

Abstract
Objective
Heterozygous variants in KCNQ2 or, more rarely, KCNQ3 genes are responsible for early‐onset developmental/epileptic disorders characterized by heterogeneous clinical presentation and course, genetic transmission, and prognosis. While familial forms mostly include benign epilepsies with seizures starting in the neonatal or early‐infantile period, de novo variants in KCNQ2 or KCNQ3 have been described in sporadic cases of early‐onset encephalopathy (EOEE) with pharmacoresistant seizures, various age‐related pathological EEG patterns, and moderate/severe developmental impairment. All pathogenic variants in KCNQ2 or KCNQ3 occur in heterozygosity. The aim of this work was to report the clinical, molecular, and functional properties of a new KCNQ3 variant found in homozygous configuration in a 9‐year‐old girl with pharmacodependent neonatal‐onset epilepsy and non‐syndromic intellectual disability.
Methods
Exome sequencing was used for genetic investigation. KCNQ3 transcript and subunit expression in fibroblasts was analyzed with quantitative real‐time PCR and Western blotting or immunofluorescence, respectively. Whole‐cell patch‐clamp electrophysiology was used for functional characterization of mutant subunits.
Results
A novel single‐base duplication in exon 12 of KCNQ3 (NM_004519.3:c.1599dup) was found in homozygous configuration in the proband born to consanguineous healthy parents; this frameshift variant introduced a premature termination codon (PTC), thus deleting a large part of the C‐terminal region. Mutant KCNQ3 transcript and protein abundance was markedly reduced in primary fibroblasts from the proband, consistent with nonsense‐mediated mRNA decay. The variant fully abolished the ability of KCNQ3 subunits to assemble into functional homomeric or heteromeric channels with KCNQ2 subunits.
Significance
The present results indicate that a homozygous KCNQ3 loss‐of‐function variant is responsible for a severe phenotype characterized by neonatal‐onset pharmacodependent seizures, with developmental delay and intellectual disability. They also reveal difference in genetic and pathogenetic mechanisms between KCNQ2‐ and KCNQ3‐related epilepsies, a crucial observation for patients affected with EOEE and/or developmental disabilities.
Keywords: early‐onset epileptic encephalopathy, homozygous loss‐of‐function variant, intellectual disability, KCNQ3, next‐generation sequencing, nonsense‐mediated mRNA decay
Key Points.
Heterozygous variants in KCNQ2 or, more rarely, KCNQ3 genes are responsible for early‐onset developmental/epileptic disorders
We describe a patient with severe epilepsy and intellectual disability carrying a homozygous frameshift loss‐of‐function variant in KCNQ3
Familial members who are heterozygous carriers of the KCNQ3 variant are unaffected
The present results highlight difference in genetic and pathogenetic mechanisms between KCNQ2‐ and KCNQ3‐related epilepsies
1. INTRODUCTION
Next‐generation sequencing (NGS) technologies have revolutionized diagnostic procedures in developmental disorders, intellectual disability (ID), and pediatric epilepsies, allowing early identification of the molecular defects in an ever‐growing number of human disease‐causing genes.1 Early genetic diagnosis is critical for prognostic assessment, genetic counseling, and, in some cases, personalized treatment attempts.2
Pathogenic variants in KCNQ2 and KCNQ3 genes coding for Kv7.2 and Kv7.3 neuronal voltage‐gated potassium (K+) channel subunits cause early‐onset epilepsies with wide phenotypic heterogeneity.3, 4, 5 At the benign end of this spectrum is benign familial neonatal seizures (BFNS), an autosomal dominant self‐limiting neonatal epilepsy with seizures beginning in otherwise healthy infants in the first days of life and spontaneously disappearing in the following months, with mostly normal neurocognitive development and unremarkable neuroimaging.3, 6, 7 On the other hand, sporadic cases of early‐onset epileptic encephalopathy (EOEE) with cognitive disability, various age‐related pathological EEG patterns such as suppression‐burst/multifocal epileptic activity/hypsarrhythmia,8, 9 and distinct neuroradiological features have been more recently described in association to KCNQ2 variants.10, 11
When compared to KCNQ2, pathogenic variants in KCNQ3 have been more rarely described, mostly in families affected with familial forms of benign epilepsies with seizures starting in the neonatal (BFNS)6, 12, 13 or early‐infantile (benign familial infantile seizures, BFIS) period.14, 15 Fewer than twenty families with BFNS and three families with BFIS with a heterozygous KCNQ3 pathogenic variant have been reported to date.16 In addition, de novo variants in KCNQ3 have been described in few children with EOEE,17, 18, 19, 20 ID apparently without epilepsy,21, 22 and cortical visual impairment.23
Notably, all pathogenic variants in KCNQ2 and KCNQ3, except one,24 occur in heterozygosity. In the present manuscript, we report the clinical, molecular, and functional properties of a new KCNQ3 variant found in homozygous configuration in a 9‐year‐old girl with pharmacodependent neonatal‐onset epilepsy and non‐syndromic ID; the variant, a single‐base duplication in exon 12 of KCNQ3 (chr8:g.133150233dup in GRCh37; NM_004519.3:c.1599dup), is predicted to result in a frameshift p.(Phe534Ilefs*15) which could lead to either mRNA degradation by the nonsense‐mediated mRNA decay (NMD) machinery25 and/or a truncation of a large part of the C‐terminus. Ex vivo results showed that KCNQ3 transcript levels were markedly reduced in primary fibroblasts from the proband when compared to those from the unaffected noncarrier brother, and in vitro studies revealed that the variant fully abolished the ability of KCNQ3 subunits to assemble into functional homomeric or heteromeric channels with KCNQ2 subunits. The present results provide the first clinical, genetic, and functional evidence for a severe phenotype associated with a homozygous loss‐of‐function (LoF) variant in KCNQ3 and highlight previously unrecognized difference in genetic and pathogenetic mechanisms between KCNQ2‐ and KCNQ3‐related epilepsies.
2. MATERIALS AND METHODS
2.1. Informed patient consent
Written informed consent was obtained from all study participants or from parents/authorized legal guardians. The study was performed within the framework of the Genetique des Anomalies du Développement (GAD) collection performed at Equipe Inserm U1231 of the Université de Bourgogne in Dijon (FR), approved by institutional review board (no DC2011‐1332).
2.2. Exome sequencing
Singleton exome sequencing was performed using an Agilent CRE capture kit (Agilent Technologies) and a HiSeq 4000 sequencer (Illumina); exome data analysis and variant filtering were performed as previously described26 using the following annotation databases and software versions: dbsnp 149, clinvar 2016_11_03, cosmic COSMICv79, omim 2016_11_29, refseq_annotation 2016_11_27, FASTQC 2015_12_15, TRIMMOMATIC 0.35, BWA_0.7.12, PICARD_2.0.1, GATK_3.5, samtools_1.2, IGVTools_2.3.67, reference genome GRCh37/hg19, refseq 2015_07_30. Reads alignment resulted in coverage of at least 10× for 96.5% of the bases on target and an average sequencing depth of 116.01×.
2.3. Cell culture
Chinese hamster ovary (CHO) cells were grown in Dulbecco's Modified Eagle's Medium (DMEM) containing 10% fetal bovine serum (FBS), 2 mmol/L L‐glutamine, penicillin (50 U/mL), and streptomycin (50 μg/mL) in a humidified atmosphere at 37°C with 5% CO2. Primary fibroblasts were obtained from punch skin biopsies. Tissue was cut into small pieces (1 × 3 mm), and cells were cultivated in a fibroblast growth medium consisting of AmnioMax (Gibco, Thermo Fisher) supplemented with 20% FBS. For long‐term culture, fibroblasts were maintained in DMEM supplemented with 10% FBS.
2.4. Mutagenesis and heterologous expression of KCNQ2 or KCNQ3 cDNAs
The variant of interest was engineered in KCNQ3 human cDNA cloned into pcDNA3.1 (variant 1; NM_004519.3; 872 aa) by QuikChange site‐directed mutagenesis (Agilent Technologies). Channel subunits were expressed in CHO cells by transient transfection using Lipofectamine 2000 (Invitrogen);27, 28 when two or more cDNAs were cotransfected, their molar ratio was modified such that total cDNA in the transfection mixture was kept constant at 3 μg. Enhanced green fluorescent protein (1 μg; Clontech) was used as transfection marker.
2.5. Western blot experiments
KCNQ3 subunits in total protein lysates from CHO cells obtained 24 hours post‐transfection were analyzed by Western blotting on 8% SDS‐PAGE using two primary rabbit anti‐KCNQ3 polyclonal antibodies: (a) the first directed against a C‐terminal epitope (rat aa 668‐686; accession number O88944; C‐KCNQ3) (clone APC‐051, dilution 1:1000; Alomone Labs) and (b) the second raised against an N‐terminal epitope (rat aa 1‐71; N‐KCNQ3) (PA1‐930; dilution 1:1000; Thermo Scientific). Both antibodies also recognized human KCNQ3 subunits. Following exposure to primary antibodies, filters were incubated with HRP‐conjugated anti‐rabbit secondary antibodies (NA934V; dilution 1:5,000; GE Healthcare) and reactive bands visualized by chemiluminescence. Data acquisition and analysis were performed with the Gel Doc Imaging System (Bio‐Rad) and ImageLab software (version 4.1; Bio‐Rad), respectively.
2.6. RNA isolation, reverse transcription, and quantitative PCR
Isolation and purification of RNA was performed using the TriReagent (Sigma). 1 μg of total RNA was retrotranscribed with the High Capacity cDNA RT Kit (Applied Biosystem, Thermo Fisher Scientific). For quantitative PCR, cDNA was amplified with the TaqMan Gene Expression assay in a 7500 Fast Real‐Time PCR System thermocycler (Applied Biosystems, Thermo Fisher Scientific). Commercially available probes were used to amplify KCNQ1, KCNQ2, KCNQ3, KCNQ4, and KCNQ5 mRNAs (Applied Biosystem TaqMan gene expression, codes KCNQ1: hs00923522_m1; KCNQ2: hs01548339_m1; KCNQ3: hs01120412_m1; KCNQ4: hs00542548_m1; KCNQ5: hs01068536_m1). The comparative ΔΔCT method was used to quantify transcript abundance using the ubiquitin‐conjugating enzyme (UBC; hs05002522_g1) gene as control.29 Three separate experiments, each in triplicate, were performed for each probe.
2.7. Immunofluorescence
Cells were fixed with 4% paraformaldehyde in PBS for 10 minutes at room temperature (RT). After permeabilization with 0.1% Tween‐20 for 5 minutes and blocking with 0.5% BSA for 1 hour at RT, cells were incubated overnight at 4°C with the N‐KCNQ3 antibody (1:300), followed by a 1 hour incubation with donkey anti‐rabbit Cy3‐conjugated secondary antibody (Applied Biosystems, Thermo Fisher Scientific) at RT. Nuclei were visualized using Hoechst 33258 (1:5000) in PBS. Coverslips were mounted in Fluoromount G (eBioscience, Hatfield, Hertfordshire, UK); images were acquired with a Zeiss inverted LSM 700 confocal laser scanning microscope (Carl Zeiss) and analyzed with ImageJ (NIH). Slides in which the primary antibody was omitted were used as controls in all experiments.
2.8. Whole‐cell electrophysiology
Macroscopic current recordings from transiently transfected CHO cells, as well as data processing and analysis, were performed as described.27, 28 Currents from CHO cells were recorded at room temperature with the whole‐cell configuration of the patch‐clamp technique, using glass micropipettes of 3‐5 MΩ resistance. The extracellular solution contained (in mmol/L) the following: 138 NaCl, 5.4 KCl, 2 CaCl2, 1 MgCl2, 10 glucose, and 10 HEPES, pH 7.4 with NaOH. The pipette (intracellular) solution contained (in mmol/L) the following: 140 KCl, 2 MgCl2, 10 EGTA, 10 HEPES, and 5 Mg‐ATP, pH 7.3‐7.4 with KOH. The pCLAMP software (version 10.0.2) was used for data acquisition and analysis. Linear cell capacitance (C) and series‐resistance (RS) calculation were performed as described previously.30 In the experiments with tetraethylammonium, currents were activated by 3‐second voltage ramps from −80 mV to +40 mV at 0.08 Hz frequency. Fast solution exchanges (<1 second) were achieved by means of a cFlow 8 flow controller attached to a cF‐8VS eight‐valve switching apparatus (Cell MicroControls).
2.9. Statistics
Data are expressed as the mean ± SEM. Data reported in Table 1 are average values of at least 9 individual measurements, recorded in at least 3 separate experimental sessions (transfections). Statistically significant differences between the data were evaluated with Student's t test (P < .05).
Table 1.
Biophysical and pharmacological properties of currents recorded in CHO cells transfected with the indicated plasmid combinations
| cDNA transfected (µg) | n | V ½ (mV) | K (mV/efold) | Current density (pA/pF at 0 mV) | Blockade by 3 mmol/L TEA (%) | |
|---|---|---|---|---|---|---|
| Nontransfected | ‐ | 10 | ‐ | ‐ | 0.5 ± 0.1 | ‐ |
| KCNQ3 | 3 | 9 | −41.9 ± 1.5*, ** | 8.2 ± 0.9*, ** | 11.5 ± 4.8*, ** | 8.0 ± 2.1*, ** |
| KCNQ3 p.(Phe534Ilefs*15) | 3 | 9 | ‐ | ‐ | 0.3 ± 0.1 | ‐ |
| KCNQ2 + pcDNA3 | 1.5 + 1.5 | 11 | −21.7 ± 1.9 | 13.2 ± 0.8 | 21.7 ± 5.1* | ‐ |
| KCNQ2 | 3 | 13 | −23.0 ± 1.5 | 12.0 ± 0.5 | 42.2 ± 9.7 | 94.0 ± 1.0** |
| KCNQ2 + KCNQ3 | 1.5 + 1.5 | 23 | −35.1 ± 1.6 | 13.0 ± 0.7 | 117.6 ± 15.1 | 56.1 ± 6.6 |
| KCNQ2 + KCNQ3 p.(Phe534Ilefs*15) | 1.5 + 1.5 | 20 | −23.9 ± 1.9** | 15.4 ± 1.5 | 17.5 ± 2.5** | 90.0 ± 1.5** |
| KCNQ2 +KCNQ3 + pcDNA3 | 1.5 + 0.75 + 0.75 | 13 | −27.5 ± 1.0** | 13.0 ± 0.7 | 33.6 ± 6.9** | 62.0 ± 4.3* |
| KCNQ2 + KCNQ3 + KCNQ3 p.(Phe534Ilefs*15) | 1.5 + 0.75 + 0.75 | 24 | −29.5 ± 1.8** | 12.8 ± 0.7 | 39.6 ± 6.1** | 62.2 ± 3.2* |
P < 0.05 vs KCNQ2 (3 µg).
P < 0.05 vs KCNQ2 + KCNQ3 (1.5 + 1.5 µg).
3. RESULTS
3.1. Clinical features
The proband (individual II‐3; Figure 1A) is a French 9‐year‐old female originary from Morocco born to consanguineous healthy parents after an uneventful pregnancy and delivery, apart from a transient prematurity risk at 35 weeks. She was delivered at term (38 weeks 4 days), and birth parameters were normal: 3040 g weight, 46.5 cm length, and 34 cm occipito‐frontal head circumference (OCF). At the age of 2 days, she presented with both focal (affecting either the left or right hemi‐body) and generalized convulsions associated with hypotonia, cyanosis, and clonic movements of the four limbs. Biochemical and metabolic screening was noncontributive. Initial neurological examination and tonus were normal. The first electroencephalograms (EEGs), performed in the following days, revealed electrical seizures characterized by central and temporal slow waves prevailing on the right side not always associated with clinical manifestations. Biotin, pyridoxine, and folinic acid were ineffective. At the age of 2 months, convulsions were controlled with phenytoin and vigabatrin; ocular contact was poor; and the tonus was insufficient. At 7 months of age, sodium valproate monotherapy was effective in controlling seizures; a marked strabismus was noted requiring specialized management including botulinum toxin injection. Interruption of sodium valproate treatment at the age of 3‐4 years resulted in seizure recurrence during late night, including febrile episodes, with left hemispheric spikes and waves recorded on the EEG; valproate therapy was therefore reintroduced. Since then, she exhibited rare tonic‐clonic seizures, and all subsequent EEG recordings were normal, the last one at age 6 years. She is currently treated with sodium valproate with good response, and her epilepsy shows the characteristics of a pharmacodependent epilepsy.31 Brain MRI performed at day 15 revealed a suspected mild cortical dysplasia of the right frontal and temporal lobes, but a further MRI at age 6 years and 5 months was normal, as well as a brain CT scan. The proband acquired head control at age 6‐7 months, could sit unsupported at age 12 months, and walked independently around age 30‐36 months. At age 3 years, she could speak 2 words and required speech therapy, including Makaton technique while verbal language was insufficient; she attended specialized educational institution at age 5 years and started to produce short rudimentary word associations since age 6 years, with about 10 words in her vocabulary; sentences were still incompletely produced at age 8 years. At age 6.5 years, her psychomotor development was estimated around 22 months. She did not exhibit behavioral disturbances. At last examination (age 9 years), no abnormal morphological features were noted and neurological examination was normal apart from divergent strabismus increased in superior gaze. She has a moderate intellectual disability with poor vocabulary and little autonomy in daily life; she still attends a medical institute with little schooling abilities. Growth charts showed regular evolution of length, weight, and OFC between median and −1 SD. Familial history revealed that a maternal uncle who had mild cognitive disabilities with some degree of learning (reading and writing) difficulties also suffered from transient neonatal seizures, requiring a specialized pediatric follow‐up in the first 6 months of life. However, he has a milder phenotype than his niece as he could achieve a relatively good autonomy in daily life. All other members of the family had normal psychomotor and cognitive development with no history of seizures.
Figure 1.
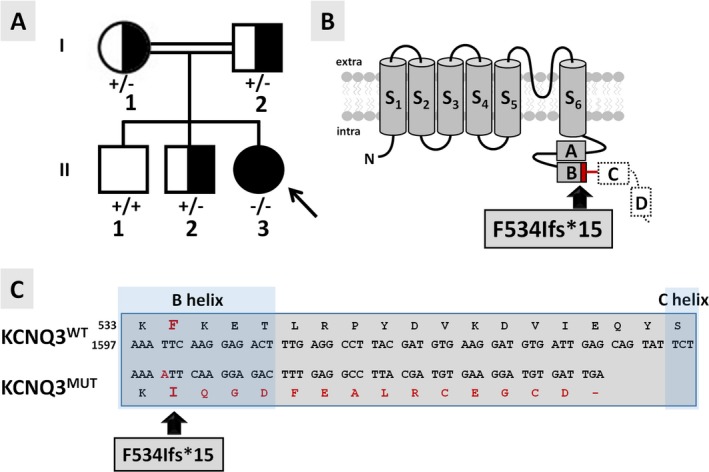
Pedigree of the investigated family and topological models of the mutant KCNQ3 subunit. A, Pedigree of the family investigated. ʻ + ʼ indicates the wild‐type KCNQ3 allele; ʻ−ʼ indicates the mutant KCNQ3 p.(Phe534Ilefs*15) allele. The arrow indicates the proband. B, Schematic topology of a KCNQ3 subunit: S1‐S6 refer to the six transmembrane segments, while boxes labeled from A to D depict the four α‐helical regions in the intracellular C‐terminus. The p.(Phe534Ilefs*15) variant located in the helix B is indicated by the arrow. The aa sequence deleted in the mutant KCNQ3 protein is indicated by a dashed line. The red line indicates the amino acid sequence altered by the frameshift variant. C, Partial alignments of the primary sequences of KCNQ3 and KCNQ3 p.(Phe534Ilefs*15, indicated as KCNQ3MUT) subunits. The B and C helices are highlighted with darker blue boxes.
3.2. Genetic data
Array‐CGH, KCNQ2‐targeted sequencing, and gene panel sequencing in the proband (individual II‐3) were normal; exome sequencing was therefore performed. The analysis on OMIM morbid genes responsible for mental retardation associated or not with epileptic encephalopathy highlighted the presence of two rare heterozygous variants, one in HERC2, associated with an autosomal recessive form of mental retardation (MIM 615516), and one in FRAS1, associated with autosomal recessive Fraser syndrome 1 (MIM 219000). Both variants were not retained as causative because the mode of inheritance was not compatible with the genotype of our patient. A variant of DYNC1H1, gene associated with Mental Retardation autosomal dominant 13 (MIM 614563), was not retained as causal as this variant has been reported 77 times in the healthy population (gnomAD 2.1.1). No further variants were retained using a sporadic mode of inheritance. The homozygosity analysis obtained through HomozygosityMapper32 using default parameters revealed relatively small regions of homozygosity in chr1, chr9, chr10, chr14, chr16, and chr22 compatible with the consanguinity between the parents (the 2 grandmothers of the proband are sisters, see expanded pedigree shown as Figure S1). Within these regions, we identified a rare variant of the DMBT1 gene at the homozygous state, not retained as causal because it was found at the homozygous state in 7 individuals in the healthy population. Outside the regions of homozygosity, the analysis on OMIM morbid genes at the recessive state retained an homozygous variant of the KCNQ3 gene absent from public databases as the only candidate compatible with the clinical presentation of the proband. This variant (ClinVar submission number SUB5837801) is a homozygous single‐base duplication (chr8:g133150233dup in GRCh37, NM 004519.3:c.1599dup) in exon 12 (135/138 reads detected the variant) which results in a shift in the open reading frame p.(Phe534Ilefs*15) and the occurrence of a premature termination codon (PTC) at position 549, possibly leading to the synthesis of a truncated protein deleted of a large part of the C‐terminal region (Figure 1B,C). The parents and one of the brothers (individuals I‐1, I‐2, and II‐2, respectively) were heterozygous carriers for the KCNQ3 variant; the eldest brother (individual II‐1) carried two copies of the wild‐type allele (Figure 1A); and the maternal uncle was unavailable for genetic analysis.
3.3. The KCNQ3 mutant allele is expressed at lower levels when compared to healthy control
Premature termination codons often result in mRNA instability and precocious degradation by nonsense‐mediated mRNA decay (NMD).25 KCNQs expression was previously reported in human fibroblasts33; therefore, the effect of the NM_004519.3:c.1599dup variant on KCNQ3 transcript levels was evaluated in primary fibroblasts from the proband (II‐3, carrying two copies of the mutant allele) and a control member of the family (II‐1, carrying two copies of the wild‐type allele), using qRT‐PCR. In control cells, the Ct values for the different KCNQs are the following: KCNQ1: 36; KCNQ2: 28; KCNQ3: 28; KCNQ4: 32; and KCNQ5: 31; all these values are considerably higher than that of UBC, the housekeeping gene used as comparator (Ct value of 25), consistent with the described low expression levels for these transcripts.33
As shown in Figure 2A, in cells from the proband, KCNQ3 transcript levels were reduced to ~22% when compared to those in control cells; a quantitatively similar decrease in transcript levels was also observed for KCNQ4, whereas those for KCNQ2 were markedly increased. No significant difference was instead observed when comparing KCNQ1 and KCNQ5 transcript abundance between proband and control primary fibroblasts.
Figure 2.
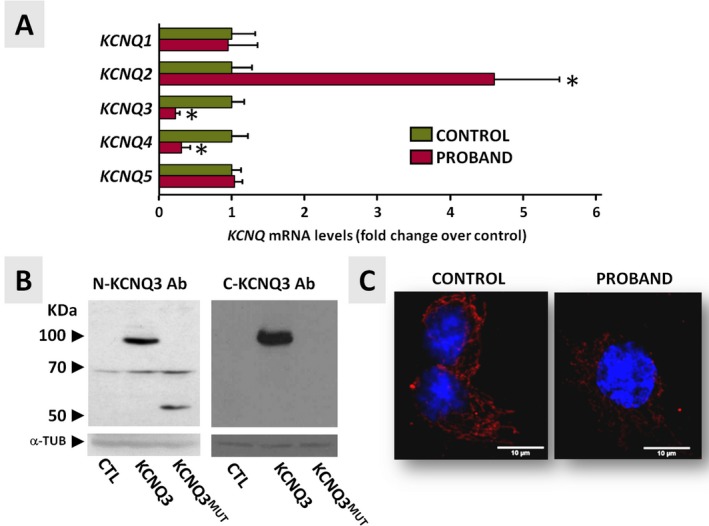
KCNQ transcript and protein expression profile in primary fibroblasts from the proband (individual II‐3) and healthy brother (individual II‐1). A, KCNQ1‐5 qRT‐PCR data from primary fibroblasts. Data are expressed as cycle threshold values for each KCNQ transcript normalized to that of UBC; after normalization, data from control fibroblasts were expressed as one (green bars), and data from proband fibroblasts were expressed relative to controls (red bars). Asterisks indicate values statistically different (P < 0.05) from respective controls. B, Western blot performed on protein lysates from transiently transfected CHO cells using N‐ and C‐KCNQ3 antibodies. CHO cells were transfected with wild‐type (KCNQ3) or mutant (KCNQ3MUT) KCNQ3 cDNA, and total lysates were analyzed with N‐KCNQ3 (left panel) or C‐KCNQ3 (right panel) antibodies. α‐tubulin (α‐TUB) served as a loading control. C, Confocal images of fibroblasts from the proband (individual II‐1) and the healthy brother (individual II‐1) stained with N‐KCNQ3 primary antibodies (in red) and a nuclear marker (Hoechst, in blue)
To investigate whether such decrease in KCNQ3 mRNA levels also led to changes in KCNQ3 protein expression, immunofluorescence experiments using N‐KCNQ3 primary antibodies were performed in primary fibroblasts from the proband and the noncarrier, unaffected brother. These antibodies were validated in Western blots on lysates from CHO cells transiently transfected with wild‐type KCNQ3 or KCNQ3 p.(Phe534Ilefs*15)‐encoding plasmids (KCNQ3MUT; Figure 2B, left panel). In these experimental groups, N‐KCNQ3 antibodies specifically recognized a ~100 kDa or a ~60 kDa protein band, respectively. These values are consistent with the molecular masses expected for wild‐type or mutant KCNQ3 proteins, respectively. Western blot experiments also revealed that, in this heterologous cellular system, the mutant protein was expressed at lower levels when compared to the wild‐type KCNQ3 protein (Figure 2B, left panel). As expected, C‐KCNQ3 antibodies targeting a C‐terminal epitope located downstream the predicted premature termination site introduced by the variant, while detecting the 100kDa band corresponding to the wild‐type KCNQ3 protein, failed to recognize any specific signal in cells transfected with the mutant plasmid (Figure 2B, right panel). Western blot experiments performed in primary fibroblasts from proband and control samples with both antibodies failed to detect specific signals corresponding to either wild‐type or mutant KCNQ3 proteins, possibly because of the low abundance of the endogenous protein in these cells (data not shown). Immunochemistry experiments using N‐KCNQ3 antibodies performed in primary fibroblasts from the proband (individual II‐3) and the wild‐type brother (individual II‐1) (Figure 2C, right and left panels, respectively) revealed a KCNQ3‐specific signal in both groups, although the intensity was lower in the former when compared to the latter. Notably, in fibroblasts from both individuals, the immunofluorescent signal was mainly cytosolic, showing a subcellular distribution consistent with an endoplasmic reticulum localization.
3.4. Functional and pharmacological characterization of homomeric and heteromeric channels carrying KCNQ3 p.(Phe534Ilefs*15) mutant subunits
Previously shown data obtained in primary fibroblasts from the proband suggest that the p.(Phe534Ilefs*15) truncating variant led to a significant decrease in KCNQ3 transcript and protein levels, a result consistent with NMD.25 However, transcript expression pattern in fibroblasts might not parallel that in neurons; moreover, a significant, although reduced in abundance, fraction of KCNQ3 protein was still detected in fibroblasts. Therefore, electrophysiological recordings in transiently transfected CHO cells were carried out to evaluate the effects prompted by the p.(Phe534Ilefs*15) variant on KCNQ3 channel function.
Heterologous expression of wild‐type KCNQ3 subunits led to the appearance of voltage‐dependent K+‐selective currents characterized by a slow time course of activation and deactivation and a threshold for current activation around −50 mV.5, 7 By contrast, no currents could be recorded in cells transfected with the KCNQ3 p.(Phe534Ilefs*15) plasmid, consistent with the variant causing a complete loss‐of‐function (LoF) effect (Figure 3A and Table 1).
Figure 3.
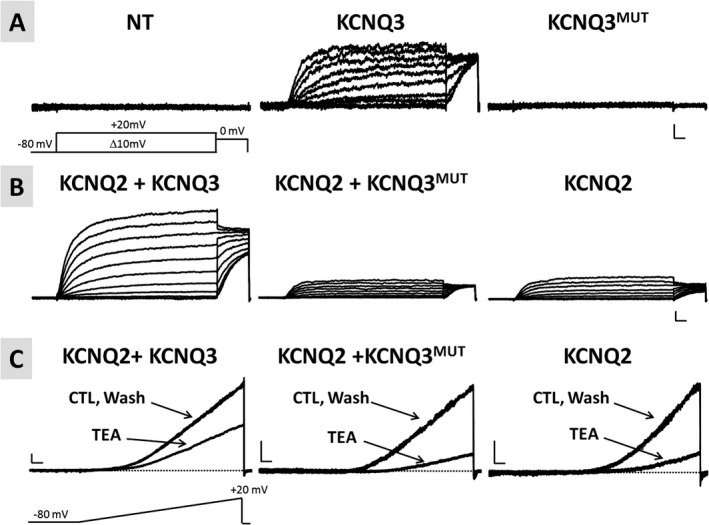
Functional characterization of homomeric or heteromeric channels incorporating KCNQ3 p.(Phe534Ilefs*15) subunits. A, Representative current traces from nontransfected cells (NT; left panel) or from cells transfected with either wild‐type KCNQ3‐ (KCNQ3; middle panel) or KCNQ3 p.(Phe534Ilefs*15)‐encoding plasmids (KCNQ3MUT; right panel) in response to the voltage protocol shown. Horizontal scale bar: 100 ms; vertical scale bar: 2 pA/pF. B, Representative current traces recorded in cells expressing the indicated subunits, in response to the same voltage protocol shown in A. Horizontal scale bar: 100 ms; vertical scale bar: 20 pA/pF. C, Representative current traces from cells expressing the indicated subunits in response to the indicated voltage ramp protocol before TEA exposure (CTL, control), during TEA exposure (TEA, 3 mmol/L), and upon drug washout (Wash). Horizontal scale bar: 200 ms; vertical scale bar: 10 pA/pF
In adult neuronal cells, KCNQ3 subunits assemble into heteromeric channels with KCNQ2 to form IKM.5 To investigate the functional consequences of mutant KCNQ3 subunits when coexpressed with KCNQ2 subunits, CHO cells were cotransfected with KCNQ2 and KCNQ3 cDNAs at a 1:1 ratio (to mimic the genetic balance of the noncarrier Individual II‐1) or with KCNQ2 and KCNQ3 p.(Phe534Ilefs*15) at 1:1 ratio (to mimic the genetic balance of the proband, Individual II‐3). Coexpression of wild‐type KCNQ3 subunits and KCNQ2 subunits markedly increased current size when compared to KCNQ2‐ or KCNQ3‐only expressing cells (Table 1); in addition, KCNQ2/3 heteromeric channels display a significant leftward shift in the activation V 1/2 and a decrease in current sensitivity to blockade by TEA (Table 1).5, 28, 34 By contrast, coexpression of KCNQ3 p.(Phe534Ilefs*15) with KCNQ2 subunits failed to enhance current density and to modify the V 1/2 (Figure 3B and Table 1). Currents recorded in KCNQ2 + KCNQ3 p.(Phe534Ilefs*15)‐transfected cells displayed a sensitivity to blockade by 3 mmol/L TEA higher than that of KCNQ2/KCNQ3‐transfected cells and identical to that of KCNQ2‐only transfected cells (Figure 3C, Table 1). These results suggest that mutant KCNQ3 subunits fail to form functional heteromeric channels with KCNQ2 subunits.
To replicate in vitro genetic combination occurring in the family members who are heterozygous carriers of the mutant allele (Individuals I‐1, I‐2, and II‐2), functional studies were also carried out upon transfection of KCNQ2, KCNQ3, and KCNQ3 p.(Phe534Ilefs*15) plasmids in a 1:0.5:0.5 cDNA ratio; for these experiments, cells transfected with an identical (1:0.5:0.5) cDNA ratio of KCNQ2 + KCNQ3+pcDNA plasmids served as controls. The results obtained suggest that the presence of an halved dose of functional/wild‐type KCNQ3 allele is sufficient to generate currents displaying the biophysical and pharmacological properties of KCNQ2/KCNQ3 heteromers, although with a reduced density when compared to that recorded in cells transfected with a full dose of KCNQ3 (1:1 KCNQ2:KCNQ3 cDNA ratio). Notably, the pharmacological and functional properties of the currents recorded in cell transfected with KCNQ2, KCNQ3, and KCNQ3 p.(Phe534Ilefs*15) plasmids (1:0.5:0.5 cDNA ratio) were identical to KCNQ2 + KCNQ3+pcDNA‐expressing cells (1:0.5:0.5 cDNA ratio) (Table 1).
4. DISCUSSION
4.1. Epilepsy and ID caused by a novel homozygous KCNQ3 frameshift variant: clinical and ex vivo results
We herein report the clinical, ex vivo, and in vitro results from a family carrying the novel frameshift p.(Phe534Ilefs*15) variant in KCNQ3 (NM_004519.3:c.1599dup). The proband is a 9‐year‐old girl diagnosed with neonatal‐onset and pharmacosensitive seizures and non‐syndromic ID; she was found to be a homozygous carrier for this variant. While our study was in progress, another family with three siblings affected with neonatal‐onset seizures (reported as pharmacosensitive in one of them) and ID of variable severity due to a different homozygous frameshift variant in KCNQ3 (c.1220_1221delCT; p.(Ser407Phefs*27)) has been described in a large cohort of children suffering from epileptic encephalopathy.24 In this family, where no functional analyses were performed, neonatal seizures or neurodevelopmental problems did not occur in consanguineous parents or extended family members who were heterozygous carriers for the KCNQ3 mutant allele. One of the affected sibling in Kothur et al.24 exhibited severe developmental delay, while the two others presented with mild ID; since genetic analysis was only limited to a restricted epileptic encephalopathy gene panel, it is unknown whether additional genetic defects, potentially enabled by consanguinity, could account for the more severe phenotype in this particular sibling.
PTC‐containing mRNAs often undergo NMD when the PTC is located upstream the ~50‐55th nucleotide before the last exon‐intron junction,35 as it occurs with the presently described variant. NMD represents a quality control mechanism to avoid production of truncated proteins with potentially deleterious effects.25, 36 KCNQ3 transcript levels in primary fibroblasts33 from the proband were markedly decreased when compared to those from the unaffected noncarrier brother II‐1, a result consistent with NMD; the reduction in KCNQ3 mutant transcript levels was accompanied by an increase in KCNQ2, a reduction of KCNQ4, and no change in KCNQ1 or KCNQ5 transcript levels. It remains to be determined whether these changes are due to compensatory effects or uncover a more complex coordination of gene expression. In this regard, whether the transcriptional repressor RE1 silencing transcription factor (REST), which has been shown to regulate KCNQ2 and KCNQ337 as well as KCNQ438 expression, participates in this coordination is currently unknown; moreover, it should be highlighted that conditional and selective ablation of Kcnq2 or Kcnq3 in cortical mouse tissue also modifies the expression of other members of the Kcnq subfamily.39 Notably, immunofluorescence experiments also revealed a reduced intensity of the KCNQ3 signal in primary fibroblasts from the proband; in these experiments, the KCNQ3 signal showed a diffuse cytoplasmic staining consistent with endoplasmic reticulum‐Golgi localization with no remarkable difference in subcellular localization between fibroblasts from the proband or the control brother. A diffuse staining pattern on both cell surface and intracellular components was also detected for KCNQ3 in rat40 and human41 hippocampal and cortical pyramidal neurons.
4.2. Functional consequences of the KCNQ3 p.(Phe534Ilefs*15) variant
Our results indicate that transcript and protein levels encoded by the mutant KCNQ3 allele are detectable, prompting investigation of the functional consequences of the p.(Phe534Ilefs*15) variant on KCNQ3 subunit function. The results obtained clearly suggest the KCNQ3 p.(Phe534Ilefs*15) protein is fully nonfunctional; indeed, homomeric expression of mutant subunits failed to generate detectable voltage‐gated K+ currents. Moreover, mutant KCNQ3 subunits did not incorporate into functional heteromeric channels when coexpressed with KCNQ2 subunits, a result consistent with the variant‐induced deletion of a significant portion of the long C‐terminus where critical domains responsible for homomeric and heteromeric subunit assembly and plasmembrane trafficking have been identified.4, 42, 43 Furthermore, the presence of an halved dose of wild‐type KCNQ3 protein, an experimental condition mimicking in vitro the genetic combination of the family members who are heterozygous carriers of the mutant KCNQ3 allele, was sufficient to generate currents displaying the biophysical and pharmacological properties of KCNQ2/Q3 heteromers, although with a reduced density when compared to that recorded in cells transfected with a full dose of KCNQ3. Currents recorded in cells transfected with KCNQ2, KCNQ3, and mutant KCNQ3 cDNAs were identical to those in cells transfected with the same cDNA amount of KCNQ2 and KCNQ3 cDNAs only, arguing in favor of the inability of the protein encoded by the mutant allele to heteromerize and interfere with the function of wild‐type subunits. This functional result strongly suggests that haploinsufficiency is the main molecular mechanism for the severe disease in the affected proband; this is in sharp contrast to KCNQ2‐related EOEE pathogenesis, where mutant subunits carrying heterozygous missense variants are functional and heteromerize with wild‐type subunits, thereby poisoning channel function by a dominant‐negative mechanism.27, 44 Additional pathogenic mechanisms for KCNQ2 EOEE include changes in subcellular localization,45 and/or in calmodulin‐46, 47 or phosphatidylinositol 4,5‐bisphosphate48‐dependent regulation.
4.3. Clinical spectrum and genetic mechanisms of KCNQ3‐related diseases
Heterozygous pathogenic variants in KCNQ3 have been associated with neonatal‐onset epilepsies showing broad clinical heterogeneity and diverse genetic transmission mechanisms.16 These range from relatively benign familial phenotypes with seizures starting in the neonatal (BFNS)6, 12, 13 or early‐infantile (BFIS)14, 15 period, to sporadic cases with severe clinical presentations characterized by developmental disabilities with or without refractory seizures17, 19, 21, 22, 49 or by cortical visual impairment.23
Notably, in both familial and sporadic cases, KCNQ3 pathogenic variants are all single missense variants, either with autosomal dominant inheritance or arising de novo, respectively (34/34; www.rikee.org).11 A notable exception is a recently described EOEE patient who carries two missense variants in compound heterozygosity, each inherited from an asymptomatic parent.20 Interestingly, no heterozygous pathogenic frameshift KCNQ3 variant has ever been associated with a human phenotype, whereas haploinsufficiency due to heterozygous frameshift variants in KCNQ2 is a frequent cause of BFNS;16, 50 indeed, frameshift/deletion variants account for 36% (39/108) of BFNS‐causing variants in the KCNQ2 gene11, 13, 51, 52 (www.rikee.org). Notably, in both families where KCNQ3 homozygous frameshift variants were identified, that is, the presently described family and the one reported by Kothur et al.24, heterozygous carriers of the KCNQ3 frameshift variant are unaffected, with no seizures or psychomotor/cognitive impairment. Instead, no homozygous frameshift variant in KCNQ2 has ever been described in humans, as minimal KCNQ2 residual activity is probably essential under penalty of potential lethality. Although several potential mechanisms may account for the more severe clinical consequences associated with KCNQ2 variants when compared to KCNQ3 ones, the fact that, in both rodents and human brains,53, 54 the ratio of KCNQ3 to KCNQ2 expression is low at birth and increases during postnatal development provides a plausible explanation; notably, deletion of Kcnq2, but not of Kcnq3, from cortical pyramidal neurons in mice is sufficient for the development of aberrant EEG activity and leads to death by the third week of life.39
Noteworthy, about 10 nonsense or frameshift variants leading to KCNQ3 truncation spread throughout the gene are reported in public databases such as ClinVar55or gnomAD.56Among these variants, only one, found in an individual in the gnomAD’s non‐neuro‐samples from individuals who were not ascertained for having a neurological condition in a neurological case/control study, was interpreted as pathogenic (although the phenotype is unknown), while the others were associated with an uncertain clinical significance.
5. CONCLUSIONS
This is the first study exploring the functional consequences of a novel KCNQ3 homozygous LoF variant responsible for a severe phenotype characterized by neonatal‐onset pharmacodependent seizures, with developmental delay and ID. Ex vivo and in vitro experiments revealed a decrease in transcript abundance proportional to variant expression levels and an impaired ability of mutant subunits to assemble into functional homomeric or heteromeric channels with KCNQ2. A lesser degree of channel dysfunction occurs when a single copy of the mutant allele is present, a result possibly contributing to the lack of neurodevelopmental phenotype in heterozygous carriers. The described LoF mechanism allows to hypothesize that, in close analogy to LoF variants found in KCNQ2‐EOEE‐affected patients,50, 57 KCNQ activators such as retigabine may be useful precision medicines to counteract the channel dysfunction triggered by the herein described novel KCNQ3 variant as well as similar variants that may be identified in the future.
CONFLICT OF INTEREST
The authors declare no competing financial interests. The Authors confirm that they have read the Journal's position on issues involved in ethical publication and affirm that this report is consistent with those guidelines.
Supporting information
ACKNOWLEDGMENTS
We are deeply indebted to: Dr. Thomas J. Jentsch, Department of Physiology and Pathology of Ion Transport, Leibniz‐Institut für Molekulare Pharmakologie (FMP), Berlin (Germany) for sharing KCNQ2 and KCNQ3 cDNAs. The present work was also supported by the following projects: the Telethon Foundation (grant number GGP15113) and the Italian Ministry for University and Research (MIUR) (PRIN 2017ALCR7C) to MT; MIUR (Scientific Independence of Researchers Project 2014 RBSI1444EM and PRIN 2017YH3SXK), PRIN (2017YH3SXK), and the University of Naples “Federico II” and Compagnia di San Paolo within the STAR Program “Sostegno Territoriale alle Attività di Ricerca” (project number 6‐CSP‐UNINA‐120) to FM; and the Italian Ministry of Health (Project GR‐2016‐02363337) and MIUR (PRIN 2017ALCR7C) to MVS. We thank the family members who participated to the study.
Lauritano A, Moutton S, Longobardi E, et al. A novel homozygous KCNQ3 loss‐of‐function variant causes non-syndromic intellectual disability and neonatal‐onset pharmacodependent epilepsy. Epilepsia Open. 2019;4:464–475. 10.1002/epi4.12353
Lauritano, Moutton, and Longobardi contributed equally.
REFERENCES
- 1. Moller RS, Dahl HA, Helbig I. The contribution of next generation sequencing to epilepsy genetics. Expert Rev Mol Diagn. 2015;15:1531–8. [DOI] [PubMed] [Google Scholar]
- 2. Demarest ST, Brooks‐Kayal A. From molecules to medicines: the dawn of targeted therapies for genetic epilepsies. Nat Rev Neurol. 2018;14:735–45. [DOI] [PubMed] [Google Scholar]
- 3. Jentsch TJ KCNQ potassium channels: physiology and role in disease. Nat Rev Neurosci. 2000;1:21–30. [DOI] [PubMed] [Google Scholar]
- 4. Soldovieri MV, Miceli F, Taglialatela M. Driving with no brakes: molecular pathophysiology of Kv7 potassium channels. Physiology (Bethesda). 2011;26:365–76. [DOI] [PubMed] [Google Scholar]
- 5. Wang HS, Pan Z, Shi W, Brown BS, Wymore RS, Cohen IS, et al. KCNQ2 and KCNQ3 potassium channel subunits: molecular correlates of the M‐channel. Science. 1998;282:1890–3. [DOI] [PubMed] [Google Scholar]
- 6. Singh NA, Westenskow P, Charlier C, Pappas C, Leslie J, Dillon J, et al. KCNQ2 and KCNQ3 potassium channel genes in benign familial neonatal convulsions: expansion of the functional and mutation spectrum. Brain. 2003;126:2726–37. [DOI] [PubMed] [Google Scholar]
- 7. Soldovieri MV, Castaldo P, Iodice L, Miceli F, Barrese V, Bellini G, et al. Decreased subunit stability as a novel mechanism for potassium current impairment by a KCNQ2 C terminus mutation causing benign familial neonatal convulsions. J Biol Chem. 2006;281:418–28. [DOI] [PubMed] [Google Scholar]
- 8. Weckhuysen S, Ivanovic V, Hendrickx R, VanCoster R, Hjalgrim H, Møller RS, et al. Extending the KCNQ2 encephalopathy spectrum: clinical and neuroimaging findings in 17 patients. Neurology. 2013;81(19):1697–703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Millichap JJ, Miceli F, De Maria M, Keator C, Joshi N, Tran B, et al. Infantile spasms and encephalopathy without preceding neonatal seizures caused by KCNQ2 R198Q, a gain‐of‐function variant. Epilepsia. 2017;58:e10–e15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Weckhuysen S, Mandelstam S, Suls A, Audenaert D, Deconinck T, Claes L, et al. KCNQ2 encephalopathy: emerging phenotype of a neonatal epileptic encephalopathy. Ann Neurol. 2012;71:15–25. [DOI] [PubMed] [Google Scholar]
- 11. Joshi N, Taglialatela M, Weckhuysen S, Nesbitt G, Cooper EC. An informatics infrastructure for KCNQ2 encephalopahty research including a patient registry, database, curation platform, and website. Annual Meeting of the American Epilepsy Society. 2016;Abstract:3.329.
- 12. Charlier C, Singh NA, Ryan SG, Lewis TB, Reus BE, Leach RJ, et al. A pore mutation in a novel KQT‐like potassium channel gene in an idiopathic epilepsy family. Nat Genet. 1998;18:53–5. [DOI] [PubMed] [Google Scholar]
- 13. Soldovieri MV, Boutry‐Kryza N, Milh M, Doummar D, Heron B, Bourel E, et al. Novel KCNQ2 and KCNQ3 mutations in a large cohort of families with benign neonatal epilepsy: first evidence for an altered channel regulation by syntaxin‐1A. Hum Mutat. 2014;35:356–67. [DOI] [PubMed] [Google Scholar]
- 14. Heron SE, Ong YS, Yendle SC, McMahon JM, Berkovic SF, Scheffer IE, et al. Mutations in PRRT2 are not a common cause of infantile epileptic encephalopathies. Epilepsia. 2013;54:e86–e89. [DOI] [PubMed] [Google Scholar]
- 15. Zara F, Specchio N, Striano P, Robbiano A, Gennaro E, Paravidino R, et al. Genetic testing in benign familial epilepsies of the first year of life: clinical and diagnostic significance. Epilepsia. 2013;54:425–36. [DOI] [PubMed] [Google Scholar]
- 16. Miceli F, Soldovieri MV, Joshi N, Weckhuysen S, Cooper EC, Taglialatela M. KCNQ3‐related disorders In: Pagon RA, Adam MP, Ardinger HH, Wallace SE, Amemiya A, Bean L, Bird TD, Ledbetter N, Mefford HC, Smith R, Stephens K. editors. GeneReviews® [Internet], Seattle (WA): University of Washington, Seattle, 2017. [Google Scholar]
- 17. Allen AS, Berkovic SF, Cossette P,Delanty N, Dlugos D, Eichler EE, et al. De novo mutations in epileptic encephalopathies. Nature. 2013;501:217–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Miceli F, Striano P, Soldovieri MV, Fontana A, Nardello R, Robbiano A, et al. A novel KCNQ3 mutation in familial epilepsy with focal seizures and intellectual disability. Epilepsia. 2015;56:e15–e20. [DOI] [PubMed] [Google Scholar]
- 19. Grozeva D, Carss K, Spasic‐Boskovic O, Tejada M‐I, Gecz J, Shaw M, et al. Targeted next‐generation sequencing analysis of 1,000 individuals with intellectual disability. Hum Mutat. 2015;36:1197–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Ambrosino P, Freri E, Castellotti B, Soldovieri MV, Mosca I, Manocchio L, et al. Compound heterozygous variants in early onset encephalopathy reveal additive contribution of C‐terminal residues to PIP2‐dependent K+ channel gating. Mol Neurobiol. 2018;55:7009–24. [DOI] [PubMed] [Google Scholar]
- 21. Rauch A, Wieczorek D, Graf E, Wieland T, Endele S, Schwarzmayr T, et al. Range of genetic mutations associated with severe non‐syndromic sporadic intellectual disability: an exome sequencing study. Lancet. 2012;380:1674–82. [DOI] [PubMed] [Google Scholar]
- 22. McRae JF, Clayton S, Fitzgerald TW, Kaplanis J, Prigmore E, Rajan D, et al. Prevalence and architecture of de novo mutations in developmental disorders. Nature. 2017;542:433–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Bosch D, Boonstra FN, de Leeuw N, Pfundt R, Nillesen WM, de Ligt J, et al. Novel genetic causes for cerebral visual impairment. Eur J Hum Genet. 2016;24:660–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kothur K, Holman K, Farnsworth E, Ho G, Lorentzos M, Troedson C, et al. Diagnostic yield of targeted massively parallel sequencing in children with epileptic encephalopathy. Seizure. 2018;59:132–40. [DOI] [PubMed] [Google Scholar]
- 25. Lykke‐Andersen S, Jensen TH. Nonsense‐mediated mRNA decay: an intricate machinery that shapes transcriptomes. Nat Rev Mol Cell Biol. 2015;16:665–77. [DOI] [PubMed] [Google Scholar]
- 26. Thevenon J, Duffourd Y, Masurel‐Paulet A, Lefebvre M, Feillet F, El Chehadeh‐Djebbar S, et al. Diagnostic odyssey in severe neurodevelopmental disorders: toward clinical whole‐exome sequencing as a first‐line diagnostic test. Clin Genet. 2016;89:700–7. [DOI] [PubMed] [Google Scholar]
- 27. Miceli F, Soldovieri MV, Ambrosino P, Barrese V, Migliore M, Cilio MR, et al. Genotype‐phenotype correlations in neonatal epilepsies caused by mutations in the voltage sensor of K(v)7.2 potassium channel subunits. Proc Natl Acad Sci USA. 2013;110:4386–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Miceli F, Soldovieri MV, Ambrosino P, De Maria M, Migliore M, Migliore R, et al. Early‐onset epileptic encephalopathy caused by gain‐of‐function mutations in the voltage sensor of Kv7.2 and Kv7.3 potassium channel subunits. J Neurosci. 2015;35:3782–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Lanzafame M, Botta E, Teson M, Fortugno P, Zambruno G, Stefanini M, et al. Reference genes for gene expression analysis in proliferating and differentiating human keratinocytes. Exp Dermatol. 2015;24:314–6. [DOI] [PubMed] [Google Scholar]
- 30. Soldovieri MV, Cilio MR, Miceli F, Bellini G, Miraglia del Giudice E, Castaldo P, et al. Atypical gating of M‐type potassium channels conferred by mutations in uncharged residues in the S4 region of KCNQ2 causing benign familial neonatal convulsions. J Neurosci. 2007;27:4919–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Rosati A, De Masi S, Guerrini R. Antiepileptic drug treatment in children with epilepsy. CNS Drugs. 2015;29:847–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Seelow D, Schuelke M, Hildebrandt F, Nürnberg P. HomozygosityMapper–an interactive approach to homozygosity mapping. Nucleic Acids Res. 2009;37:W593–599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Poulet C, Künzel S, Büttner E, Lindner D, Westermann D, Ravens U. Altered physiological functions and ion currents in atrial fibroblasts from patients with chronic atrial fibrillation. Physiol Rep. 2016;4.pii:e12681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Hadley JK, Noda M, Selyanko AA, Wood IC, Abogadie FC, Brown DA. Differential tetraethylammonium sensitivity of KCNQ1‐4 potassium channels. Br J Pharmacol. 2000;129:413–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Hwang J, Sato H, Tang Y, Matsuda D, Maquat LE. UPF1 association with the cap‐binding protein, CBP80, promotes nonsense mediated mRNA decay at two distinct steps. Mol Cell. 2010;39:396–409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Hug N, Longman D, Cáceres JF. Mechanism and regulation of the nonsense‐mediated decay pathway. Nucleic Acids Res. 2016;44:1483–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Mucha M, Ooi L, Linley JE, Mordaka P, Dalle C, Robertson B, et al. Transcriptional control of KCNQ channel genes and the regulation of neuronal excitability. J Neurosci. 2010;30:13235–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Iannotti FA, Barrese V, Formisano L, Miceli F, Taglialatela M. Specification of skeletal muscle differentiation by repressor element‐1 silencing transcription factor (REST)‐regulated Kv7.4 potassium channels. Mol Biol Cell. 2013;24:274–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Soh H, Pant R, LoTurco JJ, Tzingounis AV. Conditional deletions of epilepsy‐associated KCNQ2 and KCNQ3 channels from cerebral cortex cause differential effects on neuronal excitability. J Neurosci. 2014;34:5311–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Shah MM, Mistry M, Marsh SJ, Brown DA, Delmas P. Molecular correlates of the M‐current in cultured rat hippocampal neurons. J Physiol. 2002;544:29–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Cooper EC, Aldape KD, Abosch A, Barbaro NM, Berger MS, Peacock WS, et al. Colocalization and coassembly of two human brain M‐type potassium channel subunits that are mutated in epilepsy. Proc Natl Acad Sci USA. 2000;97:4914–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Haitin Y, Attali B. The C‐terminus of Kv7 channels: a multifunctional module. J. Physiol. 2008;586:1803–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Choveau FS, Zhang J, Bierbower SM, Sharma R, Shapiro MS. The role of the carboxyl terminus helix C‐D linker in regulating KCNQ3 K+ current amplitudes by controlling channel trafficking. PLoS ONE. 2015;10:e0145367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Orhan G, Bock M, Schepers D, Ilina EI, Reichel SN, Löffler H, et al. Dominant‐negative effects of KCNQ2 mutations are associated with epileptic encephalopathy. Ann Neurol. 2014;75:382–94. [DOI] [PubMed] [Google Scholar]
- 45. Abidi A, Devaux JJ, Molinari F, Alcaraz G, Michon FX, Sutera‐Sardo J, et al. A recurrent KCNQ2 pore mutation causing early onset epileptic encephalopathy has a moderate effect on M current but alters subcellular localization of Kv7 channels. Neurobiol Dis. 2015;80:80–92. [DOI] [PubMed] [Google Scholar]
- 46. Ambrosino P, Alaimo A, Bartollino S, Manocchio L, De Maria M, Mosca I, et al. Epilepsy‐causing mutations in Kv7.2 C‐terminus affect binding and functional modulation by calmodulin. Biochim Biophys Acta. 2015;1852:1856–66. [DOI] [PubMed] [Google Scholar]
- 47. Kim EC, Zhang J, Pang W, Wang S, Lee KY, Cavaretta JP, et al. Reduced axonal surface expression and phosphoinositide sensitivity in Kv7 channels disrupts their function to inhibit neuronal excitability in Kcnq2 epileptic encephalopathy. Neurobiol Dis. 2018;118:76–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Soldovieri MV, Ambrosino P, Mosca I, De Maria M, Moretto E, Miceli F, et al. Early‐onset epileptic encephalopathy caused by a reduced sensitivity of Kv7.2 potassium channels to phosphatidylinositol 4,5‐bisphosphate. Sci Rep. 2016;6:38167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Sands TT, Miceli F, Lesca G, Beck AE, Sadleir LG, Arrington DK, et al. Autism and developmental disability caused by KCNQ3 gain‐of‐function variants. Ann Neurol. 2019;86:181–92. [DOI] [PubMed] [Google Scholar]
- 50. Millichap JJ, Park KL, Tsuchida T, Ben‐Zeev B, Carmant L, Flamini R, et al. (2016) KCNQ2 encephalopathy: features, mutational hot spots, and ezogabine treatment of 11 patients. Neurol Genet. 2016;2:e96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Goldberg‐Stern H, Kaufmann R, Kivity S, Afawi Z, Heron SE. Novel mutation in KCNQ2 causing benign familial neonatal seizures. Pediatr Neurol. 2009;41:367–70. [DOI] [PubMed] [Google Scholar]
- 52. Grinton BE, Heron SE, Pelekanos JT, Zuberi SM, Kivity S, Afawi Z, et al. Familial neonatal seizures in 36 families: Clinical and genetic features correlate with outcome. Epilepsia. 2015;56:1071–80. [DOI] [PubMed] [Google Scholar]
- 53. Hadley JK, Passmore GM, Tatulian L, Al‐Qatari M, Ye F, Wickenden AD, et al. Stoichiometry of expressed KCNQ2/KCNQ3 potassium channels and subunit composition of native ganglionic M channels deduced from block by tetraethylammonium. J Neurosci. 2003;23:5012–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Kanaumi T, Takashima S, Iwasaki H, Itoh M, Mitsudome A, Hirose S. Developmental changes in KCNQ2 and KCNQ3 expression in human brain: possible contribution to the age‐dependent etiology of benign familial neonatal convulsions. Brain Dev. 2008;30:362–9. [DOI] [PubMed] [Google Scholar]
- 55. Landrum MJ, Lee JM, Benson M, Brown GR, Chao C, Chitipiralla S, et al. ClinVar: improving access to variant interpretations and supporting evidence. Nucleic Acids Res. 2018;46:D1062–D1067. (retrieved from https://www.ncbi.nlm.nih.gov/clinvar/?term=KCNQ3%5Bgene%5D). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Karczewski KJ, Francioli LC, Tiao G,Cummings BB, Alföldi J, Wang Q, et al. Variation across 141,456 human exomes and genomes reveals the spectrum of loss-of-function intolerance across human proteincoding genes. bioRxiv 531210; doi: 10.1101/531210. (retrieved from https://gnomad.broadinstitute.org/gene/ENSG00000184156). [DOI] [Google Scholar]
- 57. Weckhuysen S, Ivanovic V, Hendrickx R, Van Coster R, Hjalgrim H, Moller RS, et al. Extending the KCNQ2 encephalopathy spectrum: clinical and neuroimaging findings in 17 patients. Neurology. 2013;81:1697–703. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials