

Abstract
WHIM syndrome is a rare combined primary immunodeficiency disease named by acronym for the diagnostic tetrad of Warts, Hypogammaglobulinemia, Infections and Myelokathexis. Myelokathexis is a unique form of non-cyclic severe congenital neutropenia caused by accumulation of mature and degenerating neutrophils in the bone marrow; monocytopenia and lymphopenia, especially B lymphopenia, also commonly occur. WHIM syndrome is usually caused by autosomal dominant mutations in the G protein-coupled chemokine receptor CXCR4 that impair desensitization, resulting in enhanced and prolonged G protein- and β-arrestin-dependent responses. Accordingly, CXCR4 antagonists have shown promise as mechanism-based treatments in Phase 1 clinical trials. This review is based on analysis of all 105 published cases of WHIM syndrome and covers current concepts, recent advances, unresolved enigmas and controversies, and promising future research directions.
Keywords: chemokine, CXCL12, CXCR4, CXCR2, myelokathexis, human papillomavirus, plerixafor
I. Historical background
Myelokathexis was first described as a new type of severe congenital neutropenia in 1964 by Krill and colleagues from the University of Cincinnati School of Medicine and Wolf Zuelzer from Wayne State University School of Medicine [1,2]. Remarkably, their independent but complementary findings were based on studies of the same 9-year-old girl suffering from recurrent bacterial infections, published by the New England Journal of Medicine as two single case reports six weeks apart, without cross-references [1,2]. Both papers described the same distinctive hematopathology: 1) full myeloid maturation; 2) a hypercellular bone marrow with increased granulocyte reserve (increased myeloid to erythroid [M:E] ratio with a ‘shift to the right’); and 3) numerous dysmorphic bone marrow neutrophils having cytoplasmic hypervacuolation and hyperlobulated pyknotic nuclear lobes connected by long thin strands (Figure 1). Neutrophils could be mobilized to the blood by acute infections and endotoxin challenge. In Zuelzer’s classic and astute interpretation: ‘The observations presented indicate an increased rate of intramedullary cell death and reduced viability and functional activity of the mature granulocytes in the bone marrow and suggest that the retention of cells damaged at the site of their origin was the mechanism responsible both for the “shift to the right” within the marrow and for the peripheral granulocytopenia. The term “myelokathexis” (kathexis, retention) is proposed for this mechanism [2].’
Figure 1.
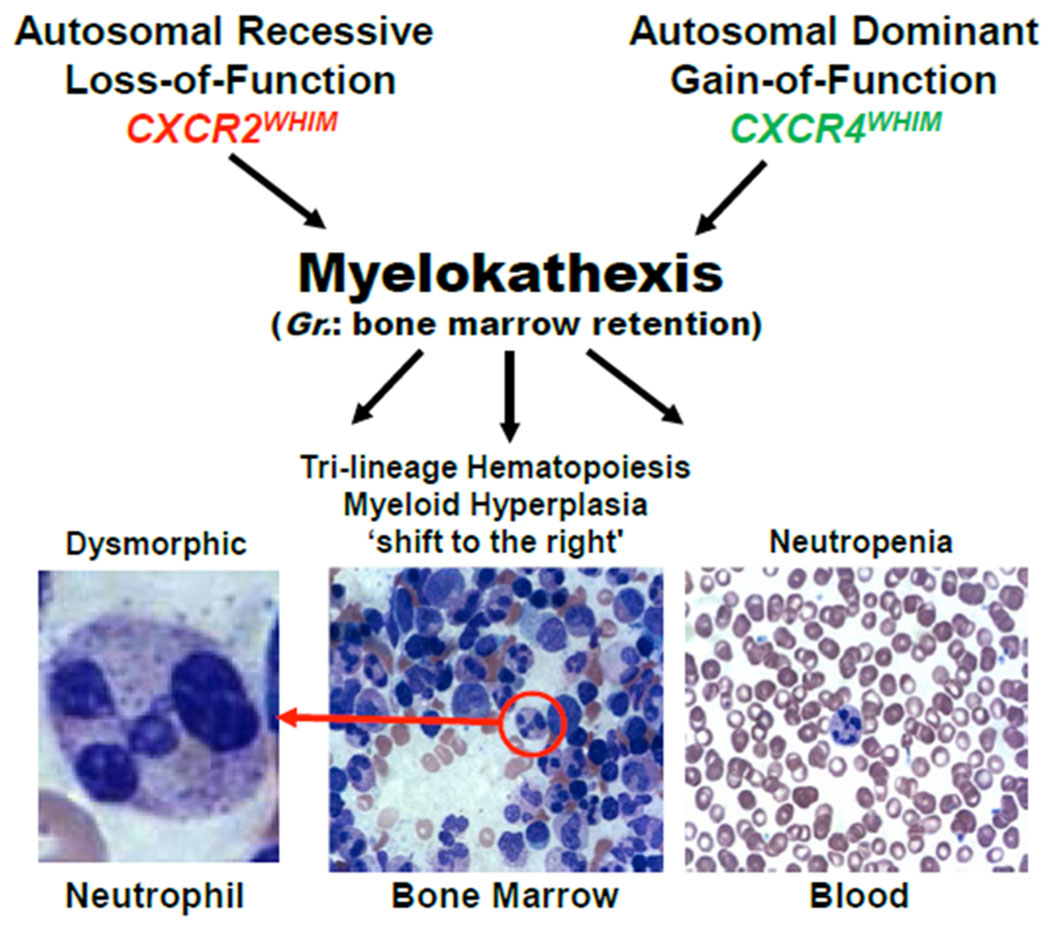
Defining hematologic features of myelokathexis, the pathognomonic syndromic feature of WHIM syndrome: neutropenia, myeloid hyperplasia with a shift to the right, and a high frequency of degenerating dysmorphic neutrophils in bone marrow. Eleven different CXCR4 mutations (see Figure 3) and one CXCR2 mutation (p.H323fs329X) have been reported in WHIM patients. The figure is a modification of Figure 1 in Reference 43. Gr., Greek. In some patients and in WHIM model mice, the neutrophils are not dysmorphic, but the other features are present.
By 1990 several other myelokathexis cases had been reported, including in several kindreds [3–11]. An autosomal dominant pattern of monogenic inheritance was suspected, and phenotype analysis found that myelokathexis commonly occurred as a syndrome together with treatment-refractory warts, hypogammaglobulinemia and recurrent infections [9]. These observations were codified by Wetzler and colleagues in the acronym WHIM, and the Online Mendelian Inheritance in Man (OMIM) catalog accepted WHIM syndrome as a new disease entity in 1990 (listed as OMIM #193670). In 2003 George Diaz from Mt. Sinai School of Medicine performed gene linkage analysis of DNA from WHIM pedigrees and discovered three different causal gain-of-function mutations in CXCR4 [12]. Greater than 95% of genotyped myelokathexis cases have CXCR4 mutations, which has oriented research into mechanism-based treatment and cure strategies.
Previously, from 1993-1996, CXCR4 (CD184) had been discovered independently multiple other times, first as an orphan G protein-coupled receptor (GPCR) superfamily member cloned under various aliases [13–17], then as an HIV coreceptor named fusin isolated using an unbiased genetic screen (Supplemental Table 1) [18–26]. CXCR4 was originally reported to be a neuropeptide Y receptor; however, this has not held up, and its homology to known chemokine receptors ultimately directed investigators to its correct identification as a receptor for the chemokine CXCL12 [25–28]. Since then, several other non-chemokine ligands for CXCR4 have been identified, including HMGB1, MIF and CD74 [29]; however, the biological significance of CXCL12 agonism is best-validated. Human CXCR4 is most closely related to two other chemokine receptors CXCR1 and CXCR2 (~37% amino acid identity), and it is encoded by a 2-exon gene on human chromosome 2: a 103 bp 5’ exon and a 1,563 bp 3’ exon separated by a 2,132 bp intron between codons 5 and 6 of a 352-codon open reading frame [30]. CXCR4 is now recognized as an essential multitasking receptor important in diverse aspects of development [31–35], cancer [36–39], immune homeostasis and infectious, inflammatory and immunologically-mediated disease [40,41]. On January 23, 2019 a search of PubMed Titles and Abstracts using the query term ‘CXCR4’ returned 11,173 published papers, including 1124 reviews. It is the most intensely studied of all chemokine receptors and one of the best understood GPCRs, from its 3-dimensional structure to its role in human disease. Of these, 110 papers, including 28 reviews, matched the search term ‘WHIM syndrome’, almost all of which were included in the list of 116 papers including 29 reviews [29,42–48] that matched the search term ‘myelokathexis’, which demonstrates statistically that myelokathexis is almost pathognomonic for WHIM syndrome.
WHIM syndrome is typically classified as a severe congenital neutropenia, yet most patients have multiple leukocyte deficiencies [42,49,50], even panleukopenia [51,52], and therefore it might also be classified as a severe combined immunodeficiency. B lymphopenia is particularly severe and this probably accounts in part for hypogammaglobulinemia [53]. Some patients also have developmental defects of the cardiovascular, urogenital and nervous systems, although only the cardiovascular defects appear to be clinically significant [42,54–57]. Paradoxically, WHIM syndrome behaves as a relatively benign immunodeficiency. Infections are not usually invasive or life-threatening, and patients survive into adulthood, thanks in part to the ability of the host response to acute infections to mobilize leukocytes to the blood.
Remarkably, Krill et al and Zuelzer’s original myelokathexis patient was spontaneously cured of WHIM syndrome as an adult by a second genetic event known as chromothripsis (‘chromosome shattering’ followed by complex chromosomal rearrangements) [58]. The mechanism involved deletion of the CXCR4 WHIM allele and 163 other genes on one copy of chromosome 2 in a single hematopoietic stem cell (HSC), which fortuitously acquired a selective and benign growth advantage resulting in repopulation of her bone marrow with chromothriptic non-WHIM myeloid cells [58–61]. She is now followed by us at the National Institutes of Health (NIH) and is referred to as WHIM-09.
II. Epidemiology
We identified 105 individuals in the biomedical literature through January 31, 2019, who were reported to have a diagnosis of myelokathexis or WHIM syndrome or a germline CXCR4 mutation. This expands the list of 60 cases aggregated in a classic review published in 2012 by Beaussant-Cohen and colleagues, who estimated the frequency of WHIM syndrome in France to be ~0.23 cases per 1,000,000 live births [42]. Many of the patients have been reported multiple times (Supplemental Appendix); a minority have been reported to French and US neutropenia registries [42,62–64].
We mined data reported for all 105 patients across 207 clinical, genetic, hematologic, immunologic, cellular and molecular parameters. The cases included 62 females, 41 males and 2 individuals whose sex was not reported, distributed among 6 infants, 47 children (ages 1-18), 48 adults, and 4 individuals whose age was not reported (Supplemental Table 2). The broad and inclusive search strategy we used enabled identification of the classic WHIM tetrad due to CXCR4 mutations in 38% of the 105 cases, as well as phenotypic and genotypic variants (57% and 5%, respectively) of the classic presentation (Supplemental Table 3). The great majority of cases were from Western Europe, especially Italy and France, and the United States. Race information was available for only 44 of the 105 reported cases, including 32 Caucasians, 10 Asians and 2 Hispanics. One report which focused on HPV seroprevalence in Africa alluded to 13 Nigerian females with a ‘history or signs of WHIM as a genetic disorder’, but without providing diagnostic evidence or any other information [65]. These cases are therefore not included with the 105 aggregated here, and no other cases of African ancestry have been described yet. One case was from South Africa, but the race was not specified [66].
III. Genetics
Gene linkage analysis of 7 WHIM pedigrees originally established CXCR4 on human chromosome 2q22.1 as the disease gene for WHIM syndrome [12,67]. Germline CXCR4 mutations have not been reported in any other disease. Forty-seven of the 105 cases occurred de novo; the other 58 were distributed among 21 pedigrees, the largest of which had 6 affected members. Eighty-six of the 105 cases were reported to have specific CXCR4 mutations and 3 lacked CXCR4 mutations and remain genetically undefined. Genotypic information was lacking for the other 14 cases, most of which were reported before CXCR4 was discovered as the disease gene [12]. Two siblings from Slovenia who presented with myelokathexis and recurrent infections but not warts or hypogammaglobulinemia were found to have an autosomal recessive loss-of-function (ERK phosphorylation, chemotaxis) mutation in CXCR2, designated p.H323fs329X [68]. This is the second and only other mutation in the chemokine system known to cause a Mendelian conditioncoincidentally the same one. Consistent with this, in mouse mixed bone marrow chimeras transplanted with Cxcr2−/− and Cxcr2+/+ cells, Cxcr2−/− neutrophils are preferentially retained in the bone marrow and cannot be dislodged by transient blockade of Cxcr4 [69]. Thus, CXCR2 signaling is thought to counter-regulate CXCR4 with respect to promoting hematopoietic stem cell and neutrophil egress from the bone marrow, with CXCR2 promoting egress and CXCR4 inhibiting it (Figure 2) [70,71]. The CXCR2 mechanism is thought to involve CXCL1 and CXCL2 produced by endothelial cells which direct neutrophils to cross endothelium and enter the blood. Mouse neutrophils lacking both receptors are constitutively mobilized, indicating a dominant effect for CXCR4.
Figure 2.
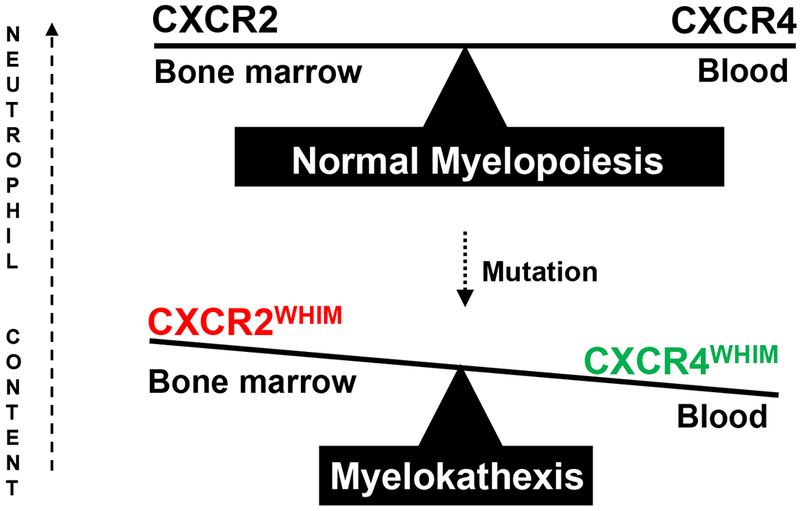
Genetic mechanisms of myelokathexis. The balance of CXCR2 release signals and CXCR4 retention signals determines the rate of neutrophil egress from bone marrow to blood and the distribution of neutrophils between the two compartments. Thus, both loss of function CXCR2 mutations and gain of function CXCR4 mutations cause myelokathexis. CXCR4 mutations account for >95% of genotyped cases of WHIM syndrome, whereas a CXCR2 mutation has been identified in two siblings.
All eleven known CXCR4 WHIM mutations as well as the CXCR2 myelokathexis mutation are located in the genomic region that encodes the C-terminal domain of the receptors (Figure 3). All but one of the CXCR4 mutations truncate this domain, four by premature termination and six by frameshifts that introduce 3 to 24 additional new amino acids [12,43]. The CXCR2 mutation is also a frameshift. The only myelokathexis-associated missense mutation is CXCR4 p.E343K, which involves a single amino acid substitution and a charge change [72]. By far the most common WHIM mutation is CXCR4 > T (p.Arg334Ter, also written informally as R334X), which accounts for 52% of genotyped cases in the literature, including both de novo and familial cases (Table 1). CXCR4 p.S338X accounts for 15 of the genotyped cases [73]. The remaining 29 genotyped cases are distributed among 10 mutations, 8 of which were observed in only 1-3 cases each. Interestingly, acquired CXCR4 WHIM-like mutations, including R334X, also occur in ~28% of cases of Waldenstrom’s macroglobulinemia, a plasma cell cancer, and are associated with poorer prognosis and worse treatment responses [74,75].
Figure 3.

CXCR4 mutations in WHIM syndrome are located in the C-terminus of the receptor. The 7-transmembrane domain crystal structure of CXCR4 is shown on the left with stars demarcating locations of WHIM mutations. The sequence of the C-tail of the mutant receptors is shown on the right compared to the wild-type sequence. Neo-sequence imposed by frameshift mutations is underlined. The circled amino acid is a missense mutation. The figure is modified and updated from Figure 2 in Reference [43].
Table 1.
Genotype-phenotype relationships among all 105 reported cases in the WHIM syndrome spectrum literature. The first 12 entries are CXCR4 genotypes, where the mutant allele is named according to the amino acid changes. W, warts; H, hypogammaglobulinemia; I, recurrent infections; M, myelokathexis; fs, frameshifted mutant allele; X, termination; +, wild type allele.
| WHIM Phenotype Penetrance (n) | Total (n) | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| WHIM | WIM | HIM | WM | HM | IM | W | M | none | |||
| GENOTYPE | +/R334X | 26 | 5 | 10 | 1 | 2 | 0 | 0 | 2 | 1 | 47 |
| +/S338X | 3 | 1 | 4 | 0 | 1 | 5 | 1 | 0 | 0 | 15 | |
| +/G323fs343X | 1 | 1 | 1 | 0 | 1 | 0 | 0 | 1 | 1 | 6 | |
| +/S339Cfs342X | 4 | 0 | 0 | 0 | 0 | 2 | 0 | 0 | 0 | 6 | |
| +/E343K | 1 | 0 | 0 | 0 | 0 | 2 | 0 | 1 | 0 | 4 | |
| +/G336X | 2 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 2 | |
| +/E343X | 1 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 2 | |
| +/S319fs343X | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | |
| +/L329Qfs341X | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | |
| +/S341Pfs365X | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | |
| +/S324fs344X | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | |
| +/+ | 2 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 3 | |
| CXCR2fs/fs | 0 | 0 | 0 | 0 | 0 | 2 | 0 | 0 | 0 | 2 | |
| Not determined | 5 | 1 | 2 | 0 | 2 | 4 | 0 | 0 | 0 | 14 | |
| Total (n) | 47 | 9 | 19 | 1 | 6 | 15 | 1 | 4 | 3 | 105 | |
CXCR4 WHIM mutations occur in the context of a gene that is exceptionally highly conserved in vertebrate phylogeny and in a region of the gene that is even more highly conserved than the gene as a whole [72]. In particular, the 19 C-terminal amino acids in CXCR4 deleted by the R334X mutation are identical from human to bird and differ by only a single amino acid between human and zebrafish [72]. In contrast, almost all other chemokine and chemokine receptor genes are highly divergent across vertebrate phylogeny with the notable and telling exceptions of genes for the sole known CXCR4 chemokine ligand CXCL12 and the atypical CXCL12 receptor ACKR3 (previously known as RDC1 and CXCR7) [26,76–80]. Accordingly, Cxcl12, Cxcr4 and Ackr3, but not any of the other ~45 chemokine or 23 chemokine receptor genes, are essential for life, as demonstrated by perinatal lethal phenotypes in the corresponding gene knockout mice [28,31,81–83]. Not surprisingly, no humans lacking CXCL12, CXCR4 or ACKR3 have been identified. Cxcl12 and Cxcr4 knockout mice are phenocopies of abnormal hematologic, cardiovascular and cerebellar development, with death occurring most likely from cardiovascular malformations (ventricular septal defect, abnormal gastric vascular development). The fatal phenotype of Ackr3 knockout mice is also cardiovascular, but pathologically distinct: atresia of all four cardiac valves [82,83].
IV. Clinical Manifestations
A. Phenotypes
Where it has been looked for, myelokathexis has almost always been found in patients with germline gain-of-function CXCR4 mutations. Exceptions include an aborted fetus with a CXCR4 G323fs343X mutation [42] and an unaffected chimeric father with R334X somatic and gonadal mosaicism [12,56]. The latter indicates that myelokathexis requires expression of the WHIM mutation specifically in hematopoietic cells.
Mild thrombocytopenia may also occur in many untreated WHIM patients; however, anemia is not a phenotype. Hypogammaglobulinemia was mentioned in 70% of the reported cases. When present, it is usually relatively mild and may involve one or several subclasses of immunoglobulin (Supplemental Table 3 and Table 1). With the exception of HPV, those WHIM patients who have severe CD4+ T lymphopenia do not present with opportunistic infections seen in HIV infection, idiopathic CD4+ T lymphopenia or other types of CD4+ T cell deficiency, and WHIM patients with even the most severe levels of neutropenia (ANC ~50 cells/microliter) rarely present with invasive bacterial infections (only 5 cases of sepsis and 3 of meningitis reported among the 105 patients). Instead, the most common infectious problems in WHIM syndrome are HPV-induced pathology lesions and recurrent infections of the oto-sino-pulmonary tract and skin caused by common extracellular Gram-positive bacteria, which were reported in 58% and 90% of the 105 patients, respectively [29]. Recurrence may result in important chronic sequelae such as hearing loss and bronchiectasis. This pattern somewhat resembles common variable immunodeficiency disease (CVID) patients, who have a selective defect in B cell development associated with antibody deficiency. An important distinction is that CVID patients classically suffer from acute and chronic gastroenteropathy that may be infectious or non-infectious, whereas in WHIM syndrome infectious enterocolitis is episodic and uncommon and inflammatory bowel disease has not been reported [84,85]. Gingivitis and tooth loss may also be problems in WHIM syndrome. Urogenital tract infections and infections with intracellular bacterial pathogens, fungi and protozoans have been documented anecdotally and are not clearly more common in WHIM patients than in the general population (Supplemental Table 4).
HPVs are a large group of structurally-related viruses that infect the basal cell layer of stratified squamous epithelium. Infection stimulates cell proliferation resulting in a broad spectrum of changes ranging from benign hyperplasia to dysplasia to invasive carcinoma. HPVs can be differentiated by tissue tropism (skin or mucous membranes), associated diseases (latent infections, skin warts, Epidermodysplasia Verruciformis, anogenital warts, condyloma acuminata, mucosal intraepithelial lesions), and their oncogenic potential (low-risk versus high-risk genital HPV types). The unusual susceptibility of WHIM patients to HPV is highlighted by a recent report of 83 novel HPV types discovered by metagenomic analysis of skin swab or wart biopsy samples from patients with immunodeficiency, most of whom were WHIM patients [86]. The viruses typically coinfected the same area of skin, in some cases even the same wart, and an unusually high proportion of the novel types were members of the gamma genus of HPV, which are not typically isolated from common warts in healthy individuals [86]. Remarkably, one patient was documented to have 17 different types of HPV recovered from eczematoid lower leg skin lesions in the absence of warts [87].
The warts in WHIM patients may be flat or exophytic and most commonly involve the hands and feet. In adults, anogenital disease is also common and may progress to condyloma acuminata and neoplasia [42]. In this regard, two WHIM patients have died with active HPV-associated disease, one with HPV+ head and neck squamous cell carcinoma [87], the other with HPV+ vulvar cancer [42]. Six additional cases of HPV-related squamous cell carcinoma andmany cases of precancerous genital lesions that have required surgical management have also been reported [42,44,50,67,87–89]. Only about half the cases had HPV status defined, and none had the exact HPV types defined. Treatment-refractory warts also occur in patients with inherited and acquired T cell deficiencies but are not typically associated with other forms of immunodeficiency that affect only the neutrophil lineage [90,91].
Only 10 of the 105 cases we found were described to have problems with oral and/or genital infections with herpes simplex virus, and 9 patients were reported to have varicella-zoster virus infections, including 3 with severe varicella and 6 with zoster. Anecdotal cases of infection with rubella, Epstein-Barr virus (EBV), influenza, Molluscum contagiosum virus, rubeola and Trichodysplasia spinulosum virus were also noted in the WHIM literature. Three cases of B cell lymphoma were recorded among the 105 patients, including two that were defined as EBV+ [92–94]; however, no other forms of EBV-related lymphoproliferative disease have been reported.
Growth and development in WHIM patients are usually normal. However, several non-immunologic developmental phenotypes have been reported in a small subset of patients, including cardiovascular, urogenital and cerebellar defects. The cardiovascular defects are the most clinically significant, especially Tetralogy of Fallot (ToF), a severe cardiovascular malformation that has a prevalence in the general population of only 1 in 3000, yet has occurred in 4 out of the 105 reported WHIM patients, all from different pedigrees (Supplemental Table 5) [44,50,55]. Approximately 20% of ToF cases in the general population are syndromic, and ~75% of these have 22q11.2 deletion syndrome, caused by mutations in the T-box transcription factor TBX1 [95]. Among these are cases of DiGeorge Syndrome, the only other primary immunodeficiency disease besides WHIM syndrome that is strongly linked to ToF [96]. Interestingly, Cxcl12, Cxcr4 and Ackr3 knockout mice also have cardiovascular defects, as mentioned previously, and Cxcl12 and Cxcr4 knockout mice commonly have ventricular septal defect, which has been reported as a stand-alone cardiovascular phenotype in WHIM patients but also occurs as one of multiple malformations that comprise ToF [44,50,55,56]. Non-immunologic developmental defects have not been reported in WHIM model mice [97]. Consistent with a cerebellar phenotype in Cxcr4−/− mice, human cerebellar structural changes have been associated with mild functional abnormalities in a few patients [98].
Interestingly, WHIM syndrome patients usually appear quite well in between infectious events, and comorbidities rarely include reports of immunologically-mediated disease. There are one report of diabetes mellitus (Type 1) [99], 4 reports of possible allergic reactions (IgG, Levaquin, strawberries and vancomycin) [9,67,100] and one report of asthma [101]. There are no other descriptions of autoimmunity, food allergy or allergy to common aero-allergens. In addition, there are no cases reported of myocardial infarction, angina, hypertension or stroke. This apparent comorbidity deficit could be due to a reporting bias because of the focus in the papers on WHIM phenotypes, or in part to the relatively narrow age distribution of the cohort (only 9 patients over age 40). Alternatively, and more interestingly, WHIM immunodeficiency might paradoxically benefit patients by protecting against immunologically-mediated and inflammatory disease. Moreover, apart from HPV-associated squamous cell carcinoma, EBV+ B cell lymphoma and two cases of EBV-negative cutaneous lymphoma, only two other defined cancers were reported among the 105 WHIM patients: a melanoma and a basal cell carcinoma [50,67]. There were no pediatric cases of cancer, and no cases of common epithelial cancers (colon, breast, lung).
B. Diagnosis and Differential Diagnosis
WHIM syndrome is a clinicopathologic diagnosis, not a genetic diagnosis, and myelokathexis is a hematopathologic diagnosis [45]. The clinical problems that bring WHIM patients to medical attention are almost always recurrent infections and problems controlling warts, both of which may occur in many other types of immunodeficiencies. In four cases, the presenting manifestation was ToF. A blood sample showing severe neutropenia will ordinarily lead directly and quickly to a specific diagnosis of myelokathexis by bone marrow biopsy or indirectly by finding a CXCR4 mutation [102]. Nevertheless, diagnosis may be delayed [44], sometimes even into adulthood, by the time required to establish and recognize a pattern of treatment-refractory warts and/or recurrent infections, especially since the causative pathogens are frequently not defined in WHIM patients, and since defined pathogens are usually not distinctive by being either classic opportunists or invasive. Diagnosis may also be delayed if the absolute neutrophil count is determined only during acute infections when patients most commonly seek medical attention, since, as mentioned previously, acute infection can mobilize neutrophils from the bone marrow of WHIM patients [1,2,44].
By definition, a diagnosis of WHIM syndrome can be certain when the diagnostic tetrad is present. However, the following proposed classification scheme is needed to accommodate the genotype and phenotype variation that has been observed: 1) complete WHIM syndrome with a CXCR4 mutation (only 38% of the 105 cases considered in this review); 2) idiopathic complete WHIM syndrome (lacking a CXCR4 mutation; 3% of the 105 cases); 3) incomplete WHIM syndrome with a CXCR4 mutation (myelokathexis without warts and/or hypogammaglobulinemia and/or recurrent infection; 57% of cases); and 4) incomplete WHIM syndrome without a CXCR4 mutation (2% of cases). Marked phenotypic heterogeneity may occur for the same genotype even within the same family (Table 1) [72]. Genetic and environmental factors that may account for this have not yet been defined.
When neutropenia and a CXCR4 mutation are both present, a diagnosis of “WHIM syndrome spectrum” is reasonable even when a bone marrow examination is not possible or does not satisfy all four criteria for myelokathexis. G6PC3 deficiency and Cohen syndrome are two other rare types of severe congenital neutropenia that may in some cases present like WHIM syndrome with myeloid hyperplasia and recurrent bacterial infections; however, both may be easily differentiated by other clinical manifestations and by genotyping [103–105]. Cohen Syndrome is caused by truncating autosomal recessive mutations in VPS13B (vacuolar protein sorting 13 homolog B) encoding a Golgi protein thought to be involved in intracellular protein glycosylation, sorting and trafficking [106]. Interestingly, features of myelokathexis and possibly other Cohen syndrome manifestations are phenocopied in border collies with the Trapped Neutrophil Syndrome, which is caused by mutations in the canine homologue of VPS13B [107]. Unlike WHIM syndrome, neutropenia in Cohen syndrome is usually milder and myeloid hyperplasia is usually left-shifted [108]. To date there have been no clear-cut published examples of acquired myelokathexis.
C. Natural History
Forty-six percent of the 105 WHIM cases reported in the literature were adults and there are 21 pedigrees, supporting the notion that the disease generally behaves as a relatively ‘benign’ immunodeficiency with relatively low mortality. The two oldest WHIM patients recorded in the literature were age 73 and 78 [42]. Nevertheless, Beaussant-Cohen et al have provided estimates that life expectancy may be reduced in WHIM patients primarily because of excess cancer mortality [42]. Updating this point, 9 deaths were reported among the 48 adults in the 105 patients we reviewed; there were no pediatric deaths. The aborted fetus with a CXCR4 mutation from a WHIM kindred [42] was found to have a double aortic arch.
Seven of the 9 deaths had been diagnosed with one or more cancers (Supplemental Table 6), including 2 cases of unspecified cancer; four cases of lymphoma [92,93,109]; one case of HPV+ head and neck squamous cell carcinoma (HNSCC) deceased at age 46; and one case of HPV+ vulvar cancer deceased at age 41 [42,87,92]. One patient had both an EBV+ B cell lymphoma and HPV+ HNSCC [87,94]. A total of 16 cancers were reported in 13 patients, all but one of which were among the 48 adults. Eight cancers were HPV-related SCC (6 anogenital SCC and 2 HNSCC), and two were EBV+ B cell lymphomas. Thus, 63% of cancers reported in WHIM patients were related to viral etiologies, either HPV or EBV. The remaining six cancers included one unspecified B cell lymphoma, two EBV-negative primary cutaneous lymphomas (one B cell, one T cell), and a single case each of basal cell carcinoma, melanoma and undefined cancer. Ten cases of bronchiectasis were reported, all in adults, but no patients were reported to have died of chronic lung disease. As recognition of the disease expands and aggressive treatment becomes standardized and improved, survival in WHIM syndrome is likely to increase further from its already very high level for a combined immunodeficiency, but at the expense of further refining knowledge of the natural history.
V. Molecular Pathogenesis
A. The Classic Model
Like all GPCRs, the structure of CXCR4 contains 7 transmembrane helices, an extracellular N-terminal domain and an intracellular C-terminal domain [110–115]. After activation, GPCRs are negatively regulated by powerful desensitization mechanisms, including physical internalization of activated receptor from the plasma membrane by clathrin-mediated endocytosis, destined for recycling or degradation [116]. The classic desensitization mechanism involves a GPCR kinase (GRK), which phosphorylates specific serines and threonines distributed along the C-terminal domain of CXCR4, to which β-arrestin is recruited to drive receptor internalization [117–119]. Impaired internalization prolongs receptor residence time on the cell surface and is thought to contribute to amplification and prolongation of receptor signaling. This has been observed for multiple WHIM receptor variants expressed in cell lines using multiple signaling assays, including calcium flux, chemotaxis, Akt/protein kinase B and ERK phosphorylation, and direct G protein activation [12,72,73,120,121]. Thus, paradoxically, a loss of structure from WHIM mutations leads to a gain-of-function by stabilizing the mutant receptor on the cell surface.
Probing this general model mechanistically using phosphosite-specific CXCR4 antibodies, Mueller et al uncovered a hierarchical organization of CXCR4 phosphorylation events which may explain in part how small structural changes from WHIM mutations are leveraged into large functional effects in CXCR4 [122]. In particular, CXCL12 was found to induce long-lasting GRK2/3-dependent phosphorylation of a serine 346/347 phosphosite which preceded less stable phosphorylation at serine 324/325 and serine 338/339 phosphosites. WHIM mutants lacking the serine 346/347 phosphosite as well as the E343K missense WHIM mutant all had impaired phosphorylation at the intact proximal serine 324/325 and serine 338/339 sites and impaired CXCL12-induced receptor internalization. Lagane et al have reported that, contrary to the classical model, the S338X WHIM mutation does not abrogate β-arrestin-2 binding to CXCR4, but instead unexpectedly enhances and prolongs β-arrestin-2-dependent signaling, which is important for the chemotactic response [123]. The SHSK motif in intracellular loop 3 of this WHIM receptor was important for β arrestin-2-dependent signaling, but not coupling to G proteins, and disrupting it normalized chemotactic signaling. Cases of WHIM syndrome known to lack a CXCR4 mutation still have augmented agonist-induced CXCR4 signaling in neutrophils associated with impaired receptor downregulation, but the mechanism appears to involve deficient GRK expression and function [73].
B. Unanswered Questions
Defective receptor downregulation may not be the whole explanation for the molecular pathogenesis of WHIM syndrome. CXCR4 downregulation is too slow (30-60 minutes) and too insensitive to CXCL12 stimulation to directly explain the increase in WHIM receptor signaling that can be observed by real-time calcium flux assay of transfected cell systems within seconds after stimulation with very low concentrations of CXCL12, suggesting that another mechanism, for example more efficient coupling to G protein, may also contribute to the observed gain-of-function. The two most common WHIM variants, R334X and S338X, have been tested and shown to have enhanced G protein activation when stimulated with CXCL12, but only in transfection systems [73]. Additional studies are needed to evaluate this using primary cells from patients.
Secondly, since WHIM syndrome is an autosomal dominant condition, both the wild type and WHIM alleles could be and probably are co-expressed in primary patient-derived cells. Therefore, since the crystal structure of CXCR4 is a homodimer [110,111], both the WHIM and wild type receptors could coexist as independent monomers and/or as homodimers and/or as heterodimers in primary cells, potentially conferring novel and unpredictable effects on receptor expression levels, effector coupling and signaling potential. The stoichiometry of the different forms could vary according to cell type and among different patients, potentially contributing to the phenotypic heterogeneity that is observed even for the same mutation in different members of the same pedigree. There is biochemical evidence that WHIM variant receptors can in fact heterodimerize with the wild type receptor [123]. Going one step further, it has recently been reported that CXCR4 activation results in receptor nanoclustering that may be important for signaling. Thus, effects of WHIM mutations on larger order signalosomes must now also be considered [124].
A full explanation of the underlying mechanism will require detailed comparisons of receptor stoichiometry, dimerization, signaling and internalization using primary cells from multiple WHIM patients and healthy matched controls, a difficult analysis to perform. Of note, CXCR4 antibodies have revealed impaired internalization of the epitope in CXCL12-stimulated primary cells suggesting that normal internalization of any coexpressed wild-type receptor may be inhibited in trans by expression of the WHIM receptor [120]. It is difficult to test this using primary cells from patients with nonsense mutations since the receptor sizes are so similar. However, WHIM receptors with frame-shift mutations provide an opportunity to generate WHIM receptor-specific antibodies as investigational tools to begin to investigate stoichiometry. Studies of coexpressed differentially tagged wild-type and WHIM receptors in transfection systems would also be relevant and possibly insightful.
It is important to note that classic CXCR4 signaling by the WHIM receptor is typically no more than double the magnitude for the wild type receptor in assays tested to date in vitro [12,120], and that hemizygous Cxcr4+/o mice have no overt phenotype, unlike Cxcr4−/− mice. Thus, it must be considered that something more than just a relatively modest quantitative change in CXCR4 signaling capacity through conventional pathways may be involved in the extreme hematologic phenotypes of WHIM syndrome.
VI. Immunopathogenesis
A. Overview
Understanding the mechanism of combined immunodeficiency in WHIM syndrome requires first delineating which leukocyte subsets are deficient in the blood for each patient and then defining for each deficient subset whether the cause is decreased production, increased clearance or cell death, abnormal sequestration or a combination of these mechanisms; whether the abnormalities are leukocyte intrinsic, autonomous or extrinsic; and whether mature leukocyte effector functions are also affected. In addition, a pathway must be defined for how CXCR4 mutation leads to each leukocyte abnormality and an assessment of how the abnormality results in the infections that occur. Very little of this has been accomplished to date, in part because 1) the disease is rare, 2) panleukopenia is severe making it sometimes difficult to obtain enough cells for functional studies, 3) other than bone marrow, immune organs are not readily accessible for analysis, 4) leukocyte dynamics are difficult to measure in patients in vivo, 5) there is substantial phenotypic heterogeneity, and 6) the “WHIM” mouse model does not fully phenocopy the human disease.
Regarding this last point, the mouse model is a knock-in of the second most common human WHIM mutation CXCR4 S338X into mouse Cxcr4 (Table 2) [97]. It develops severe B lymphopenia, as do patients, but the absolute neutrophil count is only reduced by ~50%, not 90-95% as in most patients. The dysmorphic neutrophil features characteristic of myelokathexis have been described in detail in 3 patients with the S338X mutation but are not found in the S338X knock-in mice [50,55,97]. Moreover, specific assays of apoptosis did not reveal increased levels in bone marrow neutrophils from the knock-in mice [97]. The model is also limited by the inability of mice to be infected by HPV, as well as the absence of hypogammaglobulinemia, spontaneous infections and congenital malformations. Still, significant progress has been made in understanding WHIM immunopathogenesis by combining both patient and mouse model investigations and may continue to be made in the future with challenge studies using relevant WHIM pathogens, including MmuPV1, a mouse-specific papillomavirus.
Table 2.
Effect of mutations in the CXCR2 and CXCR4 signaling systems.
| Genotype | Phenotype | ||||
|---|---|---|---|---|---|
| Receptor Signaling | Early Mortality | Immune System | Cardiovascular Development | Cerebellar Development | |
| Human CXCR4+/WHIM | Increased; impaired desensitization | 0% | Panleukopenia Myelokathexis Low Igs Impaired vaccine responses Recurrent infections Persistent HPV disease HPV and EBV associated cancer |
Tetralogy of Fallot (n=4) | Abnormal vermis (n=5) |
| Mouse Cxcr4+/WHIM | Increased; impaired desensitization | 0% | Panleukopenia (B>T~N), reversed by AMD3100 No myelokathexis Normal Ig levels Low cellularity in spleen due to low B and T lymphocytes No spontaneous infections No warts or spontaneous cancers |
na | na |
| Mouse Cxcr4+/o | Decreased | 0% | normal | na | na |
| Mouse Cxcr4−/− | None | 100% | Impaired BM myelopoiesis Impaired B cell lymphopoiesis Neutrophilia |
VSD Abnormal gastric and renal vascularization | Disorganized granule cell layer |
| Mouse Cxcl2−/− | None | 100% | Impaired BM myelopoiesis Impaired B cell lymphopoiesis |
VSD Abnormal gastric and renal vascularization | Disorganized granule cell layer |
| Mouse Ackr3−/− | Impaired due to defective Cxcl12 gradient formation | 70-95% | Impaired marginal zone B cell development | Cardiomegaly, VSD, semilunar valve atresia, lymphatic sac dilatation | Normal |
| Human CXCR2fs/fs | Absent CXCL2-dependent chemotaxis, ERK phosphorylation | 0% | Myelokathexis | na | na |
| Mouse Cxcr2−/− | None | 0% | Complete ko, SPF: neutrophilia, myeloid hyperplasia, extramedullary myelopoiesis Complete ko, GF: myeloid hyperplasia, normal ANC Mixed WT/ Cxcr2−/− bone marrow chimeras: Cxcr2−/− Myelokathexis |
na | na |
B, B cells; T, T cells; N, neutrophils; Ig, immunoglobulin; BM, bone marrow; VSD, ventricular septal defect; n, number of patients; na, not available/reported; SPF, specific pathogen-free; GF, germ-free; WT, wild type; ANC, absolute neutrophil count in blood
At the most basic level, WHIM syndrome can be understood as a multisystem and combined immunodeficiency disease consistent with 1) broad expression of CXCR4 by all subsets of both hematopoietic stem and progenitor cells and mature leukocytes (www.immgen.org) as well as by many non-hematopoietic cell types, including endothelial and epithelial cells and fibroblasts; 2) broad expression of CXCL12, including at high levels in all primary and secondary immune organs; and 3) the close (but imperfect) correspondence between the most profoundly affected cell types (myeloid cells, B cells) and organs (heart, brain) in WHIM syndrome patients and Cxcr4 knockout mice [29,41].
B. Myelokathexis and Innate Immunity
Our current molecular understanding of myelokathexis was anticipated in part by pioneering studies of the role of CXCL12 and CXCR4 in normal HSC homeostasis by Lapidot and colleagues [125–128]. HSCs home from blood to bone marrow niches in a Cxcr4-dependent manner in response to local production of Cxcl12 by CAR cells (Cxcl12-associated reticular cells) [129]. This suggested that the gain-of-function WHIM allele might simply shift the normal equilibrium of all CXCR4-expressing cells to pathologically favor sequestration in CXCL12-rich storage areas resulting in panleukopenia.
i. Neutrophils
The classic descriptions and detailed studies of neutrophils in the first WHIM patient, now known as WHIM-09, anticipated the molecular model and remain the most extensive and detailed clinical investigations into myelokathexis carried out to date. [1,2] A central sequestration mechanism of neutropenia was supported by 1) the lack of detectable leukoagglutinins; 2) the failure of splenectomy, high-dose corticosteroid therapy and experimental epinephrine challenge (to reveal the marginated pool) to reverse neutropenia; and 3) the presence of bone marrow hypercellularity and myeloid hyperplasia with a marked shift to the right (high M:E ratio). This bone marrow picture has subsequently been consistently observed in most other WHIM patients (Supplemental Table 7). The inference that the central pathogenic mechanism involved retention and sequestration of mature neutrophils in bone marrow was originally based on 1) the high proportion of mature neutrophils in the bone marrow (~25% of nucleated cells), which were even more abundant than band forms; 2) the absence of bands from the blood; 3) the normal appearance of the rest of myeloid development through the band stage both in terms of cell distribution and morphology; and 4) the rapid and transient increase of the ANC caused by mobilization of mature neutrophils, not bands, after challenging the patient intravenously with Pseudomonas and Salmonella endotoxins [1,2]. Experimental reversibility of neutropenia aligned with the association of marked, transient and non-cyclic neutrophilia (as high as 28,700 total white blood cells/microliter with 92% neutrophils) in the patient during episodes of acute bacterial infection.
Krill et al found that accelerated clearance of neutrophils from the blood might also contribute to myelokathexis in the patient [1]. In particular, they performed P32-tagged di-isopropylfluorophosphate (DFP32)-labeled leukocyte kinetic studies, which showed that labeled leukocytes from WHIM-09 were cleared at a similar markedly accelerated rate from the blood after transfer back into either the patient or into a ‘compatible’ previously non-transfused healthy adult recipient, whereas the healthy individual’s labeled cells were cleared with much slower kinetics when transferred into WHIM-09. Calculations revealed massive decreases in the total, circulating and marginated granulocyte pools in the blood but only a 50% reduction of the blood granulocyte turnover rate for the patient compared to archival healthy adult norms. This also suggested that the problem was neutrophil-autonomous, which was further supported by the inability of passive transfer of patient serum to induce neutropenia in a hematologically normal 6-month-old infant recipient. The clearance studies were limited, however, by not defining how the cells were cleared and by their anecdotal nature, as they were never attempted subsequently in other WHIM patients.
The discovery of CXCR4 as the disease gene in WHIM syndrome allowed development of zebrafish [130] and mouse [97] models by genetic knock-in of mutant human alleles. In both cases neutropenia was recapitulated. Neutropenia could also be selectively phenocopied in congenic lethally irradiated wild-type mice transplanted with WHIM mouse HSCs in competition with wild-type HSCs, confirming an HSC-autonomous effect of the WHIM allele in driving neutropenia [58,60]. Furthermore, in a human experiment of nature, the chromothriptic event in WHIM-09 purged the myeloid lineage, but not the lymphoid lineage, of the CXCR4 WHIM allele, but not the wild-type allele, causing a transition from neutropenia to neutrophilia without improving the patient’s severe lymphopenia or hypogammaglobulinemia [58]. This event, essentially an acquired myeloid-specific selective knockout of the CXCR4 WHIM allele, suggests that the WHIM allele on hematopoietic cells alone may promote leukopenia and that the hematologic effects of the WHIM allele may be myeloid lineage-autonomous [58,60,61]. This is also consistent with a chimeric R334X individual in a WHIM pedigree from Japan who lacks the mutation selectively in hematopoietic cells and is hematologically normal. In addition, transducing the WHIM allele R334X into human CD34+ progenitor cells enhanced homing to mouse bone marrow from blood compared to cells transduced with wild-type CXCR4 [131].
Meanwhile, clinical studies with the CXCR4-specific small molecule antagonist AMD3100 (developed first by Genzyme, which was acquired by Sanofi; trade and market names Mozobil and plerixafor) [132,133] showed that blocking CXCR4 signaling mobilizes most subtypes of leukocytes [49,53]. This has been confirmed in wild-type mice [134], although the provenance of the cells is not settled, with indirect and direct mobilization studies suggesting bone marrow as the most important source and direct imaging studies indicating that plerixafor does not mobilize bone marrow neutrophils but rather increases the ANC by mobilizing neutrophils from the marginal pool of the lung and by inhibiting neutrophil return from blood to bone marrow [70,134–136].
At the functional level, Zuelzer’s original report described an absence of spontaneous bone marrow neutrophil motility in vitro and reduced spontaneous blood neutrophil motility, suggesting a primary motility defect in neutrophils (Supplemental Table 8) [2]. Consistent with this, a Rebuck window test of neutrophil migration in vivo using typhoid vaccine as an irritant on denuded forearm skin was markedly abnormal with only small numbers of degenerating neutrophils appearing in a delayed manner on the window. A similar result was revealed by tail wounding in a zebrafish model of WHIM syndrome [130]. The original neutrophil motility assays for WHIM-09 have not been reattempted in subsequent patients with the exception of local skin challenge with vaccines, which was found to be defective for three out of four patients using either mumps or typhoid vaccine [3,4]. Instead, follow-up ex vivo studies of blood neutrophil motility in WHIM patients have commonly but not invariably found normal responses to most chemoattractants and selective hyper-responsiveness to the CXCR4 agonist CXCL12, which is not a stimulus that could have been tested by Krill and Zuelzer in 1964 [9,56,120]. The defective Rebuck skin window test in WHIM-09 and other WHIM patients may have been more a consequence of neutropenia than an intrinsic inability of neutrophils to migrate from blood to tissue. In most other WHIM patients for whom neutrophil functional studies have been reported, phagocytosis was reported to be normal, as were superoxide production and bacterial killing capacity.
An alternative explanation for impaired spontaneous bone marrow neutrophil motility in WHIM-09 was included in Zuelzer’s original report: a high proportion of bone marrow neutrophils (~2/3) had morphologic features consistent with cell death, and a high percentage failed to exclude trypan blue [2]. Studies in seven other WHIM patients have confirmed an increased proportion of apoptotic neutrophils, which have been shown to have an increased rate of apoptotic death when cultured ex vivo and a suggestion of decreased bcl-x expression [100]. Whether CXCR4 signaling drives neutrophil apoptosis ex vivo has not been demonstrated, since neither constitutive signaling by the receptor nor the presence of CXCL12 in the ex vivo system nor the effect of CXCL12 depletion have been evaluated. On the contrary, addition of CXCL12 has been reported in three patients to reduce spontaneous neutrophil apoptosis ex vivo [56,137]. This is the opposite of what would be expected if increased CXCR4 WHIM receptor signaling accelerates neutrophil apoptosis. G-CSF addition has also been reported to reduce WHIM neutrophil apoptosis ex vivo [137]. Additional experiments are clearly needed to clarify this, including work with CXCR4 antagonists, particularly since wild-type CXCR4 signaling has been considered to have anti-apoptotic effects in various systems.
An attractive model of myelokathexis has been proposed that couples pro-adhesive, egress and apoptotic mechanisms. In the model, enhanced neutrophil CXCR4 signaling capacity conferred by the WHIM allele biases the cells towards retention in the bone marrow. Cells with the lowest CXCR4 levels egress best in response to CXCR2 signaling, but later than normal. As they senesce in the periphery, CXCR4 levels increase which drives the cells back to the CXCL12-rich bone marrow where they die and are cleared by macrophages [138] [139]. This model is consistent with the discovery of loss-of-function CXCR2 mutations and gain-of-function CXCR4 mutations in myelokathexis patients, as described in the Genetics Section. The prediction from the model is that the rates of neutrophil egress from bone marrow, homing to bone marrow and residence time in the periphery should all be reduced, which is consistent with the neutrophil kinetics in WHIM-09 published by Krill and colleagues when she still had WHIM syndrome [1].
. There is strong evidence that hypoxic induction of hypoxia-inducible factor-1 (HIF-1) upregulates both CXCL12 in endothelial cells and CXCR4 in cancer cells and may be an important mechanism in both metastasis and tissue regeneration [140,141], as well as evidence that hypoxia equips neutrophils for prolonged survival, for example in inflammatory microenvironments [142]. However, clear evidence for HIF-1 regulation of CXCR4 triggered by maturation, senescence or hypoxia in either normal or WHIM neutrophils is currently lacking. CXCR4 has also been implicated with CXCR2 as a molecular timer in a cell-autonomous aging mechanism that controls diurnal compartmentalization of neutrophils between blood and tissue [143].
Accelerated neutrophil senescence with induction of high levels of CXCR4 expression might explain why all of nine WHIM patients entered into plerixafor dose escalation studies (described in detail in a later section) achieved substantially lower maximal neutrophil mobilization responses compared to both healthy control subjects and to their own lymphocyte responses [49,53,63]. The relatively poor response to plerixafor raises the question of whether WHIM patients might have a general hyporesponsiveness to neutrophil-mobilizing agents, such as epinephrine, lipopolysaccharide and G-CSF. This would require comparative studies in multiple patients with healthy controls, which have not been performed. Alternatively, the high levels of neutrophilia that have been documented for many WHIM patients during acute infections suggest that the sluggish mobilization response of neutrophils to plerixafor may be an unexplained idiosyncrasy of the drug rather than a direct effect of the mutation or a result of degeneration per se. Additional studies are needed that carefully compare migration responses of neutrophils harvested from the blood and bone marrow of the same patient at the same time to further address the role of apoptosis in driving neutropenia.
Importantly, recent studies of long-term low-dose plerixafor treatment in WHIM patients reported a marked improvement in the proportion of apoptotic-appearing neutrophils in the bone marrow and a return of the myeloid:erythroid ratio to normal; neutropenia also was improved but did not normalize at the doses used [49,87]. Additional studies are needed to quantitate the effect of CXCR4 blockade on the rate of mature neutrophil senescence and to compare the mobilization potency and efficacy of G-CSF and plerixafor in WHIM patients, as well as to optimize plerixafor dosing. In addition, the factors that override CXCR4-dependent neutrophil retention in the bone marrow during infection and after endotoxin administration, which may include G-CSF, need to be defined. The relationship between neutrophil dynamics in vivo and CXCR4 expression dynamics also needs to be established. Finally, the effect of the WHIM mutation on neutrophil adhesion needs to be investigated at the molecular level, as this is the most likely terminal effector mechanism explaining myelokathexis and may reveal another therapeutic target.
Myelokathexis clearly highlights the concept of the neutrostat and underscores the point that the impact of leukopenia on host defense cannot be generalized and is instead strongly dependent on context and mechanism [144]. For example, studies in the chemotherapy literature have defined 500 neutrophils/microliter as a useful threshold below which patients acquire a high risk of developing life-threatening invasive bacterial infections [145], and in AIDS 200 CD4+ T cells/microliter plays the same clinical role for susceptibility to most opportunistic infections and AIDS-related cancers. However, these benchmarks occur in settings where the total body mass of the corresponding cell type is reduced and where cell function may be compromised. In WHIM syndrome, where neutrophil development is not blocked, and total body neutrophil mass and neutrophil effector function may not be compromised, the impact of severe neutropenia on invasive infection susceptibility should be and is relatively low compared to other types of congenital and acquired neutropenia. Nevertheless, there have been no studies defining the relationship of the absolute neutrophil count and infection susceptibility in WHIM patients. Conversely, as suggested previously, this defect could conceivably protect against some non-infectious neutrophil-mediated acute and chronic inflammatory diseases such as chronic obstructive lung disease and may help WHIM patients who develop bronchiectasis from repeated infection, although this has not been studied.
ii. Natural killer cells
Relatively little is known about NK cells in WHIM syndrome [46]. Normal NK cell development requires CXCR4 signaling [146], and NK cell egress from bone marrow is regulated by the balance of retention and egress signals from CXCR4 and sphingosine-1 phosphate receptor 5, respectively [147]. NK cell development is not affected by WHIM mutations; however, circulating NK cell levels are reduced in a subset of WHIM patients [49,88,148]. Since NK cells express CXCR4, this may be a direct effect of the WHIM mutation on trafficking, retention or positioning. Consistent with this, plerixafor treatment increases NK cell levels in the blood in WHIM patients [49], as well as in wild-type rhesus macaques [149] and mice [147]. However, another possible factor is neutropenia per se, which has been reported to impair NK cell maturation and function [150]. This issue has not been studied in the context of WHIM syndrome. In WHIM mice, although NK cell development doesn’t seem to be disrupted, the total NK cell numbers are increased in bone marrow and lymph node but reduced in spleen and blood [147]. NK cell function and the role of NK cells in recurrent infections and HPV pathogenesis in WHIM patients are undefined.
C. Adaptive Immunity
The original studies of Krill et al and Zuelzer documented marked lymphopenia in WHIM-09 but did not study this phenotype or include it in the definition of myelokathexis [1,2]. At the time there were no simple methods to distinguish B and T cells, so the discovery of B lymphopenia as a severe and strongly penetrant WHIM phenotype came later in other patients [4]. The circulating levels of T cells may also be reduced in WHIM patients but usually less severely and less consistently than B cells and neutrophils [42,44,56,121,151,152]. B lymphopenia is also the most prominent hematologic phenotype in WHIM mice [97]. Nevertheless, hypogammaglobulinemia is not severe or invariant in patients and does not occur in WHIM mice. Moreover, WHIM patients do not have a strong susceptibility to infection with encapsulated organisms or bacteremia as do patients with severe B cell deficiency associated with severely impaired immunoglobulin production.
i. Humoral Immunity
The high impact of WHIM mutations on B cell distribution is consistent with the fact that all stages of developing B cells normally express CXCR4 and at significantly higher levels than for most other leukocyte subsets (www.immgen.org). A relevant historical point here is that CXCL12 was first identified by direct purification from bone marrow stromal cells as a pre-B-cell growth-stimulating factor (PBSF), which together with stromal cell-derived factor-1 (SDF-1) was one of its two original names [27,153]. Moreover, Cxcr4 knockout mice have defective bone marrow myelopoiesis and B lymphopoiesis but T lymphopoiesis is relatively intact [81]. In bone marrow, CXCR4 signaling in B cells induces B cell motility and the gradual loss of CXCR4 results in reduced motility followed by egress of immature B cells [154].
There is no discernable block in B cell development in either WHIM patients or WHIM model mice [97]. In WHIM mice, mature B cells are abundant in bone marrow, but there are fewer pro/pre and immature B cells. This was not attributed to increased apoptosis (Figure 4) [97]. B cells are also abundant in the bone marrow of WHIM patients and can be efficiently mobilized to the blood by plerixafor, suggesting that B lymphopenia, like neutropenia, may be caused at least in part by B cell retention in the bone marrow [49,53,155]. Consistent with this, plerixafor injection in wild-type mice markedly increases blood levels of both naive and memory B cells while concurrently depleting these cells from bone marrow [134]. Splenectomized wild-type mice given plerixafor have much higher absolute lymphocyte counts in the blood after plerixafor administration than sham-operated mice, suggesting the spleen may be a storage site and not a source for plerixafor-mobilized cells, although there are other possible explanations [134]. Consistent with this, an asplenic WHIM patient treated with plerixafor had by far the strongest lymphocyte and B cell mobilization response to plerixafor that we have seen [87].
Figure 4.
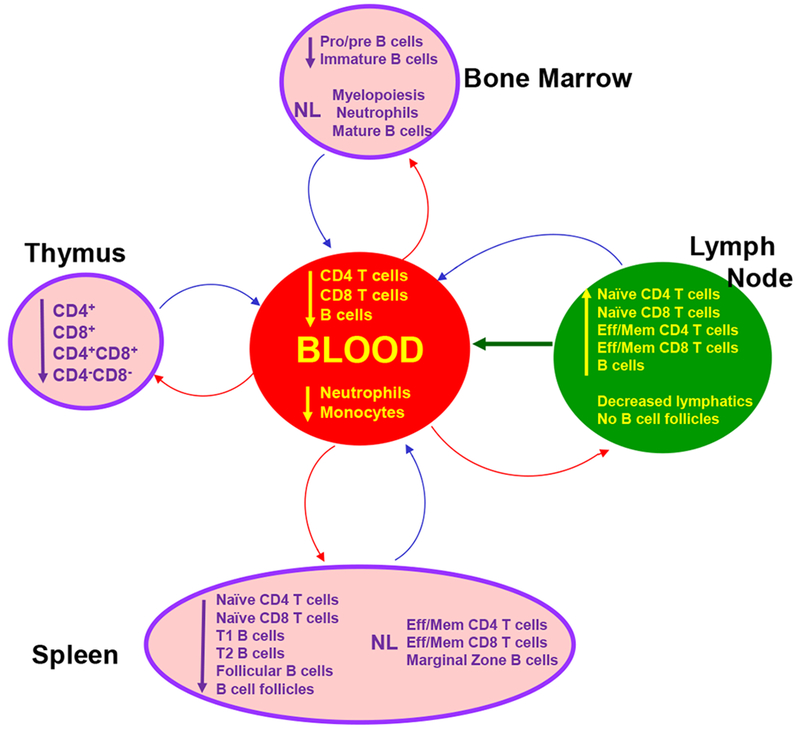
Leukocyte distribution in immune organs is distorted in WHIM model mice [97]. A corresponding detailed assessment of leukocyte distribution is unavailable in WHIM patients. Eff/Mem, effector/memory. The figure is reproduced from [204].
The paucity of pathologic studies of immune organs is a major weakness in WHIM syndrome research. There is rarely a clinical indication for biopsy or excision and autopsies are rarely performed. Only three patients have been reported to have had a lymph node biopsy or excision. In two patients this was performed following diphtheria toxoid vaccination and was reported to show few follicles and few plasma cells [4]. Pathologic examination of the spleen of patient WHIM-09 removed at age 9 revealed lymphoid hyperplasia and a two-fold-increase in weight [1]. In WHIM mice, the spleen is also enlarged. Splenic architecture is normal but the size and number of follicles are markedly decreased, associated with a 45% reduction in cellularity which involves all major splenocyte subsets [97].
Extramedullary hematopoiesis also occurs in WHIM mice and is associated with increased levels of CXCL12 in spleen and increased trafficking of hematopoietic stem and progenitor cells from blood to spleen [156]. In the peritoneal cavity, the B cell compartment is defective, with an expanded B1a subset, which is derived from fetal liver precursor cells, and reduced content of bone marrow-derived B2 and B1b subsets [97].
Consistent with B lymphopenia as a major effect of gain-of-function WHIM mutations, targeted deletion of Cxcr4 in B cells causes premature egress of B cell precursors from bone marrow [157]. The cells localize within splenic follicles which reduces mature B cell content in the primary follicles and marginal zones of the spleen and induces formation of aberrant B cell follicles ectopically in intestinal lamina propria and Peyer’s patch. As a functional consequence, T-independent antibody responses are blunted [157].
Many WHIM patients present with hypogammaglobulinemia, with defects in one or more subclasses of antibody [42,50,56,57,67,148,151,158]. How some WHIM patients with severe B lymphopenia manage to avoid hypogammaglobulinemia has not been explained. Detailed B cell functional studies performed in a few patients have documented restricted immunoglobulin heavy chain variable region diversity, impaired class switching and poor or unsustained responses to vaccines [42,48–50,57,88,148]. In particular, HPV-specific antibody titers were lower and less durable in a 12-year old WHIM patient compared to healthy controls vaccinated with the quadrivalent HPV vaccine, Gardasil® [159]. In a second study, reduced B cell Ig gene class switching was observed after immunization [148]. The mechanism was not defined and could include defects in germinal center trafficking of lymphocytes and organization of germinal center light and dark zones, which are known to be regulated in part by CXCR4 (and CXCR5) [160] and to be distorted in WHIM mice, as well as intrinsic defects in B cell activation [161].
In contrast to patients, hypogammaglobulinemia has not been reported in WHIM mice [97]. In fact, IgG and IgM levels are elevated, while IgA levels are comparable to wild-type controls. Furthermore, immunization of WHIM mice has been reported to increase absolute numbers and the proportion of antigen-specific functional plasma cells (PCs) in the spleen and lymph node germinal centers [162]. Nevertheless, although immature PCs accumulate in the bone marrow early after immunization, antigen-specific PCs fail to accumulate in the bone marrow, and antigen-specific antibody responses are not durable [162].
At the molecular level, CXCR4 normally colocalizes with and costimulates the IgD-B cell receptor (BCR) resulting in actin remodeling and PI3K/AKT and ERK signaling; B cells lacking IgD show reduced chemotactic responses to CXCL12 stimulation [163]. In normal B cells CXCL12 stimulation upregulates the activation marker CD69, and this is enhanced in cells from WHIM patients and WHIM mice. Moreover, B cells from unstressed WHIM patients and mice are hyperactivated at baseline and, like WHIM neutrophils, are hypersusceptible to apoptosis. In this regard, lowering the dose of immunogen has been reported to paradoxically improve and prolong antibody responses in WHIM mice [161]. Additional studies are needed to define B cell responses in larger numbers of WHIM patients across a broad panel of vaccines and to test whether CXCR4 blockade and immunogen dose variation might be combined to elicit durable and protective responses in these patients, as suggested by studies in WHIM mice. In addition, the question of how WHIM mutations affect B cell effector function remains fundamentally unanswered.
The precise contribution of hypogammaglobulinemia relative to neutropenia to infection risk is undefined in WHIM syndrome. As previously mentioned, there is not a strong signal of infection with encapsulated bacteria, and documented bacteremia is uncommon. Of the 105 cases we reviewed, there were only 8 total life-threatening infections (4 infants, 1 adult, 3 age unreported), including 5 reports of sepsis and 3 reports of meningitis. Moreover, many WHIM patients suffering from recurrent infections do not have hypogammaglobulinemia. Nevertheless, in those that do, anecdotal evidence suggests that Ig supplementation early in life may reduce the frequency of respiratory tract infections [57]. Of note, low-dose, long-term twice daily plerixafor injections in five WHIM patients durably raised the circulating B cell levels in a biphasic manner and reduced infection frequency but had only modest if any effects on immunoglobulin levels [49,87]. Vaccine responses were not assessed. It will be interesting to test whether delivering plerixafor by continuous infusion might stabilize B cell counts in the normal range in blood and improve immunoglobulin levels and vaccine responses but attributing any clinical benefit to this rather than to reduced neutropenia will be impossible since plerixafor has pleotropic immunologic effects including reversing neutropenia. WHIM-09 provides compelling though anecdotal information on this point since her warts cleared and recurrent infections stopped as her neutropenia, but not her severe hypogammaglobulinemia, was corrected by chromothripsis [58]. She is also asplenic, creating a further complication of interpretation from this single case.
ii. Cellular Immunity
Naive, effector, regulatory and memory T cell subsets may all be reduced in the blood of WHIM patients, and CD8+ T cells tend to be more affected than CD4+ T cells [49,56,87,121,151,152,164,165]. T lymphocytes are abundant in primary and secondary lymphoid organs and can be mobilized to the blood by plerixafor treatment in healthy humans [166], rhesus macaques [149], wild-type mice [134] and WHIM patients [53], suggesting a primary CXCR4-dependent distribution problem. There does not appear to be an absolute block to T cell development [97]. However, in limited studies the WHIM mutation has been associated with distorted maturation and a narrowed T cell repertoire [48]. A recent study has suggested that recent thymic emigrant output is affected [44]. Apart from a 30% reduction in thymic cellularity, circulating T cell numbers and thymic architecture, and frequency and spatial distribution of thymocytes were all reported to be normal in WHIM mice [97]. This is somewhat surprising since Cxcr4 has been reported to modulate multiple stages of T cell development, including survival and proliferation of T cell precursors in the bone marrow; homing of Cxcr4+ thymic progenitors to the thymus from the blood [167,168]; migration of immature thymocytes from the corticomedullary junction to the cortex [169]; stimulation of TCR £]-chain selection [170]; expansion of double-negative (DN) thymocytes [171,172]; and Notch-dependent development of DN cells into double positive (DP) thymocytes [171]. A plausible justification for these processes might be redundancy in the roles of the main thymic chemokine receptors, CCR7, CCR9 and CXCR4 [167]. Thymic dendritic cells also express Cxcr4 and Cxcl12, which enhance survival by increasing the ratio of the survival factor Bcl2 to the pro-apoptotic factor Bax [173]. Too few lymph nodes have been excised and examined in sufficient pathologic detail from patients to draw any firm conclusions about the distribution of specific subsets. As mentioned previously, this was performed in two patients following diphtheria toxoid vaccination and reportedly showed a low number of follicles [4]. In contrast to thymus, the axillary and inguinal lymph nodes display increased cellularity in WHIM mice, especially for naive CD4+ and CD8+ T cell subsets [97].
As with B cells, functional defects have been reported for T cells from WHIM patients. CXCR4 has been reported to normally localize to the immunologic synapse of CD4+ T cells and to physically associate with and activate the TCR, enhancing ZAP-70 (protein tyrosine kinase zeta-associated protein) binding to the TCR ITAM (immunoreceptor tyrosine-based activation motif) domain. This results in prolonged activation of the mitogen-activated protein kinase, ERK, increased intracellular calcium ion flux, increased activity of the transcription factor AP-1 and secretion of cytokines [174]. In addition, CXCR4 costimulation induces F-actin polymerization, which enhances the number and stability of microclusters formed by the adaptor molecule SLP-76 [175]. Activated ZAP-70 phosphorylates two tyrosine residues on SLP-76, resulting in enhanced proliferation [176] and upregulation of T cell activation molecules, including CD69 and CD25 [177]. Costimulation also results in enhanced production of the cytokines IFN-γ, IL-10 and IL-4 [174,177]. Thus, it might be anticipated that gain-of-function WHIM variants of CXCR4 would have augmented costimulatory activity. However, this does not appear to be the case, and T cells from WHIM patients are unable to form long-lasting immunologic synapses with antigen-presenting cells [178]. This may contribute to delayed immunoglobulin gene class switching. Along with a weakened immunological synapse, inhibitory mechanisms may also operate to hinder CXCR4-mediated costimulation of T cells. In some patients, WHIM T cells appeared to proliferate normally and to produce cytokines in response to mitogenic stimulation or TCR cross-linking with anti-CD3 plus costimulation with anti-CD28 [178]. T lymphopenia in WHIM syndrome might also result in part from defective homeostatic proliferation, which is partially regulated by CXCR4 [179]. However, CXCR4 does not appear to play a role in self-renewal of CD8+ memory T cells upon antigen rechallenge [179].
The clinical impact of cellular immunodeficiency in the WHIM syndrome is relatively mild. As mentioned previously, EBV+ lymphomas have occurred in a few patients [109]; however, EBV-dependent lymphoproliferative disease has otherwise not been reported. Nor have there been reports of mycobacterial infections, toxoplasmosis, CMV disease, candidiasis or Pneumocystis jirovecii infection. As mentioned previously, HSV and VZV have caused significant infections in only ~10% of patients.
The problem pathogen, HPV, is normally restricted to infecting basal cells of and mature keratinocytes of the epidermis which provides a type of immunological sanctuary slowing clearance even in healthy individuals. In WHIM patients cutaneous and anogenital warts are particularly resistant, and several cases of HPV-associated squamous cell cancer have been reported. HPV disease can occur in WHIM patients who lack hypogammaglobulinemia and T lymphopenia and who have normal in vitro T cell proliferative responses to mitogens (Table 1). In this regard, Balabanian and colleagues have published a series of papers describing a non-immunological autocrine CXCL12-CXCR4/ACKR3-dependent mechanism in keratinocytes for HPV-mediated cell transformation driven by the viral oncogenes E6 and E7 [73,180,181]. They found that CXCL12 and CXCR4 are co-expressed by keratinocytes in warts from both healthy individuals and WHIM patients, but not by non-lesional keratinocytes. E6 and E7 from oncogenic HPV16- and HPV18-immortalized keratinocytes induced CXCL12, CXCR4 and ACKR3 expression. CXCL12 signaling in immortalized keratinocytes expressing wild type CXCR4 induced cell motility and survival, whereas cells expressing the S338X WHIM CXCR4 variant became transformed. The mechanism involved stabilization of E6 and E7 and could be reversed by CXCR4 antagonists. Despite these insights, the precise role of keratinocyte CXCR4 signaling in keratinocyte biology and in HPV-associated lesions is still unsettled [182]. Future research may benefit from focusing on the role of the receptor in potentially modulating innate anti-viral mechanisms within keratinocytes, including viral DNA pattern recognition receptors/sensors (e.g. AIM2, TLR3 and TLR7) which may activate inflammasomes to produce caspase-1 for generation of IL-1β, as well as activated the Type I interferon pathway [183].
The importance of monocyte-derived cells in HPV susceptibility was suggested by the case of WHIM-09, whose warts resolved as the WHIM allele was purged from her myeloid cells, but not other cells, by chromothripsis [58]. CD8+ T cells are known to limit HPV pathogenesis and tumorigenesis [184–186]. However, the specific role of T cells in HPV susceptibility in WHIM syndrome is not known. This has been approached experimentally in a transgenic mouse model of squamous cell cancer induced by the expression of the HPV16 early region oncogenes E6 and E7 driven by the K14 keratinocyte promoter [187]. In this model, plerixafor treatment reduced keratinocyte hyperproliferation, immune cell infiltration and HPV-induced epidermal neoplasia.
Plasmacytoid dendritic cells (pDC) may also contribute to HPV control by producing IFN-α [188]. Production of pDCs and trafficking of pDCs in tissues are both regulated by CXCR4, and in WHIM patients, pDCs have been reported to be significantly reduced in the circulation and from WHIM patient warts, as well as to be incapable of secreting IFN-α upon exposure to HSV-1 and the TLR-9 agonist CpG [189]. Consistent with this, the Type 1 interferon-responsive gene MxA is ordinarily detectable in warts from healthy individuals but was not detected in warts from a WHIM patient. In contrast, mature DCs from WHIM patients have been reported to produce normal amounts of interleukin-12 (p70) compared with healthy donors [189], and both Langerhans cells and CD1a+ DCs were present in WHIM warts [189].
VII. Patient Management
A. Treatment
i. Overview
There is no standardized consensus treatment for WHIM syndrome (Table 3). Best practices are aimed at treating infections and warts, correcting neutropenia and supplementing immunoglobulin deficiency when present. Most infections respond well to oral antibiotics, and patients rarely need to be hospitalized and rarely die of acute bacterial infections. Patients with frequent infections are often treated with prophylactic antibiotics. Chronic sequelae of repeated infection, including hearing loss and bronchiectasis, and HPV-associated neoplasia can be difficult to manage.
Table 3.
Outcomes after immune modulation therapy in WHIM patients. Ig, supplemental immunoglobulin (IV or subcutaneous route of administration); HSC, hematopoietic stem cell. Adverse events are those attributed to the treatment.
| Clinical Response (n) | |||||
|---|---|---|---|---|---|
| Treatment | n | Improved | Not Improved | Not Mentioned | Adverse events (n) |
| G-CSF | 59 | 24 | 9 | 28 | thrombocytopenia (3) myelofibrosis (1) bone pain |
| GM-CSF | 4 | 3 | - | 1 | myelofibrosis (1) bone pain |
| Ig | 33 | 6 | 1 | 21 | allergic reaction (1) |
| Plerixafor | 11 | 11 (Hematologic) 5 (Clinical) |
0 | - | None observed |
| HSC Transplantation | 4 | 4 (Cured) | 0 | - | bronchitis flare (1) mild skin GVHD (1) |
ii. Vaccines
Inactivated vaccinations have not been associated with unusual complications and should be given on schedule. Live vaccinations should be considered on a case by case basis to balance individual risk and benefit, especially if the CD4+ T cell count is <200 cells/microliter. However, no complications of vaccination have been reported. Instead, as described in the section on humoral immunity, vaccine responses have been reported to be weak or unsustained [42,49,50,57,88,148,164]. WHIM patients have been diagnosed with influenza infection after receiving the vaccine [72]. However, it is not known whether the frequency of vaccine failure or the intensity of disease exceeds those in the general population. It is unlikely that HPV vaccination will substantially affect wart incidence or clearance in WHIM patients, since there are over 250 known types of HPV but only nine high-risk types represented in the broadest vaccine. Whether established HPV disease burden might be affected by HPV vaccination in WHIM patients is unknown.
iii. Leukocyte-mobilizing agents
Patients with ANC < 500 cells/microliter and recurrent infections are often advised to receive G-CSF (G-CSF/filgrastim, Neupogen; Amgen, Inc), the standard of care for severe congenital neutropenia (SCN). Most types of SCN involve defective myeloid production and therefore rely on all functions of the drug: stimulation of survival, proliferation, differentiation, and function of neutrophil precursors and production and bone marrow release of mature neutrophils. In contrast, myelokathexis may only require and primarily take advantage of the neutrophil-mobilizing activity of the drug. The mechanism is complex but appears to involve cleavage and inactivation of CXCL12 by neutrophil elastase and selective downregulation of CXCR4 on bone marrow neutrophils [125]. Many patients can be treated with daily low-dose G-CSF. This strategy helps to promote compliance by avoiding bone pain, which is a common and sometimes extremely disabling side-effect associated with higher doses [190]. Several WHIM patients have developed thrombocytopenia, in at least one case due to severe myelofibrosis [86], during treatment with G-CSF. The precise efficacy of G-CSF for reducing infection frequency and severity has not been defined by clinical trials in the disease and could be quite different from other types of SCN. Moreover, defining efficacy for G-CSF in WHIM syndrome must first resolve an ethical dilemma regarding study design as to whether a placebo ought to be used at all as a control for experimental treatment in a specific type of SCN when G-CSF is already the standard of care for SCN as a disease class.
Although immunoglobulin replacement and G-CSF treatment improve hypogammaglobulinemia and neutropenia and are thought to be clinically beneficial, some treated patients still suffer recurrent infections and persistent warts resulting in substantial morbidity and in some cases premature mortality, possibly because G-CSF does not significantly affect monocytopenia or lymphopenia. To address this unmet need, two potent specific CXCR4 antagonists have been entered into clinical trials, plerixafor (also known as AMD3100 and Mozobil®, Sanofi) [132,133] and X4P-001 (Mavorixafor; X4-Pharma, Inc.) [191].
Plerixafor is a small molecule bicyclam initially developed as an HIV entry inhibitor [192–194]. It failed further development for that indication because most strains of HIV in patients can use CCR5 for entry and because it is only bioavailable parenterally and has a short half-life of ~5 hours [195]. The drug was then repurposed for single dose HSC mobilization and is currently approved by the US Food and Drug Administration for use in combination with G-CSF in patients with non-Hodgkin’s lymphoma and multiple myeloma to mobilize and collect HSCs for autologous bone marrow transplantation after chemotherapy [194,196].
As of January 2019, plerixafor was listed as the study agent in 157 clinical trials on www.clinicaltrials.gov, mostly in the areas of HSC mobilization and cancer. Three trials were listed for WHIM syndrome, including two phase 1 open label trials (ClinicalTrials.gov identifiers: and ) and a phase 3 randomized double-blind non-inferiority comparison trial with G-CSF sponsored by the National Institute of Allergy and Infectious Diseases () [49,53,87,155]. In the phase 1 trials, the absolute numbers of neutrophils, lymphocytes and monocytes were transiently increased in the blood after administration of plerixafor in a dose-dependent manner in both healthy individuals and 11 WHIM patients (Figure 5). Conversely, the frequencies of neutrophils and CD19+ B cells in the bone marrow were reduced, suggesting mobilization from that site. Long-term (6 months to ~5 years), open label low-dose twice daily plerixafor administration in 5 patients was associated with durable mobilization responses to blood for most leukocyte subsets tested, lower infection frequency and wart burdens, stabilization of HPV16+ head and neck squamous cell carcinoma in one patient, improved myelokathexis, and improved quality of life [53,87]. Immunoglobulin levels did not increase. One patient who harbored 17 different types of HPV on G-CSF had no detectable HPV after chronic treatment with plerixafor [87]. Two of the patients treated with high-dose G-CSF developed myelofibrosis, anemia and severe thrombocytopenia, which all markedly improved or resolved after discontinuing the drug and switching to plerixafor [87]. No serious adverse events attributable to plerixafor were noted in this limited study. The phase 3 plerixafor versus G-CSF trial is fully enrolled with 20 patients and will be completed in 2020. Of interest regarding long term safety, mice lacking a single copy of Cxcr4 (Cxcr4+/o mice) have normal longevity and hematopoiesis [60].
Figure 5.
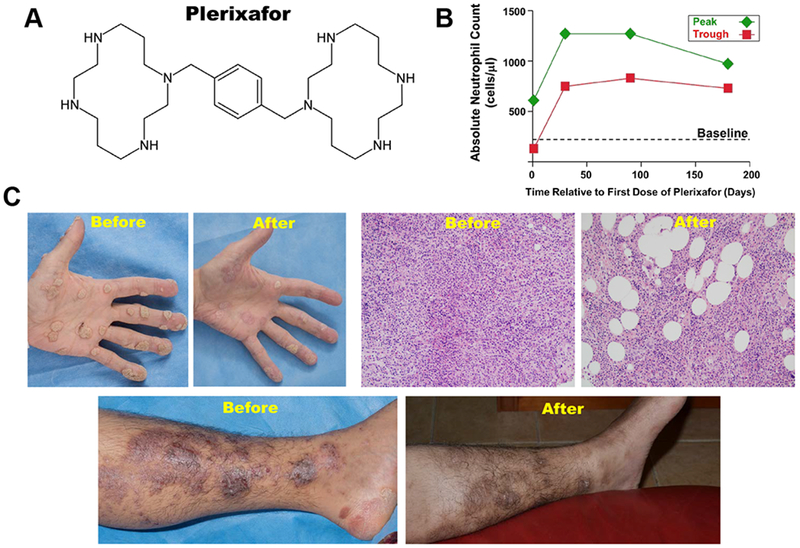
CXCR4 antagonist plerixafor has shown promise in Phase 1 trials in patients with WHIM syndrome. A, Structure of the bicyclam plerixafor. B, Plerixafor durably increases the absolute neutrophil count in WHIM patients. This figure is reproduced from Reference [49]. C, Plerixafor treatment of WHIM patients was associated with clinical improvement. Photographs show clinical manifestation from two patients before and approximately 6 months after twice daily treatment with plerixafor, including partial regression of a heavy wart burden on the hand, reduced myelokathexis in bone marrow and resolved chronic inflammatory lower leg lesions. The ‘before’ photograph of the leg in panel C is reproduced from Reference [87]; the after photograph of the leg shows discoloration at sites of previous inflammation, probably due to hemosiderin deposition that had not yet cleared. See details in Reference [87].
Plerixafor’s short half-life, high cost and need for parenteral administration are acceptable limitations for HSC mobilization but impose significant challenges for insurance coverage and compliance in a chronic disease like WHIM syndrome. One WHIM patient in Germany is receiving the drug paid for by insurance, whereas all other patients currently receive the drug through clinical trials. X4P-001 (Mavorixafor) is an allosteric CXCR4 antagonist and has the advantages of oral bioavailability and a longer half-life for once-a-day dosing [191]. In a phase 1 clinical trial consisting of eight WHIM patients (), X4P-001 was well-tolerated and increased the ANC and ALC in a dose-dependent manner [191]. A placebo-controlled Phase 3 trial is now recruiting patients.
Many other CXCR4 blocking agents have been identified and are in development for various indications. In a phase 2 pilot study, the orally bioavailable small molecule CXCR4 inhibitor Burixafor (TaiGen Biotechnology Co.) in combination with G-CSF mobilized stem cells into peripheral blood in patients with multiple myeloma, non-Hodgkin’s lymphoma or Hodgkin’s lymphoma () [197]. The oligonucleotide NOX-A12 is a CXCL12-specific inhibitor that has a longer half-life than plerixafor and also mobilizes lymphocytes and other leukocyte subsets in healthy controls [198]. Balixafortide is a cyclopeptide inhibitor of CXCR4 that has been utilized in metastatic breast cancer therapy in a phase 3 trial (). The small-molecule inhibitor Chalcone 4 is also specific to CXCL12 and inhibits CXCR4-CXCL12 interaction [199]. Recombinant single-domain anti-CXCR4 llama antibody fragments known as nanobodies have been developed that are potent inhibitors of WHIM receptor signaling [200]. These therapies require further investigation to assess their hematological effects and safety and efficacy in generating vaccine responses and reducing HPV-induced wart burden and recurrent bacterial infection frequency during prolonged administration in WHIM patients.
B. Cure Strategies
i. Allotransplantation
To date, all four WHIM patients who have received allogeneic HSC transplantation (HSCT) have achieved durable and full donor chimerism, no longer receive immunosuppression and are clinically cured [152,201–203]. The patients were transplanted at ages 3, 4, 8 and 20. Mild acute GVHD (skin, grade I, stage I) occurred in one patient, who received a haploidentical HSC transplant from her father matched at 7 or 8 loci [201]. The other patients received matched unrelated donor HSC transplants. In addition to durable reconstitution of αβ T cells, a transient increase in the proportion and number of γδ T cells was observed in one patient, attributed to transient inhibition of £]-selection in the thymus post-transplantation [201]. The application of HSCT more generally in WHIM syndrome is limited by the availability of immunologically-matched donors and by an unfavorable risk-benefit ratio in patients who may have an acceptable non-disabling infection burden [59].
ii. Gene Therapy
Since WHIM syndrome is a gain-of-function disorder, gene therapy will require silencing or correcting the disease allele. Two observations suggest WHIM allele inactivation may be superior to correction and that WHIM syndrome may be particularly amenable to cure by gene therapy: 1) patient WHIM-09 was cured by spontaneous chromothriptic deletion of the WHIM allele in HSCs (Figure 6), and 2) in an attempt to recapitulate WHIM-09’s cure experimentally, Cxcr4+/o HSCs displayed a marked long-term advantage over Cxcr4+/+ and Cxcr4+/WHIM HSCs for supporting blood chimerism after transplantation in the context of both lethally irradiated and non-conditioned congenic recipients [58–60]. The mechanism appears to involve enhanced mature leukocyte release to the blood and possibly enhanced HSC proliferation. The power of CRISPR/Cas9 gene editing is currently being used to test this approach in preclinical models.
Figure 6.
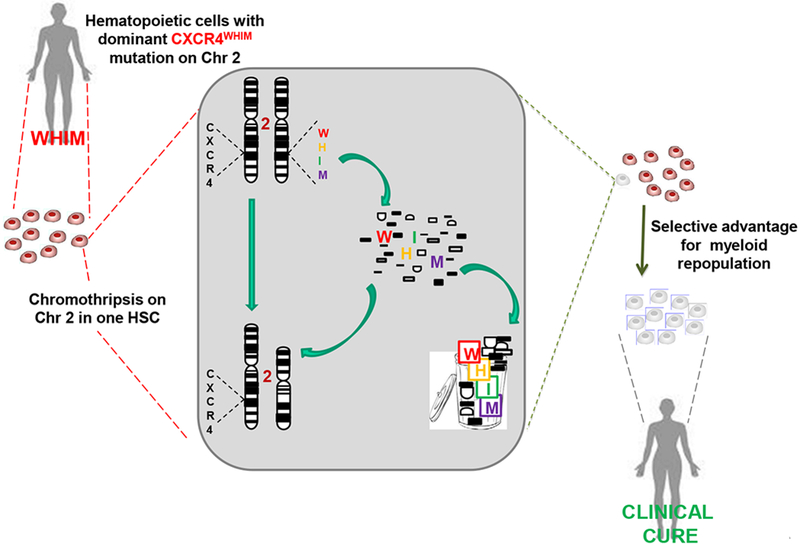
Chromothriptic cure of WHIM patient WHIM-09. Chromothripsis, a Greek neologism that means chromosome shattering, affected one copy of chromosome 2 in a single hematopoietic stem cell from the patient. The WHIM copy of CXCR4 and 163 other genes on that chromosome were deleted and the remaining non-deleted shattered fragments were rejoined to form a derivative chromosome. The cell clonally expanded and selectively replaced the myeloid lineage with non-WHIM cells, resulting in clinical cure. The lymphoid lineage was unaffected by the event, so that the patient is a hematologic chimeric of WHIM lymphoid cells and chromothriptic non-WHIM myeloid cells. The patient is estimated to have lived >25 years since the chromothriptic event. This case report suggests that WHIM allele inactivation may be superior to WHIM allele correction as a gene therapy strategy. Consistent with this, Cxcr4 haploinsufficiency markedly enhanced hematopoietic reconstitution of donor HSCs and corrected leukopenia in unconditioned transplanted WHIM model mice. The figure is modified from Reference [58].
VIII. Conclusions
The study of WHIM syndrome is providing unique insights into diverse aspects of both innate and adaptive immunity. It has identified important new questions regarding host defense mechanisms, HPV pathogenesis and development of the cardiovascular system. Although understanding the protean hematologic and non-hematopoietic phenotypes that occur has been focused at the molecular level by discovery of causal CXCR4 mutations, delineating precise mechanisms still remains a challenge due to the rarity of the disease. Nevertheless, the emergence of CXCR4 antagonists as potential mechanism-based therapy, gene editing and transplantation together give WHIM patients new hope for safe and effective treatment and even cure.
Supplementary Material
Acknowledgements
This work was supported by the Division of Intramural Research of the National Institute of Allergy and Infectious Diseases.
Footnotes
Publisher's Disclaimer: This Author Accepted Manuscript is a PDF file of a an unedited peer-reviewed manuscript that has been accepted for publication but has not been copyedited or corrected. The official version of record that is published in the journal is kept up to date and so may therefore differ from this version.
Conflict of Interest Statement
PMM is a member of the Scientific Advisory Board of X4-Pharma. PMM, J-LG and DHM are listed as inventors on a patent application disclosing a method of enhancing hematopoietic stem cell engraftment by CXCR4 knockdown. There are no other stated conflicts of interest.
References
- 1.Krill CE, Smith HD, Mauer AM. Chronic Idiopathic Granulocytopenia. New England Journal of Medicine. 1964;270:973–9. [DOI] [PubMed] [Google Scholar]
- 2.Zuelzer WW. “MYELOKATHEXIS”--A NEW FORM OF CHRONIC GRANULOCYTOPENIA. REPORT OF A CASE. N Engl J Med 1964;270:699–704. [DOI] [PubMed] [Google Scholar]
- 3.O’Regan S, Newman AJ, Graham RC. “Myelokathexis”. Neutropenia with marrow hyperplasia. Am J Dis Child. 1977;131:655–8. [DOI] [PubMed] [Google Scholar]
- 4.Mentzer WC, Johnston RB, Baehner RL, Nathan DG. An unusual form of chronic neutropenia in a father and daughter with hypogammaglobulinaemia. Br J Haematol 1977;36:313–22. [DOI] [PubMed] [Google Scholar]
- 5.Bohinjec J Myelokathexis: chronic neutropenia with hyperplastic bone marrow and hypersegmented neutrophils in two siblings. Blut. 1981;42:191–6. [DOI] [PubMed] [Google Scholar]
- 6.Bassan R, Viero P, Minetti B, Comotti B, Barbui T. Myelokathexis: a rare form of chronic benign granulocytopenia. Br J Haematol 1984;58:115–7. [DOI] [PubMed] [Google Scholar]
- 7.Plebani A, Cantù-Rajnoldi A, Collo G, Allavena P, Biolchini A, Pirelli A, et al. Myelokathexis associated with multiple congenital malformations: immunological study on phagocytic cells and lymphocytes. Eur J Haematol 1988;40:12–7. [DOI] [PubMed] [Google Scholar]
- 8.Rassam SM, Roderick P, al-Hakim I, Hoffbrand AV. A myelokathexis-like variant of myelodysplasia. Eur J Haematol 1989;42:99–102. [DOI] [PubMed] [Google Scholar]
- 9.Wetzler M, Talpaz M, Kleinerman ES, King A, Huh YO, Gutterman JU, et al. A new familial immunodeficiency disorder characterized by severe neutropenia, a defective marrow release mechanism, and hypogammaglobulinemia. Am J Med 1990;89:663–72. [DOI] [PubMed] [Google Scholar]
- 10.Ganser A, Ottmann OG, Erdmann H, Schulz G, Hoelzer D. The effect of recombinant human granulocyte-macrophage colony-stimulating factor on neutropenia and related morbidity in chronic severe neutropenia. Ann Intern Med 1989;111:887–92. [DOI] [PubMed] [Google Scholar]
- 11.Ohtake M, Kobayashi M, Watanabe N, Nagai Y, Kato S, Konnai Ikuo, et al. A clinical report of the first case of myelokathexis in Japan. J Jpn Pediatr Soc 1988;92:160–165. [Google Scholar]
- 12.Hernandez PA, Gorlin RJ, Lukens JN, Taniuchi S, Bohinjec J, Francois F, et al. Mutations in the chemokine receptor gene CXCR4 are associated with WHIM syndrome, a combined immunodeficiency disease. Nat Genet 2003;34:70–4. [DOI] [PubMed] [Google Scholar]
- 13.Herzog H, Hort YJ, Shine J, Selbie LA. Molecular cloning, characterization, and localization of the human homolog to the reported bovine NPY Y3 receptor: lack of NPY binding and activation. DNA Cell Biol 1993;12:465–71. [DOI] [PubMed] [Google Scholar]
- 14.Federsppiel B, Melhado IG, Duncan AM, Delaney A, Schappert K, Clark-Lewis I, et al. Molecular cloning of the cDNA and chromosomal localization of the gene for a putative seven-transmembrane segment (7-TMS) receptor isolated from human spleen. Genomics. 1993;16:707–12. [DOI] [PubMed] [Google Scholar]
- 15.Jazin EE, Yoo H, Blomqvist AG, Yee F, Weng G, Walker MW, et al. A proposed bovine neuropeptide Y (NPY) receptor cDNA clone, or its human homologue, confers neither NPY binding sites nor NPY responsiveness on transfected cells. Regul Pept 1993;47:247–58. [DOI] [PubMed] [Google Scholar]
- 16.Nomura H, Nielsen BW, Matsushima K. Molecular cloning of cDNAs encoding a LD78 receptor and putative leukocyte chemotactic peptide receptors. Int Immunol 1993;5:1239–49. [DOI] [PubMed] [Google Scholar]
- 17.Loetscher M, Geiser T, O fReilly T, Zwahlen R, Baggiolini M, Moser B. Cloning of a human seven-transmembrane domain receptor, LESTR, that is highly expressed in leukocytes. J Biol Chem 1994;269:232–7. [PubMed] [Google Scholar]
- 18.Feng Y, Broder CC, Kennedy PE, Berger EA. HIV-1 entry cofactor: functional cDNA cloning of a seven-transmembrane, G protein-coupled receptor. Science. 1996;272:872–7. [DOI] [PubMed] [Google Scholar]
- 19.Berger EA, Murphy PM, Farber JM. Chemokine receptors as HIV-1 coreceptors: roles in viral entry, tropism, and disease. Annu Rev Immunol 1999;17:657–700. [DOI] [PubMed] [Google Scholar]
- 20.Zhang L, Huang Y, He T, Cao Y, Ho DD. HIV-1 subtype and second-receptor use. Nature. 1996;383:768. [DOI] [PubMed] [Google Scholar]
- 21.Connor RI, Sheridan KE, Ceradini D, Choe S, Landau NR. Change in coreceptor use correlates with disease progression in HIV-1--infected individuals. J Exp Med. 1997;185:621–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Björndal A, Deng H, Jansson M, Fiore JR, Colognesi C, Karlsson A, et al. Coreceptor usage of primary human immunodeficiency virus type 1 isolates varies according to biological phenotype. J Virol 1997;71:7478–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Scarlatti G, Tresoldi E, Bjorndal A, Fredriksson R, Colognesi C, Deng HK, et al. In vivo evolution of HIV-1 co-receptor usage and sensitivity to chemokine-mediated suppression. Nat Med 1997;3:1259–65. [DOI] [PubMed] [Google Scholar]
- 24.Bazan HA, Alkhatib G, Broder CC, Berger EA. Patterns of CCR5, CXCR4, and CCR3 usage by envelope glycoproteins from human immunodeficiency virus type 1 primary isolates. J Virol 1998;72:4485–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Oberlin E, Amara A, Bachelerie F, Bessia C, Virelizier J-L, Arenzana-Seisdedos F, et al. The CXC chemokine SDF-1 is the ligand for LESTR/fusin and prevents infection by T-cell-line-adapted HIV-1. Nature. 1996;382:833–5. [DOI] [PubMed] [Google Scholar]
- 26.Bleul CC, Farzan M, Choe H, Parolin C, Clark-Lewis I, Sodroski J, et al. The lymphocyte chemoattractant SDF-1 is a ligand for LESTR/fusin and blocks HIV-1 entry. Nature. 1996;382:829–33. [DOI] [PubMed] [Google Scholar]
- 27.Tashiro K, Tada H, Heilker R, Shirozu M, Nakano T, Honjo T. Signal sequence trap: a cloning strategy for secreted proteins and type I membrane proteins. Science. 1993;261:600–3. [DOI] [PubMed] [Google Scholar]
- 28.Nagasawa T, Hirota S, Tachibana K, Takakura N, Nishikawa S, Kitamura Y, et al. Defects of B-cell lymphopoiesis and bone-marrow myelopoiesis in mice lacking the CXC chemokine PBSF/SDF-1. Nature. 1996;382:635–8. [DOI] [PubMed] [Google Scholar]
- 29.Heusinkveld LE, Yim E, Yang A, Azani AB, Liu Q, Gao J-L, et al. Pathogenesis, diagnosis and therapeutic strategies in WHIM syndrome immunodeficiency. Expert Opin Orphan Drugs. 2017;5:813–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wegner SA, Ehrenberg PK, Chang G, Dayhoff DE, Sleeker AL, Michael NL. Genomic organization and functional characterization of the chemokine receptor CXCR4, a major entry co-receptor for human immunodeficiency virus type 1. J Biol Chem 1998;273:4754–60. [DOI] [PubMed] [Google Scholar]
- 31.Zou Y-R, Kottmann AH, Kuroda M, Taniuchi I, Littman DR. Function of the chemokine receptor CXCR4 in haematopoiesis and in cerebellar development. Nature. 1998;393:595–9. [DOI] [PubMed] [Google Scholar]
- 32.Yang S, Edman LC, Sanchez-Alcaniz JA, Fritz N, Bonilla S, Hecht J, et al. Cxcl12/Cxcr4 signaling controls the migration and process orientation of A9-A10 dopaminergic neurons. Development. 2013;140:4554–64. [DOI] [PubMed] [Google Scholar]
- 33.Abe P, Mueller W, Schutz D, MacKay F, Thelen M, Zhang P, et al. CXCR7 prevents excessive CXCL12-mediated downregulation of CXCR4 in migrating cortical interneurons. Development. 2014;141:1857–63. [DOI] [PubMed] [Google Scholar]
- 34.Ivins S, Chappell J, Vernay B, Suntharalingham J, Martineau A, Mohun TJ, et al. The CXCL12/CXCR4 Axis Plays a Critical Role in Coronary Artery Development. Developmental Cell. 2015;33:455–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Doitsidou M, Reichman-Fried M, Stebler J, Koprunner M, Dorries J, Meyer D, et al. Guidance of primordial germ cell migration by the chemokine SDF-1. Cell. 2002;111:647–59. [DOI] [PubMed] [Google Scholar]
- 36.Guo F, Wang Y, Liu J, Mok SC, Xue F, Zhang W. CXCL12/CXCR4: a symbiotic bridge linking cancer cells and their stromal neighbors in oncogenic communication networks. Oncogene. 2016;35:816–26. [DOI] [PubMed] [Google Scholar]
- 37.Burger JA. CXCR4: a key receptor in the crosstalk between tumor cells and their microenvironment. Blood. 2006;107:1761–7. [DOI] [PubMed] [Google Scholar]
- 38.Zhao H, Guo L, Zhao H, Zhao J, Weng H, Zhao B. CXCR4 over-expression and survival in cancer: A system review and meta-analysis. Oncotarget [Internet]. 2015. [cited 2019 Feb 25];6 Available from: http://www.oncotarget.com/fulltext/3217 [DOI] [PMC free article] [PubMed]
- 39.Müller A, Homey B, Soto H, Ge N, Catron D, Buchanan ME, et al. Involvement of chemokine receptors in breast cancer metastasis. Nature. 2001;410:50–6. [DOI] [PubMed] [Google Scholar]
- 40.Murphy PM, Heusinkveld L. Multisystem multitasking by CXCL12 and its receptors CXCR4 and ACKR3. Cytokine. 2018;109:2–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Janssens R, Struyf S, Proost P. Pathological roles of the homeostatic chemokine CXCL12. Cytokine & Growth Factor Reviews. 2018;44:51–68. [DOI] [PubMed] [Google Scholar]
- 42.Beaussant Cohen S, Fenneteau O, Plouvier E, Rohrlich P-S, Daltroff G, Plantier I, et al. Description and outcome of a cohort of 8 patients with WHIM syndrome from the French Severe Chronic Neutropenia Registry. Orphanet Journal of Rare Diseases. 2012;7:71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.McDermott DH, Murphy PM. WHIM syndrome: Immunopathogenesis, treatment and cure strategies. Immunol Rev 2019;287:91–102. [DOI] [PubMed] [Google Scholar]
- 44.Dotta L, Notarangelo LD, Moratto D, Kumar R, Porta F, Soresina A, et al. Long term outcome of WHIM syndrome in 18 patients: high risk of lung disease and HPV-related malignancies. J Allergy Clin Immunol Pract 2019;7(5):1568–77. [DOI] [PubMed] [Google Scholar]
- 45.Al Ustwani O, Kurzrock R, Wetzler M. Genetics on a WHIM. Br J Haematol 2014;164:15–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Majumdar S, Murphy P. Adaptive Immunodeficiency in WHIM Syndrome. International Journal of Molecular Sciences. 2018;20:3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kawai T, Malech HL. WHIM syndrome: congenital immune deficiency disease. Curr Opin Hematol 2009;16:20–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gulino AV. Altered leukocyte response to CXCL12 in patients with warts hypogammaglobulinemia, infections, myelokathexis (WHIM) syndrome. Blood. 2004;104:444–52. [DOI] [PubMed] [Google Scholar]
- 49.McDermott DH, Liu Q, Velez D, Lopez L, Anaya-O fBrien S, Ulrick J, et al. A phase 1 clinical trial of long-term, low-dose treatment of WHIM syndrome with the CXCR4 antagonist plerixafor. Blood. 2014;123:2308–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Tassone L, Notarangelo LD, Bonomi V, Savoldi G, Sensi A, Soresina A, et al. Clinical and genetic diagnosis of warts, hypogammaglobulinemia, infections, and myelokathexis syndrome in 10 patients. Journal of Allergy and Clinical Immunology. 2009;123:1170–1173.e3. [DOI] [PubMed] [Google Scholar]
- 51.Latger-Cannard V, Bensoussan D, Bordigoni P. The WHIM syndrome shows a peculiar dysgranulopoiesis: myelokathexis. British Journal of Haematology. 2006;132:669–669. [DOI] [PubMed] [Google Scholar]
- 52.Palm MD, Tyring SK, Rady PL, Tharp MD. Human papillomavirus typing of verrucae in a patient with WHIM syndrome. Arch Dermatol 2010;146:931–2. [DOI] [PubMed] [Google Scholar]
- 53.McDermott DH, Liu Q, Ulrick J, Kwatemaa N, Anaya-O fBrien S, Penzak SR, et al. The CXCR4 antagonist plerixafor corrects panleukopenia in patients with WHIM syndrome. Blood. 2011;118:4957–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.McDermott DH. Warts, Hypogammaglobulinemia, Infections, and Myelokathexis Syndrome. Stiehm fs Immune Deficiencies [Internet]. Elsevier; 2014. [cited 2019 Feb 18]. p. 711–9. Available from: https://linkinghub.elsevier.com/retrieve/pii/B9780124055469000352 [Google Scholar]
- 55.Badolato R, Dotta L, Tassone L, Amendola G, Porta F, Locatelli F, et al. Tetralogy of Fallot is an Uncommon Manifestation of Warts, Hypogammaglobulinemia, Infections, and Myelokathexis Syndrome. The Journal of Pediatrics. 2012;161:763–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Taniuchi S, Yamamoto A, Fujiwara T, Hasui M, Tsuji S, Kobayashi Y. Dizygotic twin sisters with myelokathexis: mechanism of its neutropenia. Am J Hematol 1999;62:106–11. [DOI] [PubMed] [Google Scholar]
- 57.Badolato R, Donadieu J, the WHIM Research Group. How I treat warts, hypogammaglobulinemia, infections, and myelokathexis syndrome. Blood. 2017;130:2491–8. [DOI] [PubMed] [Google Scholar]
- 58.McDermott DH, Gao J-L, Liu Q, Siwicki M, Martens C, Jacobs P, et al. Chromothriptic Cure of WHIM Syndrome. Cell. 2015;160:686–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.McDermott DH, Gao J-L, Murphy PM. Chromothriptic cure of WHIM syndrome: Implications for bone marrow transplantation. Rare Diseases. 2015;3:e1073430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Gao J-L, Yim E, Siwicki M, Yang A, Liu Q, Azani A, et al. Cxcr4-haploinsufficient bone marrow transplantation corrects leukopenia in an unconditioned WHIM syndrome model. Journal of Clinical Investigation. 2018;128:3312–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Liu Q, Li Z, Y Yang A, Gao J-L, S Velez D, J Cho E, et al. Mechanisms of Sustained Neutrophilia in Patient WHIM-09, Cured of WHIM Syndrome by Chromothripsis. J Clin Immunol 2018;38:77–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Skokowa J, Dale DC, Touw IP, Zeidler C, Welte K. Severe congenital neutropenias. Nature Reviews Disease Primers. 2017;3:17032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Dale DC, Bolyard AA, Schwinzer BG, Pracht G, Bonilla MA, Boxer L, et al. The Severe Chronic Neutropenia International Registry: 10-Year Follow-up Report. Supportive Cancer Therapy. 2006;3:220–31. [DOI] [PubMed] [Google Scholar]
- 64.Sicre de Fontbrune F, Moignet A, Beaupain B, Suarez F, Galicier L, Socie G, et al. Severe chronic primary neutropenia in adults: report on a series of 108 patients. Blood. 2015;126:1643–50. [DOI] [PubMed] [Google Scholar]
- 65.Aminu M, Gwafan JZ, Oguntayo OA, Ella EE, Koledade AK, Inabo IH. Seroprevalence of human papillomavirus immunoglobulin G antibodies among women presenting at the reproductive health clinic of a university teaching hospital in Nigeria. International Journal of Women’s Health. 2014;479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Goddard EA, Hughes EJ, Beatty DW. A case of immunodeficiency characterized by neutropenia, hypogammaglobulinaemia, recurrent infections and warts. Clin Lab Haematol 1994;16:297–302. [DOI] [PubMed] [Google Scholar]
- 67.Gorlin RJ, Gelb B, Diaz GA, Lofsness KG, Pittelkow MR, Fenyk JR. WHIM syndrome, an autosomal dominant disorder: clinical, hematological, and molecular studies. Am J Med Genet 2000;91:368–76. [PubMed] [Google Scholar]
- 68.Auer PL, Teumer A, Schick U, O fShaughnessy A, Lo KS, Chami N, et al. Rare and low-frequency coding variants in CXCR2 and other genes are associated with hematological traits. Nat Genet 2014;46:629–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Eash KJ, Greenbaum AM, Gopalan PK, Link DC. CXCR2 and CXCR4 antagonistically regulate neutrophil trafficking from murine bone marrow. J Clin Invest 2010;120:2423–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Martin C, Burdon PCE, Bridger G, Gutierrez-Ramos JC, Williams TJ, Rankin SM. Chemokines acting via CXCR2 and CXCR4 control the release of neutrophils from the bone marrow and their return following senescence. Immunity. 2003;19:583–93. [DOI] [PubMed] [Google Scholar]
- 71.Hoggatt J, Singh P, Tate TA, Chou B-K, Datari SR, Fukuda S, et al. Rapid Mobilization Reveals a Highly Engraftable Hematopoietic Stem Cell. Cell. 2018;172:191–204.e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Liu Q, Chen H, Ojode T, Gao X, Anaya-O fBrien S, Turner NA, et al. WHIM syndrome caused by a single amino acid substitution in the carboxy-tail of chemokine receptor CXCR4. Blood. 2012;120:181–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Balabanian K WHIM syndromes with different genetic anomalies are accounted for by impaired CXCR4 desensitization to CXCL12. Blood. 2005;105:2449–57. [DOI] [PubMed] [Google Scholar]
- 74.Hunter ZR, Xu L, Yang G, Zhou Y, Liu X, Cao Y, et al. The genomic landscape of Waldenstrom macroglobulinemia is characterized by highly recurring MYD88 and WHIM-like CXCR4 mutations, and small somatic deletions associated with B-cell lymphomagenesis. Blood. 2014;123:1637–46. [DOI] [PubMed] [Google Scholar]
- 75.Roccaro AM, Sacco A, Jimenez C, Maiso P, Moschetta M, Mishima Y, et al. C1013G/CXCR4 acts as a driver mutation of tumor progression and modulator of drug resistance in lymphoplasmacytic lymphoma. Blood. 2014;123:4120–31. [DOI] [PubMed] [Google Scholar]
- 76.Valentin G, Haas P, Gilmour D. The chemokine SDF1a coordinates tissue migration through the spatially restricted activation of Cxcr7 and Cxcr4b. Curr Biol 2007;17:1026–31. [DOI] [PubMed] [Google Scholar]
- 77.Naumann U, Cameroni E, Pruenster M, Mahabaleshwar H, Raz E, Zerwes H-G, et al. CXCR7 functions as a scavenger for CXCL12 and CXCL11. PLoS ONE. 2010;5:e9175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Miao Z, Luker KE, Summers BC, Berahovich R, Bhojani MS, Rehemtulla A, et al. CXCR7 (RDC1) promotes breast and lung tumor growth in vivo and is expressed on tumor-associated vasculature. Proc Natl Acad Sci USA. 2007;104:15735–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Bachelerie F, Graham GJ, Locati M, Mantovani A, Murphy PM, Nibbs R, et al. New nomenclature for atypical chemokine receptors. Nat Immunol 2014;15:207–8. [DOI] [PubMed] [Google Scholar]
- 80.Balabanian K, Lagane B, Infantino S, Chow KYC, Harriague J, Moepps B, et al. The chemokine SDF-1/CXCL12 binds to and signals through the orphan receptor RDC1 in T lymphocytes. J Biol Chem 2005;280:35760–6. [DOI] [PubMed] [Google Scholar]
- 81.Ma Q, Jones D, Borghesani PR, Segal RA, Nagasawa T, Kishimoto T, et al. Impaired B-lymphopoiesis, myelopoiesis, and derailed cerebellar neuron migration in CXCR4- and SDF-1-deficient mice. Proceedings of the National Academy of Sciences. 1998;95:9448–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Gerrits H, van Ingen Schenau DS, Bakker NEC, van Disseldorp AJM, Strik A, Hermens LS, et al. Early postnatal lethality and cardiovascular defects in CXCR7-deficient mice. genesis. 2008;46:235–45. [DOI] [PubMed] [Google Scholar]
- 83.Sierro F, Biben C, Martinez-Munoz L, Mellado M, Ransohoff RM, Li M, et al. Disrupted cardiac development but normal hematopoiesis in mice deficient in the second CXCL12/SDF-1 receptor, CXCR7. Proceedings of the National Academy of Sciences. 2007;104:14759–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Uzzan M, Ko HM, Mehandru S, Cunningham-Rundles C. Gastrointestinal Disorders Associated with Common Variable Immune Deficiency (CVID) and Chronic Granulomatous Disease (CGD). Curr Gastroenterol Rep 2016;18:17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Alimchandani M, Lai J-P, Aung PP, Khangura S, Kamal N, Gallin JI, et al. Gastrointestinal histopathology in chronic granulomatous disease: a study of 87 patients. Am J Surg Pathol 2013;37:1365–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Pastrana DV, Peretti A, Welch NL, Borgogna C, Olivero C, Badolato R, et al. Metagenomic Discovery of 83 New Human Papillomavirus Types in Patients with Immunodeficiency. Imperiale MJ, editor. mSphere [Internet]. 2018. [cited 2019 Feb 18];3 Available from: http://msphere.asm.org/lookup/doi/10.1128/mSphereDirect.00645-18 [DOI] [PMC free article] [PubMed]
- 87.McDermott DH, Pastrana DV, Calvo KR, Pittaluga S, Velez D, Cho E, et al. Plerixafor for the Treatment of WHIM Syndrome. New England Journal of Medicine. 2019;380:163–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Tarzi MD, Jenner M, Hattotuwa K, Faruqi AZ, Diaz GA, Longhurst HJ. Sporadic case of warts, hypogammaglobulinemia, immunodeficiency, and myelokathexis syndrome. Journal of Allergy and Clinical Immunology. 2005;116:1101–5. [DOI] [PubMed] [Google Scholar]
- 89.Beynon DWG, Lopes A, Daras B, Monaghan JM. Radical vulvectomy and groin node dissection in a patient with chronic neutropenia-maintenance of leucocyte count using granulocyte colony-stimulating factor. Int J Gynecol Cancer. 1993;3:405–7. [DOI] [PubMed] [Google Scholar]
- 90.Leiding JW, Holland SM. Warts and all: Human papillomavirus in primary immunodeficiencies. Journal of Allergy and Clinical Immunology. 2012;130:1030–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Sri JC, Dubina MI, Kao GF, Rady PL, Tyring SK, Gaspari AA. Generalized verrucosis: A review of the associated diseases, evaluation, and treatments. Journal of the American Academy of Dermatology. 2012;66:292–311. [DOI] [PubMed] [Google Scholar]
- 92.Imashuku S, Miyagawa A, Chiyonobu T, Ishida H, Yoshihara T, Teramura T, et al. Epstein-Barr virus-associated T-lymphoproliferative disease with hemophagocytic syndrome, followed by fatal intestinal B lymphoma in a young adult female with WHIM syndrome. Annals of Hematology. 2002;81:470–3. [DOI] [PubMed] [Google Scholar]
- 93.Yoshii Y, Kato T, Ono K, Takahashi E, Fujimoto N, Kobayashi S, et al. Primary cutaneous follicle center lymphoma in a patient with WHIM syndrome. J Eur Acad Dermatol Venereol 2016;30:529–30. [DOI] [PubMed] [Google Scholar]
- 94.Chae KM, Ertle JO, Tharp MD. B-cell lymphoma in a patient with WHIM syndrome. J Am Acad Dermatol 2001;44:124–8. [DOI] [PubMed] [Google Scholar]
- 95.Momma K Cardiovascular anomalies associated with chromosome 22q11.2 deletion syndrome. Am J Cardiol 2010;105:1617–24. [DOI] [PubMed] [Google Scholar]
- 96.Kobayashi D, Sallaam S, Humes RA. Tetralogy of Fallot with complete DiGeorge syndrome: report of a case and a review of the literature. Congenit Heart Dis 2013;8:E119–126. [DOI] [PubMed] [Google Scholar]
- 97.Balabanian K, Brotin E, Biajoux V, Bouchet-Delbos L, Lainey E, Fenneteau O, et al. Proper desensitization of CXCR4 is required for lymphocyte development and peripheral compartmentalization in mice. Blood. 2012;119:5722–30. [DOI] [PubMed] [Google Scholar]
- 98.Galli J, Pinelli L, Micheletti S, Palumbo G, Notarangelo LD, Lougaris V, et al. Cerebellar involvement in warts Hypogammaglobulinemia immunodeficiency myelokathexis patients: neuroimaging and clinical findings. Orphanet J Rare Dis 2019;14:61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Takaya J, Fujii Y, Higashino H, Taniuchi S, Nakamura M, Kaneko K. A case of WHIM syndrome associated with diabetes and hypothyroidism. Pediatr Diabetes. 2009;10:484–6. [DOI] [PubMed] [Google Scholar]
- 100.Aprikyan AA, Liles WC, Park JR, Jonas M, Chi EY, Dale DC. Myelokathexis, a congenital disorder of severe neutropenia characterized by accelerated apoptosis and defective expression of bcl-x in neutrophil precursors. Blood. 2000;95:320–7. [PubMed] [Google Scholar]
- 101.Siedlar M, Rudzki Z, Strach M, Trzyna E, Pituch-Noworolska A, B.aut-Szlosarczyk A, et al. Familial occurrence of warts, hypogammaglobulinemia, infections, and myelokathexis (WHIM) syndrome. Arch Immunol Ther Exp (Warsz). 2008;56:419–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Aghamohammadi A, Abolhassani H, Puchalka J, Greif-Kohistani N, Zoghi S, Klein C, et al. Preference of Genetic Diagnosis of CXCR4 Mutation Compared with Clinical Diagnosis of WHIM Syndrome. Journal of Clinical Immunology. 2017;37:282–6. [DOI] [PubMed] [Google Scholar]
- 103.Banka S, Newman WG. A clinical and molecular review of ubiquitous glucose-6-phosphatase deficiency caused by G6PC3 mutations. Orphanet Journal of Rare Diseases. 2013;8:84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.McDermott DH, De Ravin SS, Jun HS, Liu Q, Priel DAL, Noel P, et al. Severe congenital neutropenia resulting from G6PC3 deficiency with increased neutrophil CXCR4 expression and myelokathexis. Blood. 2010;116:2793–802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Boztug K, Appaswamy G, Ashikov A, Schaffer AA, Salzer U, Diestelhorst J, et al. A syndrome with congenital neutropenia and mutations in G6PC3. N Engl J Med 2009;360:32–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Kolehmainen J, Black GCM, Saarinen A, Chandler K, Clayton-Smith J, Traskelin A-L, et al. Cohen syndrome is caused by mutations in a novel gene, COH1, encoding a transmembrane protein with a presumed role in vesicle-mediated sorting and intracellular protein transport. Am J Hum Genet 2003;72:1359–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Shearman JR, Wilton AN. A canine model of Cohen syndrome: Trapped Neutrophil Syndrome. BMC Genomics. 2011;12:258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Kivitie-Kallio S, Rajantie J, Juvonen E, Norio R. Granulocytopenia in Cohen syndrome. Br J Haematol 1997;98:308–11. [DOI] [PubMed] [Google Scholar]
- 109.Chae KM, Ertle JO, Tharp MD. B-cell lymphoma in a patient with WHIM syndrome. J Am Acad Dermatol 2001;44:124–8. [DOI] [PubMed] [Google Scholar]
- 110.Wu B, Chien EYT, Mol CD, Fenalti G, Liu W, Katritch V, et al. Structures of the CXCR4 Chemokine GPCR with Small-Molecule and Cyclic Peptide Antagonists. Science. 2010;330:1066–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Handel TM. The Structure of a CXCR4:Chemokine Complex. Frontiers in Immunology [Internet]. 2015. [cited 2019 Feb 19];6 Available from: http://journal.frontiersin.org/Article/10.3389/fimmu.2015.00282/abstract [DOI] [PMC free article] [PubMed]
- 112.Wescott MP, Kufareva I, Paes C, Goodman JR, Thaker Y, Puffer BA, et al. Signal transmission through the CXC chemokine receptor 4 (CXCR4) transmembrane helices. Proceedings of the National Academy of Sciences. 2016;113:9928–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Kufareva I, Stephens BS, Holden LG, Qin L, Zhao C, Kawamura T, et al. Stoichiometry and geometry of the CXC chemokine receptor 4 complex with CXC ligand 12: Molecular modeling and experimental validation. Proceedings of the National Academy of Sciences. 2014;111:E5363–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Ziarek JJ, Getschman AE, Butler SJ, Taleski D, Stephens B, Kufareva I, et al. Sulfopeptide Probes of the CXCR4/CXCL12 Interface Reveal Oligomer-Specific Contacts and Chemokine Allostery. ACS Chemical Biology. 2013;8:1955–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Qin L, Kufareva I, Holden LG, Wang C, Zheng Y, Zhao C, et al. Crystal structure of the chemokine receptor CXCR4 in complex with a viral chemokine. Science. 2015;347:1117–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Busillo JM, Benovic JL. Regulation of CXCR4 signaling. Biochim Biophys Acta. 2007;1768:952–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Futahashi Y, Komano J, Urano E, Aoki T, Hamatake M, Miyauchi K, et al. Separate elements are required for ligand-dependent and -independent internalization of metastatic potentiator CXCR4. Cancer Sci 2007;98:373–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Cheng ZJ, Zhao J, Sun Y, Hu W, Wu YL, Cen B, et al. beta-arrestin differentially regulates the chemokine receptor CXCR4-mediated signaling and receptor internalization, and this implicates multiple interaction sites between beta-arrestin and CXCR4. J Biol Chem 2000;275:2479–85. [DOI] [PubMed] [Google Scholar]
- 119.Balabanian K, Levoye A, Klemm L, Lagane B, Hermine O, Harriague J, et al. Leukocyte analysis from WHIM syndrome patients reveals a pivotal role for GRK3 in CXCR4 signaling. Journal of Clinical Investigation [Internet]. 2008. [cited 2018 Nov 7]; Available from: http://www.jci.org/articles/view/33187 [DOI] [PMC free article] [PubMed]
- 120.McDermott DH, Lopez J, Deng F, Liu Q, Ojode T, Chen H, et al. AMD3100 is a potent antagonist at CXCR4R334X, a hyperfunctional mutant chemokine receptor and cause of WHIM syndrome. Journal of Cellular and Molecular Medicine. 2011;15:2071–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Liu Q, Pan C, Lopez L, Gao J, Velez D, Anaya-O fBrien S, et al. WHIM Syndrome Caused by Waldenstrom fs Macroglobulinemia-Associated Mutation CXCR4 L329fs. Journal of Clinical Immunology. 2016;36:397–405. [DOI] [PubMed] [Google Scholar]
- 122.Mueller W, Schutz D, Nagel F, Schulz S, Stumm R. Hierarchical Organization of Multi-Site Phosphorylation at the CXCR4 C Terminus. Klein R, editor. PLoS ONE. 2013;8:e64975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Lagane B, Chow KYC, Balabanian K, Levoye A, Harriague J, Planchenault T, et al. CXCR4 dimerization and -arrestin-mediated signaling account for the enhanced chemotaxis to CXCL12 in WHIM syndrome. Blood. 2008;112:34–44. [DOI] [PubMed] [Google Scholar]
- 124.Martinez-Munoz L, Rodriguez-Frade JM, Barroso R, Sorzano COS, Torreno-Pina JA, Santiago CA, et al. Separating Actin-Dependent Chemokine Receptor Nanoclustering from Dimerization Indicates a Role for Clustering in CXCR4 Signaling and Function. Mol Cell. 2018;70:106–119.e10. [DOI] [PubMed] [Google Scholar]
- 125.Petit I, Szyper-Kravitz M, Nagler A, Lahav M, Peled A, Habler L, et al. G-CSF induces stem cell mobilization by decreasing bone marrow SDF-1 and up-regulating CXCR4. Nat Immunol 2002;3:687–94. [DOI] [PubMed] [Google Scholar]
- 126.Peled A, Kollet O, Ponomaryov T, Petit I, Franitza S, Grabovsky V, et al. The chemokine SDF-1 activates the integrins LFA-1, VLA-4, and VLA-5 on immature human CD34(+) cells: role in transendothelial/stromal migration and engraftment of NOD/SCID mice. Blood. 2000;95:3289–96. [PubMed] [Google Scholar]
- 127.Peled A, Grabovsky V, Habler L, Sandbank J, Arenzana-Seisdedos F, Petit I, et al. The chemokine SDF-1 stimulates integrin-mediated arrest of CD34(+) cells on vascular endothelium under shear flow. J Clin Invest 1999;104:1199–211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Peled A, Petit I, Kollet O, Magid M, Ponomaryov T, Byk T, et al. Dependence of human stem cell engraftment and repopulation of NOD/SCID mice on CXCR4. Science. 1999;283:845–8. [DOI] [PubMed] [Google Scholar]
- 129.Sugiyama T, Kohara H, Noda M, Nagasawa T. Maintenance of the hematopoietic stem cell pool by CXCL12-CXCR4 chemokine signaling in bone marrow stromal cell niches. Immunity. 2006;25:977–88. [DOI] [PubMed] [Google Scholar]
- 130.Walters KB, Green JM, Surfus JC, Yoo SK, Huttenlocher A. Live imaging of neutrophil motility in a zebrafish model of WHIM syndrome. Blood. 2010;116:2803–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Kawai T, Choi U, Whiting-Theobald NL, Linton GF, Brenner S, Sechler JMG, et al. Enhanced function with decreased internalization of carboxy-terminus truncated CXCR4 responsible for WHIM syndrome. Experimental Hematology. 2005;33:460–8. [DOI] [PubMed] [Google Scholar]
- 132.Hatse S, Princen K, Bridger G, De Clercq E, Schols D. Chemokine receptor inhibition by AMD3100 is strictly confined to CXCR4. FEBS Letters. 2002;527:255–62. [DOI] [PubMed] [Google Scholar]
- 133.Fricker SP, Anastassov V, Cox J, Darkes MC, Grujic O, Idzan SR, et al. Characterization of the molecular pharmacology of AMD3100: a specific antagonist of the G-protein coupled chemokine receptor, CXCR4. Biochem Pharmacol 2006;72:588–96. [DOI] [PubMed] [Google Scholar]
- 134.Liu Q, Li Z, Gao J-L, Wan W, Ganesan S, McDermott DH, et al. CXCR4 antagonist AMD3100 redistributes leukocytes from primary immune organs to secondary immune organs, lung, and blood in mice: Leukocyte signaling. European Journal of Immunology. 2015;45:1855–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Devi S, Wang Y, Chew WK, Lima R, A-Gonzalez N, Mattar CNZ, et al. Neutrophil mobilization via plerixafor-mediated CXCR4 inhibition arises from lung demargination and blockade of neutrophil homing to the bone marrow. The Journal of Experimental Medicine. 2013;210:2321–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.De Filippo K, Rankin SM. CXCR4, the master regulator of neutrophil trafficking in homeostasis and disease. Eur J Clin Invest 2018;48 Suppl 2:e12949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Sanmun D, Garwicz D, Smith CIE, Palmblad J, Fadeel B. Stromal-derived factor-1 abolishes constitutive apoptosis of WHIM syndrome neutrophils harbouring a truncating CXCR4 mutation. Br J Haematol 2006;134:640–4. [DOI] [PubMed] [Google Scholar]
- 138.Rankin SM. The bone marrow: a site of neutrophil clearance. Journal of Leukocyte Biology. 2010;88:241–51. [DOI] [PubMed] [Google Scholar]
- 139.Weisel KC, Bautz F, Seitz G, Yildirim S, Kanz L, Mohle R. Modulation of CXC chemokine receptor expression and function in human neutrophils during aging in vitro suggests a role in their clearance from circulation. Mediators Inflamm 2009;2009:790174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Ceradini DJ, Kulkarni AR, Callaghan MJ, Tepper OM, Bastidas N, Kleinman ME, et al. Progenitor cell trafficking is regulated by hypoxic gradients through HIF-1 induction of SDF-1. Nat Med 2004;10:858–64. [DOI] [PubMed] [Google Scholar]
- 141.Staller P, Sulitkova J, Lisztwan J, Moch H, Oakeley EJ, Krek W. Chemokine receptor CXCR4 downregulated by von Hippel-Lindau tumour suppressor pVHL. Nature. 2003;425:307–11. [DOI] [PubMed] [Google Scholar]
- 142.Walmsley SR, Cadwallader KA, Chilvers ER. The role of HIF-1alpha in myeloid cell inflammation. Trends Immunol. 2005;26:434–9. [DOI] [PubMed] [Google Scholar]
- 143.Adrover JM, Del Fresno C, Crainiciuc G, Cuartero MI, Casanova-Acebes M, Weiss LA, et al. A Neutrophil Timer Coordinates Immune Defense and Vascular Protection. Immunity. 2019;50:390–402.e10. [DOI] [PubMed] [Google Scholar]
- 144.Smith E, Zarbock A, Stark MA, Burcin TL, Bruce AC, Foley P, et al. IL-23 is required for neutrophil homeostasis in normal and neutrophilic mice. J Immunol 2007;179:8274–9. [DOI] [PubMed] [Google Scholar]
- 145.Boxer LA. How to approach neutropenia. Hematology Am Soc Hematol Educ Program. 2012;2012:174–82. [DOI] [PubMed] [Google Scholar]
- 146.Noda M, Omatsu Y, Sugiyama T, Oishi S, Fujii N, Nagasawa T. CXCL12-CXCR4 chemokine signaling is essential for NK-cell development in adult mice. Blood. 2011;117:451–8. [DOI] [PubMed] [Google Scholar]
- 147.Mayol K, Biajoux V, Marvel J, Balabanian K, Walzer T. Sequential desensitization of CXCR4 and S1P5 controls natural killer cell trafficking. Blood. 2011;118:4863–71. [DOI] [PubMed] [Google Scholar]
- 148.Mc Guire PJ, Cunningham-Rundles C, Ochs H, Diaz GA. Oligoclonality, impaired class switch and B-cell memory responses in WHIM syndrome. Clinical Immunology. 2010;135:412–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Kean LS, Sen S, Onabajo O, Singh K, Robertson J, Stempora L, et al. Significant mobilization of both conventional and regulatory T cells with AMD3100. Blood. 2011;118:6580–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Jaeger BN, Donadieu J, Cognet C, Bernat C, Ordonez-Rueda D, Barlogis V, et al. Neutrophil depletion impairs natural killer cell maturation, function, and homeostasis. The Journal of Experimental Medicine. 2012;209:565–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Alapi K, Erd?s M, Kovacs G, Marodi L. Recurrent CXCR4 sequence variation in a girl with WHIM syndrome. European Journal of Haematology. 2007;78:86–8. [DOI] [PubMed] [Google Scholar]
- 152.Moens L, Frans G, Bosch B, Bossuyt X, Verbinnen B, Poppe W, et al. Successful hematopoietic stem cell transplantation for myelofibrosis in an adult with warts-hypogammaglobulinemia-immunodeficiency-myelokathexis syndrome. Journal of Allergy and Clinical Immunology. 2016;138:1485–1489.e2. [DOI] [PubMed] [Google Scholar]
- 153.Nagasawa T, Kikutani H, Kishimoto T. Molecular cloning and structure of a pre-B-cell growth-stimulating factor. Proc Natl Acad Sci USA. 1994;91:2305–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Beck TC, Gomes AC, Cyster JG, Pereira JP. CXCR4 and a cell-extrinsic mechanism control immature B lymphocyte egress from bone marrow. The Journal of Experimental Medicine. 2014;211:2567–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Dale DC, Bolyard AA, Kelley ML, Westrup EC, Makaryan V, Aprikyan A, et al. The CXCR4 antagonist plerixafor is a potential therapy for myelokathexis, WHIM syndrome. Blood. 2011;118:4963–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Freitas C, Wittner M, Nguyen J, Rondeau V, Biajoux V, Aknin M-L, et al. Lymphoid differentiation of hematopoietic stem cells requires efficient Cxcr4 desensitization. The Journal of Experimental Medicine. 2017;214:2023–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Nie Y, Waite J, Brewer F, Sunshine M-J, Littman DR, Zou Y-R. The Role of CXCR4 in Maintaining Peripheral B Cell Compartments and Humoral Immunity. The Journal of Experimental Medicine. 2004;200:1145–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Shin DW, Park SN, Kim S-M, Im K, Kim J-A, Hong KT, et al. WHIM Syndrome With a Novel CXCR4 Variant in a Korean Child. Annals of Laboratory Medicine. 2017;37:446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Handisurya A, Schellenbacher C, Reininger B, Koszik F, Vyhnanek P, Heitger A, et al. A quadrivalent HPV vaccine induces humoral and cellular immune responses in WHIM immunodeficiency syndrome. Vaccine. 2010;28:4837–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Allen CDC, Ansel KM, Low C, Lesley R, Tamamura H, Fujii N, et al. Germinal center dark and light zone organization is mediated by CXCR4 and CXCR5. Nature Immunology. 2004;5:943–52. [DOI] [PubMed] [Google Scholar]
- 161.Roselli G, Martini E, Lougaris V, Badolato R, Viola A, Kallikourdis M. CXCL12 Mediates Aberrant Costimulation of B Lymphocytes in Warts, Hypogammaglobulinemia, Infections, Myelokathexis Immunodeficiency. Frontiers in Immunology [Internet]. 2017. [cited 2018 Oct 9];8 Available from: http://journal.frontiersin.org/article/10.3389/fimmu.2017.01068/full [DOI] [PMC free article] [PubMed]
- 162.Biajoux V, Natt J, Freitas C, Alouche N, Sacquin A, Hemon P, et al. Efficient Plasma Cell Differentiation and Trafficking Require Cxcr4 Desensitization. Cell Reports. 2016;17:193–205. [DOI] [PubMed] [Google Scholar]
- 163.Becker M, Hobeika E, Jumaa H, Reth M, Maity PC. CXCR4 signaling and function require the expression of the IgD-class B-cell antigen receptor. Proceedings of the National Academy of Sciences. 2017;114:5231–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Gulino AV. WHIM syndrome: a genetic disorder of leukocyte trafficking. Curr Opin Allergy Clin Immunol. 2003;3:443–50. [DOI] [PubMed] [Google Scholar]
- 165.Saettini F, Notarangelo LD, Biondi A, Bonanomi S. Neutropenia, hypogammaglobulinemia, and pneumonia: A case of WHIM syndrome. Pediatrics International. 2018;60:318–9. [DOI] [PubMed] [Google Scholar]
- 166.Lundqvist A, Smith AL, Takahashi Y, Wong S, Bahceci E, Cook L, et al. Differences in the Phenotype, Cytokine Gene Expression Profiles, and In Vivo Alloreactivity of T Cells Mobilized with Plerixafor Compared with G-CSF. The Journal of Immunology. 2013;191:6241–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Calderon L, Boehm T. Three chemokine receptors cooperatively regulate homing of hematopoietic progenitors to the embryonic mouse thymus. Proceedings of the National Academy of Sciences. 2011;108:7517–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Robertson P, Means TK, Luster AD, Scadden DT. CXCR4 and CCR5 mediate homing of primitive bone marrow.derived hematopoietic cells to the postnatal thymus. Experimental Hematology. 2006;34:308–19. [DOI] [PubMed] [Google Scholar]
- 169.Plotkin J, Prockop SE, Lepique A, Petrie HT. Critical role for CXCR4 signaling in progenitor localization and T cell differentiation in the postnatal thymus. J Immunol 2003;171:4521–7. [DOI] [PubMed] [Google Scholar]
- 170.Trampont PC, Tosello-Trampont A-C, Shen Y, Duley AK, Sutherland AE, Bender TP, et al. CXCR4 acts as a costimulator during thymic ƒÀ-selection. Nature Immunology. 2010;11:162–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Janas ML, Varano G, Gudmundsson K, Noda M, Nagasawa T, Turner M. Thymic development beyond ƒÀ-selection requires phosphatidylinositol 3-kinase activation by CXCR4. The Journal of Experimental Medicine. 2010;207:247–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Ara T, Itoi M, Kawabata K, Egawa T, Tokoyoda K, Sugiyama T, et al. A Role of CXC Chemokine Ligand 12/Stromal Cell-Derived Factor-1/Pre-B Cell Growth Stimulating Factor and Its Receptor CXCR4 in Fetal and Adult T Cell Development in Vivo. The Journal of Immunology. 2003;170:4649–55. [DOI] [PubMed] [Google Scholar]
- 173.Hernandezlopez C, Valencia J, Hidalgo L, Martinez V, Zapata A, Sacedon R, et al. CXCL12/CXCR4 signaling promotes human thymic dendritic cell survival regulating the Bcl-2/Bax ratio. Immunology Letters. 2008;120:72–8. [DOI] [PubMed] [Google Scholar]
- 174.Kumar A, Humphreys TD, Kremer KN, Bramati PS, Bradfield L, Edgar CE, et al. CXCR4 Physically Associates with the T Cell Receptor to Signal in T Cells. Immunity. 2006;25:213–24. [DOI] [PubMed] [Google Scholar]
- 175.Smith X, Schneider H, Kohler K, Liu H, Lu Y, Rudd CE. The chemokine CXCL12 generates costimulatory signals in T cells to enhance phosphorylation and clustering of the adaptor protein SLP-76. Sci Signal. 2013;6:ra65. [DOI] [PubMed] [Google Scholar]
- 176.Molon B, Gri G, Bettella M, Gomez-Mouton C, Lanzavecchia A, Martinez-A C, et al. T cell costimulation by chemokine receptors. Nature Immunology. 2005;6:465–71. [DOI] [PubMed] [Google Scholar]
- 177.Nanki T, Lipsky PE. Cutting Edge: Stromal Cell-Derived Factor-1 Is a Costimulator for CD4+ T Cell Activation. The Journal of Immunology. 2000;164:5010–4. [DOI] [PubMed] [Google Scholar]
- 178.Kallikourdis M, Trovato AE, Anselmi F, Sarukhan A, Roselli G, Tassone L, et al. The CXCR4 mutations in WHIM syndrome impair the stability of the T-cell immunologic synapse. Blood. 2013;122:666–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Chaix J, Nish SA, Lin W-HW, Rothman NJ, Ding L, Wherry EJ, et al. Cutting Edge: CXCR4 Is Critical for CD8+ Memory T Cell Homeostatic Self-Renewal but Not Rechallenge Self-Renewal. The Journal of Immunology. 2014;193:1013–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Meuris F, Carthagena L, Jaracz-Ros A, Gaudin F, Cutolo P, Deback C, et al. The CXCL12/CXCR4 Signaling Pathway: A New Susceptibility Factor in Human Papillomavirus Pathogenesis. PLoS Pathog. 2016;12:e1006039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Chow KYC, Brotin E, Ben Khalifa Y, Carthagena L, Teissier S, Danckaert A, et al. A pivotal role for CXCL12 signaling in HPV-mediated transformation of keratinocytes: clues to understanding HPV-pathogenesis in WHIM syndrome. Cell Host Microbe. 2010;8:523–33. [DOI] [PubMed] [Google Scholar]
- 182.Bollag WB, Hill WD. CXCR4 in epidermal keratinocytes: crosstalk within the skin. J Invest Dermatol 2013;133:2505–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Westrich JA, Warren CJ, Pyeon D. Evasion of host immune defenses by human papillomavirus. Virus Res 2017;231:21–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Komdeur FL, Prins TM, van de Wall S, Plat A, Wisman GBA, Hollema H, et al. CD103+ tumor-infiltrating lymphocytes are tumor-reactive intraepithelial CD8+ T cells associated with prognostic benefit and therapy response in cervical cancer. Oncoimmunology. 2017;6:e1338230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Kim TJ, Jin H-T, Hur S-Y, Yang HG, Seo YB, Hong SR, et al. Clearance of persistent HPV infection and cervical lesion by therapeutic DNA vaccine in CIN3 patients. Nature Communications [Internet]. 2014. [cited 2018 Oct 25];5 Available from: http://www.nature.com/articles/ncomms6317 [DOI] [PMC free article] [PubMed]
- 186.Diniz MO, Sales NS, Silva JR, Ferreira LCS. Protection against HPV-16-Associated Tumors Requires the Activation of CD8+ Effector Memory T Cells and the Control of Myeloid-Derived Suppressor Cells. Molecular Cancer Therapeutics. 2016;15:1920–30. [DOI] [PubMed] [Google Scholar]
- 187.Meuris F, Gaudin F, Aknin M-L, Hemon P, Berrebi D, Bachelerie F. Symptomatic Improvement in Human Papillomavirus-Induced Epithelial Neoplasia by Specific Targeting of the CXCR4 Chemokine Receptor. Journal of Investigative Dermatology. 2016;136:473–80. [DOI] [PubMed] [Google Scholar]
- 188.Bontkes HJ, Ruizendaal JJ, Kramer D, Meijer CJLM, Hooijberg E. Plasmacytoid dendritic cells are present in cervical carcinoma and become activated by human papillomavirus type 16 virus-like particles. Gynecologic Oncology. 2005;96:897–901. [DOI] [PubMed] [Google Scholar]
- 189.Tassone L, Moratto D, Vermi W, De Francesco M, Notarangelo LD, Porta F, et al. Defect of plasmacytoid dendritic cells in warts, hypogammaglobulinemia, infections, myelokathexis (WHIM) syndrome patients. Blood. 2010;116:4870–3. [DOI] [PubMed] [Google Scholar]
- 190.Weston B, Axtell RA, Todd RF, Vincent M, Balazovich KJ, Suchard SJ, et al. Clinical and biologic effects of granulocyte colony stimulating factor in the treatment of myelokathexis. The Journal of Pediatrics. 1991;118:229–34. [DOI] [PubMed] [Google Scholar]
- 191.Dale D, Bolyard AA, Dick E, Kelley ML, Makaryan V, Johnson R, et al. X4P-001: A Novel Molecularly-Targeted Oral Therapy for Whim Syndrome. Blood. 2017;130:995.28646116 [Google Scholar]
- 192.De Clercq E The AMD3100 story: The path to the discovery of a stem cell mobilizer (Mozobil). Biochemical Pharmacology. 2009;77:1655–64. [DOI] [PubMed] [Google Scholar]
- 193.De Clercq E AMD3100/CXCR4 Inhibitor. Frontiers in Immunology [Internet]. 2015. [cited 2018 Sep 19];6 Available from: http://journal.frontiersin.org/Article/10.3389/fimmu.2015.00276/abstract
- 194.De Clercq E The bicyclam AMD3100 story. Nat Rev Drug Discov 2003;2:581–7. [DOI] [PubMed] [Google Scholar]
- 195.Hendrix CW, Collier AC, Lederman MM, Schols D, Pollard RB, Brown S, et al. Safety, pharmacokinetics, and antiviral activity of AMD3100, a selective CXCR4 receptor inhibitor, in HIV-1 infection. J Acquir Immune Defic Syndr 2004;37:1253–62. [DOI] [PubMed] [Google Scholar]
- 196.Liu T, Li X, You S, Bhuyan SS, Dong L. Effectiveness of AMD3100 in treatment of leukemia and solid tumors: from original discovery to use in current clinical practice. Experimental Hematology & Oncology [Internet]. 2015. [cited 2018 Oct 11];5 Available from: http://ehoonline.biomedcentral.com/articles/10.1186/s40164-016-0050-5 [DOI] [PMC free article] [PubMed]
- 197.Gayatri S, Nabil H, Bita J, Sharon F, Loretta P, Fengshuo L, et al. A Phase II, Open-Label Pilot Study to Evaluate the Hematopoietic Stem Cell Mobilization of TG-0054 Combined with G-CSF in 12 Patients with Multiple Myeloma, Non-Hodgkin Lymphoma or Hodgkin Lymphoma - an Interim Analysis. Blood. :515. [Google Scholar]
- 198.Vater A, Sahlmann J, Kroger N, Zollner S, Lioznov M, Maasch C, et al. Hematopoietic Stem and Progenitor Cell Mobilization in Mice and Humans by a First-in-Class Mirror-Image Oligonucleotide Inhibitor of CXCL12. Clinical Pharmacology & Therapeutics. 2013;94:150–7. [DOI] [PubMed] [Google Scholar]
- 199.Hachet-Haas M, Balabanian K, Rohmer F, Pons F, Franchet C, Lecat S, et al. Small Neutralizing Molecules to Inhibit Actions of the Chemokine CXCL12. Journal of Biological Chemistry. 2008;283:23189–99. [DOI] [PubMed] [Google Scholar]
- 200.de Wit RH, Heukers R, Brink HJ, Arsova A, Maussang D, Cutolo P, et al. CXCR4-Specific Nanobodies as Potential Therapeutics for WHIM syndrome. Journal of Pharmacology and Experimental Therapeutics. 2017;363:35–44. [DOI] [PubMed] [Google Scholar]
- 201.Kawahara Y, Oh Y, Kato T, Zaha K, Morimoto A. Transient Marked Increase of γδ T Cells in WHIM Syndrome After Successful HSCT. Journal of Clinical Immunology. 2018;38:553–5. [DOI] [PubMed] [Google Scholar]
- 202.Krivan G, Erd.s M, Kallay K, Benyo G, Toth A, Sinko J, et al. Successful umbilical cord blood stem cell transplantation in a child with WHIM syndrome. European Journal of Haematology. 2010;84:274–5. [DOI] [PubMed] [Google Scholar]
- 203.Bhar S, Yassine K, Martinez C, Sasa GS, Naik S, Jr DM, et al. Allogeneic Stem Cell Transplantation in a Pediatric Patient with Whim Syndrome. 126:5528. [Google Scholar]
- 204.Murphy PM, McDermott DH. Unexpected developments in immune organs in WHIM syndrome. Blood. 2012;119:5610–2. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.