

Abstract
Rift Valley Fever Phlebovirus (RVFV), genus Phlebovirus, family Phenuiviridae, order Bunyavirales, has a single-stranded, negative-sense RNA genome, consisting of L, M and S segments. Here, we report the establishment of a strand-specific, quantitative reverse transcription (RT)-PCR assay system that can selectively distinguish between the genomic and antigenomic RNAs of each of the three viral RNA segments produced in RVFV-infected cells. To circumvent the obstacle of primer-independent cDNA synthesis during RT, we used a tagged, strand-specific RT primer, carrying a non-viral ‘tag’ sequence at the 5’ end, which ensured the strand-specificity through the selective amplification of only the tagged cDNA in the real-time PCR assay. We used this assay system to examine the kinetics of intracellular accumulation of genomic and antigenomic viral RNAs in mammalian cells infected with the MP-12 strain of RVFV. The genomic RNA copy numbers, for all three viral RNA segments, were higher than that of their corresponding antigenomic RNAs throughout the time-course of infection, with a notable exception, wherein the M segment genomic and antigenomic RNAs exhibited similar copy numbers at specific times post-infection. Overall, this assay system could be a useful tool to gain an insight into the mechanisms of RNA replication and packaging in RVFV.
Keywords: Rift Valley fever Phlebovirus, Strand-specific quantitative PCR, RNA replication
1. Introduction
Viruses belonging to the Order Bunyavirales carry segmented RNA genomes that can cause serious and important diseases in both human and domestic animals. Rift Valley fever phlebovirus (RVFV), a member of the genus Phlebovirus, family Phenuiviridae, order Bunyavirales, is a mosquito-borne virus that can cause severe disease in humans and ruminants. RVFV is endemic in sub-Saharan Africa but has spread to Madagascar and the Arabian Peninsula (Balkhy and Memish, 2003; Linthicum et al., 2016; Madani et al., 2003; Pepin et al., 2010). Human infection occurs from the bite of infected mosquitos or from direct transmission of the virus from infected animal tissues or blood. Human disease manifestations include transient incapacitating febrile illness, encephalitis, vision loss and hemorrhagic fever(AI-Hazmi et al., 2005; Alrajhi et al., 2004; Deutman and Klomp, 1981; Ikegami and Makino, 2011; Linthicum etal.,2016; Pepin et al., 2010; Peters et al., 1989). RVFV has the potential to spread to any area of the world, including North America, by naturally occurring mosquito populations (Gargan et al., 1988; House et al., 1992; Linthicum et al., 2016; Rolin et al., 2013). One of the hallmarks of diseases caused by RVFV is high mortality in young ruminants and high rates of abortion in pregnant animals called “abortion storms” causing an enormous negative impact on the economy of affected regions. The lack of anti-viral treatments and licensed vaccines for use in humans or domestic animals is of great concern.
RVFV carries three genomic RNA segments, L, M, and S (Fig. 1). L and M segments are of negative-sense polarity, whereas S segment is of ambisense polarity. The RNA genome segments are encapsidated by the nucleocapsid protein, N, and also bound to the L protein, the RNA-dependent RNA polymerase, forming the ribonucleoprotein (RNP) complexes. In virusinfected cells, three different viral RNA species are produced for each of the three viral RNA segments, which include the genomic RNA, its complementary antigenomic RNA and cognate mRNAs. Immediately after infection, the incoming genomic L and M RNAs serve as templates for the synthesis of their respective antigenomic RNAs and mRNAs. The L mRNA encodes L protein and the M mRNA encodes two accessory proteins, NSm and the 78-kDa protein, along with two major envelope glycoproteins, Gn and Gc. The RVFV S segment uses an ambisense strategy for gene expression. The incoming genomic S RNA serves as the template for the synthesis of N mRNA encoding the N protein, while antigenomic S RNA serves as the template for the synthesis of NSs mRNA, which encodes NSs, a nonstructural protein, which is a major viral virulence factor (Bouloy and Weber, 2010; Gaudreault et al., 2019; Gauliard et al., 2006; Hornak et al., 2016; Ikegami, 2012; Ikegami and Makino, 2011; Sun et al., 2018). A host-derived cap structure is added to the 5’-end of all viral mRNAs by a cap-snatching mechanism, but viral mRNAs lack the poly(A) sequence at the 3’-end (Hopkins and Cherry, 2013). The three viral RNA segments have noncoding regions (NCRs) that flank the open reading frames (ORFs). In the antigenomic sense, L, M and S segments have a relatively shorter 5’ NCRs than their 3’ NCRs, which vary in length, including 107 nt for L RNA, 271 nt for M RNA and 34 nt for S RNA. These NCRs carry signals necessary for viral RNA synthesis and viral RNA packaging (Gauliard et al., 2006; Murakami et al., 2012). The 3’ NCRs of antigenomic L segment and M segment also carry transcription termination signals for L mRNA and M mRNA, respectively. The S segment carries an intergenic NCR, which harbors the transcription termination signals for N and NSs mRNAs, between the N and NSs ORFs (Albarino et al., 2007; Ikegami et al., 2007; Lara et al., 2011).
Figure 1. RVFV RNA species and coding strategy.
Schematic showing all RNA species for each viral segment. Full length genomic segments are negative sense and serve as templates for the generation of complementary antigenomic RNAs. The S segment has an ambisense coding strategy wherein, the N mRNA is transcribed from the genomic RNA and NSs mRNA is transcribed from the antigenomic RNA. The genomic strands of L and M segments are used as templates to generate L and M mRNAs, respectively. The dashed lines indicate the 3’-ends of L and M mRNAs compared to the full-length antigenomic segments.
An insight into the underlying rules and mechanisms that drive viral RNA replication is valuable for understanding the regulation of virus replication and the pathogenic potential of the virus. This knowledge is also critical for the development of antiviral drugs and the design of strategies for live attenuated vaccines. Although it is well-established that L and N proteins drive viral RNA synthesis in the cytoplasm of cells infected with bunyaviruses(Bouloy and Weber, 2010; Gaudreault et al., 2019; Hornak et al., 2016; Ikegami, 2012; Ikegami and Makino, 2011; Sun et al., 2018), our knowledge about the mechanisms of bunyavirus RNA replication is limited. In fact, due to the absence of an assay system that can accurately distinguish between genomic and antigenomic viral RNA segments, the accumulation kinetics of replicating viral RNA segments and their relative abundance during infection have not been reported for any bunyaviruses. A lack of such an assay system has also hindered the advancement of our knowledge about other aspects of the viral replication cycle, for example, clarifying the mechanism of viral RNA packaging into virus particles.
In this study, we have developed a strand-specific RT-qPCR assay that accurately distinguishes between the genomic and antigenomic RNAs of RVFV L, M and S segments. Using this assay system, we examined the accumulation kinetics of the genomic and antigenomic viral RNAs in RVFV-infected cells. Our study represents the first quantitative analysis of the kinetics of intracellular accumulation of genomic and antigenomic viral RNA segments in bunyaviruses.
2. Materials and Methods
2.1. Cell culture and virus infection.
Vero E6 cells (kidney epithelial cells from African green monkeys) were cultured in Dulbecco’s Modified Eagle Medium (DMEM), supplemented with 5% fetal bovine serum (FBS) and 1% penicillin-streptomycin. Huh7 cells (human hepatocellular carcinoma cells) were cultured in DMEM supplemented with 10% FBS and 1% kanamycin. RVFV MP-12 strain was generated by a reverse genetics system (Ikegami et al., 2006) and passaged once in Vero E6 cells. The virus titers were determined by plaque assay in Vero E6 cells. For virus infection, Vero E6 and Huh7 cells, with 90% confluency in 6-well plates, were inoculated with MP-12 at a MOI of 3. After virus absorption at 37°C for 1 h, cells were washed three times with phosphate buffered saline (PBS) and fresh medium was added to the wells.
2.2. Intracellular RNA extraction from virus-infected cells.
Virus-infected cells were lysed at indicated time points by directly adding 1 ml of Trizol reagent (Invitrogen) to the wells. Total RNA was extracted according to the manufacturer’s instructions and re-suspended in 30 µl RNase-free water. The concentration of total RNA was determined by the use of spectrophotometry and adjusted to 100 ng/µl for cDNA synthesis.
2.3. Plasmid construction.
Plasmids containing the T7 promoter sequence with MP-12 genomic RNA segments pProT7-L(−), pProT7-M(−), pProT7-S(−) and antigenomic RNA segments pProT7-L(+), pProT7- M(+), pProT7-S(+) were reported previously (Ikegami et al., 2006). For the generation of a plasmid expressing L mRNA, we used the pProT7-L (+) plasmid as the PCR DNA template. We produced a PCR product by using a forward primer containing Spe I site (5’-TCGAAAACTAGTAGCCCGCCAGGAAG-3’), and a reverse primer containing the end of the transcription termination signal of L RNA (Ikegami et al., 2007) with the Not I site (5’-TCGAATGCGGCCGCGTAGCACTATGCTAGTATC-3’). The purified PCR product replaced a 2.7-kb-long Spe I-Not I region, which contains a portion of the L segment ORF and the entire 3’-NCR, of pProT7-L (+), resulting in the pProT7-L-mRNA like plasmid. The pProT7-M-mRNA like plasmid was similarly generated using a 1.2-kb-long Xba I-Not I region, which contains a portion of the M segment ORF and the entire 3’-NCR, of pProT7-M (+), that was replaced by a purified PCR product. This PCR product was obtained by using a forward primer containing Xba I (5’-CTAGTCTCTAGAGATCACAGACTTTGATGGC-3’) and a reverse primer containing the end of the transcription termination signal for M segment (Ikegami et al., 2007) along with the Not I site (5’-TCGAATGCGGCCGCCACCCCAAATTACAAC-3’) and the use of the pProT7-M(+) plasmid as a PCR DNA template. Sequence analysis confirmed that both plasmids had the expected sequences and lacked viral sequences after the transcription termination sites.
2.4. In-vitro RNA synthesis.
Plasmids were linearized by Not I digestion and subjected to in-vitro transcription reactions at 37°C for 4 h by using the MEGAscript T7 transcription kit (Applied Biosystems/Invitrogen). The samples were then incubated with 2 units of Turbo DNase for 15 min at 37°C, and RNA was extracted using phenol/chloroform and precipitated using ethanol. To ensure complete removal of residual DNAs, the samples were subjected to a second round of DNase treatment and RNA purification using RNeasy Mini Protocol for RNA Cleanup and On-Column DNase digestion (Qiagen). The on-column DNase treatment was extended to 30 min at room temperature. The concentrations of purified RNA transcripts were determined by using spectrophotometry and quality of RNAs were examined by agarose gel electrophoresis. The molecular copies of synthetic RNAs were calculated from the total molecular weight and length of each RNA segment. At 1 ng of RNA, the copy numbers of full-length L, M, and S segment were 2.925 × 108 copies, 4.820 × 108 copies, and 1.108 × 109 copies respectively. The stocks of in-vitro synthesized viral RNAs were subsequently serially diluted to form standard curves used for our RT-qPCR studies.
2.5. Reverse Transcription.
cDNA synthesis of viral RNAs by using unmodified RT primers (Supplementary Table S1) was performed by using the Superscript-III first strand synthesis system (Invitrogen). Briefly, 100 ng of in-vitro synthesized RNA was mixed with 2 μM of strand-specific RT primer. The mixture was incubated at 65°C for 5 min and then cooled to 4°C. After addition of the reaction buffer and enzyme mixture, cDNA synthesis was carried out by incubating the sample at 50°C for 50 min, terminated by heating at 85°C for 5 min, cooled to 4°C, and then treated with RNase H at 37°C for 20 min. RT of viral RNA by using tagged RT primers (Supplementary Table S3) was carried out in a similar method, except that 10 pg of in-vitro synthesized RNA or 100 ng of total RNA from virus-infected cells were used as the source of RNA samples. cDNA synthesis was performed by incubating the samples at 55°C, instead of 50°C, for 50 min. We used 500 ng of total RNA from virus-infected cells to detect antigenomic RNA segments at the 2 h time-point because RNA levels were lower than the detection limit early in infection with 100 ng total RNA. Accordingly, the resulting copy numbers for the antigenomic RNA segments were adjusted by dividing by a factor of 5 and plotted with the data obtained for the other time-points using 100 ng total RNA. The cDNAs were purified using an on-column PCR purification kit (Qiagen). To test for cDNA synthesis caused by RNA self-priming, RT reactions were carried out in the absence of an RT primer.
2.6. Standard PCR.
PCRs were performed by using the SsoAdvanced™ Universal SYBR® Green Supermix (Bio Rad). For PCR of cDNAs generated by using unmodified RT primers or no RT primers, 2 µl of cDNA was added to the PCR master mix containing the viral strand-specific PCR primers (Supplementary Table S2). For cDNAs generated by tagged RT primers and purified by on-column purification, 1 µl of cDNA was added to the PCR master mix containing the non-viral tagged sequence as a forward primer and a viral strand-specific PCR reverse primer (Supplementary Table S4). For both PCRs, thermocycling conditions were as follows: 95°C for 30 sec, 35 cycles of 95°C for 15 sec and 60°C for 20 sec. PCR products were analyzed by gel electrophoresis. Gel Images are scanned, cropped, and assembled by AlphaEase FC Software.
2.7. Quantitative PCR.
The real time qPCR assays were prepared using the SsoAdvanced™ Universal SYBR® Green Supermix. Total RNAs from virus-infected cells were subjected to RT using tagged strand-specific RT primers as stated before. After cDNA purification, 1 µl of cDNA was added to the PCR master mix containing the non-viral tagged sequence as a forward primer and a viral strand-specific PCR reverse primer (Supplementary Table S4). Thermocycling conditions were the same as standard PCR, followed by melting curve analysis. The assays were performed by the CFX96 Touch Real-time PCR detection system and analyzed using the provided software (BioRad CFX Manager 3.1).
3. Results
3.1. Strand-specific RT-PCR assay design using unmodified primers fails to selectively amplify target viral RNA segments
Our initial approach for quantifying RVFV genomic and antigenomic segments was to synthesize cDNAs by using RT primers, each of which specifically bind to the 3’ NCR of each viral RNA segment (Fig. 1), and subsequent amplification of the cDNA products by PCR. Antigenomic L segment and M segment have the same orientation as L mRNA and M mRNA, respectively (Fig. 1). As transcription of both mRNAs is terminated at the transcription termination site(Ikegami et al., 2007; Lara et al., 2011) located within the 3’ NCR of these antigenomic segments, both mRNAs lack sequences present downstream of the transcription termination sites. To determine the abundances of antigenomic L and M segments, we designed RT primers that bind downstream of their transcription termination sites such that the RT primers would not bind to L and M mRNAs and only amplify full-length antigenomic segments. Genomic S segment and antigenomic S segment serve as templates for N mRNA and NSs mRNA, respectively (Fig. 1). As RT primers used for the cDNA synthesis of genomic S segment and antigenomic S segment had the same sequence as N mRNA and NSs mRNA, respectively, they would not bind to these mRNAs. Supplementary Table 1 lists the RT primers used for each viral segment. Our initial experimental design used the PCR primer sets reported by others (Maquart et al., 2014; Naslund et al., 2008; Wilson et al., 2013) (Supplementary Table S2).
To test whether this experimental design worked, we generated full-length genomic and antigenomic viral RNAs for each segment by using in-vitro transcription. After DNase treatment of the samples, these RNAs were subjected to cDNA synthesis by using RT primers, each of which binds to genomic L, M and S segment, and subsequent PCR amplification by using the PCR primer sets reported by others(Maquart et al., 2014; Naslund et al., 2008; Wilson et al., 2013). Surprisingly, we detected PCR products of the expected sizes from target genomic segments, whereas we also detected PCR products of the same sizes from corresponding antigenomic segments (Fig. 2A). We performed similar experiments, in which in-vitro synthesized genomic L, M and S segment, antigenomic L, M and S segment, and L and M mRNAs, were subjected to cDNA synthesis by using RT primers, each of which designed to bind to each full-length antigenomic segment. Amplification of the cDNAs by using the reported PCR primers resulted in generation of the PCR products of the expected sizes from both antigenomic and genomic segment RNAs and an unexpected PCR product of ~500 bp from L mRNA (Fig. 2B). These data showed that this experimental design was unable to selectively amplify target RNAs. To exclude the possibility of incomplete DNA digestion after in-vitro transcription leading to the generation of the PCR products from low levels of residual DNAs, the RNA samples were directly subjected to the PCR reaction. No PCR products were generated (Supplementary Fig. S1), demonstrating absence of DNA contamination in the RNA samples.
Figure 2. Standard RT-PCR using unmodified primers does not display strand-specificity.
(A) 100 ng of in-vitro synthesized RNAs corresponding to the genomic (G) and antigenomic (AG) segments of L, M, and S RNAs were used for cDNA synthesis using unmodified RT primers specific for genomic L, M, and S segments respectively. The corresponding cDNAs were subjected to PCR by using unmodified PCR primer sets, specific for genomic L (lanes 2-4), M (lanes 5-7) and S (lanes 8-10) segments. NT represents the "no template" control for RT-PCR analysis (lanes 2, 5 and 8). (B) 100 ng of in-vitro synthesized RNAs corresponding to the genomic (G), antigenomic (AG) segments of L, M, and S RNAs, and cognate mRNAs (mRNA) of L and M segments were used for cDNA synthesis using unmodified RT primers specific for antigenomic L, M, and S segments respectively. The corresponding cDNAs were subjected to PCR by using an unmodified PCR primer set, specific for antigenomic L (lanes 2-5), M (lanes 6-9) and S (lanes 10-12) segments. Lane 1 in (A) and (B) represents the DNA size marker, and the location of the markers having 100 and 200 base pairs (bp) bands are indicated by arrows. The PCR products were analyzed by agarose gel electrophoresis. NT represents the no template control for RT-PCR analysis (lanes 2, 6 and 10).
3.2. Primer-independent cDNA synthesis prevents selective amplification of target viral RNAs
Multiple studies have reported a phenomenon known as self-primed reverse transcription, where cDNA can be generated in the absence of an RT primer(Beiter et al., 2007; Craggs et al., 2001; Feng et al., 2012; Haist et al., 2015; Lanford and Chavez, 1999; Lim et al., 2013; Peyrefitte et al., 2003; Plaskon et al., 2009; Tuiskunen et al., 2010; Vashist et al., 2012). The self-priming in the absence of the RT primer is most probably due to the intrinsic ability of the RNA itself or the presence of small RNA fragments in RNA preparations that can serve as primers. To test whether self-primed reverse transcription occurred in this assay system, we performed cDNA synthesis from synthetic genomic and antigenomic viral segments along with RNAs isolated from RVFV-infected cells in the absence of RT primers. The samples were then subjected to PCR. In all samples, we detected PCR products of the same sizes as those generated by amplification of the correct cDNAs by using PCR primer sets (Fig. 3), suggesting that cDNA was synthesized in the absence of the RT primer from these viral RNAs. Therefore, we concluded that cDNAs produced by false-priming, possibly due to RNA self-priming, underwent PCR-mediated amplification, preventing strand-specificity of the assay system.
Figure 3. Primer-independent cDNA synthesis of viral RNA impairs standard RT-PCR strand-specificity.
100 ng of in-vitro synthesized RNAs corresponding to the genomic (G) and antigenomic (AG) segments of L, M, and S RNAs and intracellular RNAs from RVFV-infected cells (MP-12 IC) were used for cDNA synthesis without the use of specific RT primers. (A) After cDNA synthesis, the samples underwent PCR using unmodified PCR primer sets, specific for genomic L (lanes 2-4), M (lanes 5-7), and S (lanes 8-10) segments. (B) Experiments were done similar to (A), except that the samples underwent PCR using unmodified PCR primer sets, specific for antigenomic L (lanes 2-4), M (lanes 5-7), and S (lanes 8-10) segments. Lane 1 in (A) and (B) represents the DNA size marker, and the location of the markers having 100 and 200 base pairs (bp) bands are indicated by arrows. The PCR products were analyzed by agarose gel electrophoresis.
3.3. Strand-specific RT-PCR using ‘tagged’ RT primers selectively amplifies the target viral RNA segment.
To establish a strand-specific RT-qPCR assay, we modified our assay by increasing the annealing temperature during reverse transcription from 50°C to 55°C and by using ‘tagged’ RT primers (Supplementary Table S3). Increasing the annealing temperature in the reverse transcription step has been shown to reduce non-specific primer binding (Feng et al., 2012; Lanford and Chavez, 1999; Lim et al., 2013). The tagged primer method has been used in numerous studies to overcome false-priming issues(Bannister et al., 2010; Brennan et al., 2014; Chatterjee et al., 2012; Kawakami et al., 2011; Lim et al., 2013; Peyrefitte et al., 2003; Plaskon et al., 2009; Strydom and Pietersen, 2018; Tuiskunen et al., 2010; Vashist et al., 2012). In this modified assay, cDNA synthesis was performed by using a tagged RT primer complementary to each of the target RNA segments (Fig. 4). Each tagged RT primer had a non-viral tag sequence, modified from Brennan et al., (Brennan et al., 2014) at the 5’-end. Hence, cDNAs that are synthesized by using the tagged RT primer, but not those synthesized by false-priming, carry the ‘tag’ sequence at the 5’-end. The tagged RT primer for the antigenomic M segment and that for the antigenomic L segment were designed not to bind to M mRNA and L mRNA, respectively. The tag sequence of the RT primer was used as the forward primer (5’-GGCCGTCATGGTGGCGAATA-3’) and a strand-specific primer was used as the reverse primer in PCR. Use of the tag sequence as the forward primer ensures that only primer-driven tagged cDNA, but not those generated by false-priming, gets amplified during PCR.
Figure 4. Schematic diagram of modified RT-qPCR with tagged RT primers and modified PCR primer sets.
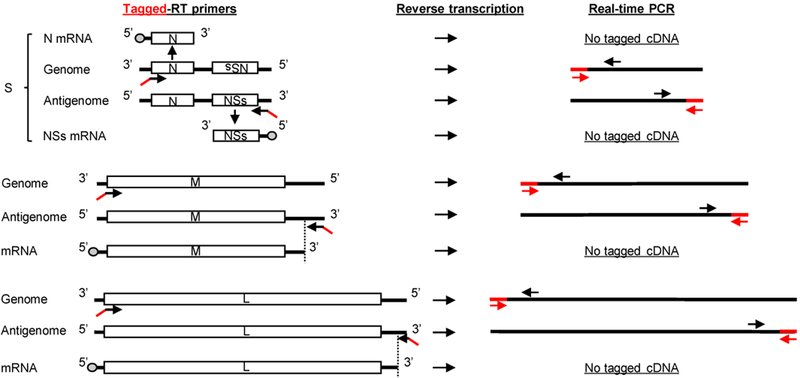
Left panel shows schematic diagram of RVFV RNAs and binding sites of tagged RT primers to viral RNA segments. cDNA synthesis is performed using a tagged RT primer complementary to either genomic or antigenomic viral RNA segments. The tagged non-viral sequence, shown in red, is attached to the 5’-end of each strand-specific RT primer. For genomic segments, the tagged genomic RT primers bind to the 3’-end of these RNAs. Note that the tagged genomic RT primer binds to the 3’-end of genomic S segment, but not N mRNA, as the RT primer is in the same orientation as N mRNA. The same principle is applied for the tagged RT primer for antigenomic S segment, which does not bind to NSs mRNA. The tagged RT primers for antigenomic M and L segments bind downstream of the transcription termination sites of these segments. The dashed lines indicate the 3’-ends of L and M mRNAs compared to the full-length antigenomic segments. The right panel outlines the PCR of the cDNA products by using the forward PCR primer (red arrows), which is the tag portion of the RT primer, and a strand-specific reverse PCR primer (black arrows). Use of the tagged portion as a forward PCR primer ensures that only primer-driven tagged cDNAs undergo amplification.
To determine whether a combination of forward tag PCR primer and strand-specific reverse PCR primer amplifies cDNAs synthesized by false-priming, we performed cDNA synthesis from in-vitro synthesized genomic and antigenomic viral segments and RNAs from RVFV-infected cells in the absence of the RT primers. The samples were then subjected to PCR amplification by using a forward tag PCR primer and strand-specific reverse PCR primer for each of the genomic and antigenomic viral segments (Supplementary Table S4). No PCR products were generated (Supplementary Fig. S2A and S2B), demonstrating that a combination of the forward tag PCR primer and the strand-specific reverse PCR primer did not amplify cDNAs produced by false-priming.
We next tested whether use of the tagged RT primer for cDNA synthesis and subsequent PCR using forward tag PCR primer and strand-specific reverse PCR primer sets lead to selective amplification of the target RNA. We used in-vitro synthesized genomic and antigenomic L, M and S segments and L and M mRNAs as source RNAs. As residual RT primer present during PCR amplification can also cause false-priming (Craggs et al., 2001; Feng et al., 2012; Lim et al., 2013; Peyrefitte et al., 2003; Vashist et al., 2012), we purified the cDNAs after reverse transcription to remove residual tagged RT primers and reverse transcriptase, and then subjected them to PCR amplification using the forward tag PCR primer and strand-specific reverse PCR primer for each target RNA (Supplementary Table S4). We detected PCR products of the expected sizes only from the target RNA templates (Fig. 5), demonstrating selective amplification of the target RNAs.
Figure 5. Validation of a strand-specific RT-qPCR using modified tagged RT primers and PCR primer sets.

(A) 10 pg of in-vitro synthesized RNAs corresponding to the genomic (G) and antigenomic (AG) segments of L, M, and S RNAs were used for cDNA synthesis using tagged RT primers specific for genomic L, M, and S segments. The corresponding cDNAs were subjected to PCR by using PCR primer sets with the ‘tag’ sequence as the forward primer and a segment-specific reverse primer that selectively amplifies genomic L (lanes 2-4), M (lanes 5-7) and S (lanes 8-10) segments. NT represents the "no template" control for RT-PCR analysis (lanes 2, 5 and 8). (B) 10 pg of in-vitro synthesized RNAs corresponding to the genomic (G), antigenomic (AG) segments of L, M, and S RNAs, and cognate mRNAs (mRNA) of L and M segments were used for cDNA synthesis by using tagged RT primers specific for antigenomic L, M, and S segments. The corresponding cDNAs were subjected to PCR using PCR primer sets with the ‘tag’ sequence as the forward primer and a segment-specific reverse primer that selectively amplifies antigenomic L (lanes 2-5), M (lanes 6-9) and S (lanes 10-12) segments. NT represents the "no template" control for RT-PCR analysis (lanes 2, 6 and 10). Lane 1 in (A) and (B) represents the DNA size marker, and the location of the markers having 100 and 200 base pairs (bp) bands are indicated by arrows. The PCR products were analyzed by agarose gel electrophoresis.
3.4. Establishment of strand-specific RT-qPCR assays for genomic and antigenomic RVFV RNAs.
As the experimental approach described above successfully amplified the target viral RNAs, we proceeded to further validate and establish the RT-qPCR assay and examined its sensitivity and selectivity. To determine absolute copy number for target viral RNAs that are detectable in this assay, we performed an RT-qPCR assay by using known copy numbers of in-vitro transcribed RNA and established standard curves for each viral segment, where 103 to 1010 copies of genomic and antigenomic L and M segments and 104 to 1011 copies of genomic and antigenomic S segments showed strong linearity with 0.992 or better correlation coefficient (R2) values (Fig. 6 and Table 1).
Figure 6. Standard curves for genomic and antigenomic segments.
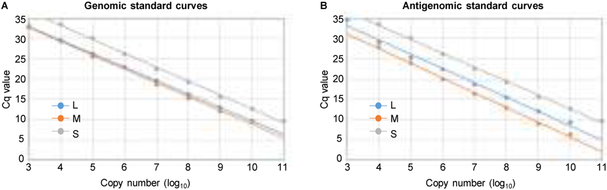
Standard curves for (A) genomic and (B) antigenomic viral segments were generated by plotting the quantification cycle (Cq) value against the input of in-vitro transcribed RNA of known copy numbers. In-vitro synthesized genomic RNA segments (A) and antigenomic RNA segments (B) were serially diluted ten-fold, (1010 to 103 copies) for L (blue) and M (orange) segments and (1011 to 104 copies) for S segment (grey) and were used to generate standard curves.
Table 1.
Validation parameters for RT-qPCR
| Sensitivity (Copies) |
Linear Regression | ||||
|---|---|---|---|---|---|
| Genome | Slope | Intercept |
Amplification efficiency (%) |
R2 value | |
| L segment | 103 | −3.329 | 44.471 | 99.7 | 0.998 |
| M segment | 103 | −3.408 | 45.288 | 96.5 | 0.996 |
| S segment | 104 | −3.446 | 47.207 | 95.1 | 0.998 |
| Antigenome | |||||
| L segment | 103 | −3.535 | 45.480 | 91.8 | 0.992 |
| M segment | 104 | -−3.645 | 44.609 | 88.1 | 0.997 |
| S segment | 104 | −3.222 | 44.611 | 104.3 | 0.995 |
We next tested whether this assay was capable of quantifying target viral RNAs in the presence of RNAs in the opposite sense. We prepared RNA samples, where 104 - 108 copies of target genomic or antigenomic L or M segments were mixed with 106 copies of their opposite-strand RNA and 105 - 109 copies of target genomic or antigenomic S segment were mixed with 107 copies of their opposite-strand RNA, and used them as RNA sources for cDNA synthesis. After cDNA synthesis using specific tagged RT primers for the target RNA, we purified cDNAs and performed qPCR. The amounts of all target RNAs were accurately determined under this experimental condition, demonstrating that this strand-specific RT-qPCR assay was able to measure the amounts of the target RNAs in the presence of up to ~100-fold excess amounts of their opposite-strand RNAs (Fig. 7). Taken together, these data demonstrate that the modified strand-specific RT-qPCR assay was effective for overcoming false priming and allowed us to distinguish between all of the genomic and antigenomic RVFV RNAs.
Figure 7. Demonstration of strand-specific RT-qPCR in the presence of opposite-sense RNA transcripts.
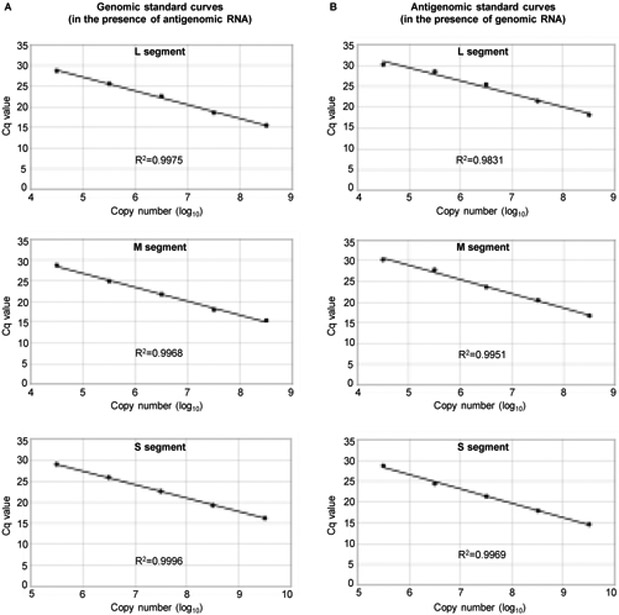
(A) Genomic standard curves were generated by synthesizing cDNAs from indicated copy number of in-vitro transcribed genomic L (top panel), M (middle panel), and S (bottom panel) segments by using a tagged RT primer in the presence of 106 copies of antigenomic L, 106 copies of antigenomic M and 107 copies of antigenomic S segments, respectively. The cDNAs were subjected to PCR by using primer sets with the tag sequence as the forward primer and a specific reverse PCR primer designed specifically for amplifying genomic L, M and S segments. (B). Antigenomic standard curves were generated by synthesizing cDNAs from the indicated copy numbers of in-vitro transcribed antigenomic L (top panel), M (middle panel), and S (bottom panel) segments using a tagged RT primer in the presence of 106 copies of genomic L, 106 copies of genomic M and 107 copies of genomic S segments, respectively. The cDNAs were subjected to PCR using primer sets with the tag sequence as the forward primer and a specific reverse PCR primer designed specifically for amplifying antigenomic L, M and S segments. The linear regression (R2) value is displayed on each graph.
3.5. Kinetics of viral RNA accumulation in RVFV-infected cells
We used this newly established strand-specific RT-qPCR assay to investigate the intracellular accumulation kinetics of RVFV RNAs following infection in Vero E6 cells and Huh7 cells, a hepato-cellular carcinoma cell line (Nakabayashi et al., 1982), at a multiplicity of infection (MOI) of 3. We determined the amounts of intracellular genomic and antigenomic L, M and S segments at 2, 4, 6, 8, 12, 16, and 20 h post-infection (p.i.) (Fig. 8).
Figure 8. Accumulation kinetics of genomic and antigenomic viral RNA segments in RVFV-infected cells.
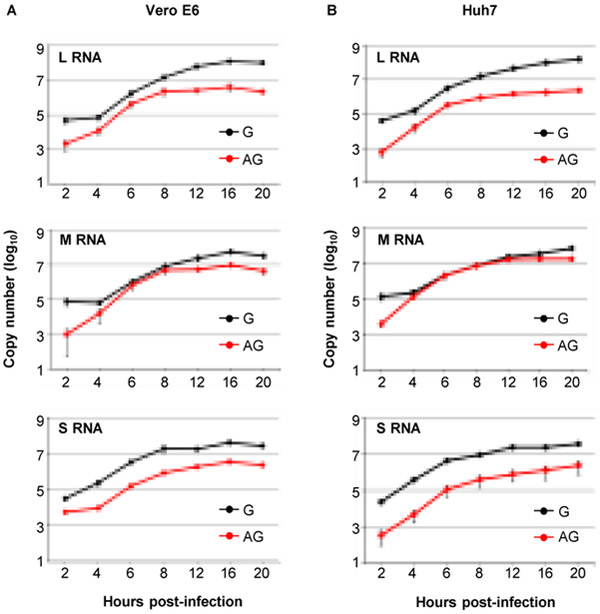
(A) Vero E6 and (B) Huh7 cells were infected with RVFV at an MOI of 3. Total intracellular RNAs were extracted in triplicate at the indicated times p.i. Total RNAs were subjected to strand-specific RT-qPCR and the copy numbers for each viral segment, which were determined with synthetic viral RNA as a reference standard, are plotted. Error bars represent the mean (+/− the standard deviation) of triplicate experiments.
In both cell lines, accumulation of all three genomic RNA segments were substantially higher than their antigenomic counterparts throughout the course of infection, with the exception of M segment at 6-8 h p.i., in Vero E6 cells and 4-12 h p.i. in Huh7 cells, showing similar levels of accumulation for genomic and antigenomic RNAs. Accumulation of antigenomic M segment in both cell lines were higher relative to antigenomic L and S segment after 4 h p.i., being more prominent in Huh7 cells (Supplementary Fig. S3B). We also noted that early in infection (4-6 h p.i.), genomic S segment copy numbers were higher compared to genomic L and M segments (Fig. 8 and Supplementary Fig. S3A).
4. Discussion
The present study reported the development of a RT-qPCR assay that determines the amounts of genomic and antigenomic L, M and S segments of RVFV; this assay represents the first system that can distinguish and measure the accumulation of all genomic and antigenomic viral RNA segments in any bunyaviruses. Several methods have been used to detect and quantify the different viral RNA species generated during RVFV infection, including radiolabeling, Northern blot, in-situ hybridization, and RT-qPCR assays (Brennan et al., 2014; Garcia et al., 2001; Gauliard et al., 2006; Ikegami et al., 2005; Maquart et al., 2014; Naslund et al., 2008; Wichgers Schreur and Kortekaas, 2016; Wilson et al., 2013). Limitations of these studies include limited sensitivity, different probe efficiencies, and the inability to distinguish antigenomic RNA from mRNA and genomic RNA from antigenomic RNA. Multiple groups have reported the use of RT-qPCR approaches to quantitate RVFV RNA load, however these assays were not designed to specifically target individual replicative RNA species(Garcia et al., 2001; Maquart et al., 2014; Naslund et al., 2008; Wilson et al., 2013). Additionally, these assays do not take into account cDNA synthesized due to false priming.
The assay developed in this study incorporated the combined use of the tagged RT primer and the forward tag and the strand-specific reverse PCR primers, which eliminated amplification of cDNAs generated by false-priming (Fig. 5). This modified assay excluded amplification of viral mRNAs (Fig. 5B) and accurately measured between 103 to 1010 copies of the genomic and antigenomic L and M segments and between 104 to 1011 copies of the genomic and antigenomic S segments in samples (Fig. 6). The assay was also able to accurately determine the amounts of target viral RNAs present in up to ~100-fold excess amounts of their opposite-strand RNAs (Fig. 7). Brennan et al., reported a similar RT-qPCR assay, where tagged RT primer and sets of forward tag and strand-specific reverse PCR primers were used to determine the amounts of the genomic and antigenomic RVFV M and S segments, however their assay did not include the measuring method for the genomic and antigenomic L segments (Brennan et al., 2014). Our RT-qPCR assay design benefited from their study; we used the same tag sequence used in their tagged RT primers, except that our tag sequence had an extra ‘A’ at the 3’-end, and used the same strand-specific reverse PCR primer for the genomic-sense S RNA. Because the transcription termination signal for L mRNA is located very close to the 3’- end of the antigenomic L RNA(lkegami et al., 2007; Lara et al., 2011), it was necessary to determine an appropriate binding site of the tagged RT primer for cDNA synthesis of antigenomic L segment, but not for cDNA synthesis of L mRNA, within ~20 nt from the 3’-end of antigenomic L segment. After testing several tagged RT primer candidates, we were able to identify an appropriate tagged RT primer for antigenomic L segment.
Like other past studies that have reported self-primed reverse transcription in various viruses (Beiter et al., 2007; Craggs et al., 2001; Feng et al., 2012; Haist et al., 2015; Lanford and Chavez, 1999; Lim et al., 2013; Peyrefitte et al., 2003; Plaskon et al., 2009; Tuiskunen et al., 2010; Vashist et al., 2012), our data suggested synthesis of cDNAs in the absence of an RT primer from in-vitro synthesized RVFV RNAs as well as intracellular RNAs obtained from RVFV-infected cells (Fig. 3). Because the 5’- and 3’-ends of both genomic and antigenomic RVFV RNA segments have a stretch of short complementary sequences that form a double-stranded panhandle structure(Gaudreault et al., 2019; Hornak et al., 2016; Ikegami, 2012; Sun et al., 2018), a likely mechanism of primer-independent cDNA synthesis in RVFV RNAs is that reverse transcriptase recognizes the panhandle structure and initiates cDNA synthesis.
By using our newly developed RT-qPCR assay, we examined the accumulation kinetics of replicating RVFV RNAs in Vero E6 and Huh7 cells (Fig. 8); the former is widely used for studies of RVFV replication and preparation of virus stocks (Ikegami et al., 2006) and the latter, a hepato-cellular carcinoma cell line (Nakabayashi et al., 1982), was selected because one of the major target organs of RVFV in mammals is the liver (Gommet et al., 2011; Terasaki and Makino, 2015). To our knowledge, this is the first study demonstrating the accumulation kinetics of replicating viral RNA species in bunyavirus-infected cells. We noted similar accumulation kinetics of viral RNAs in both cell lines. The amounts of the genomic RNA segments were always higher than corresponding antigenomic RNA segments throughout the infection, except that the amounts of genomic and antigenomic M segments were similar at 6-8 h p.i. in Vero cells and at 4-12 h p.i. in Huh7 cells. Our finding of similar levels of the genomic and antigenomic M segments in RVFV-infected cells are a marked contrast to accumulation of viral RNAs in cells infected with positive-stranded RNAs, where abundances of genomic RNAs always exceed antigenomic RNAs. Currently, it is unclear why only M segment showed similar accumulations of genomic and antigenomic RNAs at certain times p.i.
In both cell lines, genomic S RNA, but not genomic L and M RNAs, rapidly accumulated from 2 to 4 h p.i. (Fig. 8 and Supplementary Fig. S3A). Antigenomic S RNA is packaged efficiently into RVFV particles, and we previously reported that the incoming antigenomic S RNA serves as template for the synthesis of NSs mRNA immediately following RVFV infection, leading to NSs expression early in infection (Ikegami et al., 2005). Expression of NSs immediately after infection would be critical for efficient virus replication in mammalian hosts, as the incoming RVFV RNPs trigger RIG-I-mediated antiviral innate immune signaling(Bouloy et al., 2001; Ly and Ikegami, 2016; Terasaki and Makino, 2015). Rapid accumulation of genomic S segment from 2 to 4 h p.i. suggest that the incoming antigenomic S segment also serves as the template for genomic S RNA replication. This results in efficient genomic S RNA accumulation, which was higher than the accumulation of genomic L and M segments from 4-6 h p.i. (Supplementary Fig. 3A). Efficient accumulation of genomic S RNA, which serves as the template for N mRNA, during the early phase of infection may lead to efficient accumulation of N protein early in infection resulting in optimal virus replication.
In conclusion, our strand-specific RT-qPCR assay determined the amounts of genomic and antigenomic RVFV RNAs with accuracy and specificity, allowing us to determine the accumulation kinetics of these viral RNAs in infected cells. Overall, our assay system will be a valuable tool to examine the replication mechanisms of RVFV.
Supplementary Material
Highlights:
Self-primed reverse transcription impairs strand-specific detection of RVFV RNAs.
A strand-specific RT-qPCR assay system for detection of replicative RNAs of RVFV.
Quantitative analysis of the replication kinetics of RVFV RNAs in infected cells.
A methodology to examine the mechanisms of RVFV RNA replication and packaging.
Acknowledgements
This work was supported by Public Health Service grants, AI114657, AI117445, and AI127984 from the NIH.
Footnotes
Competing Interests
The author(s) declare no competing interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Al-Hazmi A, Al-Rajhi AA, Abboud EB, Ayoola EA, Al-Hazmi M, Saadi R, Ahmed N, 2005. Ocular complications of Rift Valley fever outbreak in Saudi Arabia. Ophthalmology 112, 313–318. [DOI] [PubMed] [Google Scholar]
- Albarino CG, Bird BH, Nichol ST, 2007. A shared transcription termination signal on negative and ambisense RNA genome segments of Rift Valley fever, sandfly fever Sicilian, and Toscana viruses. J Virol 81, 5246–5256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alrajhi AA, Al-Semari A, Al-Watban J, 2004. Rift Valley fever encephalitis. Emerg Infect Dis 10, 554–555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balkhy HH, Memish ZA, 2003. Rift Valley fever: an uninvited zoonosis in the Arabian peninsula. Int J Antimicrob Agents 21, 153–157. [DOI] [PubMed] [Google Scholar]
- Bannister R, Rodrigues D, Murray EJ, Laxton C, Westby M, Bright H, 2010. Use of a highly sensitive strand-specific quantitative PCR to identify abortive replication in the mouse model of respiratory syncytial virus disease. Virol J 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beiter T, Reich E, Weigert C, Niess AM, Simon P, 2007. Sense or antisense? False priming reverse transcription controls are required for determining sequence orientation by reverse transcription-PCR. Analytical biochemistry 369, 258–261. [DOI] [PubMed] [Google Scholar]
- Bouloy M, Janzen C, Vialat P, Khun H, Pavlovic J, Huerre M, Haller O, 2001. Genetic evidence for an interferon-antagonistic function of Rift Valley fever virus nonstructural protein NSs. J Virol 75, 1371–1377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouloy M, Weber F, 2010. Molecular biology of rift valley Fever virus. The open virology journal 4, 8–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brennan B, Welch SR, Elliott RM, 2014. The consequences of reconfiguring the ambisense S genome segment of Rift Valley fever virus on viral replication in mammalian and mosquito cells and for genome packaging. PLoS pathogens 10, e1003922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chatterjee SN, Devhare PB, Lole KS, 2012. Detection of negative-sense RNA in packaged hepatitis E virions by use of an improved strand-specific reverse transcription-PCR method. J Clin Microbiol 50, 1467–1470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Craggs JK, Ball JK, Thomson BJ, Irving WL, Grabowska AM, 2001. Development of a strand-specific RT-PCR based assay to detect the replicative form of hepatitis C virus RNA. Journal of virological methods 94, 111–120. [DOI] [PubMed] [Google Scholar]
- Deutman AF, Klomp HJ, 1981. Rift Valley fever retinitis. Am J Ophthalmol 92, 38–42. [DOI] [PubMed] [Google Scholar]
- Feng L, Lintula S, Ho TH, Anastasina M, Paju A, Haglund C, Stenman UH, Hotakainen K, Orpana A, Kainov D, Stenman J, 2012. Technique for strand-specific gene-expression analysis and monitoring of primer-independent cDNA synthesis in reverse transcription. BioTechniques 52, 263–270. [DOI] [PubMed] [Google Scholar]
- Garcia S, Crance JM, Billecocq A, Peinnequin A, Jouan A, Bouloy M, Garin D, 2001. Quantitative real-time PCR detection of Rift Valley fever virus and its application to evaluation of antiviral compounds. J Clin Microbiol 39, 4456–4461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gargan TP 2nd, Clark GG, Dohm DJ, Turell MJ, Bailey CL, 1988. Vector potential of selected North American mosquito species for Rift Valley fever virus. Am J Trop Med Hyg 38, 440–446. [DOI] [PubMed] [Google Scholar]
- Gaudreault NN, Indran SV, Balaraman V, Wilson WC, Richt JA, 2019. Molecular aspects of Rift Valley fever virus and the emergence of reassortants. Virus genes 55, 1–11. [DOI] [PubMed] [Google Scholar]
- Gauliard N, Billecocq A, Flick R, Bouloy M, 2006. Rift Valley fever virus noncoding regions of L, M and S segments regulate RNA synthesis. Virology 351, 170–179. [DOI] [PubMed] [Google Scholar]
- Gommet C, Billecocq A, Jouvion G, Hasan M, Zaverucha do Valle T, Guillemot L, Blanchet C, van Rooijen N, Montagutelli X, Bouloy M, Panthier JJ, 2011. Tissue tropism and target cells of NSs-deleted rift valley fever virus in live immunodeficient mice. PLoS neglected tropical diseases 5, e1421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haist K, Ziegler C, Botten J, 2015. Strand-Specific Quantitative Reverse Transcription-Polymerase Chain Reaction Assay for Measurement of Arenavirus Genomic and Antigenomic RNAs. PLoS One 10, e0120043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hopkins K, Cherry S, 2013. Bunyaviral cap-snatching vs. decapping: recycling cell cycle mRNAs. Cell cycle 12, 3711–3712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hornak KE, Lanchy JM, Lodmell JS, 2016. RNA Encapsidation and Packaging in the Phleboviruses. Viruses 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- House JA, Turell MJ, Mebus CA, 1992. Rift Valley fever: present status and risk to the Western Hemisphere. Ann N Y Acad Sci 653, 233–242. [DOI] [PubMed] [Google Scholar]
- Ikegami T, 2012. Molecular biology and genetic diversity of Rift Valley fever virus. Antiviral Res 95, 293–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikegami T, Makino S, 2011. The pathogenesis of Rift Valley fever. Viruses 3, 493–519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikegami T, Won S, Peters CJ, Makino S, 2005. Rift Valley fever virus NSs mRNA is transcribed from an incoming anti-viral-sense S RNA segment. Journal of Virology 79, 12106–12111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikegami T, Won S, Peters CJ, Makino S, 2006. Rescue of infectious Rift Valley fever virus entirely from cDNA, analysis of virus lacking the NSs gene, and expression of a foreign gene. Journal of Virology 80, 2933–2940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikegami T, Won S, Peters CJ, Makino S, 2007. Characterization of Rift Valley fever virus transcriptional terminations. J Virol 81, 8421–8438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawakami E, Watanabe T, Fujii K, Goto H, Watanabe S, Noda T, Kawaoka Y, 2011. Strand-specific real-time RT-PCR for distinguishing influenza vRNA, cRNA, and mRNA. Journal of virological methods 173, 1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lanford RE, Chavez D, 1999. Strand-Specific rTth RT-PCR for the Analysis of HCV Replication. Methods in molecular medicine 19, 471–481. [DOI] [PubMed] [Google Scholar]
- Lara E, Billecocq A, Leger P, Bouloy M, 2011. Characterization of wild-type and alternate transcription termination signals in the Rift Valley fever virus genome. J Virol 85, 12134–12145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim SM, Koraka P, Osterhaus AD, Martina BE, 2013. Development of a strand-specific real-time qRT-PCR for the accurate detection and quantitation of West Nile virus RNA. Journal of virological methods 194, 146–153. [DOI] [PubMed] [Google Scholar]
- Linthicum KJ, Britch SC, Anyamba A, 2016. Rift Valley Fever: An Emerging Mosquito-Borne Disease. Annual review of entomology 61, 395–415. [DOI] [PubMed] [Google Scholar]
- Ly HJ, Ikegami T, 2016. Rift Valley fever virus NSs protein functions and the similarity to other bunyavirus NSs proteins. Virol J 13, 118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madani TA, Al-Mazrou YY, Al-Jeffri MH, Mishkhas AA, Al-Rabeah AM, Turkistani AM, Al-Sayed MO, Abodahish AA, Khan AS, Ksiazek TG, Shobokshi O, 2003. Rift Valley fever epidemic in Saudi Arabia: epidemiological, clinical, and laboratory characteristics. Clin Infect Dis 37, 1084–1092. [DOI] [PubMed] [Google Scholar]
- Maquart M, Temmam S, Heraud JM, Leparc-Goffart I, Cetre-Sossah C, Dellagi K, Cardinale E, Pascalis H, 2014. Development of real-time RT-PCR for the detection of low concentrations of Rift Valley fever virus. Journal of virological methods 195, 92–99. [DOI] [PubMed] [Google Scholar]
- Murakami S, Terasaki K, Narayanan K, Makino S, 2012. Roles of the coding and noncoding regions of rift valley Fever virus RNA genome segments in viral RNA packaging. J Virol 86, 4034–4039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakabayashi H, Taketa K, Miyano K, Yamane T, Sato J, 1982. Growth of Human Hepatoma-Cell Lines with Differentiated Functions in Chemically Defined Medium. Cancer Res 42, 3858–3863. [PubMed] [Google Scholar]
- Naslund J, Lagerqvist N, Lundkvist A, Evander M, Ahlm C, Bucht G, 2008. Kinetics of Rift Valley Fever Virus in experimentally infected mice using quantitative real-time RT-PCR. Journal of virological methods 151, 277–282. [DOI] [PubMed] [Google Scholar]
- Pepin M, Bouloy M, Bird BH, Kemp A, Paweska J, 2010. Rift Valley fever virus(Bunyaviridae: Phlebovirus): an update on pathogenesis, molecular epidemiology, vectors, diagnostics and prevention. Veterinary research 41, 61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters CJ, Liu CT, Anderson GW Jr., Morrill JC, Jahrling PB, 1989. Pathogenesis of viral hemorrhagic fevers: Rift Valley fever and Lassa fever contrasted. Rev Infect Dis 11 Suppl 4, S743–749. [DOI] [PubMed] [Google Scholar]
- Peyrefitte CN, Pastorino B, Bessaud M, Tolou HJ, Couissinier-Paris P, 2003. Evidence for in vitro falsely-primed cDNAs that prevent specific detection of virus negative strand RNAs in dengue-infected cells: improvement by tagged RT-PCR. Journal of virological methods 113, 19–28. [DOI] [PubMed] [Google Scholar]
- Plaskon NE, Adelman ZN, Myles KM, 2009. Accurate strand-specific quantification of viral RNA. PLoS One 4, e7468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rolin AI, Berrang-Ford L, Kulkarni MA, 2013. The risk of Rift Valley fever virus introduction and establishment in the United States and European Union. Emerging microbes & infections 2, e81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strydom E, Pietersen G, 2018. Development of a strand-specific RT-PCR to detect the positive sense replicative strand of Soybean blotchy mosaic virus. Journal of virological methods 259, 39–44. [DOI] [PubMed] [Google Scholar]
- Sun Y, Li J, Gao GF, Tien P, Liu W, 2018. Bunyavirales ribonucleoproteins: the viral replication and transcription machinery. Critical reviews in microbiology 44, 522–540. [DOI] [PubMed] [Google Scholar]
- Terasaki K, Makino S, 2015. Interplay between the Virus and Host in Rift Valley Fever Pathogenesis. J Innate Immun 7, 450–458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuiskunen A, Leparc-Goffart I, Boubis L, Monteil V, Klingstrom J, Tolou HJ, Lundkvist A, Plumet S, 2010. Self-priming of reverse transcriptase impairs strand-specific detection of dengue virus RNA. The Journal of general virology 91, 1019–1027. [DOI] [PubMed] [Google Scholar]
- Vashist S, Urena L, Goodfellow I, 2012. Development of a strand specific real-time RT-qPCR assay for the detection and quantitation of murine norovirus RNA. Journal of virological methods 184, 69–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wichgers Schreur PJ, Kortekaas J, 2016. Single-Molecule FISH Reveals Non-selective Packaging of Rift Valley Fever Virus Genome Segments. PLoS pathogens 12, e1005800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson WC, Romito M, Jasperson DC, Weingartl H, Binepal YS, Maluleke MR, Wallace DB, van Vuren PJ, Paweska JT, 2013. Development of a Rift Valley fever real-time RT-PCR assay that can detect all three genome segments. Journal of virological methods 193, 426–431. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.