

Summary
Mental illness exerts a major burden on human health, yet evidence‐based treatments are rudimentary due to a limited understanding of the underlying pathologies. Clinical studies point to roles for the immune system in psychiatric diseases, while basic science has revealed that the brain has an active and multi‐cellular resident immune system that interacts with peripheral immunity and impacts behavior. In this perspective, we highlight evidence of immune involvement in human psychiatric disease and review data from animal models that link immune signaling to neuronal function and behavior. We propose a conceptual framework for linking advances in basic neuroimmunology to their potential relevance for psychiatric diseases, based on the subtypes of immune responses defined in peripheral tissues. Our goal is to identify novel areas of focus for future basic and translational studies that may reveal the potential of the immune system for diagnosing and treating mental illnesses
Keywords: brain, human, neuroimmunology, psychiatry
Introduction: proposing a new framework
Psychiatric diseases massively impact human health 1 but lack reliable diagnostic and prognostic biomarkers, molecularly targeted therapies and definitive treatments. The dire combination of severe disease burden and limited treatment efficacy makes it critical to increase our mechanistic understanding of psychiatric disease etiology. Causal connections between the immune system and brain function have been hypothesized and studied for many years, and an abundant literature has identified correlations between measures of immune function and psychiatric diseases in humans, yet the meaning of these associations remains largely mysterious.
How does the immune system contribute to causing or modifying psychiatric disease? Answering this question has been challenging due to several distinct properties of the central nervous sytem (CNS) compared to other organs. First, the CNS environment differs from the blood and other peripheral tissues due to its network properties, regional complexity, distinct metabolic demands and a blood–brain barrier that restricts exposure to the circulation. The result is a homeostatic immune set‐point and dynamic range quite different from other tissues, leading to the common view that the brain is ‘immune privileged’. In addition, while the classic immune triggers in peripheral tissues – viruses, damage and pathogen‐associated molecular patterns – have been studied since the time of van Leeuwenhoek's ‘animalcules’, the relevant triggers that modulate immune tone in the brain remain undiscovered. Consequently, potential links between the immune system and psychiatrically relevant triggers such as stress and brain trauma are poorly understood.
However, a greater problem is that our understanding of the neuroimmunology of psychiatric diseases is not up to date with recent advances in basic immunology. Clinical studies measure inflammatory markers that are sensitive but not necessarily specific or relevant to the brain environment [e.g. interleukin (IL)‐6, C‐reactive protein (CRP)]. The notion of regulatory, adaptive and stimulus‐specific immune responses has not gained sufficient traction. It is now clear that there is a complex immune environment in the brain. The resident immune cells of the brain, known as microglia, can both promote synapses and engulf them, as well as directly influence neurotransmission. Immune modulators such as tumor necrosis factor (TNF), transforming growth factor (TGF)‐β, IL‐33 and many others are made and tightly regulated within the brain parenchyma, where they act on microglia, neurons and other glia. Within the brain meninges, there are immune cells in both the myeloid and lymphoid lineages that may influence brain function.
There has been great progress in identifying these varied cellular constituents of the brain's immune system. As such, there is an opportunity to understand more clearly their roles in the healthy brain and in psychiatric diseases. In this perspective paper, we will first highlight studies associating immune dysfunction with human psychiatric disease. We will then review the immune landscape of the brain, with attention to data from animal models that links tissue‐resident and subtype‐specific immune signaling to neuronal function and behavior. We propose that a conceptual framework linking advances in basic neuroimmunology to their potential relevance for psychiatric diseases will reveal novel areas of focus for future basic and translational studies.
The immune system and psychiatric diagnoses: what human studies tell us
A broad human literature has implied a role for immune dysfunction in psychiatric diseases (reviewed in 2, 3, 4, 5, 6). These data strongly argue that brain–immune interactions are important; however, specific molecular underpinnings have not yet emerged. Which immune cells and cytokines are preferentially relevant in different psychiatric diagnoses? What brain regions are affected and how? Are brain–immune connections unfolding primarily in the brain or the periphery? Below we present vignettes from clinical literature that highlight evidence for immune dysfunction, but also make clear the need for more specific investigations that can target the questions above.
The evidence for immune dysfunction in a psychiatric syndrome is perhaps best exemplified in the schizophrenia literature. Schizophrenia is characterized by severely disordered thinking (including hallucinations and delusional beliefs) and cognitive deficits, and affects 1% of the population worldwide. It has its roots in neurodevelopment 7, 8 and is correlated with thinning of the cortical gray matter where synapses reside 9. There are epidemiological links between viral infections, autoimmune diseases and schizophrenia diagnosis 10. Altered blood cytokines and circulating immune cell abnormalities have also been identified, but with incompletely controlled confounders 11, 12. Critically, a large genomewide association study of schizophrenia 13 prominently implicated the major histocompatibility complex class 1 (MHC1) locus, which contains some ~250 immune genes, providing the first evidence of a possible causal association. These data intersect with years of evidence from rodent models that the MHC1 pathway regulates synaptic refinement (reviewed in 14 and discussed in the ‘Signaling molecules regulating CNS immunity’ section below). Another gene in the MHC locus – complement C4 protein – accounts for a small but significant increase in schizophrenia risk, correlated with the presence of gain‐of‐function alleles 15. These data intersect with molecular studies in mouse, suggesting that the complement system promotes pruning of synapses by brain microglia. This convergence of human genetics with a specific molecular mechanism may be consistent with Irwin Feinberg's ‘pruning hypothesis’ of excess synaptic pruning in schizophrenia 8, 16. Although caution must be taken in interpreting still preliminary findings, this cross‐modal observation provides a clear foothold for future research.
Autism spectrum disorder (ASD) is another example in which a potential role of the immune system has been much debated. ASD is characterized by social and language deficits that present in early childhood, although the literature on several fronts seems to indicate that the aberrant neurodevelopment begins much earlier, in early to mid‐fetal stages 17.The genetics of autism are complex, but so far the evidence of immune involvement is less clear than for schizophrenia 17. Some epidemiological data suggest links between maternal infections during pregnancy and later risk for autism 18, 19. There is some clinical evidence of alterations in immune function in autism, both in peripheral blood (decreased TGF‐β1 and increased IL‐1β) and in brain gene expression 20, 21, reviewed in 5. How these postnatal findings might relate mechanistically to deficits in mid‐gestation is not yet clear.
Gestational immune insults have been studied in rodents using a maternal immune activation model (MIA) of behavioral disturbance that is sometimes described as a mouse model of autism and schizophrenia. The model requires injection of the viral mimic poly(I:C) into pregnant mouse dams at embryonic day 12·5. The timing matters, suggesting that there is a sensitive period in mid‐fetal development for this type of immune disruption. The MIA model generates some social deficits reminiscent of autism symptoms. We would argue, however, that findings should be interpreted with caution, for several reasons. First, data from different groups has been challenging to standardize 22. Secondly, poly(I:C) in mice is an imperfect phenocopy of human anti‐viral immune responses. Most importantly, and not limited to studies of neuroimmunity, there is strong reason for caution when relying on mouse behavior as a model for human behavioral diseases. Conversely, some studies of the MIA model have provided relevant insight into how type III immune responses modulate embryonic brain development (see ‘Signaling molecules regulating CNS immunity’ section below). Thus, there are opportunities to improve current animal models to reflect the construct validity most consistent with the clinical data (mid‐gestational infection), while being cautious about over‐reliance on face validity (mouse behavior).
Studies of anxiety and mood disorders anchored in the field of psychoneuroimmunology have produced a similarly intriguing but incomplete picture. Type I interferons used clinically induce depressive symptoms responsive to antidepressant therapy 23, 24, 25. Some preliminary immunological biomarkers in major depressive disorder (MDD) have been described, including increased CD4/CD8 T cell ratios and serum IL‐6 26. Among mood and anxiety disorders, post‐traumatic stress disorder (PTSD) is perhaps the most mechanistically accessible – for example, acute stress responses can be elicited and characterized in clinical studies 27. Chronic PTSD is correlated with increased IL‐6, IL‐1β and TNF‐α 28. It is important to note that many of these same cytokines are known to cause an identifiable pattern of ‘sickness behavior’ in animal models (see ‘Signaling molecules regulating CNS immunity’ section, below). This model has yielded insight into how cytokines acutely affect brain function, but at the same time it is clear that sickness behavior in humans is not the same thing as depression, a chronic and episodic condition with strong social triggers. Distinguishing acute and chronic effects of immune function in the brain is an ongoing challenge. If some aspect of psychiatric disorders is indeed due to excess or dysregulated inflammation, it is possible that defining the homeostatic role of regulatory immune responses in the brain (see ‘The interface between peripheral and CNS immunity’ section below) could yield insight into why some individuals are more resilient to stressors associated with mood and anxiety disorders.
Complex as it is, one key theme is that the intersection of immune dysfunction and psychiatric disease may inform how environmental factors contribute to disease risk. In most mental illness, social stressors are a major contributor to disease. In addition to the infectious correlations described above, other immune activators such as traumatic brain injury (TBI) may be risk factors for suicide 29, schizophrenia 30, 31 and dementia 32. In ‘The interface between peripheral and CNS immunity’ section below we will discuss how to conceptualize the impact of TBI in terms of type II immune responses in the brain. We also review the notion that stress may generate a unique type of immune response. It is our hope that a framework focused on specific subtypes of immune dysfunction will encourage new hypotheses that can advance clinical questions in a meaningful way.
An overview of the brain and its immune environment: diverse cells mediate co‐ordinated responses
The brain is arguably the most regionally complex tissue in the body. Its neurons are supported by an almost equal number of glial cells that shape and support neuronal function. These include astrocytes that provide metabolic support and promote synapse formation, oligodendrocytes that generate the protective myelin sheath and microglia, the resident macrophages of the brain. Neurons form precise intracellular connections termed synapses. Electrical signals determined by the connectivity and strength of these synapses drive local and brain‐wide oscillations that determine communication between brain regions. Variables at all these levels ultimately generate behavior. As different brain regions mediate unique functions (e.g. movement, higher‐order cognition) these regions are non‐redundant. Thus, the ability to compensate for damage or tissue loss is extremely limited. Preserving the pattern and strength of synaptic connections between neurons is essential to all brain function.
For many years, the idea that immune signaling could contribute to brain function gained little traction, based on the notion that the brain is ‘immune privileged’. Indeed, at first pass this appears to be the case. Early studies demonstrated reduced clearance of tumors injected in the brain relative to other organs, suggesting suppressed cellular immune responses 33. Immune entry into the brain is tightly regulated by the blood brain barrier (BBB), a neurovascular unit that controls movement of ions, molecules and cells between the blood and CNS 34. The brain is protected from trauma, encased in bone and floating within a protective cushion of cerebrospinal fluid (CSF). This CSF is held in place by a meningeal barrier similar to the pleural cavities around peripheral organs.
Although access to the brain is tightly regulated, it is not static. Permeability can be altered during physiological states such as sleep 35, as well as during injury, stress or pathology. As such, much is still unknown about how immune information can cross brain barriers. Nonetheless, there is a full complement of tissue‐resident innate and adaptive lymphocytes in the brain meninges and in the vasculature (36, Fig. 1). In addition, there is evidence that lymphocyte‐derived cytokines impact neural circuit activity and behavior. In ‘Signaling molecules regulating CNS immunity’ below, we discuss the tissue‐resident immunity of the brain, and we also review evidence that peripheral immune signals may cross barriers to affect brain function.
Figure 1.
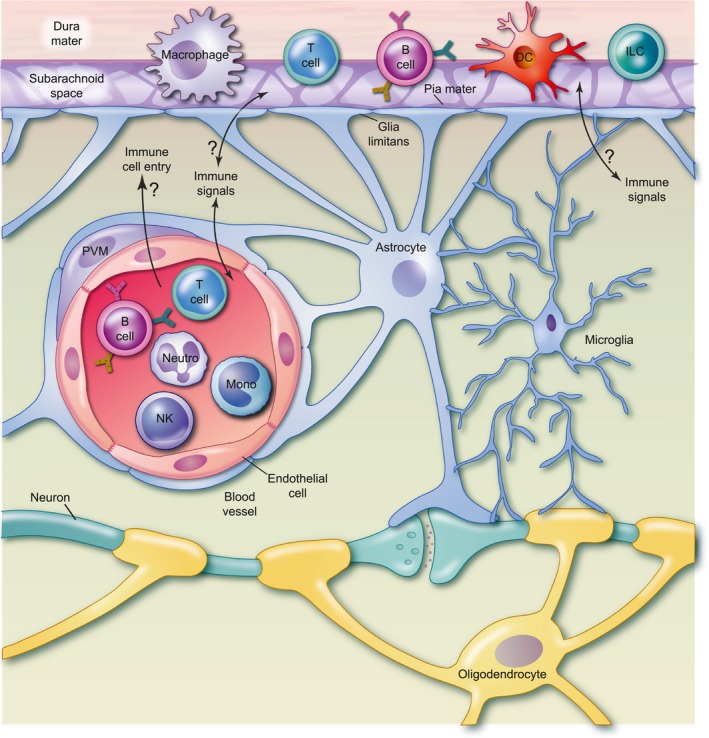
Immune cells of the brain. A diverse complement of immune cells resides in and around the brain. Lymphoid and myeloid lineages including T lymphocytes, B lymphocytes, innate lymphoid cells (ILCs), macrophages and dendritic cells (DCs) are resident to the three meningeal layers surrounding the brain parenchyma. They are found embedded in the dura, a fibrous protective layer, and in the subarachnoid space, where cerebrospinal fluid (CSF) circulates. Immune cells may also contact the pia mater, a delicate meningeal layer immediately adjacent to the brain parenchyma. Their precise localizations within these structures remain incompletely understood and may be dynamic, represented here conceptually by cell type icons that span the meningeal layers. Perivascular macrophages (PVM) surround blood vessels that allow a dynamic population of circulating immune cells to travel through the brain, including myeloid (monocytes, granulocytes) and lymphoid [T, B, natural killer (NK) cells] populations. Microglia are the resident immune cells of the brain parenchyma. They interact directly with neurons, astrocytes and oligodendrocytes. Although physically proximal, these three immune compartments (meninges, circulation and parenchyma) are distinct, and separated by barriers that actively mediate the flow of immune molecules and cells. The circulation is separated from the brain by the blood brain barrier, composed of tight junctions between specialized brain endothelial cells, the endothelial basement membrane, and surrounded by astrocyte foot processes that form the glia limitans, which also creates a barrier from the meninges. Signaling between meningeal, vascular and parenchymal immune cell compartments is tonically active but tightly regulated. It remains poorly understood how immune signals are communicated across central nervous system (CNS) barriers. Furthermore, although largely absent during homeostasis, circulating immune cells can enter the brain parenchyma from the circulation in disease states. Whether they do so in psychiatric diseases, and how this might contribute to their pathogenesis, remains largely to be discovered.
Tissue‐resident immunity of the brain: cellular players
It is increasingly clear that the immune system plays important homeostatic roles which are best appreciated by studying the tissue under homeostatic conditions 37. As discussed in ‘The immune system and psychiatric diagnoses: what human studies tell us’ section, there is clear evidence of correlations between immune perturbation and psychiatric disease. However, when compared to neurological diseases such as multiple sclerosis and neurodegeneration, there is no discrete ‘lesion’ that can be studied to determine etiology. Thus, there is much to be learned by defining the homeostatic roles of the immune system in the developing and adult brain.
Microglia, resident immune cells of the brain
Much immunity is local, regulated at the level of tissue resident macrophages, which are in homeostasis with their resident tissue 38, 39 and directly respond to tissue cues to promote remodeling. Microglia are highly ramified brain‐resident macrophages that comprise approximately 10% of brain cells. Their dynamic processes physically contact neurons and glia, surveying the entire volume of the brain every few hours 40, 41. Like macrophages in other tissues, they are phagocytic and broadly sensitive to immune signals. However, microglia also have unique attributes, such as the expression of many platelet genes implicated in hemostasis 42 and a very long lifespan (4·2 years in humans 43.) Microglia originate from primitive hematopoietic progenitors in the yolk sac and arrive in the fetal head as the neural plate begins to form. These colonize the brain parenchyma for life, with little or no contribution from circulating cells 44, 45, 46. Microglia dynamically engage with neurons and glia, and are strongly programmed by the brain environment (including tonic TGF‐β signaling 47, 48, 49, 50 ) to adopt a brain‐specific state 51. Not surprisingly, they perform critical local immune functions during development, homeostasis and in disease states.
One aspect of microglia especially relevant to psychiatric disease is their role in the refinement of neural circuit circuits through effects on synapse numbers. The pruning hypothesis of schizophrenia postulates that the physiological decrease in synapse numbers observed in the adolescent human brain 52represents a period of ‘pruning’ of excess synaptic connections, and that over‐pruning leads to the progressive loss in prefrontal cortical gray matter observed in schizophrenic patients 9. Synapse loss has also been reported in human MDD 53. Defective pruning during development has been associated with abnormal circuit function in several brain regions 54, 55, 56 and is implicated in Rett's syndrome 57, 58, 59, 60, 61. During early postnatal development of the visual thalamus, microglia were shown to engulf complement tagged synapses, a candidate molecular mechanism for inappropriate pruning in psychiatric diseases. Although it is not clear whether its role in development extends beyond this early, non‐experience dependent synapse engulfment, complement deficiency has been strongly implicated in synapse loss during neurodegeneration and viral infection 62, 63, 64.
A human genetics study recently found that complement pathway gain of function is positively correlated with schizophrenia (see ‘The immune system and psychiatric diagnoses: what human studies tell us’ section), and that complement protein C4 also participates in developmental synapse pruning of the visual system in mouse 65. Could complement dependent phagocytosis by microglia underlie the pruning hypothesis? Complement and microglia are implicated in synapse pruning in psychiatrically relevant brain regions such as the hippocampus. Microglial engulfment of synapses, however, has yet to be clearly shown in prefrontal cortex during a time window compatible with developmental synapse loss in schizophrenia. The nature of microglial engulfment is also not fully understood – although synaptic material is clearly observed in phagocytic vesicles inside microglia, it remains to be seen whether synapse engulfment is an active microglia‐driven process. Live imaging studies have demonstrated a ‘nibbling’ of synaptic material termed trogocytosis 66, that is hypothesized to promote stabilization of synapses. In adult motor cortex, dendritic spine formation rather than elimination is defective after depletion of microglia, raising the possibility of multiple synaptic functions for microglia 67. In the search for new treatments, better tools and molecular candidates are essential to resolve the role of microglia at synapses.
Other known microglial functions have not been deeply studied in the context of psychiatric diseases but hold great promise as contributors. For example, microglia appear to play a role in myelination and the health of the brain's myelinating cells, oligodendrocytes. Mice depleted of microglia using genetic and pharmacological strategies show abnormal myelination during development, and have abnormal numbers of oligodendrocyte precursors in adulthood 68. Multiple groups have identified a transcriptionally distinct microglial subset during development that is associated with white matter, and could support normal myelination, potentially by secretion of insulin‐like growth factor 1 (IGF1) and/or phagocytosis of oligodendrocyte lineage cells 69, 70, 71. As white matter abnormalities have been reported in diverse psychiatric diseases, this is a promising candidate mechanism for further study. Microglia also participate actively in phagocytic clearance of apoptotic neurons 72, 73. Although neuronal cell death is observed in many psychiatric diseases, particularly schizophrenia, the role of this microglial clearance function, which is critical to normal brain development, remains unexplored.
In summary, direct relationships between established microglial functions and psychiatric disease remain theoretical but compelling. As the only immune cells within the brain parenchyma, microglia should continue to be a major focus in the search for links between immunity and behavior. Several studies in mouse suggest that microglia may directly affect behavior. For example, absence of homeobox protein Hox‐B8 (Hoxb8) or progranulin in microglia leads to obsessive–compulsive disorder (OCD)‐like symptoms in rodents, such as excessive grooming 74, 75, while absence of Cx3cr1 leads to ASD‐like symptoms 76. A recent study also observed a sex‐specific role for microglial phagocytosis in male social play 77, setting the stage for further study of molecular mechanisms by which microglia cause psychiatric symptoms.
Other cell types
Astrocytes are implicated in the pathogenesis of many psychiatric diseases, particularly through their contributions to synapse formation, function and elimination. Although not immune cells, it is clear that astrocytes are also essential to the immune response. Similar to stromal cells in other tissues, astrocytes are critical to the structural integrity of healthy brain tissue and the formation of glial scars after injury. Glial scars are essential to protecting brain tissue from further damage, but excessive glial scarring by reactive astrocytes leads to a fibrotic condition classically thought to impair tissue regeneration (reviewed in 78). Similar to non‐professional phagocytes in other tissues, astrocytes have also been shown to engulf synapses in early postnatal development 79. Astrocytes can both secrete and sense immune signals. For example, microglial‐secreted inflammatory mediators (IL‐α, TNF, C1q) can induce a reactive astrocyte state that is associated with neurodegenerative diseases, characterized by decreased production of synaptogenic factors and impaired phagocytosis 80. Further, astrocytes can produce cytokines that affect the function of microglia and other brain cells. This is relevant both during homeostasis 54 and in response to inflammation 81. As discussed in the sections below, astrocytes and many other cell types, including neurons and endothelial cells, may directly sense immune signals. Decoding this multi‐cellular response is an important area for future study.
Signaling molecules regulating CNS immunity
TNF
Nearly all brain cells can respond to tissue resident immune signals. TNF‐α, a cytokine made by multiple local cell types and associated with type 1 immunity, also plays important physiological roles in the brain that appear to be distinct from its roles as an inflammatory mediator in the circulation. Several studies using TNF receptor (TNFR) and TNF‐deficient animals show that, in development, TNF signaling restricts neurogenesis 82, 83, 84 and dendrite maturation 85, possibly even acting through mother's milk on offspring to impact adult behavior 86. The TNF receptor superfamily member Tnfrsf12a is induced in neurons in response to visual experience and is involved in activity‐dependent synaptic refinement 87. A separate series of studies in adult hippocampus demonstrate that TNF promotes activity‐dependent synaptic strengthening through AMPA receptor (α‐amino‐3‐hydroxy‐5‐methyl‐4‐isoxazolepropionic acid) trafficking 88, 89, 90 and impacts cognition 91. Together, these studies suggest that TNF‐α physiologically impacts brain function by restricting synaptic plasticity and stabilizing synapses.
Major histocompatibility complex
Interestingly, signaling through the MHC‐1 pathway has also been shown to restrict brain plasticity. Loss of MHC‐1 or its receptor pirB lead to defects in synaptic refinement in the visual system 92, 93, presumably through neuron‐to‐neuron communication. Although it is unclear exactly how MHC‐1 deficiency causes synaptic defects at the molecular level, genetic links between the MHC1 locus and schizophrenia make this a worthy target for future study.
TNF and MHC1 both have the general effect of restricting synapse plasticity during homeostasis. To what extent should these be considered ‘co‐opted’ (e.g. non‐immune) roles of these signaling pathways? This probably depends on whether signaling is primarily between neurons, or also includes microglia and other immune cells. Additional insight will come from defining how these physiological roles in neural circuits may be engaged in adaptive or maladaptive ways when homeostasis is perturbed. Such approaches could have clear implications for understanding gene–environment interactions in brain diseases.
IL‐1 family members
Emerging data shows that IL‐1 family members produced locally in the brain play important homeostatic roles, in addition to their more well‐studied roles in the context of inflammation. We recently found that the IL‐1 family cytokine IL‐33 made by astrocytes physiologically regulates microglial synapse elimination 59. This homeostatic function is distinct from its reparative role in the context of injury 94, 95, Alzheimer's disease and ischemic stroke 95, 96, 97. Another IL‐1 family member, IL‐18, is also locally expressed in the brain, including in the emotion and threat regulating centers in the basolateral amygdala and has been proposed to mediate behavioral responses to chronic stress 98. While both IL‐33 and IL‐18 seem to have physiological as well as inflammatory roles, IL‐1 itself has largely been studied for its roles in response to immune activation, as occurs with lipopolysaccharide (LPS) injection. IL‐1β or LPS lead to sickness behavior – a response to infection that includes social withdrawal, anorexia and anhedonia 99. Sickness behavior has been used by some as a rodent model for depression. Although of unclear construct validity, it has yielded important insights into how cytokines affect brain function. IL‐1β can be made by activated macrophages, although its relevant sources in the brain or periphery in the context of sickness behavior are not clear. However, analysis of its downstream targets has yielded compelling recent data on its potential mechanisms of action. This highlights multiple cellular targets of IL‐1 signaling in the brain, particularly endothelial cells, which are required for IL‐1's effects on sickness behavior by permitting leukocyte infiltration 100, 101. Other studies show that IL‐1 induction is responsible for impaired hippocampal neurogenesis and impaired spatial learning 81. These data indicate an important role of this cytokine in brain–peripheral interactions in the context of inflammation.
In summary, several tissue resident signals and cytokines have fundamental physiological and pathological roles in regulating cognition, memory, neurogenesis and behavior. These alterations are commonly observed in psychiatric diseases, and so are appealing pathways for future study. Below we discuss how distinct subtypes of immune responses may provide additional levels of immune regulation in the brain.
The interface between peripheral and CNS immunity
Lymphocyte functional heterogeneity is central to the specificity of the immune response. Phenotypically distinct subsets of lymphocytes respond to unique immune triggers and secrete specific cytokines that co‐ordinate downstream immune responses 102, 103. Data from lymphocyte‐deficient animals has suggested that lymphocytes may be physiologically relevant to brain function, although with somewhat conflicting behavioral outcomes 104, 105, 106. Recent advances reinvigorate the question of whether specific subsets of lymphocytes may contribute to heterogeneous immune responses in the brain. One is the identification of lymphocytes in the meningeal spaces of the brain and the discovery of a brain lymphatic system that could mediate communication between innate and adaptive immune responses 107, 108, 109. Another is evidence that specific lymphocyte‐derived cytokines impact brain development, neural function and behavior (Fig. 2). A third is the identification of new channels for immune–CNS communication such as the gut–brain axis and infiltration of circulating cells into the brain. Below, we highlight recent examples of this concept and its implications for understanding brain–peripheral interactions.
Figure 2.
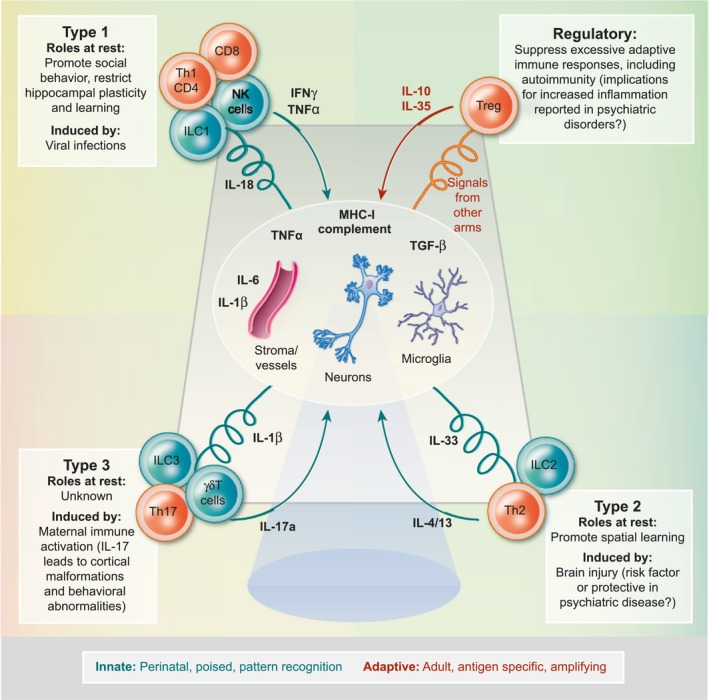
Immune responses in neuronal function and behavior. Local, tissue resident immunity of the brain (oval) is regulated by interactions between microglia, neurons, vessels and stromal cells (including astrocytes and oligodendrocytes), via tissue resident cytokines and signals. Under conditions of immune perturbation, homeostasis is restored by engagement of defined immune responses that modulate local immunity via release of lymphocyte derived cytokines. Instances in which these cytokines have been implicated in behavior and cognition are highlighted, as well as their potential implications for understanding psychiatric diseases (with inspiration from 37, 136, 137).
Type 1 immunity and social behavior
Type I immunity is classically elicited by tumors and intracellular microbes, in response to cytokines such as IL‐12/‐15/‐18. In the periphery, these cytokines recruit and activate lymphocytes [natural killer (NK), innate lymphoid cells (ILC)1 and CD4 T cells] which then cause the release of effector cytokines such as interferon (IFN)‐γ and TNF. However, there is evidence that these cytokines have behaviorally relevant roles. A recent study demonstrated a physiological role for the canonical type 1 effector cytokine IFN‐γ in promoting social behavior, using rodent loss of function models in a three‐chamber social interaction test 110. Another study indicated that absence of IFN‐γ promotes hippocampal plasticity and cognition 111. Mechanistically, some studies suggest that IFN‐γ acts directly on inhibitory interneurons to modulate function 110, 112, thus leading to increased circuit excitability. However, other mechanisms, including developmental roles, are also possible. Is this role of IFN‐γ an accidental side effect of endogenous viral protection systems 112, or does it play an adaptive role in linking immunity to behavior? Further defining the mechanistic links between these different behavioral observations in models of IFN‐γ could be relevant to understanding the epidemiological links between viral infections and psychiatric diseases.
Type II immunity – a focus on traumatic brain injury
Type II responses, classically elicited by large extracellular parasites and allergens, are in fact responsive to a diverse array of stimuli that signal via receptors that recognize tissue damage 103. These responses lead to alternative macrophage activation, mediated by cytokines such as Il‐4 and IL‐13 and the epidermal growth factor receptor (EGFR) ligand amphiregulin. Type II responses are typically delayed, rather than acute, consistent with a chronic process of tissue repair that occurs after an injury. Evidence for a physiologic role of type II immunity in brain function comes from studies of IL‐4 knock‐out animals, which have impaired spatial learning which is partially rescued by transplant of wild‐type T cells 104. Type II innate lymphoid cells are required for efficient repair after spinal cord injury, as is the upstream cytokine IL‐33 94, 113, 114, which also has physiological roles as discussed in the ‘Tissue‐resident immunity of the brain: cellular players’ section; as discussed in that section, traumatic brain injury is a risk factor for schizophrenia as well as behavioral disturbances, including suicidality and impulsivity. It is interesting to speculate that type II remodeling functions could be relevant links between brain immunity and behavior. Thus, defining how type II responses induced after brain injury impact behaviorally relevant brain circuitry could provide important mechanistic insights and therapeutic targets.
Type III immunity in neurodevelopment
Type III responses are classically triggered by extracellular bacteria (both commensals and invasive pathogens), and impact tissues via effector cytokines such as IL‐17A and IL‐22. Physiological roles for IL‐17 have not been demonstrated in the mammalian brain, although IL‐17 has been proposed as a neuromodulator in Caenorhabditis elegans 115. However, recent work suggests that IL‐17 is a critical mediator of the neurodevelopmental abnormalities associated with maternal immune activation by the viral mimic poly(I:C). The authors found that IL‐17 produced by maternal Th17 cells caused cortical malformations reminiscent of human autism cases 116, as well as social behavioral defects mediated by the maternal microbiome, which promotes Th17 cell differentiation 117, 118 . Other studies implicate the broadly proinflammatory cytokine IL‐6 in the behavioral effects of maternal immune activation (MIA) 119, 120 via IL‐6 derived from the placenta 119. Resolving the different contributions of these cytokines in brain development will be essential areas of study. Pregnancy, which is a powerful modulator of immune responses, will be an important variable to be considered. However, we would suggest that pursuing signaling pathways from well‐defined cellular sources and with clear targets (e.g. IL‐17) are likely to be more straightforward. Studying signals that are comparatively pleiotropic and can be produced by and impact multiple cell types (e.g. IL‐6) will require carefully targeted genetic approaches.
Regulatory immune responses and their connection to stress
In all the data described above, the role of the regulatory arm of the immune system in psychiatric disease remains understudied. Regulatory T cells (Treg) are present in most organs, including in the brain meninges. They play key roles in preventing autoimmunity and dampening excessive immune responses via effector cytokines (e.g. IL‐10, IL‐35), acting as a sponge for the proinflammatory cytokine IL‐2 and inhibiting dendritic cell activation. An immense clinical literature, briefly surveyed in the ‘The immune system and psychiatric diagnoses: what human studies tell us’ section, suggests that many psychiatric disorders can be associated with markers of increased inflammation. To what extent might stress generically suppress immune responses, perhaps via dysfunction of meningeal or peripheral Tregs? Do Tregs secrete cytokines that could impact the function of microglia or other cells? Exploring the sensitivity or function of Tregs in meninges, gut or circulation could provide mechanistic clues and markers that exceed the sensitivity of measuring transient bursts of short‐lived inflammatory cytokines in peripheral blood. Alternately, could some forms of stress or trauma generate a unique subtype of immune response (‘Type X’) distinct from established categories? Answering this question requires further study, but is greatly facilitated by our growing ability to understand complex cellular and network immune responses via ‘omic’ approaches.
The gut–brain axis in modulation of immune tone
A discussion of immune–brain interactions is incomplete without considering the direct neuronal links between brain and periphery. The peripheral nervous system includes sensory, motor and autonomic nerves. It is outside the blood–brain barrier and thus a key potential modulator of neuroimmunity. The sympathetic and parasympathetic nervous systems control most autonomic functions, including digestion and secretions. Of these peripheral connections, the gut has emerged as a key link between peripheral and central immunity. Second only to the brain in its number of neurons, the gut also has an active immune system. Recent data suggest that type II immune responses in the gut are responsive to signals from the autonomic nervous system. Norepinephrine (the major mediator of sympathetic ‘fight or flight’ responses) can suppress ILC2 function and type II immunity. Conversely, the neuropeptide neuromedin U, which is produced by cholinergic parasympathetic neurons, can promote type II remodeling responses 121, 122. Acute stress is a state of sympathetic activation and hyperarousal in response to immediate threat. It is intriguing to speculate that suppressing energetically costly remodeling functions during acute stress could be beneficial in the short term, but detrimental to learning and cognition in the long term. Investigating this hypothesis could reveal immune links between acute stress and the development of chronic stress disorders such as PTSD.
The gut microbiome is an increasingly recognized modulator of peripheral immune responses and, by extension, the brain 123. Gut microbiota may modulate autism symptoms 124, and their relevance to psychiatric diseases can cytokine‐dependent 118 or act directly through bacterial metabolites. For example, specific spore‐forming bacteria in both mouse and human gut directly promote serotonin biosynthesis by host enterochromaffin cells 125. The human commensal Bacteriodes is protective against behavioral deficits seen in the MIA model 124. Finally, bacterial species that reduce gamma‐glutamyl amino acids may be responsible for some of the benefits of the ketogenic diet, widely used in intractable epilepsy and, despite weaker evidence, recommended by some for treatment of autism 126. While much remains to be determined about direct versus indirect effects of the microbiome on the brain, defining the underlying mechanisms represents an exciting area for further study with many potential therapeutic implications.
What are the physical and anatomical links between gut and brain? One candidate is the parasympathetic vagus nerve, which consists of 80% afferent fibers and is one of the major pathways transmitting signals from gut to brain 127. Data from vagal nerve transection studies indicate that IL‐1‐mediated sickness behavior requires intact connections between the parasympathetic nervous system and brain (reviewed in 128, 129). Clearly, the vagus is poised to sense signals from gut immune cells and the microbiota. Vagal stimulation has demonstrated efficacy in the management of treatment‐resistant depression 130. This raises interesting questions as to what extent this could involve modulation of immune responses or its impact on the brain.
Breaching of boundaries
Another variation on brain immune responses occurs when normal boundaries between brain and periphery are breached. In homeostasis, microglia constitute nearly all brain resident immune cells at rest and are not replaced from the blood. However, circulating myeloid cells commonly enter the brain in inflammatory diseases such as multiple sclerosis (MS), malignancy and stroke. Animal models show that infiltrating macrophages have clear effects on disease pathogenesis. They are known to exacerbate clinical symptoms in an MS model, and can even cause disease when infiltrating cells are deficient for Tgfbr2 50, 131. In most cases infiltrating cells are associated with structural brain damage and disappear after disease resolution, probably by apoptosis 131. Recent evidence, however, points to the unexpected long‐term residence of brain‐engrafted peripheral myeloid cells after pediatric bone marrow transplantation, in schizophrenia and in AD 42, 132, 133, 134. Despite this, bone marrow‐derived macrophages cannot adopt a true microglial identity. Instead, they show transcriptional overlap with microglia from models of neuroinflammation and neurodegeneration, including abnormal expression of genes associated with phagocytosis and antigen presentation. Whether they exert meaningful effects on psychiatric disease pathogenesis requires further study, but represents an exciting potential treatment target.
Conclusions: linking bench to bedside in future work
Work in recent years makes it clear that defining the complexity of neuroimmune cross‐talk has clear implications for understanding both homeostasis and disease. Ongoing work should continue to focus on defining basic mechanisms of immune function in homeostasis, in valid animal models of neuropsychiatric disease or after defined perturbations that are known risk factors for psychiatric diagnoses. Understanding this complex cellular cross‐talk will benefit greatly from high‐dimensional data that define more effectively the immune composition in the meninges, brain parenchyma and intracerebral vasculature and its response to challenges.
As the basic science moves towards translation, new challenges will arise. First, what should we be measuring in clinical populations? Measurements of individual pan‐inflammatory cytokines in peripheral blood have so far not led to reliable biomarkers. Much more needs to be understood about how immune processes happening centrally manifest in peripheral tissues. Might the chromatin profile or other definable features of circulating immune cells provide clues as to the ‘immune history’ of the body or brain? Could we one day biopsy the cervical lymph nodes draining meningeal lymphatics as a proxy for meningeal immunity? Could imaging biomarkers such as translocator protein (TSPO) 135, but much more targeted, be used to visualize immune processes within the human brain with molecular precision? All these are possibilities that close collaboration between basic scientists and clinicians could one day make feasible.
Secondly, and most importantly, how can we move towards new treatment targets for these devastating illnesses? In addition to advances in basic science, some clues could arise from defining the immune impact of current clinical therapies. With future advances in basic mechanisms, it is possible that immune modulation could be used to promote brain repair and cure the long‐term effects of trauma. It is conceivable that cellular therapies and antibody‐based treatments such as those being developed for cancer and rheumatoid arthritis could one day be the standard of care for mental illnesses. It is probable that our current symptom‐based diagnostic categories will be completely rewritten as the mechanistic heterogeneity of these diseases is revealed. Defining the links between immune function and the brain will yield novel and unexpected avenues to diagnosis and treatment.
Acknowledgements
Sincere thanks to Drs Mariko Bennett and Ari Molofsky for helpful discussions, feedback and comments on the manuscript. The authors acknowledge funding from NIMH (K08 MH112120‐01 to F. C. B. and DP2MH116507 and R01MH119349 to A. V. M.), the Pew Charitable Trusts, the Brain and Behavior Research Foundation and the Burroughs Wellcome fund. We regret that many excellent references were not cited due to space constraints.
OTHER ARTICLES PUBLISHED IN THIS REVIEW SERIES
Neuroimmune interactions: how the nervous and immune systems influence each other. Clinical and Experimental Immunology 2019, 197: 276–277.
Neuroimmunology – the past, present and future. Clinical and Experimental Immunology 2019, 197: 278–293.
Depressive symptoms in inflammatory bowel disease: an extraintestinal manifestation of inflammation? Clinical and Experimental Immunology 2019, 197: 308–318.
From early adversities to immune activation in psychiatric disorders: the role of the sympathetic nervous system. Clinical and Experimental Immunology 2019, 197: 319–328.
References
- 1. Organisation Mondiale de la Santé (OMS). The global burden of disease 2004, update . World Health Organ 2004; 146 10.1038/npp.2011.85. [DOI] [Google Scholar]
- 2. Hodes GE, Kana V, Menard C, Merad M, Russo SJ. Neuroimmune mechanisms of depression. Nat Neurosci 2015; 18:1386–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Miller AH, Raison CL. The role of inflammation in depression: from evolutionary imperative to modern treatment target. Nat Rev Immunol 2016; 16:22–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Estes ML, McAllister AK. Immune mediators in the brain and peripheral tissues in autism spectrum disorder. Nat Rev Neurosci 2015; 16:469–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Onore C, Careaga M, Ashwood P. The role of immune dysfunction in the pathophysiology of autism. Brain Behav Immun 2012; 26:383–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Hou R, Baldwin DS. A neuroimmunological perspective on anxiety disorders. Hum Psychopharmacol 2011. 10.1002/hup.1259. [DOI] [PubMed] [Google Scholar]
- 7. Insel TR. Rethinking schizophrenia. Nature 2010; 468:187–93. [DOI] [PubMed] [Google Scholar]
- 8. Feinberg I. Neurodevelopmental model of schizophrenia. Biol Psychiatry 1992; 32:212–3. [DOI] [PubMed] [Google Scholar]
- 9. Vita A, De Peri L, Deste G, Sacchetti E. Progressive loss of cortical gray matter in schizophrenia: a meta‐analysis and meta‐regression of longitudinal MRI studies. Transl Psychiatry 2012; 2:e190–e190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Benros ME, Nielsen PR, Nordentoft M, Eaton WW, Dalton SO, Mortensen PB. Autoimmune diseases and severe infections as risk factors for schizophrenia: a 30‐year population‐based register study. Am J Psychiatry 2011; 168:1303–10. [DOI] [PubMed] [Google Scholar]
- 11. Miller BJ, Buckley P, Seabolt W, Mellor A, Kirkpatrick B. Meta‐analysis of cytokine alterations in schizophrenia: clinical status and antipsychotic effects. Biol Psychiatry 2011; 70:663–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Potvin S, Stip E, Sepehry AA, Gendron A, Bah R, Kouassi E. Inflammatory cytokine alterations in schizophrenia: a systematic quantitative review. Biol Psychiatry 2008; 63:801–8. [DOI] [PubMed] [Google Scholar]
- 13. Schizophrenia Working Group of the Psychiatric Genomics Consortium . Biological insights from 108 schizophrenia‐associated genetic loci. Nature 2014; 511:421–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Elmer BM, McAllister AK. Major histocompatibility complex class I proteins in brain development and plasticity. Trends Neurosci 2012; 35:660–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Sekar A, Bialas AR, de Rivera H et al Schizophrenia risk from complex variation of complement component 4. Nature 2016; 530:177–183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Feinberg I.. Adolescence and mental illness. Science (New York, NY) 1987; 236:507–8. [PubMed] [Google Scholar]
- 17. Jeremy Willsey A, State MW. Autism spectrum disorders: from genes to neurobiology. Curr Opin Neurobiol 2015;92–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. al‐Haddad BJS, Jacobsson B, Chabra S et al Long‐term risk of neuropsychiatric disease after exposure to infection in utero. JAMA Psychiatry 2019. 10.1001/jamapsychiatry.2019.0029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Atladóttir HOÓ, Thorsen P, Østergaard L et al Maternal infection requiring hospitalization during pregnancy and autism spectrum disorders. J Autism Dev Disord 2010; 40:1423–30. [DOI] [PubMed] [Google Scholar]
- 20. Masi A, Quintana DS, Glozier N, Lloyd AR, Hickie IB, Guastella AJ. Cytokine aberrations in autism spectrum disorder: a systematic review and meta‐analysis. Mol Psychiatry 2015; 20:440–6. [DOI] [PubMed] [Google Scholar]
- 21. Voineagu I, Wang X, Johnston P et al Transcriptomic analysis of autistic brain reveals convergent molecular pathology. Nature 2011; 474:380–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Knuesel I, Chicha L, Britschgi M et al Maternal immune activation and abnormal brain development across CNS disorders. Nat Rev Neurol 2014; 10:643–60. [DOI] [PubMed] [Google Scholar]
- 23. Hauser P, Khosla J, Aurora H et al A prospective study of the incidence and open‐label treatment of interferon‐induced major depressive disorder in patients with hepatitis C. Mol Psychiatry 2002; 7:942–7. [DOI] [PubMed] [Google Scholar]
- 24. Musselman DL, Lawson DH, Gumnick JF et al Paroxetine for the prevention of depression induced by high‐dose interferon alfa. N Engl J Med 2001; 344:961–6. [DOI] [PubMed] [Google Scholar]
- 25. Dieperink E, Ho SB, Thuras P, Willenbring ML. A prospective study of neuropsychiatric symptoms associated with interferon‐alpha‐2b and ribavirin therapy for patients with chronic hepatitis C. Psychosomatics 2003; 44:104–12. [DOI] [PubMed] [Google Scholar]
- 26. Zorrilla EP, Luborsky L, McKay JR et al The relationship of depression and stressors to immunological assays: a meta‐analytic review. Brain Behav Immun 2001; 15:199–226. [DOI] [PubMed] [Google Scholar]
- 27. O'Donovan A, Slavich GM, Epel ES, Neylan TC. Exaggerated neurobiological sensitivity to threat as a mechanism linking anxiety with increased risk for diseases of aging. Neurosci Biobehav Rev 2013;96–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Passos IC, Vasconcelos‐Moreno MP, Costa LG et al Inflammatory markers in post‐traumatic stress disorder: a systematic review, meta‐analysis, and meta‐regression. Lancet Psychiatry 2015; 2:1002–12. [DOI] [PubMed] [Google Scholar]
- 29. Fazel S, Wolf A, Pillas D, Lichtenstein P, Långström N. Suicide, fatal injuries, and other causes of premature mortality in patients with traumatic brain injury. JAMA Psychiatry 2014; 71:326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. AbdelMalik P, Husted J, Chow EWC, Bassett AS. Childhood head injury and expression of schizophrenia in multiply affected families. Arch Gen Psychiatry 2003; 60:231–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Molloy C, Conroy RM, Cotter DR, Cannon M. Is traumatic brain injury a risk factor for schizophrenia? A meta‐analysis of case‐controlled population‐based studies. Schizophr Bull 2011; 37:1104–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Gardner RC, Burke JF, Nettiksimmons J, Kaup A, Barnes DE, Yaffe K. Dementia risk after traumatic brain injury vs nonbrain trauma: the role of age and severity. JAMA Neurol 2014; 71:1490–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Murphy JB, Sturm E. Conditions determining the transplantability of tissues in the brain. J Exp Med 1923; 38:183–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Daneman R, Prat A. The blood–brain barrier. Cold Spring Harb Perspect Biol 2015; 7:a020412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Zhang SL, Yue Z, Arnold DM, Artiushin G, Sehgal A. A circadian clock in the blood–brain barrier regulates xenobiotic efflux. Cell 2018; 173:130–139.e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Korin B, Ben‐Shaanan TL, Schiller M et al High‐dimensional, single‐cell characterization of the brain's immune compartment. Nat Neurosci 2017; 20:1300–9. [DOI] [PubMed] [Google Scholar]
- 37. Chovatiya R, Medzhitov R. Stress, inflammation, and defense of homeostasis. Mol Cell 2014; 54:281–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Lavin Y, Winter D, Blecher‐Gonen R et al Tissue‐resident macrophage enhancer landscapes are shaped by the local microenvironment. Cell 2014; 159:1312–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Gosselin D, Skola D, Coufal NG et al An environment‐dependent transcriptional network specifies human microglia identity. Science 2017; 356:eaal3222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Nimmerjahn A, Kirchhoff F, Helmchen F. Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo – resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science 2005; 308:1314–9. [DOI] [PubMed] [Google Scholar]
- 41. Davalos D, Grutzendler J, Yang G et al ATP mediates rapid microglial response to local brain injury in vivo. Nat Neurosci 2005; 8:752–8. [DOI] [PubMed] [Google Scholar]
- 42. Bennett FC, Bennett ML, Yaqoob F et al A combination of ontogeny and CNS environment establishes microglial identity. Neuron 2018; 98:1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Réu P, Khosravi A, Bernard S et al The lifespan and turnover of microglia in the human brain. Cell Rep 2017; 20:779–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Li Q, Barres BA. Microglia and macrophages in brain homeostasis and disease. Nat Rev Immunol 2018; 18:225–42. [DOI] [PubMed] [Google Scholar]
- 45. Gomez Perdiguero E, Klapproth K, Schulz C et al Tissue‐resident macrophages originate from yolk‐sac‐derived erythro‐myeloid progenitors. Nature 2015; 581:547–551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Ginhoux F, Greter M, Leboeuf M et al Fate mapping analysis reveals that adult microglia derive from primitive macrophages. Science 2010; 330:841–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Gosselin D, Link VM, Romanoski CE et al Environment drives selection and function of enhancers controlling tissue‐specific macrophage identities. Cell 2014; 159:1327–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Gosselin D, Skola D, Coufal NG et al An environment‐dependent transcriptional network specifies human microglia identity. Science (New York, NY) 2017; 1617:eaal3222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Butovsky O, Jedrychowski MP, Moore CS et al Identification of a unique TGF‐β‐dependent molecular and functional signature in microglia. Nat Neurosci 2014; 17:131–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Lund H, Pieber M, Parsa R et al Fatal demyelinating disease is induced by monocyte‐derived macrophages in the absence of TGF‐β signaling. Nat Immunol 2018; 19:1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Matcovitch‐Natan O, Winter DR, Giladi A et al Microglia development follows a stepwise program to regulate brain homeostasis. Science 2016; 353:aad8670. [DOI] [PubMed] [Google Scholar]
- 52. Huttenlocher PR, Dabholkar AS. Regional differences in synaptogenesis in human cerebral cortex. J Comp Neurol 1997; 387:167–78. [DOI] [PubMed] [Google Scholar]
- 53. Kang HJ, Voleti B, Hajszan T et al Decreased expression of synapse‐related genes and loss of synapses in major depressive disorder. Nat Med 2012; 18:1413–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Vainchtein ID, Chin G, Cho FS et al Astrocyte‐derived interleukin‐33 promotes microglial synapse engulfment and neural circuit development. Science 2018; 359:1269–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Schafer DP, Lehrman EK, Kautzman AG et al Microglia sculpt postnatal neural circuits in an activity and complement‐dependent manner. Neuron 2012; 74:691–705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Paolicelli RC, Bolasco G, Pagani F et al Synaptic pruning by microglia is necessary for normal brain development. Science (New York, NY) 2011; 333:1456–58. [DOI] [PubMed] [Google Scholar]
- 57. Schafer DP, Heller CT, Gunner G et al Microglia contribute to circuit defects in Mecp2 null mice independent of microglia‐specific loss of Mecp2 expression. eLife 2016; 5:185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Schafer DP, Lehrman EK, Kautzman AG et al Microglia sculpt postnatal neural circuits in an activity and complement‐dependent manner. Neuron 2012; 74:691–705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Vainchtein ID, Chin G, Cho FS et al Astrocyte‐derived interleukin‐33 promotes microglial synapse engulfment and neural circuit development. Science (New York, NY) 2018; 359:1269–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Paolicelli RC, Bolasco G, Pagani F et al Synaptic pruning by microglia is necessary for normal brain development. Science 2011; 333:1456–58. [DOI] [PubMed] [Google Scholar]
- 61. Weinhard L, di Bartolomei G, Bolasco G et al Microglia remodel synapses by presynaptic trogocytosis and spine head filopodia induction. Nat Commun 2018; 9:1228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Shi Q, Colodner KJ, Matousek SB et al Complement C3‐deficient mice fail to display age‐related hippocampal decline. J Neurosci 2015; 35:13029–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Hong S, Beja‐Glasser VF, Nfonoyim BM. Complement and microglia mediate early synapse loss in Alzheimer mouse models. Science 2016; 352:712–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Vasek MJ, Garber C, Dorsey D et al A complement‐microglial axis drives synapse loss during virus‐induced memory impairment. Nature 2016; 534:538–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Sekar A, Bialas AR, de Rivera H et al Schizophrenia risk from complex variation of complement component 4. Nature 2016; 530:177–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Weinhard L, di Bartolomei G, Bolasco G et al Microglia remodel synapses by presynaptic trogocytosis and spine head filopodia induction. Nat Commun 2018; 9:1228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Parkhurst CN, Yang G, Ninan I et al Microglia promote learning‐dependent synapse formation through brain‐derived neurotrophic factor. Cell 2013; 155:1596–609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Hagemeyer N, Hanft K‐M, Akriditou M‐A et al Microglia contribute to normal myelinogenesis and to oligodendrocyte progenitor maintenance during adulthood. Acta Neuropathol 2017; 134:441–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Wlodarczyk A, Holtman IR, Krueger M et al A novel microglial subset plays a key role in myelinogenesis in developing brain. EMBO J 2017; 36:3292–308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Li Q, Cheng Z, Zhou L et al Developmental heterogeneity of microglia and brain myeloid cells revealed by deep single‐cell RNA sequencing. Neuron 2019; 101:207–223.e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Hammond TR, Dufort C, Dissing‐Olesen L et al Single‐cell RNA sequencing of microglia throughout the mouse lifespan and in the injured brain reveals complex cell‐state changes. Immunity 2019; 50:253–71.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Ji R, Tian S, Lu HJ et al TAM receptors affect adult brain neurogenesis by negative regulation of microglial cell activation. J Immunol 2013; 191:6165–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Fourgeaud L, Través PG, Tufail Y et al TAM receptors regulate multiple features of microglial physiology. Nature 2016; 532:240–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Chen S‐K, Tvrdik P, Peden E et al Hematopoietic origin of pathological grooming in Hoxb8 mutant mice. Cell 2010; 141:775–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Krabbe G, Minami SS, Etchegaray JI et al Microglial NFκB‐TNFα hyperactivation induces obsessive‐compulsive behavior in mouse models of progranulin‐deficient frontotemporal dementia. Proc Natl Acad Sci USA 2017; 114:5029–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Zhan Y, Paolicelli RC, Sforazzini F et al Deficient neuron‐microglia signaling results in impaired functional brain connectivity and social behavior. Nat Neurosci 2014; 17:400–6. [DOI] [PubMed] [Google Scholar]
- 77. VanRyzin JW, Marquardt AE, Argue KJ et al Microglial phagocytosis of newborn cells is induced by endocannabinoids and sculpts sex differences in juvenile rat social play. Neuron 2019; 102:435–49.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Pekny M, Pekna M. Astrocyte reactivity and reactive astrogliosis: costs and benefits. Physiol Rev 2014; 94:1077–98. [DOI] [PubMed] [Google Scholar]
- 79. Chung WS, Clarke LE, Wang GX et al Astrocytes mediate synapse elimination through MEGF10 and MERTK pathways. Nature 2013; 504:394–400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Liddelow SA, Guttenplan KA, Clarke LE et al Neurotoxic reactive astrocytes are induced by activated microglia. Nature 2017; 541:481–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Garber C, Vasek MJ, Vollmer LL, Sun T, Jiang X, Klein RS. Astrocytes decrease adult neurogenesis during virus‐induced memory dysfunction via IL‐1. Nat Immunol 2018; 19:151–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Chen Z, Palmer TD. Differential roles of TNFR1 and TNFR2 signaling in adult hippocampal neurogenesis. Brain Behav Immun 2013; 30:45–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Iosif RE, Ekdahl CT, Ahlenius H et al Development/plasticity/repair tumor necrosis factor receptor 1 is a negative regulator of progenitor proliferation in adult hippocampal neurogenesis. J Neurosci 2006; 26:9703–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Dellarole A, Morton P, Brambilla R et al Neuropathic pain‐induced depressive‐like behavior and hippocampal neurogenesis and plasticity are dependent on TNFR1 signaling. Brain Behav Immun 2014; 41:65–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Golan H, Levav T, Mendelsohn A, Huleihel M. Involvement of tumor necrosis factor alpha in hippocampal development and function. Cereb Cortex 2004; 14:97–105. [DOI] [PubMed] [Google Scholar]
- 86. Liu B, Zupan B, Laird E et al Maternal hematopoietic TNF, via milk chemokines, programs hippocampal development and memory. Nat Neurosci 2014; 17:97–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Cheadle L, Tzeng CP, Kalish BT et al Visual experience‐dependent expression of Fn14 is required for retinogeniculate refinement. Neuron 2018; 99:525–539.e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Stellwagen D, Malenka RC. Synaptic scaling mediated by glial TNF‐α. Nature 2006; 440:1054–9. [DOI] [PubMed] [Google Scholar]
- 89. Eric C. Beattie RCM, Stellwagen D, Morishita W et al Control of synaptic strength by glial TNFα. Science 2002; 295:2282–85. [DOI] [PubMed] [Google Scholar]
- 90. Stellwagen D, Beattie EC, Seo JY, Malenka RC. Cellular/molecular differential regulation of AMPA receptor and GABA receptor trafficking by tumor necrosis factor. J Neurosci 2005; 25:3219–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Baune BT, Wiede F, Braun A, Golledge J, Arolt V, Koerner H. Cognitive dysfunction in mice deficient for TNF‐ and its receptors. Am J Med Genet Part B Neuropsychiatr Genet 2008; 147B:1056–64. [DOI] [PubMed] [Google Scholar]
- 92. Huh GS, Boulanger LM, Du H, Riquelme PA, Brotz TM, Shatz CJ. Functional requirement for class I MHC in CNS development and plasticity. Science 2000; 290:2155–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Syken J, Grandpre T, Kanold PO, Shatz CJ. PirB restricts ocular‐dominance plasticity in visual cortex. Science 2006; 313:1795–1800. [DOI] [PubMed] [Google Scholar]
- 94. Gadani SP, Walsh JT, Smirnov I, Zheng J, Kipnis J. The glia‐derived alarmin IL‐33 orchestrates the immune response and promotes recovery following CNS injury. Neuron 2015; 85:703–9. [DOI] [PubMed] [Google Scholar]
- 95. Luo Y, Zhou Y, Xiao W et al Interleukin‐33 ameliorates ischemic brain injury in experimental stroke through promoting Th2 response and suppressing Th17 response. Brain Res 2015; 1597:86–94. [DOI] [PubMed] [Google Scholar]
- 96. Zhang SR, Piepke M, Chu HX et al IL‐33 modulates inflammatory brain injury but exacerbates systemic immunosuppression following ischemic stroke. JCI Insight 2018; 3: e121560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Fu AKY, Hung K‐W, Yuen MYF et al IL‐33 ameliorates Alzheimer's disease‐like pathology and cognitive decline. Proc Natl Acad Sci USA 2016; 113:E2705–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Kim T‐K, Kim J‐E, Choi J et al Local interleukin‐18 system in the basolateral amygdala regulates susceptibility to chronic stress. Mol Neurobiol 2016; 29:201–12. [DOI] [PubMed] [Google Scholar]
- 99. Wang A, Huen SC, Luan HH et al Opposing effects of fasting metabolism on tissue tolerance in bacterial and viral inflammation. Cell 2016; 166:1512–25.e12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Liu X, Yamashita T, Chen Q et al Interleukin 1 type 1 receptor restore: a genetic mouse model for studying interleukin 1 receptor‐mediated effects in specific cell types. J Neurosci 2015; 35:2860–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Liu X, Nemeth DP, McKim DB, Popovich PG, Bilbo SD, Quan N. Cell‐type‐specific interleukin 1 receptor 1 signaling in the brain regulates distinct neuroimmune activities. Immunity 2019; 50:317–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Annunziato F, Romagnani C, Romagnani S. The 3 major types of innate and adaptive cell‐mediated effector immunity. J Allergy Clin Immunol 2015; 135:626–35. [DOI] [PubMed] [Google Scholar]
- 103. Pulendran B, Artis D. New Paradigms in Type 2 Immunity. n.d. [DOI] [PMC free article] [PubMed]
- 104. Derecki NC, Cardani AN, Yang CH et al Regulation of learning and memory by meningeal immunity: a key role for IL‐4. J Exp Med 2010; 207:1067–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Cushman J, Lo J, Huang Z, Wasserfall C, Petitto JM. Neurobehavioral changes resulting from recombinase activation gene 1 deletion. Clin Diagn Lab Immunol 2003; 10:13–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Kipnis J, Cohen H, Cardon M, Ziv Y, Schwartz M. T cell deficiency leads to cognitive dysfunction: Implications for therapeutic vaccination for schizophrenia and other psychiatric conditions. Proc Natl Acad Sci 2004; 101:8180–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Aspelund A, Antila S, Proulx ST et al A dural lymphatic vascular system that drains brain interstitial fluid and macromolecules. J Exp Med 2015; 212:991–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Absinta M, Ha S‐K, Nair G et al Human and nonhuman primate meninges harbor lymphatic vessels that can be visualized noninvasively by MRI. eLife 2017; 6:e29738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Louveau A, Smirnov I, Keyes TJ et al Structural and functional features of central nervous system lymphatic vessels. Nature 2015; 523:337–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Filiano AJ, Xu Y, Tustison NJ et al Unexpected role of interferon‐γ in regulating neuronal connectivity and social behaviour. Nature 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Monteiro S, Ferreira FM, Pinto V et al Absence of IFNγ promotes hippocampal plasticity and enhances cognitive performance. Transl Psychiatry 2016; 6:e707–e707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Cavanaugh SE, Holmgren AM, Rall GF. Homeostatic interferon expression in neurons is sufficient for early control of viral infection. J Neuroimmunology 2015; 279:11–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Gadani SP, Smirnov I, Smith AT, Overall CC, Kipnis J. Characterization of meningeal type 2 innate lymphocytes and their response to CNS injury. J Exp Med 2017; 214:285–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Pomeshchik Y, Kidin I, Korhonen P et al Interleukin‐33 treatment reduces secondary injury and improves functional recovery after contusion spinal cord injury. Brain Behav Immun 2015; 44:68–81. [DOI] [PubMed] [Google Scholar]
- 115. Chen C, Itakura E, Nelson GM et al IL‐17 is a neuromodulator of Caenorhabditis elegans sensory responses. Nature 2017; 542:43–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Stoner R, Chow ML, Boyle MP et al Patches of disorganization in the neocortex of children with autism. N Engl J Med 2014; 370:1209–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Choi GB, Yim YS, Wong H, Kim S, Kim H, Kim SV, Hoeffer CA, Littman DR, Huh JR. The maternal interleukin‐17a pathway in mice promotes autism‐like phenotypes in offspring. Science 2016; 351:933–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Kim S, Kim H, Yim YS et al Maternal gut bacteria promote neurodevelopmental abnormalities in mouse offspring. Nature 2017; 549:528–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Wu W‐L, Hsiao EY, Yan Z, Mazmanian SK, Patterson PH. The placental interleukin‐6 signaling controls fetal brain development and behavior. Brain Behav Immun 2017; 62:11–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Wei H, Chadman KK, McCloskey DP et al Brain IL‐6 elevation causes neuronal circuitry imbalances and mediates autism‐like behaviors. Biochim Biophys Acta Mol Basis Dis 1822; 2012:831–42. [DOI] [PubMed] [Google Scholar]
- 121. Klose CSN, Mahlakõiv T, Moeller JB et al The neuropeptide neuromedin U stimulates innate lymphoid cells and type 2 inflammation. Nature 2017; 549:282–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Moriyama S, Brestoff JR, Flamar A‐L et al β2‐adrenergic receptor‐mediated negative regulation of group 2 innate lymphoid cell responses. Science 2018; 359:1056–61. [DOI] [PubMed] [Google Scholar]
- 123. Pronovost GN, Hsiao EY. Perinatal Interactions between the microbiome, immunity, and neurodevelopment. Immunity 2019; 50:18–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Hsiao EY, McBride SW, Hsien S et al Microbiota modulate behavioral and physiological abnormalities associated with neurodevelopmental disorders. Cell 2013; 155:1451–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Yano JM, Yu K, Mazmanian SK et al Indigenous bacteria from the gut microbiota regulate host serotonin biosynthesis. Cell 2015; 161:264–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Olson CA, Vuong HE, Yano JM, Liang QY, Nusbaum DJ, Hsiao EY. The gut microbiota mediates the anti‐seizure effects of the ketogenic diet. Cell 2018; 173:1728–41.e13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Bonaz B, Bazin T, Pellissier S. The vagus nerve at the interface of the microbiota–gut–brain axis. Front Neurosci 2018; 12:49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Kelley KW, Bluthé R‐M, Dantzer R et al Cytokine‐induced sickness behavior. Brain Behav Immun 2003; 17:112–8. [DOI] [PubMed] [Google Scholar]
- 129. Dantzer R. Cytokine, sickness behavior, and depression. Immunol Allergy Clin North Am 2009; 29:247–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Aaronson ST, Sears P, Ruvuna F et al A 5‐year observational study of patients with treatment‐resistant depression treated with vagus nerve stimulation or treatment as usual: comparison of response, remission, and suicidality. Am J Psychiatry 2017; 174:640–8. [DOI] [PubMed] [Google Scholar]
- 131. Ajami B, Bennett JL, Krieger C, McNagny KM, Rossi FMV. Infiltrating monocytes trigger EAE progression, but do not contribute to the resident microglia pool. Nat Neurosci 2011; 14:1142–9. [DOI] [PubMed] [Google Scholar]
- 132. Shemer A, Grozovski J, Tay TL et al Engrafted parenchymal brain macrophages differ from microglia in transcriptome, chromatin landscape and response to challenge. Nat Commun 2018; 9:5206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Cronk JC, Filiano AJ, Louveau A et al Peripherally derived macrophages can engraft the brain independent of irradiation and maintain an identity distinct from microglia. J Exp Med 2018; 215:1627–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Cai HQ, Catts VS, Webster MJ et al Increased macrophages and changed brain endothelial cell gene expression in the frontal cortex of people with schizophrenia displaying inflammation. Mol Psychiatry 2018. 10.1038/s41380-018-0235-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135. Narayanaswami V, Dahl K, Bernard‐Gauthier V, Josephson L, Cumming P, Vasdev N. Emerging PET radiotracers and targets for imaging of neuroinflammation in neurodegenerative diseases: outlook beyond TSPO. Mol Imaging 2018; 17:153601211879231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Orihara K, Nakae S, Pawankar R, Saito H. Role of regulatory and proinflammatory T‐cell populations in allergic diseases. World Allergy Organ J 2008; 1:9–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137. Kotas ME, Locksley RM. Why innate lymphoid cells? Immunity 2018; 48:1081–90. [DOI] [PMC free article] [PubMed] [Google Scholar]