

We show that genotype-by-environment interaction can be inferred from an analysis without environmental data in a large sample.
Abstract
Genotype-by-environment interaction (GEI) is a fundamental component in understanding complex trait variation. However, it remains challenging to identify genetic variants with GEI effects in humans largely because of the small effect sizes and the difficulty of monitoring environmental fluctuations. Here, we demonstrate that GEI can be inferred from genetic variants associated with phenotypic variability in a large sample without the need of measuring environmental factors. We performed a genome-wide variance quantitative trait locus (vQTL) analysis of ~5.6 million variants on 348,501 unrelated individuals of European ancestry for 13 quantitative traits in the UK Biobank and identified 75 significant vQTLs with P < 2.0 × 10−9 for 9 traits, especially for those related to obesity. Direct GEI analysis with five environmental factors showed that the vQTLs were strongly enriched with GEI effects. Our results indicate pervasive GEI effects for obesity-related traits and demonstrate the detection of GEI without environmental data.
INTRODUCTION
Most human traits are complex because they are affected by many genetic and environmental factors as well as potential interactions between them (1, 2). Despite the long history of effort (3–5), there has been limited success in identifying genotype-by-environment interaction (GEI) effects in humans (5–8). This is likely because many environmental exposures are unknown or difficult to record during the life course and because the effect sizes of GEI are small, given the polygenic nature of most human traits (9–11), so that the sample sizes of most previous studies are not large enough to detect the small GEI effects. For model complex traits such as body mass index (BMI), GEI analyses have been limited to GEI tests at known BMI loci (12–14) or estimation of GEI variance captured by all common SNPs (15, 16).
GEI effect of a genetic variant on a quantitative trait could lead to differences in variance of the trait among groups of individuals with different variant genotypes (Fig. 1, A and B, and note S1). GEI can therefore be inferred from a variance quantitative trait locus (vQTL) analysis (17), although there are other explanations for an observed vQTL such as direct effect on phenotypic dispersion [e.g., induced by selection (18)], epistasis (17), and phantom vQTL (19, 20). Unlike the classical QTL analysis that tests the allelic substitution effect of a variant on the mean of a phenotype (Fig. 1C), vQTL analysis tests the allelic substitution effect on the trait variance (Fig. 1, B or D). In comparison to the analyses that perform direct GEI tests, vQTL analysis is more flexible because it does not require measures of environmental factors and thus can be performed in a very large sample where the environmental factors are unknown, unavailable, or incomplete (21). Of course, the vQTL test is less powerful than the direct GEI test if the corresponding environmental factor has been measured on all the genotyped individuals in the sample (17). Although there had been empirical evidence for the genetic control of phenotypic variance in livestock for decades (22, 23), it was not until recent years that genome-wide vQTL analysis was applied in humans (17, 24, 25), and only a handful of vQTLs have been identified for a limited number of traits [e.g., the FTO locus for BMI (25)] owing to small effect sizes of vQTLs. The availability of data from large biobank-based genome-wide association studies (GWAS) (26, 27) provides an opportunity to interrogate the genome for vQTLs for a range of phenotypes in cohorts with unprecedented sample size.
Fig. 1. Schematic of the differences in mean or variance among genotype groups in the presence of GEI, QTL, and vQTL effects.
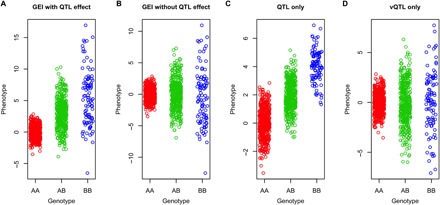
The phenotypes of 1000 individuals were simulated on the basis of a genetic variant (MAF = 0.3) with (A) both QTL and GEI effects, (B) GEI effect only (no QTL effect), (C) QTL effect only (no GEI or vQTL effect), or (D) vQTL only (no QTL effect).
On the other hand, statistical methods for vQTL analysis are not entirely mature (21). There have been a series of classical nonparametric methods (28), originally developed to detect violation of the homogeneous variance assumption in linear regression model, which can be used to detect vQTLs, including the Bartlett’s test (29), the Levene’s test (30, 31), and the Fligner-Killeen (FK) test (32). Recently, more flexible parametric models have been proposed, including the double generalized linear model (DGLM) (33–35) and the likelihood ratio test for variance effect (LRTV) (19). In addition, it has been suggested that the transformation of phenotype that alters phenotype distribution also has an influence on the power and/or false-positive rate (FPR) of a vQTL analysis (24, 36).
In this study, we calibrated the most commonly used statistical methods for vQTL analysis by extensive simulations. We then used the best performing method to conduct a genome-wide vQTL analysis for 13 quantitative traits in 348,501 unrelated individuals using the UK Biobank (UKB) data (26). We further investigated whether the detected vQTLs are enriched for GEI by conducting a direct GEI test for the vQTLs with five environmental factors (or covariates).
RESULTS
Evaluation of the vQTL methods by simulation
We used simulations to quantify the FPR and power (i.e., true-positive rate) for the vQTL methods and phenotype processing strategies (Methods). We first simulated a quantitative trait based on a simulated single-nucleotide polymorphism (SNP), i.e., a single-SNP model, under a number of different scenarios, namely, (i) five different distributions for the random error term (i.e., individual-specific environment effect) and (ii) four different types of SNP with or without the effect on mean or variance (Methods). We used the simulated data to compare the four most widely used vQTL methods, namely, Bartlett’s test (29), Levene’s test (30, 31), FK test (32), and DGLM (33–35). We observed no inflation in FPR for the Levene’s test under the null (i.e., no vQTL effect) regardless of the skew or kurtosis of the phenotype distribution or the presence or absence of SNP effect on the mean (fig. S1A). These findings are in line with the results from previous studies (24, 28, 37) that the Levene’s test is robust to the distribution of the phenotype. The FPR of the Bartlett’s test or DGLM was inflated if the phenotype distribution was skewed or heavy-tailed (fig. S1A). The FK test seemed to be robust to kurtosis but vulnerable to skewness of the phenotype distribution (fig. S1A). Because the Levene’s test performed the best in the simulations, for this test, we investigated the impact of nonlinear transformations of the phenotype by considering logarithm [log(y)], square (y2), cube (y3), and rank-based inverse-normal transformation (RINT) and found that these nonlinear transformations could result in inflated FPR (fig. S1B).
To simulate more complex scenarios, we used a multiple-SNP model with two covariates (age and sex) and different numbers of SNPs (Fig. 2). The results were similar to those described above, although the power of the Levene’s test decreased with an increase of the number of causal SNPs (Fig. 2A). Nonlinear transformations led to an inflated FPR when the variance explained by a QTL effect (i.e., SNP effect on mean) was relatively large and a loss of power of vQTL detection when the per-QTL variance explained was relatively small, although logarithm transformation did not seem to affect the power (Fig. 2B). These results also suggested that pre-adjusting the phenotype by covariates slightly increased the power (Fig. 2B). On the basis of the results of these simulations, we used the Levene’s test, a one-way analysis of variance (ANOVA) to test for absolute deviations from the medians (Methods), for real data analysis, with the phenotypes pre-adjusted for covariates without any nonlinear transformation.
Fig. 2. Evaluation of the statistical methods and phenotype processing strategies for vQTL analysis by simulation.
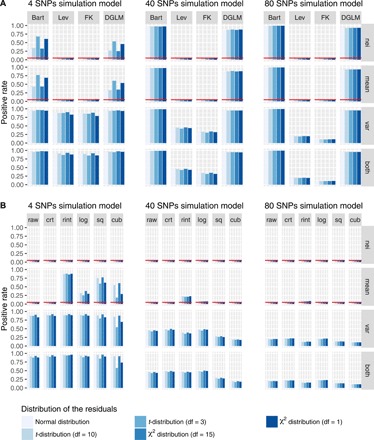
Phenotypes of 10,000 individuals were simulated on the basis of the different number of causal SNPs (i.e., 4, 40, or 80), two covariates (i.e., sex and age), and one error term in a multiple-SNP model (Methods). The SNP effects were simulated under four scenarios: (i) effect on neither mean nor variance (nei), (ii) effect on mean only (mean), (iii) effect on variance only (var), or (iv) effect on both mean and variance (both). The error term was generated from five different distributions: normal distribution, t-distribution with df = 10 or 3, or χ2 distribution with df = 15 or 1. (A) Four statistical test methods, i.e., the Bartlett’s test (Bart), the Levene’s test (Lev), the Fligner-Killeen test (FK), and the DGLM, were used to detect vQTLs. (B) The Levene’s test was used to analyze phenotypes processed using six strategies, i.e., raw phenotype (raw), raw phenotype adjusted for covariates (adj), RINT after adj (rint), logarithm transformation after adj (log), square transformation after adj (sq), and cube transformation after adj (cub). Positive rate is defined as the number of vQTLs with P < 0.05 divided by the total number of tests across 1000 simulations, which is the FPR under the null (“nei” and “mean”) and power under the alternative (“var” and “both”). The red horizontal line represents an FPR of 0.05.
Genome-wide vQTL analysis for 13 UKB traits
We performed a genome-wide vQTL analysis using the Levene’s test with 5,554,549 genotyped or imputed common variants on 348,501 unrelated individuals of European ancestry for 13 quantitative traits in the UKB (Methods; table S1A and fig. S2A) (26). For each trait, we pre-adjusted the phenotype for age and the first 10 principal components (PCs; derived from SNP data) and standardized the residuals to z scores in each gender group (Methods). This process removed not only the effects of age and the first 10 PCs on the phenotype but also the differences in mean and variance between the two genders. We excluded individuals with adjusted phenotypes more than 5 SDs from the mean and removed SNPs with minor allele frequency (MAF) smaller than 0.05 to avoid potential false-positive associations due to the coincidence of a low-frequency variant with an outlier phenotype (see fig. S3A for an example). We acknowledge that this process could potentially result in a loss of power, but this can be compensated for by the use of a very large sample (n ~ 350,000).
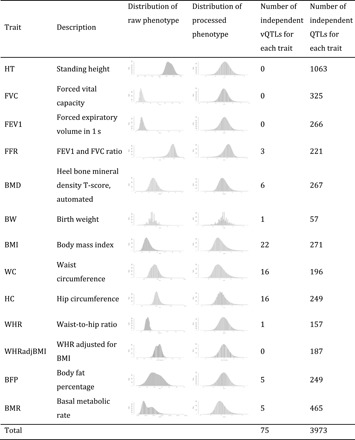
With an experiment-wise significant threshold of 2.0 × 10−9 [i.e., 1 × 10−8/5.0, with 1 × 10−8 being a more stringent genome-wide significant threshold recommended by recent studies (38, 39) and 5.0 being the effective number of independent traits (note S4)], we identified 75 vQTLs [independent to linkage disequilibrium (LD) r2 < 0.01 within trait] across the nine traits (Fig. 3, Table 1, and table S2A). There was no vQTL for height, consistent with the observation in a previous study (25). We identified more than 15 vQTLs for each of the three obesity-related traits, i.e., BMI, waist circumference (WC), and hip circumference (HC) (Table 1). The 75 vQTLs were located at 41 near-independent loci after excluding one of each between-trait pair of top vQTL SNPs (i.e., the SNP with lowest vQTL P value at each vQTL association peak) with LD r2 > 0.01, suggesting that some of the loci were associated with the phenotypic variance of multiple traits. For example, the FTO locus was associated with the phenotypic variance of WC, HC, BMI, body fat percentage (BFP), and basal metabolic rate (BMR) (fig. S3B), and the vQTL associations were likely to be driven by a shared causal variant with pleiotropic vQTL effects on multiple traits (fig. S3C). For the lung function–related traits, there was no significant vQTL for forced expiratory volume in 1 s (FEV1) and forced vital capacity (FVC) but there were three vQTLs for the FEV1/FVC ratio (FFR). There was no evidence for an effect of MAF on vQTL test statistic at the 41 independent loci (fig. S3D), consistent with the observation in a previous study (25).
Fig. 3. Manhattan plots of genome-wide vQTL analysis for 13 traits in the UKB.
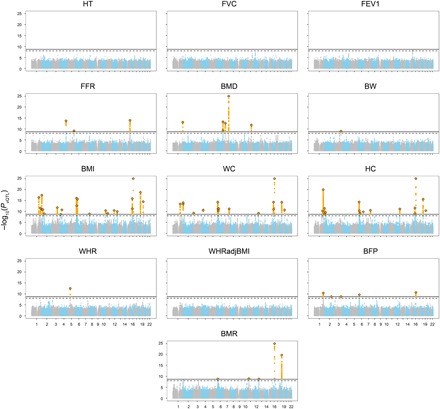
For each of the 13 traits (see Table 1 for full names of the traits), test statistics [−log10(PvQTL)] of all common (MAF ≥ 0.05) SNPs from the vQTL analysis are plotted against their physical positions. The dashed line represents the genome-wide significance level 1.0 × 10−8, and the solid line represents the experiment-wise significance level 2.0 × 10−9. For graphical clarity, SNPs with PvQTL < 1 × 10−25 are omitted, SNPs with PvQTL < 2.0 × 10−9 are color-coded in orange, the top vQTL SNP for each locus is represented by a diamond, and the remaining SNPs on odd and even chromosomes are color-coded in gray and blue, respectively.
Table 1. The number of experiment-wise significant vQTLs or QTLs for the 13 UKB traits.
The Levene’s test assesses the difference in variance among three genotype groups free from the assumption about additivity (i.e., the vQTL effect of carrying two copies of the effect allele is not assumed to be twice that carrying one copy). We found two vQTLs (i.e., rs141783576 and rs10456362) potentially showing nonadditive genetic effect on the variance of HC and BMR, respectively (table S2A).
To demonstrate the vulnerability of vQTL analysis to nonlinear transformations in real data, we performed genome-wide vQTL analysis for height squared and cubed. There was no genome-wide significant vQTL for height squared but one genome-wide significant vQTL for height cubed, which was very likely to be driven by a strong QTL signal for height [PQTL(Height) = 4.35 × 10−150] (fig. S3, E and F), consistent with our simulation results that nonlinear transformations could inflate the vQTL test statistics in the presence of a strong QTL signal (Fig. 2B and fig. S1B). Although we have not applied any nonlinear transformation to the UKB traits, some of them are nonlinear functions of other traits, i.e., BMI (= WT/HT2), FFR (= FEV1/FVC), and WHR (= WC/HC). We therefore explored whether the BMI, FFR, and WHR vQTLs were driven by the nonlinear functions by testing the variance effects of the BMI, FFR, and WHR vQTLs on 1/HT2, 1/FVC, and 1/HC, respectively. There were 26 tests in total, none of which reached the experiment-wise significance level (i.e., 2.0 × 10−9) used to claim vQTLs in this study and 23 of which had a P value larger than 0.05 (table S2B), suggesting that the BMI, FFR, and WHR vQTLs were not driven by the nonlinear functions. Although the variance effect of an FFR vQTL (rs56077333) on 1/FVC was significant after correcting for 26 tests (P = 5.11 × 10−6; table S2B), the effect of rs56077333 on the variance of 1/FVC was not large enough to drive the vQTL signal for FFR and rs56077333 has a known GEI effect on lung function (see below for more details).
GWAS analysis for the 13 UKB traits
To investigate whether the SNPs with effects on variance also have effects on mean, we performed GWAS (or genome-wide QTL) analyses for the 13 UKB traits described above. We identified 3973 QTLs at an experiment-wise significance level (i.e., PQTL < 2.0 × 10−9) for the 13 traits in total, a much larger number than that of the vQTLs (Fig. 4 and Table 1). Among the 75 vQTLs, the top vQTL SNPs at nine loci did not pass the experiment-wise significance level in the QTL analysis (table S2A). For example, the CCDC92 locus showed a significant vQTL effect but no significant QTL effect on WC (table S2A and fig. S3G), whereas the FTO locus showed both significant QTL and vQTL effects on WC (fig. S3G). For the 66 vQTLs with both QTL and vQTL effects, the vQTL effects were all in the same directions as the QTL effects, meaning that, for any of these SNPs, the genotype group with a larger phenotypic mean also tends to have a larger phenotypic variance than the other groups. For the nine loci with vQTL effects only, it is equivalent to a scenario where a QTL has a GEI effect with no (or a substantially reduced) effect on average across different levels of an environmental factor (Fig. 1B).
Fig. 4. Manhattan Sunset plot of genome-wide vQTL and QTL analyses for WC in the UKB.
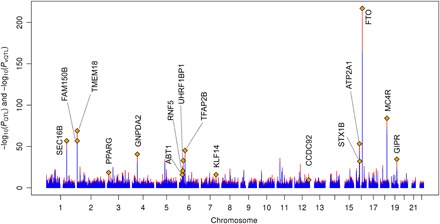
Test statistics [−log10(P values)] of all common SNPs from vQTL (red bars) and QTL (blue bars) analysis are plotted against their physical positions. The top vQTL SNP is represented by an orange diamond, and the name of the nearest protein-coding gene is indicated for each significant vQTL locus (PvQTL < 2.0 × 10−9).
vQTL and GEI
To further investigate whether the associations between vQTLs and phenotypic variance can be explained by GEI, we performed a direct GEI test based on an additive genetic model with an interaction term between a top vQTL SNP and one of five environmental factors/covariates in the UKB data (Methods). The five environmental factors/covariates are sex, age, physical activity (PA), sedentary behavior (SB), and ever smoking (note S5, fig. S2B, and table S1B). We observed 16 vQTLs showing a significant GEI effect with at least one of five environmental factors after Bonferroni correction for multiple tests [P < 1.33 × 10−4 = 0.05/(75 × 5); Fig. 5A and table S2C].
Fig. 5. Enrichment of GEI effects among the 75 vQTLs compared with a random set of QTLs.
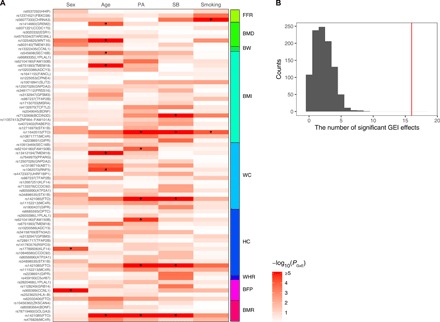
Five environmental factors/covariates, i.e., sex, age, physical activity (PA), sedentary behavior (SB), and smoking, were used in the GEI analysis. (A) Heatmap plot of GEI test statistics [−log10(PGEI)] for the 75 top vQTL SNPs. “*” denotes significant GEI effects after Bonferroni correction [PGEI < 1.33 × 10−4 = 0.05/(75 × 5)]. (B) Distribution of the number of significant GEI effects for the 75 top QTL SNPs randomly selected from all the top QTL SNPs with 1000 repeats (mean, 2.25; SD, 1.49). The red line represents the number of significant GEI effects for the 75 top vQTL SNPs (i.e., 16).
To test whether the GEI effects are enriched among vQTLs in comparison with the same number of QTLs, we performed GEI test for the 75 top GWAS SNPs randomly selected from all the QTLs and repeated the analysis 1000 times. Of the 75 top SNPs with QTL effects, the number of SNPs with significant GEI effects was 2.25, averaged from the 1000 repeated samplings with an SD of 1.49 (Fig. 5B), significantly lower than the number (16) observed for the vQTLs (the difference is larger than 9 SDs, equivalent to P = 1.4 × 10−20). This result shows that SNPs with vQTL effects are much more enriched with GEI effects compared to those with QTL effects. To exclude the possibility that the GEI signals were driven by phenotype processing (e.g., the adjustment of phenotype for sex and age), we repeated the GEI analyses using raw phenotype data without covariate adjustment; the results remain largely unchanged (fig. S5).
DISCUSSION
In this study, we leveraged the genetic effects associated with phenotypic variability to infer GEI. We calibrated the most commonly used vQTL methods by simulation. We found that the FPR of the Levene’s test was well calibrated across all simulation scenarios, whereas the other methods showed an inflated FPR if the phenotype distribution was skewed or heavy-tailed under the null hypothesis (i.e., no vQTL effect), although the Levene’s test appeared to be less powerful than the other methods particularly when the per-variant vQTL effect was small (Fig. 2 and fig. S1). Parametric bootstrap or permutation procedures have been proposed to reduce the inflation in the test statistics of DGLM and LRTv, both of which are expected to be more powerful than the Levene’s test (19, 37), but bootstrap and permutation are computationally inefficient and thus are not practically applicable to biobank data such as the UKB. We observed inflated FPR for the Levene’s test in the absence of vQTL effects but in the presence of QTL effects if the phenotype was nonlinearly transformed (e.g., logarithm transformation or RINT). We therefore recommend the use of the Levene’s test in practice without nonlinear transformation of the phenotype. In addition, a very recent study by Young et al. (40) developed an efficient algorithm to perform a DGLM analysis and proposed a method [called dispersion effect test (DET)] to remove confounding in vQTL associations (identified by DGLM) due to QTL effects. We showed by simulation that, when the number of simulated causal variants was relatively large (note that the DET test is not applicable to oligogenic traits), the Young et al. method (DGLM followed by DET) performed similarly as the Levene’s test, with differences depending on how the phenotype was processed (fig. S6).
We demonstrated in the analysis of the UKB data that a number of vQTLs (with enriched GEI effects) can be detected by an appropriate analytical strategy in a very large sample. Traits with a larger number of vQTLs detected at the experiment-wise significance level tended to have a higher genomic inflation factor (defined as the mean or median χ2 statistic divided by its expected value) even after excluding the top vQTLs as well as SNPs in LD with them (fig. S4), consistent with a polygenic model of variance effect (41, 42), suggesting a large number of vQTLs with small variance effects yet to be discovered in larger samples in the future.
There are several vQTLs for which the GEI effect has been reported in previous studies. The first example is the interaction effect of the CHRNA5-A3-B4 locus (rs56077333) with smoking for lung function (as measured by FFR, i.e., FEV1/FVC), PvQTL = 1.1 × 10−14 and PGEI(smoking) = 4.6 × 10−25 (table S3A). The CHRNA5-A3-B4 gene cluster is known to be associated with smoking and nicotine dependence (43–45). However, results from recent GWAS (46–48) do not support the association of this locus with lung function. We hypothesize that the effect of the CHRNA5-A3-B4 locus on lung function depends on smoking (table S3A) (49). The vQTL signal at this locus remained (PvQTL = 5.2 × 10−12) after adjusting the phenotype for array effect, which was reported to affect the QTL association signal at this locus (26). The second example is the interaction of the WNT16-CPED1 locus with age for bone mineral density (BMD) [rs10254825: PvQTL = 2.0 × 10−45 and PGEI(age) = 1.2 × 10−7]. The WNT16-CPED1 locus is one of the strongest BMD-associated loci identified from GWAS (50, 51). We observed a genotype-by-age interaction effect at this locus for BMD (table S3B), in line with the results from previous studies that the effect of the top SNP at WNT16-CPED1 on BMD in humans (52) and the knockout effect of Wnt16 on bone mass in mice (53) are age dependent. The third example is the interaction of the FTO locus with PA and SB for obesity-related traits [PvQTL < 1 × 10−10 for BMI, WC, HC, BFP, and BMR; PGEI(PA) = 1.3 × 10−10 for BMI, 1.4 × 10−7 for WC, 5.3 × 10−7 for HC, and 2.6 × 10−7 for BMR]. The FTO locus was one of the first loci identified by the GWAS of obesity-related traits (54), although subsequent studies (55, 56) show that IRX3 and IRX5 (rather than FTO) are the functional genes responsible for the GWAS association. The top associated SNP at the FTO locus is not associated with PA, but its effect on BMI decreases with the increase of PA level (12, 57), consistent with the interaction effects of the FTO locus with PA or SB for obesity-related traits identified in this study (table S3, C and D). In addition, 5 of the 22 BMI vQTLs were in LD (r2 > 0.5), with the variants (identified by a recently developed multiple-environment GEI test) showing significant interaction effects at a false discovery rate (FDR) of <5% (corresponding to P < 1.16 × 10−3) with at least 1 of 64 environmental factors for BMI in the UKB (58).
It should be noted that GEI is sufficient but not necessary to generate a vQTL. For the vQTLs that did not show a direct GEI effect in our GEI analysis, we cannot distinguish whether they are due to undetected GEI or direct effects on phenotypic dispersion, although GEI is a more likely explanation because of the enrichment of GEI (Fig. 5); hence, these traits and loci are candidates for follow-up studies to identify putative environmental risk factors that may be amendable to lifestyle modification. We also explored two other interpretations of the observed vQTLs, i.e., “phantom vQTLs” (19, 20) and epistasis (genotype-by-genotype interaction) (17). If the underlying causal QTL is not well imputed or not well tagged by a genotyped/imputed variant, then the untagged variation at the causal QTL will inflate the vQTL test statistic, potentially leading to a spurious vQTL association, i.e., the so-called phantom vQTL. We showed by theoretical deviations that the Levene’s test statistic due to the phantom vQTL effect was a function of sample size, effect size of the causal QTL, allele frequency of the causal QTL, allele frequency of the phantom vQTL, and LD between the causal QTL and the phantom vQTL (note S6 and fig. S7A). From our deviations, we computed the numerical distribution of the expected phantom vQTL F-statistics, given a number of parameters including the sample size (n = 350,000), variance explained by the causal QTL (q2 = 0.005, 0.01, or 0.02), and MAFs of the causal QTL and the phantom vQTL (MAF = 0.05 to 0.5). The result showed that, for a causal QTL with q2 < 0.005 and MAF > 0.05, the largest possible phantom vQTL F-statistic was smaller than 2.69 (corresponding to a P value of 6.8 × 10−2; fig. S7, B to D). This explains why there were thousands of genome-wide significant QTLs but no significant vQTL for height (Fig. 3 and Table 1). This result also suggests that the vQTLs detected in this study are very unlikely to be phantom vQTLs because the estimated variance explained by their QTL effects were all smaller than 0.005, except for rs10254825 at the WNT16 locus on BMD (q2 = 0.014) (fig. S7E). However, our numerical calculation also indicated that, for a QTL with MAF > 0.3 and q2 < 0.02, the largest possible phantom vQTL F-statistic was smaller than 5.64 (corresponding to a P value of 3.6 × 10−3), suggesting that rs10254825 is also unlikely to be a phantom vQTL. Note that we used the variance explained estimated at the top GWAS SNP to approximate q2 of the causal QTL so that q2 was likely to be underestimated because of imperfect tagging. However, considering the extremely high imputation accuracy for common variants (59), the strong LD between the causal QTLs and the GWAS top SNPs observed in a previous simulation study based on whole-genome sequence data (38), and the overestimation of variance explained by the GWAS top SNPs because of winner’s curse, the underestimation in causal QTL q2 is likely to be small. In addition, we reran the vQTL analysis, with the phenotype adjusted for the top GWAS variants within 10 Mb of the top vQTL SNP; the vQTL signals after this adjustment were highly concordant with those without adjustment (fig. S7F). We further showed that there was no evidence for epistatic interactions between the top vQTL SNPs and any other SNP located more than 10 Mb away or on a different chromosome (fig. S7G). Note that we did not perform epistatic test for SNP pairs within 10 Mb to avoid possible spurious epistatic signals caused by LD (60).
In conclusion, we systematically quantified the FPR and power for four commonly used vQTL methods by extensive simulations and demonstrated the robustness of the Levene’s test. We also showed that, in the presence of QTL effects, the Levene’s test statistic could be inflated if the phenotype was nonlinearly transformed. We implemented the Levene’s test as part of the OSCA software package (http://cnsgenomics.com/software/osca) (61) for efficient genome-wide vQTL analysis. We applied OSCA-vQTL to 13 quantitative traits in the UKB and identified 75 vQTL (at 41 near-independent loci) associated with 9 traits, 9 of which did not show a significant QTL effect. As a proof of principle, we performed GEI analyses in the UKB with five environmental factors/covariates and demonstrated the enrichment of GEI effects among the detected vQTLs. Hence, the vQTL trait-loci combinations we have identified could be investigated for as-yet-undetermined but measurable environmental risk factors generating GEI, as these factors could be amenable to lifestyle change interventions. We further derived the theory to compute the expected “phantom vQTL” test statistic due to untagged causal QTL effect and showed by numerical calculation that our observed vQTLs were very unlikely to be driven by imperfectly tagged QTL effects. Our theory is also consistent with the observation of pervasive phantom vQTLs for molecular traits with large-effect QTLs [e.g., DNA methylation (20)]. However, the conclusions from this study may only be applicable to quantitative traits of polygenic architecture. We caution vQTL analysis for binary or categorical traits, or molecular traits (e.g., gene expression or DNA methylation), for which the methods need further investigation.
METHODS
Simulation study
We used a DGLM (33–35) to simulate the phenotype based on two models with simulated SNP data in a sample of 10,000 individuals, i.e., a single-SNP model and multiple-SNP model with two covariates (i.e., age and sex). The single-SNP model can be written as
and the multiple-SNP model can be expressed as
where y is a simulated phenotype; w or wk is a standardized SNP genotype, i.e., , with x being the genotype indicator variable coded as 0, 1, or 2, generated from binomial(2, f) and f being the MAF generated from uniform(0.01, 0.5); cj is a standardized covariate with c1 (sex) generated from binomial(1, 0.5) and c2 (age) generated from uniform(20, 60); e is an error term with mean 0 and variance . To simulate the error term with different levels of skewness and kurtosis, we generated e from five different distributions, including normal distribution, t-distribution with df = 10 or 3, and χ2 distribution with df = 15 or 1. β (ϕ) is the effect on mean (variance) generated from N(0,1) if exists, 0 otherwise. Log(σ2) is the intercept of the second linear model, which was set to 0. We rescaled the different components to control the variance explained, i.e., 0.1 and 0.9 for the genotype component and error term, respectively, in the single-SNP model, and 0.2, 0.4, and 0.4 for the covariate component, genotype component, and error term, respectively, in the multiple-SNP model. We simulated the SNP effects in four different scenarios: (i) effect on neither mean nor variance (nei), (ii) effect on mean only (mean), (iii) effect on variance only (var), or (iv) effect on both mean and variance (both). We simulated only one causal SNP in the single-SNP model and 4, 40, or 80 causal SNPs in the multiple-SNP model.
We performed vQTL analyses using the simulated phenotype and SNP data to compare four vQTL methods, including the Bartlett’s test (29), the Levene’s test (31), the FK test (32), and the DGLM (note S2). We also performed the Levene’s test with six phenotype process strategies, including raw phenotype (raw), raw phenotype adjusted for covariates (adj), RINT after adj (rint) (note S3), logarithm transformation after adj (log), square transformation after adj (sq), and cube transformation after adj (cub). We repeated the simulation 1000 times and calculated the FPR and power at P < 0.05 at a single-SNP level.
The UKB data
The full release of the UKB data consisted of genotype and phenotype data for ~500,000 participants across the United Kingdom (26). The genotype data were cleaned and imputed to the Haplotype Reference Consortium (59) and UK10K (62) reference panels by the UKB team. Genotype probabilities from imputation were converted to hard-call genotypes using PLINK2 (--hard-call 0.1) (63). We excluded genetic variants with MAF < 0.05, Hardy-Weinberg equilibrium test P value < 1 × 10−5, missing genotype rate > 0.05, or imputation INFO score < 0.3 and retained 5,554,549 variants for further analysis.
We identified a subset of individuals of European ancestry (n = 456,422) by projecting the UKB PCs onto those of the 1000 Genome Project (1KGP) (64). We then removed one of each pair of individuals with SNP-derived (based on HapMap 3 SNPs) genomic relatedness >0.05 using GCTA-GRM (65) and retained 348,501 unrelated European individuals for further analysis.
We selected 13 quantitative traits for our analysis (table S1A and fig. S2A). We adjusted the raw phenotype values for age and the first 10 PCs, excluded from the analysis phenotype values that were more than 5 SDs from the mean, and then standardized to z scores with mean 0 and variance 1 in each gender group.
Genome-wide vQTL analysis
The genome-wide vQTL analysis was conducted using the Levene’s test implemented in the software tool OSCA (http://cnsgenomics.com/software/osca) (61). The Levene’s test used in the study [also known as the median-based Levene’s test or the Brown-Forsythe test (31)] is a modified version of the original Levene’s test (30) developed in 1960 that is essentially a one-way ANOVA test of the variable , where yij is the phenotype of the jth individual in the ith group and is the median of the ith group. The Levene’s test statistic
approximately follows an F distribution with k − 1 and n − k degrees of freedom under the null hypothesis, where n is the total sample size, k is the number of groups (k = 3 in vQTL analysis), ni is the sample size of the ith group, i.e., , and .
The experiment-wise significance level was set to 2.0 × 10−9, which is the genome-wide significance level (i.e., 1 × 10−8) (38, 39) divided by the effective number of independent traits (i.e., 5.00 for our 13 traits). The effective number of independent traits was estimated on the basis of the phenotypic correlation matrix (note S4) (66). To determine the number of near-independent vQTLs, we performed an LD clumping analysis for each trait using PLINK2 (--clump option with parameters --clump-p1 2.0e-9 --clump-p2 2.0e-9 --clump-r2 0.01 and --clump-kb 5000) (63). To visualize the results, we generated the Manhattan and regional association plots using the ggplot2 package in R.
GWAS analysis
The GWAS (or genome-wide QTL) analysis was conducted using PLINK2 (63) (--assoc option) using the same data as used in the vQTL analysis (note that the phenotype had been pre-adjusted for covariates and PCs). The other analyses, including LD clumping and visualization, were performed using the same pipelines as those for genome-wide vQTL analysis described above.
GEI analysis
Five environmental factors/covariates (i.e., sex, age, PA, SB, and smoking) were used for the direct GEI tests. Sex was coded as 0 or 1 for female or male. Age was an integer number ranging from 40 to 74. PA was assessed by a three-level categorical score (i.e., low, intermediate, and high) based on the short form of the International Physical Activity Questionnaire (IPAQ) guideline (67). SB was an integer number defined as the combined time (hours) spent driving, non–work-related computer using, or TV watching. The smoking factor “ever smoked” was coded as 0 or 1 for never or ever smoker. More details about the definition and derivation of environmental factor PA, SB, and smoking can be found in note S5, fig. S2B, and table S1B.
We performed a GEI analysis to test the interaction effect between the top vQTL SNP and one of the five environmental factors based on the following model
where y is the phenotype, μ is the mean term, xg is the mean-centered SNP genotype indicator, and xE is the mean-centered environmental factor. We used a standard ANOVA analysis to test for βgE and applied a stringent Bonferroni-corrected threshold 1.33 × 10−4 [i.e., 0.05/(75 × 5)] to claim a significant GEI effect.
Supplementary Material
Acknowledgments
Funding: This research was supported by the Australian Research Council (DP160101343 and DP160101056, FT180100186), the Australian National Health and Medical Research Council (1078037, 1078901, 1113400, 1107258, and 1083656), and the Sylvia & Charles Viertel Charitable Foundation. Author contributions: J.Y. and A.F.M. conceived the study. J.Y., H.W., and A.F.M. designed the experiment. F.Z. developed the software tool. H.W. performed simulations and data analyses under the assistance or guidance from J.Y., J.Z., Y.W., K.E.K., A.X., and M.Z. J.E.P., M.E.G., N.R.W., and P.M.V. provided critical advice that significantly improved the experimental design and/or interpretation of the results. P.M.V., N.R.W., and J.Y. contributed resources and funding. H.W. and J.Y. wrote the manuscript with the participation of all authors. Competing interests: The authors declare that they have no competing interests. Data and material availability: This study makes use of data from the UKB (project ID: 12514). A full list of acknowledgments of this data set can be found in note S7. The individual-level genotype and phenotype data used in this study can be provided by the UKB (http://www.ukbiobank.ac.uk/) pending scientific review and a completed material transfer agreement (requests for these data should be submitted to the UKB). The vQTL summary statistics of all SNPs for the 13 UKB traits are available at the OSCA website (http://cnsgenomics.com/software/osca). All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/5/8/eaaw3538/DC1
Note S1. Theoretical derivation of vQTL as a consequence of GEI
Note S2. The Bartlett’s test, the FK test, and the DGLM test
Note S3. Rank-based inverse-normal transformation
Note S4. The effective number of independent traits
Note S5. Definitions of the three environmental factors—PA, SB, and smoking
Note S6. Expected inflation in the Levene’s test statistic due to phantom vQTL effect
Note S7. Acknowledgments
Fig. S1. Evaluation of statistical methods and phenotype processing strategies for the vQTL analysis by simulation based on a single-SNP model.
Fig. S2. Phenotypic correlations among 13 quantitative traits, and PA and SB measures in the UKB.
Fig. S3. Genome-wide vQTL and QTL analyses for 13 traits in the UKB.
Fig. S4. Quantile-quantile plots of vQTL associations for the 13 UKB traits.
Fig. S5. Enrichment of GEI effects among the 75 vQTLs compared with a random set of QTLs using the raw phenotypic values.
Fig. S6. Comparison of the Young et al. method with the Levene’s test by vQTL simulation.
Fig. S7. Excluding two alternative explanations for vQTL signals: Phantom vQTLs and epistasis.
Table S1. Descriptive summary of (A) the quantitative traits and (B) the environmental data used in this study from the UKB.
Table S2. Seventy-five experiment-wise significant vQTLs for nine UKB traits.
Table S3. GEI examples.
REFERENCES AND NOTES
- 1.D. S. Falconer, T. F. C. Mackay, Introduction to Quantitative Genetics (Longman, ed. 4, 1996). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.M. Lynch, B. Walsh, Genetics and Analysis of Quantitative Traits (Sinauer Associates, 1998). [Google Scholar]
- 3.Garrod A. E., The incidence of alkaptonuria: A study in chemical individuality. Lancet 160, 1616–1620 (1902). [Google Scholar]
- 4.J. Haldane, Heredity and Politics (WW Norton & Co., 1938). [Google Scholar]
- 5.Kraft P., Hunter D., Integrating epidemiology and genetic association: The challenge of gene-environment interaction. Philos. Trans. R. Soc. Lond. B Biol. Sci. 360, 1609–1616 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Thomas D., Gene–environment-wide association studies: Emerging approaches. Nat. Rev. Genet. 11, 259–272 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Aschard H., Lutz S., Maus B., Duell E. J., Fingerlin T. E., Chatterjee N., Kraft P., van Steen K., Challenges and opportunities in genome-wide environmental interaction (GWEI) studies. Hum. Genet. 131, 1591–1613 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.McAllister K., Mechanic L. E., Amos C., Aschard H., Blair I. A., Chatterjee N., Conti D., Gauderman W. J., Hsu L., Hutter C. M., Jankowska M. M., Kerr J., Kraft P., Montgomery S. B., Mukherjee B., Papanicolaou G. J., Patel C. J., Ritchie M. D., Ritz B. R., Thomas D. C., Wei P., Witte J. S., Current challenges and new opportunities for gene-environment interaction studies of complex diseases. Am. J. Epidemiol. 186, 753–761 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yang J., Lee T., Kim J., Cho M. C., Han B. G., Lee J. Y., Lee H. J., Cho S., Kim H., Ubiquitous polygenicity of human complex traits: Genome-wide analysis of 49 traits in Koreans. PLOS Genet. 9, e1003355 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Shi H., Kichaev G., Pasaniuc B., Contrasting the genetic architecture of 30 complex traits from summary association data. Am. J. Hum. Genet. 99, 139–153 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Maier R. M., Visscher P. M., Robinson M. R., Wray N. R., Embracing polygenicity: A review of methods and tools for psychiatric genetics research. Psychol. Med. 48, 1055–1067 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kilpeläinen T. O., Qi L., Brage S., Sharp S. J., Sonestedt E., Demerath E., Ahmad T., Mora S., Kaakinen M., Sandholt C. H., Holzapfel C., Autenrieth C. S., Hyppönen E., Cauchi S., He M., Kutalik Z., Kumari M., Stančáková A., Meidtner K., Balkau B., Tan J. T., Mangino M., Timpson N. J., Song Y., Zillikens M. C., Jablonski K. A., Garcia M. E., Johansson S., Bragg-Gresham J. L., Wu Y., van Vliet-Ostaptchouk J. V., Onland-Moret N. C., Zimmermann E., Rivera N. V., Tanaka T., Stringham H. M., Silbernagel G., Kanoni S., Feitosa M. F., Snitker S., Ruiz J. R., Metter J., Larrad M. T. M., Atalay M., Hakanen M., Amin N., Cavalcanti-Proença C., Grøntved A., Hallmans G., Jansson J. O., Kuusisto J., Kähönen M., Lutsey P. L., Nolan J. J., Palla L., Pedersen O., Pérusse L., Renström F., Scott R. A., Shungin D., Sovio U., Tammelin T. H., Rönnemaa T., Lakka T. A., Uusitupa M., Rios M. S., Ferrucci L., Bouchard C., Meirhaeghe A., Fu M., Walker M., Borecki I. B., Dedoussis G. V., Fritsche A., Ohlsson C., Boehnke M., Bandinelli S., van Duijn C. M., Ebrahim S., Lawlor D. A., Gudnason V., Harris T. B., Sørensen T. I. A., Mohlke K. L., Hofman A., Uitterlinden A. G., Tuomilehto J., Lehtimäki T., Raitakari O., Isomaa B., Njølstad P. R., Florez J. C., Liu S., Ness A., Spector T. D., Tai E. S., Froguel P., Boeing H., Laakso M., Marmot M., Bergmann S., Power C., Khaw K. T., Chasman D., Ridker P., Hansen T., Monda K. L., Illig T., Järvelin M. R., Wareham N. J., Hu F. B., Groop L. C., Orho-Melander M., Ekelund U., Franks P. W., Loos R. J. F., Physical activity attenuates the influence of FTO variants on obesity risk: A meta-analysis of 218,166 adults and 19,268 children. PLOS Med. 8, e1001116 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Abadi A., Alyass A., du Pont S. R., Bolker B., Singh P., Mohan V., Diaz R., Engert J. C., Yusuf S., Gerstein H. C., Anand S. S., Meyre D., Penetrance of polygenic obesity susceptibility loci across the body mass index distribution. Am. J. Hum. Genet. 101, 925–938 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nagpal S., Gibson G., Marigorta U., Pervasive modulation of obesity risk by the environment and genomic background. Genes 9, 411 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yang J., Manolio T. A., Pasquale L. R., Boerwinkle E., Caporaso N., Cunningham J. M., de Andrade M., Feenstra B., Feingold E., Hayes M. G., Hill W. G., Landi M. T., Alonso A., Lettre G., Lin P., Ling H., Lowe W., Mathias R. A., Melbye M., Pugh E., Cornelis M. C., Weir B. S., Goddard M. E., Visscher P. M., Genome partitioning of genetic variation for complex traits using common SNPs. Nat. Genet. 43, 519–525 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Robinson M. R., English G., Moser G., Lloyd-Jones L. R., Triplett M. A., Zhu Z., Nolte I. M., van Vliet-Ostaptchouk J. V., Snieder H.; LifeLines Cohort Study, Esko T., Milani L., Mägi R., Metspalu A., Magnusson P. K. E., Pedersen N. L., Ingelsson E., Johannesson M., Yang J., Cesarini D., Visscher P. M., Genotype-covariate interaction effects and the heritability of adult body mass index. Nat. Genet. 49, 1174–1181 (2017). [DOI] [PubMed] [Google Scholar]
- 17.Pare G., Cook N. R., Ridker P. M., Chasman D. I., On the use of variance per genotype as a tool to identify quantitative trait interaction effects: A report from the Women’s Genome Health Study. PLOS Genet. 6, e1000981 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Metzger B. P., Yuan D. C., Gruber J. D., Duveau F., Wittkopp P. J., Selection on noise constrains variation in a eukaryotic promoter. Nature 521, 344–347 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cao Y., Wei P., Bailey M., Kauwe J. S. K., Maxwell T. J., A versatile omnibus test for detecting mean and variance heterogeneity. Genet. Epidemiol. 38, 51–59 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ek W. E., Rask-Andersen M., Karlsson T., Enroth S., Gyllensten U., Johansson A., Genetic variants influencing phenotypic variance heterogeneity. Hum. Mol. Genet. 27, 799–810 (2018). [DOI] [PubMed] [Google Scholar]
- 21.Rönnegård L., Valdar W., Recent developments in statistical methods for detecting genetic loci affecting phenotypic variability. BMC Genet. 13, 63 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Van Vleck L. D., Variation of milk records within paternal-sib groups. J. Dairy Sci. 51, 1465–1470 (1968). [Google Scholar]
- 23.Hill W. G., Mulder H. A., Genetic analysis of environmental variation. Genet. Res. 92, 381–395 (2010). [DOI] [PubMed] [Google Scholar]
- 24.Struchalin M. V., Dehghan A., Witteman J. C., van Duijn C., Aulchenko Y. S., Variance heterogeneity analysis for detection of potentially interacting genetic loci: Method and its limitations. BMC Genet. 11, 92 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yang J., Loos R. J., Powell J. E., Medland S. E., Speliotes E. K., Chasman D. I., Rose L. M., Thorleifsson G., Steinthorsdottir V., Mägi R., Waite L., Smith A. V., Yerges-Armstrong L. M., Monda K. L., Hadley D., Mahajan A., Li G., Kapur K., Vitart V., Huffman J. E., Wang S. R., Palmer C., Esko T., Fischer K., Zhao J. H., Demirkan A., Isaacs A., Feitosa M. F., Luan J., Heard-Costa N. L., White C., Jackson A. U., Preuss M., Ziegler A., Eriksson J., Kutalik Z., Frau F., Nolte I. M., Van Vliet-Ostaptchouk J. V., Hottenga J. J., Jacobs K. B., Verweij N., Goel A., Medina-Gomez C., Estrada K., Bragg-Gresham J. L., Sanna S., Sidore C., Tyrer J., Teumer A., Prokopenko I., Mangino M., Lindgren C. M., Assimes T. L., Shuldiner A. R., Hui J., Beilby J. P., McArdle W. L., Hall P., Haritunians T., Zgaga L., Kolcic I., Polasek O., Zemunik T., Oostra B. A., Junttila M. J., Grönberg H., Schreiber S., Peters A., Hicks A. A., Stephens J., Foad N. S., Laitinen J., Pouta A., Kaakinen M., Willemsen G., Vink J. M., Wild S. H., Navis G., Asselbergs F. W., Homuth G., John U., Iribarren C., Harris T., Launer L., Gudnason V., O'Connell J. R., Boerwinkle E., Cadby G., Palmer L. J., James A. L., Musk A. W., Ingelsson E., Psaty B. M., Beckmann J. S., Waeber G., Vollenweider P., Hayward C., Wright A. F., Rudan I., Groop L. C., Metspalu A., Khaw K. T., van Duijn C. M., Borecki I. B., Province M. A., Wareham N. J., Tardif J. C., Huikuri H. V., Cupples L. A., Atwood L. D., Fox C. S., Boehnke M., Collins F. S., Mohlke K. L., Erdmann J., Schunkert H., Hengstenberg C., Stark K., Lorentzon M., Ohlsson C., Cusi D., Staessen J. A., Van der Klauw M. M., Pramstaller P. P., Kathiresan S., Jolley J. D., Ripatti S., Jarvelin M. R., de Geus E. J., Boomsma D. I., Penninx B., Wilson J. F., Campbell H., Chanock S. J., van der Harst P., Hamsten A., Watkins H., Hofman A., Witteman J. C., Zillikens M. C., Uitterlinden A. G., Rivadeneira F., Zillikens M. C., Kiemeney L. A., Vermeulen S. H., Abecasis G. R., Schlessinger D., Schipf S., Stumvoll M., Tönjes A., Spector T. D., North K. E., Lettre G., McCarthy M. I., Berndt S. I., Heath A. C., Madden P. A., Nyholt D. R., Montgomery G. W., Martin N. G., McKnight B., Strachan D. P., Hill W. G., Snieder H., Ridker P. M., Thorsteinsdottir U., Stefansson K., Frayling T. M., Hirschhorn J. N., Goddard M. E., Visscher P. M., FTO genotype is associated with phenotypic variability of body mass index. Nature 490, 267–272 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bycroft C., Freeman C., Petkova D., Band G., Elliott L. T., Sharp K., Motyer A., Vukcevic D., Delaneau O., O’Connell J., Cortes A., Welsh S., Young A., Effingham M., McVean G., Leslie S., Allen N., Donnelly P., Marchini J., The UK Biobank resource with deep phenotyping and genomic data. Nature 562, 203–209 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Collins F. S., Varmus H., A new initiative on precision medicine. N. Engl. J. Med. 372, 793–795 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Conover W. J., Johnson M. E., Johnson M. M., A comparative study of tests for homogeneity of variances, with applications to the outer continental shelf bidding data. Technometrics 23, 351–361 (1981). [Google Scholar]
- 29.Bartlett M. S., Properties of sufficiency and statistical tests. Proc. R. Soc. Lond. A 160, 113–126 (1937). [Google Scholar]
- 30.H. Levene, Robust tests for equality of variances, in Contributions to Probability and Statistics: Essays in Honor of Harold Hotelling (Stanford Univ. Press, 1960), pp. 278–292. [Google Scholar]
- 31.Brown M. B., Forsythe A. B., Robust tests for the equality of variances. J. Am. Stat. Assoc. 69, 364–367 (1974). [Google Scholar]
- 32.Fligner M. A., Killeen T. J., Distribution-free two-sample tests for scale. J. Am. Stat. Assoc. 71, 210–213 (1976). [Google Scholar]
- 33.Rönnegård L., Felleki M., Fikse F., Mulder H. A., Strandberg E., Genetic heterogeneity of residual variance - estimation of variance components using double hierarchical generalized linear models. Genet. Sel. Evol. 42, 8 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rönnegård L., Valdar W., Detecting major genetic loci controlling phenotypic variability in experimental crosses. Genetics 188, 435–447 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Smyth G. K., Generalized linear models with varying dispersion. J. R. Stat. Soc. B. Methodol. 47–60 (1989). [Google Scholar]
- 36.Sun X., Elston R., Morris N., Zhu X., What is the significance of difference in phenotypic variability across SNP genotypes? Am. J. Hum. Genet. 93, 390–397 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.R. W. Corty, W. Valdar, Mean-variance QTL mapping on a background of variance heterogeneity. bioRxiv 276980 [Preprint]. 16 March 2018. 10.1101/276980 [DOI]
- 38.Wu Y., Zheng Z., Visscher P. M., Yang J., Quantifying the mapping precision of genome-wide association studies using whole-genome sequencing data. Genome Biol. 18, 86 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pulit S. L., de With S. A., de Bakker P. I., Resetting the bar: Statistical significance in whole-genome sequencing-based association studies of global populations. Genet. Epidemiol. 41, 145–151 (2017). [DOI] [PubMed] [Google Scholar]
- 40.Young A. I., Wauthier F. L., Donnelly P., Identifying loci affecting trait variability and detecting interactions in genome-wide association studies. Nat. Genet. 50, 1608–1614 (2018). [DOI] [PubMed] [Google Scholar]
- 41.Yang J., Weedon M. N., Purcell S., Lettre G., Estrada K., Willer C. J., Smith A. V., Ingelsson E., O'Connell J. R., Mangino M., Mägi R., Madden P. A., Heath A. C., Nyholt D. R., Martin N. G., Montgomery G. W., Frayling T. M., Hirschhorn J. N., McCarthy M. I., Goddard M. E., Visscher P. M.; GIANT Consortium , Genomic inflation factors under polygenic inheritance. Eur. J. Hum. Genet. 19, 807–812 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bulik-Sullivan B. K., Loh P.-R., Finucane H. K., Ripke S., Yang J.; Schizophrenia Working Group of the Psychiatric Genomics Consortium, Patterson N., Daly M. J., Price A. L., Neale B. M., LD score regression distinguishes confounding from polygenicity in genome-wide association studies. Nat. Genet. 47, 291–295 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Saccone S. F., Hinrichs A. L., Saccone N. L., Chase G. A., Konvicka K., Madden P. A., Breslau N., Johnson E. O., Hatsukami D., Pomerleau O., Swan G. E., Goate A. M., Rutter J., Bertelsen S., Fox L., Fugman D., Martin N. G., Montgomery G. W., Wang J. C., Ballinger D. G., Rice J. P., Bierut L. J., Cholinergic nicotinic receptor genes implicated in a nicotine dependence association study targeting 348 candidate genes with 3713 SNPs. Hum. Mol. Genet. 16, 36–49 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Thorgeirsson T. E., Geller F., Sulem P., Rafnar T., Wiste A., Magnusson K. P., Manolescu A., Thorleifsson G., Stefansson H., Ingason A., Stacey S. N., Bergthorsson J. T., Thorlacius S., Gudmundsson J., Jonsson T., Jakobsdottir M., Saemundsdottir J., Olafsdottir O., Gudmundsson L. J., Bjornsdottir G., Kristjansson K., Skuladottir H., Isaksson H. J., Gudbjartsson T., Jones G. T., Mueller T., Gottsäter A., Flex A., Aben K. K. H., de Vegt F., Mulders P. F. A., Isla D., Vidal M. J., Asin L., Saez B., Murillo L., Blondal T., Kolbeinsson H., Stefansson J. G., Hansdottir I., Runarsdottir V., Pola R., Lindblad B., van Rij A. M., Dieplinger B., Haltmayer M., Mayordomo J. I., Kiemeney L. A., Matthiasson S. E., Oskarsson H., Tyrfingsson T., Gudbjartsson D. F., Gulcher J. R., Jonsson S., Thorsteinsdottir U., Kong A., Stefansson K., A variant associated with nicotine dependence, lung cancer and peripheral arterial disease. Nature 452, 638–642 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Fowler C. D., Lu Q., Johnson P. M., Marks M. J., Kenny P. J., Habenular α5 nicotinic receptor subunit signalling controls nicotine intake. Nature 471, 597–601 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Repapi E., Sayers I., Wain L. V., Burton P. R., Johnson T., Obeidat M., Zhao J. H., Ramasamy A., Zhai G., Vitart V., Huffman J. E., Igl W., Albrecht E., Deloukas P., Henderson J., Granell R., McArdle W. L., Rudnicka A. R.; Wellcome Trust Case Control Consortium, Barroso I., Loos R. J., Wareham N. J., Mustelin L., Rantanen T., Surakka I., Imboden M., Wichmann H. E., Grkovic I., Jankovic S., Zgaga L., Hartikainen A. L., Peltonen L., Gyllensten U., Johansson A., Zaboli G., Campbell H., Wild S. H., Wilson J. F., Gläser S., Homuth G., Völzke H., Mangino M., Soranzo N., Spector T. D., Polasek O., Rudan I., Wright A. F., Heliövaara M., Ripatti S., Pouta A., Naluai A. T., Olin A. C., Torén K., Cooper M. N., James A. L., Palmer L. J., Hingorani A. D., Wannamethee S. G., Whincup P. H., Smith G. D., Ebrahim S., McKeever T. M., Pavord I. D., MacLeod A. K., Morris A. D., Porteous D. J., Cooper C., Dennison E., Shaheen S., Karrasch S., Schnabel E., Schulz H., Grallert H., Bouatia-Naji N., Delplanque J., Froguel P., Blakey J. D.; NSHD Respiratory Study Team, Britton J. R., Morris R. W., Holloway J. W., Lawlor D. A., Hui J., Nyberg F., Jarvelin M. R., Jackson C., Kähönen M., Kaprio J., Probst-Hensch N. M., Koch B., Hayward C., Evans D. M., Elliott P., Strachan D. P., Hall I. P., Tobin M. D., Genome-wide association study identifies five loci associated with lung function. Nat. Genet. 42, 36–44 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hancock D. B., Eijgelsheim M., Wilk J. B., Gharib S. A., Loehr L. R., Marciante K. D., Franceschini N., van Durme Y. M. T. A., Chen T. H., Barr R. G., Schabath M. B., Couper D. J., Brusselle G. G., Psaty B. M., van Duijn C. M., Rotter J. I., Uitterlinden A. G., Hofman A., Punjabi N. M., Rivadeneira F., Morrison A. C., Enright P. L., North K. E., Heckbert S. R., Lumley T., Stricker B. H. C., O’Connor G. T., London S. J., Meta-analyses of genome-wide association studies identify multiple loci associated with pulmonary function. Nat. Genet. 42, 45–52 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wain L. V., Shrine N., Artigas M. S., Shrine N., Artigas M. S., Erzurumluoglu A. M., Noyvert B., Bossini-Castillo L., Obeidat M., Henry A. P., Portelli M. A., Hall R. J., Billington C. K., Rimington T. L., Fenech A. G., John C., Blake T., Jackson V. E., Allen R. J., Prins B. P., Campbell A., Porteous D. J., Jarvelin M. R., Wielscher M., James A. L., Hui J., Wareham N. J., Zhao J. H., Wilson J. F., Joshi P. K., Stubbe B., Rawal R., Schulz H., Imboden M., Probst-Hensch N. M., Karrasch S., Gieger C., Deary I. J., Harris S. E., Marten J., Rudan I., Enroth S., Gyllensten U., Kerr S. M., Polasek O., Kähönen M., Surakka I., Vitart V., Hayward C., Lehtimäki T., Raitakari O. T., Evans D. M., Henderson A. J., Pennell C. E., Wang C. A., Sly P. D., Wan E. S., Busch R., Hobbs B. D., Litonjua A. A., Sparrow D. W., Gulsvik A., Bakke P. S., Crapo J. D., Beaty T. H., Hansel N. N., Mathias R. A., Ruczinski I., Barnes K. C., Bossé Y., Joubert P., van den Berge M., Brandsma C. A., Paré P. D., Sin D. D., Nickle D. C., Hao K., Gottesman O., Dewey F. E., Bruse S. E., Carey D. J., Kirchner H. L., Jonsson S., Thorleifsson G., Jonsdottir I., Gislason T., Stefansson K., Schurmann C., Nadkarni G., Bottinger E. P., Loos R. J., Walters R. G., Chen Z., Millwood I. Y., Vaucher J., Kurmi O. P., Li L., Hansell A. L., Brightling C., Zeggini E., Cho M. H., Silverman E. K., Sayers I., Trynka G., Morris A. P., Strachan D. P., Hall I. P., Tobin M. D., Genome-wide association analyses for lung function and chronic obstructive pulmonary disease identify new loci and potential druggable targets. Nat. Genet. 49, 416–425 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kaur-Knudsen D., Nordestgaard B. G., Bojesen S. E., CHRNA3 genotype, nicotine dependence, lung function and disease in the general population. Eur. Respir. J. 40, 1538–1544 (2012). [DOI] [PubMed] [Google Scholar]
- 50.Estrada K., Styrkarsdottir U., Evangelou E., Hsu Y. H., Duncan E. L., Ntzani E. E., Oei L., Albagha O. M. E., Amin N., Kemp J. P., Koller D. L., Li G., Liu C. T., Minster R. L., Moayyeri A., Vandenput L., Willner D., Xiao S. M., Yerges-Armstrong L. M., Zheng H. F., Alonso N., Eriksson J., Kammerer C. M., Kaptoge S. K., Leo P. J., Thorleifsson G., Wilson S. G., Wilson J. F., Aalto V., Alen M., Aragaki A. K., Aspelund T., Center J. R., Dailiana Z., Duggan D. J., Garcia M., Garcia-Giralt N., Giroux S., Hallmans G., Hocking L. J., Husted L. B., Jameson K. A., Khusainova R., Kim G. S., Kooperberg C., Koromila T., Kruk M., Laaksonen M., Lacroix A. Z., Lee S. H., Leung P. C., Lewis J. R., Masi L., Mencej-Bedrac S., Nguyen T. V., Nogues X., Patel M. S., Prezelj J., Rose L. M., Scollen S., Siggeirsdottir K., Smith A. V., Svensson O., Trompet S., Trummer O., van Schoor N. M., Woo J., Zhu K., Balcells S., Brandi M. L., Buckley B. M., Cheng S., Christiansen C., Cooper C., Dedoussis G., Ford I., Frost M., Goltzman D., González-Macías J., Kähönen M., Karlsson M., Khusnutdinova E., Koh J. M., Kollia P., Langdahl B. L., Leslie W. D., Lips P., Ljunggren Ö., Lorenc R. S., Marc J., Mellström D., Obermayer-Pietsch B., Olmos J. M., Pettersson-Kymmer U., Reid D. M., Riancho J. A., Ridker P. M., Rousseau F., lagboom P. E. S., Tang N. L. S., Urreizti R., van Hul W., Viikari J., Zarrabeitia M. T., Aulchenko Y. S., Castano-Betancourt M., Grundberg E., Herrera L., Ingvarsson T., Johannsdottir H., Kwan T., Li R., Luben R., Medina-Gómez C., Th Palsson S., Reppe S., Rotter J. I., Sigurdsson G., van Meurs J. B. J., Verlaan D., Williams F. M. K., Wood A. R., Zhou Y., Gautvik K. M., Pastinen T., Raychaudhuri S., Cauley J. A., Chasman D. I., Clark G. R., Cummings S. R., Danoy P., Dennison E. M., Eastell R., Eisman J. A., Gudnason V., Hofman A., Jackson R. D., Jones G., Jukema J. W., Khaw K. T., Lehtimäki T., Liu Y., Lorentzon M., McCloskey E., Mitchell B. D., Nandakumar K., Nicholson G. C., Oostra B. A., Peacock M., Pols H. A. P., Prince R. L., Raitakari O., Reid I. R., Robbins J., Sambrook P. N., Sham P. C., Shuldiner A. R., Tylavsky F. A., van Duijn C. M., Wareham N. J., Cupples L. A., Econs M. J., Evans D. M., Harris T. B., Kung A. W. C., Psaty B. M., Reeve J., Spector T. D., Streeten E. A., Zillikens M. C., Thorsteinsdottir U., Ohlsson C., Karasik D., Richards J. B., Brown M. A., Stefansson K., Uitterlinden A. G., Ralston S. H., Ioannidis J. P. A., Kiel D. P., Rivadeneira F., Genome-wide meta-analysis identifies 56 bone mineral density loci and reveals 14 loci associated with risk of fracture. Nat. Genet. 44, 491–501 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kemp J. P., Morris J. A., Medina-Gomez C., Forgetta V., Warrington N. M., Youlten S. E., Zheng J., Gregson C. L., Grundberg E., Trajanoska K., Logan J. G., Pollard A. S., Sparkes P. C., Ghirardello E. J., Allen R., Leitch V. D., Butterfield N. C., Komla-Ebri D., Adoum A. T., Curry K. F., White J. K., Kussy F., Greenlaw K. M., Xu C., Harvey N. C., Cooper C., Adams D. J., Greenwood C. M. T., Maurano M. T., Kaptoge S., Rivadeneira F., Tobias J. H., Croucher P. I., Ackert-Bicknell C. L., Bassett J. H. D., Williams G. R., Richards J. B., Evans D. M., Identification of 153 new loci associated with heel bone mineral density and functional involvement of GPC6 in osteoporosis. Nat. Genet. 49, 1468–1475 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Medina-Gomez C., Kemp J. P., Estrada K., Eriksson J., Liu J., Reppe S., Evans D. M., Heppe D. H. M., Vandenput L., Herrera L., Ring S. M., Kruithof C. J., Timpson N. J., Zillikens M. C., Olstad O. K., Zheng H. F., Richards J. B., St. Pourcain B., Hofman A., Jaddoe V. W. V., Smith G. D., Lorentzon M., Gautvik K. M., Uitterlinden A. G., Brommage R., Ohlsson C., Tobias J. H., Rivadeneira F., Meta-analysis of genome-wide scans for total body BMD in children and adults reveals allelic heterogeneity and age-specific effects at the WNT16 locus. PLOS Genet. 8, e1002718 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Movérare-Skrtic S., Henning P., Liu X., Nagano K., Saito H., Börjesson A. E., Sjögren K., Windahl S. H., Farman H., Kindlund B., Engdahl C., Koskela A., Zhang F. P., Eriksson E. E., Zaman F., Hammarstedt A., Isaksson H., Bally M., Kassem A., Lindholm C., Sandberg O., Aspenberg P., Sävendahl L., Feng J. Q., Tuckermann J., Tuukkanen J., Poutanen M., Baron R., Lerner U. H., Gori F., Ohlsson C., Osteoblast-derived WNT16 represses osteoclastogenesis and prevents cortical bone fragility fractures. Nat. Med. 20, 1279–1288 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Frayling T. M., Timpson N. J., Weedon M. N., Zeggini E., Freathy R. M., Lindgren C. M., Perry J. R. B., Elliott K. S., Lango H., Rayner N. W., Shields B., Harries L. W., Barrett J. C., Ellard S., Groves C. J., Knight B., Patch A. M., Ness A. R., Ebrahim S., Lawlor D. A., Ring S. M., Ben-Shlomo Y., Jarvelin M. R., Sovio U., Bennett A. J., Melzer D., Ferrucci L., Loos R. J. F., Barroso I., Wareham N. J., Karpe F., Owen K. R., Cardon L. R., Walker M., Hitman G. A., Palmer C. N. A., Doney A. S. F., Morris A. D., Smith G. D.; The Wellcome Trust Case Control Consortium, Hattersley A. T., McCarthy M. I., A common variant in the FTO gene is associated with body mass index and predisposes to childhood and adult obesity. Science 316, 889–894 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Smemo S., Tena J. J., Kim K.-H., Gamazon E. R., Sakabe N. J., Gómez-Marín C., Aneas I., Credidio F. L., Sobreira D. R., Wasserman N. F., Lee J. H., Puviindran V., Tam D., Shen M., Son J. E., Vakili N. A., Sung H. K., Naranjo S., Acemel R. D., Manzanares M., Nagy A., Cox N. J., Hui C. C., Gomez-Skarmeta J. L., Nóbrega M. A., Obesity-associated variants within FTO form long-range functional connections with IRX3. Nature 507, 371–375 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Claussnitzer M., Dankel S. N., Kim K.-H., Quon G., Meuleman W., Haugen C., Glunk V., Sousa I. S., Beaudry J. L., Puviindran V., Abdennur N. A., Liu J., Svensson P. A., Hsu Y. H., Drucker D. J., Mellgren G., Hui C. C., Hauner H., Kellis M., FTO obesity variant circuitry and adipocyte browning in humans. N. Engl. J. Med. 373, 895–907 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Loos R. J., Yeo G. S., The bigger picture of FTO—The first GWAS-identified obesity gene. Nat. Rev. Endocrinol. 10, 51–61 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Moore R., Casale F. P., Bonder M. J., Horta D.; BIOS Consortium, Franke L., Barroso I., Stegle O., A linear mixed-model approach to study multivariate gene–environment interactions. Nat. Genet. 51, 180–186 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.McCarthy S., Das S., Kretzschmar W., Delaneau O., Wood A. R., Teumer A., Kang H. M., Fuchsberger C., Danecek P., Sharp K., Luo Y., Sidore C., Kwong A., Timpson N., Koskinen S., Vrieze S., Scott L. J., Zhang H., Mahajan A., Veldink J., Peters U., Pato C., van Duijn C., Gillies C. E., Gandin I., Mezzavilla M., Gilly A., Cocca M., Traglia M., Angius A., Barrett J. C., Boomsma D., Branham K., Breen G., Brummett C. M., Busonero F., Campbell H., Chan A., Chen S., Chew E., Collins F. S., Corbin L. J., Smith G. D., Dedoussis G., Dorr M., Farmaki A. E., Ferrucci L., Forer L., Fraser R. M., Gabriel S., Levy S., Groop L., Harrison T., Hattersley A., Holmen O. L., Hveem K., Kretzler M., Lee J. C., McGue M., Meitinger T., Melzer D., Min J. L., Mohlke K. L., Vincent J. B., Nauck M., Nickerson D., Palotie A., Pato M., Pirastu N., McInnis M., Richards J. B., Sala C., Salomaa V., Schlessinger D., Schoenherr S., Slagboom P. E., Small K., Spector T., Stambolian D., Tuke M., Tuomilehto J., van den Berg L., van Rheenen W., Volker U., Wijmenga C., Toniolo D., Zeggini E., Gasparini P., Sampson M. G., Wilson J. F., Frayling T., de Bakker P. I., Swertz M. A., McCarroll S., Kooperberg C., Dekker A., Altshuler D., Willer C., Iacono W., Ripatti S., Soranzo N., Walter K., Swaroop A., Cucca F., Anderson C. A., Myers R. M., Boehnke M., McCarthy M., Durbin R., Haplotype Reference Consortium , A reference panel of 64,976 haplotypes for genotype imputation. Nat. Genet. 48, 1279–1283 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Wood A. R., Tuke M. A., Nalls M. A., Hernandez D. G., Bandinelli S., Singleton A. B., Melzer D., Ferrucci L., Frayling T. M., Weedon M. N., Another explanation for apparent epistasis. Nature 514, E3–E5 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Zhang F., Chen W., Zhu Z., Zhang Q., Nabais M. F., Qi T., Deary I. J., Wray N. R., Visscher P. M., McRae A. F., Yang J., OSCA: A tool for omic-data-based complex trait analysis. Genome Biol. 20, 107 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.The UK10K Consortium, Walter K., Min J. L., Huang J., Crooks L., Memari Y., McCarthy S., Perry J. R., Xu C., Futema M., Lawson D., Iotchkova V., Schiffels S., Hendricks A. E., Danecek P., Li R., Floyd J., Wain L. V., Barroso I., Humphries S. E., Hurles M. E., Zeggini E., Barrett J. C., Plagnol V., Richards J. B., Greenwood C. M., Timpson N. J., Durbin R., Soranzo N., The UK10K project identifies rare variants in health and disease. Nature 526, 82–90 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Chang C. C., Chow C. C., Tellier L. C., Vattikuti S., Purcell S. M., Lee J. J., Second-generation PLINK: Rising to the challenge of larger and richer datasets. Gigascience 4, 7 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.1000 Genomes Project Consortium, Abecasis G. R., Altshuler D., Auton A., Brooks L. D., Durbin R. M., Gibbs R. A., Hurles M. E., McVean G. A., A map of human genome variation from population-scale sequencing. Nature 467, 1061–1073 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Yang J., Lee S. H., Goddard M. E., Visscher P. M., GCTA: A tool for genome-wide complex trait analysis. Am. J. Hum. Genet. 88, 76–82 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Bretherton C. S., Widmann M., Dymnikov V. P., Wallace J. M., Bladé I., The effective number of spatial degrees of freedom of a time-varying field. J. Climate 12, 1990–2009 (1999). [Google Scholar]
- 67.IPAQ Research Committee, Guidelines for Data Processing and Analysis of the International Physical Activity Questionnaire (IPAQ)-Short and Long Forms (IPAQ Research Committee, 2005). [Google Scholar]
- 68.Beasley T. M., Erickson S., Allison D. B., Rank-based inverse normal transformations are increasingly used, but are they merited? Behav. Genet. 39, 580–595 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Peng B., Yu R. K., Dehoff K. L., Amos C. I., Normalizing a large number of quantitative traits using empirical normal quantile transformation. BMC Proc. 1 (suppl. 1), S156 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Craig C. L., Marshall A. L., Sjorstrom M., Marshall A. L., Sjöström M., Bauman A. E., Booth M. L., Ainsworth B. E., Pratt M., Ekelund U., Yngve A., Sallis J. F., Oja P., International physical activity questionnaire: 12-country reliability and validity. Med. Sci. Sports Exerc. 35, 1381–1395 (2003). [DOI] [PubMed] [Google Scholar]
- 71.Wray N. R., Allele frequencies and the r2 measure of linkage disequilibrium: Impact on design and interpretation of association studies. Twin Res. Hum. Genet. 8, 87–94 (2005). [DOI] [PubMed] [Google Scholar]
- 72.Chapman J. M., Cooper J. D., Todd J. A., Clayton D. G., Detecting disease associations due to linkage disequilibrium using haplotype tags: A class of tests and the determinants of statistical power. Hum. Hered. 56, 18–31 (2003). [DOI] [PubMed] [Google Scholar]
- 73.Spencer C. C., Su Z., Donnelly P., Marchini J., Designing genome-wide association studies: Sample size, power, imputation, and the choice of genotyping chip. PLOS Genet. 5, e1000477 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Giambartolomei C., Vukcevic D., Schadt E. E., Franke L., Hingorani A. D., Wallace C., Plagnol V., Bayesian test for colocalisation between pairs of genetic association studies using summary statistics. PLOS Genet. 10, e1004383 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Zhu Z., Zhang F., Hu H., Bakshi A., Robinson M. R., Powell J. E., Montgomery G. W., Goddard M. E., Wray N. R., Visscher P. M., Yang J., Integration of summary data from GWAS and eQTL studies predicts complex trait gene targets. Nat. Genet. 48, 481–487 (2016). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/5/8/eaaw3538/DC1
Note S1. Theoretical derivation of vQTL as a consequence of GEI
Note S2. The Bartlett’s test, the FK test, and the DGLM test
Note S3. Rank-based inverse-normal transformation
Note S4. The effective number of independent traits
Note S5. Definitions of the three environmental factors—PA, SB, and smoking
Note S6. Expected inflation in the Levene’s test statistic due to phantom vQTL effect
Note S7. Acknowledgments
Fig. S1. Evaluation of statistical methods and phenotype processing strategies for the vQTL analysis by simulation based on a single-SNP model.
Fig. S2. Phenotypic correlations among 13 quantitative traits, and PA and SB measures in the UKB.
Fig. S3. Genome-wide vQTL and QTL analyses for 13 traits in the UKB.
Fig. S4. Quantile-quantile plots of vQTL associations for the 13 UKB traits.
Fig. S5. Enrichment of GEI effects among the 75 vQTLs compared with a random set of QTLs using the raw phenotypic values.
Fig. S6. Comparison of the Young et al. method with the Levene’s test by vQTL simulation.
Fig. S7. Excluding two alternative explanations for vQTL signals: Phantom vQTLs and epistasis.
Table S1. Descriptive summary of (A) the quantitative traits and (B) the environmental data used in this study from the UKB.
Table S2. Seventy-five experiment-wise significant vQTLs for nine UKB traits.
Table S3. GEI examples.