

Patient mutation–specific BRAF inhibitors alter full-length kinase conformations and promote interactions with GTP-loaded RAS.
Abstract
Oncogenic BRAF mutations initiate tumor formation by unleashing the autoinhibited kinase conformation and promoting RAS-decoupled proliferative RAF-MEK-ERK signaling. We have engineered luciferase-based biosensors to systematically track full-length BRAF conformations and interactions affected by tumorigenic kinase mutations and GTP loading of RAS. Binding of structurally diverse αC-helix-OUT BRAF inhibitors (BRAFi) showed differences in specificity and efficacy by shifting patient mutation–containing BRAF reporters from the definitive opened to more closed conformations. Unexpectedly, BRAFi engagement with the catalytic pocket of V600E-mutated BRAF stabilized an intermediate and inactive kinase conformation that enhanced binary RAS:RAF interactions, also independently of RAF dimerization in melanoma cells. We present evidence that the interference with RAS interactions and nanoclustering antagonizes the sequential formation of drug-induced RAS:RAF tetramers. This suggests a previously unappreciated allosteric effect of anticancer drug-driven intramolecular communication between the kinase and RAS-binding domains of mutated BRAF, which may further promote paradoxical kinase activation and drug resistance mechanisms.
INTRODUCTION
There are two reasons why small-molecule protein kinase inhibitors are among the most intensively pursued classes of anticancer therapeutics. On the one hand, protein kinases adopt central roles in proliferation and survival signaling, and on the other hand, all kinases contain a highly conserved adenosine triphosphate (ATP)–binding pocket that enables the selective targeting by synthetic chemical lead compounds (1). Primarily, the oncogenic potential of kinase pathways is dependent on continuous upstream activation or intrinsic constitutive kinase activity, which is essential for survival and proliferation of the cancer cell. Besides deregulation of upstream pathways, defined mutations are sufficient to convert the kinase into a cancer driver, which is then susceptible to the appropriate kinase inhibitor. Deregulation of components of the RAS-RAF-ERK (extracellular signal–regulated kinase) signaling pathway has frequently been implicated in tumor formation (2–4). At the plasma membrane, the RAS-GTPase (guanosine triphosphatase) family members HRAS, NRAS, and KRAS function as molecular switches linking upstream receptor stimulation with downstream kinase activation. RAS cycles between guanosine diphosphate (GDP)–bound inactive and guanosine triphosphate (GTP)–bound active states (5). However, in human cancers, recurrent oncogenic point mutations at the codon position G12, G13, or Q61 eliminate this regulation cycle and cause constitutive RAS activation. This leads to uncontrolled RAS signaling to a multitude of effectors, including the binary protein:protein interaction (PPI) with different kinases such as RAF and PI3K (phosphatidylinositol 3-kinase) (4).
The RAF kinases (ARAF, BRAF, and CRAF/RAF1) are the key regulators of mitogen-activated protein kinase (MAPK) signaling. In the absence of a stimulus, the full-length RAF kinase (=wild type) adopts a closed conformation, and the regulatory N terminus inhibits the catalytic C terminus. Physiological RAF activation depends on PPI with GTP-bound RAS via the RAS-binding domain (RBD) of RAF. The consequences are membrane recruitment, RAF dimerization, phosphorylation, and the subsequent release of the autoinhibitory RAF configuration, leading to a shift to an opened and active RAF conformation (4, 6–8).
Oncogenic mutations in RAF kinases, particularly in BRAF, which is the most frequently mutated oncogene in the RAF kinase family (1, 2), lead to regulation-uncoupled phosphorylation events. Other human disorders besides cancer are associated with BRAF or CRAF mutations; examples are a group of developmental defects referred to as RASopathies such as the Noonan and Leopard syndromes (4). The most common gain-of-function mutation in BRAF is the substitution of V600 by a glutamic acid residue (E) and is found in around 60% of all melanoma (9). Selective inhibitors of BRAFV600E have been approved for the treatment of metastatic melanomas that express BRAFV600E [vemurafenib (PLX4032) and dabrafenib (GSK2118436A)] and show profound clinical responses in patients (9, 10). However, the duration of the antitumor response is variable, and the efficacy of the inhibitor is limited due to the onset of drug resistance. This depends primarily on the genetic nature of the melanoma-causing mutations (9, 11–13). Structurally diverse ATP-competitive BRAF inhibitors (BRAFi) have the potential to reactivate the MAPK pathway and cancer cell proliferation in a RAS-dependent manner (9, 11, 12, 14–16). What further complicates the analyses of RAF kinase drug efficacies is a collection of additional oncogenic mutations that have been identified in BRAF and CRAF (4, 17, 18). Their mechanisms of action are not fully understood, but it is assumed that they are related to (i) alterations of kinase conformations, (ii) enhancement of RAF kinase domain dimerization, (iii) mimicking of kinase autophosphorylation, or (iv) the ordering of the hydrophobic R spine (4, 6, 7, 19).
The complexity of RAF regulation, missing full-length structures, the high frequency of disease mutations, and thus the attractiveness as a therapeutic target make it highly desirable to develop new means for monitoring changes in RAF activities, conformations, and interactions. There is also a need to track modes of anticancer drug:RAF interactions in vivo that would have benefits for the understanding of temporal drug efficacies and drug resistance mechanism. Noninvasive cell-based reporter assays for systematically studying the regulation, mode of action, and inhibition of full-length RAF isoforms and their carcinogenic mutations are needed. Biochemical assays that involve disruption of cells or tissues are the modus operandi for investigations of RAS:RAF complexes and relevant GTPase and kinase activities (20, 21). These biochemical approaches have an impact on membrane-anchored complexes such as RAS dimers and might hinder the detailed characterization of physiological and more efficient perturbation of pathological RAS:RAF functions, which involves dimers/tetramers and RAS nanoclustering (4, 22).
Here, we set out to generate a reporter platform to test dose-dependent effects of approved kinase inhibitors or lead molecules on RAF conformations, binary RAF interactions, and tetrameric RAS:RAF complexes. We implemented a modular RAS:RAF reporter toolbox including full-length and mutated RAS and RAF protein isoforms. We integrated defined cancer patient mutations for tailored drug discovery efforts to be performed directly in the cell/model organism of choice. This is relevant for the differentially classified BRAF mutations that affect kinase activity–related conformations and regulatory RAS interactions. First, we endorse the opened and closed RAF kinase conformation model by direct and dynamic in vivo (= in intact cells) recordings using full-length and mutated kinase conformation reporters. Second, we unveiled unexpected allosteric effects of mutation-specific anticancer drugs on the molecular interactions of the full-length BRAF(V600E) oncoprotein with implications for the architecture of the tetrameric RAS:RAF complex. Thus, we showed the molecular details how specific BRAFi [such as paradox breaker (PB); in clinical trials for patients with mutant BRAF] still elevate RAS:RAF complexes in melanoma cells. Overall, we showed that the extendable RAF-centered and RAS-engaged reporter platform is an asset for identifying molecular details of pathological protein functions in the cell of choice. It provides new means to determine efficacies of lead molecules by precise analyses of full-length kinase conformations, activities, and regulatory binary interactions in intact cells.
RESULTS
Determination of BRAF conformation dynamics in intact cells
Small-molecule protein kinase inhibitors are applied for molecularly targeted cancer therapy (16, 23). In particular, deregulated kinase activities of the RAS-RAF-MEK-ERK signaling axis emanating from RAF and MEK are the target of ATP-competitive and allosteric kinase inhibitors (24). Biochemical evidence underlines that RAF kinase activation is reflected by alterations of its autoinhibited conformation. In its inactive and autoinhibited state, the N-terminal RAF region contacts and inhibits the C-terminal kinase domain (4, 25). We sought to engineer a genetically encoded reporter to quantify intramolecular conformational changes of RAF. Such a full-length kinase reporter should be applicable for noninvasive recordings of opened and closed kinase conformations upon mutation, BRAFi exposure, or upstream activation of the RAS-RAF-MEK-ERK pathway. As a starting point, we fused full-length BRAF N-terminally with fragment 1 (F[1]-) and C-terminally with fragment 2 (-F[2]) of the Renilla luciferase (Rluc)–based protein-fragment complementation assay (PCA) to generate the reporter hybrid protein F[1]-BRAF-F[2] (26, 27). For simplification, we termed the intramolecular Rluc-PCA biosensor from now on kinase conformation (KinCon) reporter. In the absence of a stimulus, wild-type RAF kinases adopt the closed conformation; the N terminus inhibits the C-terminal kinase domain activity through binding. Upstream RAS activation, RAS mutations, RAF mutations, and/or mutation-specific cancer drugs may affect reporter conformations by interconverting closed, intermediate, and opened kinase conformations, which is reflected by an increase or decrease of Rluc-PCA–emitted bioluminescence signals (Fig. 1A). To test this assumption, we generated, besides the wild-type BRAF KinCon reporter, a mutant form with the amino acid substitution V600E, representing one of the most recurrent oncogenic human disease mutations (9, 28). This amino acid exchange orders the activation loop (A loop) and stabilizes the R-spine of the BRAF kinase domain to create the catalytically active BRAF.
Fig. 1. Dynamics of the BRAF KinCon reporters.
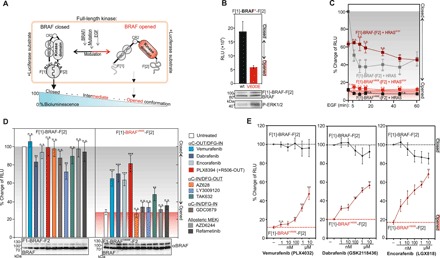
(A) Schematic depiction of the intramolecular Rluc-PCA–based BRAF kinase conformation reporter (KinCon reporter). Upstream RAS activation (EGF), RAS mutations, RAF mutations, or mutation-specific cancer drugs modulate opened, intermediate, or closed full-length RAF kinase conformations, resulting in an increase or decrease of Rluc-PCA–emitted cellular bioluminescence. (B) BRAF conformations (alterations of Rluc-PCA bioluminescence) were measured using transiently transfected HEK293 cells. Immunoblotting shows BRAF, F[1]-BRAF-F[2], and F[1]-BRAFV600E-F[2] expression levels and P-ERK1/2 levels (representative experiment; ±SD). RLU, relative light units. (C) Time-dependent effects of EGF (200 ng/ml) on BRAF conformations in the presence of hemagglutinin (HA)–tagged HRAS variants (±SEM; n = 4 independent experiments). (D) Impact of indicated BRAFi and MEKi on shown BRAF KinCon reporters (±SEM; n = 9 independent experiments; 3-hour treatments, 1 μM, HEK293 cells). (E) Dose-dependent recordings of BRAF conformations upon indicated BRAFi exposure for 3 hours. RLU signals have been normalized on the twofold elevated BRAFV600E KinCon reporter expressions. n = 8, 6, and 6 independent experiments are shown for vemurafenib, dabrafenib, and encorafenib, respectively (±SEM; amalgamated data from 24- and 48-hour reporter expressions). Student’s t test was used to evaluate statistical significance. Confidence levels: *P < 0.05, **P < 0.01, and ***P < 0.001. wt, wild-type; n.s., not significant.
Following transient expression of BRAF and BRAFV600E KinCon reporters in human embryonic kidney (HEK) 293 cells, we observed substantially elevated bioluminescence signals with the wild-type BRAF reporter when compared to the mutated BRAFV600E. Further, it was evident that the BRAFV600E KinCon reporter is catalytically active by causing elevated downstream phosphorylation of ERK1/2 (Fig. 1B). These data underline that, independent of RAS binding and activation, the V600E mutation is sufficient to convert the full-length BRAF KinCon reporter into a definitive opened and thus activated conformation. To confirm that the KinCon reporter can be used for kinetic studies of conformational kinase reorganizations in vivo, we activated endogenous epidermal growth factor receptors (EGFRs). Following time-dependent treatments with the epidermal growth factor (EGF) peptide, we observed an immediate (5 min) and distinct 40 to 60% reduction of the bioluminescence signal with the wild-type BRAF KinCon reporter in the presence of coexpressed wild-type HRAS. The drop in emitted bioluminescence emphasizes that the EGF-initiated GTP-RAS formation and BRAF interaction shift the wild-type BRAF KinCon reporter to the opened kinase conformation. Coexpression of HRASG12V was sufficient to convert BRAF directly into this intermediate and active conformation, making it therefore less stimulation responsive for EGF (Fig. 1C). Analyses using the BRAF-V600E KinCon reporter revealed that the already opened and active BRAFV600E conformation can be opened slightly further by EGF-mediated RAS activation (Fig. 1C). These data affirm the generation of a dynamic RAF KinCon reporter reflecting GTP-RAS controlled BRAF conformations.
Given that the tested BRAF KinCon reporters reflect opened (V600E) and closed (wild-type) BRAF conformations, we tested the dose- and time-dependent impact of BRAFi binding. We assumed that selective BRAFi binding into the catalytic pocket of mutated BRAF might have the potential to affect full-length kinase conformations. Therefore, we subjected BRAF KinCon reporters to treatments with an assortment of RAF inhibitor (RAFi) and MEK inhibitor (MEKi). In addition to the allosteric MEKi AZD6244 and refametinib, we used effective RAFi vemurafenib, dabrafenib, encorafenib, PLX8394, AZ628, LY3009120, TAK632, and GDC0879 (16, 29, 30). These RAFi differ in their chemical structures and can be classified according to the following kinase conformations: αC-helix-IN/DFG-IN, αC-helix-IN/DFG-OUT, or αC-helix-OUT/DFG-IN (Fig. 1D) (16). The three clinically used BRAFi vemurafenib, dabrafenib, and encorafenib differ in their abilities to stabilize the αC helix towards an OUT position. They are classified as BRAF-V600E–selective inhibitors with reduced affinities for an interacting second RAF protomer (negative allostery) (16). Other RAFi block both monomeric and dimeric RAF forms (e.g., TAK632, LY3009120; αC-IN). The PB PLX8394 (αC-OUT) has the feature to reduce the dimerization potential of inhibited BRAF mutants (16, 29).
BRAFi alter BRAF-V600E conformations
First, we tested the impact of these kinase inhibitors on the BRAF KinCon reporters. We observed that all αC-helix-OUT BRAFi significantly elevated bioluminescence signals of the BRAFV600E KinCon reporter (expressed at levels below the endogenous BRAF), underlining a shift to the more closed kinase conformation. With TAK632, we observed a slight shift of BRAFV600E KinCon to a more closed state. With the tested MEKi, we detected no major influence on BRAF reporter conformations (Fig. 1D). Further, we observed that the αC-helix-IN/DFG-OUT inhibitor LY3009120 significantly disrupted the autoinhibited BRAF conformation, as it had been demonstrated recently in a study with wild-type BRAF (31). To validate the observations of driving BRAFV600E back to the closed conformation, we profiled the conformation change of the three U.S. Food and Drug Administration (FDA)–approved BRAFi [vemurafenib (PLX4032), dabrafenib (GSK2118436A), and encorafenib (LGX818)] in intact HEK293 cells in a dose- and time-dependent manner (transient overexpression of the reporter for 24 and/or 48 hours). We determined the efficacies of BRAFi in experiments with the wild-type and the BRAFV600E-based BRAF KinCon reporters. For the comparison of BRAF KinCon dynamics, we normalized the emitted bioluminescent signals of wild-type BRAF on the approximately twofold higher expressed F[1]-BRAFV600E-F[2] reporter. In dose-dependent drug exposure experiments with these BRAFi, we observed a distinct pattern of shifting BRAFV600E to the closed kinase conformation. The αC-helix-OUT BRAFi vemurafenib, dabrafenib, and encorafenib showed a similar pattern of shifting BRAFV600E to a more closed state; vemurafenib showed the weakest effect (Fig. 1E). Also, wild-type BRAF KinCon reporters were affected at higher concentrations.
Correlations between BRAF conformations and activities
We have also included the PB PLX8394 in dose-dependency experiments to record conformations and downstream signaling (16, 29, 32). In detail, we characterized the impact of nanomolar PLX8394 doses, different patient-specific mutations, and timing on conformations and downstream BRAF KinCon reporter activities. PLX8394 was responsible for a significant shift to the closed BRAF-V600E conformation at low nanomolar concentrations (Fig. 2A). PLX8394 (with the concentration of 10 μM) further promoted a more closed conformation of the V600E and also of the wild-type KinCon BRAF reporters. These experiments indicate that all tested αC-helix-OUT BRAFi (vemurafenib, dabrafenib, encorafenib, and PLX8394) stabilized the mutated BRAF KinCon reporter, dependent on the dose used, in closed and intermediate enzyme conformations.
Fig. 2. Correlations of BRAF conformations and activities.
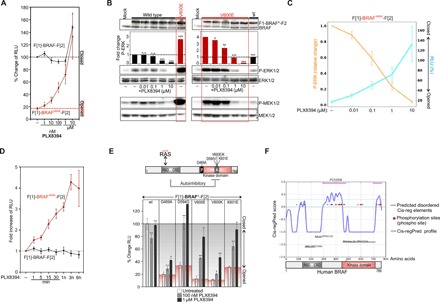
(A) Dose-dependent recordings of BRAF conformations upon PLX8394 treatment (3 hours; n = 4 independent experiments are shown; ±SEM). (B) Dose-dependent determination of P-ERK1/2 levels immediately after PLX8394 treatment (1-hour treatments; quantification from n = 4 independent experiments; ±SEM). (C) Dose-dependent correlations of BRAF-V600E KinCon reporter–dependent P-ERK/P-MEK activities and BRAF-V600E conformations upon PLX8394 exposure. (D) Time-dependent effect of PLX8394 on BRAF KinCon conformations (HEK293 cells; ±SEM from n = 4 independent experiments). (E) Schematic depiction of the modular structure of BRAF; patient mutations in the A-loop and P-loop are indicated (RBD, RAS-binding domain; CRD, cysteine-rich domain). The expression normalized values for BRAF KinCon reporter conformations in % of RLU and the impact of PLX8394 on indicated wild-type and mutant BRAF conformations (±SEM from n = 4 independent samples; representative of at least n = 3 independent experiments) are shown. (F) The cis-regulatory prediction (Cis-regPred) profile of BRAF is indicated. Pink lines at the top show the coordinates of both cis-regulatory elements (CREs). Student’s t test was used to evaluate statistical significance. Confidence levels: *P < 0.05, **P < 0.01, and ***P < 0.001.
Next, we validated KinCon reporter activities using conventional downstream readouts for kinase activations (P-ERK1/2 and P-MEK1/2). In dose-dependency experiments with encorafenib, dabrafenib, and PLX8394, we confirmed the direct correlations of ERK1/2 and P-MEK1/2 phosphorylations and opened kinase conformations using overexpressed KinCon reporters (Fig. 2B and fig. S1, A to C). To underline that the αC-helix-OUT BRAFi–initiated conformational changes directly correlate with BRAF inhibition, we display the dose-dependent and inverse correlation of P-ERK and BRAF-V600E conformations [in relative light units (RLU)] (with the PB PLX8394; Fig. 2C). Further, we compared the structurally related compounds TAK632 (in purple) with LY3009120 and AZ628 (in gray) and observed a slightly different BRAF-binding mode, which might be relevant for the observed TAK632 effect on the mutated BRAF conformation (fig. S1D). In this context, we validated the mutation-specific BRAFi by analyzing BRAF-V600E activities in A375 melanoma cells through determination of P-ERK levels downstream of BRAF* (the asterisk indicates mutation; fig. S1E). We combined data shown in Figs. 1E and 2A to compare the dose-dependent impact of the four αC-helix-OUT inhibitors on the closing of the BRAF-V600E KinCon reporters. We directly compared in fig. S1F the fitted BRAF-V600E closing curves upon BRAFi exposure and listed equipotencies for pairs of BRAFi in the graph and efficacies in the table. Next, we validated our KinCon BRAF reporters using a collection of defined mutations.
First, we integrated 14-3-3 binding site mutations of S365 and S729 (4). We assumed that the loss of these phosphorylation sites should prevent BRAF from engaging a closed conformation. We showed that single S365A and S729A mutations promoted the opened kinase conformation (fig. S2A). Second, we validated the impact of vemurafenib exposure on BRAF KinCon conformations. We present clear evidence that the drug-driven closing of the KinCon reporters is prevented in the presence of the S365A and S729A mutations and in the presence of V600E. This underlines an involvement of both serine residues in the drug-driven closing of BRAF conformations (fig. S2B). Third, we tested whether previously reported gatekeeper mutations, which affect ATP-competitive inhibitor binding (33), have an impact on the drug-induced closing of the BRAF-V600E KinCon reporters. We identified significant differences between the four tested αC-helix-OUT inhibitors. Following integration of the T529M and T529I gatekeeper mutations, the closing effect of vemurafenib and PLX8394 was abolished. However, encorafenib and dabrafenib still initiated the closing of the full-length BRAF-V600E KinCon reporter, which contained both gatekeeper mutations (fig. S2C). Comparison of structures of these BRAFi and the ATP-binding pocket provide an explanation. Dabrafenib and encorafenib carry a large hydrophobic group (t-butyl and isopropyl, respectively) that is able to fill the area occupied by T529 that is vacated by 529 reorientation in the case of T529I/M gatekeeper mutations in the mutated BRAF-V600E. Vemurafenib and PLX8394 exhibit a loss of affinity as the fluorine substituent in a roughly analogous position is unable to compensate and hence a void is likely present in the area vacated by T529 upon reorientation (fig. S2D).
To confirm that 24- and 48-hour overexpression of the reporter does not account for the BRAFi-triggered conformation changes, we transiently overexpressed the KinCon hybrid proteins for only 6 hours before we measured the consequence of BRAFi exposure (1-hour treatments). At expression levels not detectable in immunoblotting, we observed a BRAFi-initiated shift of the BRAFV600E KinCon reporter to the closed conformation (fig. S3A). Using a different experimental setup, we imaged the dose-dependent impact of BRAFi on shifting the BRAFV600E KinCon reporter to the closed conformation with detached HEK293 cells in a 1536-well plate (fig. S3B). Again, the four BRAF mutation–specific αC-helix-OUT BRAFi (vemurafenib, dabrafenib, encorafenib, and PLX8394) showed different efficacies in driving BRAF back into its more closed state.
To get an idea of the timing of BRAFi actions, we subjected PLX8394 to time-dependent measurements of BRAF KinCon changes. We observed an immediate shift of the BRAFV600E reporter to the closed conformation. The wild-type BRAF KinCon reporter was not affected (Fig. 2D). Our data indicated that these αC-helix-OUT BRAFi–initiated conformation changes of mutated BRAF directly correlated with kinase activities. They corroborate the suitability of KinCon reporters to profile receptor- and drug-controlled dynamics of BRAF conformations using full-length and oncogenic kinase variants in vivo. We confirmed the αC-helix-OUT BRAFi effect on mutant BRAF conformations using stable colon cancer cell lines (SW480; KRASG12V positive) expressing the KinCon reporter at expression levels far below the endogenous BRAF. Again, vemurafenib, dabrafenib, encorafenib, PLX8394, and TAK632 converted BRAFV600E to the closed conformations. This time, effects of αC-helix-OUT BRAFi on the wild-type BRAF reporter were evident. In the KRASG12V-positive SW480 cells, all of the αC-helix-OUT BRAFi elevated wild-type BRAF conformations. We assume that KRASG12V-activated BRAF KinCon reporter become target for a αC-helix-OUT BRAFi–mediated conformation change (due to the very low KinCon reporter expression levels; fig. S3C).
KinCon profiling of BRAF patient mutations and BRAFi
BRAF is mutated in ~8% of all cancers, with the kinase-activating V600E mutation found in about one-half of all melanomas (17). However, so far, nearly 300 distinct missense mutations of BRAF have been identified in tumor samples and cancer cell lines (17, 34). Most of the mutations occur in the A loop or in the phosphate-binding loop (Fig. 2E; a selection of frequent human BRAF mutations is shown). The biochemistry of altered BRAF proteins varies substantially with regard to activity, interactions, and responsiveness to inhibition (4, 35). We used a site-directed mutagenesis approach to generate different BRAF KinCon reporters harboring the patient-specific mutations G469A, D594G, V600E/K, and K601E. First, we overexpressed each reporter in HEK293 cells to measure KinCon-emanating bioluminescence. We present the expression-corrected bioluminescence signals of the tested KinCon reporters. We observed that all tested patient mutation–containing BRAF reporters showed reduced bioluminescence signals underlining a mutation-driven shift to a definitive opened BRAF kinase conformation (Fig. 2E; represented by the white bars). In addition, we noticed that phosphotransferase-inactivating BRAF mutations such as D594G promoted opening of the BRAF conformation. One explanation might be related to the high dimerization potential of the D594G, as it has been demonstrated previously (12, 36). In this context, it is of interest that following exposure to the PB PLX8394 (100 nM, 1 μM), all mutated BRAF KinCon reporters responded to the treatment by showing a dose-dependent shift to the closed kinase conformation (Fig. 2E, gray and black bars). These data underline that, besides V600E, other BRAF mutations promote the opened conformation of the full-length kinase.
Further, we applied a computational prediction program designed to predict cis-regulatory elements (CREs) in the full-length BRAF kinase (37). In doing so, we actually identified two stretches of CREs, which both may contribute to a more complex mode of autoinhibitory BRAF conformations that are affected by all tested patient mutations (Fig. 2, E and F).
Quantification of GTP-controlled RAS:RAF complexes in intact cells
The observation that αC-helix-OUT BRAFi affects the conformations of full-length and mutated BRAF kinases encouraged us to analyze the consequences on BRAF-emanating molecular interactions. We assume that differences in the conformations correlate with binding affinities for RAS. This assumption has been recently reinforced by a study showing that class 3 categorized cancer-associated BRAF mutants with impaired kinase activity bind more tightly to GTP-loaded RAS than wild-type BRAF (35). Further, it has been demonstrated that BRAFi induce RAF kinase domain dimerization, which enhanced RAS:RAF interactions (11, 38, 39). Recently, an impact of RAFi on closed wild-type RAF conformations and RAS interactions has also been described (31). We set out to unveil the mechanistic details and the sequence of RAFi-driven complex formation of tetrameric and RAFi-engaged RAS:RAF complexes by focusing on the hotspot V600E mutation in BRAF.
To compare the consequence of RAFi occupancy on molecular kinase interactions, we initiated immunoprecipitations (IPs) of mutated and GTP-loaded RAS either upon overexpression (fig. S4A) or by coprecipitating endogenous GTP-RAS (Fig. 3A) with all tested RAFi. To exclude secondary effects and feedback inhibition, we used 1 μM RAFi doses in treatments of up to 60 min. We observed that inhibitors of the first and second generation elevate RAF dimerization in both tested cell systems (Fig. 3A and fig. S4A), with one exception. In biochemical assays (IPs), we showed with the PB PLX8394 (that occupies the ATP-binding cleft in a way that is inhibitory to forming RAF dimers) that interactions of BRAF-V600E with CRAF were abolished. However, we observed that, in the absence of detectable dimer formation, complexes with GTP-bound RAS were significantly elevated (Fig. 3A and fig. S4A). We would like to point out that by using the chosen biochemical approach of IPs, we analyze the drug-affected associations of multimeric protein complexes. We cannot easily extract the critical molecular details how far RAF dimers, RAS nanoclustering (in intact cells), and drug concentrations affect specific binary interactions such as RAS:RAF and RAF:RAF. Therefore, it is necessary to apply other means to understand the mechanistic details of how BRAFi elevate binary complexes emanating from RAS or RAF and preferentially directly in the intact cell.
Fig. 3. Quantification of GTP-controlled and RAFi-affected RAS:RAF complexes.
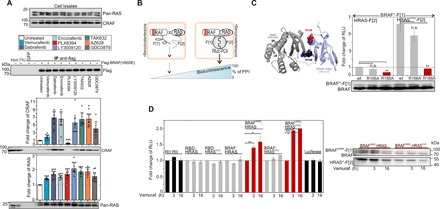
(A) IPs of transiently overexpressed and BRAFV600E using flag-tagged antibodies from SW480 cells following 1-hour RAFi exposure (quantification is shown from n = 6 independent experiments; ±SEM). (B) Illustration of the Rluc-PCA biosensor strategy to quantify PPIs of RAS:BRAF in vivo; fragments 1 and 2 of Rluc-PCA (-F[1] and -F[2]). Elevated PPIs induce complementation of Rluc-PCA fragments (PPI reporter). (C) Structure illustration indicates the localization of R188 and R166 in the RAS:RBD(BRAF) binding interface [Protein Data Bank (PDB): 4G0N and 2L05]. Following coexpression of Rluc-PCA fragment tagged pairs of full-length RAS isoforms/mutants and full-length BRAF, we determined GTP-dependent PPIs (±SEM; n = 4 independent experiments). Following coexpression of Rluc-PCA fragment tagged pairs of RAS isoforms/mutants and full-length BRAF, we determined GTP-dependent PPIs (±SD; representative experiment of n = 3 independent experiments). (D) Following coexpression of indicated luciferase and PPI reporter constructs, we treated cells for 3 or 16 hours with 10 μM vemurafenib. The PPI analyses of n = 4 independent experiments (HEK293; ±SEM) are shown. We have normalized the PPI values on the untreated PPI signals of each tested protein pair. Student’s t test was used to evaluate statistical significance. Confidence levels: *P < 0.05, **P < 0.01, and ***P < 0.001.
Given that the RAS dimerization in nanoclusters at the plasma membrane is critical for physiological and pathological RAF kinase activation (22), we aimed to develop a biosensor platform to characterize and perturbate molecular interactions of tetrameric RAS:RAF complexes without cell disruption in intact cells. We designed and implemented a full-length kinase reporter platform to systematically analyze and perturb (i) binary interactions with the upstream-located molecular switches HRAS, KRAS, and NRAS; (ii) RAF dimerization; and (iii) the implications of BRAFi occupancy without cell lysis. Under physiological conditions, RAF activation is initiated by nanoclustered and GTP-bound RAS at the plasma membrane, leading to RAF kinase recruitment, change of RAF conformation/phosphorylation, RAF dimerization, and sequential phosphorylation and activation of downstream kinases such as MEK and ERK (fig. S4B) (4). The desired feature of a cell-based reporter to study the allosteric consequence of BRAFi binding to RAF would be met by one that could capture GTP-dependent complex formation of full-length and activated RAS:RAF. Such a cell-based reporter system would be applicable for studying the dynamics of complex formation and lead molecule (BRAFi) interactions. We engineered a genetically encoded reporter to directly quantify oncogenic PPIs of RAS:BRAF in the living cell. We applied the Rluc-based PCA to analyze dynamics of RAF kinase PPIs with three members of the RAS GTPase family using different cell systems. For simplification, we termed the intermolecular Rluc-PCA biosensor from now on “PPI reporter” (Fig. 3B). As a starting point, we fused the previously described Rluc-PCA fragments (26) to the well-defined RBD of RAF, delivering RBD-F[1]. In addition, we generated expression constructs of the C-terminally tagged RAS isoforms H-, N-, and K-RAS with the Rluc-PCA fragment 2 (-F[2]). We integrated the oncogenic mutation G12V into all RAS isoforms to obtain the GTPase-deficient and thereby constitutively active GTP-bound RAS variants. Following coexpression of PCA-tagged protein pairs, we observed significantly elevated PPIs between RBD and all GTP-bound RAS isoforms when compared to the wild-type RAS interaction experiments (fig. S4C). This observation underlines that the Rluc-PCA–based PPI reporters can be subjected to accurately quantify GTP-dependent RBD:RAS interactions in intact cells. No significant PPIs between RBD and the control proteins (PKA RII subunits and Max) were detectable (fig. S4C). The differences in PPIs between RBD and the RAS isoforms resulted from vast differences in the expression levels of the Rluc-PCA–tagged GTPases H-, N-, and K-RAS (fig. S4D).
Next, we tested full-length BRAF using the Rluc-PCA PPI reporter system. Despite the size of full-length human BRAF with 766 amino acids, we detected the PPI of BRAF-F[1] with wild-type HRAS-F[2] (Fig. 3C). Complex formation was significantly elevated upon coexpression of GTP-loaded HRAS (Fig. 3C) and also other RAS variants (fig. S4E). G12V mutations had no substantial impact on HRAS-F[2] expression levels. Again, the expression levels of the GTPases explain the PPI differences between H-, N-, and K-RAS and BRAF (fig. S4D). To prove that the RBD of BRAF is responsible for the cellular complex formation of BRAF-F[1]:HRAS-F[2], we introduced mutations to perturb the RBD:HRAS interface. The conserved residue R188 is located in the RBD of the BRAF:RAS interface and is required for RAF membrane recruitment and RAS-dependent activation (Fig. 3C) (40). In addition to the R188A amino acid substitution, we also generated a second construct for PPI measurements, the R166A PPI reporter mutant, which is, according to our structure predictions, located at the PPI interface. Compared to the wild-type reporter, the BRAFR188A PPI reporter mutant showed decreased complex formation with wild-type and G12V-mutated HRAS. These data underline that the RAS:RAF PPI reporter reports full-length PPIs, which are formed via the RBD(BRAF):HRASGTP interface (Fig. 3C). So far, primarily either the RBD:RAS or interacting kinase complexes were studied using biochemical techniques (21, 41, 42). IP studies are the technique of choice for studying dynamics of full-length RAS:RAF complex formation, which actually specify only minor fractions of affinity-isolated cellular complexes. With our PPI reporter in hand, it is now possible to quantify oncogenic, binary, and exclusively full-length RAS:BRAF interactions directly in the appropriate cell setting.
BRAFi binding promotes BRAFV600E complexes with RAS
The fact that FDA-approved BRAFi affect BRAF conformations (KinCon reporter; see Figs. 1 and 2) prompted us to test the consequence of BRAF inhibition on complex formation with RAS using the PPI reporters directly in the living cell setting. Our goal was to track allosteric consequences of full-length kinase inhibition on full-length RAS:BRAF interactions in cells. Again, we focused our efforts on the most recurrent oncogenic disease mutation, the BRAFV600E variant (9). We evaluated allosteric consequences of BRAFi on particular PPIs using our in vivo RAS:RAF PPI reporter platform. At first, we integrated the V600E mutant of BRAF into the PPI analyses. Following transient overexpression of indicated proteins, we observed that GTP loading of HRAS elevated BRAF:RAS interactions. Also, mutated BRAFV600E-emanating interactions with wild-type and mutant HRAS were evident, despite the differences of the reporter expression levels (fig. S5A). Next, we tested the consequences of small-molecule phosphotransferase inhibitors (RAFi) on RAS:RAF complex formation. For this, we subjected an assortment of different PPI reporters and the full-length luciferase to time-dependent treatments with the BRAFV600E kinase inhibitor vemurafenib (43). We observed explicit elevations of PPIs in experiments analyzing BRAFV600E-emanating binary protein complexes: Following treatments of cells for 3 and 16 hours with vemurafenib, we noticed that not only the BRAFV600E interaction with wild-type HRAS but also the interaction with GTP-bound HRAS was significantly elevated (Fig. 3D). We detected no significant impact of vemurafenib exposure on the bioluminescence signal of kinase complexes without the V600E mutation, control PPIs, and full-length Rluc (Fig. 3D). This observation is in agreement with previous studies, showing an elevating effect on RAS:BRAF complex formation of 1 μM vemurafenib exposure in BRAF IPs from BRAFV600E mutant SW1736 cells (thyroid carcinoma) (44). To confirm that long-term BRAFi treatments affect RAS:RAF interaction, we tested overexpressed RAS:RAF complexes in IP assays. We confirmed that 16 hours of vemurafenib treatment elevated complex formation between BRAFV600E and HRASG12V (fig. S5B). These results underline that the selective binding of vemurafenib to BRAFV600E elevates complex formation with RAS, preferentially with the GTP-bound form.
To determine whether this complex formation–enhancing effect is a general property of vemurafenib, we set out to test the drug effect on the BRAF interactions with KRASG12V, NRASG12V, and HRASG12V. This time, we analyzed the dose-dependent effect using 100 nM and 1 μM vemurafenib. In contrast to the interactions with RBD, we observed an elevation of complex formation of BRAFV600E with all GTP-bound RAS isoforms in a dose-dependent manner (fig. S5C). These data underline that the enhancing effect of vemurafenib on RAS:BRAF complex formation can be extended to the family members H-, K-, and N-RAS. Next, we asked the question whether the other BRAF mutation–specific αC-helix-OUT inhibitors with different chemical structures show the same effect on boosting RAS:RAF PPIs. In addition to vemurafenib, we tested two further αC-helix-OUT inhibitors, dabrafenib and encorafenib, respectively, which all show antitumor activity in patients harboring metastatic melanoma with BRAFV600E mutation.
To study the dose-dependent impact on defined binary RAS:RAF interactions, we overexpressed several combinations of our PPI reporter pairs, BRAFV600E:HRASG12V, BRAF:HRASG12V, and RBD:HRASG12V. In experiments performed in parallel, we treated the cells for 3 hours with increasing concentrations of the indicated BRAFi. Following measurements of PPIs in intact HEK293 cells, we normalized the obtained bioluminescence signals on the interaction of RBD:HRASG12V serving as the control experiment.
We observed that all three drugs significantly elevated BRAFV600E:HRASG12V interactions in a dose-dependent manner (Fig. 4A). In the case of vemurafenib, low micromolar concentrations of the drug also elevated wild-type BRAF:HRASG12V interactions (Fig. 4A). These experiments reflect the suitability of the PPI reporter system to precisely record and compare consequences of BRAFi binding on BRAF:HRAS complexes in the low nanomolar range. It underlines that binding of αC-OUT inhibitors to mutated kinase complexes allosterically affects complex formation of full-length BRAF with GTP-bound RAS.
Fig. 4. RAFi elevate mutated RAS:BRAF complexes.
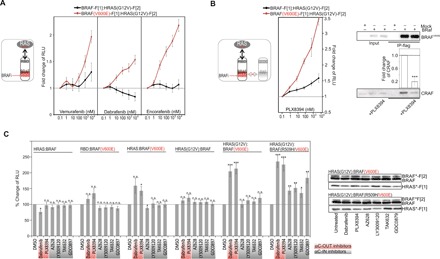
(A) Dose-dependent treatments of HEK293 cells with increasing concentrations of dabrafenib, encorafenib, and vemurafenib (3 hours) after transient coexpression of indicated PPI reporters. The obtained bioluminescence signals have been normalized on the interaction of RBD:HRASG12V and on the signals obtained with 0.1 nM drug treatment. The PPI analyses of n = 7 independent experiments (HEK293; ±SEM) are shown. (B) Following coexpression of indicated PPI reporters, we treated HEK293 cells with increasing concentrations of PLX8394 (3 hours). The obtained bioluminescence signals were normalized on the interaction of RBD:HRASG12V and on the signals obtained with 0.1 nM drug treatment. The PPI analyses of n = 4 independent experiments (HEK293; ±SEM) are shown. Co-IP of flag-tagged BRAFV600E following exposure to 1 μM PLX8394 (HRASG12V coexpression). Quantification from n = 3 independent experiments is shown (±SEM). (C) Following coexpression of indicated PPI reporters (HEK293), we analyzed the impact of indicated RAFi on cellular PPI using the Rluc-PCA as readout. The PPI analyses of at least n = 7 independent experiments (HEK293; ±SEM) are shown. Student’s t test was used to evaluate statistical significance. Confidence levels: *P < 0.05, **P < 0.01, and ***P < 0.001.
Does this phenomenon involve RAF dimers? It is a well-known fact that mutation-specific BRAFi reactivate MAPK signaling in cells with oncogenic RAS mutations and/or elevated upstream pathways (11–13). One strategy to avoid paradoxical MAPK activation was the development of PB [e.g., PLX8394 (16)], which overcomes molecular mechanisms of drug resistance such as BRAFi-driven BRAF:CRAF dimerization (29). We demonstrated that the PB of RAF dimers is capable of elevating mutated RAS:BRAF complexes. Using the Rluc-PCA PPI reporters as readouts, we observed that PLX8394 increased BRAFV600E:HRASG12V complexes in a dose-dependent manner (Fig. 4B) and more pronounced than the BRAFi encorafenib, vemurafenib, and dabrafenib (Fig. 4A). The wild-type BRAF interaction with HRASG12V was also affected at low nanomolar doses. Independent of the PPI reporter system, we confirmed that PLX8394 exposure significantly reduced interactions between BRAFV600E and CRAF (Fig. 4B, right panel). Next, we subjected all used BRAFi to analyses of binary BRAF complexes. Further, we integrated the RAF dimerization–defective R509H (15, 42) mutant into the analyses. The enhancing effect of vemurafenib on distinct BRAF:RAS complexes was evident with and without the R509H mutation (in the presence of the V600E mutation; fig. S5D). We observed that dabrafenib and PLX8394 selectively elevated BRAFV600E-emanating interactions primarily with GTP-loaded HRAS. In experiments with the RAF dimerization–deficient mutant BRAFR509H/V600E, we also observed PPI-elevating effects of αC-IN inhibitors following 60-min BRAFi exposures (1 μM; Fig. 4C). We would like to underline that, using the PPI reporter, we quantify and describe dynamics of binary PPI in vivo. This is distinct from IP results reflecting the direct/indirect formation of multimeric protein complexes with associated RAS and RAF (Fig. 3A). The PCA data further support the notion that primarily the αC-OUT inhibitors (approved and in clinical trials) have the potential to elevate binary BRAFV600E interactions with RAS and that this is also independent of RAF dimerization. In this context, it is of interest that, by using stable KinCon cell lines, we observed—when compared to αC-OUT inhibitors—a minor but significant impact of αC-IN RAFi on mutant BRAF conformations (fig. S3C). In summary, we confirmed that all tested αC-OUT inhibitors that actually had an impact on the open BRAFV600E conformation also elevated mutated RAS:BRAF interactions.
PB PLX8394 elevates RAS:RAF complexes
To gain mechanistic insight, we kept the focus on the approved αC-OUT inhibitors (vemurafenib, encorafenib, and dabrafenib) and the promising αC-OUT inhibitor from clinical trials [PB PLX8394 (NCT02428712)]. Thus, we analyzed the consequence of RBD mutations and PB PLX8394 binding on RAS and RAF interactions along with RAF dimer formation in intact cells (as outlined in the schematic of Fig. 5A). We asked the question whether it is the RBD-mediated RAS:BRAF interaction that is elevated following BRAFi exposure. We used the RAS:RAF binding–deficient R188A mutant in Rluc-PCA measurements with BRAFi. Vemurafenib and PLX8394 specifically elevated complex formation of GTP-RAS:BRAFV600E with modest effects on the residual GTP-RAS:BRAFR188A/V600E interaction (Fig. 5B).
Fig. 5. Impact of mutations and BRAFi on RAS:RAF complex formation.
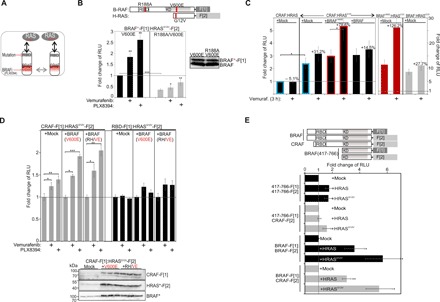
(A) Schematic depiction of analyzed PPIs. (B) Used reporter constructs are indicated. Impact of vemurafenib or PLX8394 exposure (3 hours, 1 μM) on indicated PPIs measured using the Rluc-PCA as readout (n = 4 independent experiments; ±SD). (C) Impact of vemurafenib exposure (3 hours, 1 μM) on indicated CRAF:RAS and BRAF:HRAS interactions in the presence or absence of HA-tagged BRAF or BRAFV600E (±SEM; n = 3 independent experiments). (D) Impact of vemurafenib and PLX8394 exposure (3 hours, 1 μM) on indicated RAF:RAS and RBD:HRAS interactions in the presence or absence of BRAFV600E and BRAFR509H/V600E. The PPI analyses of n = 4 independent experiments (HEK293, ±SEM) are shown. We have normalized the PPI RLUs on the untreated PPI signals. (E) Following coexpression of indicated Rluc-PCA fragment-tagged protein pairs and HRAS variants, we determined PPIs (±SEM from n = 4 independent experiments). The RLU values have been normalized on the expression of F[2]-tagged hybrid proteins. Student’s t test was used to evaluate statistical significance. Confidence levels: *P < 0.05, **P < 0.01, and ***P < 0.001.
BRAFi-induced RAS:RAF complex formation—directly mediated through RBD of RAF—might be relevant for paradoxical MAPK reactivation, which has been observed following BRAFi exposure. In this regard, it has also been shown that kinase-impaired BRAF mutants elevate CRAF activities, thereby promoting tumor formation (4, 12). Thus, we wanted to reveal the molecular mechanism of how vemurafenib and PLX8394 inhibitor binding to BRAFV600E affects complex formation of defined RAS and CRAF combinations. As a starting point, we extended the RAS:RAF PPI reporter platform to include CRAF (Fig. 5C). GTP-loaded HRAS showed elevated PPI of CRAF:HRAS (blue frame). Coexpression of flag-tagged BRAF or BRAFV600E had no major effect on the basal CRAF:HRASG12V interactions. However, after treatment with vemurafenib, we observed a substantial increase of CRAF:HRASG12V complexes in the presence of BRAFV600E (red frame); in contrast, BRAF coexpression combined with vemurafenib treatment had no major effect (Fig. 5C; expression of reporter constructs shown in fig. S6A). On the basis of these observations, we assumed that vemurafenib-initiated complex formation of BRAFV600E with GTP-bound RAS somehow elevates the formation of CRAF:HRASG12V complexes. These experiments with the CRAF PPI reporter have been repeated to test different combinations of coexpressed BRAF mutants along with vemurafenib and PB PLX8394 exposure. We observed an elevating effect of both drugs, vemurafenib and PLX8394, on CRAF:RASG12V complex formation in the presence of coexpressed BRAFV600E. An impact of BRAFi in the used concentration on CRAF:HRASG12V (in the absence of overexpressed BRAF) cannot be neglected. In parallel experiments analyzing RBD:RASG12V interactions, the BRAF:drug-driven impact on GTP-RAS interaction was minor (Fig. 5D). These data suggest that BRAFi binding to BRAF mutants further promotes CRAF:RAS interactions, possibly involving cooperative PPIs (including RAF dimers, with vemurafenib) or, in case of the PB PLX8394, also independent of RAF dimerization. This was a quite unexpected observation for this lead molecule, which has been designed to block kinase activities and kinase dimerization (29) and is undergoing clinical trials.
Next, we tested BRAFi-driven complex formation in a different cell setting using the melanoma cell line A375 (contains BRAFV600E) and short-term reporter expressions. In these experiments, we confirmed that the BRAFi vemurafenib and PLX8394 elevate RAS:RAF interactions at expression levels of the PPI reporter hybrid proteins far below the endogenous RAS and BRAF proteins (fig. S6B). This goes well along with the initial observations from the IP studies showing that PLX8394 elevates HRAS:BRAF complexes despite of reducing RAF dimers (Fig. 3A and fig. S4A). Next, we generated stable PPI reporter cell lines to test this observation in another cell setting. Time- and dose-dependent effects of PLX8394 exposure on the complex formation in stable RBD-F[1]:HRASG12V-F[2], BRAFV600E-F[1]:HRASG12V-F[2], and BRAF-F[1]:HRASG12V-F[2] SW480 cell lines were analyzed. We observed that a one-time exposure of PLX8394 (1 μM) elevated mutated BRAF:RAS complexes over a time frame of 48 hours (fig. S6C).
Tagging of RAS at the C terminus may interfere with the membrane recruitment of the complex and nanoclustering. We repeated these experiments with N-terminally tagged HRAS constructs. Independently of tagging HRAS at the C or N terminus with the Rluc-PCA fragments, we observed vemurafenib- and PLX8394-dependent elevations of the defined RAS:BRAFV600E complexes (fig. S6D). A further evidence for the specific elevation of these RAS:RAF complexes stems from experiments with N-terminally tagged BRAF constructs. Independently of BRAF tagging (at the C or N terminus with the Rluc-PCA fragments), we observed GTP (G12V mutation)–, vemurafenib-, and PLX8394-dependent elevations of the defined RAS:BRAF complexes (figs. S6D and S7, A and B). On the basis of the finding presented, we propose that the tested αC-helix-OUT BRAFi (vemurafenib, dabrafenib, and encorafenib) have the potential to alter RAF conformations and elevate binary interactions with RAS and, in case of the PB PLX8394, also independently of RAF dimerization.
Perturbation of tetrameric RAS:RAF complexes in vivo
BRAFi binding into the ATP-binding pocket leads to allosterically mediated elevations of GTP-controlled RAS:RAF complexes, suggesting a drug-initiated mode of intramolecular communication between RAS-binding and kinase domains. We hypothesize that the underlying mechanism involves the complex formation with nanoclustered RAS dimers in the plasma membrane, which we aimed to analyze in the context of tetrameric RAS:RAF complex formation. Membrane-anchored RAS nanoclustering (or dimerization) is necessary for spatial and temporal controlled RAF activation. RAS molecules engage distinct organizations in the plasma membrane, diffuse, and dimerize. Creating high-enough local concentrations and proper orientations of RAS would favor the relatively weak PPI between two RAS molecules, which involves the α4-α5 interface (22, 45). Further, it has been shown that, besides the interaction of RAF with GTP-bound RAS, the dimerization of RAF kinases promotes catalytic activity (16, 46).
In contrast to wild-type BRAF, mutant BRAFV600E monomers are active in the absence of RAS binding and dimerization (15). Following capturing of complex formation of BRAFV600E:RASGTP (Figs. 4 and 5), we set out to determine the interlinkage of full-length RAF kinase dimerization and BRAFi-driven RAS:BRAFV600E complex formation (12). At this point, we avoided tagging of RAS molecules by PCA fragments to circumvent possible influences on RAS structure and orientation at the plasma membrane. These critical parameters need to be reflected in analyses of immunoisolated RAS:RAF complexes from cell lysates. Therefore, we refined our unique cell-based biosensor system by integrating a protocol to analyze membrane-recruited and RAS-controlled RAF dimers.
First, we advanced the BRAF PPI reporter platform to analyze RAF dimerization. Besides full-length BRAF and CRAF, we tested reporter constructs that exclusively contain the BRAF kinase domain (BRAF417–766) and therefore lack the N-terminal RBD. We observed that complexes of full-length BRAF and CRAF dimers are formed primarily in the presence of coexpressed HRAS and HRASG12V. Complexes containing truncated BRAF hybrid proteins were slightly affected by RAS coexpression (Fig. 5E). These data further underline that cellular RAF dimer formation depends on RAS abundance, GTP loading, and RAS:RAF interactions (47). These experiments illustrate that, for examining the exact mode of RAS:RAF complex formation, cellular experimental setups using full-length kinase constructs are mandatory.
Because of missing druggable binding pockets and picomolar affinities for GTP, it is challenging to block RAS functions. However, recently, a synthetic high-affinity binding protein for HRAS and KRAS has been identified. The so-called NS1 monobody blocks RAS signaling and thus RAS-initiated oncogenic transformation. The underlying mechanism of NS1 action is the blockade of HRAS dimers and HRAS nanoclusters in the plasma membrane (Fig. 6A) (45). To test whether RAS dimerization blockade affects BRAFi-initiated macromolecular RAS:RAF complex formation, we generated expression constructs consisting of the N-terminal Venus-YFP (yellow fluorescent protein) variant fused to the NS1 monobody. We also created a RAS binding–deficient NS1 mutant with the tryptophan mutations W75A and W77A in the binding interface (NS1neg; Fig. 6B). First, we confirmed selective interactions of RAS PPI reporter hybrid proteins and endogenous RAS in IPs (fig. S7C). Next, we analyzed the subcellular localization of hemagglutinin (HA)–tagged HRAS variants with NS1 and NS1neg. As expected, we observed exclusive membrane recruitment of NS1 in the presence of HA-HRAS (Fig. 6C, top panel). The NS1:HRAS interaction, as indicated in the crystal structure, is opposite of the interaction site for RAF binding (Fig. 6B). NS1 binding to HRAS does not affect GTP loading or HRAS membrane recruitment. NS1 prevents the dimerization and subsequent nanoclustering of oncogenic HRAS at the plasma membrane (45). In the first experiments, we tested the impact of both NS1 coexpression and BRAFi exposure on HRAS:BRAF complex formation. We observed that NS1 binding to HRAS does not have an obvious reducing effect on HRAS:BRAF interactions. In this experimental setting, NS1 coexpression rather had a stabilizing effect on HRAS-F[2] expressions. Also, the BRAFi (PLX8394)–driven complex formation of HRAS:BRAFV600E was not affected (fig. S7D). Next, we implemented the NS1 monobody approach to validate our reporter system for tracking tetrameric RAS:RAF complexes. First, we showed that HRASG12V also elevated CRAF:BRAFV600E complexes (Fig. 6C). Using this setting, we tested the impact of NS1 coexpression on GTPase-driven RAF dimerization. In contrast to the RAS:RAF PPI reporter experiments (fig. S7D), we observed this time that NS1 coexpression significantly prevented the RAS-triggered complex formation of BRAFV600E:CRAF dimers (Fig. 6C, bottom panel; fig. S8A for reporter expression levels). We confirmed these observations independently using IPs in the presence of NS1 and NS1neg monobodies (fig. S8B). These data indicate that the GTP-dependent RAS interactions and nanoclusters are a prerequisite for the formation of tetrameric RAS:RAF complexes that involves the BRAFV600E mutant.
Fig. 6. Perturbation of tetrameric RAS:RAF complexes in intact cells.
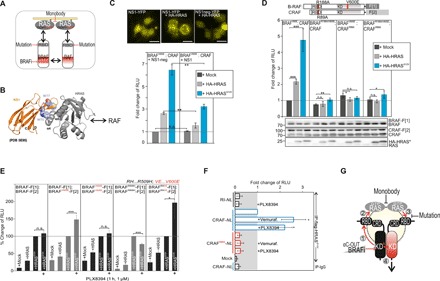
(A) Illustration of strategies on how to interfere with tetrameric RAS:RAF complexes. (B) Structure illustration indicates the localization of W75 and W77 in the RAS:NS1 binding interface (PDB: 5E95). (C) Localization study of YFP-tagged NS1 and NS1neg in the absence or presence of HA-HRAS in HEK293 cells. Scale bars, 25 μm. RAF dimerization analyses using indicated PPI reporters, NS1 variants, and upon coexpression of HRAS variants (n = 3; ±SEM). (D) RAF dimerization analyses using indicated PPI reporter constructs and coexpression of HRAS variants (n = 3; ±SEM). (E) PPI reporter analyses of transiently expressed reporter protein pairs with and without indicated HRAS variants and following 1-hour treatment with PLX8394. Quantifications from n = 5 independent experiments are shown (±SEM). (F) IP of flag-tagged HRASG12V in the presence of wild-type and RAS-binding deficient (R88A) CRAF-NanoLuc (NL) variants from BRAF-V600E–positive melanoma cells A375. The PKA RI subunit serves as unrelated prey. Quantifications from n = 4 independent experiments are shown (±SEM). Student’s t test was used to evaluate statistical significance. Confidence levels: *P < 0.05, **P < 0.01, and ***P < 0.001. (G) Systematic analyses using the full-length BRAF reporter platform unveiled the sequence of reorganizations of macromolecular kinase complexes upon αC-OUT BRAFi binding to the catalytic kinase pocket of mutated BRAF (left protomer, e.g., V600E mutation = *). Dose-dependent αC-OUT BRAFi binding to mutant BRAF promotes intermediate RAF conformations (1), thereby elevating BRAF*:RASGTP interactions (2) and endorsing the formation of RAS nanocluster/dimer–organized RASGTP:BRAF*:RAF:RASGTP tetramers (3), which may lead to the activation of a complexed second RAF protomer (4). RAS-specific monobodies and RBD:RAS interface mutations counteract BRAFi-promoted RAS:RAF complexes.
To determine the molecular requirements for RASGTP-dependent formation of tetrameric RAS:RAF complexes in the intact cell setting, we evaluated the structural details of the RAS:RAF binding interface. Again, we used the mutation R188A in the RBD of BRAF, which we have validated in Fig. 3C, to perturb the RBD:RAS interface and RAS:RAF interaction. This time, we also combined it with experiments using the corresponding mutation in the CRAF reporter construct (≙ R89A). We validated complex formation using wild-type and mutated RAS and RAF variants. We showed, using the PPI reporter as readout of cellular protein complexes, that disruption of the RAF interaction with RAS (using single-point mutations in either CRAF or BRAF) is sufficient to prevent the formation of RAF dimer, thereby underlining the existence of tetrameric RASGTP:CRAF:BRAFV600E:RASGTP complexes (Fig. 6D). We confirmed the existence of tetramers in IP experiments showing RAS-dependent RAF dimers and their reduction upon perturbation of each of the RBD:RAS binding interfaces by mutation (fig. S8C). Last, we tested the impact of PLX8394 exposure on binary RAF complexes. By using the in vivo PPI reporter and coexpression of nanoclustered HRAS variants, we show clear evidence that, despite reduced affinities for binary BRAF complexes, the PB elevated nanocluster-organized BRAF dimers, which are components of tetrameric GTP-RAS2:BRAF*2 complexes (Fig. 6E). To validate this observation that the PB also elevates RAS:CRAF interactions, we initiated LUMIER experiments of BRAFV600E-expressing A375 melanoma cell lines overexpressing HRASG12V and CRAF tagged with NanoLuc (48). In these LUMIER experiments, we showed that both vemurafenib and PLX8394 exposures elevate CRAF:HRASG12V complexes in IPs using flag-tagged HRAS (Fig. 6F). These data point out that even without kinase dimerization, BRAFi have the potential to promote activated and tetrameric RAS:RAF complexes. The implementation of the cell-based reporter system made it possible to unveil this unexpected molecular mechanism of drug treatment.
In summary, the systematic analyses of drug-mediated alterations of kinase conformations and interactions provide evidence that αC-helix-OUT BRAFi binding allosterically elevates GTPase-configured RAS:RAF tetramers by promoting “intermediate” kinase conformations. The data suggest that the sequential formation of these BRAFi-induced RAS:RAF complexes may occur also decoupled from RAF dimerization. Besides targeting binary RAS:RAF interactions, the interference with RAS nanoclustering may reduce drug-driven complex formation of tetrameric RAS:RAF, both of which should be considered to be implemented in polypharmacology approaches for targeted inhibition of deregulated RAF kinase signaling.
DISCUSSION
Structurally diverse BRAFi show significant therapeutic responses leading to regression of human tumors harboring BRAFV600E mutation. The benefit of FDA-approved αC-helix-OUT BRAFi [vemurafenib (PLX4032) and dabrafenib (GSK2118436A)] is the selectivity and high efficacy for inhibiting mutant BRAF activities, thereby avoiding or reducing subsequent and proliferative MEK-ERK activation, for example, in melanoma. However, the observed therapeutic effect is often temporary due to the occurrence of drug resistance, which is in part based on altered PPIs and on kinase-activating scaffolding functions of inhibited kinase protomers. Tumors become insensitive to the treatments due to different mechanisms that lead to reactivation of MAPK signaling. Such a reactivation of ERK signaling involves the drug-triggered formation of RAF dimers along with the creation of additional feedback circuits (9, 11–16). These observations highlight the importance to systematically dissect the underlying molecular mechanisms of RAS-RAF-MEK-ERK signaling related to changes in kinase conformations and physical effector interactions directly in the cellular setting (16). We generated and implemented a unique cell-based reporter platform to noninvasively profile RAF kinase conformations (KinCon reporters), RAF dimerization, and perturbations of tetrameric interactions between RAF variants and RAS (PPI reporters). In this way, we tracked efficacies of defined bioactive small molecules along with approved drugs on kinase conformations and RAS:RAF PPIs. By this means, we unveiled the sequence of events of drug-driven RAS:RAF complex formation that, unexpectedly, proceeds also independent of RAF dimerization.
As a starting point, we generated an intramolecular KinCon reporter to track full-length kinase conformation dynamics directly in intact cells. Activation of endogenous EGFR by EGF is sufficient to immediately initiate a RAS-dependent transition of the wild-type reporter to a definitive opened kinase conformation (Fig. 1C). We showed that the BRAF patient mutations display a similar opening effect on the full-length BRAF conformation. We present direct evidence that, independent of RAS engagement, all tested patient mutations of BRAF converted the autoinhibitory kinase into opened kinase conformations (Figs. 1B and 2E). Next, the suitability of the KinCon reporters to profile and compare target specificity of kinase inhibitors was demonstrated by analyzing a collection of BRAF mutants using the full-length RAF kinase as a flexible reporter framework. By this means, we unveiled an unexpected allosteric effect of mutation-specific anticancer drugs (FDA-approved: vemurafenib, dabrafenib, and encorafenib) and one lead molecule (PLX8394) in clinical trials on mutated BRAF conformations. We observed that the engagement of all αC-helix-OUT BRAFi with the catalytic pocket of patient mutation–containing BRAF reporters stabilized more closed intermediate full-length kinase conformations. In the case of BRAFV600E, the KinCon kinase activities inversely correlated with full-length BRAF conformations (Fig. 2C). However, we detected these opening/closing effects with BRAF KinCon reporters harboring both phosphotransferase-activating (G469A, V600E/K, and K601E) and phosphotransferase-inactivating mutations (D594G). It is feasible that the kinase-dimer formation potential of BRAF mutants [which is high for D594G (12, 36)] is relevant for KinCon reporter dynamics (mutation-driven “opening” and drug-driven “closing”). Also, BRAFV600E forms RAS-dependent dimers with other RAF protomers. However, direct BRAFV600E downstream signaling does not rely on an intact interface (15, 36, 47, 49). These observations now promote research efforts to systematically correlate the kinase dimerization potential, drug-driven kinase conformations, and the dynamics of quaternary RAS:RAF complexes using an extended collection of less frequent BRAF mutants.
The notable transformations of mutated and full-length BRAF kinase conformations upon specific BRAFi engagement with the ATP-binding pocket prompted us to explore feasible consequences of conformation changes on RAF-emanating binary protein interactions. We focused on PPI dynamics emanating from BRAFV600E. For this purpose, we generated an intermolecular PPI reporter for quantifying changes in the interactions of full-length RAS:RAF complexes in vivo. With this extendable full-length RAS:RAF PPI reporter toolbox, we characterized the mechanistic details for drug-driven RAS:RAF complex formation. In addition to GTP-dependent recordings of cellular BRAF, CRAF dimerization and KRAS, NRAS, and HRAS interactions, we found that, selectively, αC-helix-OUT BRAFi binding elevated RAF:RAS complexes (Figs. 3 to 5). We demonstrated that BRAFi binding into the catalytic cleft of the mutant BRAF C-terminal kinase domain allosterically elevated complex formation of mutated RAS:BRAF, which is directly mediated by the regulatory N-terminal RBD. We schematically depict the successive consequences of BRAFi binding on (i) full-length and mutated RAF conformation, (ii) elevated RAS interactions, and (iii) promotion of tetramerization in Fig. 6G.
These alterations of RAF:RAS complexes are a specific feature of the tested αC-helix-OUT BRAF mutation–specific inhibitors vemurafenib, dabrafenib, encorafenib, and PLX8394. Apparently, these BRAFi promote intermediate BRAF conformations that display higher affinity for RAS and then the fully activated and opened BRAF mutants. Recently, Jin and colleagues (31) published that selected pan-RAFi disrupt autoinhibitory conformations of wild-type BRAF. Our data confirmed that the pan-RAFi LY3009120 reduces autoinhibited wild-type BRAF conformations (Fig. 1D and fig. S3C). On the basis of the available data, we assume that pan-RAFi binding to wild-type BRAF (their study) and αC-helix-OUT BRAFi binding to BRAF mutants (our study) favor an intermediate kinase conformation, which may display higher affinities for GTP-loaded RAS (fig. S8, E and F). This working model also provides a feasible explanation for higher RASGTP affinities of different class 3 BRAF mutants with different conformations and impaired kinase activities (35).
The implementation of PBs into this study of mutated BRAF complexes unveiled an efficient autoinhibitory effect on all patient mutation–initiated opened BRAF conformations. It was quite unexpected for us to observe that the PLX8394-driven formation of BRAF*:RASGTP occurred also independently of RAF dimerization. Only recently, the potency of PLX8394 has been demonstrated by reducing the cell growth in certain lung carcinoma cells, which actually depends on noncanonical BRAF mutant activities. This includes the BRAF mutant G469A (32), which we have shown to be affected in the BRAFi profiling experiments (Fig. 2E). We analyzed the consequence of binding of αC-helix-OUT BRAFi to the full-length BRAFV600E oncoprotein, which has implications for the architecture of the tetrameric RAS:RAF complex. We admit that, for a full understanding of the underlying mechanistic details of drug-driven RAS:RAF complex formation, full-length structures are required. However, we show that the RAS functions related to RAS nanoclusters along with the existence of functionally important RAS dimers (22, 50) are relevant for the observed phenomenon of drug-driven complex formation of RAS:RAF tetramers. As predicted, we confirmed in co-IPs that the PB PLX8394 disrupts kinase dimers. However, we showed that mutation-specific RAS:RAF interactions were somehow elevated with PLX8394 (Fig. 3A and figs. S4A and S6F). It was the implementation of the cell-based reporter system that unveiled the molecular details of this unexpected observation. We adapted our reporter studies for recordings of plasma membrane–organized RAS:RAF tetramers. The reporter studies with intact RAS nanoclusters unveiled the mechanistic details of how BRAFi elevate tetramers with and without binary kinase dimerization. We have validated our cell-based reporter platform using defined RAS mutations along with RAS-specific monobodies, which were used to antagonize RASGTP-organized and BRAFi-BRAF*–promoted RAS:RAF:RAF:RAS tetramers (Fig. 6). The systematic implementation of the full-length kinase reporter platform facilitated the deciphering of the sequential events of the formation of these multilayered RAF complexes (Fig. 6G).
Despite the size of full-length BRAF and CRAF, the different KinCon reporters are functional phosphotransferases, and inhibition using the αC-helix-OUT BRAFi is reflected by the correlation of conformational change and ERK1/2 phosphorylation (Fig. 2 and fig. S1). RAF contains autoinhibited sequence elements, which explain conformational reorganization upon activation. This fact underlines that the integration of other kinases into the KinCon reporter system is reasonable for measuring kinase conformation dynamics. Such a modular KinCon reporter platform opens new means to systematically analyze—in the case of RAF signaling—the nearly 300 distinct missense mutations of BRAF, which have been identified in cancer cell lines or tumor cells (17). We assume that extensive analyses of KinCon:drug interactions will foster and accelerate a more rational design of effective kinase drugs in the context of personalized medicine by selecting the right drug for BRAF-specific patient mutations. However, it should not be forgotten that the kinase needs to be tagged at both termini to obtain a direct readout for kinase conformations. This might hamper kinase activities, kinase compartmentalization, and reporter expression. For every KinCon reporter, correlations between conformations and activities need to be performed. Another drawback might be the size of the used Rluc fragments for tagging. The implementation of smaller and brighter luciferase PCAs might be a feasible alternative (51).
Our reporter studies underline that BRAFi interactions with the highly dynamic and activity-relevant αC helix are allosterically coupled with conformational changes of the full-length autoinhibited kinase (4, 49). We assume that the spatially conserved hydrophobic spines of kinases, termed catalytic and regulatory spines, change their alignment, which may contribute to the observed alterations of RAF KinCon and RAF PPIs with RAS (52). Other kinase conformations and kinase:drug interactions need to be tested to evaluate the possibility that ATP-competitive kinase inhibitors cause alterations of conformations/molecular interactions, which are relevant for off-target effects. On the basis of these observations, we speculate that, upon BRAFi (vemurafenib, dabrafenib, encorafenib, and PLX8394) treatments, the proliferation of cancer cell subpopulations showing elevated GTP-RAS activities or levels will be favored (53). Using the PPI reporter platform, we managed to quantify binary complexes in the in vivo setting. Similar to the KinCon biosensor, the tagging might affect protein function and molecular interactions. Nevertheless, we validated PPI using binding interface mutations showing clear evidence that even with expression levels below the endogenous protein of interest, we show dynamics of binary complex formation (see Figs. 3C and 5C and figs. S3, A and C, and S5A), which cannot be observed using conventional biochemical assays such as IPs.
The BRAFi-induced elevation of RAS:RAF complexes might be one additional explanation for the phenomenon of drug resistance through receptor tyrosine kinase (RTK) or RAS up-regulation (14, 16). We assume that preventing the drug-driven recruitment of BRAF into RAS complexes might be a feasible strategy to reduce cooperative RAF activation, which leads to paradoxical ERK activation. Note that this reporter setup can be subjected to evaluate or screen lead small molecules, which directly or allosterically interfere with this complex in the appropriate cell setting.
This study showed one example how the binding of a small molecule into the catalytic cleft of an enzyme reshapes kinase-regulating molecular interactions. We assume that this unexpected phenomenon of a drug-driven allosteric regulation of PPIs might be relevant for efficacies (function and off-target function) of other lead molecules targeting autoinhibited enzymes. The rational generation of a bidirectional RAF reporter platform exemplifies that systematic investigations of full-length oncokinase:drug and oncokinase:regulator interactions in an intact cell setting are possible. The realization that opened and closed states of autoinhibited enzymes can be tracked opens new opportunities for engineering novel interaction and conformation reporters for “undruggable,” novel, or neglected drug targets.
MATERIALS AND METHODS
Experimental design
We aimed to engineer a genetically encoded reporter platform to quantify (i) molecular interactions of kinases with regulatory proteins, (ii) with bioactive small molecules along with (iii) accompanying intramolecular kinase conformational. The full-length kinase reporters should be applicable for noninvasive recordings of open and closed kinase conformations and molecular protein interactions upon mutation, drug exposure, or upstream activation of the appropriate signaling pathway.
Reagents
BRAFi used are as follows: PLX4032 (vemurafenib; MedChemExpress, #HY-12057), LGX818 (encorafenib; MedChemExpress, #HY-15605), GSK2118436A (dabrafenib; Selleckchem, #S2807), PLX8394 (MedChemExpress, #HY-18972), AZ628 (Selleckchem, #S2746), GDC0879 (Selleckchem, #S1104), LY3009120 (Selleckchem, #S7842), and TAK632 (Selleckchem, #S7291). MEKi used are as follows: AZD6244 (selumetinib; Selleckchem, #S1008), BAY86-9766 (refametinib; MedChemExpress, #HY-102156), and benzyl-coelenterazine (NanoLight, #301).
Cell culture and antibodies
HEK293, A375, and SW480 cells were grown in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum (FBS). Transient transfections were performed with TransFectin reagent (Bio-Rad, #1703352) or jetPRIME (Polyplus). Primary antibodies used were mouse anti-Rluc antibodies (Chemi-Con, #MAB4400 versus Rluc-F[2] and #MAB4410 versus Rluc-F[1]), mouse anti–HA-tag (Covance, #MMS-10P), mouse anti-FLAG (Sigma-Aldrich, #F3165), mouse anti-V5 (Invitrogen, #R9302), mouse anti-BRAF [Santa Cruz Biotechnology, sc-5284 (#F-7) and sc-166 (#C-19)], rabbit anti-CRAF (Cell Signaling Technology, #9422), mouse anti-RAS (Thermo Fisher Scientific, #MA1-012X), mouse anti-GFP (green fluorescent protein) (Roche, #11814460001), mouse anti-MEK1/2 (Cell Signaling Technology, #4684S), rabbit anti–P-MEK1/2 (Ser217/Ser221) (Cell Signaling Technology, #9154), rabbit anti–P-ERK1/2 (Cell Signaling Technology, #9101), rabbit anti-ERK1/2 (Cell Signaling Technology, #4696S), and mouse anti-PKA RIIβ (BD Biosciences, #61062).
Expression constructs
PPI reporter
The Rluc-PCA–based hybrid proteins RIIβ-F[1] and RIIβ-F[2] (NM_001030020.1) were designed as previously described (26). Rluc-PCA fusions of RAS and RAF were generated using an analogous cloning approach. Following polymerase chain reaction (PCR) amplification of the human RAS genes (HRas: NM_005343.3, Addgene plasmid #39503; KRas: NM_004985.4 and NRas: NM_002524.4) or RAF genes (BRaf: NM_004333.4, Addgene plasmid #40775; CRaf: NM_002880.3), we fused them C/N-terminally with either -F[1] or -F[2] of the Rluc-PCA (pcDNA3.1 backbone vector). KRas and NRas were provided by the laboratory of P. Crespo. A site-directed mutagenesis approach was used to generate the RAS(G12V), BRAF (R166A, R188A, G469A, R509H, D594G, V600E, V600K, and K601E), and CRAF (R89A) amino acid substitutions.
KinCon reporter
The Rluc-PCA–based hybrid proteins F[1]-BRAF*-F[2] and F[1]-CRAF-F[2] were generated using the identical cloning approach. Following PCR amplification of the human BRAF or CRAF gene, we fused the protein-coding region N-terminally with F[1]- and C-terminally with -F[2] of the Rluc. We inserted interjacent 10–amino acid linkers.
Monobody
NS1 and NS1neg monobodies were generated by inserting the NS1 coding sequence C-terminally of the Venus-YFP coding sequence using Not I and Xba I (45).
Peptide-tagged constructs
V5-, flag-, and HA-tagged constructs were constructed by inserting the corresponding Flag/HA/V5 tag sequence to the N terminus of the RAF or RAS coding sequence. The resulting constructs were then inserted into the multiple cloning site of pcDNA3.1 (Not I:Xba I).
Luciferase PCA analyses
Indicated cells were grown in DMEM supplemented with 10% FBS. We transiently overexpressed indicated versions of the Rluc-PCA–based reporter in 96-, 24-, or 12-well plate formats. Twenty-four or 48 hours after transfection, we initiated the drug exposure experiments. We partially removed the growth medium and added BRAFi or MEKi with the final concentrations as indicated in the figure legends. To measure the dose-dependent effect of the lead molecules on the intramolecular Rluc-PCA reporter, we treated cells with different concentrations and for different time frames. For the luciferase-PCA measurements, the growth medium was carefully removed and the cells were washed with phosphate-buffered saline (PBS). Cell suspensions were transferred to 96-well plates and subjected to luminescence analysis using the SpectraMax L Microplate Reader (Molecular Devices). Luciferase luminescence signals were integrated for 10 s following addition of the Rluc substrate benzyl-coelenterazine (NanoLight, #301). Dose-dependent effects of BRAFi exposure on BRAF conformation reporter–emanating luminescence signals were measured simultaneously in 1536-well plate format. Detached HEK293 cells (0.5 × 106 cells) transiently expressing indicated conformation reporters were each treated for 1 hour with increasing concentrations of BRAFi in 1.5-ml test tubes. Immediately after BRAFi exposure (1 hour), the cells were centrifuged and the pellets were resuspended in 25 μl of PBS and 0.125 μl of benzyl-coelenterazine; 7.5 μl of cell suspension was transferred to each well of a 1536-well plate and the luminescence was captured for 30 min on the Fusion imaging platform (Bio-Rad).
Immunoprecipitation
Following 48 hours of transient overexpression of indicated flag-tagged or YFP-tagged expression constructs, we treated the cells with 1 μM or 10 μM vemurafenib, encorafenib, dabrafenib, or PLX8394 for 1, 3, or 16 hours. Subsequent to PBS washing steps, we homogenized them using a syringe with 15 strikes [standard lysis buffer: 10 mM sodium phosphate (pH 7.2), 150 mM NaCl, 0.5% Triton X-100 supplemented with standard protease inhibitors and phosphatase inhibitors]. We clarified the lysate (13,000 rpm, 20 min) and performed IPs using Protein A/G mixtures and 2 μg of control and anti–flag-tagged or GFP antibodies for 3 hours at 4°C. Resin-associated proteins were washed four times with standard lysis buffer and eluted with Laemmli sample buffer.
ERK1/2 and P-MEK1/2 phosphorylation
Following overexpression of indicated KinCon constructs, we directly determined the phosphorylation status of ERK1/2 and/or MEK1/2. We treated HEK293 cells with indicated BRAFi, exchanged the medium, and added the Laemmli sample buffer.
Stable cell lines
SW480 cells were grown in DMEM supplemented with 10% FBS.Transient transfection was performed with TransFectin reagent (Bio-Rad, #1703350). Forty-eight hours after transfection, the growth medium was exchanged and zeocin (Invitrogen, #R25001) and/or hygromycin (Thermo Fisher Scientific, #10687-010) with a final concentration of 250 μg/ml were added as selection markers. The growth medium supplemented with zeocin and/or hygromycin was exchanged every day for 5 days. Cells were trypsinized and split 1:2 on two cell culture dishes to remove the dead cells. Cells were checked daily, and growth medium supplemented with zeocin and/or hygromycin was exchanged. Stable clones with a diameter of ~1 mm were selected and transferred to each well of a 24-well plate. They were grown to confluency and transferred to 12-well plates. Cells (0.5 × 106) of each clone were selected for Rluc-PCA measurement to control the successful overexpression of the reporter in the cell line.
Imaging
HEK293 cells were seeded in low density (50,000 cells per well) on a μ-Slide four-well chambered coverslip (ibidi, #80426) in DMEM supplemented with 10% FBS. The cells were cotransfected with differentially tagged variants of HRAS and NS1-VenusYFP using the TransFectin reagent (Bio-Rad, #1703352). Twenty-four hours after transfection, the cells were fixed for 10 min using 4% paraformaldehyde in PBS. The localizations of NS1 and NS1neg were recorded using an Axiovert 200M microscope (Carl Zeiss) with a 63-fold magnification. Acquisition was controlled with the VisiView4.0 software. The images were recorded over an exposure time of 200 ms and processed with the ImageJ software.
In silico CRE prediction
The amino acid sequence of BRAF was processed by the Cis-regPred server (http://aimpred.cau.ac.kr) for the presence of CREs (37).
LUMIER experiments
A375 melanoma cell lines (BRAFV600E positive) were transiently transfected with CRAF-NL (NanoLuc) and flag-tagged HRAS-G12V constructs using jetPRIME transfection reagent. Clarified cell extracts were generated and incubated with control and anti-flag antibodies (2 μg per sample) and chilled on ice for 1 hour. After addition of protein G–Sepharose beads, samples were incubated for 2 hours at 4°C on an overhead shaker. Immunoisolated complexes were washed three times with lysis buffer and three times with PBS.Probes were transferred to 96-well white-walled plates and subjected to bioluminescence analysis using the PHERAstar FSX luminometer. NL bioluminescence signals were integrated for 10 s following addition of luciferase substrate (48).
Statistics
Data were examined for Gaussian distribution using the Kolmogorov-Smirnov normality test. Non-Gaussian–distributed data were analyzed using the nonparametric Mann-Whitney U test. Unpaired Student’s t tests were used to evaluate statistical significance. Values are expressed as means ± SEM or means ± SD, as indicated. Significance was set at the 95% confidence level and ranked as *P < 0.05, **P < 0.01, and ***P < 0.001.
Supplementary Material
Acknowledgments
We thank K. Bister and A. Shaw for comments on the manuscript; V. Bachmann and J. Fleischmann for cloning support; P. Tschaikner for helping with the imaging; S. Beiler, J. Insole, and A. Reintjes for technical support; and G. Reiter for management support. Funding: This work was supported by grants from the Austrian Science Fund (FWF; P22608, P27606, P30441, to E.S. and SFB-F44) and the U.S. NIH (grant R01 CA212608 to S.K. and grant DK54441 to S.S.T.). Author contributions: R.R., J.E.M., O.T.-Q., F.E., A.R., P.R., A.F., R.G.H., and E.S. performed the experiments. R.R., J.E.M., F.E., O.T.-Q., P.R., S.K., S.S.T., J.T., and E.S. analyzed the results. E.S. conceived the project and wrote the manuscript. Competing interests: S.K. is listed as an inventor of issued and pending patents on the monobody technology filed by the University of Chicago and Novartis AG (U.S. Patent 9512199 B2, EU patent application EP2598529A2, and related applications). Aspects of the present study (KinCon reporter) are subject of pending patent applications (J.E.M., E.S.; University of Innsbruck). The authors declare no other competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. The KinCon and PPI vectors can be provided by the corresponding author (E.S.), pending scientific review and a completed material transfer agreement. Requests for the KinCon and PPI vectors should be submitted to eduard.stefan@uibk.ac.at (University of Innsbruck). Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/5/8/eaav8463/DC1
Table S1. Used expression constructs, materials, and cell lines.
Fig. S1. BRAF KinCon reporter activities and BRAF profiling.
Fig. S2. Impact of defined kinase mutations on BRAF KinCon reporter dynamics.
Fig. S3. BRAF KinCon reporter dynamics.
Fig. S4. Complex formation of RAS:BRAF and PPI reporter dynamics.
Fig. S5. Impact of vemurafenib on indicated PPIs.
Fig. S6. Impact of BRAFi on binary RAS:RAF interactions.
Fig. S7. PPI reporter dynamics and NS1 monobody IPs.
Fig. S8. Validation of PPI dynamics and impact of RAFi on RAF conformations.
REFERENCES AND NOTES
- 1.Fleuren E. D. G., Zhang L., Wu J., Daly R. J., The kinome ‘at large’ in cancer. Nat. Rev. Cancer 16, 83–98 (2016). [DOI] [PubMed] [Google Scholar]
- 2.Davies H., Bignell G. R., Cox C., Stephens P., Edkins S., Clegg S., Teague J., Woffendin H., Garnett M. J., Bottomley W., Davis N., Dicks E., Ewing R., Floyd Y., Gray K., Hall S., Hawes R., Hughes J., Kosmidou V., Menzies A., Mould C., Parker A., Stevens C., Watt S., Hooper S., Wilson R., Jayatilake H., Gusterson B. A., Cooper C., Shipley J., Hargrave D., Pritchard-Jones K., Maitland N., Chenevix-Trench G., Riggins G. J., Bigner D. D., Palmieri G., Cossu A., Flanagan A., Nicholson A., Ho J. W. C., Leung S. Y., Yuen S. T., Weber B. L., Seigler H. F., Darrow T. L., Paterson H., Marais R., Marshall C. J., Wooster R., Stratton M. R., Futreal P. A., Mutations of the BRAF gene in human cancer. Nature 417, 949–954 (2002). [DOI] [PubMed] [Google Scholar]
- 3.Wellbrock C., Karasarides M., Marais R., The RAF proteins take centre stage. Nat. Rev. Mol. Cell Biol. 5, 875–885 (2004). [DOI] [PubMed] [Google Scholar]
- 4.Lavoie H., Therrien M., Regulation of RAF protein kinases in ERK signalling. Nat. Rev. Mol. Cell Biol. 16, 281–298 (2015). [DOI] [PubMed] [Google Scholar]
- 5.Pylayeva-Gupta Y., Grabocka E., Bar-Sagi D., RAS oncogenes: Weaving a tumorigenic web. Nat. Rev. Cancer 11, 761–774 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Durrant D. E., Morrison D. K., Targeting the Raf kinases in human cancer: The Raf dimer dilemma. Br. J. Cancer 118, 3–8 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cseh B., Doma E., Baccarini M., “RAF” neighborhood: Protein-protein interaction in the Raf/Mek/Erk pathway. FEBS Lett. 588, 2398–2406 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Desideri E., Cavallo A. L., Baccarini M., Alike but different: RAF paralogs and their signaling outputs. Cell 161, 967–970 (2015). [DOI] [PubMed] [Google Scholar]
- 9.Lito P., Rosen N., Solit D. B., Tumor adaptation and resistance to RAF inhibitors. Nat. Med. 19, 1401–1409 (2013). [DOI] [PubMed] [Google Scholar]
- 10.Girotti M. R., Saturno G., Lorigan P., Marais R., No longer an untreatable disease: How targeted and immunotherapies have changed the management of melanoma patients. Mol. Oncol. 8, 1140–1158 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hatzivassiliou G., Song K., Yen I., Brandhuber B. J., Anderson D. J., Alvarado R., Ludlam M. J. C., Stokoe D., Gloor S. L., Vigers G., Morales T., Aliagas I., Liu B., Sideris S., Hoeflich K. P., Jaiswal B. S., Seshagiri S., Koeppen H., Belvin M., Friedman L. S., Malek S., RAF inhibitors prime wild-type RAF to activate the MAPK pathway and enhance growth. Nature 464, 431–435 (2010). [DOI] [PubMed] [Google Scholar]
- 12.Heidorn S. J., Milagre C., Whittaker S., Nourry A., Niculescu-Duvas I., Dhomen N., Hussain J., Reis-Filho J. S., Springer C. J., Pritchard C., Marais R., Kinase-dead BRAF and oncogenic RAS cooperate to drive tumor progression through CRAF. Cell 140, 209–221 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Poulikakos P. I., Zhang C., Bollag G., Shokat K. M., Rosen N., RAF inhibitors transactivate RAF dimers and ERK signalling in cells with wild-type BRAF. Nature 464, 427–430 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nazarian R., Shi H., Wang Q., Kong X., Koya R. C., Lee H., Chen Z., Lee M.-K., Attar N., Sazegar H., Chodon T., Nelson S. F., McArthur G., Sosman J. A., Ribas A., Lo R. S., Melanomas acquire resistance to B-RAF(V600E) inhibition by RTK or N-RAS upregulation. Nature 468, 973–977 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Poulikakos P. I., Persaud Y., Janakiraman M., Kong X., Ng C., Moriceau G., Shi H., Atefi M., Titz B., Gabay M. T., Salton M., Dahlman K. B., Tadi M., Wargo J. A., Flaherty K. T., Kelley M. C., Misteli T., Chapman P. B., Sosman J. A., Graeber T. G., Ribas A., Lo R. S., Rosen N., Solit D. B., RAF inhibitor resistance is mediated by dimerization of aberrantly spliced BRAF(V600E). Nature 480, 387–390 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Karoulia Z., Gavathiotis E., Poulikakos P. I., New perspectives for targeting RAF kinase in human cancer. Nat. Rev. Cancer 17, 676–691 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Holderfield M., Deuker M. M., McCormick F., McMahon M., Targeting RAF kinases for cancer therapy: BRAF-mutated melanoma and beyond. Nat. Rev. Cancer 14, 455–467 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zebisch A., Staber P. B., Delavar A., Bodner C., Hiden K., Fischereder K., Janakiraman M., Linkesch W., Auner H. W., Emberger W., Windpassinger C., Schimek M. G., Hoefler G., Troppmair J., Sill H., Two transforming C-RAF germ-line mutations identified in patients with therapy-related acute myeloid leukemia. Cancer Res. 66, 3401–3408 (2006). [DOI] [PubMed] [Google Scholar]
- 19.Kim J., Ahuja L. G., Chao F.-A., Xia Y., McClendon C. L., Kornev A. P., Taylor S. S., Veglia G., A dynamic hydrophobic core orchestrates allostery in protein kinases. Sci. Adv. 3, e1600663 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Peng S. B., Henry J. R., Kaufman M. D., Lu W. P., Smith B. D., Vogeti S., Rutkoski T. J., Wise S., Chun L., Zhang Y., Van Horn R. D., Yin T., Zhang X., Yadav V., Chen S. H., Gong X., Ma X., Webster Y., Buchanan S., Mochalkin I., Huber L., Kays L., Donoho G. P., Walgren J., McCann D., Patel P., Conti I., Plowman G. D., Starling J. J., Flynn D. L., Inhibition of RAF isoforms and active dimers by LY3009120 leads to anti-tumor activities in RAS or BRAF mutant cancers. Cancer Cell 28, 384–398 (2015). [DOI] [PubMed] [Google Scholar]
- 21.Castro A. F., Rebhun J. F., Quilliam L. A., Measuring Ras-family GTP levels in vivo—Running hot and cold. Methods 37, 190–196 (2005). [DOI] [PubMed] [Google Scholar]
- 22.Ambrogio C., Köhler J., Zhou Z.-W., Wang H., Paranal R., Li J., Capelletti M., Caffarra C., Li S., Lv Q., Gondi S., Hunter J. C., Lu J., Chiarle R., Santamaría D., Westover K. D., Jänne P. A., KRAS dimerization impacts MEK inhibitor sensitivity and oncogenic activity of mutant KRAS. Cell 172, 857–868.e15 (2018). [DOI] [PubMed] [Google Scholar]
- 23.Zhang J., Yang P. L., Gray N. S., Targeting cancer with small molecule kinase inhibitors. Nat. Rev. Cancer 9, 28–39 (2009). [DOI] [PubMed] [Google Scholar]
- 24.Samatar A. A., Poulikakos P. I., Targeting RAS-ERK signalling in cancer: Promises and challenges. Nat. Rev. 13, 928–942 (2014). [DOI] [PubMed] [Google Scholar]
- 25.Cutler R. E. Jr., Stephens R. M., Saracino M. R., Morrison D. K., Autoregulation of the Raf-1 serine/threonine kinase. Proc. Natl. Acad. Sci. U.S.A. 95, 9214–9219 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Röck R., Bachmann V., Bhang H.-e. C., Malleshaiah M., Raffeiner P., Mayrhofer J. E., Tschaikner P. M., Bister K., Aanstad P., Pomper M. G., Michnick S. W., Stefan E., In-vivo detection of binary PKA network interactions upon activation of endogenous GPCRs. Sci. Rep. 5, 11133 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bachmann V. A., Mayrhofer J. E., Ilouz R., Tschaikner P., Raffeiner P., Röck R., Courcelles M., Apelt F., Lu T.-W., Baillie G. S., Thibault P., Aanstad P., Stelzl U., Taylor S. S., Stefan E., Gpr161 anchoring of PKA consolidates GPCR and cAMP signaling. Proc. Natl. Acad. Sci. U.S.A. 113, 7786–7791 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wan P. T. C., Garnett M. J., Roe S. M., Lee S., Niculescu-Duvaz D., Good V. M., Project C. G., Jones C. M., Marshall C. J., Springer C. J., Barford D., Marais R., Mechanism of activation of the RAF-ERK signaling pathway by oncogenic mutations of B-RAF. Cell 116, 855–867 (2004). [DOI] [PubMed] [Google Scholar]
- 29.Zhang C., Spevak W., Zhang Y., Burton E. A., Ma Y., Habets G., Zhang J., Lin J., Ewing T., Matusow B., Tsang G., Marimuthu A., Cho H., Wu G., Wang W., Fong D., Nguyen H., Shi S., Womack P., Nespi M., Shellooe R., Carias H., Powell B., Light E., Sanftner L., Walters J., Tsai J., West B. L., Visor G., Rezaei H., Lin P. S., Nolop K., Ibrahim P. N., Hirth P., Bollag G., RAF inhibitors that evade paradoxical MAPK pathway activation. Nature 526, 583–586 (2015). [DOI] [PubMed] [Google Scholar]
- 30.Adelmann C. H., Ching G., Du L., Saporito R. C., Bansal V., Pence L. J., Liang R., Lee W., Tsai K. Y., Comparative profiles of BRAF inhibitors: The paradox index as a predictor of clinical toxicity. Oncotarget 7, 30453–30460 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jin T., Lavoie H., Sahmi M., David M., Hilt C., Hammell A., Therrien M., RAF inhibitors promote RAS-RAF interaction by allosterically disrupting RAF autoinhibition. Nat. Commun. 8, 1211 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Okimoto R. A., Lin L., Olivas V., Chan E., Markegard E., Rymar A., Neel D., Chen X., Hemmati G., Bollag G., Bivona T. G., Preclinical efficacy of a RAF inhibitor that evades paradoxical MAPK pathway activation in protein kinase BRAF-mutant lung cancer. Proc. Natl. Acad. Sci. U.S.A. 113, 13456–13461 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Whittaker S., Kirk R., Hayward R., Zambon A., Viros A., Cantarino N., Affolter A., Nourry A., Niculescu-Duvaz D., Springer C., Marais R., Gatekeeper mutations mediate resistance to BRAF-targeted therapies. Sci. Transl. Med. 2, 35ra41 (2010). [DOI] [PubMed] [Google Scholar]
- 34.Dankner M., Rose A. A. N., Rajkumar S., Siegel P. M., Watson I. R., Classifying BRAF alterations in cancer: New rational therapeutic strategies for actionable mutations. Oncogene 37, 3183–3199 (2018). [DOI] [PubMed] [Google Scholar]
- 35.Yao Z., Yaeger R., Rodrik-Outmezguine V. S., Tao A., Torres N. M., Chang M. T., Drosten M., Zhao H., Cecchi F., Hembrough T., Michels J., Baumert H., Miles L., Campbell N. M., de Stanchina E., Solit D. B., Barbacid M., Taylor B. S., Rosen N., Tumours with class 3 BRAF mutants are sensitive to the inhibition of activated RAS. Nature 548, 234–238 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Roring M., Herr R., Fiala G. J., Heilmann K., Braun S., Eisenhardt A. E., Halbach S., Capper D., von Deimling A., Schamel W. W., Saunders D. N., Brummer T., Distinct requirement for an intact dimer interface in wild-type, V600E and kinase-dead B-Raf signalling. EMBO J. 31, 2629–2647 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yeon J. H., Heinkel F., Sung M., Na D., Gsponer J., Systems-wide identification of cis-regulatory elements in proteins. Cell Syst. 2, 89–100 (2016). [DOI] [PubMed] [Google Scholar]
- 38.Holderfield M., Merritt H., Chan J., Wallroth M., Tandeske L., Zhai H., Tellew J., Hardy S., Hekmat-Nejad M., Stuart D. D., McCormick F., Nagel T. E., RAF inhibitors activate the MAPK pathway by relieving inhibitory autophosphorylation. Cancer Cell 23, 594–602 (2013). [DOI] [PubMed] [Google Scholar]
- 39.Karoulia Z., Wu Y., Ahmed T. A., Xin Q., Bollard J., Krepler C., Wu X., Zhang C., Bollag G., Herlyn M., Fagin J. A., Lujambio A., Gavathiotis E., Poulikakos P. I., An integrated model of RAF inhibitor action predicts inhibitor activity against oncogenic BRAF signaling. Cancer Cell 30, 485–498 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Marais R., Light Y., Paterson H. F., Mason C. S., Marshall C. J., Differential regulation of Raf-1, A-Raf, and B-Raf by oncogenic ras and tyrosine kinases. J. Biol. Chem. 272, 4378–4383 (1997). [DOI] [PubMed] [Google Scholar]
- 41.Rebocho A. P., Marais R., ARAF acts as a scaffold to stabilize BRAF:CRAF heterodimers. Oncogene 32, 3207–3212 (2013). [DOI] [PubMed] [Google Scholar]
- 42.Lavoie H., Thevakumaran N., Gavory G., Li J. J., Padeganeh A., Guiral S., Duchaine J., Mao D. Y. L., Bouvier M., Sicheri F., Therrien M., Inhibitors that stabilize a closed RAF kinase domain conformation induce dimerization. Nat. Chem. Biol. 9, 428–436 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zhang W., BRAF inhibitors: The current and the future. Curr. Opin. Pharmacol. 23, 68–73 (2015). [DOI] [PubMed] [Google Scholar]
- 44.Sos M. L., Levin R. S., Gordan J. D., Oses-Prieto J. A., Webber J. T., Salt M., Hann B., Burlingame A. L., McCormick F., Bandyopadhyay S., Shokat K. M., Oncogene mimicry as a mechanism of primary resistance to BRAF inhibitors. Cell Rep. 8, 1037–1048 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Spencer-Smith R., Koide A., Zhou Y., Eguchi R. R., Sha F., Gajwani P., Santana D., Gupta A., Jacobs M., Herrero-Garcia E., Cobbert J., Lavoie H., Smith M., Rajakulendran T., Dowdell E., Okur M. N., Dementieva I., Sicheri F., Therrien M., Hancock J. F., Ikura M., Koide S., O’Bryan J. P., Inhibition of RAS function through targeting an allosteric regulatory site. Nat. Chem. Biol. 13, 62–68 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Rajakulendran T., Sahmi M., Lefrancois M., Sicheri F., Therrien M., A dimerization-dependent mechanism drives RAF catalytic activation. Nature 461, 542–545 (2009). [DOI] [PubMed] [Google Scholar]
- 47.Freeman A. K., Ritt D. A., Morrison D. K., Effects of Raf dimerization and its inhibition on normal and disease-associated Raf signaling. Mol. Cell 49, 751–758 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hall M. P., Unch J., Binkowski B. F., Valley M. P., Butler B. L., Wood M. G., Otto P., Zimmerman K., Vidugiris G., Machleidt T., Robers M. B., Benink H. A., Eggers C. T., Slater M. R., Meisenheimer P. L., Klaubert D. H., Fan F., Encell L. P., Wood K. V., Engineered luciferase reporter from a deep sea shrimp utilizing a novel imidazopyrazinone substrate. ACS Chem. Biol. 7, 1848–1857 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Thevakumaran N., Lavoie H., Critton D. A., Tebben A., Marinier A., Sicheri F., Therrien M., Crystal structure of a BRAF kinase domain monomer explains basis for allosteric regulation. Nat. Struct. Mol. Biol. 22, 37–43 (2015). [DOI] [PubMed] [Google Scholar]
- 50.Zhou Y., Hancock J. F., Ras nanoclusters: Versatile lipid-based signaling platforms. Biochim. Biophys. Acta 1853, 841–849 (2015). [DOI] [PubMed] [Google Scholar]
- 51.Dixon A. S., Schwinn M. K., Hall M. P., Zimmerman K., Otto P., Lubben T. H., Butler B. L., Binkowski B. F., Machleidt T., Kirkland T. A., Wood M. G., Eggers C. T., Encell L. P., Wood K. V., NanoLuc complementation reporter optimized for accurate measurement of protein interactions in cells. ACS Chem. Biol. 11, 400–408 (2016). [DOI] [PubMed] [Google Scholar]
- 52.Hu J., Ahuja L. G., Meharena H. S., Kannan N., Kornev A. P., Taylor S. S., Shaw A. S., Kinase regulation by hydrophobic spine assembly in cancer. Mol. Cell. Biol. 35, 264–276 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Callahan M. K., Rampal R., Harding J. J., Klimek V. M., Chung Y. R., Merghoub T., Wolchok J. D., Solit D. B., Rosen N., Abdel-Wahab O., Levine R. L., Chapman P. B., Progression of RAS-mutant leukemia during RAF inhibitor treatment. N. Engl. J. Med. 367, 2316–2321 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/5/8/eaav8463/DC1
Table S1. Used expression constructs, materials, and cell lines.
Fig. S1. BRAF KinCon reporter activities and BRAF profiling.
Fig. S2. Impact of defined kinase mutations on BRAF KinCon reporter dynamics.
Fig. S3. BRAF KinCon reporter dynamics.
Fig. S4. Complex formation of RAS:BRAF and PPI reporter dynamics.
Fig. S5. Impact of vemurafenib on indicated PPIs.
Fig. S6. Impact of BRAFi on binary RAS:RAF interactions.
Fig. S7. PPI reporter dynamics and NS1 monobody IPs.
Fig. S8. Validation of PPI dynamics and impact of RAFi on RAF conformations.