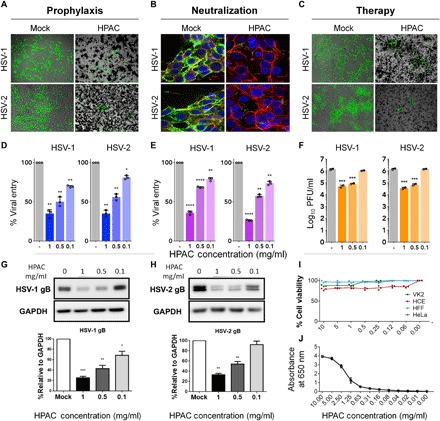
(A) Fluorescence imaging of green fluorescent protein (GFP)–HSV-1– and GFP–HSV-2–infected human corneal epithelial cells (HCEs) or HeLa cells treated with HPAC (1 mg/ml) prophylactically. (B) GFP HSV-1 and HSV-2 viruses were neutralized with HPAC (1 mg/ml) before its application to HeLa cells, and the viral entry was measured. Blue, 4′,6-diamidino-2-phenylindole; red, phalloidin staining of actin; green, GFP virus. (C) HCEs and HeLa cells were infected with HSV-1 and HSV-2, respectively, for a period of 2 hours before the addition of mock phosphate-buffered saline (PBS) or HPAC at 1 mg/ml. Twenty-four hours after infection, fluorescence images were taken to understand the extent of viral spread in HPAC-treated samples compared to mock. Green, 17 GFP HSV-1 or HSV-2 333 GFP virus. Viral entry for prophylactic (D) and neutralization treatments (E) was quantified using a β-galactosidase reporter virus. (F) Intracellular viral load for HPAC therapeutic treatment was quantified using a plaque assay. PFU, plaque-forming units. Representative immunoblots of HSV-1 (G) or HSV-2 gB (H) protein and human glyceraldehyde-3-phosphate dehydrogenase (GAPDH) from infected HCEs or HeLa cells treated with varying concentrations of HPAC. (I) Toxicity of HPAC in vitro was evaluated by MTT [3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide] assay after the incubation of multiple concentrations of HPAC with HCEs, HeLa cells, VK2s (vaginal epithelial cells), and HFFs (human foreskin fibroblasts) for 24 hours. (J) Optical density measurements were performed by serially diluting HPAC in a 96-well plate and measuring the optical absorbance at 650 nm. Lower absorbance corresponds to a clearer solution. Data are presented as means ± SD. Significance to mock-treated cells was determined by one-way analysis of variance (ANOVA) followed by Dunnett’s multiple comparisons test (n = 3 replicates). *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001.
HPAC traps HSV particles
To determine the underlying mechanism of HPAC in inhibiting HSV-1 and HSV-2, we incubated green fluorescent protein (GFP) virus with HPAC for a period of 30 min. Following incubation, HPAC was separated from non-neutralized virus in the supernatant and overlaid on HeLa cells. Six hours after infection, centrifuged HPAC pellet (fig. S1) and HeLa cells infected with supernatant virus (Fig. 1C, right) were fixed and imaged. Given the highly porous and active nature of HPAC’s surface, we hypothesized that it would be able to efficiently trap the virus in its nanopores. As expected, HPAC neutralization contained significantly lower virus compared to mock-neutralized virus. In addition, we performed an entry assay similar to the one explained in the previous section using a β-galactosidase reporter virus. The results from the entry assay confirmed that HPAC efficiently bound the virus and restricted viral entry into cells. This confirms that HPAC at a concentration of 1 mg/ml inhibits virus entry by trapping the virus in its nanopores (Fig. 1, B and E).
HPAC treatment shows therapeutic efficacy
To further investigate the antiviral potential of HPAC treatment, we first infected cells with HSV-1 and HSV-2 at 0.1 MOI and then therapeutically treated them with HPAC. Cells were imaged (Fig. 1C), and the cell lysates that were collected from these samples at 24 hpi (hours post infection) were used as inoculants for plaque assays to determine intracellular viral load. We observed that the number of plaques for HPAC-treated samples was significantly fewer than that for mock-treated samples (Fig. 1F). Supernatants collected from this experiment were used to determine the released virus load (fig. S2). Immunoblotting for the presence of HSV-1 and HSV-2 glycoprotein B (gB) (viral protein) indicated a significant reduction (>50%) in gB bands when cells were treated with HPAC (0.5 mg/ml) compared to mock-treated controls (Fig. 1G). Furthermore, the same experimental setup was implemented again, this time using the cell lysates.
HPAC is biocompatible and nontoxic
To determine the viability of cells in the presence of HPAC, an MTT [3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide] assay was conducted on human corneal epithelial cells (HCEs), human foreskin fibroblasts (HFFs), vaginal epithelial cells (VK2s), and HeLa cell lines. Evidently, no significant toxic effect or loss in cell viability was observed when HPAC was used at concentrations as high as 10 mg/ml (Fig. 1I). All the concentrations used showed cell viability greater than 75%, proving no cytotoxic effects in HCEs, HFFs, VK2s, and HeLa cell lines when HPAC is used at a size of about 1 μm.
Furthermore, we wanted to understand whether the addition of HPAC to ocular tissue causes visual obstruction. In this regard, we performed an indirect measure by analyzing optical densities of HPAC at various concentrations within the visual spectrum (400 to 700 nm). We observed that the optical density of HPAC was near zero under the concentration of 0.5 mg/ml for all measurable wavelengths (Fig. 1J and fig. S3).
HPAC treatment does not induce host cytokine response
While the ability of HPAC to trap viruses was evident from entry assays, we sought to further probe its effect on host cytokine response. Previous studies on nanoparticles have shown that their prolonged surface attachment could instigate cytokine production and, in turn, create disease-like effects (33–36). In addition, many nanoparticle- and microparticle-based antivirals inhibit viral replication/entry through up-regulation of cytokines such as interferon-α (IFN-α), IFN-β, and tumor necrosis factor–α (TNF-α) (34). To evaluate this potential effect with HPAC, we probed for common host-triggered antiviral markers through quantitative reverse transcription polymerase chain reaction (PCR). HCEs and HFFs treated with HPAC for a period of 24 hours did not elicit this response when compared to mock-treated or HSV-1/2–infected samples (figs. S4 and S5). While these results do not support the notion that HPAC treatment elevates host immune response, it does indicate that repetitive HPAC application does not result in interferon elevation.
HPAC encapsulates drugs with high loading efficiency
Next, we sought to test whether HPAC particles can improve efficacy or generate synergy when combined with a U.S. Food and Drug Administration (FDA)–approved treatment such as ACV. Given their surface-active properties, HPAC particles (1 mg/ml) were incubated with ACV (100 μg) solution for a period of 12 hours to enable drug loading. At the end of incubation, HPAC particles were centrifuged, and the loading concentration was determined via the following standard formula
To our surprise, we found greater than 99% drug loading of HPAC when incubated with ACV (Fig. 2A). A negligible amount of ACV was detected in the supernatant of the HPAC-drug mix, indicating that almost 100% of ACV was loaded into the HPAC particles forming a DECON particle (37, 38).
Fig. 2. ACV release from DECON is triggered by the addition of virus.
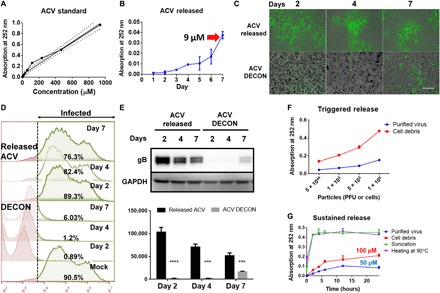
ACV (100 μg) dissolved in dimethyl sulfoxide (DMSO) was added to 1 ml of HPAC (1 mg/ml) and incubated overnight to estimate drug loading. (A) ACV standard curve generated by ultraviolet (UV) absorbance at 252 nm. (B) ACV release from HPAC was measured by dispersing DECON in minimum essential medium (MEM) at 1 mg/ml dilution for 7 days, and readings were taken every day to estimate the amount of ACV release from DECON. (C) DECON was dispersed in seven individual tubes containing MEM at 1 mg/ml dilution and incubated at 37°C. Each day for 7 days, a single tube was taken and centrifuged at 14,000g for 5 min. Two microliters of the supernatant was used to estimate ACV concentration via a UV spectrometer. Five hundred microliters of the supernatant was overlaid on HSV-1–infected HCEs to test the antiviral efficacy. DECON pellet was washed and dispersed in fresh 1-ml MEM before adding it to the HSV-1–infected cells. Representative fluorescence images of HSV-1–infected cells treated with the supernatant (top) or DECON pellet (bottom) on days 2, 4, and 7. Green represents 17 GFP HSV-1 virus. Scale bar (similar for all images), 100 μm. (D) Flow cytometry was conducted on the aforementioned experiment. Briefly, HCEs infected with HSV-1 17 GFP virus were overlaid with either released ACV or ACV-loaded DECON taken on either day 2, 4, or 7. Twenty-four hours after infection, cells were washed, resuspended, and fixed with 4% paraformaldehyde before they were analyzed through a BD Accuri C6 flow cytometer. Noninfected (GFP-negative) cells were used as negative control and infected-nontreated (GF-positive) cells were used as positive control. The panel on the right side indicated in green color represents the number of cells infected in each treatment group. X-axis indicates fluorescence. Cells to right of dashed line show fluorescence exceeding that of negative controls. (E) Representative immunoblots (top) and quantification (bottom) from samples treated with supernatant or DECON pellet on respective days. ***P < 0.001; ****P < 0.0001. (F) Varying concentrations of purified virus and cell debris were added to fresh DECON (1 mg/ml) to estimate whether they triggered ACV release from DECON. Samples were incubated at 37°C for 15 min before they were centrifuged, and UV absorbance readings were recorded at 252 nm on the supernatants to estimate ACV release from DECON. (G) Burst and sustained release profiles for DECON. DECON dispersed at 1 mg/ml in MEM was either sonicated, heated at 90°C for 5 min, or incubated with purified virus or cell debris. ACV release was estimated using the supernatants from these samples for a period of 24 hours.
After multiple washes, we tested DECON particles for their drug release characteristics when incubated in phosphate-buffered saline (PBS) or minimum essential medium (MEM) alone. Unexpectedly, DECON particles did not release any significant amount of ACV into the surrounding media over an incubation period of 48 hours. To further test sustained drug release, we incubated DECON particles with 1 ml of MEM for a period of 7 days, and samples of the released drug were collected every day via centrifugation. Supernatants collected from the samples were analyzed for the presence of ACV followed by their addition to HCEs infected with HSV-1. Similarly, the pellets were resuspended in MEM and added to HSV-1–infected cells. If a significant amount of ACV was released into the MEM over a period of 7 days, then there would be a quantifiable antiviral activity associated with the supernatant or the pellet. To our surprise, while ultraviolet (UV) measurement showed only a release of 9 μM ACV into the media over a period of 7 days (Fig. 2B), the overlaid supernatant showed minimal antiviral activity in HSV-1–infected HCEs [Fig. 2, C (top), D, and E]. However, the media containing the pellet of DECON particles showed potent antiviral activity [Fig. 2, C (bottom), D, and E]. This surprised us because although DECON was not passively diffusing ACV into its surrounding media, it showed a potent antiviral activity when added to HSV-1–infected cells.
Drug release in DECON is triggered by the addition of virus
In the previous section, we made the observation that DECON did not passively release drug into the surrounding media. This led us to hypothesize that DECON was interacting with the virus or cellular surface, triggering the release of the drug. To test this hypothesis, we conducted a series of experiments involving the addition of purified virus and cell debris to DECON. First, we added increasing amounts of cell debris and purified virus to the DECON. Unexpectedly, their addition triggered the release of ACV (UV absorption at 252 nm) in a dose-dependent manner (Fig. 2F). In the previous section, we noted that HPAC is able to trap viruses in its pores and attach to cells; it is for this reason that the cellular and viral components could competitively bind the surface of DECON, dislodging ACV from its pores. We also observed that heating DECON to 90°C for 5 min or sonicating it for 20 s released most of the ACV (Fig. 2G).
To further investigate whether DECON releases ACV over a period of time in a sustained fashion, we incubated lower concentrations of purified virus and cell debris with DECON for a period of 24 hours. As expected, the addition of virus and cell components was able to release significant amounts of ACV by 24 hours (Fig. 2G). Incubation with purified virus triggered the release of approximately 50 μM ACV over a period of 24 hours, a concentration recommended for potent antiviral activity in vitro.
DECON particles restrict HSV infection in vitro at low concentration
From the previous section, we were able to understand that DECON does not release the trapped drug passively; however, it releases ACV in a sustained manner when incubated with virus or cells. To comprehensively test the efficacy of drug release and resulting antiviral activity of DECON particles, we used a prophylactic and therapeutic model of treatment on HCEs followed by HSV-1 infection in vitro.
During prophylactic treatment, we observed no viral protein or viral replication in DECON-treated cells compared to HPAC alone or mock-treated samples at 0.1 mg/ml concentration (Fig. 3, A to C). Furthermore, HCEs treated with DECON did not show any signs of toxicity and were well tolerated. The antiviral efficacy of DECON particles was tested at various concentrations starting from 0.05 to 0.5 mg/ml, and we found that DECON had an IC90 at 0.08 mg/ml. DECON addition was as effective as ACV alone even when the drug administration was carried out 24 hours after initial HSV infection (fig. S6). Last, we tested the efficacy of DECON on other Herpesviridae viruses including HSV-2, pseudorabies virus (PRV), and bovine herpes virus (BHV) (Fig. 3, D to F). ACV-loaded DECON was able to curb viral replication by multiple log10-fold.
Fig. 3. Prophylactic or therapeutic use of DECON protects from herpes infections in vitro.
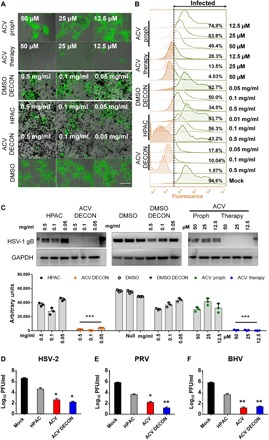
(A) Fluorescent images showing extent of HSV-1 infection (green) in HCEs treated with either ACV-loaded DECON, DMSO-loaded DECON, HPAC alone, mock DMSO, prophylactically added ACV, or therapeutically added ACV. Scale bar (similar for all images), 100 μm. (B) Flow cytometry conducted on the samples at 24 hpi showing the extent of cells infected with HSV-1 GFP. Cells to right of dashed line show fluorescence exceeding that of negative controls. (C) Representative immunoblots (top) and quantification (bottom) for samples showing HSV-1 gB protein in comparison with GAPDH for HSV-1–infected HCEs at 24 hpi. (D to F) Plaque assay data showing extent of virus inhibition by DECON in comparison to ACV, HPAC, and mock DMSO for HSV-2, PRV, and BHV for intracellular virus collected 24 hpi. Data are presented as means ± SD. Significance to mock-treated cells was determined by one-way ANOVA followed by Dunnett’s multiple comparisons test (n = 3 replicates).
Single topical dose of DECON on alternate days restricts HSV-1 replication in vivo
Next, we looked at the in vivo efficacy of DECON treatment in mice while also optimizing the dosing and administration. Since DECON sticks to the cells and releases drug upon viral infection, we hypothesized that when DECON is administered in vivo, alternate days of treatment would suffice to restrict viral replication. To compare the efficacy of DECON with existing topical antivirals, we used TFT as control along with HPAC and mock PBS treated in a murine model of ocular infection. Treatments were started day 1 after infection and administered every alternate day for 11 days. Our results showed that except for DECON, none of the other topical treatments were able to restrict viral replication (Fig. 4A). Note that the concentration of DECON used in this experiment was as low as 0.1 mg/ml, which had low to no visual obstruction, as shown in Fig. 4B. The reduction in viral replication was observed by plaque assays conducted using ocular washes collected on days 2, 4, and 7 after infection (Fig. 4C). Up to 20% of TFT, 60% of HPAC, and 80% of mock-treated mice succumbed to death by day 15 (Fig. 5D). DECON treatment was observed to be very effective where the mice showed no signs of blepharitis or corneal keratitis or any disease score during the period of infection (Fig. 4E). It is pertinent to note that murine eyes treated with DECON were healthy and did not show any signs of local tissue inflammation when compared to those infected with HSV-1 (Fig. 4F and fig. S7). Draining lymph nodes collected from these mice showed that mice treated with DECON did not elicit any germinal response (increase in lymph node size) in the presence or absence of infection (Fig. 4, G and H). Furthermore, no decline in the corneal sensitivity was observed for mice treated with DECON (Fig. 4I). These results indicate that ACV delivery through our novel DECON platform can reduce the number of times these topical antivirals need to be administered.
Fig. 4. Alternate-day ocular dosing with DECON curbs HSV-1 in a murine model of ocular infection.
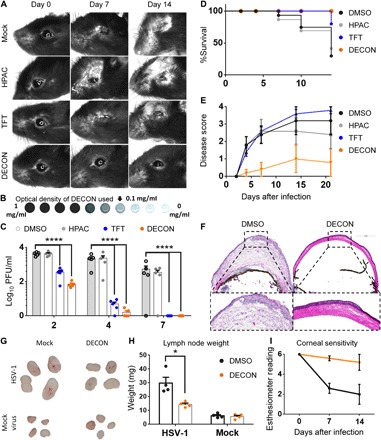
C57BL/6 mice (four groups; n = 5) were infected with HSV-1 McKrae on day 0. ACV-loaded DECON, TFT, HPAC, or DMSO was topically administered on alternate days for a period of 11 days. (A) Stereoscope images of the ocular region taken on days 0, 7, and 14 for mice infected and treated as stated above. (B) Representative image of the DECON concentration used for this set of experiments (top) is shown. (C) Ocular washes were collected on days 2, 4, and 7 and analyzed for the presence of virus through plaque assays. (D) Kaplan-Meier survival curves for the infected and treated mice. (E) Disease scores (0 to 4; 4 being severe) taken on days 2, 4, 7, 10, 14, and 21 were scored in a blinded fashion. (F) Mice were euthanized on day 21, and their eyes were frozen in OPT (Optimal Cutting Temperature) medium for histology. Ten-micrometer sections of the eye for all the groups were taken and stained with hematoxylin and eosin stain. (G and H) Draining lymph nodes isolated from mice either mock-infected or HSV-1–infected and either mock-treated or DECON-treated were photographed and weighed. (I) Corneal sensitivity measured using a manual esthesiometer in mice (n = 5 per treatment group). Lower scores indicate loss in corneal sensitivity. (Photo credit: Tejabhiram Yadavalli, University of Illinois at Chicago)
Fig. 5. Topical vaginal application of DECON on alternate days is as effective as daily systemic ACV dosing.
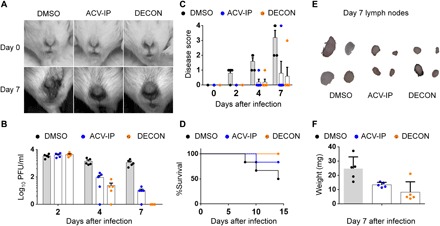
Naïve BALB/c mice (three groups; n = 5 per group) were primed with medroxyprogesterone before intravaginal HSV-2 infection. Starting at day 1 after infection, mice were treated with either topical DECON (alternate days) or systemic ACV (5 mg/kg; every day) by intraperitoneal (IP) injections. (A) Representative stereoscope images of the murine genital region 0 and 7 days after infection. (B) Vaginal swabs collected on days 2, 4, and 7 were overlaid on Vero cells to estimate the extent of virus production through plaque assays. (C) Disease scores were given in a blinded fashion from 0 to 4, with 4 being severe. (D) Kaplan-Meier survival curves for the infected and treated mice. (E) Draining lymph nodes for the infected and treated mice were collected on day 21, washed, photographed, and weighed (F). (Photo credit: Tejabhiram Yadavalli, University of Illinois at Chicago)
Intravaginal DECON administration is as effective as systemic ACV therapy during HSV-2 infection
With the success of topical ocular therapy with DECON, we next investigated whether this delivery platform can be effective for vaginal HSV-2 infections as well. Naïve BALB/c mice were primed with medroxyprogesterone 5 days before intravaginal HSV-2 infection. Starting at day 1 after infection, mice were treated with either topical DECON or systemic ACV (5 mg/kg) by intraperitoneal injections. While systemic ACV was administered every day, DECON was administered intravaginally every other day. The genital area was imaged using a stereoscope on days 0 and 7 to visualize the extent of damage done to the site of infection. All the mice had equal amount of infection on day 2 after infection; however, we saw a steady reduction in viral titers on days 4 and 7 after infection for ACV-treated and ACV-loaded DECON–treated mice (Fig. 5B). This was observed by the phenotype of infection and analyzed by plaque assays from vaginal swabs. Furthermore, we observed only slight inflammation for the ACV- and DECON-treated mice compared to mock-treated mice, which had severe inflammation and scarring of the vaginal and surrounding anogenital area (Fig. 5C). In the mock-treated group, 60% of mice succumbed to death by day 14 after infection, while 20% of mice treated with systemic ACV and none of the DECON-treated mice succumbed to death (Fig. 5D). The lymph nodes collected for DECON- and ACV-treated mice on day 14 after infection showed little to no increase in size, while mock-treated mice had predominantly large and inflamed lymph nodes (Fig. 5, E and F). In conclusion, this confirms that alternate-day treatment with ACV-loaded DECON is as beneficial as a daily dose of systemic ACV to curb HSV-2 infection in the genital region.
DISCUSSION
Activated charcoal is highly porous in nature and has a surface area far greater than any other nanoparticle or microparticle known to materials science (39, 40). Furthermore, carbonization at very high temperatures in the presence of steam activates every pore present on the surface of charcoal (41, 42). On average, the iodine absorption coefficient of activated charcoal is greater than 1, suggesting that it can absorb more iodine (or other effluent molecules) than its own weight (41, 43). Given this property, activated charcoal has been widely used in various industries including clinics and emergency medical departments. Clinically, it is used as emergency medication to treat drug overdose since it adsorbs specific drugs in large amounts, thereby preventing their absorption in the human gut (44, 45). Their role as scavenging systems for poison ingestion has proven to be a lifesaving emergency treatment (45). Activated charcoal is also currently available as a prescription-free dietary supplement for relieving gastric trouble and bloating of the stomach, although their efficacy has not been scientifically tested (46). Furthermore, owing to their toxin-scavenging properties, their use as gut cleansers in the health drink industry has also seen an upsurge (47).
Given activated charcoal’s unique surface properties and its applications in cosmetics, we wanted to investigate not only its antiviral potential but also its possible use as a drug delivery platform. HPAC is generally regarded as safe for human use by the FDA and adverse effects due to their exposure have seldom been reported. Their TC50 value is considered well above 1 g/kg body weight for adults and 0.5 g/kg body weight for infants. This accounts for a TC50 value greater than 50 mg/ml of HPAC for human use (48). Pro-kidney health benefits also make activated charcoal an attractive platform for drug delivery, especially in cases where renal failure has been reported after the prolonged use of a drug such as ACV (30–32, 49, 50).
Few studies in the past have reported the use of activated charcoal in the treatment of STDs. A patent by The Procter & Gamble Company claimed a method of preventing pregnancy by administering a safe and effective amount of activated charcoal to the subject in need. They also claimed that activated charcoal could be used to prevent the transmission of STDs. However, their claim was limited to preventing bacterial infections such as Chlamydia trachomatis and Neisseria gonorrhoeae (51). In another study by Tominaga et al. (52), an unblinded prospective randomized controlled 10-day trial was designed to study 64 women with bacterial vaginosis. The study was divided into two groups where one group was treated with 100 mg of chloramphenicol and the other group was treated with transvaginal tampons dipped in 10% w/v activated charcoal solution. While both treatments successfully reduced the amount of vaginal secretion and showed significant improvements in the magnitude of malodor, 27 of 32 (84%) women from the group treated with chloramphenicol completely lost vaginal flora (lactobacilli). However, only 3.1% of those treated with activated charcoal lost vaginal flora, indicating a potentially promising treatment for this infection without adverse effects (53, 54).
We tested HPAC as a prophylactic and therapeutic treatment against both HSV-1 and HSV-2 infection. We found that HPAC was able to efficiently restrict viral entry into cells when added prophylactically (added before infection) to the cell surface. We further observed that when added therapeutically (added after infection), HPAC showed potent inhibition in viral spread and replication. Furthermore, we analyzed the efficiency of HPAC in (i) limiting cell-to-cell syncytial fusion using a virus-free transfection assay and (ii) trapping the virus using a neutralization entry assay. Results from these studies showed that HPAC not only was able to restrict syncytia formation but also trapped virus efficiently in its nanopores.
To test synergistic benefits as well as drug loading potential, we incubated ACV with HPAC (DECON). HPAC was able to efficiently absorb ~100% of the drug, leaving no traces of ACV in free solution. HPAC showed slight signs of drug release from its nanopores over an incubation period of 7 days. While the drug loading efficiency was promising for HPAC, drug release studies initially damped our enthusiasm regarding its applicability as a drug delivery agent. We then tested our DECON particles for antiviral activity and, to our surprise, saw that DECON exhibited antiviral efficacy whether added prophylactically or therapeutically to HSV-infected cells (both HSV-1 and HSV-2). In contrast, prophylactically added ACV did not elicit a robust antiviral response, unlike that observed when added therapeutically. The robust antiviral activity seen from DECON may be a result of its charged porous surface interacting with the cell’s surface, inducing an active exchange of ions (Na+, K+, Ca+, Cl−, and OH−) and resulting in sustained or slow release of ACV from its pores. Alternatively, given that HPAC efficiently traps HSV, we showed that the addition of virus to DECON-treated cells triggers ACV release from its pores when virus is captured in them (fig. S8).
These results by DECON prompted us to perform antiviral efficacy studies in vivo in a murine model of ocular (HSV-1) and genital (HSV-2) infection. Typical anti-HSV studies administer compounds two to six times a day to achieve effective response. However, to show that DECON particles can effectively stick and deliver drugs over a prolonged period of time, we topically administered DECON on alternate days. As expected, we saw significant differences with reduced infection upon DECON treatment. For the ocular model of HSV-1 infection, groups treated with HPAC and TFT on alternate days did not show any significant reduction in viral replication when compared to mock-treated groups. Furthermore, DECON treatment during vaginal HSV-2 infection showed therapeutic efficacy equal to or better than systemically administered ACV. Note that while topical administration of DECON was effective when administered on alternate days, ACV treatment was effective only when administered every day for a period of 7 days. In conclusion, our drug delivery model (fig. S8) hypothesizes that when HSV (fig. S8, step 1 or 5) infects corneal/vaginal epithelium, the presence of DECON (fig. S8, step 2) helps capture the virus and releases the encapsulated drug (fig. S8, step 3), providing a dual protection (fig. S8, step 4) to the treated cells, while nontreated tissue gets infected (fig. S8, step 6), leading to viral shedding (fig. S8, step 7), which, in turn, causes inflammation and immune cell infiltration into the infected tissue (fig. S8, step 8).
MATERIALS AND METHODS
HPAC produced via carbonization of coconut shell at 1300°C, in the size range of 1 μm, was procured from US Research Nanomaterials Inc., TX, USA. Accordingly, aqueous solutions (10 and 1 mg/ml) were prepared by mixing required amounts of HPAC to sterile PBS. These HPAC powders are reported to have an iodine adsorption of 1380 mg/g and a specific surface area of 1360 m2/g. ACV was purchased from Selleckchem at 99% purity (50 mg) and used at a dilution of 50 mM in dimethyl sulfoxide (DMSO).
Cells and viruses
Chinese hamster ovary (CHO-K1) cells were provided by P. G. Spear (Northwestern University). CHO-K1 cells were passaged in Ham’s F12 medium (Gibco/BRL, Carlsbad, CA, USA) supplemented with 10% fetal bovine serum (FBS) and 1% penicillin and streptomycin (P/S) (Sigma-Aldrich). HeLa cells were provided by B. P. Prabhakar (University of Illinois at Chicago), which were passaged in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% FBS and 1% P/S. African green monkey kidney (Vero) cells and HCEs were provided by P. G. Spear (Northwestern University). Human VK2/E6 cells and HFFs were obtained from the American Type Culture Collection. Vero, VK2, and HFF cells were passaged in DMEM supplemented with 10% FBS and P/S. The HCE cell line (RCB1834 HCE-T) was obtained from K. Hayashi (National Eye Institute) and passaged in MEM (Gibco/BRL, Carlsbad, CA, USA) supplemented with 10% FBS and 1% P/S.
HSV-1 (17 GFP), HSV-2 (333 strain), HSV-2 GFP (333 strain, GFP variant), and β-galactosidase–expressing HSV-1 (gL86) and HSV-2 (333)gJ− used in this study were provided by P. G. Spear’s laboratory at Northwestern University. Virus stocks were propagated and titered on Vero cells and stored at −80°C. PRV and BHV were provided by G. Smith and R. Longnecker, respectively, from Northwestern University. Purification of virus was performed using a sucrose gradient method. Briefly, Vero cells were infected with HSV at an MOI of 0.1. Three days after infection, supernatants from these infected cells were collected and stored at 4°C while sucrose gradients were prepared. Sucrose gradient was prepared, carefully layering 10, 15, 30, and 60% aqueous sucrose solutions in a centrifuge tube. The virus supernatants were carefully poured on top of the sucrose gradient and spun at 26,000g for 1 hour at 4°C. A thin white virus layer that formed between the 30 and 60% sucrose gradient was carefully removed using a micropipette and poured into a separate centrifuge tube. This virus layer was washed two to three times with distilled water using high-speed centrifugation. After the final wash, the pellet was resuspended in PBS and titrated on Vero cells by a plaque assay.
MTT assay
MTT viability assay on HCE VK2, HFF, and HeLa cell lines using various concentrations of HPAC was performed after 24 hours of incubation. Briefly, cells were plated at a density of 1 × 104 per well in a 96-well plate overnight. The following morning, concentrations starting at 10 mg/ml were twofold serially diluted and added to cell monolayers in whole media for a period of 24 hours. At the end of incubation, MTT (0.5 mg/ml in whole media) was added to cells and incubated for a period of 3 hours to allow crystal formation. Acidified isopropanol (1% glacial acetic acid v/v) was added to cells to dissolve the formazan crystals. Dissolved violet crystals were transferred to a new 96-well plate and analyzed by a microplate reader (Tecan GENious Pro) at 492 nm. MTT assay was conducted under cell-free HPAC conditions, and their values were subtracted from the cell-mediated conditions to account for signal generated through oxidative degradation of MTT by HPAC alone.
Treatment models
HPAC was dispersed in Opti-MEM at 10 mg/ml concentration and used as stock for all the experiments. DECON was dispersed in Opti-MEM at 1 mg/ml concentration and used as stock for all the experiments.
Prophylaxis
HPAC or DECON was added to a monolayer of cells at the required concentration 30 min before infection. Cell monolayer was washed twice with PBS before whole media with required HSV infection were added.
Neutralization
HPAC or mock at the required concentration was incubated with HSV-1 or HSV-2 for a period of 30 min in an Eppendorf tube and mixed continuously in a tube holder before the contents were centrifuged at 10,000g. The supernatant was carefully removed without disturbing the pellet and added onto a monolayer of cells.
Viral entry assay
β-Galactosidase–expressing viruses HSV-1 gL86 and HSV-2 (333)gJ− at an MOI of 10 were used in this study. Cells were plated at a density of 1 × 104 in 96-well plates overnight before use. Neutralization or prophylactic treatment using HPAC at concentrations of 1, 0.5, and 0.1 mg/ml was conducted before the cells were infected with the virus at 37°C. HSV-1 strain gL86 and HSV-2 strain gJ− were allowed to infect cell monolayers for 6 hours, after which the cells were washed with PBS twice and 100 μl of soluble substrate o-nitrophenyl-β-d-galactopyranoside (3 mg/ml) was added to the cells along with 0.5% Nonidet P-40 in PBS. Enzymatic activity was measured by a microplate reader (Tecan GENious Pro) at 405/600 nm.
Immunoblotting
Cells were scraped and incubated with 100 μl of radioimmunoprecipitation assay buffer with protease phosphatase inhibitor [Halt Protease and Phosphatase Inhibitor Cocktail (100×); Thermo Fisher Scientific] for a period of 30 min on ice. Whole-cell protein extracts (supernatant) were collected by centrifuging the mixture at 13,500 rpm on a benchtop refrigerated (4°C) centrifuge for 15 min. Protein samples were quantified (Thermo Fisher Scientific Bicinchoninic Acid Kit), normalized, and then denatured in NuPAGE LDS Sample Buffer (Invitrogen, NP00007) and β-mercaptoethanol by heating them to 80°C for 10 min. The denatured protein samples were allowed to cool, and equal amounts of protein were added to 4 to 12% SDS–polyacrylamide gel electrophoresis loading gels and run at a constant speed at 70 V for 3 hours. The protein from the gel was then transferred to a nitrocellulose membrane using an iBlot 2 dry transfer instrument (Thermo Fisher Scientific, USA). Nitrocellulose membrane was blocked in 5% nonfat milk in tris-buffered saline and 0.1% Tween 20 (TBST) for 1 hour at room temperature. After the blocking step, membranes were incubated with anti–HSV-1 and anti–HSV-2 gB mouse monoclonal antibody (Abcam, 6506) or anti-GAPDH (glyceraldehyde-3-phosphate dehydrogenase) (Proteintech, 10494-1-AP) antibody at dilutions of 1:1000 overnight at 4°C. The following day, the blots were washed multiple times with TBST before the addition of horseradish peroxidase–conjugated secondary immunoglobulin G antibody at dilutions of 1:10,000 at room temperature. Protein bands were visualized on an ImageQuant LAS 4000 imager (GE Healthcare Life Sciences) by the addition of SuperSignal West Pico maximum sensitivity substrate (Pierce, 34080). The density of the bands was quantified using ImageQuant TL image analysis software (version 7). GAPDH was measured as a loading control.
Plaque assay
Cells infected with virus mentioned in the previous section were suspended in 500 μl of Opti-MEM and sonicated using a probe sonication system at 70% amplitude for 30 s. These sonicated cells were used as inoculants for a plaque assay to determine the total amount of infectious viruses released from the infected cells. In a typical experiment, Vero cells were plated at a seating density of 5 × 104 per well in a 24-well plate. Upon confluency, the cell monolayers were washed with PBS, and supernatants (mentioned above) were used as inoculants of infection for a period of 2 hours. After completion of 2 hours, the cells were washed twice with PBS, and DMEM mixed with 0.5% methylcellulose was added. The plates were incubated for 72 hours at 37°C and 5% CO2 before they were fixed with methanol and stained with crystal violet to determine the extent of plaque formation. Plaques were counted using a microscope with 2× objective, and total plaque count was tallied with each other to determine the efficacy of activated charcoal treatment.
Quantitative PCR assay
RNA from cells was extracted using TRIzol (Life Technologies) according to the manufacturer’s described protocol. The thus obtained RNA was quantified using NanoDrop (Thermo Fisher Scientific, USA) and equilibrated for all samples with Molecular Biology Grade water (Corning, USA) before they were reverse-transcribed into complementary DNA (cDNA) using the High-Capacity cDNA Reverse Transcription Kit (Applied Biosystems, Foster City, CA). Equal amounts of cDNA were analyzed via real-time quantitative PCR using Fast SYBR Green Master Mix on QuantStudio 7 Flex system (Applied Biosystems). The primers used in this study were as follows: GAPDH forward, 5′-TCCACTGGCGTCTTCACC-3′; GAPDH reverse, 5′-GGCAGAGATGATGACCCTTTT-3′; IFN-α forward, 5′-GATGGCAACCAGTTCCAGAAG-3′; IFN-α reverse, 5′-AAAGAGGTTGAAGATCTGCTGGAT-3′; IFN-β forward, 5′-CTCCACTACAGCTCTTTCCAT-3′; IFN-β reverse, 5′-GTCAAAGTTCATCCTGTCCTT-3′; TNF-α forward, 5′-AGCCCATGTTGTAGCAAACCC-3′; TNF-α reverse, 5′-GGACCTGGGAGTAGATGAGGT-3′.
Fluorescent virus infection model
Mock, neutralization, and prophylactic treatments similar to those mentioned in earlier sections were conducted with an MOI of 50 using GFP fluorescent viruses. The infection was allowed to occur for a period of 90 min at 4°C followed by a brief incubation of 15 min at 37°C. This process allowed the virus to remain on the cell surface rather than enter the cell. After 15 min of incubation, the cells were fixed using 4% paraformaldehyde followed by nuclear (4′,6-diamidino-2-phenylindole) and actin (rhodamine-phalloidin) staining. The cells were visualized to understand the extent of infection reaching the cell surface. Five images per sample were procured and quantitatively analyzed for GFP fluorescence per image. The quantitative data would directly translate to the total amount of viral infection in a given area. The fluorescent intensity of all of the channels was kept constant, and the images were analyzed using MetaMorph Microscopy Automation and Image Analysis Software to quantitatively determine total intensity from the green channel.
Flow cytometry
Cells infected with GFP virus were collected 24 hpi using Hanks’-based, enzyme-free cell dissociation buffer (Gibco, 13150). Cells were washed once with PBS before they were fixed using 4% parafolmaldehyde (Electron Microscopy Sciences, USA) and suspended in fluorescence-activated cell sorting (FACS) buffer (PBS and 5% FBS). Cell suspensions were filtered through a 70-μm mesh, and cells were resuspended in FACS buffer before analyzing them using an in-house flow cytometer (BD Accuri C6 Plus). A total of 4 × 104 cells were collected for each sample, and the analysis was performed using FlowJo (version 10).
ACV standard curve
Increasing amounts of ACV (Selleckchem, USA) dissolved in DMSO was suspended in PBS to make a concentration gradient. NanoDrop UV Spectrophotometer was used to determine the absorption peak of ACV at 252 nm. Typically, 2 μl of the desired solution was dropped onto the analysis port of the NanoDrop before closing the lid and starting the measurement. Each measurement was conducted thrice with three individual drops, and three separate replicates were used to determine the absorbance of every ACV concentration. The readings were noted down manually and entered into GraphPad Prism software to prepare the concentration versus absorbance curve.
Drug loading studies
ACV (100 μg), dissolved in 50 μl of DMSO, was added to HPAC (1 mg/ml) and incubated at room temperature overnight on a 3D rocker (BioRocker 3D, Denville, USA). The mixture was centrifuged at 14,000g for 15 min to separate the drug-loaded HPAC particles from the free drug supernatant. Supernatant was analyzed for the amount of ACV using the UV spectrophotometer, as mentioned above. Three individual loading experiments were conducted to analyze the drug loading efficiency, and as-prepared DECON particles were stored at 4°C for further characterization.
Drug release studies
Passive release
Seven individual vials containing DECON particles (1 mg/ml) diluted in 1 ml of MEM were incubated at 37°C for up to 7 days. Every day, a single vial was removed and centrifuged at 14,000g for 15 min. The supernatant (2 μl) was used to analyze the ACV concentration using a UV spectrophotometer, as mentioned above. MEM was used as blank for these studies.
Triggered release
For active drug release studies, purified virus (sucrose gradient purification) and cell debris (50 million HCEs sonicated in 1 ml of MEM) were used. Increasing concentrations of purified virus and cell debris were added to DECON (1 mg/ml) in MEM and incubated for a period of 15 min at 37°C before the mixtures were spun at 14,000g, and the supernatants were analyzed using the UV spectrophotometer for the presence of ACV. In parallel, DECON (1 mg/ml in MEM) was heated to 90°C using a preheated heat block for a period of 5 min before the samples were centrifuged and the supernatant was analyzed for ACV. In addition, DECON (1 mg/ml in MEM) was sonicated using a probe sonicator (Fisher Scientific, USA) at 30% amplitude for 40 s separated by a 5-s pause. The sample was then centrifuged at 14,000g before the supernatant was analyzed for ACV using a UV spectrophotometer.
Sustained release
A total of 5 × 104 PFU (plaque-forming units) of purified virus (18) or equivalent cell debris was added to 100 μl of DECON (1 mg/ml in MEM) and incubated at 37°C for a period of 24 hours. At set time intervals, 2 μl of supernatant from the vial was removed and analyzed for the concentration of ACV using a UV spectrophotometer.
In vivo ocular HSV-1 infection and treatment
C57BL/6 mice, bred and housed at the university biological resource laboratory, were used for the ocular model of murine HSV-1 infection. Standard feed and water were provided to the mice with a 12-hour light/12-hour dark cycle with no more than five mice per cage. On day 0, 6- to 8-week-old mice were anesthetized, as described previously, before the application of a topical anesthetic (proparacaine hydrochloride, 0.5%). Corneal epithelial debridement was performed using a 30-gauge needle followed by the application of 5 × 105 PFU HSV-1 (McKrae) to the eye. Topical treatments were performed on days 1, 3, 5, 7, and 9 after infection, while ocular washes and mice pictures (Carl Zeiss Stereoscope) were collected on days 2, 4, 7, and 10 after infection.
Corneal sensitivity recording
Corneal sensitivity of mouse eyes was measured by a manual esthesiometer (12/100 mm; Luneau SAS, France). The esthesiometer consisting of a nylon filament 6 cm in length is applied to the center of the mouse cornea, and the pressure exerted was measured as blink response. At the highest length, if the mice blinked, it was considered most responsive, and the absence of a blink at the shortest length was considered least responsive. The length of the nylon filament was reduced 0.5 cm at a time to record the blink response. The measurement was taken in triplicate at 10-s intervals. The mice that did not respond were given an arbitrary score of 0. A high score indicates high/normal corneal sensitivity, while a low score corresponds to the absence or loss of corneal sensitivity.
In vivo vaginal infection and treatment
Naïve 4-week-old female BALB/c mice were purchased from the Jackson Laboratory (Bar Harbor, USA) and housed in the university biological resource laboratory for a period of 1 week for acclimatization before each one was subcutaneously injected with 2 mg of medroxyprogesterone (Depo-Provera). On day 5 after injection, mice were intravaginally infected with 1 × 106 PFU HSV-2 (333 strain). Similar to the ocular model of infection, DECON was applied topically (intravaginally) on alternate days, while ACV (50 mg/ml in PBS) was administered via an intraperitoneal injection. PBS was applied intravaginally as mock treatment for the control group. Vaginal swabs were collected using a Calgiswab (Calcium Alginate Mini-tip Urethro-Genital Swab, Puritan) dipped in Opti-MEM (Gibco, USA). Images of the anogenital region were taken on day 0 and day 7 after infection using a Carl Zeiss stereoscope at 7.5× magnification.
Experiments involving animals were performed under a UIC (University of Illinois at Chicago)–approved protocol ACC 14-091. The mice were monitored for weight loss, and disease scores were recorded in a blinded fashion for 14 days. Sick mice were euthanized according to the Institutional Animal Care and Use Committee protocol followed by the collection of ocular/vaginal tissue and lymph nodes. Ocular wash and vaginal swabs were used to assess viral titers using a plaque assay.
Statistical analysis
GraphPad Prism software (version 4.0) was used for statistical analysis of each group. P < 0.05 and P < 0.001 were considered as the significant differences among mock-treated and treated groups.
CONCLUSIONS
In this study, we report a unique use of activated charcoal in topical drug delivery. In parallel, we show HPAC as an inhibitor of HSV-1 and HSV-2 viral entry and its subsequent effects on viral replication through prophylactic, neutralization, and therapeutic treatment models. Through immunoblotting techniques and fluorescent imaging, we confirm effective virus trapping of the virus by HPAC. Our treatments using DECON show a promising path toward a cost-effective way of drug delivery. Given that activated charcoal and ACV are FDA-approved products for use in humans and rated generally regarded as safe, we envision widespread development of DECON-based ACV and other topical treatments in the near future. The therapeutic model shown here against genital herpes would also pave the way for future DECON lubricants or gels that prevent and/or treat viral transmission during sexual intercourse. Furthermore, the affordable cost, facile synthesis, and known safety benefits would encourage usage in other dermatology indications and development of unique vanity products that simultaneously provide health and beauty benefits especially for patients with HSV who suffer from frequent outbreaks.
Supplementary Material
Acknowledgments
Funding: This work was supported by grants from the NIH (R01 EY024710, RO1 AI139768, and RO1 EY029426) to D.S. A.A. was supported by an F30EY025981 grant from the National Eye Institute, NIH. Competing interests: D.S. and T.Y. are inventors on a provisional patent application related to this work filed by the University of Illinois at Chicago on 9 April 2019. The authors declare no other competing interests. Author contributions: T.Y. and D.S. designed the experiments. T.Y., J.A., A.A., R.S., D.J., N.T., J.H., and L.K. performed in vitro experiments. T.Y., J.A., R.S., and A.A. performed in vivo experiments. T.Y., J.A., L.K., and D.S. wrote and revised the manuscript. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/5/8/eaax0780/DC1
Fig. S1. HPAC strongly binds to HSV-1 GFP virus.
Fig. S2. Extracellular virus–based plaque assay for HPAC therapy.
Fig. S3. HPAC does not cause a loss in visual acuity by blocking light.
Fig. S4. HPAC does not elevate interferons in HCEs.
Fig. S5. HPAC does not elevate interferons in HFFs.
Fig. S6. DECON is effective when added 24 hours after infection.
Fig. S7. DECON protects the murine cornea from HSV-1 infection.
Fig. S8. Graphical abstract showing DECON protecting cells against viral infection.
Fig. S9. Full-length blots for the Western blot shown in Fig. 1 (G and H).
Fig. S10. Full-length blots for the Western blot shown in Fig. 3D.
REFERENCES AND NOTES
- 1.Agelidis A. M., Shukla D., Cell entry mechanisms of HSV: What we have learned in recent years. Future Virol. 10, 1145–1154 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ruderfer D., Krilov L. R., Herpes simplex viruses 1 and 2. Pediatr. Rev. 36, 86–90 (2015). [DOI] [PubMed] [Google Scholar]
- 3.Patel R., Genital herpes. Sex. Transm. Infect. 93, 444 (2017). [DOI] [PubMed] [Google Scholar]
- 4.Farooq A. V., Shukla D., Herpes simplex epithelial and stromal keratitis: An epidemiologic update. Surv. Ophthalmol. 57, 448–462 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Coleman J. L., Shukla D., Recent advances in vaccine development for herpes simplex virus types I and II. Hum. Vaccin. Immunother. 9, 729–735 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.De Clercq E., Schinazi R. F., Editorial overview: Antiviral strategies. Curr. Opin. Virol. 18, v–vi (2016). [DOI] [PubMed] [Google Scholar]
- 7. Unfortunate synergy between HIV and genital herpes. Can. Fam. Physician 62, 146 (2016). [PMC free article] [PubMed] [Google Scholar]
- 8.R. Emerson, S. Saltzman, “Is activated charcoal really the secret to perfect skin (and teeth)?”; www.allure.com/gallery/activated-charcoal-beauty-products [accessed 2 May 2019].
- 9.Herpetic Eye Disease Study Group , Oral acyclovir for herpes simplex virus eye disease: Effect on prevention of epithelial keratitis and stromal keratitis. Arch. Ophthalmol. 118, 1030–1036 (2000). [PubMed] [Google Scholar]
- 10.Whitley R. J., Gnann J. W. Jr., Acyclovir: A decade later. N. Engl. J. Med. 327, 782–789 (1992). [DOI] [PubMed] [Google Scholar]
- 11.Durai R. D., Drug delivery approaches of an antiviral drug: A comprehensive review. Asian J. Pharm. 9, 1–12 (2015). [Google Scholar]
- 12.Yaldiz M., Solak B., Kara R. O., Cosansu N., Erdem M. T., Comparison of famciclovir, valaciclovir, and brivudine treatments in adult immunocompetent patients with herpes zoster. Am. J. Ther. 25, e626–e634 (2016). [DOI] [PubMed] [Google Scholar]
- 13.Muharemagic D., Labib M., Ghobadloo S. M., Zamay A. S., Bell J. C., Berezovski M. V., Anti-Fab aptamers for shielding virus from neutralizing antibodies. J. Am. Chem. Soc. 134, 17168–17177 (2012). [DOI] [PubMed] [Google Scholar]
- 14.Brandt C. R., Peptide therapeutics for treating ocular surface infections. J. Ocul. Pharmacol. Ther. 30, 691–699 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jaishankar D., Buhrman J. S., Valyi-Nagy T., Gemeinhart R. A., Shukla D., Extended release of an anti–heparan sulfate peptide from a contact lens suppresses corneal herpes simplex virus-1 infection. Invest. Ophthalmol. Vis. Sci. 57, 169–180 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jaishankar D., Yakoub A. M., Yadavalli T., Agelidis A., Thakkar N., Hadigal S., Ames J., Shukla D., An off-target effect of BX795 blocks herpes simplex virus type 1 infection of the eye. Sci. Transl. Med. 10, eaan5861 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Agelidis A. M., Hadigal S. R., Jaishankar D., Shukla D., Viral activation of heparanase drives pathogenesis of herpes simplex virus-1. Cell Rep. 20, 439–450 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yadavalli T., Agelidis A., Jaishankar D., Mangano K., Thakkar N., Penmetcha K., Shukla D., Targeting herpes simplex virus-1 gD by a DNA aptamer can be an effective new strategy to curb viral infection. Mol. Ther. Nucleic Acids 9, 365–378 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yadavalli T., Shukla D., Role of metal and metal oxide nanoparticles as diagnostic and therapeutic tools for highly prevalent viral infections. Nanomedicine 13, 219–230 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Trigilio J., Antoine T. E., Paulowicz I., Mishra Y. K., Adelung R., Shukla D., Tin oxide nanowires suppress herpes simplex virus-1 entry and cell-to-cell membrane fusion. PLOS ONE 7, e48147 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Antoine T. E., Hadigal S. R., Yakoub A. M., Mishra Y. K., Bhattacharya P., Haddad C., Valyi-Nagy T., Adelung R., Prabhakar B. S., Shukla D., Intravaginal zinc oxide tetrapod nanoparticles as novel immunoprotective agents against genital herpes. J. Immunol. 196, 4566–4575 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Antoine T. E., Mishra Y. K., Trigilio J., Tiwari V., Adelung R., Shukla D., Prophylactic, therapeutic and neutralizing effects of zinc oxide tetrapod structures against herpes simplex virus type-2 infection. Antiviral Res. 96, 363–375 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mitra A. K., Anand B. S., Duvvuri S., Drug delivery to the eye. Adv. Organ Biol. 10, (2005). [Google Scholar]
- 24.Hui A., Contact lenses for ophthalmic drug delivery. Clin. Exp. Optom. 100, 494–512 (2017). [DOI] [PubMed] [Google Scholar]
- 25.Mainardes R. M., Urban M. C. C., Cinto P. O., Khalil N. M., Chaud M. V., Evangelista R. C., Gremião M. P. D., Colloidal carriers for ophthalmic drug delivery. Curr. Drug Targets 6, 363–371 (2005). [DOI] [PubMed] [Google Scholar]
- 26.Loftsson T., Stefánsson E., Cyclodextrins in ocular drug delivery: Theoretical basis with dexamethasone as a sample drug. J. Drug Deliv. Sci. Technol. 17, 3–9 (2007). [Google Scholar]
- 27.Le Bourlais C., Acar L., Zia H., Sado P. A., Needham T., Leverge R., Ophthalmic drug delivery systems—Recent advances. Prog. Retin. Eye Res. 17, 33–58 (1998). [DOI] [PubMed] [Google Scholar]
- 28.H. Crosby, “Make your own natural eyeshadow or liner out of charcoal”; https://yumuniverse.com/make-your-own-natural-eyeshadow-or-liner-out-of-charcoal/ [accessed 2 May 2019].
- 29.Melissandre, “New in cosmetics: Activated charcoal”; www.inno-foodproducts-brainbox.com/2017/07/18/new-in-cosmetics-activated-charcoal/ [accessed 2 May 2019].
- 30.Ali B. H., Alza’abi M., Ramkumar A., Al-Lawati I., Waly M. I., Beegam S., Nemmar A., Brand S., Schupp N., The effect of activated charcoal on adenine-induced chronic renal failure in rats. Food Chem. Toxicol. 65, 321–328 (2014). [DOI] [PubMed] [Google Scholar]
- 31.Shimoishi K., Anraku M., Kitamura K., Tasaki Y., Taguchi K., Hashimoto M., Fukunaga E., Maruyama T., Otagiri M., An oral adsorbent, AST-120 protects against the progression of oxidative stress by reducing the accumulation of indoxyl sulfate in the systemic circulation in renal failure. Pharm. Res. 24, 1283–1289 (2007). [DOI] [PubMed] [Google Scholar]
- 32.Yildiz C., Ozsurekci Y., Gucer S., Cengiz A. B., Topaloglu R., Acute kidney injury due to acyclovir. CEN Case Rep. 2, 38–40 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Akhtar J., Shukla D., Viral entry mechanisms: Cellular and viral mediators of herpes simplex virus entry. FEBS J. 276, 7228–7236 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Potter T. M., Neun B. W., Rodriguez J. C., Ilinskaya A. N., Dobrovolskaia M. A., Analysis of pro-inflammatory cytokine and type II interferon induction by nanoparticles. Methods Mol. Biol. 1682, 173–187 (2018). [DOI] [PubMed] [Google Scholar]
- 35.Lee M.-Y., Yang J.-A., Jung H. S., Beack S., Choi J. E., Hur W., Koo H., Kim K., Yoon S. K., Hahn S. K., Hyaluronic acid-gold nanoparticle/interferon α complex for targeted treatment of hepatitis C virus infection. ACS Nano 6, 9522–9531 (2012). [DOI] [PubMed] [Google Scholar]
- 36.Orłowski P., Kowalczyk A., Tomaszewska E., Ranoszek-Soliwoda K., Węgrzyn A., Grzesiak J., Celichowski G., Grobelny J., Eriksson K., Krzyzowska M., Antiviral activity of tannic acid modified silver nanoparticles: Potential to activate immune response in herpes genitalis. Viruses 10, 524 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tsuchiya T., Levy G., Relationship between effect of activated charcoal on drug absorption in man and its drug adsorption characteristics in vitro. J. Pharm. Sci. 61, 586–589 (1972). [DOI] [PubMed] [Google Scholar]
- 38.Jain S., Vyas R. K., Pandit P., Dalai A. K., Adsorption of antiviral drug, acyclovir from aqueous solution on powdered activated charcoal: Kinetics, equilibrium, and thermodynamic studies. Desalination Water Treat. 52, 4953–4968 (2014). [Google Scholar]
- 39.Bayly G. R., Ferner R. E., Activated charcoal for drug overdose. Prescr. J. 35, 12–17 (1995). [Google Scholar]
- 40.Sellers E. M., Khouw V., Dolman L., Comparative drug adsorption by activated charcoal. J. Pharm. Sci. 66, 1640–1641 (1977). [DOI] [PubMed] [Google Scholar]
- 41.Erb F., Gairin D., Leroux N., Activated charcoals: Properties—Experimental studies. J. Toxicol. Clin. Exp. 9, 235–248 (1989). [PubMed] [Google Scholar]
- 42.Zacaroni L. M., Magriotis Z. M., das Graças Cardoso M., Santiago W. D., Mendonça J. G., Vieira S. S., Nelson D. L., Natural clay and commercial activated charcoal: Properties and application for the removal of copper from cachaça. Food Control 47, 536–544 (2015). [Google Scholar]
- 43.Reyerson L. H., Cameron A. E., The sorption of bromine and iodine by activated charcoal. J. Phys. Chem. 40, 233–237 (1935). [Google Scholar]
- 44.Jürgens G., Groth Hoegberg L. C., Graudal N. A., The effect of activated charcoal on drug exposure in healthy volunteers: A meta-analysis. Clin. Pharmacol. Ther. 85, 501–505 (2009). [DOI] [PubMed] [Google Scholar]
- 45.Lipscomb D. J., Widdop B., Studies with activated charcoal in the treatment of drug overdosage using the pig as an animal model. Arch. Toxicol. 34, 37–46 (1975). [DOI] [PubMed] [Google Scholar]
- 46.S. Podolsky, “Is it safe to take charcoal pills for gas and bloating?”; https://health.clevelandclinic.org/is-it-safe-to-take-charcoal-pills-for-gas-and-bloating/ [accessed 2 May 2019].
- 47.M. Oaklander, “Charcoal juice is now a thing”; http://time.com/3678820/charcoal-juice-health-benefits/ [accessed 2 May 2019].
- 48.Chyka P. A., Seger D., Krenzelok E. P., Vale J. A.; American Academy of Clinical Toxicology; European Association of Poisons Centres and Clinical Toxicologists , Position paper: Single-dose activated charcoal. Clin. Toxicol. 43, 61–87 (2005). [DOI] [PubMed] [Google Scholar]
- 49.Meng J.-b., Zheng X., Zhang G., Fang Q., Oral acyclovir induced acute renal failure. World J. Emerg. Med. 2, 310–313 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Fleischer R., Johnson M., Acyclovir nephrotoxicity: A case report highlighting the importance of prevention, detection, and treatment of acyclovir-induced nephropathy. Case Rep. Med. 2010, 602783 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.R. Sunberg, S. Hunt, Activated carbon for preventing pregnancy and sexually transmitted disease. U.S. Patent US10/064,710 (2002).
- 52.Tominaga K., Sato S., Hayashi M., Activated charcoal as an effective treatment for bacterial vaginosis. Pers. Med. Univ. 1, 54–57 (2012). [Google Scholar]
- 53.Abe H., New era in personalized medicine. Pers. Med. Univ. 5, 1–2 (2016). [Google Scholar]
- 54.Javed A., Parvaiz F., Manzoor S., Bacterial vaginosis: An insight into the prevalence, alternative regimen treatments and its associated resistance patterns. Microb. Pathog. 127, 21–30 (2019). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/5/8/eaax0780/DC1
Fig. S1. HPAC strongly binds to HSV-1 GFP virus.
Fig. S2. Extracellular virus–based plaque assay for HPAC therapy.
Fig. S3. HPAC does not cause a loss in visual acuity by blocking light.
Fig. S4. HPAC does not elevate interferons in HCEs.
Fig. S5. HPAC does not elevate interferons in HFFs.
Fig. S6. DECON is effective when added 24 hours after infection.
Fig. S7. DECON protects the murine cornea from HSV-1 infection.
Fig. S8. Graphical abstract showing DECON protecting cells against viral infection.
Fig. S9. Full-length blots for the Western blot shown in Fig. 1 (G and H).
Fig. S10. Full-length blots for the Western blot shown in Fig. 3D.