


Abstract
Group II introns are self-splicing mobile genetic retroelements. The spliced intron RNA and the intron-encoded protein (IEP) form ribonucleoprotein particles (RNPs) that recognize and invade specific DNA target sites. The IEP is a reverse transcriptase/maturase that may bear a C-terminal endonuclease domain enabling the RNP to cleave the target DNA strand to prime reverse transcription. However, some mobile introns, such as RmInt1, lack the En domain but nevertheless retrohome efficiently to transient single-stranded DNA target sites at a DNA replication fork. Their mobility is associated with host DNA replication, and they use the nascent lagging strand as a primer for reverse transcription. We searched for proteins that interact with RmInt1 RNPs and direct these RNPs to the DNA replication fork. Co-immunoprecipitation assays suggested that DnaN (the β-sliding clamp), a component of DNA polymerase III, interacts with the protein component of the RmInt1 RNP. Pulldown assays, far-western blots and biolayer interferometry supported this interaction. Peptide binding assays also identified a putative DnaN-interacting motif in the RmInt1 IEP structurally conserved in group II intron IEPs. Our results suggest that intron RNP interacts with the β-sliding clamp of the DNA replication machinery, favouring reverse splicing into the transient ssDNA at DNA replication forks.
INTRODUCTION
Group II introns are self-splicing RNAs and mobile genetic retroelements identified in the genome of organelles (mitochondria and chloroplasts of lower eukaryotes and plants), bacteria and archaea (1–4). Group II introns are absent from the nuclear genomes of eukaryotes (5–8), but are thought to be the predecessor of the spliceosome (9) and to have played an important role in the evolution of several features of eukaryotic cell organization.
Group II introns consist of a catalytically active intron RNA (ribozyme) with a conserved secondary structure consisting of six interacting domains (DI–DVI) (10–12), and a multifunctional intron-encoded protein (IEP) encoded within DIV. Most IEPs have an N-terminal reverse transcriptase domain (RT) and an RNA-binding domain (X) that has been associated with RNA splicing or maturase activity. Some IEPs have DNA-binding (D) and endonuclease (En) domains after the X domain. The RT domain contains several blocks of conserved amino acids present in retroviral RTs (RT 1-7) with an additional conserved block (RT 0) and insertions between other conserved sequence blocks (RT 2a, 3a, 4a and 7a) (13,14).
Group II intron mobility is mediated by ribonucleoprotein particles (RNPs) consisting of the IEP and the spliced intron RNA (15,16). Several studies of RNPs assembled in vitro and in vivo have suggested that these particles are formed by an IEP dimer bound to a single lariat RNA (13,15,17–19), but a recent study of the cryo-EM structure of the RNP of Ll.LtrB from Lactococcus lactis indicated that the IEP bound as a monomer to a single RNA molecule (20). RNPs can promote intron insertion into specific DNA target sites (retrohoming) and at much lower frequency into sites resembling the normal intron site (ectopic transposition or retrotransposition). The RNP recognizes the target site through both its IEP and RNA components, through base-pairing to the target DNA (21,22). The RNA then reverse splices into the top strand of the DNA target. Introns with En domain-containing IEPs use this domain to cleave the bottom strand of the DNA target, generally 9 nt downstream from the insertion site. The 3′ end generated is used for target DNA-primed reverse transcription (TPRT) of the inserted intron RNA (2,23). Once the cDNA has been synthesized, various host factors complete intron integration into the target site. These factors mediate the degradation of intron RNA, intron second-strand synthesis, the removal of DNA overhangs and ligation (24–27). The screening of a battery of Escherichia coli mutants identified several enzymes with effects on intron mobility (24), such as RNases I and E, which are capable of degrading intron RNA, and exonuclease III (XthA), which may degrade the cDNA. RNase H1 and Pol I may be involved in the elimination of the intron RNA after reverse transcription, and DNA polymerases involved in replication and repair, such as mutD (Pol III) and Pol II, IV and V, seem to be involved in second-strand DNA synthesis.
Introns with IEPs devoid of En domains, such as the RmInt1 intron of Sinorhizobium meliloti, cannot cut the bottom strand of the DNA during retrohoming. Their retrotransposition is linked to host replication, as they use the nascent lagging strand at DNA replication forks (RNA primers or Okazaki fragments) to prime reverse transcription (28). The genes encoding the host factors required for the retrohoming of En- introns have also been identified (29). They encode proteins involved in DNA processing (xthA4, radA and dnaK), RNA processing (rne and rnhB) and Mg2+ transport (corA1). The only gene from this list that seems to be required for efficient RmInt1 retrohoming is xthA4. Mutations of any of the other genes leads to higher rates of retrohoming.
RmInt1 displays a bias for retrohoming into the lagging strand template during chromosome replication, probably because these introns reverse splice into the transient ssDNA at DNA replication forks, raising questions about possible interactions between RmInt1 RNPs and host factors, particularly the host replication machinery (30). Such interactions have been described for other mobile genetic elements, such as the Tn7 transposon and several insertion sequences (IS) (31,32). Tn7 uses two pathways to select its target site. In one of these pathways, a sequence-specific DNA-binding protein directs transposition into a single site within the bacterial chromosome. In the other, a transposon-encoded TnsE protein interacts physically with the β-sliding clamp of the DNA replication machinery (31). Transposases from many IS families displaying an orientation bias with respect to chromosomal DNA replication have been shown to interact with the β-sliding clamp (32). In this study, we investigated the host factors capable of physical interaction with the RmInt1 RNPs that might account for the DNA strand orientation bias displayed by this intron during retrohoming.
MATERIALS AND METHODS
Bacterial strains, media and growth conditions
Sinorhizobium meliloti strain RMO17 (RmInt1 intron-less strain) was grown at 28°C on TY or defined minimal medium (MM) (33). The E. coli DH5α strain was used for the cloning and maintenance of plasmid constructs. The Rosetta-gami (DE3) pLysS strain was used for protein production. Escherichia coli strains were routinely grown at 37°C on Luria-Bertani (LB) medium (34). Antibiotics were added to the medium as required, at the following concentrations: kanamycin at 200 μg/ml for S. meliloti and at 50 μg/ml for E. coli, ampicillin at 200 μg/ml, tetracycline at 10 μg/ml and gentamicin at 50 μg/ml for S. meliloti and 10 μg/ml for E. coli.
Plasmid constructs
For the construction of pMalFlagIEP, a NotI fragment containing the IEP tagged with 3xFlag was obtained by the NotI digestion of pCEP4FlagIEP (35) and inserted into the pMAL-c5X vector (New England Biolabs) digested with NotI enzyme and dephosphorylated. We obtained pMALFlagIEPΔORF in a similar manner, but using a NotI fragment from pCEP4FlagIEPΔORF (35).
pGmS4S2095-Fmal is a derivative of pGmS4S2095 (36) in which the IEP has been replaced by the fusion protein MBP-FlagIEP (MF-IEP) sequence in the form of a SpeI-AatII fragment from pMALFlagIEP. pGmS4S-Fmal is a derivative of pGmS4S (36) in which the IEP has been replaced by the MF-IEP sequence as a SpeI-AatII fragment from pMALFlagIEP. This plasmid produces the fusion protein MF-IEP and the ΔORF of RmInt1, under the control of the Sym promoter. pGmS-Fmal was constructed from pGmS4S-Fmal by digestion with NdeI and AvrII to remove the FlagIEP and ΔORF. Flag was then added back to the plasmid as a polymerase chain reaction (PCR) fragment amplified with the oligomers 5′-ACAAGGATGACGATGACAAGACTATAGGCCAATTCGCCCTATA-3′ (GmS-Flg) and CTTGTCATCGTCATCCTTGT (GmS4S-Flg).
For the construction of p16N, dnaN was amplified from S. meliloti RMO17 by PCR with the primers FwdnaN (5′-CTCATATGCGTATTACTCTCGAGCG-3′) and RvdnaN (5′-CTGGATCCTTACACGCGCATCGGCATCA-3′). The amplicon was inserted into the pGEMT-easy vector (Promega) and sequenced. It was then inserted into pET16b (Novagen) as an NdeI/BamHI fragment, to form p16N.
Retrohoming assays in Sinorhizobium meliloti RMO17
Retrohoming assays were performed as previously described (36). We used pGmS4S2095-Fmal, which expresses the fusion protein MF-IEP and a retargeted RmInt1-ΔORF flanked by short exons (−20/+5) under the control of pSyn promoter, as a control in assays of the biological activity of IEP mutants. All the mutants tested were derivatives of this plasmid.
RT activity assay
Reverse transcriptase activity was assayed with poly(rA)-oligo(dT)18 as follows: 1 μg protein was added to 10 μl of reaction mixture (50 mM Tris–HCl pH 7.5, 10 mM KCl, 25 mM MgCl2, 5 mM 1.4-Dithiothreitol (DTT)) with 1 μg poly(rA)-oligo(dT)18 or poly(rA), and 5 μCi of [α-32P]dTTP (800 Ci/mmol; Perkin Elmer). The reaction mixture was incubated for 10 min at 37°C, and 8 μl of reaction product was then spotted onto Whatman DE81 filters, washed four times (5 min each) with 2 × Saline Sodium Citrate (SSC) and counted in a Liquid Scintillation Analyser (Beckman-Coulter).
LC-MS/MS and protein identification
MS analysis was performed at the Proteomics Service of the Instituto de Parasitología y Biomedicina ‘López-Neyra’ (CSIC, Granada). Protein samples were subjected to sodium dodecyl sulphate-polyacrylamide gelelectrophoresis (SDS-PAGE) in a 4% acrylamide gel for 10 min. The gel lane was cut into 10 slices and subjected to in-gel tryptic manual digestion. The resulting peptides were fractionated with an Easy n-LC II chromatography system (Proxeon) in line with an Amazon Speed ETD mass spectrometer (Bruker Daltonics). In brief, peptides in 0.1% formic acid and 3% acetonitrile were injected onto an EASY trap column (2 cm, ID100 μm, 5 μm, C18; Thermo Fisher Scientific). After washing with 0.1% (v/v) formic acid, peptides were resolved on an Acclaim PepMap100, 15 cm × 75 μm, 3 μm, 100 A, C18 reverse-phase analytical column (Thermo Fisher Scientific) over a 120 min organic gradient from 5 to 40% B with six gradient segments (5–40% solvent B over 120 min, 40–90% B over 5 min, 90–100% B over 2 min, held at 100% B for 10 min and then reduced to 0% B over 1 min and held at 0% for 2 min) with a flow rate of 300 nl.min−1. Solvent A was 0.1% formic acid in water and solvent B was 0.1% formic acid in acetonitrile.
Peptides were ionized by a CaptiveSpray Ion Source (Bruker Daltonics) at 1300 V. Tandem mass spectra were acquired with an Amazon Speed ETD Ion Trap mass spectrometer controlled by TrapControl software v7.2 (Bruker-Daltonics), operated in AutoMS acquisition mode. The ion trap was set to analyse the survey scans in the mass range m/z 400–1400 in Enhanced Resolution MS mode and the top ten multiply charged ions in each duty cycle were selected for MS/MS in UltraScan MS/MS mode. Fragmentation conditions in the Amazon were as follows: exclusion of simply charged ions, active exclusion after two spectra and active release time of 0.5 min, threshold Absolute 20 000 and MS/MS fragmentation amplitude 60%. The raw data files were processed with DataAnalysis software v4.3 (Bruker Daltonics) and used to search the UniProtTrembl database with Mascot 2.4 (Matrix Science) and ProteinScape v4.0 (Bruker Daltonics). Peptide precursor mass tolerance was set at 0.5 Da, and MS/MS tolerance was set at 0.5 Da. The search criteria included carbamidomethylation of cysteine (+57.02 Da) as a fixed modification and oxidation of methionine (+15.99 Da) as a variable modification. Searches were performed with a maximum of two missed cleavages for tryptic digestion. The reverse database search option was enabled and all peptide data were filtered to ensure a false discovery rate (FDR) ≤ 2%.
FLAG immunoprecipitation
We harvested 200 ml of bacterial culture with an OD600 of 0.6. The cells were collected by centrifugation and the pellet was washed once with 24 ml of 0.1% sarkosyl in TE buffer and twice with 20 mM Tris–HCl pH 7.4, 200 mM NaCl, 1 mM ethylenediaminetetraacetic acid (EDTA), 1 mM DTT. The cells were again collected by centrifugation, and the resulting pellet was resuspended in 8 ml of lysis buffer (50 mM Tris–HCl pH 7.4, 150 mM NaCl, 1 mM EDTA, 1% Triton X-100) plus proteinase inhibitors and incubated for 30 min at 4°C with gentle shaking. Cells were lysed by sonication, with three pulses of 10 s each, and centrifuged at 12 000 × g for 10 min at 4°C. The pellet was discarded and 100 μl of Anti-FLAG M2-Agarose Affinity Gel (prepared according to the manufacturer's instructions) was added to the supernatant, which was then incubated overnight at 4°C with gentle shaking. Samples were then centrifuged for 30 s at 8200 × g and the supernatant was discarded. The resin was washed three times with 0.5 ml of wash buffer (50 mM Tris–HCl, pH 7.4, 150 mM NaCl). For elution, the resin was incubated with 100 μl of wash buffer supplemented with 150 ng/μl of 3× Flag peptide for 30 min at 4°C with gentle shaking, and then centrifuged at 8200 × g. The supernatant containing the proteins was stored at −20°C.
For Flag immunoprecipitation, we mixed 15 μl of RNPs with 185 μl of lysis buffer and 30 μl of ANTI-FLAG M2-Agarose Affinity Gel and incubated the mixture for 2 h at 4°C with gentle shaking. Negative controls were treated in the same way, but with 15 μl of Milli-Q water replacing the RNPs. After incubation, samples were centrifuged for 30 s at 8200 × g and the supernatant was discarded. The resin was washed three times with 0.5 ml of wash buffer and incubated again in 0.5 ml of lysis buffer containing 8 μg of His-DnaN for 2 h at 4°C, with gentle shaking. Samples were washed five times with wash buffer. Proteins bound to ANTI-FLAG-M2-Agarose were eluted with 100 μl of wash buffer supplemented with 150 μg/μl 3× Flag peptide for 30 min at 4°C, with gentle shaking.
Pulldown assays
Rosetta-gami (DE3) pLysS cells were used to produce the proteins for the assays. Cells were grown in LB supplemented with 0.2% glucose plus appropriate antibiotics at 37°C until an OD600 of 0.5 was attained. They were then induced with 0.2 mM isopropyl-β-D-thiogalactopyranoside (IPTG) and cultured at 18°C overnight. Aliquots (2 ml) were centrifuged and the pellets were resuspended in 1 ml of buffer (50 mM Tris–HCl pH 6.8, 100 mM NaCl, 10 mM CaCl2 and proteinase inhibitors for proteins containing a His tag, or 20 mM Tris–HCl pH 7.4, 200 mM NaCl, 1 mM EDTA, 1 mM DTT and proteinase inhibitors for MBP fusions) and incubated on ice for 30 min. Samples were sonicated with three pulses of 10 s each, and were then centrifuged at 12 000 × g for 15 min at 4°C. For MBP fusion proteins, 80 μl of amylose resin was added to the supernatant, which was then incubated for 1 h at 4°C. The resin was washed three times with the buffer used to resuspend the MBP fusion proteins and was then added to the supernatant of His-tagged proteins, and the resulting mixture was incubated for 3 h at 4°C with gentle shaking. During this incubation, 100 units of RNase A/T1 were added to the mixture for the RNase-treated sample. The resin was washed four times and resuspended in 100 μl of SDS sample buffer. The binding of His-tagged proteins to MBP-fusion proteins was assessed by western blotting with horseradish peroxidase (HRP)-conjugated anti-His antibodies.
Peptide binding assay
Assays were performed in a volume of 50 μl, with 700 μg of streptavidin-coated magnetic beads (Sigma Aldrich) in 50 mM Tris–HCl, 100 mM NaCl, 5% glycerol, pH 7.5. We mixed 440 μM biotinylated peptides with the beads, which were then incubated for 30 min and washed three times. We then added His-tagged DnaN (4 μM), incubated 10 min and washed the beads four times with the same buffer. Reactions were stopped by adding 1% SDS and the reaction mixture was subjected to SDS-PAGE and western blotting with anti-His tag antibodies. Signals were detected with a ChemiDoc system and quantified with Quantity One software (Bio-Rad).
Western and far-western blotting
Western blots were performed with an ECL kit (GE Healthcare), according to the manufacturer's instructions. Membranes were blocked with a blocking solution containing 3% skimmed milk powder. Antibodies were used at the following dilutions: monoclonal HRP-conjugated anti-polyHis 1:200000 (Sigma A7058); anti-Flag 1:100000 (Sigma F1804) and anti-mouse IgG-HRP conjugate 1:250000 (Sigma A9044).
Far-western blots were performed as described elsewhere (37). We subjected lysates (10 μg of total protein) of Rosetta-gami (DE3) pLysS cells expressing the fusion protein MBP-FlagIEP from pMALFlagIEP to SDS-PAGE, and the resulting bands were blotted onto PVDF (GE Healthcare) membranes. The proteins were denatured and renatured on the membranes in AC buffer (100 mM NaCl, 20 mM Tris (pH 7.6), 0.5 mM EDTA, 10% glycerol, 0.1% Tween-20, 2% skimmed milk powder and 1 mM DTT), by gradually decreasing the concentration of guanidine-HCl. The membrane was incubated in AC buffer containing 6 M guanidine-HCl for 30 min at room temperature, and then with AC buffer containing 3 M guanidine-HCl for 30 min at room temperature, followed by AC buffer plus 1 M guanidine-HCl and two incubations with AC buffer containing 0.1 M guanidine-HCl and no guanidine-HCl AC, both at 4°C, for 30 min and 1 h, respectively. The membrane was then blocked by incubation with 5% skimmed milk powder in PBST for 1 h at room temperature. The membrane was incubated with His-DnaN (total 5 μg, 1 μg.ml-1) in buffer (100 mM NaCl, 20 mM Tris (pH 7.6), 0.5 mM EDTA, 10% glycerol, 0.1% Tween-20, 2% skimmed milk powder and 1 mM DTT) for 2 h at room temperature. The unbound protein was removed by three washes in PBST, for 10 min each. The membrane was then incubated with monoclonal HRP-conjugated anti-polyHis- antibodies (1:200000) (Sigma A7058) in 3% skimmed milk powder in PBST buffer for 1 h at room temperature. It was then washed six times, for 5 min each, with PBST buffer and chemiluminescence detection was performed with an ECL kit (GE Healthcare), according to the manufacturer’s instructions.
Protein purification
MF-IEP and the mutants generated were purified as follows: pMalFlagIEP or mutants derived from this plasmid were used to transform Rosetta-gami (DE3) pLysS cells, which were cultured to an optical density (OD600) of 0.5 at 37°C in 1 l of LB medium supplemented with chloramphenicol (50 μg/ml), ampicillin (100 μg/ml) and 0.2% glucose. Protein production was then induced by adding IPTG to a final concentration of 0.3 mM and incubating at 20°C overnight. Bacterial cells were harvested by centrifugation at 5000 × g for 15 min at 4°C and resuspended in 25 ml of binding buffer (20 mM Tris–HCl, 200 mM NaCl, 1 mM EDTA, 1 mM DTT pH 7.4). They were then lysed by three passages through a 1000 PSIG French press and the resulting lysates were clarified by centrifugation at 38 000 × g for 1 h at 4°C. The supernatant was applied to an MBPTrap HP affinity column (GE Healthcare) equilibrated with binding buffer and washed with five column volumes. Proteins were eluted with elution buffer (10 mM maltose in binding buffer) and subjected to SDS-PAGE. The eluate containing the protein was dialysed against 20 mM Tris–HCl, 200 mM NaCl, 1 mM EDTA, 1 mM DTT, 10% glycerol pH 7.4 at 4°C. Dialysed proteins were passed through a size exclusion column HiLoad Superdex 75 equilibrated with the same buffer used for dialysis and the peak corresponding to monomer was collected and frozen at −80°C.
For DnaN purification, the p16N plasmid was used to transform Rosetta-gami (DE3) pLysS cells. The cells were cultured to an OD600 of 0.5 in 1 l of LB medium supplemented with 100 μg/ml ampicillin and 50 μg/ml chloramphenicol at 37°C. Protein production was induced at 20°C, by adding IPTG to a final concentration of 0.3 mM. After 18–20 h of growth, the cells were harvested by centrifugation at 5000 × g for 15 min at 4°C. They were then resuspended in 25 ml of buffer A (25 mM sodium phosphate buffer pH 7, 0.5 M NaCl, 5% glycerol) and lysed by three passages through a 1000 PSIG French press. The lysate was centrifuged at 38 000 x g for 1 h at 4°C. The supernatant was loaded onto a HisTrap™ HP 5 ml column (GE Healthcare) equilibrated with buffer A, which was then washed with three column volumes of the same buffer. The column was then washed with three column volumes of 5% buffer B (25 mM sodium phosphate buffer pH 7, 0.5 M NaCl, 5% glycerol and 1 M imidazole) in buffer A. Proteins were eluted along a gradient running from 5% buffer B to 100% buffer B over a period of 15 min. Fractions were subjected to SDS-PAGE and those containing DnaN were pooled, dialysed overnight against buffer D (50 mM Tris–HCl pH 8, 50 mM NaCl, 1 mM EDTA, 1 mM dithiothreitol (DTT) and 10% glycerol) and frozen at −80°C.
Biolayer interferometry
The association (kon) and dissociation (koff) rate constants for β-clamp binding to MF-IEP were determined with a BLItz system (ForteBio, Pall, Spain) (38).The β-clamp protein (at a concentration of 0.5 μM) was immobilized on His-tagged biosensors (Forte Bio) previously hydrated with 10× kinetic sample buffer (ForteBio, the 10× kinetic sample buffer as supplied by the manufacturer consisted of 10 mM sodium phosphate, 150 mM NaCl, Tween 20 (0.02%), albumin (0.1%) and sodium azide (0.05%) at pH 7.4). Various amounts of MF-IEP protein (typically 0.5, 1, 1.5, 2 and 3 μM) were used in the association steps. The response was monitored every 0.2 s. Each sensorgram was compared with the reference signal obtained for buffer alone at the start of each experiment.
The general scheme for association/dissociation reactions for both proteins was: 30 s of initial baseline conditions with the 10× kinetic buffer; 120 (or 240) s of DnaN (β-clamp) protein loading onto the biosensor; 30 s of baseline with 10× kinetics buffer; 120 (or 240) s of MF-IEPs (MF-IEP, or mutants MF-IEP104 and MF-IEP108) association with the biosensor (previously loaded with β-clamp); and 120 (or 240) s of MF-IEP dissociation from the biosensor. The interferometric response during the association step, R(t) (measured in resonance units, RU) and used to evaluate the kinetics of the DnaN-MF-IEP interaction is given by: 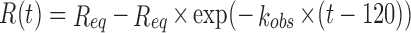 , where Req is the steady-state, or equilibrium, response obtained at infinite time. The number 120 (or 240) in the equation above corresponds to the time when the association step between DnaN-biosensor-bound and MF-IEP in the solution begins (see above). As experimental conditions correspond to a pseudo-first-order regime, the value of the kobs is given by: kobs= kon × [MF-IEP]+ koff, where kon (M-1 s-1) and koff (s-1) are the association and dissociation rate constants of the binding reaction, and [MF-IEP] is the concentration of the protein (wild-type or mutants) in the solution. From the values of kon (M-1 s-1) and koff (s-1), we thus obtained an apparent equilibrium association constant, KA, such that KA = kon/koff. Kinetic curves describing the association step between MF-IEP and DnaN-biosensor-bound were initially fitted to the sensorgrams with BLItz Pro version 1.2 software provided by the manufacturer, using a single exponential curve. Successively, to further improve the residuals of the fittings of the sensorgrams, we employed the software Kaleidagraph (Synergy software) and analysed data at all MF-IEP concentrations, using two exponential curves:
, where Req is the steady-state, or equilibrium, response obtained at infinite time. The number 120 (or 240) in the equation above corresponds to the time when the association step between DnaN-biosensor-bound and MF-IEP in the solution begins (see above). As experimental conditions correspond to a pseudo-first-order regime, the value of the kobs is given by: kobs= kon × [MF-IEP]+ koff, where kon (M-1 s-1) and koff (s-1) are the association and dissociation rate constants of the binding reaction, and [MF-IEP] is the concentration of the protein (wild-type or mutants) in the solution. From the values of kon (M-1 s-1) and koff (s-1), we thus obtained an apparent equilibrium association constant, KA, such that KA = kon/koff. Kinetic curves describing the association step between MF-IEP and DnaN-biosensor-bound were initially fitted to the sensorgrams with BLItz Pro version 1.2 software provided by the manufacturer, using a single exponential curve. Successively, to further improve the residuals of the fittings of the sensorgrams, we employed the software Kaleidagraph (Synergy software) and analysed data at all MF-IEP concentrations, using two exponential curves: 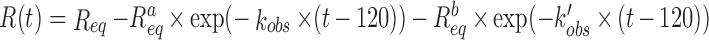 ; with this equation we are assuming that the equilibrium response, Req, is attained with the fastest kobs. Furthermore, the use of this equation indicates that the binding between MF-IEP and DnaN does not follow a simple kinetic 1:1 model. Fittings using Kaleidagraph had, in all sensorgrams, regression coefficients larger than 0.999 and the residuals did not show any tendency. The uncertainty in the reported kobs at each concentration is the fitting error to the double exponential equation. Finally, we note that in interferometry, a ‘jump’ (discontinuity) may occur between the response units at the start of the association or dissociation step. These jumps are due to the changes in the refractive index detected by the biosensor (39) at the beginning of these steps. Such discontinuities are taken into account by the manufacturer's software during curve fitting. Control experiments were also carried out with MF alone, in the same range of concentrations as used for the MF-IEP protein.
; with this equation we are assuming that the equilibrium response, Req, is attained with the fastest kobs. Furthermore, the use of this equation indicates that the binding between MF-IEP and DnaN does not follow a simple kinetic 1:1 model. Fittings using Kaleidagraph had, in all sensorgrams, regression coefficients larger than 0.999 and the residuals did not show any tendency. The uncertainty in the reported kobs at each concentration is the fitting error to the double exponential equation. Finally, we note that in interferometry, a ‘jump’ (discontinuity) may occur between the response units at the start of the association or dissociation step. These jumps are due to the changes in the refractive index detected by the biosensor (39) at the beginning of these steps. Such discontinuities are taken into account by the manufacturer's software during curve fitting. Control experiments were also carried out with MF alone, in the same range of concentrations as used for the MF-IEP protein.
Sequence and structural alignments
Structure-based sequence alignments were generated with T-COFFEE (40), as previously described (41). Structural alignments were generated manually in Coot, with the secondary structure matching algorithm (42). The figures depicting the structures were drawn with PyMOL (The PyMOL Molecular Graphics System, Version 2.0 Schrödinger, LLC).
Reconstitution of RmInt1 RNP particles in vitro
RmInt1-ΔORF RNP particles were reconstituted from in vitro-synthesized, gel-purified RmInt1-ΔORF precursor intron RNA (908 nt) and amylose affinity-purified MF-IEP, as previously described (43). Briefly, 350 nM precursor intron RNA in 80 mM MOPS pH 7.5 was denatured by heating at 90°C for 2 min and then renatured by incubation at 50°C for 2 min, followed by slow cooling at room temperature for 1 min. A self-splicing reaction was induced at 50°C by adding 500 mM NH4Cl and 100 mM MgCl2. After 5 h, 3.5 μM purified MF-IEP protein was added to the spliced RNA and the resulting complexes were incubated at 30°C for 2 h. The ribonucleoprotein particles (RNPs) were diluted in 450 mM NaCl, 5 mM MgCl2, 40 mM Tris–HCl pH 7.5 buffer and pelleted by ultracentrifugation in a Beckman 50 Ti rotor at 50 000 rpm for 18 h at 4°C. Finally, the RNP preparations were kept in 5 mM Tris–HCl pH 7.5, 0.5 mM DTT, 50% glycerol at −80°C. The sedimentation process was monitored by collecting fractions along the tube volume. Fractions were desalted and concentrated with Amicon YM-30 filter units. The protein content of each fraction and the pellet was evaluated by western blotting with an anti-FLAG primary antibody. The presence of intron RNA was demonstrated by primer extension and the resolution of products on electrophoresis in 6% polyacrylamide gels.
RESULTS
Functionality of the RmInt1 IEP fusion protein
Like other group II intron-encoded proteins, the RmInt1 IEP has a very high isoelectric point (9.83), resulting in low solubility (44). We fused a 3xFlag N-terminally tagged IEP to the C-terminal part of maltose binding protein (MBP) to facilitate purification and increase RmInt1 IEP solubility (44). We first assessed the functionality of the fusion (MBP-Flag-IEP) protein (hereafter referred as to MF-IEP) in retrohoming, by comparing intron insertion frequencies between the intron donor plasmid, pGmS4S2095, which expresses a wild-type IEP, and a retargeted RmInt1-ΔORF flanked by short exons (−20/+5) under the control of the Psyn promoter, with an equivalent intron construction (pGmS4S2095-Fmal) but harbouring the IEP fusion protein (MF-IEP). The retargeted intron inserted into the lacZ antisense strand in nucleotide position 2095 (2095as) on the lagging template strand (LAG) (36). Retrohoming assays were performed in S. meliloti strain RMO17, with a lacZ gene inserted close to the chromosomal origin of replication (36). The plasmid carrying MF-IEP displayed 51% the level of retrohoming displayed by the wild-type IEP (Figure 1). The MF-IEP is, thus, a functional protein, and can be used to identify the host proteins interacting with RmInt1 RNPs.
Figure 1.
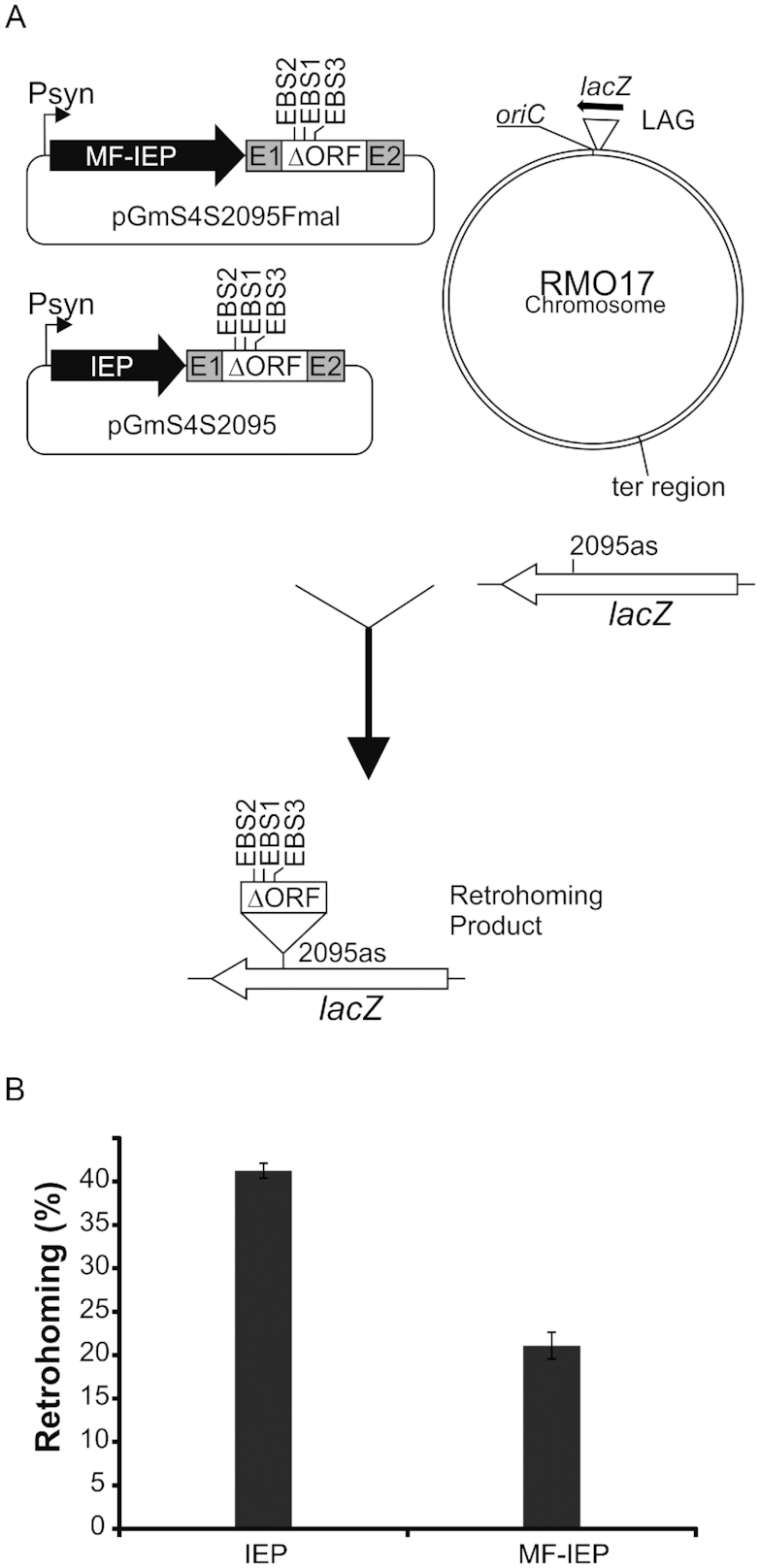
In vivo intron mobility assay. (A) Schematic of the assay showing intron donor plasmids (pGmS4S2095Fmal and pGmS4S2095), recipient lacZ gene and retrohoming product. LAG denotes that lagging strand DNA is used as primer for reverse transcription of the inserted intron RNA. In the lacZ gene the intron insertion site is shown (2095as). Psyn, syn promoter; EBS1,2,3, exon binding sites modified to recognize target 2095as into lacZ gene; E1, E2 short exons −20/+5; MF-IEP (MBP-Flag-IEP protein); IEP (wt IEP). (B) Comparison of retrohoming efficiencies between an intron expressing the wt IEP and the same intron expressing the MF-IEP fusion protein. Results are expressed as percentage of white colonies. Error bars are standard deviations (SDs) of three experiments.
Identification of proteins interacting with RmInt1 RNPs
Lysates of S. meliloti cells harbouring the intron donor plasmid, pGmS4S-Fmal, which expresses the MF-IEP, and the wild-type RmInt1-ΔORF, or the negative control, pGmS-Fmal, which expresses only the MBP-Flag (MF) protein, were subjected to immunoprecipitation with an anti-Flag antibody. Lariat intron RNA was detected in the eluted fraction of cells harbouring pGmS4S-Fmal, but not in that of cells harbouring the control plasmid. The cells harbouring pGmS4S-Fmal therefore produced RmInt1 RNPs. The proteins in the eluted complex were analysed by liquid chromatography-tandem mass spectrometry (MS), and were identified with a false discovery rate (FDR) <2% and with at least two peptides (Table 1). Interestingly, DnaN (also call β-sliding clamp), a component of DNA polymerase III involved in DNA replication, was detected only in the eluted complexes from cells harbouring pGmS4S-Fmal. We explored this interaction further, as we considered it potentially relevant for mediating the putative association of RmInt1 with the host DNA replication machinery.
Table 1.
RmInt1 RNP proteins identified by Flag immunoprecipitation
| Protein | Score | #Peptides | SC(%) |
|---|---|---|---|
| DNA polymerase III subunit beta (DnaN) | 280.72 | 5 | 14.80 |
| Ribosomal protein S2 | 111.01 | 3 | 13.3 |
| S-adenosylmethionine synthetase | 85.55 | 2 | 6.10 |
| Heat-shock protein Hsp20 | 72.89 | 2 | 13.7 |
SC: sequence coverage.
RmInt1 IEP interacts with DnaN
We investigated the possible interaction of RmInt1 RNPs with DnaN using three different techniques: MBP pulldown assays, far-western blotting and biolayer interferometry. For pulldown assays, the culture supernatants of E. coli cells overexpressing the MF-IEP and RmInt1 ΔORF (pMalFlagIEPΔORF) or just the fusion protein MF-IEP (pMalFlagIEP) were assayed against the supernatants of cells overexpressing His-tagged S. meliloti DnaN (p16N). DnaN was present in both samples (Figure 2A), but not in the control (supernatants of cells overexpressing only the MBP-Flag protein (MF)), suggesting that DnaN and RmInt1 RNPs may interact via the IEP component. We ensured that no endogenous group II intron RNA was associated with the MF-IEP (pMalFlagIEP) expressed in E. coli, by performing a pulldown assay including RNase A/T1 treatment during the incubation of resin-bound MF-IEP with DnaN. In this sample, DnaN was also recovered, providing further support for the notion that the interaction involves the IEP component of the RNPs (Figure 2A). The interaction of the IEP with DnaN was also confirmed by far-western blotting (Figure 2B), with the detection of a specific signal for His-DnaN at the same molecular weight as the MF-IEP. Consistent with these results, biolayer interferometry (Figure 2C) showed that the MF-IEP binds DnaN with an apparent affinity of 0.4 ± 0.2 μM, assuming a 1:1 kinetic pathway. The binding of the MF-IEP in solution (from 300 to 540 s) to the biosensor loaded with His-tagged DnaN, and its dissociation from the biosensor-bound β-clamp (from 540 to 780 s) following biosensor immersion in a solution containing only the buffer was demonstrated by the exponential behaviour of the curves obtained at increasing amounts of MF-IEP used (Figure 2C). By contrast, the control MF protein displayed no significant binding to the biosensor-bound DnaN. Together, these results indicate that the RmInt1 IEP binds to the DnaN protein.
Figure 2.

Interaction of the RmInt1 IEP with DnaN. (A) Pulldown assays of MF-IEP without the ΔORF ribozyme of RmInt1 (MF-IEP), MF-IEP treated with RNase A/T1 (MF-IEP + RNase), MF-IEP and RmInt1 ΔORF ribozyme (IEP+ΔORF), and MBP fused to the Flag epitope (MF) as a control. Note that the MBP and MF proteins behave similarly. All protein samples were assayed against supernatants containing His-tagged Sinorhizobium meliloti DnaN. The proteins retained were subjected to SDS-PAGE and stained with Coomassie blue (Coomassie), and a western blot was performed with antibodies against the His tag (western), demonstrating the presence of His-tagged DnaN. (B) Far-western blot. Total protein (10 μg) from cells overproducing MF-IEP (MF-IEP) or MBP (control) was subjected to SDS-PAGE and stained with Coomassie blue (Coomassie) or subjected to far western blotting with His-tagged DnaN replacing the primary antibody and detection with antibodies against the His-tag, revealing the presence of His-tagged DnaN at the same molecular weight as MF-IEP. (C) Binding of biosensor-bound DnaN to MF-IEP. Biolayer interferometry results for the MF-IEP fusion protein (top) and for MF (bottom). (D) Flag immunoprecipitation of active RNPs containing the MF-IEP protein. His-DnaN was recovered in the immunopreciptate obtained with anti-Flag antibodies only when RNPs were used (RNPs). No His-DnaN was retained by the anti-Flag antibodies (Beads).
We then investigated whether active RNPs interact with DnaN using in vitro reconstituted RNPs in Flag immunoprecipitation assays. Active RmInt1-ΔORF RNP particles were reconstituted from precursor intron RNA synthesized in vitro and purified MF-IEP, as previously described (43). The presence of MF-IEP and intron lariat RNA in the RNP pellet was confirmed by primer extension and western blotting, and the functionality of these elements was assessed in endonuclease activity assays (Supplementary Figure S1). The reconstituted RNPs were bound to anti-FLAG-M2 affinity gel and purified His-DnaN was added. As a negative control, anti-FLAG-M2 affinity gel without RNPs was also mixed with His-DnaN. After incubation and washing, the immunoprecipitated Flag-taggedproteins were analysed by western blotting with anti-His antibodies. DnaN was recovered in the RNP sample, with no signal detected for the control (Figure 2D). These results further indicate that active RmInt1 RNPs interact with DnaN.
Identifying a possible DnaN binding motif of the RmInt1 IEP
We fused three different overlapping fragments of the IEP (N, M, C; Figure 3A) to MBP and performed pulldown assays, to identify the part of the RmInt1 IEP responsible for interaction with DnaN (Figure 3A). These assays suggested that the putative binding region was probably present in the RT domain (RT2 to RT4) common to the N and M fragments of the IEP, because the C-terminal part of the protein did not appear to interact with DnaN.
Figure 3.
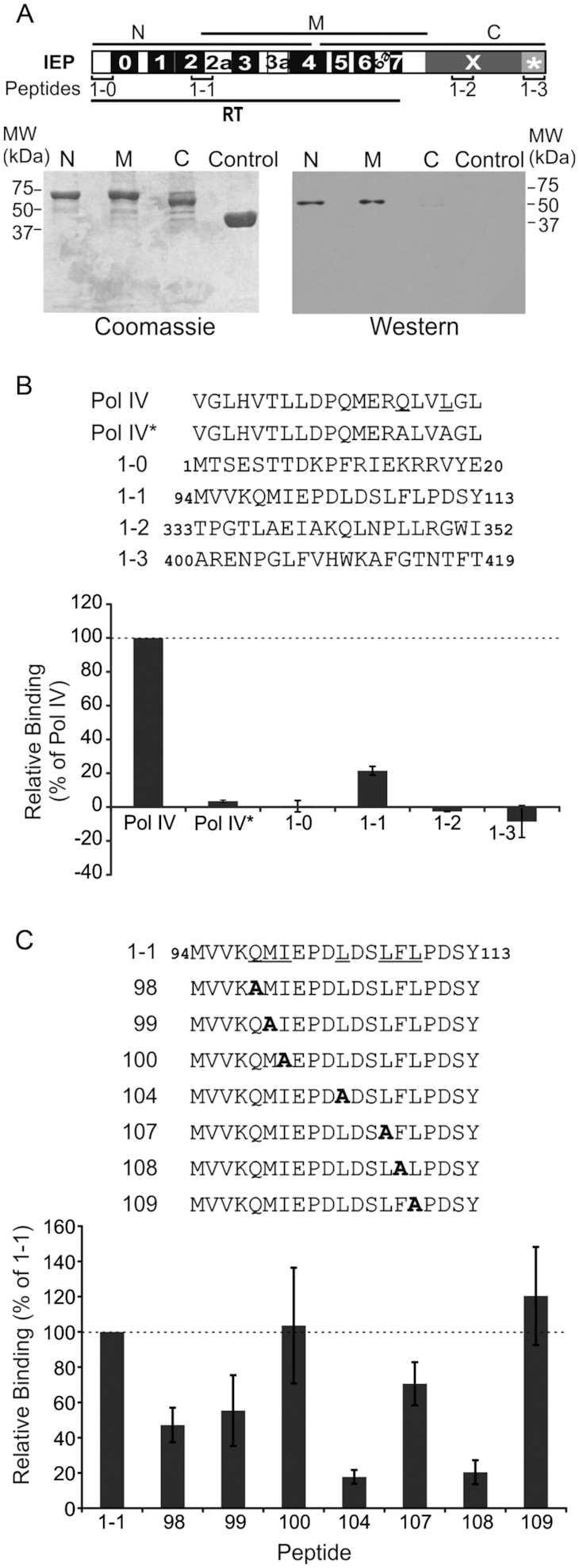
Pulldown assay performed with DnaN and various fragments of the RmInt1 IEP and peptides. (A) Diagram of the various parts of the IEP used in pulldown assays (lower panel). N, N-terminal part of IEP; M, middle region of IEP; C, C-terminal part of IEP; control, MBP. IEP domains are also shown; RT, reverse transcriptase domain with conserved RT sequence blocks (0-7); X, maturase domain; *, C-terminal tail. The positions of the peptides used in panel (B) are also shown. Bottom panel, pulldown assay performed with His-DnaN and various fragments of IEP. The proteins retained were subjected to SDS-PAGE and stained with Coomassie blue (Coomassie) or subjected to western blotting (western) with antibodies against the His-tag, revealing the presence of His-tagged DnaN in lanes N and M. (B) Peptide binding assay. Sequences of peptides from IEP and DNA polymerase IV used in the assays. Pol IV* has the same sequence as Pol IV except that the two residues underlined in the Pol IV sequence are replaced with alanines. The binding levels are expressed as a percentage relative to the amount of His-tagged DnaN retained by the Pol IV peptide (dashed line). (C) Peptide binding assay with mutant peptides. Sequences of the peptides synthesized, in which the underlined residues of peptide 1-1 have been replace with alanines. Peptides were coupled to streptavidin-coated paramagnetic beads and used to retain His-tagged DnaN. The binding levels are expressed as a percentage relative to the amount of His-tagged DnaN retained by peptide 1-1 (dashed line). In panels B and C, error bars represent standard error of the mean determined from three independent experiments.
Many proteins interacting with DnaN harbour a QLS/DLF binding motif (45), but this motif is poorly conserved and absent from a number of DnaN-interacting proteins. TnsE, a protein encoded by the Tn7 transposon, interacts with DnaN through the QLELARALFL motif (31). A similar motif, QMIEPDLDSLFL, is present in the C-terminal region of the RT2 domain of RmInt1IEP. We explored the region of the IEP binding to DnaN in more detail, by synthesizing four 20-amino-acid-long N-biotinylated peptides (Figure 3B). Peptides 1-0 and 1-3 corresponded to the first and last 20 amino acids, respectively, of the IEP. These regions have a higher predicted level of intrinsic disorder (46). Peptide 1-1 contained the QMIEPDLDSLFL motif, and peptide 1-2 contained amino-acid residues (QLNPLL) from the maturase domain resembling the DnaN-binding motif. The peptides were bound to streptavidin magnetic beads, for assessment of their ability to bind and retain His-tagged DnaN. As a positive control, we used a peptide derived from DNA polymerase IV (Pol IV). The negative control was the same peptide with two amino acids replaced with alanine residues (Pol IV*). The results are expressed as a percentage of the amount of His-tagged DnaN retained by the Pol IV peptide (Figure 3B). As expected, only peptide 1-1, carrying the QMIEPDLDSLFL motif, bound to DnaN, with retention rates 20% those for the control peptide.
We then synthesized a set of peptides similar to peptide 1-1, but with selected amino acids of the motif replaced by alanine residues. These peptides were used in pulldown assays (Figure 3C). All of the positions substituted, with the exception of I100A and L109A, appeared to be involved in the interaction, with L104A and F108A causing the strongest decrease in DnaN-retaining capacity.
In order to see if mutations L104A and F108A affect the interaction between DnaN and MF-IEP, we made mutants MF-IEP104 and MF-IEP108 (MF-IEP carrying substitution L104A and F108A, respectively) and tried to provide a quantitative measurement of the affinity between MF-IEP, MF-IEP104 and MF-IEP108 with DnaN by biolayer interferometry. For all the explored concentrations, we carried out the fitting of the association step of the sensorgrams with a double exponential curve. These results indicate that the binding between MF-IEP and DnaN does not follow a simple 1:1 kinetic mechanism. The fastest rate, kobs, was MF-IEP-concentration-dependent, while the other (slowest) rate was constant (with a value for the three species, at any of the explored concentrations, of 0.006–0.008 s-1). The pseudo-first order plots (Figure 4) for the fastest protein-concentration-dependent kobs indicate that the kon for wild-type MF-IEP was 0.082 ± 0.014 μM-1 s-1, whereas those of MF-IEP104 and MF-IEP108 were 0.044 ± 0.005 μM-1 s-1 and 0.010 ± 0.007 μM-1 s-1, respectively (as it can be easily observed from the slopes in Figure 4). Therefore, assuming the same koff for the three MF-IEP species (that is, the rate of the dissociation step from the biosensor-bound DnaN), we can conclude that the wild-type species bind faster than the other two mutants and with a larger apparent equilibrium association constant KA (that is, a larger affinity).
Figure 4.
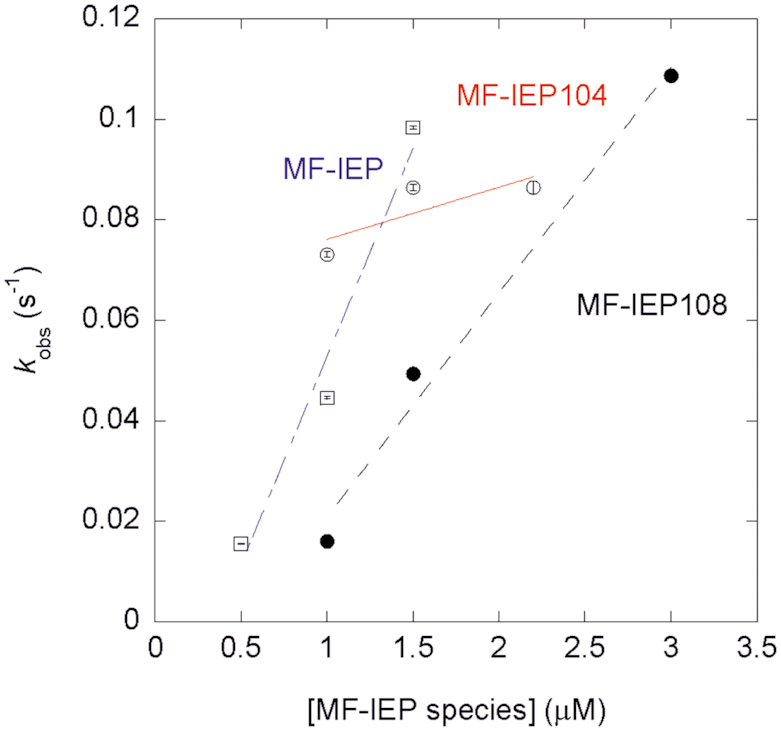
Pseudo-first order plots from biolayer interferometry for wild-type MF-IEP (blank squares and dot-and-dashed blue line), MF-IEP104 (blank circles and continuous red line) and MF-IEP108 (filled circles and dashed black line). The errors in kobs for each experimental MF-IEP species concentration are from fitting errors of the sensorgrams to a double exponential curve.
Analysis of the RmInt1 IEP DnaN binding motif in available 3D structures of IEPs
Structure-based sequence alignments of the RmInt1 IEP with the IEP of Eubacterium rectale (PDB ID: 5HHL), Roseburia intestinalis (PDB ID: 5HHJ), Geobacillus stearothermophilus (PDB ID: 6AR1) (47) and L. lactis LtrA (PDB ID: 5G2X) revealed that the QMxxxxLxxLFL motif is structurally conserved (Supplementary Figure S2). Moreover, in all available IEP 3D structures this motif points towards the protein surface (Figure 5), with the most solvent-exposed region corresponding to the C-terminal amino acids of the motif (LxxLFL), including the RmInt1 L104 and F108 analogues. Part of this solvent-exposed region forms a loop and adopts alternative conformations in Eu.re.I2/Ro.in.IEP versus LtrA. In the crystal structure of Eu.re.I2 (and correspondingly in the essentially identical structure of Ro.in.IEP), Y102 (analogous to RmInt1 L104) and F106 (analogous to RmInt1 F108) establish a network of hydrogen bonds and hydrophobic interactions with neighbouring residues located on the protein surface. In the cryoEM structure of LtrA, the density of the LxxLFL loop is not well defined suggesting flexibility, and Y122 (analogous to RmInt1 L104) and F126 (analogous to RmInt1 F108) are loosely embedded in a hydrophobic cage on the protein surface. Structural conservation of the QMxxxxLxxLFL motif and its localization in a solvent accessible region in all available structures suggest that the ability to interact with DnaN may be a general feature of group II intron IEPs.
Figure 5.
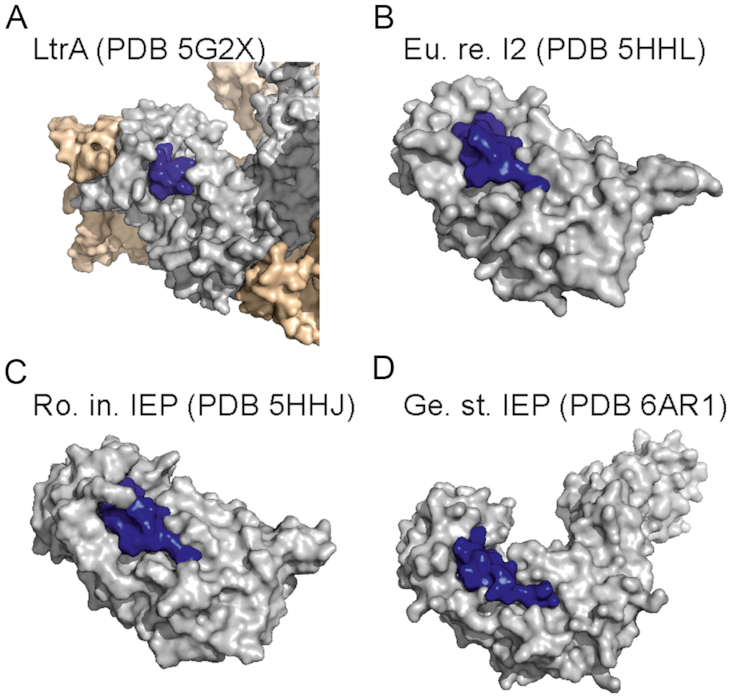
Overall structure representation of IEPs. (A) Ll.LtrB RNP. (B) Eu.re.I2. (C) Ro.in.IEP. (D) Ge.st.IEP. The surface of the IEP is depicted in grey, the QMxxxxLxxLFL motif is in blue and the intron of Ll.LtrB is in wheat colour.
Biological activities of RmInt1 IEP mutants
We assessed the importance of the interaction with DnaN for RmInt1 retrohoming, by constructing different MF-IEP mutants with the same amino-acid substitutions as were made for peptide 1-1. We analysed the retrohoming of the mutants into the lacZ gene inserted into the chromosome of S. meliloti strain RMO17 (36). Only four mutants (100, 104, 108 and 109) displayed retrohoming levels significantly lower than those of the wild-type control (Figure 6A). The 104 and 108 mutants displayed the strongest impairment, consistent with the biochemical and structural data. All the mutants produced the MF-IEP fusion protein and displayed intron RNA splicing demonstrating the production of active RNPs (Supplementary Figure S3). However, assays of RT activity revealed differences between the mutants (Figure 6B). As expected, given that the mutated amino acids belonged to the RT2 and RT2a domains, all the mutants had lower levels of RT activity than the wild-type. The greatest impairment of RT activity was observed for mutants 100, 104, 108 and 109. Despite this decrease in RT activity, mutants 100 and 109 retained significant retrohoming activity (10 and 15% wild-type levels, respectively). Interestingly, the retrohoming impairment was stronger in mutants 104 and 108 (<5% wild-type levels). One possible explanation for these results is that the lower levels of retrohoming observed for mutants 104 and 108 reflects their impaired binding to DnaN.
Figure 6.
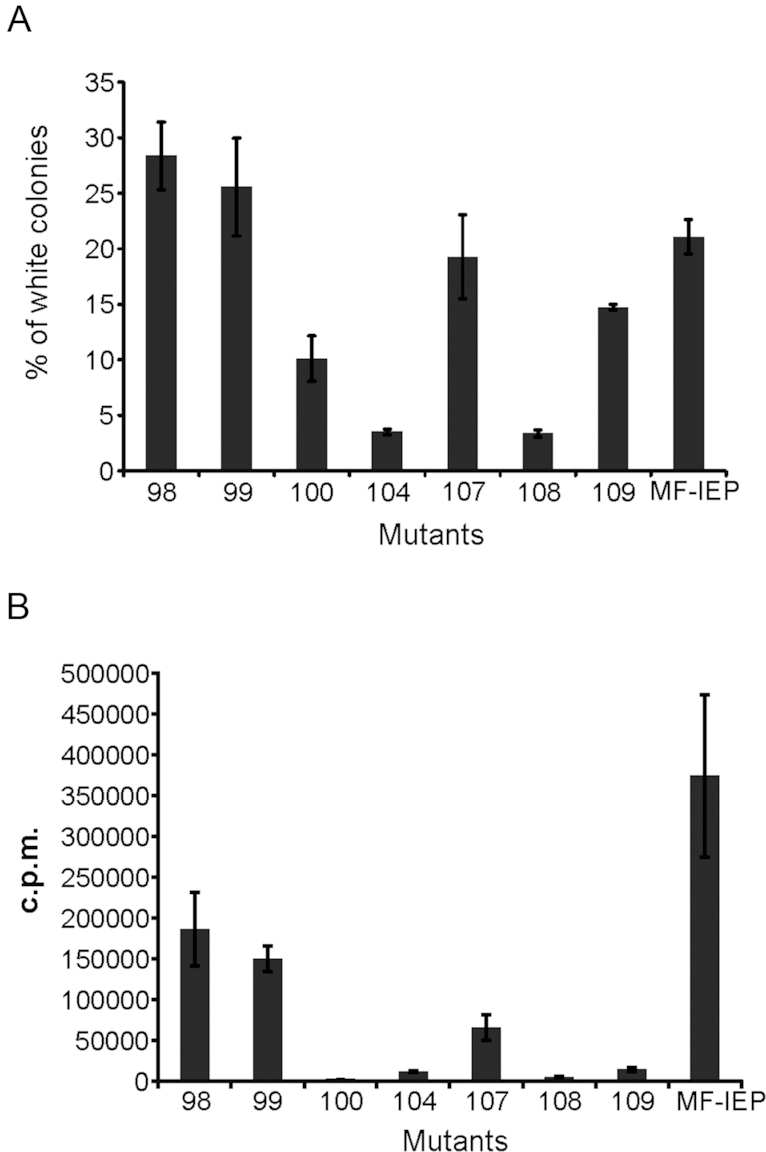
Biological activities of IEP mutants. (A) MF-IEP mutants were assayed for retrohoming into the lacZ gene inserted into the chromosome of Sinorhizobium meliloti strain RMO17. The results are expressed as the percentage of white colonies. The control is the wild-type MF-IEP with no amino-acid substitution. (B) In vitro RT activity of MF-IEP mutants.
DISCUSSION
We show here that the RmInt1 IEP interacts with DnaN (β-sliding clamp), which helps to direct intron RNPs toward the replication fork for target site selection and reverse splicing into transient single-stranded DNA.
Using the Flag epitope for immunoprecipitation, we identified a very small number of proteins that appeared to interact with RmInt1 RNPs, including the ribosomal protein S2, a component of the 30S ribosomal subunit, S-adenosylmethionine synthetase and the Hsp20 heat-shock protein. The RNA component of the RNP of the Ll.LtrB intron has been reported to interact with ribosomes via the 30S subunit (48,49), protecting the intron RNA against RNase E cleavage. The nature of the interactions of the other two proteins with the IEP remains to be clarified. Interestingly, we also found that DnaN, a component of DNA polymerase III, immunoprecipitated with the protein (IEP) component of the RmInt1 RNP. Pulldown assays, far-western blots and interferometry all revealed a direct interaction between the RmInt1 IEP and the β-sliding clamp. The interaction likely involves a motif (QMxxxxLxxLFL) more similar to that present in the TnsE domain of the Tn7 transposon than to the QLS/DLF motif present in the majority of β-sliding clamp-interacting proteins. The apparent, calculated KD (∼0.4 ∼M) was higher than the KD reported for the interaction between the δ subunit of the clamp loader complex and the β-clamp [∼0.03 μM, (50)], suggesting that the interaction with the IEP is weaker. This is not particularly surprising, because the interaction of RmInt1 RNPs with the β-clamp must not interfere with other interactions required for its normal activity. Moreover, the putative binding region of the IEP is located in the N-terminal part of the protein, whereas the binding regions of most β-sliding clamp-interacting proteins are located at the C-terminal part of the protein.
The QMxxxxLxxLFL motif is structurally conserved in group II intron En+ and En-IEPs. An analysis of available IEP 3D structures and the localization of the amino acids analogous to RmInt1 IEP L104 and F108 suggested two hypotheses for the role of these residues in regulating intron retrotransposition and, possibly, binding to DnaN. According to the first hypothesis, the analogues of L104/F108 are rigid and confer structural integrity to the QMxxxxLxxLFL motif (particularly to the LxxLFL loop portion), thereby shaping the surface of the IEP to optimize the binding and recognition of DnaN. This hypothesis is well supported by the structures of Eu.re.I2 and Ro.in.IEP, which were determined at high resolution (1.2–2.2 Å) by X-ray crystallography with a truncated IEP in the absence of the cognate intron. According to the second hypothesis, the analogues of L104/F108 are dynamic and toggle between a solvent-protected conformation (mimicked by the Eu.re.I2 and Ro.in.IEP structures) and a solvent-exposed conformation (mimicked by the LtrA structure), which brings these residues to the surface of the IEP, rendering their side-chains available for direct binding to DnaN. This hypothesis is suggested by the conformation of LxxLFL in LtrA, the structure of which was solved at medium resolution (3.8 Å) by cryoelectron microscopy with the full-length IEP bound to the cognate intron. For both these hypotheses, the deleterious effect of L104A and F108A on intron retrotransposition can be attributed to localized structural distortions of the QMxxxxLxxLFL motif caused by the mutations. The mutations are unlikely to cause the complete misfolding of the IEP, because the QMxxxxLxxLFL motif is peripheral and because we have confirmed that the mutant RNPs retain their splicing capacity.
All mutations of this binding motif decrease RT activity, but the Q98A, M99A and L107A mutants have retrohoming levels similar to that of the wild-type control, suggesting that this decrease in RT activity in vitro is overcome by RNP formation in vivo. A similar situation seems to occur for the I100A and L109A mutants, which have strongly impaired RT activity but which retain 10–15% of wild-type rehoming activity in vivo, these lower levels of retrohoming being attributable to the impairment of RT activity. Mutants 104 and 108 also displayed a strong impairment of RT activity and their retrohoming levels were the lowest observed among the mutants, possibly due to their lack of interaction with the g that the intera-clamp, the positions affected in these mutants being the most likely to play a significant role in the interaction (Figure 3C). The QMxxxxLxxLF motif thus appears to mediate the interaction of RmInt1 IEP with DnaN.
We hypothesized that RmInt1 localizes at ssDNA sites by binding to the β-clamp at the DNA replication fork, an interaction that should not interfere with other processes where DnaN is required. For example, it could take place with stalled replication forks during shifting between different host-encoded DNA polymerases, or with the DnaN that remains bound to DNA in the lagging strand behind the replication fork (Figure 7A1). We speculate that the latter situation might arise due to slow recycling after Okazaki fragment synthesis (51,52). RNP then reverse splicing into the target in its ssDNA form (Figure 7A2). The IEP then uses the nascent lagging DNA strand for reverse transcription of the intron RNA. It is possible to speculate that host DNA polymerase disengages upon encountering the highly structured intron or tightly bound IEP, leaving the nascent DNA strand in position to be used as a primer by the IEP. In this context, a host DNA polymerase would likely resume DNA synthesis after reverse transcription of the intron RNA, similarly as it occurs in a site of DNA lesion after DNA replicative polymerases disengage (53).
Figure 7.

Possible model for mobility of group II introns displaying a replication orientation bias. A1 and A2 represent the pathway used by RmInt1 (En- intron) and probably for the retrotransposition of En+ introns, which show a bias for insertion into the lagging strand template. They localize to ssDNA by interacting with g that the intera-clamp; it could take place with stalled replication forks, during the shifting between different host-encoded DNA polymerases or with the DnaN that remains bound to DNA in the lagging strand behind the replication fork (A1) and then reverse splice into its ssDNA target site (A2); DNA polymerase would disengage upon encountering the highly structured intron or tightly bound IEP, leaving the nascent DNA strand in position to be used as a primer by the IEP to reverse transcribe the inserted intron RNA. A host DNA polymerase probably resumes DNA synthesis after reverse transcription of the intron RNA. (B) It represents the pathway used by mutants of En+ introns lacking endonuclease activity and displaying an orientation replication bias toward use of the nascent leading strand DNA as a primer for reverse transcription of the inserted intron RNA. After reverse splicing into the leading strand template without cleavage of the opposite strand, the intron RNP is in a position to use the nascent leading strand from an approaching replication fork as a primer.
Introns with IEPs including an endonuclease domain, such as Ll.LtrB, retrohome via reverse splicing into double-stranded DNA with no replication orientation bias. Mutants without DNA endonuclease activity also insert into double-stranded DNA, but with a preference for insertion into the leading-strand template (54). They cannot cleave the second strand to generate a primer for reverse transcription of the inserted RNA, and, instead, probably use the nascent leading strand from an approaching replication fork as a primer for reverse transcription of the reverse-spliced intron RNA (Figure 7B). The group II intron retrotransposition (ectopic sites) pathway is mostly endonuclease-independent (55), but varies between hosts and growth conditions (56). The Ll.LtrB intron of L. lactis retrotransposes into ssDNA, using the nascent Okazaki fragment as a primer. However, in E. coli, this intron also retrotransposes into dsDNA.
The DnaN binding motif is conserved in group II intron En+ and En- IEPs. We therefore hypothesized that this interaction would be a general feature of group II introns. However, interaction with g that the intera-clamp does not necessarily impose an orientation bias for intron insertion, because there is also a g that the intera-clamp on the leading strand, but the bias toward the lagging strand template probably reflects the availability of single-stranded target sites and the preference for such targets among En- group II introns, possibly because of their limited ability to unwind double-stranded DNA. Therefore, the retrohoming and retrotransposition pathways of group II introns using lagging strand primers may be facilitated by their interaction with the replication machinery through the g that the intera-clamp component of the replisome.
Supplementary Material
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Spanish Ministerio de Economía, Industria y Competitividad including ERDF (European Regional Development Funds), Plan Nacional de I+D+i Research Grants [BIO2014-51953-P, BIO2017-82244-P to N.T.; CTQ2015-64445-R to J.L.N.]; Generalitat Valenciana [Prometeo 018/2013 to J.L.N.]; Agence Nationale de la Recherche [ANR-15-CE11-0003-01 to MM]; Agence Nationale de Recherche sur le Sida et les hépatites virales (ANRS) [ECTZ18552 to MM]; ITMO Cancer [18CN047-00 to MM]; Grenoble Instruct Center [ISBG: UMS 3518 CNRS-CEA-UJF-EMBL]; FRISBI [ANR-10-INSB-05-02]; GRAL [ANR-10-LABX-49-01]; Grenoble Partnership for Structural Biology (PSB). Funding for open access charge: Plan Nacional de I+D+i Research Grant [BIO2017-82244-P].
Conflict of interest statement. None declared.
REFERENCES
- 1. Michel F., Umesomo K., Ozeki H.. Comparative and functional anatomy of group II catalytic introns—a review. Gene. 1989; 82:5–30. [DOI] [PubMed] [Google Scholar]
- 2. Lambowitz A.M., Zimmerly S.. Mobile group II introns. Annu. Rev. Genet. 2004; 38:1–35. [DOI] [PubMed] [Google Scholar]
- 3. Toro N. Bacteria and archaea group II introns; additional mobile genetic elements in the environment. Environ. Microbiol. 2003; 5:143–151. [DOI] [PubMed] [Google Scholar]
- 4. Michel F., Ferat J.L.. Structure and activities of group II introns. Annu. Rev. Biochem. 1995; 64:435–461. [DOI] [PubMed] [Google Scholar]
- 5. Boeke J.D. The unusual phylogenetic distribution of retrotransposons: a hypothesis. Genome Res. 2003; 13:1975–1983. [DOI] [PubMed] [Google Scholar]
- 6. Pyle A.M. The tertiary structure of group II introns: implications for biological function and evolution. Crit. Rev. Biochem. Mol. Biol. 2010; 45:215–232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Cavalier-Smith T. Intron phylogeny: a new hypothesis. Trends Genet. 1991; 17:145–148. [PubMed] [Google Scholar]
- 8. Rogozin I.B., Carmel L., Csuros M., Koonin E.V.. Origin and evolution of spliceosomal introns. Biol. Direct. 2012; 7:11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Keating K.S., Toor N., Perlman P.S., Pyle A.M.. A structural analysis of the group II intron active site and implications for the spliceosome. RNA. 2010; 16:1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Lambowitz A.M., Zimmerly S.. Group II introns: mobile ribozymes that invade DNA. Cold Spring Harb. Perspect. Biol. 2011; 3:a003616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Pyle A.M. Lilley DMJ, Eckstein F. Group II introns: catalysis for splicing, genomic change and evolution. Ribozymes and RNA Catalysis. 2007; Cambridge: RSC Publishing; 201–228. [Google Scholar]
- 12. Michel F., Costa M., Westhof E.. The ribozyme core of group II introns: a structure in want of partners. Trends Biochem. Sci. 2009; 34:189–199. [DOI] [PubMed] [Google Scholar]
- 13. Blocker F.H., Mohr G., Conlan L.H., Qi L., Belfort M., Lambowitz A.M.. Domain structure and three-dimensional model of a group II intron-encoded reverse transcriptase. RNA. 2005; 11:14–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Malik H.S., Burke W.D., Eickbush T.H.. The age and evolution of non-LTR retrotransposable elements. Mol. Biol. Evol. 1999; 16:793–805. [DOI] [PubMed] [Google Scholar]
- 15. Saldanha R., Chen B., Wank H., Matsuura M., Edwards J., Lambowitz A.M.. RNA and protein catalysis in group II intron splicing and mobility reactions using purified components. Biochemistry. 1999; 38:9069–9083. [DOI] [PubMed] [Google Scholar]
- 16. Matsuura M., Noah J.W., Lambowitz A.M.. Mechanism of maturase-promoted group II intron splicing. EMBO J. 2001; 20:7259–7270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Gupta K., Contreras L.M., Smith D., Qu G., Huang T., Spruce L.A., Seeholzer S.H., Belfort M., Van Duyne G.D.. Quaternary arrangement of an active, native group II intron ribonucleoprotein complex revealed by small-angle X-ray scattering. Nucleic Acids Res. 2014; 42:5347–5360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Rambo R.P., Doudna J.A.. Assembly of an active group II intron-maturase complex by protein dimerization. Biochemistry. 2004; 43:6486–6497. [DOI] [PubMed] [Google Scholar]
- 19. Zhao C., Pyle A.M.. Crystal structures of a group II intron maturase reveal a missing link in spliceosome evolution. Nat. Struct. Mol. Biol. 2016; 23:558–565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Qu G., Kaushal P.S., Wang J., Shigematsu H., Piazza C.L., Agrawal R.K., Belfort M., Wang HW.. Structure of a group II intron in complex with its reverse transcriptase. Nat. Struct. Mol. Biol. 2016; 23:549–557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Guo H., Zimmerly S., Perlman P.S., Lambowitz A.M.. Group II intron endonucleases use both RNA and protein subunits for recognition of specific sequences in double-stranded DNA. EMBO J. 1997; 16:6835–6848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Jiménez-Zurdo J.I., García-Rodríguez F.M., Barrientos-Durán A., Toro N.. DNA target site requirements for homing in vivo of a bacterial group II intron encoding a protein lacking the DNA endonuclease domain. J. Mol. Biol. 2003; 326:413–423. [DOI] [PubMed] [Google Scholar]
- 23. Cousineau B., Smith D., Lawrence-Cavanagh S., Mueller J.E., Yang J., Mills D., Manias D., Dunny G., Lambowitz A.M., Belfort M.. Retrohoming of a bacterial group II intron: mobility via complete reverse splicing, independent of homologous DNA recombination. Cell. 1998; 94:451–462. [DOI] [PubMed] [Google Scholar]
- 24. Smith D., Zhong J., Matsuura M., Lambowitz A.M., Belfort M.. Recruitment of host functions suggests a repair pathway for late steps in group II intron retrohoming. Genes Dev. 2005; 19:2477–2487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Coros C.J., Piazza C.L., Chalamcharla V.R., Belfort M.. A mutant screen reveals RNase E as a silencer of group II intron retromobility in Escherichia coli. RNA. 2008; 14:2634–2644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Coros C.J., Piazza C.L., Chalamcharla V.R., Smith D., Belfort M.. Global regulators orchestrate group II intron retromobility. Mol. Cell. 2009; 34:250–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Yao J., Truong D.M., Lambowitz A.M.. Genetic and biochemical assays reveal a key role for replication restart proteins in group II intron retrohoming. PLoS Genet. 2013; 9:e1003469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Martínez-Abarca F., Barrientos-Durán A., Fernández-López M., Toro N.. The RmInt1 group II intron has two different retrohoming pathways for mobility using predominantly the nascent lagging strand at DNA replication forks for priming. Nucleic Acids Res. 2004; 32:2880–2888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Nisa-Martínez R., Molina-Sánchez M.D., Toro N.. Host factors influencing the retrohoming pathway of group II intron RmInt1, which has an intron-encoded protein naturally devoid of endonuclease activity. PLoS One. 2016; 11:e0162275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Lambowitz A.M., Belfort M.. Mobile bacterial group II introns at the crux of eukaryotic evolution. Microbiol. Spectrum. 2015; 3:MDNA3-0050-2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Parks A.R., Li Z., Shi Q., Owens R.M., Jin M.M., Peters J.E.. Transposition into replicating DNA occurs through interaction with the processivity factor. Cell. 2009; 138:685–695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Gómez M.J., Díaz-Maldonado H., González-Tortuero E., López de Saro F.J.. Chromosomal replication dynamics and interaction with the β sliding clamp determine orientation of bacterial transposable elements. Genome Biol. Evol. 2014; 6:727–740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Robertsen B.K., Aiman P., Darvill A.G., McNeil M., Alberstein P.. The structure of acidic extracellular polysaccharides secreted by Rhizobium leguminosarum and Rhizobium trifolii. Plant Physiol. 1981; 67:389–400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Sambrook J., Fritsch E.F., Maniatis T.. Molecular Cloning: a Laboratory Manual. 1989; 2nd ednNY: Cold Spring Harbor Laboratory Press. [Google Scholar]
- 35. Reinoso-Colacio M., García-Rodríguez F.M., García-Cañadas M., Amador-Cubero S., García Pérez J.L., Toro N.. Localization of a bacterial group II intron-encoded protein in human cells. Sci. Rep. 2015; 5:12716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. García-Rodríguez F.M., Hernández-Gutiérrez T., Díaz-Prado V., Toro N.. Use of the computer-retargeted group II intron RmInt1 of Sinorhizobium meliloti for gene targeting. RNA Biol. 2014; 11:391–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Wu Y., Li Q., Chen X.Z.. Detecting protein-protein interactions by far western blotting. Nat. Protoc. 2007; 2:3278–3284. [DOI] [PubMed] [Google Scholar]
- 38. Frenzel D., Willbolds D.. Kinetic titration series with biolayer interferometry. PLoS One. 2014; 9:e106882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Nice E.C., Catimel B.. Instrumental biosensors: new perspectives for the analysis of biomolecular interactions. Bioessays. 1999; 21:339–352. [DOI] [PubMed] [Google Scholar]
- 40. Notredame C., Higgins D.G., Heringa J.. T-Coffee: a novel method for fast and accurate multiple sequence alignment. J. Mol. Biol. 2000; 302:205–217. [DOI] [PubMed] [Google Scholar]
- 41. Marcia M., Ermler U., Peng G., Michel H.. A new structure-based classification of sulfide:quinone oxidoreductases. Proteins. 2010; 78:1073–1083. [DOI] [PubMed] [Google Scholar]
- 42. Emsley P., Cowtan K.. Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 2004; 60:2126–2132. [DOI] [PubMed] [Google Scholar]
- 43. Molina-Sánchez M.D., García-Rodríguez F.M., Toro N.. Functionality of in vitro reconstitutes group II intron RmInt1-derived ribonucleoprotein particles. Front. Mol. Biosci. 2016; 3:58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Mohr S., Ghanem E., Smith W., Sheeter D., Quin Y., King O., Polioudakis D., Iyer V.R., Hunicke-Smith S., Swamy S. et al.. Thermostable group II intron reverse transcriptase fusion proteins and their use in cDNA synthesis and next-generation RNA sequencing. RNA. 2013; 19:958–970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Dalrymple B.P., Kongsuwan K., Wijffels G., Dixon N.E., Jennings P.. A universal protein-protein interaction motif in the eubacterial DNA replication and repair systems. Proc. Natl. Acad. Sci. U.S.A. 2001; 98:11627–11632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Perkins J.R., Diboun I.D., Dessailly B.H., Lees J.G., Orengo C.. Transient protein-protein interactions: structural, functional, and network properties. Structure. 2010; 18:1233–1243. [DOI] [PubMed] [Google Scholar]
- 47. Stamos J.L., Lentzsch A.M., Lambowitz A.M.. Structure of a thermostable group II intron reverse transcriptase with template-primer and its functional and evolutionary implications. Mol. Cell. 2017; 68:926–939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Contreras L.M., Huang T., Piazza C.L., Smith D., Qu G., Gelderman G., Potraz J.P., Russell R., Belfort M.. Group II intron-ribosome association protects intron RNA from degradation. RNA. 2013; 19:1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Aseev L.V., Levandovskaya A.A., Tchufistova L.S., Skaptsova N.V., Boni I.V.. A new regulatory circuit in ribosomal protein operons: S2-mediated control of the rpsB-tsf expression in vivo. RNA. 2008; 14:1882–1894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Leu F.P., O′Donnell M.. Interplay of clamp loader subunits in opening the beta sliding clamp of Escherichia coli DNA polymerase III holoenzyme. J. Biol. Chem. 2001; 276:47185–47194. [DOI] [PubMed] [Google Scholar]
- 51. Su’etsugu M., Errington J.. The replicase sliding clamp dynamically accumulates behind progressing replication forks in Bacillus subtilis cells. Mol. Cell. 2011; 41:720–732. [DOI] [PubMed] [Google Scholar]
- 52. Moolman M.C., Krishnan S.T., Kerssemakers J.W.J., van den Berg A., Tulinski P., Depken M., Reyes-Lammothe R., Sherratt D.J., Dekker N.H.. Slow unloading leads to DNA-bound β2-sliding clamp accumulation in live Escherichia coli cells. Nat. Commun. 2014; 5:5820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Yeeles J.T.P., Poli J., Marians K.J., Pasero P.. Rescuing stalled or damaged replication forks. Cold Spring Harb. Perspect. Biol. 2013; 5:a012815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Zhong J., Lambowitz A.M.. Group II intron mobility using nascent strands at DNA replication forks to prime reverse transcription. EMBO J. 2003; 22:4555–4565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Ichiyanagi K., Beauregard A., Lawrence S., Smith D., Cousineau B., Belfort M.. Retrotransposition of the Ll.LtrB group II intron proceeds predominantly via reverse splicing intro DNA targets. Mol. Microbiol. 2002; 46:1259–1272. [DOI] [PubMed] [Google Scholar]
- 56. Coros C.J., Landthaler M., Piazza C.L., Beauregard A., Esposito D., Perutka J., Lambowitz A.M., Belfort M.. Retrotransposition strategies of the Lactococcus lactis Ll.LtrB group II intron are dictated by host identity and cellular environment. Mol. Microbiol. 2005; 56:509–524. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.