

Abstract
Tubulin genes encode a series of homologous proteins used to construct microtubules which are essential for multiple cellular processes. Neural development is particularly reliant on functional microtubule structures. Tubulin genes comprise a large family of genes with very high sequence similarity between multiple family members. Human genetics has demonstrated that a large spectrum of cortical malformations are associated with de novo heterozygous mutations in tubulin genes. However, the absolute requirement for many of these genes in development and disease has not been previously tested in genetic loss of function models. Here we directly test the requirement for Tuba1a, Tubb2a and Tubb2b in the mouse by deleting each gene individually using CRISPR-Cas9 genome editing. We show that loss of Tubb2a or Tubb2b does not impair survival but does lead to relatively mild cortical malformation phenotypes. In contrast, loss of Tuba1a is perinatal lethal and leads to significant forebrain dysmorphology. We also present a novel mouse ENU allele of Tuba1a with phenotypes similar to the null allele. This demonstrates the requirements for each of the tubulin genes and levels of functional redundancy are quite different throughout the gene family. The ability of the mouse to survive in the absence of some tubulin genes known to cause disease in humans suggests future intervention strategies for these devastating tubulinopathy diseases.
Author summary
Tubulin proteins are assembled into microtubules to provide essential cellular cytoskeletal elements. Microtubules are especially crucial for neuronal development. Multiple studies demonstrate that human malformations of cortical development are caused by genetic variants in several tubulin genes. Interestingly, most of these developmental phenotypes are the result of de novo, heterozygous variants likely to act as dominant negatives interfering with normal tubulin function(s). The roles of individual tubulin genes and the mechanism(s) leading to these malformations remain unclear. Genome sequencing efforts revealed high sequence similarity between many tubulin genes, raising the possibility of functional compensation. The requirements for many of these tubulin genes have not been previously addressed in loss of function experiments. We have generated novel deletions of several tubulin genes known to cause human disease to assess if they are required for brain development, or if the human variants act to alter function of the tubulin proteins leading to the pathogenesis. Surprisingly, our results show that some individual tubulin genes are absolutely required for survival while others are not and have much more benign cortical malformations.
Introduction
Tubulin proteins are fundamental building blocks of the cell and assemble into dynamic microtubules. Microtubules are especially crucial for cortical development where they are used in multiple cellular contexts such as the mitotic spindle, axons, dendrites, and cilia formation [1]. Mutations in tubulin genes are now known to cause multiple human cortical malformations including lissencephaly, polymicrogyria, microcephaly, dysmorphic basal ganglia, and congenital fibrosis of extraocular muscles [2]. These are collectively discussed as “tubulinopathies.” Many variants leading to malformations of cortical development have now been identified in TUBULIN, ALPHA-1A (TUBA1A) [2–18]; TUBULIN, BETA-2A (TUBB2A) [19–22]; TUBB2B [4, 5, 15, 23–29]; TUBB3 [2, 15, 30, 31]; TUBB4A [32]; and TUBB/TUBB5 [33]. In fact, for TUBA1A and TUBB2B alone, there are now 71 identified variants in cortical malformation patients [7–29, 34]. Mutations of TUBA1A and TUBB2B account for ~5% and ~1.2% of lissencephaly and related malformations of cortical development, respectively [34]. Both, but especially TUBA1A, are associated with a wide spectrum of phenotypic severity [34]. With the exception of one TUBA1A variant inherited from a mosaic parent [6] and one inherited TUBB2B variant [24], all of these variants are heterozygous, de novo changes in the identified proband. TUBB3 variants have been found to segregate as dominant traits in large pedigrees [31].
Previous work has shown that both α- and β- tubulins can be separated into different classes (isotypes) and some general characteristics about their spatiotemporal expression domains have been established [35, 36]. However, the completed genome sequences of many species, including mouse and human, have revealed the presence of more tubulin proteins than those indicated by the initial six classes. The requirements and roles in cortical development for some of the individual tubulin genes are still unknown. One possible mechanism of disease in the tubulinopathies is the production of altered tubulin monomers from the variant alleles which alter normal tubulin function(s) in any number of cellular contexts. Experiments overexpressing these pathogenic variants and/or loss of function models in mouse and cells have indeed shown defects in microtubule polymerization and heterodimer assembly, neurite extension, growth cone dynamics, neuronal migration, vesicular axon transport, and peripheral nerve regeneration, among other processes [7, 11, 15, 24, 26, 30, 31, 37–40].
Most tubulin genes exhibit high sequence homology and many are clustered in the genome. Complete genome assemblies now highlight that TUBB2A and TUBB2B are immediately adjacent to each other in both the human and mouse genome and are virtually identical (443 identical amino acids of 445 total). Similarly, TUBA1A, TUBA1B, and TUBA1C genes are also adjacent in the genome and contain high sequence homology in both human and mouse. These characteristics suggest these genes may be the product of genome duplications and the functions of each individual gene may be shared across the cluster(s).
Despite the central importance of these genes in the cytoskeleton and their relevance for human cortical malformation, relatively few genetic models of tubulin variants exist. No null alleles are published for Tuba1a, Tuba1b, Tuba1c, Tubb2a, or Tubb2b. ENU mutagenesis efforts have identified alleles in Tuba1a [7, 40, 41] and Tubb2b [42], and a mouse model of CFEOM for Tubb3 has been made [31]. More recently a null allele of Tubb3 has been produced and shown to have effects on growth cone function and axonal regeneration after induced sciatic nerve injury [37]. Here we test the requirements for Tuba1a, Tubb2a, and Tubb2b with specific genetic deletions.
Results
Null alleles of Tuba1a, Tubb2a, and Tubb2b
We used CRISPR-Cas9 genome editing to create individual deletions of Tuba1a, Tubb2a, and Tubb2b. Guide RNAs were generated to target the endonuclease to two intergenic regions flanking each tubulin gene (Fig 1A, 1D and 1G; S1 Table). Tubb2a and Tubb2b are only 49kb apart (0.02cM) in the mouse genome, suggesting that meiotic recombination between the loci will be an extremely rare event. With this consideration in mind, we first attempted to multiplex CRISPR-Cas9 editing for Tubb2a and Tubb2b with the goal of creating a single deletion of each gene as well as a simultaneous deletion of both. A mix of all eight Tubb2a and Tubb2b guides were constructed and used for blastocyst microinjection. The resulting founders were analyzed by PCR and Sanger sequencing for alterations at the Tubb2a and/or Tubb2b loci. We recovered two alleles of Tubb2a and one of Tubb2b. Tubb2aem1Rstot (hereafter referred to as Tubb2ad3963) is a 3,963bp deletion (chr13: 34,074,222–34,078,184; GRCm38/mm10) and Tubb2aem2Rstot (Tubb2ad4222) is a 4,222bp deletion (chr13: 34,074,225–34,078,446). Both independent deletions excise the entire Tubb2a open reading frame (Fig 1A–1C, S1 and S2 Figs). We also obtained one complete deletion allele of Tubb2b. Tubb2bem1Rstot (Tubb2bd4185) is a 4,185bp deletion (chr13: 34,126,374–34,130,558; Fig 1D–1F, S2 Fig).
Fig 1. Novel CRISPR/CAS9 alleles of three tubulin genes.

Deletion of Tubb2a (A-C), Tubb2b (D-F), and Tuba1a (G-I). (A,D,G) Schematics for deletions showing sites of CRISPR guide sequences (red asterisk), arrows indicate relative locations of PCR primers (red are meant to only amplify when gene deletion occurs under PCR standard conditions). (B,E,H) Sequencing chromatograms showing the results of mutant specific PCR products. Alignments to the reference genome indicate the deletions indicated for each gene. (C,F,I) PCR genotyping to specifically identify each allele. Note Tubb2a, Tubb2b, and Tuba1a are on the reverse strand of their respective chromosomes. White asterisk indicates 500 bp marker in DNA ladder.
In a parallel manner, four CRISPR guides were generated to delete Tuba1a (Fig 1G) and we recovered two independent deletions (Fig 1G–1I; S1 and S2 Figs). Tuba1aem1Rstot (Tuba1ad4304) is a 4,304 bp deletion (chr15:98,949,728–98,954,031) and Tuba1aem2Rstot (Tuba1ad4262) is a 4,262 bp deletion (chr15: 98,949,770–98,954,031). For all guides used in this study, we predicted the top ten off-target sites (S2 Table). Only one is even on the same chromosome as the desired target sequence. This potential target is 24 cM away from Tuba1a. At this genetic distance, there is virtually no chance this genomic sequence is segregating with the Tuba1a deletion allele we generated.
Loss of Tubb2a or Tubb2b leads to postnatal forebrain phenotypes
Mice heterozygous for all the deletion alleles were viable and fertile. As we hypothesized the deletion alleles would yield recessive phenotypes, we intercrossed heterozygous carriers for each. We found no reduction from Mendelian expectations in the number of live animals at weaning for any allele of Tubb2a or Tubb2b, and animals homozygous for these deletions did not appear grossly different than littermates (Table 1). Total body weight at postnatal day (P) 28 did not show any evidence of a failure to thrive upon loss of Tubb2a or Tubb2b (Fig 2A–2C, S3 Table). We collected brains for both whole mount analysis and histological examination from P28-31 animals to determine if loss of these tubulin genes caused any discernable malformations of cortical development. Total brain weight at P0 did not differ in Tubb2a or Tubb2b mutants (Fig 2D–2F, S3 Table). Whole mount imaging and initial histological analysis of the brains did not reveal any gross phenotypes (Fig 2G–2DD, S3 and S4 Figs). This included more detailed analyses of the corpus callosum (Fig 2O–2R), hippocampus (Fig 2S–2V), basal ganglia (Fig 2W–2Z) and cerebellum (Fig 2AA–2DD).
Table 1. Survival at weaning of mice with deletions of Tubb2a and Tubb2b*.
| wild-type | heterozygous | homozygous | total | p-value | |
|---|---|---|---|---|---|
| Tubb2ad3963 | 43 | 66 | 41 | 151 | 0.33 |
| Tubb2ad4222 | 24 | 42 | 20 | 87 | 0.81 |
| Tubb2bd4185 | 46 | 87 | 37 | 170 | 0.59 |
* All data here are from heterozygous intercrosses
Fig 2. General morphology and histological analysis of Tubb2a and Tubb2b deletions.

(A-C) Postnatal weights of the Tubb2a and Tubb2b mice at P28 show no sign of failure to thrive as compared to wild-type. (D-F) Brain weight at P0 is also unaffected by Tubb2a or Tubb2b deletion. Whole mount (G-J) and Nissl staining (K-DD) histological analysis of wild-type (G,K,O,S,W,AA), Tubb2ad3963/d3963 (H,L,P,T,X,BB), Tubb2ad4222/d4222 (I,M,Q,U,Y,CC) and Tubb2bd4185/d4185 (J,N,R,V,Z,DD) homozygous mice at P28-31. More detailed analysis of the corpus callosum (O-R), hippocampus (S-V), basal ganglia (W-Z), and cerebellum (AA-DD) is shown and all appear unaffected in overall size or organization. Scale bars indicate 1mm (G-J), 500 μm (K-N,AA-FF) and 200 μm (O-Z).
We performed a more comprehensive histological and immunohistochemical analysis which did reveal a number of more subtle anomalies in animals lacking tubulin genes (Figs 2–4, S3–S5 Figs). Quantification of the number of cells in the motor and somatosensory cortices as identified through Nissl staining did reveal some subtle differences (Fig 3, S3 Fig, S3 Table). The Tubb2ad3963/d3963, Tubb2ad4222/d4222 and Tubb2bd4185/d4185 homozygous mice all do appear to have slightly decreased cell numbers in both the motor (Fig 3A–3G) and somatosensory (Fig 3H–3N) cortical tissues, albeit with differing levels of statistical certainty.
Fig 4. Cortical layering deficits in Tubb2a and Tubb2b deletion mutants.

Immunohistochemistry for NeuN (A-E), CTIP2 (F-J), TBR1 (K-T) and myelin basic protein (MBP, U-II) highlights multiple phenotypes in Tubb2ad3963/d3963 (B,G,L,Q,V,AA,FF), Tubb2ad4222/d4222 (C,H,M,R,W,BB,GG) and Tubb2bd4185/d4185 (E,J,O,T,Y,DD,II) homozygous mice as compared to wild-type (A,D,F,I,K,N,P,S,U,X,Z,CC,EE,HH). (A-E) NeuN staining for differentiated neurons. (F-J) CTIP2-positive upper layer neurons. (K-T) TBR1-positive lower layer neurons. (U-II) MBP immunoreactivity indicates severe loss of axon tracts in the corpus callosum (AA, BB) and pyramidal fiber tracts (FF, GG) of the Tubb2a deletion, but not the Tubb2b deletion (DD,II). Box and asterisk in U indicate areas shown in Z and EE, respectively. Scale bars indicate 100 μm (A-T), 1 mm (U-Y), 200 μm (Z-DD) and 500 um(EE-II).
Fig 3. Quantitative analysis of cell density in the motor and somatosensory cortex in Tubb2a and Tubb2b deletions alleles.

Quantification of Nissl-positive cells/area for both the motor cortex (A-G) and somatosensory (H-N) cortex show some changes in cellular density. ANOVA p values are shown for each experiment and multiple comparison p values are shown when ANOVA p≤0.05. (Individual samples are plotted to indicate sample size, differing colors in E-G, L-N indicate littermates. n = 3 wild-type, 3 heterozygotes and 3 homozygous mutants for Tubb2ad3963; 4 wild-type, 4 heterozygotes and 4 mutants for Tubb2ad4222; and 4 wild-type, 5 heterozygotes, and 4 mutants for Tubb2bd4185).
We complemented this histological analysis with a series of immunohistochemical studies to further examine the effects of homozygous loss of Tubb2a or Tubb2b. The overall laminar organization was not disrupted in any deletion allele but we did note some subtle disruptions. NeuN is a marker of differentiated neurons and we note subtle differences in the staining as the NeuN cells seem to be more dispersed in the mutant cortex (Fig 4A–4E). CTIP2 marks layer V neurons and appears relatively unaltered in the deletion alleles (Fig 4F–4J, S5A–S5C Fig). We quantified the distribution of CTIP2-postive cells and did see subtle increases in the proportion of cells in the middle third of the cortex (Bin 2) at the expense of the dorsal third (Bin 3) consistent with a very mild migration defect of the CTIP2-positive neurons. This appears to be much less pronounced in the Tubb2b deletion mutant (S5 Fig, S4 Table). TBR1 is a marker of layer IV-VI neurons and, as with CTIP2, we observed a similar but more marked reduction in the number of cells in the middle third of the cortex (Bin 2) of the dorsomedial cortex. In the immediately adjacent parietal cortex, this migrational anomaly is again seen in the Tubb2ad3963/d3963 homozygotes but not other mutants (Fig 4K–4T, S5D–S5I Fig). Future studies will be needed to determine how significant and/or widespread these subtle anatomical differences are between the Tubb2a alleles. Tubb2bd4185/d4185 mutants also show decreased TBR1-positive cells in the middle third of the dorsomedial cortex but not the parietal cortex (Fig 4N, 4O, 4S and 4T, S5 Fig) We also used Myelin Basic Protein (MBP) to highlight axon tracts and found reduced staining in the corpus callosum and pyramidal fibers of the Tubb2a homozygotes but not in Tubb2b homozygotes (Fig 4U-II). We conclude from these data that neither Tubb2a, nor Tubb2b, are uniquely required for survival in the mouse but do result in subtle cortical malformations. However, neither loss of Tubb2a nor Tubb2b results in the severe cortical malformations seen in the most common tubulinopathy patients.
Deletion of either Tubb2a or Tubb2b leads to no change in total β-tubulin protein levels
Tubulin expression is well known to be auto-regulated in mammalian cells [43, 44]. The production of the initial portion of the tubulin polypeptide chain serves as a signal to the cell to reduce the stability of remaining tubulin mRNA [45–51]. This mechanism has implications for the ability of a cell to compensate for deletion of a single tubulin locus. We therefore considered if the amount of tubulin protein in the P0 brain was altered upon deletion of Tubb2a or Tubb2b. We are unable to definitively demonstrate the loss of TUBB2A or TUBB2B protein in our mutants as the amino acid sequences are identical at all but two amino acids precluding any antibody-based analyses such as Western immunoblotting (S6 and S7 Figs). RNA analysis of gene expression is similarly challenging due to sequence similarity (S8 and S9 Figs). Some RT-PCR primers have been used previously, but our analysis suggests these might amplify other genomic elements such as poorly annotated pseudogenes (S10 Fig). Expression of Tubb2b mRNA in mouse has been reported [26], but a further analysis of the published in situ probe shows high sequence similarity to the Tubb2a sequence, suggesting that this reported expression may, perhaps, be a combination of Tubb2a and Tubb2b (S11 Fig).
However, we were able to address the question more broadly with an antibody that recognizes multiple β-tubulins (Fig 5, S12 Fig, S5 Table). Immunoblotting with this antibody did not indicate any biologically significant reductions in the deletion mutants. (We did note an apparent increase in total β-tubulin levels in Tubb2ad4222/wt animals, which become magnified in comparison to a very small decrease in Tubb2ad4222/d4222 total β-tubulin when each is compared to wild-type, Fig 5B). We note that the high levels of tubulin in comparison to other specific protein in the cell makes precise quantification difficult with the low levels of total protein analyzed in these experiments (see below discussion in Tuba1a mutants). This suggests that loss of TUBB2A or TUBB2B is compensated by other β-tubulins expressed in the developing brain. This may explain the relatively mild phenotypes we report here in these homozygous deletion mutants.
Fig 5. Total β-tubulin protein levels in Tubb2a and Tubb2b mutants.
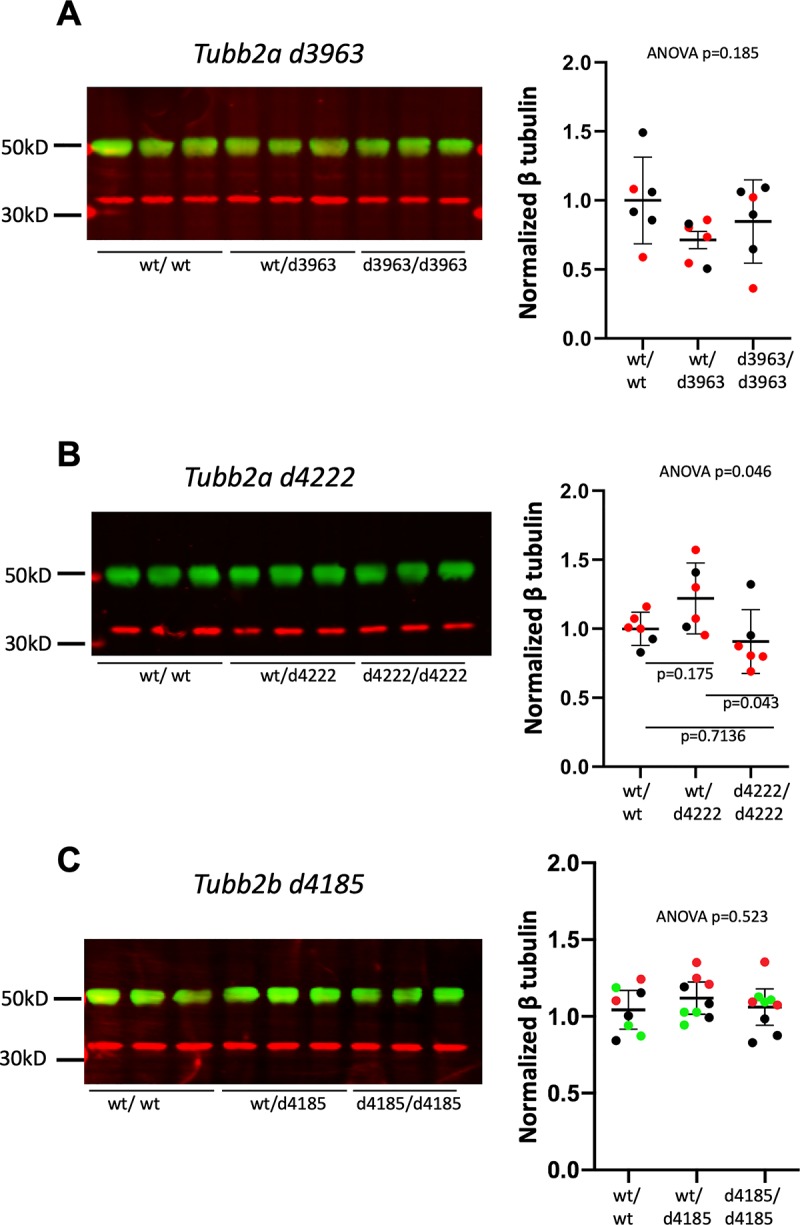
Western immunoblotting with an antibody recognizing multiple isoforms of β-tubulin for P0 brain lysates from (A) Tubb2ad3963 (B) Tubb2ad4222and (C) Tubb2bd4185 mice. Representative immunoblots for each allele are shown. Quantifications are of multiple experiments normalized to wild-type littermate controls. (A,B) Colors indicate littermates. n = 3 animals of each genotype, each set was run in duplicate. (C) Colors indicate replicate experiments from one set of littermates (Green bands indicate tubulin and red indicate GAPDH loading control for all immunoblotting experiments).
Tuba1a is required for survival and brain development
In stark contrast to our findings with Tubb2a and Tubb2b, Tuba1a is absolutely required for survival to weaning as we did not recover any homozygotes for either Tuba1a deletion allele (n = 177 total; Table 2). We do note a small reduction in heterozygote survival among the surviving animals. To begin to understand the reason for the lethality and to assess brain development in the homozygotes, we collected embryos at late organogenesis stages. A combined analysis of both Tuba1a deletion alleles in embryos from embryonic day (E)14.5-E18.5 did not reveal a significant loss of homozygous embryos during embryonic stages (Table 2).
Table 2. Survival of Tuba1a deletion mutants*.
| wild-type | heterozygous | homozygous | total | p-value | |
|---|---|---|---|---|---|
| Survival at weaning | |||||
| Tuba1ad4262 | 36 | 39 | 0 | 75 | 2.95e-08 |
| Tuba1ad4304 | 38 | 64 | 0 | 102 | 9.39e-26 |
| Tuba1a combined | 74 | 103 | 0 | 177 | 3.4e-15 |
| Embryonic survival | |||||
| Tuba1ad4262 x Tuba1ad4262 | 7 | 18 | 11 | 36 | 0.64 |
| Tuba1ad4304 x Tuba1ad4304 | 40 | 105 | 51 | 196 | 0.17 |
| Tuba1a combined | 47 | 123 | 62 | 232 | 0.18 |
* All data here are from heterozygous intercrosses
Tuba1ad4304/d4304 and Tuba1ad4262/d4262 animals had very obvious and consistent phenotypes. We noticed thoracic curvature and thoracic edema with significant hemorrhaging in the cervical region extending towards the forelimbs (Fig 6A–6C). Histological analysis of the developing forebrain revealed a striking series of forebrain malformation phenotypes. Sections from both posterior and anterior regions of the forebrain highlight enlarged lateral and third ventricles, reduced basal ganglia, as well as a widened base of the third ventricle in posterior brain regions (Fig 6D–6O). Examination of cortical morphology showed a 115–179% increase in the width of the ventricular zone at E16.5, a 50% reduction in the intermediate zone, and an approximate 30% reduction in width of the cortical plate as compared to wild-type (Fig 6P–6U; S6 Table). We also noted a number of the homozygous mutants had cleft palate (Fig 6W). This was incompletely penetrant and seen in 3/11 Tuba1ad4304/d4304 embryos (27%) and 2/16 Tuba1ad4262/d4262 (12.5%) embryos. We noted cleft palate in only one out of 52 heterozygous embryos analyzed, but we suspect this was likely due to delayed development in that particular embryo based on morphological staging. We conclude that Tuba1a is required for survival and that loss of Tuba1a leads to major cortical malformations with similarities to what is seen in the human tubulinopathy patients.
Fig 6. Loss of Tuba1a leads to perinatal lethality and cortical malformations.

Embryos homozygous for either of two Tuba1a deletions (Tuba1ad4304 B,E,H,K,N; Tuba1ad4262 C,F,I,L,O) at E16.5 have significant gross morphological phenotypes when compared to wild-type (A,D,G,J,M). Apparent hemorraghing in the cervical region is often noted in mutants (B,C) making them distinguishable upon initial embryo harvesting. Histological analysis shows enlarged ventricles with hypoplastic basal ganglia (BG) and disruptions to the ventricular zone (VZ), intermediate zone (IZ) and cortical plate (CP) in both mutants. Black boxes in D-F show highlighted areas in G-I, respectively. (Sections in D-I are in a more anterior portion of the embryo than J-O to accentuate different aspects of the cortical malformations). Quantification of the width of the VZ (P,Q), IZ (R,S) and CP (T,U) confirms an increased width of the VZ but reductions in the IZ and CP for both alleles. (n = 3 wild-type, 27 heterozygous and 6 mutant embryos for Tuba1a d4304 and 1 wild-type, 12 heterozygous, and 5 mutant embryos for Tuba1a d4262). Some mutants show cleft palate (W) resulting from a failure of the hypoplastic palatal shelves (arrowheads) to elevate and fuse as seen in wild-type embryos (V). Scale bars indicate 400 μm in D-F,J-L; 100 μm in G-I,M-O; and 200 μm in V,W.
Altered patterns of neurogenesis upon loss of Tuba1a
We have performed an initial molecular analysis of neurogenesis in the Tuba1ad4304/d4304 embryos at E14.5 and E16.5 as compared to unaffected wild-type or heterozygous littermates (Fig 7, S7 Table). We first measured proliferation rates with immunohistochemistry for phosphorylated histone H3 (pHH3) to mark actively dividing cells in M-phase and found no significant difference between wild-type and controls at E14.5 (Fig 7A–7C) but a slight 15.7% increase at E16.5 (Fig 7D–7F). We further examined neurogenesis with an EdU-Ki67 pulse chase assay in which we treated pregnant dams with EdU at E13.5 to label dividing cells, sacrificed at E14.5 and used an antibody against Ki67 to mark cells in G1, S, and G2 phases of the cell cycle at the time of sacrifice (Fig 7G–7N). This analysis indicated a 15.6% decrease in total EdU-positive, Ki67-negative cells in the mutant cortex at E14.5 (Fig 7K) suggesting that fewer overall cells were produced in the 24-hour period between EdU administration and sacrifice. Cells which are Ki67-positive and EdU-negative are actively dividing at E14.5 and were increased 56.7% (Fig 7L). Note that Ki67 marks a broader sample of the cell cycle than pHH3 which may explain why this is increased while the number of pHH3-positive cells did not appear different. Cells positive for both EdU and Ki67 were progenitors dividing at E13.5 which re-entered cell cycle and these are increased in mutants by 84.3% (Fig 7M). The quit fraction is the proportion of cells leaving the cell cycle over total labeled cells (EdU+Ki67-/EdU+) and is 2% reduced in mutants (Fig 7N). We conclude from these data that the enlarged ventricular zone we see in the homozygous Tuba1a deletion mutants is due to disrupted neuroprogenitor cell division kinetics in which some cells at E14.5 are preferentially re-entering cell cycle rather than terminally dividing.
Fig 7. Loss of Tuba1a leads to altered patterns of neurogenesis and migration.

(A-F) Immunohistochemistry for pHH3 does not indicate a difference between wild-type embryos and homozygous Tuba1a mutants at E14.5, but an increase in mutants at E16.5 (n = 3 wild-type and mutant embryos at E14.5 and n = 4 for both at E16.5). (G-R) EdU pulse chase analysis. EdU cells were labeled in vivo at E13.5 and embryos were fixed and immunostained for Ki67 and EdU at E14.5 (G-O) or E16.5 (P-R). In comparison to wild-type cells at E14.5 (n = 4 wild-type and mutant embryos), the number of Tuba1a mutant total EdU+ cells are reduced (K), EdU-Ki67+ cells are increased (L), EdU+Ki67+ cells are increased (M) and the quit fraction (EdU+Ki67-/EdU+) is decreased (N). (O) The position of EdU+ cells across the dorsal-ventral axis cortex during radial migration shows no difference in tissue distribution between wild-type and mutant. (P-R) Embryos labeled from E13.5-E16.5 show reduced radial migration (n = 3 wild-type and mutant embryos). (S-X) The number of TBR2-positive intermediate progenitors at E14.5 (S-U)does not appear to differ between wild-type and Tuba1a mutants but is increased at E16.5 (V-X; n = 4 wild-type and mutant embryos). (Y-BB) TuJI-positive differentiated neurons appear to be dispersed over a broader portion of the cortex in Tuba1a mutants at E14.5 and E16.5 (n = 3 control and mutant embryos for both). (CC-HH) Cleaved caspase 3 immunoreactivity as a marker for apoptosis is increased at E14.5 and E16.5 (n = 3 control and mutant embryos for both). Scale bars indicate 100 μm.
EdU treatment also allowed us to assess the fate of cells 24 and 72 hours after division. We divided the cortex in three equal parts and quantified the percentage of EdU cells in each third of the cortex along the dorsal-ventral axis to query their developmental trajectory as they migrate into the cortical plate. We did not see a significant change in these parameters at E14.5 (Fig 7O). However, when we administered EdU at E13.5 and sacrificed at E16.5, we noted dramatic increases in cells in the middle third of the cortex at the expense of the outer third, consistent with a failure of neurons born at E13.5 to fully survive, differentiate and/or migrate towards the pial surface (Fig 7P–7R).
We also measured the levels of TBR2-positive intermediate progenitors and observed no changes at E14.5 (Fig 7S–7U) but a slight increase at E16.5 (Fig 7V–7X; 13.5% increase). TuJI immunoreactivity of maturing neurons indicated a slight reduction in levels of differentiation and a broader dispersal of TuJI-positive cells throughout the cortical tissue in homozygous mutants at both E14.5 and E16.5 (Fig 7Y-BB). We also examined levels of apoptotic cell death with immunohistochemistry for cleaved caspase 3 and found increases at both E14.5 and E16.5 (elevated 75.9% and 866% respectively, Fig 7CC-HH). Taken together, all of these data lead us to conclude changes in neurogenesis, migration and cell death contribute to the cortical phenotypes. The ultimate cause of the lethality of these cells, and the mouse mutant embryos as a whole, remains to be determined.
A novel ENU allele of Tuba1a
In a separate experiment utilizing an ENU mutagenesis forward genetic screen to ascertain new alleles important for cortical development, we identified an additional mutant with similar brain phenotypes to those seen in the Tuba1a deletion mice. The quasimodo mutants were first identified by an abnormal curvature of the thoracic region seen in many of these mutants (Fig 8B). Histological analysis of mutants showed a significant brain malformation with lateral ventriculomegaly, reduced cortical tissue, and enlarged third ventricle (Fig 8C–8F). Whole exome sequencing of three homozygous mutants identified a large number of homozygous variants as predicted. Filtering for SNPs which were shared by all three mutants, not present in dbSNP, predicted to have a high/moderate effect on the protein (e.g., missense variants at conserved residues, premature stop codons, etc.), and having only one variant in the gene (i.e., not a highly polymorphic sequence) left a total of ten variants. A review of literature to identify known consequences for loss of function in these genes or known roles of these genes did not yield any compelling candidates (Table 3).
Fig 8. The quasimodo mutation is an allele of Tuba1a.

(A-F) Homozygous quasimodo mutants are distinguishable at E18.5 by the systemic edema (B). Histological analysis (E17.5) shows enlargement of the third ventricle (D) and reduced cortical tissue (F). (G) Sanger sequencing confirms homozygosity for a candidate SNP in a conserved DNA (H) and protein (I) sequence (SNP and coding change shown in red, red asterisk indicates premature stop codon). (J) Structure of the wild-type α/β tubulin dimer is shown above and the remaining α-tubulin structure is shown below (red = non-hydrolyzable GTP at the monomer interface). (K-P) Complementation analysis at E17.5 with the Tuba1ad4304 is consistent with quasimodo being an allele of Tuba1a as Tuba1ad4304/quas mutants have gross (L) and histological (N,P) phenotypes similar to the other Tuba1a homozygous phenotypes. The failure of alleles to complement indicates both mutations are in Tuba1a. Boxes in C,D,M,N show areas enlarged in E,F,O,P, respectively. Scale bars indicate 200 μm in C,D,M,N and 50 μm in E,F,O,P.
Table 3. Mouse exome variant analysis in Tuba1aquas homozygous mutants.
| Exome Variants | # of variants |
|---|---|
| total | 199,085 |
| Homozygous in all three animals | 982 |
| not in dbSNP | 575 |
| SNP | 324 |
| High/ moderate predicted effect | 43 |
| Single variant in gene (Rapgef4, Ckap5, Gon4l, Cope1, Fat2, Med1, Nrxn3, Glac, Ptpn21, Dnahc11) | 10 |
We then performed a parallel analysis for all heterozygous missense variants shared by the three sequenced mutants with the alternative hypothesis that the quasimodo phenotype may be an incompletely penetrant heterozygous variant. One variant from this analysis was a nonsense mutation in Tuba1a creating a premature stop codon at amino acid 215 (p.R215*). We noted that the sequence in this region of Tuba1a, Tuba1b and Tuba1c is highly similar and carefully analyzed the exome .bam files. Virtually all of the reads for this region of genomic sequence were aligned to the Tuba1a gene and virtually no reads were mapped to Tuba1b or Tuba1c. Given the phenotypic similarity to the Tuba1a deletion, we hypothesized the similarity among these genes may have confounded the automated sequence alignment and pursued Sanger sequencing with intronic primers that would specifically amplify Tuba1a. This analysis showed that the quasimodo mutants were all in fact homozygous for the Tuba1a variant creating the nonsense mutation (Fig 8G–8I). This mutation is predicted to produce a protein missing approximately half of the amino acid sequence, if that protein is even stable in the cellular context (Fig 8J). The mutant protein would be missing significant portions of the polypeptide which would normally interface with the adjacent β-tubulin monomer.
In order to confirm that the Tuba1a R215* variant is actually the causative variant in the quasimodo mutants, we performed a complementation test with the Tuba1ad4304 deletion allele. We crossed Tuba1aquas/wt with Tuba1ad4304/wt animals and in three litters (22 live pups), we recovered no Tuba1ad4304/quas live animals at weaning (Table 4). We further explored this hypothesis with embryonic dissections and recovered two Tuba1ad4304/quas embryos at E17.5. These mutants exhibited the exaggerated curvature of the thoracic region, as previously seen in Tuba1aquas/quas mice and Tuba1ad4304/d4304 mice (Figs 6B and 8L). Histological analysis showed ventriculomegaly with disrupted cortical architecture including a loss of the intermediate zone, hypoplastic basal ganglia, and cleft palate in both mutants (Fig 8M–8P). The similarity of these phenotypes to the Tuba1ad4304/d4304 and Tuba1aquas/quas mutants suggests that the Tuba1a R215* variant found in quasimodo mutant is indeed the causal variant. Moreover, this would support the above conclusion that loss of Tuba1a function results in significant cortical malformations.
Table 4. Quasimodo is an allele of Tuba1a.
| wild-type | quas/wt | d4304/wt | d4304/quas | total | p | |
|---|---|---|---|---|---|---|
| Tuba1ad4304/wt x Tuba1aquas/wt weaning | 7 | 6 | 9 | 0 | 22 | 0.04 |
|
Tuba1ad4304/wt x Tuba1aquas/wt embryos |
6 | 9 | 8 | 2 | 25 | 0.42 |
Proliferation and radial migration phenotypes in Tuba1aquas mutants
We performed a molecular analysis of neurogenesis in the Tuba1aquas mutants as we did for the Tuba1a deletion mutant (Fig 9, S7 Table). Surprisingly, although the terminal phenotypes are similar, the data suggest different underlying mechanisms. Proliferation in Tuba1aquas mutants was increased by 18.9% as measured by pHH3 immunoreactivity at E14.5 (Fig 9A–9C) but decreased 7.2% at E16.5 (Fig 9D–9F). In contrast to the Tuba1a deletions however, the EdU-Ki67 pulse chase experiments did not reveal any robust differences in patterns of neurogenesis as we saw in the Tuba1a deletion mutant (Fig 9G–9N). The EdU labeling to analyze cell migration highlighted a significant reduction in cells labeled at E13.5 which migrated to the upper third of the cortex at E14.5 and a corresponding increase in number of cells in the middle third (Fig 9O). The same patterns are seen at E16.5 with more marked changes in cellular distributions (Fig 9P–9R). This data is most consistent with a decreased radial migration by cells born on E13.5, similar to what we observed in the Tuba1a deletion mutants.
Fig 9. Altered proliferation, radial migration and apoptosis in Tuba1aquas mutants.

(A-F) Immunohistochemistry for pHH3 indicates a difference between wild-type embryos and homozygous Tuba1aquas mutants at E14.5, but not E16.5 (n = 3 wild-type and mutant embryos for each). (G-R) EdU pulse chase analysis. EdU cells were labeled in vivo at E13.5 and embryos were fixed and immunostained for Ki67 and EdU at E14.5 (G-O) or E16.5 (P-R). In comparison to wild-type cells at E14.5 (n = 4 wild-type and mutant embryos), none of the cell cycle parameters appear to differ (G-N). (O) The position of EdU+ cells across the dorsal-ventral axis cortex during radial migration shows altered migration patterns. (P-R) Embryos labeled from E13.5-E16.5 also show reduced radial migration (n = 4 wild-type and mutant embryos). (S-X) The number of TBR2-positive intermediate progenitors at E14.5 (S-U) does not appear to differ between wild-type and Tuba1aquas mutants, but is increased at E16.5 (V-X; n = 4 wild-type and mutant embryos). (Y-BB) TuJI-positive differentiated neurons appear to be dispersed over a broader portion of the cortex in Tuba1aquas mutants at E14.5 and E16.5 (n = 3 control and mutant embryos for both). (CC-HH) Apoptosis is increased at E14.5 and E16.5 (n = 3 control and mutant embryos for both). Scale bars indicate 100 μm.
We also saw no marked changes in the numbers of TBR2-positive intermediate progenitors in the Tuba1aquas mutants at E14.5 (Fig 9S–9U) but a slight 21.5% increase at E16.5 (Fig 9V–9X). TuJI immunoreactivity again showed an increased range of differentiated cells similar to the Tuba1a deletion mutants at both E14.5 and E16.5 (Fig 9Y-BB). Similar to the analysis of the Tuba1a deletion mutants, Tuba1aquas mutants show elevated levels of apoptotic cell death which is readily apparent at E14.5 and E16.5 (elevated 185% and 1721% respectively, Fig 9CC-HH). We again conclude that the Tuba1aquas phenotype is the result of disrupted neurogenesis, disrupted migration and a marked increase in cell death.
Mutations at Tuba1a locus lead to changes in α-tubulin and β-tubulin protein levels
Similar to the analysis we performed on the levels of β-tubulin protein in Tubb2a and Tubb2b mutants (Fig 5), we also analyzed total α-tubulin levels in the Tuba1a mutants described here. We used an antibody that shares similarity with multiple α-tubulin proteins at the N-terminal portion of the reported epitope (Fig S13). We find that total α-tubulin levels do not appear to be reduced in Tuba1ad4304/d4304 homozygous mutants when normalized to GAPDH levels within each litter matched set (Fig 10A). Even after five independent experiments, we could not convincingly demonstrate loss of α-tubulin even though matched samples (e.g., asterisks in Fig 10A are a set of littermates) often indicated a subtle decrease. We reasoned the very low level of protein input to our experiments may have challenged the limits of accurate and robust detection of the imaging system in embryonic brain lysate. We note GAPDH levels are extremely low at this stage of development. We ran a parallel analysis for total protein, rather than a single protein, as a loading control. REVERT total protein stain is not based on any particular protein epitope. When we analyzed REVERT staining of the transfer membrane we saw a striking decrease in total protein around the molecular weight of tubulin in mutant samples (Fig 10B). We reason this species is likely mostly tubulin protein. When we normalize the levels of total α-tubulin to total protein as measured by REVERT total protein stain, we do see a significant reduction in total α-tubulin in mutants as compared to both wild-type and Tuba1ad4304 heterozygous littermates (Fig 10B, S8 Table). We performed a similar analysis of total α-tubulin in Tuba1aquas mutants and regardless of which tubulin normalization method used, we see a dramatic loss of more than half the α-tubulin in mutant tissue (Fig 10C and 10D).
Fig 10. Total α-tubulin and β-tubulin protein levels in Tuba1a mutants.

Immunoblotting for pan-α (A-D) and pan-β (E-H) tubulin in Tuba1ad4304 (A,B,E,F) and Tuba1a quas (C,D,G,H) wild-type, heterozygous and mutant embryos. Protein levels were quantified by comparison to GAPDH loading control (A,C,E,G) and REVERT total protein stain (B,D,F,H). (A) Colors indicate results from five different technical replicates. (B-H) Colors indicate littermates (technical replicates are shown for A-C,E,G). For Tuba1ad4304, n = 3 animals of each genotype, for Tuba1aquas, wild-type n = 3, heterozygous n = 2, and mutant n = 5. (t = presumed tubulin protein, a = presumed actin protein).
The dramatic reduction in overall tubulin on the membranes stained for total protein (e.g., Fig 10B) led us to hypothesize that decreased levels of α-tubulin might have an effect on β-protein as well. This may be through an unknown mechanism to keep massive amounts of unpolymerized β-tubulin monomers from flooding the cell. To our surprise, we do indeed see a decrease in total β-tubulin upon loss of Tuba1a (Fig 10E and 10F) and in homozygous Tuba1aquas mutants (Fig 10G and 10H). This regulation of tubulin monomers is a very intriguing question to be addressed further in future research. This will likely require work in vitro with tagged versions of the tubulins of interest.
Discussion
We have for the first time demonstrated the consequences of the loss of three tubulin genes known to be important for normal mammalian cortical development: Tuba1a, Tubb2a, and Tubb2b. We also identified a new Tuba1a mouse allele with severe CNS phenotypes through an ENU mutagenesis experiment. These alleles contribute to a larger body of work indicating the importance of tubulin genes in cortical development and disease. These deletion alleles allow us to further explore the model that the human variants identified in TUBA1A, TUBB2A and TUBB2B are acting as dominant negative heterozygous mutations (e.g. [52]), but may not be truly essential for development. We find that Tubb2a and Tubb2b can be deleted from the mouse genome with no obvious effect on gross cortical development, animal survival, or overall health. Tuba1a, however, is quite different, as loss of Tuba1a is lethal in the mouse and leads to severe malformations of cortical development. Thus, other tubulin genes with high sequence similarity cannot compensate for Tuba1a but may be doing so for Tubb2a and Tubb2b (e.g., Tubb3). Together, these data show that some tubulin genes are absolutely critical for mouse CNS development and organism survival, while others are less critical.
Tubb2a and Tubb2b are highly similar and immediately adjacent in the genomes of both mice and humans, suggesting that a recent duplication event in a common ancestor may have rendered these genes redundant for each other. This may explain why a single deletion of each does not lead to major CNS phenotypes. However, we do note more subtle cortical phenotypes including changes in cell number in the motor and somatosensory cortices and in disrupted cortical lamination consistent with more mild radial migration defects. We also note a striking reduction in MBP immuno-staining in the Tubb2a deletion homozygous mice. We suspect this is a result of reduced axon tracts rather than defective myelination. This phenotype and the possible behavioral consequences of the cortical disruptions should be interrogated rigorously in future studies. We acknowledge that mouse models of human cortical malformations may not be perfect due to species differences. However, we do not believe any such differences can explain the normal presentation of these mice. Indeed, we have previously shown structural and behavioral phenotypes in a mouse Tubb2b missense allele [42, 53].
While the Tubb2a and Tubb2b deletion mutants appear to have relatively subtle phenotypes, they each may be rescued from more global malformations through functional redundancy. This model can be tested with a double mutant analysis. The genes are sufficiently close to each other within the genome such that this experiment will require creation of an independent deletion or conditional allele in cis to one of the existing mutations.
Given these findings that homozygous deletions of Tubb2a or Tubb2b do not result in catastrophic malformations of cortical development, it is perhaps surprising that there are only two homozygous coding missense variants and no homozygous loss of function variants in both TUBB2A and TUBB2B combined in the gnomAD collection of over 140,000 healthy individuals as of this writing [54]. Given the phenotypes we now report, it may be that the homozygous missense mutations lead to neurological disease which has not yet been attributed to tubulin genes, but would prevent inclusion of the affected individuals in the gnomAD and ExAC databases. The sequence similarities may also confound mapping of genomic information.
The mice heterozygous for the Tubb2a or Tubb2b deletions do not show any biologically significant loss of total beta-protein (Fig 5, S5 Table). We hypothesize other tubulins are produced at higher levels to compensate for the loss of each single deletion (e.g., Tubb3). These findings are consistent with the known data on tubulin autoregulation (see above). The reduced mRNA levels from the deletions should lead to reduced polypeptide production and, thereby, less de-stabilization of the tubulin mRNA, ultimately keeping monomer levels stable.
The TUBA1A polypeptide sequence is highly similar to TUBA1B (99.6% identical) and TUBA1C (98.2%) and three genes coding for these similar proteins are also clustered in the genome. These features suggest they may also be able to compensate for each other similar to our hypothesis regarding Tubb2a and Tubb2b. In direct contrast to our findings with Tubb2a and Tubb2b, however, loss of Tuba1a is catastrophic for the mouse embryo. The individual requirements for Tuba1b and Tuba1c remain unexplored and we will address this hypothesis with future deletion alleles. Loss of Tuba1a leads to an increased ventricular zone at E16.5 and our initial data from E14.5 and E16.5 suggests this is from a dysregulation of neural progenitor proliferation and cell cycle exit. Ultimately, failures in radial migration and/or apoptosis contribute to the phenotypes we observed. We also report an independently derived ENU-induced mutation in Tuba1a which encodes a transcript with a premature stop codon approximately halfway through the protein coding sequence. These homozygous Tuba1aquas mutants have similar cortical phenotypes to the deletion mutants, but apparently develop through different mechanisms as the cell cycle kinetics do not appear to be disrupted (Figs 7 vs. 9). However, we do also see evidence for abnormal radial migration and elevated cell death in these Tuba1aquas mutants. The significant difference in molecular phenotypes is likely to be the effect of a deletion of the gene as compared to production of a truncated polypeptide that may perturb microtubule dynamics.
The analysis of total α-tubulin protein levels in these mutants raises some other interesting points. Deletion of Tuba1a leads to decreased overall α-tubulin levels but not as dramatically as the reduction in the Tuba1aquas mutants. This is consistent with the autoregulation of tubulin and suggests that other isoforms of Tuba1a are upregulated in the deletion mutant. However, given the significant phenotypes in the Tuba1a mutant, these other isoforms are likely unable to carry out some currently unknown critical function of the TUBA1A protein. Note that the ENU variant protein in the Tuba1aquas allele is likely to create a protein lacking the amino acids needed for the anti-α-tubulin antibody we used. However, if the first several amino acids are translated from this locus as a missense mutation rather than completely missing in a deletion, this is consistent with the model of tubulin autoregulation where this initial translation leads to destabilizing existing tubulin mRNA transcripts. Given the extreme phenotype of the Tuba1aquas homozygous embryos, we hypothesize the truncated TUBA1A protein produced in these mutants is not sufficient for normal embryonic development and in fact may interfere with multiple processes of tubulin physiology.
It is not known why the paradigm of genetic compensation is so different for these two gene clusters (Tuba1a/b/c as compared to Tubb2a/b). Why does loss of one result in severe malformations of cortical development while loss of others does not compromise survival or gross brain development? Do cells express a mix of α and β tubulin monomers? Are there changes in specific gene expression over time, perhaps between neurogenesis stages and terminal differentiation? These questions remain largely unanswered although a Tubb2b-eGFP transgenic line suggests Tubb2b is expressed in both progenitors and postmitotic neurons [55]. A detailed expression analysis would be useful in generating models to explain these genetic findings. Unfortunately, this is currently quite challenging. No antibodies will distinguish between these proteins in an immunohistochemical analysis (S6 and S7 Figs). RNA expression studies are complicated by this homology as well (S8 and S9 Figs). RNA in situ hybridization probes against the untranslated regions may be able to distinguish between genes but some hypotheses about co-expression at the cellular level would be challenging with in situ hybridization-based approaches. RNA-Seq, both bulk sequencing and single cell sequencing, would potentially be useful in addressing these models at least in a preliminary way. However, our experience with the quasimodo mouse mutant exome sequencing suggests the current library preparations and/or sequence alignment algorithms are confounded by the sequence similarities between these genes, and these data sets should to be interpreted with extreme caution. We propose that a series of epitope-fusion protein alleles in the mouse would be a useful tool set to explore both expression of discrete tubulin genes and will serve as a platform for biochemical approaches to identify microtubule associated protein and other cellular interaction partners.
While the phenotypes we present here are striking malformations of cortical development, we do not yet completely know the cellular and molecular basis for these phenotypes. The histological changes in the ventricular zone, intermediate zone and cortical plate all suggest fundamental perturbations of neurogenesis and/or cellular migration. Detailed mechanistic studies are needed to determine how these processes are altered in the face of tubulin mutations. The human missense mutations are not extensively modeled in mice, with the exception of the Tubb3R262C model of CFEOM [31]. A mouse model of some of the most common mutations in Tuba1a would be helpful in both understanding the underlying mechanism(s), but also as a platform for testing potential therapeutic intervention.
Human genetics studies have identified a series of tubulin genes as crucial building blocks of the brain and tubulins have long been appreciated as important components for neuronal cell biology. As our genomic tools and mouse modeling capabilities have advanced, we now have the exciting opportunity to return to some fundamental questions about the requirement(s) and role(s) of tubulin genes in mammalian development and disease.
Materials & methods
Ethics statement
This study was carried out in strict accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health. Animals were housed at or below IACUC determined densities with AALAC-approved veterinary care and fed Autoclavable Mouse Breeder Diet 5021 (LabDiet, St. Louis, MO). The protocol was approved by the Institutional Animal Care and Use Committee of the Cincinnati Children’s Hospital Medical Center (protocol number IACUC2016-0068). All euthanasia (cervical dislocation followed by thoracotomy) and embryo harvests were performed after isoflurane sedation to minimize animal suffering and discomfort.
Mouse allele generation
CRISPR guides flanking the tubulin genes of interest were evaluated using the Fusi (Benchling.com) and Moreno-Mateos (Crisprscan.org) algorithms. Potential guide RNA (gRNA) sequences were selected and ordered as complementary oligonucleotide pairs with BbsI over-hangs (S1 Table; IDT, Coralville, IA). These were ligated into the pSpCas9(BB)-2A-GFP (px458) vector and transfected into MK4 cells at low confluence using the Lipofectamine 2000 transfection reagent (Thermo Fisher Scientific, Massachusetts). Cells were harvested 48 h after transfection and genomic DNA was isolated and used with the Surveyor mutation detection kit (IDT) in order to test gRNA cutting efficiency. As a control, cutting efficiencies of potential guides were compared with that of a previously-published mTet2 gRNA. Cas9 and gRNAs were injected into C57BL/6N zygotes (Taconic) by the CCHMC Transgenic Core. Potential founders were validated with Sanger sequencing of tail DNA and subsequently maintained on a C57BL/6J (Jackson Labs) background. pSpCas9(BB)-2 A-GFP (PX458) was a gift from Feng Zhang (Addgene plasmid #48138).
Animal husbandry
For embryo collections, noon of the day of vaginal plug detection was designated as E0.5. Embryos and brains were removed and imaged using a Zeiss Discovery.V8 stereo-microscope.
ENU mutagenesis
ENU mutagenesis was performed as previously described [56, 57] on C57BL/6J males.
Histological analysis
For adult histology, littermate animals underwent transcardial perfusion using cold heparinized phosphate buffered saline (PBS) and formalin (SIGMA) solution. Brains were dissected and fixed for 72 h in formalin at room temperature followed by immersion in 70% ethanol (for histology). Embryo samples were fixed in Bouin’s fixative solution, formalin or 4% paraformaldehyde (PFA). Samples were then paraffin embedded or sucrose-dehydrated and cryo-embedded, sectioned at 6μm for adult tissue and 10μm for embryonic tissue, and processed through hematoxylin and eosin (H&E) or Nissl staining. Sections were sealed using Cytoseal Mounting Medium (Thermo-Scientific). All adult histological samples shown are representative examples from at least 3 animals of each genotype with at least 2 slides analyzed per animal. Dot plots are used to precisely show how many measurements are made.
Immunohistochemistry
Immunohistochemistry was done accorded to standard protocols. In brief, E14.5 and E16.5 embryos and P0 brains were fixed overnight in 4% paraformaldehyde, dehydrated in 30% sucrose, cryo-embedded and sectioned at a thickness of 10 μm. P0 sections were collected at a thickness of 12 μm. Images were collected from a sample set of at least 2 slides of at least 3–5 sections for each animal and 3 control and 3 mutant embryos for each genotype at E14.5 and E16.5. P0 immunohistochemistry done on at least two animals per genotype, with 4 slides from each animal with 3–5 sections per slide. Sections for pHH3, TuJ1, NeuN, CTIP2, MBP, TBR1 were placed in boiling citrate buffer (sodium citrate 1M, citric acid 1M pH 6.0) and allowed to cool on bench top for 40 minutes. Sections for TBR2 were boiled for 4 minutes in antigen unmasking solution (Vector Labs, Burlingame, CA). Slides were blocked in 4% Normal Goat Serum in PBST for 30 minutes and then incubated overnight at 4 oC in primary antibody. Secondary antibodies were added for 1 hour at room temperature. Slides were then washed in 1X PBS and DAPI was added for 15 minutes, after which slides were sealed with ProLong Gold (Invitrogen) and imaged on a Nikon Eclipse Ti or Nikon C2 confocal microscope. Primary antibodies used were pHH3 (1:500, SIGMA), TuJ1 (1:500, SIGMA), TBR2 (1:200, Abcam ab23345), TBR1 (1:500, Abcam ab31940), CTIP2 (1:1000, Abcam ab28448), and NeuN (1:1000, Millipore clone A60). Secondary antibodies were goat anti-rabbit (1:500 Invitrogen Alexa Fluor 488) for pHH3, TBR2, TBR1, and CTIP2, and goat anti-mouse (1:500 Invitrogen Alex Fluor 488) for TuJ1 and NeuN.
MBP immunohistochemistry staining was done with 3,3′-Diaminobenzidine (DAB) labeling. 5μm paraffin sections were de-paraffinized, rehydrated in graded ethanol, and incubated with chicken anti-MBP (1:500, Aves MBP) for 2 hours at room temperature, rinsed, and incubated in biotinylated goat-anti chicken secondary antibody (1:500; Vector Laboratories, Burlingame, CA, USA) for 2 hours at room temperature. Sections were then washed in PBS and incubated in avidin-biotin-complex (ABC) solution (Vector Laboratories) for 1 hour at room temperature. Sections were then washed and visualized using DAB for 5 min followed by a PBS wash, ethanol dehydration, xylene clearance, and mounted with Cytoseal (Thermo). Images were obtained with a Zeiss Discovery.V8 Stereoscope. From each genotype, 2 animals were included. From each of these animals, 2 slides each with 3–6 sections were stained. Sample sizes for quantifications are indicated in each relevant data plot. For P0 and adult histological analysis, areas of higher magnification analysis are shown (S3 Fig). Established anatomical landmarks were used to define cortical areas.
Quantification of pHH3-positive cells was done on Imaris Image Analysis software (Bitplane, South Windsor, CT) by manually drawing a surface around the Ventricular Zone and calculating the number of positive cells. Quantification of TBR2 and CC3 was done using Nikon Elements software by manually drawing a Region of Interest (ROI) around the entire cortex and calculating immuno-positive cells with a bright spot detection function.
“Binning” data for CTIP2 and TBR1-positive cells were acquired with Nikon Elements software after drawing an ROI that extended from the VZ to the pial surface. The length of this ROI is divided by three. The three identical squares were drawn to represent the three bins. Bright spot detection tool was employed to determine the amount of immune-positive cells in each bin. GraphPad Prism was used to plot the data and perform statistical analysis.
EdU/Ki67 staining and quantification
EdU (Invitrogen) was injected at a concentration of 20mg/kg interperitoneally into E13.5 pregnant females. Embryos were harvested 24 or 72 hours later and stained as above. Ki67 primary antibody (1:200 Abcam ab15580) was added overnight at 4°C. A goat anti-rabbit secondary antibody (1:500 Invitrogen Alexa-Fluor 594) was added for 1 hour at room temperature. Slides were washed twice with 1X PBS and once with 3% BSA in PBS. EdU was labeled using the Click-iT EdU Alexa Fluor 488 Imaging Kit (Invitrogen).
Quantification was done using Imaris software package. A surface was manually created that contained the cortex from the VZ to the CP and the enclosed area was quantified. EdU positive cells and Ki67 positive cells were then identified using the Imaris “Spots” function. Cells both EdU and Ki67 positive were quantified using the “Colocalize Spots” function. Each positive cell count was normalized to the area of the section. Binning data for EdU positive cells was performed with Nikon Elements. Three identical rectangular Region of Interest bins were made for each section by dividing the width of the cortex into three equal parts. The number of cells in each bin was calculated using bright spot detection. The percentage of cells in each bin was used to determine any differences.
Western immunoblotting
Whole cell lysate from P0/P1 brains were extracted with RIPA buffer (Thermo Scientific) with a protease and phosphatase inhibitor cocktail (Calbiochem) added immediately before cell lysis. A standard fluorescent western blotting protocol was followed. 5 μg of protein lysates were electrophoresed in 4–12% gradient Tris glycine PAGE gel (Thermo Fisher Scientific) and 1X Tris -Glycine SDS running buffer (Bio-Rad) and then transferred into PVDF membrane using Tris-Glycine transfer buffer with 20% methanol. Proteins were blocked in Odyssey blocking buffer (Licor Bioscience). Antibodies used were mouse anti TUBB3 (Tuj1, 1:400, SIGMA T8660), mouse anti-pan-β-tubulin (1:1000, Millipore 05–661), anti-pan-α-tubulin (1:500, Sigma T6199) and rabbit anti-actin (1:3000, SIGMA A2066) and were incubated with membrane overnight. Species specific secondary antibodies were applied to the membrane after stringent PBS washes of the primary antibodies. Odyssey CLx Imaging System were used to detect the bands of expression and Image Studio Lite software was used to analyze the band intensity. In each western experiment, GAPDH served as loading control and intensity of tubulin was normalized against GAPDH band intensity. For lysate from embryonic brains, we used a second loading/ normalization method by normalizing against total protein stain (REVERT from Licor Bioscience using manufacturer protocols).
Statistical analysis
Analyses and data plots were generated with Prism8 (GraphPad) and plots show the mean +/- the 95% confidence interval. An ANOVA was performed for experiments with more than two comparisons. If an ANOVA p-value was ≤ 0.05, a Tukey’s multiple comparison test was performed to analyze which group(s) within the experiment were different from each other. Experiments with two sets of data were analyzed with a student’s t-test. The p-values for these experiments are shown in the relevant figure and/or supplemental data set. We report the statistical test values directly to facilitate readership and largely refrain from assigning labels of “significance” on our own.
All data presented here are available to the community in accordance with best practices for data sharing.
Supporting information
Additional CRISPR-Cas9 deletions of Tubb2a (A,C) and Tuba1a (B,D).
(TIF)
Extended PCR analysis of Tubb2a (A), Tubb2b (B), and Tuba1a (C) deletion alleles.
(PDF)
High magnification images with Hematoxylin and Eosin staining as shown for these alleles in Fig 2 for wild-type (A,E,I), Tubb2ad3964 (B,F,J), Tubb2ad4222 (C,G,K), and Tubb2bd4185 (D,H,L) homozygous mice.
(TIF)
Quantification of the CTIP2 (A-C) and TBR1 (D-I) in the Tubb2a (A,B,D,E,G,H) and Tubb2b (C,F,I) deletion mutants.
(TIF)
(DOCX)
(DOCX)
(DOCX)
(DOCX)
Analysis of Tubb2a and Tubb2b genomic DNA (A,C) and cDNA (B,D) in Tubb2a and Tubb2b mutants.
(DOCX)
(DOCX)
(DOCX)
(DOCX)
(DOCX)
(DOCX)
(DOCX)
(DOCX)
(DOCX)
(DOCX)
(DOCX)
(DOCX)
Acknowledgments
We are grateful to Yueh-Chiang Hu and the CCHMC Transgenic Animal and Genome Editing Core for advice and technical assistance, and to Bill Dobyns for extensive discussion of this work prior to publication.
Data Availability
All relevant data are within the manuscript and its Supporting Information files.
Funding Statement
Funding was provided by the Cincinnati Children's Research Foundation and National Institutes of Health (R01NS085023 to R.W.S.). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1.Alberts B. Molecular biology of the cell. Sixth edition ed. New York, NY: Garland Science, Taylor and Francis Group; 2015. 1 volume (various pagings) p. [Google Scholar]
- 2.Bahi-Buisson N, Poirier K, Fourniol F, Saillour Y, Valence S, Lebrun N, et al. The wide spectrum of tubulinopathies: what are the key features for the diagnosis? Brain. 2014;137(Pt 6):1676–700. 10.1093/brain/awu082 . [DOI] [PubMed] [Google Scholar]
- 3.Bahi-Buisson N, Poirier K, Boddaert N, Saillour Y, Castelnau L, Philip N, et al. Refinement of cortical dysgeneses spectrum associated with TUBA1A mutations. J Med Genet. 2008;45(10):647–53. 10.1136/jmg.2008.058073 . [DOI] [PubMed] [Google Scholar]
- 4.Cushion TD, Dobyns WB, Mullins JG, Stoodley N, Chung SK, Fry AE, et al. Overlapping cortical malformations and mutations in TUBB2B and TUBA1 A. Brain. 2013;136(Pt 2):536–48. 10.1093/brain/aws338 . [DOI] [PubMed] [Google Scholar]
- 5.Fallet-Bianco C, Laquerriere A, Poirier K, Razavi F, Guimiot F, Dias P, et al. Mutations in tubulin genes are frequent causes of various foetal malformations of cortical development including microlissencephaly. Acta Neuropathol Commun. 2014;2:69 10.1186/2051-5960-2-69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jansen AC, Oostra A, Desprechins B, De Vlaeminck Y, Verhelst H, Regal L, et al. TUBA1A mutations: from isolated lissencephaly to familial polymicrogyria. Neurology. 2011;76(11):988–92. 10.1212/WNL.0b013e31821043f5 . [DOI] [PubMed] [Google Scholar]
- 7.Keays DA, Tian G, Poirier K, Huang GJ, Siebold C, Cleak J, et al. Mutations in alpha-tubulin cause abnormal neuronal migration in mice and lissencephaly in humans. Cell. 2007;128(1):45–57. 10.1016/j.cell.2006.12.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Morris-Rosendahl DJ, Najm J, Lachmeijer AM, Sztriha L, Martins M, Kuechler A, et al. Refining the phenotype of alpha-1a Tubulin (TUBA1A) mutation in patients with classical lissencephaly. Clin Genet. 2008;74(5):425–33. 10.1111/j.1399-0004.2008.01093.x . [DOI] [PubMed] [Google Scholar]
- 9.Poirier K, Keays DA, Francis F, Saillour Y, Bahi N, Manouvrier S, et al. Large spectrum of lissencephaly and pachygyria phenotypes resulting from de novo missense mutations in tubulin alpha 1A (TUBA1A). Hum Mutat. 2007;28(11):1055–64. 10.1002/humu.20572 . [DOI] [PubMed] [Google Scholar]
- 10.Romaniello R, Arrigoni F, Cavallini A, Tenderini E, Baschirotto C, Triulzi F, et al. Brain malformations and mutations in alpha- and beta-tubulin genes: a review of the literature and description of two new cases. Dev Med Child Neurol. 2014;56(4):354–60. 10.1111/dmcn.12370 . [DOI] [PubMed] [Google Scholar]
- 11.Tian G, Jaglin XH, Keays DA, Francis F, Chelly J, Cowan NJ. Disease-associated mutations in TUBA1A result in a spectrum of defects in the tubulin folding and heterodimer assembly pathway. Hum Mol Genet. 2010;19(18):3599–613. 10.1093/hmg/ddq276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kumar RA, Pilz DT, Babatz TD, Cushion TD, Harvey K, Topf M, et al. TUBA1A mutations cause wide spectrum lissencephaly (smooth brain) and suggest that multiple neuronal migration pathways converge on alpha tubulins. Hum Mol Genet. 2010;19(14):2817–27. 10.1093/hmg/ddq182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lecourtois M, Poirier K, Friocourt G, Jaglin X, Goldenberg A, Saugier-Veber P, et al. Human lissencephaly with cerebellar hypoplasia due to mutations in TUBA1A: expansion of the foetal neuropathological phenotype. Acta Neuropathol. 2010;119(6):779–89. 10.1007/s00401-010-0684-z . [DOI] [PubMed] [Google Scholar]
- 14.Myers KA, Bello-Espinosa LE, Kherani A, Wei XC, Innes AM. TUBA1A Mutation Associated With Eye Abnormalities in Addition to Brain Malformation. Pediatr Neurol. 2015;53(5):442–4. 10.1016/j.pediatrneurol.2015.07.004 . [DOI] [PubMed] [Google Scholar]
- 15.Oegema R, Cushion TD, Phelps IG, Chung SK, Dempsey JC, Collins S, et al. Recognizable cerebellar dysplasia associated with mutations in multiple tubulin genes. Hum Mol Genet. 2015;24(18):5313–25. 10.1093/hmg/ddv250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Okumura A, Hayashi M, Tsurui H, Yamakawa Y, Abe S, Kudo T, et al. Lissencephaly with marked ventricular dilation, agenesis of corpus callosum, and cerebellar hypoplasia caused by TUBA1A mutation. Brain Dev. 2013;35(3):274–9. 10.1016/j.braindev.2012.05.006 . [DOI] [PubMed] [Google Scholar]
- 17.Shimojima K, Narita A, Maegaki Y, Saito A, Furukawa T, Yamamoto T. Whole-exome sequencing identifies a de novo TUBA1A mutation in a patient with sporadic malformations of cortical development: a case report. BMC Res Notes. 2014;7:465 10.1186/1756-0500-7-465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yokoi S, Ishihara N, Miya F, Tsutsumi M, Yanagihara I, Fujita N, et al. TUBA1A mutation can cause a hydranencephaly-like severe form of cortical dysgenesis. Sci Rep. 2015;5:15165 10.1038/srep15165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cushion TD, Paciorkowski AR, Pilz DT, Mullins JG, Seltzer LE, Marion RW, et al. De novo mutations in the beta-tubulin gene TUBB2A cause simplified gyral patterning and infantile-onset epilepsy. Am J Hum Genet. 2014;94(4):634–41. 10.1016/j.ajhg.2014.03.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ejaz R, Lionel AC, Blaser S, Walker S, Scherer SW, Babul-Hirji R, et al. De novo pathogenic variant in TUBB2A presenting with arthrogryposis multiplex congenita, brain abnormalities, and severe developmental delay. Am J Med Genet A. 2017;173(10):2725–30. 10.1002/ajmg.a.38352 . [DOI] [PubMed] [Google Scholar]
- 21.Lee H, Deignan JL, Dorrani N, Strom SP, Kantarci S, Quintero-Rivera F, et al. Clinical exome sequencing for genetic identification of rare Mendelian disorders. JAMA. 2014;312(18):1880–7. 10.1001/jama.2014.14604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rodan LH, El Achkar CM, Berry GT, Poduri A, Prabhu SP, Yang E, et al. De Novo TUBB2A Variant Presenting With Anterior Temporal Pachygyria. J Child Neurol. 2017;32(1):127–31. 10.1177/0883073816672998 . [DOI] [PubMed] [Google Scholar]
- 23.Amrom D, Tanyalcin I, Verhelst H, Deconinck N, Brouhard GJ, Decarie JC, et al. Polymicrogyria with dysmorphic basal ganglia? Think tubulin! Clin Genet. 2014;85(2):178–83. 10.1111/cge.12141 . [DOI] [PubMed] [Google Scholar]
- 24.Cederquist GY, Luchniak A, Tischfield MA, Peeva M, Song Y, Menezes MP, et al. An inherited TUBB2B mutation alters a kinesin-binding site and causes polymicrogyria, CFEOM and axon dysinnervation. Hum Mol Genet. 2012;21(26):5484–99. 10.1093/hmg/dds393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Guerrini R, Mei D, Cordelli DM, Pucatti D, Franzoni E, Parrini E. Symmetric polymicrogyria and pachygyria associated with TUBB2B gene mutations. Eur J Hum Genet. 2012;20(9):995–8. 10.1038/ejhg.2012.21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jaglin XH, Poirier K, Saillour Y, Buhler E, Tian G, Bahi-Buisson N, et al. Mutations in the beta-tubulin gene TUBB2B result in asymmetrical polymicrogyria. Nat Genet. 2009;41(6):746–52. 10.1038/ng.380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Laquerriere A, Gonzales M, Saillour Y, Cavallin M, Joye N, Quelin C, et al. De novo TUBB2B mutation causes fetal akinesia deformation sequence with microlissencephaly: An unusual presentation of tubulinopathy. Eur J Med Genet. 2016;59(4):249–56. 10.1016/j.ejmg.2015.12.007 . [DOI] [PubMed] [Google Scholar]
- 28.Romaniello R, Tonelli A, Arrigoni F, Baschirotto C, Triulzi F, Bresolin N, et al. A novel mutation in the beta-tubulin gene TUBB2B associated with complex malformation of cortical development and deficits in axonal guidance. Dev Med Child Neurol. 2012;54(8):765–9. 10.1111/j.1469-8749.2012.04316.x . [DOI] [PubMed] [Google Scholar]
- 29.Jamuar SS, Lam AT, Kircher M, D'Gama AM, Wang J, Barry BJ, et al. Somatic mutations in cerebral cortical malformations. N Engl J Med. 2014;371(8):733–43. 10.1056/NEJMoa1314432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Poirier K, Saillour Y, Bahi-Buisson N, Jaglin XH, Fallet-Bianco C, Nabbout R, et al. Mutations in the neuronal ss-tubulin subunit TUBB3 result in malformation of cortical development and neuronal migration defects. Hum Mol Genet. 2010;19(22):4462–73. 10.1093/hmg/ddq377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tischfield MA, Baris HN, Wu C, Rudolph G, Van Maldergem L, He W, et al. Human TUBB3 mutations perturb microtubule dynamics, kinesin interactions, and axon guidance. Cell. 2010;140(1):74–87. 10.1016/j.cell.2009.12.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Simons C, Wolf NI, McNeil N, Caldovic L, Devaney JM, Takanohashi A, et al. A de novo mutation in the beta-tubulin gene TUBB4A results in the leukoencephalopathy hypomyelination with atrophy of the basal ganglia and cerebellum. Am J Hum Genet. 2013;92(5):767–73. 10.1016/j.ajhg.2013.03.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Breuss M, Heng JI, Poirier K, Tian G, Jaglin XH, Qu Z, et al. Mutations in the beta-tubulin gene TUBB5 cause microcephaly with structural brain abnormalities. Cell Rep. 2012;2(6):1554–62. 10.1016/j.celrep.2012.11.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Di Donato NMD, Timms AE, Aldinger KA, Mirzaa GM, Bennett JT, Collins S, et al. Analysis of 17 genes detects mutations in 81% of 811 patients with lissencephaly. Genetics in Medicine. 2018; 20(11):1354–1364. 10.1038/gim.2018.8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Guo J, Qiang M, Luduena RF. The distribution of beta-tubulin isotypes in cultured neurons from embryonic, newborn, and adult mouse brains. Brain Res. 2011;1420:8–18. 10.1016/j.brainres.2011.08.066 . [DOI] [PubMed] [Google Scholar]
- 36.Leandro-Garcia LJ, Leskela S, Landa I, Montero-Conde C, Lopez-Jimenez E, Leton R, et al. Tumoral and tissue-specific expression of the major human beta-tubulin isotypes. Cytoskeleton (Hoboken). 2010;67(4):214–23. 10.1002/cm.20436 . [DOI] [PubMed] [Google Scholar]
- 37.Latremoliere A, Cheng L, DeLisle M, Wu C, Chew S, Hutchinson EB, et al. Neuronal-Specific TUBB3 Is Not Required for Normal Neuronal Function but Is Essential for Timely Axon Regeneration. Cell Rep. 2018;24(7):1865–79 e9. Epub 2018/08/16. 10.1016/j.celrep.2018.07.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bamba Y, Shofuda T, Kato M, Pooh RK, Tateishi Y, Takanashi J, et al. In vitro characterization of neurite extension using induced pluripotent stem cells derived from lissencephaly patients with TUBA1A missense mutations. Mol Brain. 2016;9(1):70 Epub 2016/07/20. 10.1186/s13041-016-0246-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Niwa S, Takahashi H, Hirokawa N. beta-Tubulin mutations that cause severe neuropathies disrupt axonal transport. EMBO J. 2013;32(10):1352–64. Epub 2013/03/19. 10.1038/emboj.2013.59 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gartz Hanson M, Aiken J, Sietsema DV, Sept D, Bates EA, Niswander L, et al. Novel alpha-tubulin mutation disrupts neural development and tubulin proteostasis. Dev Biol. 2016;409(2):406–19. 10.1016/j.ydbio.2015.11.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Furuse T, Yamada I, Kushida T, Masuya H, Miura I, Kaneda H, et al. Behavioral and neuromorphological characterization of a novel Tuba1 mutant mouse. Behav Brain Res. 2012;227(1):167–74. 10.1016/j.bbr.2011.11.002 . [DOI] [PubMed] [Google Scholar]
- 42.Stottmann RW, Donlin M, Hafner A, Bernard A, Sinclair DA, Beier DR. A mutation in Tubb2b, a human polymicrogyria gene, leads to lethality and abnormal cortical development in the mouse. Hum Mol Genet. 2013;22(20):4053–63. 10.1093/hmg/ddt255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Cleveland DW. Autoregulated control of tubulin synthesis in animal cells. Curr Opin Cell Biol. 1989;1(1):10–4. Epub 1989/02/01. . [DOI] [PubMed] [Google Scholar]
- 44.Cleveland DW, Sullivan KF. Molecular biology and genetics of tubulin. Annu Rev Biochem. 1985;54:331–65. Epub 1985/01/01. 10.1146/annurev.bi.54.070185.001555 . [DOI] [PubMed] [Google Scholar]
- 45.Bachurski CJ, Theodorakis NG, Coulson RM, Cleveland DW. An amino-terminal tetrapeptide specifies cotranslational degradation of beta-tubulin but not alpha-tubulin mRNAs. Mol Cell Biol. 1994;14(6):4076–86. Epub 1994/06/01. 10.1128/mcb.14.6.4076 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ben-Ze'ev A, Farmer SR, Penman S. Mechanisms of regulating tubulin synthesis in cultured mammalian cells. Cell. 1979;17(2):319–25. Epub 1979/06/01. 10.1016/0092-8674(79)90157-0 . [DOI] [PubMed] [Google Scholar]
- 47.Cleveland DW, Lopata MA, Sherline P, Kirschner MW. Unpolymerized tubulin modulates the level of tubulin mRNAs. Cell. 1981;25(2):537–46. Epub 1981/08/01. 10.1016/0092-8674(81)90072-6 . [DOI] [PubMed] [Google Scholar]
- 48.Gay DA, Yen TJ, Lau JT, Cleveland DW. Sequences that confer beta-tubulin autoregulation through modulated mRNA stability reside within exon 1 of a beta-tubulin mRNA. Cell. 1987;50(5):671–9. Epub 1987/08/28. 10.1016/0092-8674(87)90325-4 . [DOI] [PubMed] [Google Scholar]
- 49.Gonzalez-Garay ML, Cabral F. alpha-Tubulin limits its own synthesis: evidence for a mechanism involving translational repression. J Cell Biol. 1996;135(6 Pt 1):1525–34. Epub 1996/12/01. 10.1083/jcb.135.6.1525 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lau JT, Pittenger MF, Cleveland DW. Reconstruction of appropriate tubulin and actin gene regulation after transient transfection of cloned beta-tubulin and beta-actin genes. Mol Cell Biol. 1985;5(7):1611–20. Epub 1985/07/01. 10.1128/mcb.5.7.1611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Yen TJ, Machlin PS, Cleveland DW. Autoregulated instability of beta-tubulin mRNAs by recognition of the nascent amino terminus of beta-tubulin. Nature. 1988;334(6183):580–5. Epub 1988/08/18. 10.1038/334580a0 . [DOI] [PubMed] [Google Scholar]
- 52.Aiken J, Moore JK, Bates EA. TUBA1A mutations identified in lissencephaly patients dominantly disrupt neuronal migration and impair dynein activity. Hum Mol Genet. 2019;28(8):1227–43. Epub 2018/12/06. 10.1093/hmg/ddy416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Stottmann RW, Driver A, Gutierrez A, Skelton MR, Muntifering M, Stepien C, et al. A heterozygous mutation in tubulin, beta 2B (Tubb2b) causes cognitive deficits and hippocampal disorganization. Genes Brain Behav. 2016. 10.1111/gbb.12327 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lek M, Karczewski KJ, Minikel EV, Samocha KE, Banks E, Fennell T, et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature. 2016;536(7616):285–91. Epub 2016/08/19. 10.1038/nature19057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Breuss M, Morandell J, Nimpf S, Gstrein T, Lauwers M, Hochstoeger T, et al. The Expression of Tubb2b Undergoes a Developmental Transition in Murine Cortical Neurons. J Comp Neurol. 2015;523(15):2161–86. 10.1002/cne.23836 . [DOI] [PubMed] [Google Scholar]
- 56.Stottmann R, Beier D. ENU Mutagenesis in the Mouse. Curr Protoc Hum Genet. 2014;82:1541–0. Epub 2014/07/22. 10.1002/0471142905.hg1504s82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Stottmann RW, Moran JL, Turbe-Doan A, Driver E, Kelley M, Beier DR. Focusing forward genetics: a tripartite ENU screen for neurodevelopmental mutations in the mouse. Genetics. 2011;188(3):615–24. Epub 2011/04/26. 10.1534/genetics.111.126862 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional CRISPR-Cas9 deletions of Tubb2a (A,C) and Tuba1a (B,D).
(TIF)
Extended PCR analysis of Tubb2a (A), Tubb2b (B), and Tuba1a (C) deletion alleles.
(PDF)
High magnification images with Hematoxylin and Eosin staining as shown for these alleles in Fig 2 for wild-type (A,E,I), Tubb2ad3964 (B,F,J), Tubb2ad4222 (C,G,K), and Tubb2bd4185 (D,H,L) homozygous mice.
(TIF)
Quantification of the CTIP2 (A-C) and TBR1 (D-I) in the Tubb2a (A,B,D,E,G,H) and Tubb2b (C,F,I) deletion mutants.
(TIF)
(DOCX)
(DOCX)
(DOCX)
(DOCX)
Analysis of Tubb2a and Tubb2b genomic DNA (A,C) and cDNA (B,D) in Tubb2a and Tubb2b mutants.
(DOCX)
(DOCX)
(DOCX)
(DOCX)
(DOCX)
(DOCX)
(DOCX)
(DOCX)
(DOCX)
(DOCX)
(DOCX)
(DOCX)
Data Availability Statement
All relevant data are within the manuscript and its Supporting Information files.