


Abstract
The DNA damage response network guards the stability of the genome from a plethora of exogenous and endogenous insults. An essential feature of the DNA damage response network is its capacity to tolerate DNA damage and structural impediments during DNA synthesis. This capacity, referred to as DNA damage tolerance (DDT), contributes to replication fork progression and stability in the presence of blocking structures or DNA lesions. Defective DDT can lead to a prolonged fork arrest and eventually cumulate in a fork collapse that involves the formation of DNA double strand breaks. Four principal modes of DDT have been distinguished: translesion synthesis, fork reversal, template switching and repriming. All DDT modes warrant continuation of replication through bypassing the fork stalling impediment or repriming downstream of the impediment in combination with filling of the single-stranded DNA gaps. In this way, DDT prevents secondary DNA damage and critically contributes to genome stability and cellular fitness. DDT plays a key role in mutagenesis, stem cell maintenance, ageing and the prevention of cancer. This review provides an overview of the role of DDT in these aspects.
INTRODUCTION
Prior to each cell division, DNA has to be faithfully replicated and chromosomes equally distributed over the daughter cells, as failure to maintain DNA integrity may lead to premature ageing and cancer (1). The DNA constantly suffers from lesions arising through endogenous as well as exogenous processes. Endogenous sources of DNA damage include spontaneous deamination, reactive oxygen species (ROS), S‐adenosylmethionines, aldehydes and active enzymatic DNA processes like cytosine deamination by members of the AID/APOBEC family (2,3). Examples of exogenous sources of DNA damage include UV irradiation, γ-irradiation, tobacco smoke carcinogens, alcohol and DNA damaging chemotherapeutics. Thus, our genomes are constantly exposed to DNA damage and dedicated DNA damage response pathways protect the genome from DNA damages. The DNA damage response network can be divided into DNA damage signalling, DNA damage repair and DNA damage tolerance (DDT) pathways. Specific DNA repair pathways repair defined spectra of DNA lesions. These pathways include base excision repair, nucleotide excision repair, interstrand crosslink repair, mismatch repair, ribonucleotide removal pathway, direct damage reversal and several double strand break repair pathways (1,4). Next to DNA repair, the DDT system plays a key role during DNA replication, mainly in response to DNA damage. As not all lesions are repaired prior to DNA replication, replication forks may run into these lesions. When replication forks stall, lesions cannot be repaired canonically, as the two strands are separated and hence direct access to the repair-template is lacking.
Replicative polymerases δ and ϵ (POLδ and POLϵ) perform the bulk of DNA synthesis, operate on the lagging or leading strand, respectively (5–7), and are highly specialized in warranting precise and swift DNA synthesis (8). Furthermore, replicative polymerases POLδ and POLϵ have proofread capacity to prevent mis-incorporation of nucleotides. Due to these traits, lesions in the DNA can stall replicative polymerases, as these lesions generally do not base pair well with incoming nucleotides and therefore cannot be accommodated in the catalytic site of replicative polymerases, leading to replication fork stalling. Lesions that stall replicative polymerases include abasic sites, base adducts, intrastrand crosslinks, interstrand crosslinks, cyclobutane pyrimidine dimers (CPD) and (6–4) photoproducts (6-4PP). Furthermore, replicative polymerases can be halted by structures such as DNA–protein crosslinks, incorporated ribonucleotides, secondary DNA structures as G4 structures, fragile sites and R-loops (9–13). Replicative bypass of replication fork stalling structures and lesions requires DDT pathways. DDT is defined as the restarting of replication after replication fork stalling and filling in post replicative gaps generated upon repriming 3′ of the replication impediment. When replication forks stall, helicases and replicative polymerases are uncoupled, exposing an increased amount of single-stranded DNA. Single-stranded DNA is coated with replication protein A (RPA) protein, which activates DDT to prevent prolonged stalling and consequent fork collapse (14–17). Modes of DDT are translesion synthesis (TLS), fork reversal, template switching and repriming (18–23) (Figure 1). Repriming can be followed by post-replicative gap filling by TLS or template switching. TLS involves replicative bypass directly opposite the fork stalling entity by specialized TLS polymerases. During fork reversal, the replication fork reverts to a chicken foot like structure in order to use the nascent strand of the sister chromatid as a template to bypass fork stalling lesions. Repriming involves primer formation 3′ of the fork stalling lesion, allowing a quick restart of the fork but generating a post-replicative gap. Template switching uses the nascent sister chromatid as a template in post-replicative gap filling, in order to bypass the fork stalling lesion.
Figure 1.

Modes of DNA damage tolerance. Upon stalling of the replication fork, several modes of DDT can be activated. TLS and fork reversal can occur at the stalled fork. Alternatively, TLS or template switching can occur after repriming event. Figure adapted from (23).
TRANSLESION SYNTHESIS (TLS)
TLS is performed by specialized DNA polymerases, which insert DNA opposite of a damaged template or replication impediment. These specialized TLS polymerases generally have larger active sites and lack proofread-activity, which allows non-Watson Crick base pairs to accommodate within the catalytic site (24). Both features enable TLS polymerases to tolerate lesions that stall replicative polymerases: POLδ and POLϵ. Mammalian DNA polymerases with TLS capabilities include REV1, POLH (POLη), POLI (POLι), POLK (POLκ), POLN (POLν), POLQ (POLθ) and POLζ (consisting of catalytic subunit REV3/accessory subunit REV7) and PRIMPOL (8,18–20,25). POLH, POLI, POLK and REV1 belong to the Y-family of DNA polymerases. Most TLS polymerases use the lesion as template, REV1 on the other hand is a DNA template independent dCMP-transferase that uses its own arginine residue 324 as protein template (26). The TLS polymerase complex POLζ catalytic subunit REV3 is a B-family polymerase and can extend from mismatches left by other TLS polymerases. The A-family of DNA polymerases comprising POLQ and POLN also have TLS capability, though the role of POLN in DDT remains unknown (8,25,27). POLQ plays a role in alternative-end joining, a DNA double strand break repair pathway (28,29). In addition, POLQ is also involved in TLS (30).
Regulation of TLS by PCNA monoubiquitination
Central to the regulation of DDT is proliferating cell nuclear antigen (PCNA). PCNA is a homotrimeric DNA clamp that acts as a key processivity factor for many DNA polymerases and central hub regulating a myriad of processes during DNA replication, tolerance and repair (16,31). Regulation of TLS occurs through site-specific PCNA monoubiquitination (PCNA-Ub) on lysine 164 (K164), which promotes polymerase switching from one of the replicative POLδ or POLϵ to a TLS polymerase (Figure 2) (32–34). PCNA is monoubiquitinated by the E2/E3 ubiquitin ligase complex RAD6/RAD18. Other E3 ligases targeting PCNA K164 have been revealed, such as CRL4Cdt2 and RNF8 (35–38). Additionally, spliceosome-associated factor 3 (SART3) has also been reported to affect PCNA monoubiquitination and POLH recruitment into foci (39). Furthermore, PCNA-associated factor 15 (PAF15) is double monoubiquitinated and regulates polymerase switching through its interaction with PCNA (40,41). PAF15 is deubiquitinated upon fork stalling, this releases PCNA and facilitates interaction with TLS polymerases. Paf15 depletion through shRNA-mediated knockdown leads to increased mutagenesis upon UV exposure, likely due to an increased error-prone TLS activity. In vitro analysis using purified PCNA, POLH and PAF15 revealed an inhibition of POLH TLS when PAF15 is present (42).
Figure 2.

Translesion synthesis. TLS pathways are regulated by PCNA K164 ubiquitination or REV1. PCNA Ub can also interact with REV1 directly, as with other TLS polymerases. PAF15 is degraded upon fork stalling, which allows PCNA to interact with TLS polymerases. RPA recruits RAD6/RAD18 ligase that can ubiquitinate PCNA to recruit TLS polymerases. SPRTN binds PCNA Ub preventing PCNA deubiquitylation. After TLS is completed, the TLS polymerase replaced again by the replicative polymerase. In the REV1-dependent pathway, REV1 can recruit other TLS polymerases. Figure adapted from (23).
E3 ubiquitin ligase HECT, UBA And WWE Domain Containing 1 (HUWE1) has been shown to interact with PCNA, which is important for suppressing replication stress, though the exact mechanism remains unclear (43). FANCD2-associated nuclease 1 (FAN1) has an PCNA interaction peptide (PIP) through which it binds to PCNA-Ub (44). The FAN1 PIP is required for FAN1 foci formation and prevents fork collapse upon replication fork stalling induced by G4 stabilizing compounds. This function of FAN1 is likely independent of its function in interstrand crosslink repair, as FAN1 foci still form upon interstrand crosslink inducer MMC in cells expressing FAN1 PIP mutant.
Factors that regulate PCNA and RAD18 interaction includes BCRA1. BRCA1 is a key factor in double strand break and interstrand crosslink repair, though also regulates RAD18 (45). BRCA1 deficiency impairs RAD18 recruitment and PCNA K164 ubiquitination. Next to BRCA1, SIVA1 apoptosis inducing factor (SIVA1) was found to direct RAD6/RAD18 to ubiquitinate PCNA (46). In addition, SprT-like domain-containing protein SPRTN is involved in a feed forward loop with PCNA-Ub, as SPRTN is recruited by PCNA-Ub, and also involved in recruitment of RAD18 and TLS polymerases that requires the DNA-binding domain of SPRTN (47–52). Whether this relates to a direct effect on recruitment of TLS polymerases or through stabilization of PCNA-Ub remains to be established. Additionally, NIBRIN (NBN) that is mutated in Nijmegen breakage syndrome also affects PCNA-Ub formation through interaction with RAD18. Knockdown of NBN leads to decreased number of POLH foci, UV sensitivity and increased mutagenesis (53).
Deubiquitinase USP1 is the most investigated deubiquitinating enzyme of PCNA-Ub, surprisingly USP1 is degraded through autocleavage upon DNA damage induction by UV, but not mitomycin C (MMC), methyl methanesulfonate (MMS) or hydroxylurea (HU) (54,55). The degradation of USP1 upon UV is counterintuitive, considering that the PCNA-Ub trimer should be deubiquitinated to dissociate from the TLS polymerase once the TLS step is completed. This suggests that USP1 activity keeps PCNA deubiquitinated under DNA damage free conditions and that there is yet another USP responsible in PCNA K164 deubiquitination once TLS is completed. USP1 has been reported to interact with PCNA unloading factor ATPase family AAA domain containing 5 (ATAD5) and this interaction was considered important in the deubiquitination of PCNA-Ub (56,57). The release of TLS polymerase from PCNA-Ub is complex. PCNA-Ub can be modified by ubiquitin-like modification E3 ubiquitin/ISG15 ligase TRIM25/EFP. TRIM25/EFP ISGylates PCNA-Ub on two lysine residues, one of which is K168 (58). ISGylation of the two lysine residues was demonstrated to depend on PCNA K168. ISGylated PCNA (PCNA-ISG) in turn binds USP10, which deubiquitinates PCNA-Ub leading to the release of POLH. PCNA-ISG is then deISGylated by UBP43, enabling the switch back to a replicative polymerase. Apparently, ISGylation signals the release of TLS polymerases from PCNA.
Regulation of TLS by REV1
Next to PCNA-Ub, TLS polymerase REV1 can recruit other TLS polymerases independently of PCNA-Ub, through its C-terminal domain (59–62) (Figure 2). REV1 is ubiquitinated and interacts with Fanconi anemia associated protein 20 (FAAP20), a subunit of the Fanconi core complex, in UV-induced REV1 foci (63). The REV1 FAAP20 complex is also associated with PCNA at the replication fork, though it is not clear whether this process is PCNA K164 ubiquitination dependent. Interestingly, the knockdown of Polh in mouse embryonic fibroblasts (MEFs) also leads to a reduction of UV-induced REV1 foci, suggesting that also POLH has a recruiting function (64). The genetic characterization of Rev1 and Pcna K164 has shown that REV1 and PCNA K164 are mainly non-epistatic (65,66), indicating that there is a PCNA-K164-dependent DDT pathway and a REV1-dependent DDT pathway. Although REV1 functions independently of PCNA-Ub, it also operates in a PCNA-Ub-dependent manner as seen with increased binding efficiency with PCNA-UB over PCNA (65). Furthermore, the presence of REV1 increases PCNA-Ub levels, likely by stabilizing the RAD18 interaction with PCNA (67).
In summary, the recruitment and exclusion of potentially error-prone TLS polymerases are tightly regulated by a myriad of protein interactions and post-translational modifications. Furthermore, PCNA and REV1 seem to exert mainly non-epistatic functions regarding the regulation of DDT.
HOMOLOGY-DIRECTED DNA DAMAGE TOLERANCE
During homology-directed DDT, the newly synthesized DNA strand of the sister chromatid is used as template in order to bypass the fork stalling entity either at the ongoing replication fork or in a post-replicative gap setting. Two pathways are thought to perform homology-directed DDT, specifically fork reversal and template switching (21,67,68) (Figure 3). In mammals, fork reversal seems the dominant homology-directed pathway, whereas in yeast template switching is more dominant (69). Compared to TLS, both homology-directed DDT pathways are considered relatively error-free (70). Both homology-directed DDT pathways depend on the formation of PCNA polyubiquitination (PCNA-Ubn), where one of the Rad5 homologues SNF2 histone linker PHD RING helicase (SHPRH) or helicase-like transcription factor (HLTF) extends a Ub-K63 linked polyubiquitin moiety to PCNA-Ub. The observation that PCNA-Ubn is still formed in the absence of both SHPRH and HLTF implies the existence of at least one additional ubiquitin ligase generating PCNA-Ubn (71). For a more detailed insight into the molecular mechanisms of template switching and fork reversal, see these reviews (21,72). In mouse and human cells, the frequency of PCNA-Ubn, the main indicator of homology-directed DDT, appears quite rare as suggested by steady state levels of PCNA-Ub versus PCNA-Ubn (34,35,71,73,74). In both homology-directed DDT mechanisms, the fork stalling entity can be avoided by using the newly synthesized template of the other DNA strand.
Figure 3.
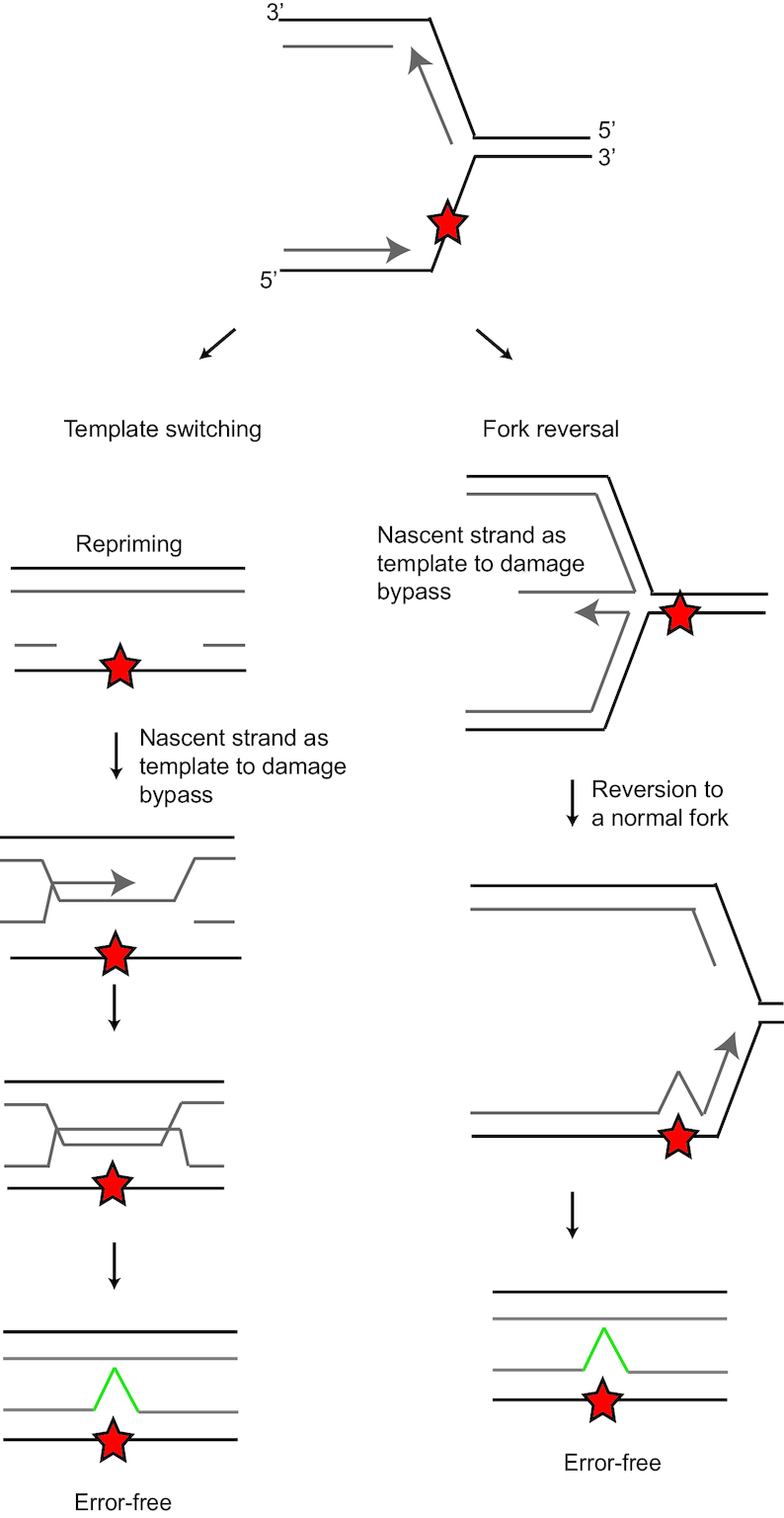
Homology-directed DNA damage tolerance. Homology-directed DDT involves template switching and fork reversal. In order to prevent use of the fork stalling entity as template, the nascent DNA of the sister chromatid is used. Template switching occurs after a priming event in a post replicative gap. After fork stalling, the replication fork can reverse. Fork reversal leads to a chicken foot intermediate structure, which allows access to the nascent DNA of the sister chromatid.
Fork reversal
During fork reversal, the replication fork regresses from a fork into a chicken foot-like DNA structure (72) (Figure 3). In the chicken foot structure, the replication machinery has the opportunity to use the newly synthesized strand of the intact sister chromatid as alternative to the damaged template. Though it has not been proven directly that fork reversal promotes damage bypass, it is an attractive model for the function of the fork reversal as fork reversal gives nascent DNA of the stalled strand direct access to the nascent homologues undamaged template. Alternatively, fork reversal may occur to stabilize the stalled fork (22). PCNA-Ubn formation facilitates fork reversal and E2 ubiquitin ligase UBC13 is needed to generate a K63-linked polyubiquitin moiety at PCNA K164 (69). Translocase zinc finger RANBP2-type containing 3 (ZRANB3) binds PCNA-Ubn and remodels the replication fork to a reversed fork and is involved in D-loop dissociation (69,74–76). Another translocase, SWI/SNF related, matrix associated, actin dependent regulator of chromatin, subfamily a like 1 (SMARCAL1) is also involved in promoting fork reversal (75,77). SMARCAL1 can evict RPA from the DNA and reanneal complementary single-stranded DNAs (78–80). In this manner, SMARCAL1 is involved in reversing stalled replication forks. Furthermore, SMARCAL1 phosphorylation by ATR prevents nuclease-mediated fork collapse (81). The phosphorylation of SMARCAL1 by ATR limits fork regression by SMARLCAL1. Other factors that are involved in fork reversal are the homologous recombination proteins BRCA2 and RAD51. These factors protect the fork form Meiotic Recombination 11 Homologue 1 (MRE-11) exonuclease activity (82–84). When forks are reversed, these structures need to be protected from MRE-11 exonuclease activity. Without protection of the reversed fork, MRE-11 degrades the newly synthesized DNA of the leading and lagging strand from 3′ to 5′ and 5′ to 3′, respectively (85). As in vitro MRE-11 only has a 3′–5′ resection activity, the 5′ to 3′ degradation of the lagging strand may involve another exonuclease whose activity depends on MRE-11 (86). In addition, RecQ like helicase (RECQL), DNA replication helicase/nuclease 2 (DNA2) and Werner syndrome RecQ like helicase (WRN) helicases are involved in promoting replication fork restart during fork reversal (22). Another report in human cells suggest that RECQL limits DNA2- and WRN-mediated degradation of the reversed fork to create the 3′ overhang in the reversed fork (87). Furthermore, PARP1 is involved in promoting fork reversal, as shown in topoisomerase inhibitor induced fork reversal (88). In addition, helicase FANCM has been implicated in fork reversal in promoting branch migration, as shown using purified FANCM (89,90). This observation has been corroborated independently in fission yeast, where FANCM orthologue Fml1 promotes fork reversal (91).
Template switching
Template switching occurs in post replicative gap filling and serves to close the single-stranded gap containing the lesion in an error-free manner (21) (Figure 3). Template switching involves formation of structures resembling double holiday junctions (92). In Saccharomyces cerevisiae, it has been demonstrated that the cohesion/condensing-like Smc5/6 complex stimulates template switching. Furthermore, template switching is needed to replicate through natural pausing sites (93). In the same model system, the helicase Pif1 was described to be key in template switching through enlarging single-stranded DNA in post-replicative gaps (94). The combined action of the E3 ubiquitin ligases Rad18- and Rad5 generates PCNA-Ubn, which is necessary for template switching. Next to PCNA-Ubn, PCNA SUMOylation mediated by Mms2 and Ubc13 has been shown to promote template switching in S. cerevisiae (95). Furthermore, PCNA SUMOylation prevents the formation of DNA double strand break formation and recombination upon fork stalling (96). Human PCNA is SUMOylated on several residues, including K164. K164 SUMOylation is not site-specific, as K164R mutation does not abrogate PCNA SUMOylation, although the level of SUMOylation was found to decrease. Rad18-deficient cells are used as PCNA-Ub free system, though there are alternative E3 ligases in higher eukaryotes that can ubiquitinate PCNA. Therefore, it remains unclear whether the PCNA K164R mutation leads to SUMOylation related defect in the human system. In line with preventing double strand break formation, PCNA SUMOylation prevents spontaneous gene conversion (97).
RESTARTING DNA SYNTHESIS BY REPRIMING
In order to start DNA synthesis, a primase is required that generates a primer for a DNA polymerase. On the lagging strand this is warranted by continuous priming by POLα. Therefore, a stalled fork does not necessarily cause a stop of replication on the lagging strand (98). On the leading strand, replication is usually continuous and hence repetitive priming by POLα is not required. Therefore, on the leading strand a stalled fork might have more impact on the progression of replication. However, also on the leading strand repriming occurs (99,100). In mammals, this task appears primarily executed by the primase and polymerase PRIMPOL (18,20,101). In contrast to POLα that produces an RNA primer and subsequently extends the primer with DNA, PRIMPOL uses mainly dNTPs for priming (20). PRIMPOL has both primase and TLS polymerase activity, though the primase activity seems to be more important for fork progression and genome stability (102,103). PRIMPOL reprimes after lesions to rapidly restart replication (18,20,101). PRIMPOL binds to single-stranded DNA–RPA filaments. At high concentrations of RPA, RPA inhibits PRIMPOL activation (Figure 4). However, at non-saturating RPA conditions, single-stranded RPA–DNA filaments do activate PRIMPOL (104,105). This model fits with the observation that Primpol-defective MEFs do not have a replicative defect upon low dose UV-C exposure, though at a higher dose they were found impaired (106). Furthermore, free RPA becomes rate limiting in high levels of DNA damage or replication fork stalling (107). These data suggest that the concentration of unbound RPA regulates PRIMPOL activation. In chicken DT40 lymphoma cells, loss of PRIMPOL leads to epigenetic instability especially at G4 stacks on the leading strand (108). The epigenetic instability refers to loss of histone marks during replication. Under steady state conditions, histone marks are the same on the DNA before and after replication of a locus as many histones are recycled and distributed over the sister chromatids, but under certain replication stresses the histone marks can be instable possibly due to defective histone recycling (109). Using the same system, also REV1 was shown to exert the same effect on the epigenome (10). In this earlier article, one of the conclusions was that G4s on the lagging strand do not affect epigenetic stability. The authors suggest that this may be due to continuous repriming. Thus, G4-induced epigenetic instability in this setting cannot be used for restriction of activity to the leading strand, due to the absence of an effect on the lagging strand. The only evidence that PRIMPOL selectively targets the leading strand is based on the analyses of the different base-to-base substitutions during somatic hypermutation of Igh genes in PrimPol-deficient B cells, where a strand-biased anti-mutagenic effect of PRIMPOL on the leading strand was observed (106,110). This anti-mutagenic effect likely relates to a preferential PRIMPOL activity on the leading strand, promoting an error-free DDT pathway such as template switching. Additional evidence for this hypothesis awaits further experimentation. At present, it remains unclear how the system targets PRIMPOL preferentially to the leading strand.
Figure 4.
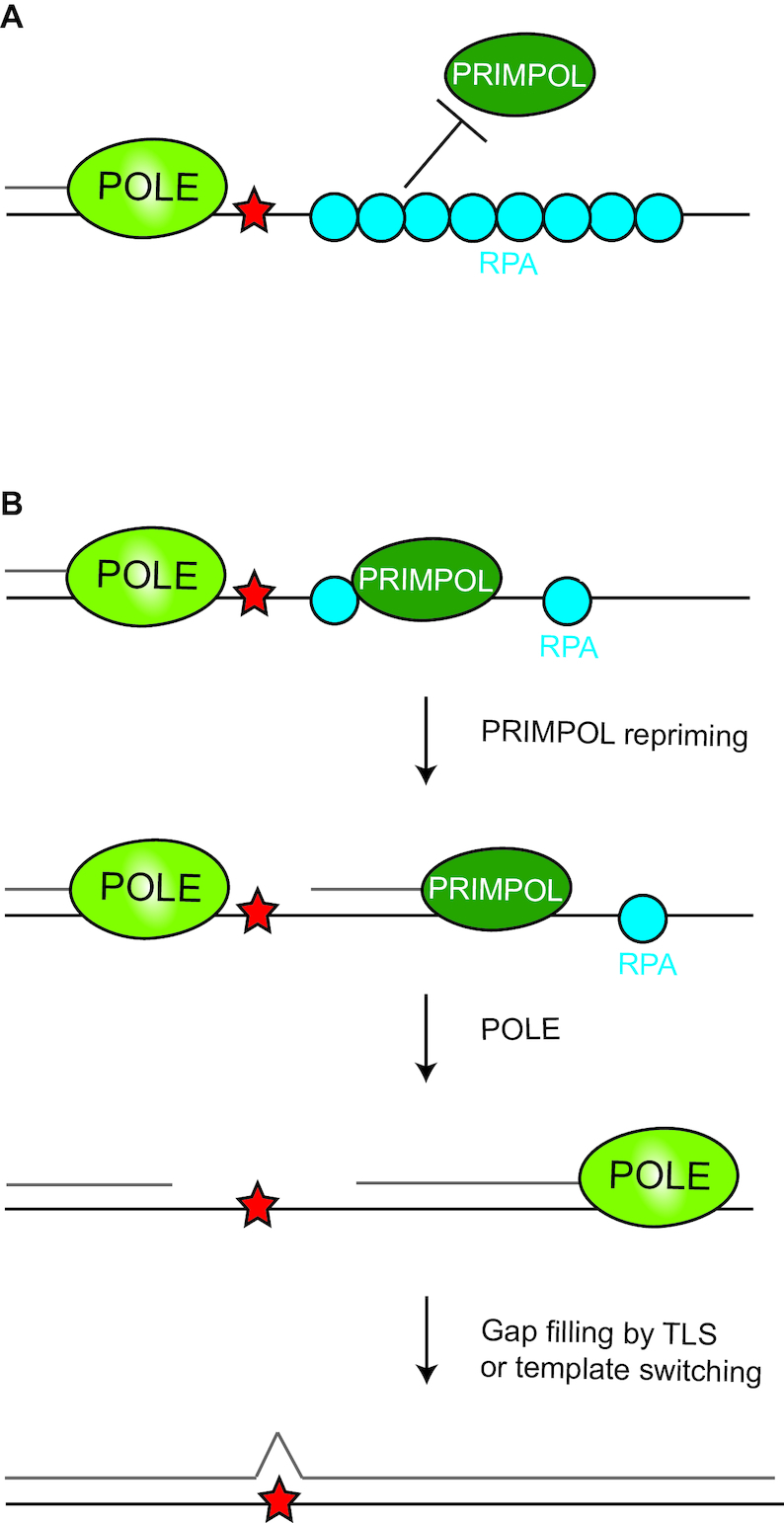
Regulation of PRIMPOL by RPA. (A) PRIMPOL is inhibited from access to single-stranded DNA by high concentration of RPA on DNA. (B) When the amount of RPA coating single-stranded DNA is low, PRIMPOL can bind single-stranded DNA and form a primer for a replicative polymerase to continue.
TRANSLESION SYNTHESIS DURING INTERSTRAND CROSSLINK REPAIR
Fanconi anemia is a disease characterized by reduced fertility, bone marrow failure induced anemia, and increased risk of acute myeloid leukemia and solid tumours (111). Furthermore, patients show increased chromosomal instability and sensitivity to interstrand crosslink inducing agents such as mitomycin C and cisplatin. This is due to disease-causing mutations found in genes associated with interstrand crosslink repair. The Fanconi anemia pathway is involved in interstrand crosslink repair. Interstrand crosslinks are covalently linked bases on opposite strands of the Watson/Crick double helix, and prohibit the melting of two DNA strands necessary for central biological processes, such as transcription and replication. Consequently, DNA interstrand crosslinks are highly toxic lesions (112). Interstrand crosslink repair appears one of the molecularly most challenging and intricate DNA repair processes. Most relevant in the context of this review, interstrand crosslink repair depends on TLS. During S phase, as demonstrated in Xenopus nuclear egg extracts, interstrand crosslinks require two converged and stalled replisomes for replication-coupled repair (113). Upon stalling of the replication fork, FANCD2 and FANCI are monoubiquitinated by the Fanconi anemia core complex. The monoubiquitination of FANCD2 is necessary for recruitment of nucleases that then make the incisions and unhook the interstrand crosslink (114) (Figure 5A). After unhooking of the crosslink, TLS is required to continue replication opposite the non-instructive, unhooked interstrand crosslink. REV1 and POLζ have been implicated in this step (115). At present, the role of PCNA-Ub is ill defined, as studies in Xenopus egg extracts could not adequately deplete PCNA to determine the reliance of interstrand crosslink repair on PCNA K164 (115). Resolving the molecular details of interstrand crosslink repair greatly benefitted from studies in nuclear Xenopus egg extracts using cisplatin-induced interstrand crosslinked plasmids.
Figure 5.

Translesion synthesis during interstrand crosslink repair. Interstrand crosslinks can be induced by cisplatin, mitomycin C, psoralen and abasic sites. Cisplatin and mitomycin C are repaired by the Fanconi anemia pathway (A), whereas psoralen and abasic site induced ICL are repaired through NEIL3-dependent pathway (B andC). Figure adapted from (23).
Of note, many mammalian studies frequently used psoralen, as they are known to induce specifically interstrand crosslinks. However, as shown in Xenopus egg extracts, these appeared to be repaired by an alternative interstrand crosslink repair pathway involving the DNA glycosylase NEIL3. NEIL3 has also been shown to repair abasic site induced interstrand crosslinks in Xenopus. Both psoralen and abasic site induced interstrand crosslinks do depend on REV1 for TLS (116) (Figure 5B and C). In the case of psoralen-induced interstrand crosslinks, NEIL3 cleavage leaves an abasic site on one strand and an unhooked ICL on the other. Subsequent TLS on both sister chromatids requires REV1. For abasic site induced interstrand crosslinks, however, NEIL3 cleavage leaves one strand intact while the other contains the abasic site. Apparently, the bypass of the abasic site involves REV1 in Xenopus.
There is also an alternative model to the converging fork model. However, in this study with mammalian cells, psoralens are used and might be repaired more by the NEIL3 pathway rather than the Fanconi pathway. The authors reveal FANCM is involved in traversing the replication fork over the interstrand crosslink (117). In the model proposed by the authors, the replication fork stalls at the interstrand crosslink and FANCM is needed to restart the replication fork on the opposite side to the interstrand crosslink. Further investigation revealed that the interaction between FANCM and PCNA is necessary for traversing of the replication fork (118). When the replication fork traverses the interstrand crosslink, the replication fork can continue. Therefore, interstrand crosslink traversing can be seen as a mode of DDT.
DNA DAMAGE TOLERANCE DEFECTS AND DISEASES
Defects in genes involved in DDT are associated with several diseases. Similar to DNA damage repair associated diseases, symptoms include cancer susceptibility, neurological disorders, stem cell defects and progeria. The following sections address these genes and their associated genetic defects in an alphabetical order (Table 1).
Table 1.
Genetic defects of genes implicated in DNA damage tolerance and their associated diseases
| Gene | Disease | Citation |
|---|---|---|
| ATAD5 | Ovarian cancer | (119) |
| ATR | Cutaneous telangiectasia and cancer syndrome, familial, Seckel syndrome | (123,124) |
| BRCA1/FANCS | Familial breast-ovarian cancer susceptibility, Fanconi anemia (complementation Group S) and prostate cancer | (127,128) |
| BRCA2/FANCD1 | Familial breast-ovarian cancer, Fanconi anemia (complementation group D1), Wilms tumour and other cancers | (130,131) |
| DNA2 | Seckel syndrome and progressive external ophthalmoplegia | (134,135) |
| HUWE1 | X-linked syndromic mental retardation turner type and colon cancer | (129–137) |
| PRIMPOL/CCDC111 | Myopia | (142) |
| RAD51/FANCR | Fanconi anemia, complementation group R, mirror movements and breast cancer | (133–145) |
| REV3 | Möbius syndrome | (149) |
| REV7/MAD2L2/FANCV | Fanconi anemia | (151) |
| SMARCAL1 | Schimke immunoosseous dysplasia | (154) |
| SPRTN | Ruijs-Aalf syndrome, hepatocellular carcinoma | (161) |
| POLH | Xeroderma Pigmentosum Variant | (167) |
| UBE2A/RAD6A | X-linked syndromic mental retardation, Nascimento type | (176) |
| UBE2B/RAD6B | Male infertility | (179) |
| WRN | Werner syndrome | (184,185) |
ATAD5—Ovarian cancer
ATAD5 has been implicated in unloading of the DNA clamp PCNA and is also involved in the deubiquitination of PCNA-Ub through interaction with USP1 (56). In humans, ATAD5 mutations have been associated as a risk factor for ovarian cancer (119). In line, imice with a heterozygous Atad5 defect tumours of diverse origins arise (120). As the genome instability observed in Atad5-deficient cells is at least partially linked to the unloading of PCNA from chromosomes, it appears that this defect is responsible for the increased genome instability and tumorigenesis (121).
ATR—Cutaneous telangiectasia and cancer syndrome, familial, Seckel syndrome
ATR is a key kinase in the DNA damage response involved in fork stability and the management of replication stress (122). In humans, ATR mutations have been associated with familial cutaneous telangiectasia and cancer syndrome and Seckel syndrome (123,124). Seckel syndrome is characterized by short stature, intellectual disability, premature ageing and many more diverse symptoms between patients. One mouse model with an Atr mutation also shows Seckel syndrome (125). These homozygous Atr impaired mice show also increased dwarfism, increased replication stress and accelerated ageing, but do not show increased tumorigenesis. All these symptoms are in common with the human symptoms of ataxia telangiectasia, with the exception of tumour predisposition. It remains unknown to what extend the fork stabilizing function of ATR contributes in preventing Seckel syndrome and cancer susceptibility.
BRCA1/FANCS—Familial breast-ovarian cancer susceptibility, Fanconi anemia (complementation group S) and prostate cancer
BRCA1 is involved in genome maintenance through its function in homologous recombination, interstrand crosslink repair and fork stability (72,111,126). Mutations in human BRCA1 contribute to familial breast and ovarian cancer, Fanconi anemia and other cancer types such as prostate cancer (127,128). Mouse models of Brca1 deficiency are embryonic lethal. But mammary gland specific knockouts and hypo-morphic non-lethal mutations show increased incidence in developing mammary tumours, in line with human phenotypes (129). It remains unknown whether and to what extent the fork stabilization of BRCA1 contributes to cancer formation.
BRCA2/FANCD1—Familial breast-ovarian cancer, Fanconi anemia (complementation group D1), Wilms tumour and other cancers
Similar to BRCA1, BRCA2 is also involved in homologous recombination, interstrand crosslink repair and fork reversal (72,111,126). In line, human BRCA2 deficiency contributes to familial breast and ovarian cancer and Fanconi anemia (130,131). Mice deficient for Brca2 are also embryonic lethal. Similar to the human pathology, conditional Brca2 alleles show increased mammary tumour incidence in mice. Also, Fanconi anemia phenotypes are reconstituted in Brca1 mutant mice, as hematopoietic stem cell defect and increased sensitivity to mitomycin C (132). It remains unknown whether and to what extend the fork stabilization of BRCA2 contributes to cancer formation.
DNA2—Seckel syndrome and progressive external ophthalmoplegia
DNA2 is a helicase and nuclease involved in fork reversal, long-patch base-excision repair and the processing of Okazaki fragments (133). Mutations in human DNA2 is associated with Seckel syndrome and progressive external ophthalmoplegia with mitochondrial DNA deletions (134,135). Progressive external ophthalmoplegia is an increasing weakness of the eye muscles, the cause of droopy eyes. Dna2 homozygous knockout model is embryonic lethal, though the heterozygous Dna2 is not lethal. Heterozygous Dna2 mice show shorter telomeres and show increased tumorigenesis, though no Seckel or ophthalmoplegia symptoms have been reported (136). It remains unclear to what extend the role of DNA2 in fork reversal contributes to Seckel syndrome and progressive external ophthalmoplegia.
HUWE1—X-linked syndromic mental retardation turner type and colon cancer
The E3-ligase HUWE1 facilitates DDT through its interaction with PCNA, thereby preventing replication stress (43). However, HUWE1 does not affect PCNA ubiquitination. Similar to UBE2A/RAD6, HUWE1 is also associated with X-linked turner syndrome (137,138). HUWE1 is lost often in colon cancer. Consistent, in Apc-deficient mice the loss of Huwe1 activity increased the tumour incidence (139). Increased tumorigenesis due to HUWE1 deficiency is likely due to a mixed effect of increased WNT signalling, genetic instability and MYC activity (43,139,140). Mouse models of Huwe1 are embryonic lethal and neuronal ablation resulted in severe neuronal differentiation defects (141).
PRIMPOL/CCDC111—Myopia
PRIMPOL is associated with leading strand repriming (106). Mutation Y89D in PRIMPOL has been associated with myopia (142). The PRIMPOL Y89D mutation leads to both primase and polymerase defects in the DT40 B-cell lymphoma (143). The mutation impairs PRIMPOL function; however, the causal relationship to myopia remains to be determined.
RAD51/FANCR—Fanconi anemia, complementation group R, mirror movements and breast cancer
RAD51 is involved in homologous recombination, fork reversal and interstrand crosslink repair (72,144). In human, RAD51 gene mutations lead to breast cancer, Fanconi anemia and mirror movements (145–147). Mirror movements are movements of limbs one side that are mirrored by the other. In mice, Rad51 deficiency leads to embryonic lethality (148). It remains unclear to what extent the role of RAD51 in fork reversal contributes to mirror movements and breast cancer.
REV3—Möbius syndrome
Surprisingly, while REV7 mutations are linked to Fanconi anemia, mutations in the catalytic subunit of the POLζ complex REV3 in humans are causal for the Möbius syndrome in an autosomal dominant fashion (149). These observations suggest that REV3 and REV7 may have some differences in activity, in addition to being part of the POLζ complex. Möbius syndrome patients display facial paralysis and eye movement defects (150). In line, in mice heterozygous Rev3 deficiency closely mimicked phenotypes observed in patients. As the main function of REV3 is TLS, it is likely that the TLS defect in REV3-defective cells leads to Möbius syndrome.
REV7/MAD2L2/FANCV—Fanconi anemia
As mentioned above, Fanconi anemia is a disease associated with defects in the interstrand crosslink repair pathway. Patients suffering from Fanconi anemia display bone marrow failure and subsequent anaemia, reduced fertility, congenital defects, an increased cancer incidence, and their cells are highly sensitive to MMC. A biallelic REV7 mutation has been implicated in the development of Fanconi anemia, and therefore REV7 was coined FANCV (151). As REV7 is a part of the POLζ complex, a REV3/REV7 heterodimer, it is likely functions as extender TLS polymerase activity opposite unhooked interstrand crosslink during the TLS synthesis step. Besides its roles in TLS, REV7 exerts an REV3-independent function by preventing end-resection and thereby promoting non-homologous end joining (152,153). However, since the non-homologous end joining pathway does not play a prominent role in interstrand crosslink repair, the Fanconi anemia phenotype of REV7 impairment likely relates to defective TLS over the interstrand crosslink.
SMARCAL1—Schimke immunoosseous dysplasia
SMARCAL1 is a translocase involved in reversing stalled replication forks to generate a chicken foot structured replication fork. Mutations in the human SMARCAL1 gene contribute to Schimke immunoosseous dysplasia (154). This syndrome is an autosomal recessive disease, characterized by T-cell immunodeficiency, dysplasia and renal dysfunction, leading to death at around 8 years (155). In line with observations in humans where the mutation alone is often not enough to cause the disease, knockout mouse models only show phenotypes of Schimke immunoosseous dysplasia upon inhibition of an RNA polymerase II inhibitor (156). These data suggest a Smarcal1 mutation predisposes to the disease but is not sufficient in causing the disease. Furthermore, differential gene expression was also found in Smarcal1-deficient MEFs and speculated that SMARCAL1 regulates gene transcription directly, and that this contributes to disease progression in combination with loss of genomic integrity. On the other hand, SMARCAL1 mutations found in patients affect SMARCAL1 ability to remodel replication forks and bind to Holliday junctions (157). This suggests that the differential gene expression in SMARCAL1-deficient cells is due to replication stress, rather than to direct regulation of transcription.
SPRTN—Ruijs-Aalf syndrome, hepatocellular carcinoma
SPRTN is recruited by PCNA-Ub and once recruited is thought to prevent PCNA deubiquitination through inhibition of USP1 (47–50). SPRTN also plays a role in resolving fork stalling DNA–protein crosslinks (158–160). In human, the SPRTN gene is associated with progeroid subtype Ruijs-Aalf syndrome and hepatocellular carcinoma (161). In mice, genetic deletion of Sprtn leads to embryonic lethality. A mouse model of Sprtn combining a hypo-morphic allele and a knockout allele also show progeroid phenotype and increased tumour incidence, in line with the human disease (162,163). It remains unclear to what extend the interaction with PCNA-Ub and SPRTN prevents Ruijs-Aalf syndrome and cancer.
POLH—Xeroderma pigmentosum variant
The first human disease linked to impaired DDT was Xeroderma Pigmentosum Variant (XP-V). Patients suffering from Xeroderma Pigmentosum (XP) are defective in nucleotide excision repair (NER) and highly sensitive to UV-induced DNA lesions (164–166). Patients suffering from the variant form of XP (XP-V) are nucleotide excision repair competent but defective in the Y-family TLS POLH gene (167). XP-V is characterized by an UV-hypersensitivity, a high skin cancer incidence due to higher mutagenicity of UV-induced DNA damage in the absence of POLH (168,169). In line with the observation that POLH can perform TLS opposite TT CPDs error-free, Polh-deficient mice show increased cancer incidence upon UV treatment (170–172). Furthermore, also heterozygous mice show an increased cancer incidence (173). These data indicate that POLH haploinsufficiency in humans might have and increased cancer incidence, due to the loss of the remaining functional allele (173). The increased mutagenesis in Polh-deficient mice seems to be related to increased compensatory POLI activity, as Polh;Poli double mutation was found to decrease mutagenesis upon UV, compared Polh single mutants (174,175). Interestingly, while Polh;Poli double mutants showed a lower mutation rate, they suffer from an increased tumour susceptibility following UV irradiation (174). This suggests, in this setting not the UV-induced point mutations, but other types of genomic instability as result of the Polh;Poli defect are the main drivers of tumorigenesis. In addition to POLI, POLQ has been reported to be involved in error-prone TLS in UV-induced lesions. Similar to Polh;Poli double mutants, Polh;Polq double mutates also show decreased point mutagenesis and increased tumorigenesis upon UV, compared to the Polh single mutant (30). Collectively, POLH-dependent TLS on UV-induced lesions leads to decreased mutagenesis and thereby contributes prevention of tumorigenesis.
UBE2A/RAD6A—X-linked syndromic mental retardation, Nascimento type
E2 ubiquitin ligase UBE2A/RAD6A is involved in PCNA monoubiquitination (35). In humans, UBE2A/RAD6A defect is associated with X-linked syndromic mental retardation (176). X-linked mental retardation syndrome is linked to a lot of different loci on the X chromosome. A mouse model of Ube2a/Rad6a shows defects in learning, memory and long-term depression, and female infertility (177,178). In contrast to infertile mouse mutants carrying a non-modifiable PcnaK164R mutation where germ cells are lacking, ovaria and oocytes develop in Ube2a/Rad6a mice. The inability of the embryo to survive the two-cell stage causes the infertility in Ube2a/Rad6a mice. It is not clear whether the symptoms are due to defective DDT, as ubiquitin ligases have many targets.
UBE2B/RAD6B—male infertility
E2 ligase UBE2B/RAD6B is involved in PCNA monoubiquitination (35). UBE2B/RAD6B mutations lead to male infertility (179). Mouse defective in Ube2b/Rad6b show male infertility, due to defects in synaptonemal complex and meiotic recombination in spermatocytes (180,181). It is not clear whether the symptoms relate to defective DDT, as ubiquitin ligases generally have many targets.
WRN—Werner syndrome
The WRN gene encodes a DNA helicase and exonuclease and is involved in homologous recombination repair, and fork reversal (182,183). In humans, aberrant mutations in the WRN gene lead to Werner syndrome (184,185). Clinical features of Werner syndrome include premature ageing, short stature and cancer predisposition (186). Werner syndrome inheritance is recessive and the average age of death is 54 years. Surprisingly, Wrn-defective mice do not recapitulate Werner syndrome symptoms of premature ageing as found in humans, though they are born at a sub-Mendelian frequency (187). However, Wrn defective mice kept for 3–5 generations do show increased ageing phenotypes, such as grey hair, osteoporosis, increased cancer susceptibility and other phenotypes of seen during normal ageing. This phenotype could also be accomplished by crossing Wrn mice to telomere maintenance factor Terc-deficient mice (188). As mice have very long telomeres compared to humans, these data suggest that the Werner syndrome might be mainly caused by defects in telomere maintenance (189). In fact, the Wrn defect leads to telomeric loss likely due to G4 stack bypass defect during replication of the G4 stack dense telomeric DNA (190). This suggests that premature ageing in this model relates to defective DDT of G4 stacks, ultimately leading to loss of telomeres.
In summary, DDT plays a key role in preventing disease. Overall, DDT defects are associated with neurological symptoms, accelerated ageing, infertility and cancer. These symptoms are shared with DNA repair defects, highlighting the critical contribution of the DDT system to the DNA damage response network. Mouse models with genetically defined DDT defects will be of value to further study the disease-related DDT defects and develop strategies that alleviate the symptoms or cure the patients.
THE ROLE OF DNA DAMAGE TOLERANCE IN MUTAGENESIS
The role of translesion synthesis in the generation of point mutations
DDT has been divided into error-prone TLS, error-free TLS and error-free homology-directed DDT, the latter includes fork reversal and template switching (191). Error-free definition in this context should be not considered literally but relatively, as error-free DDT can still contribute to mutagenesis, albeit at a lower frequency. Despite the fact that TLS is closely linked to the generation of point mutations, the existence of multiple TLS polymerases implies the existence of specificity per polymerase for the type of DNA lesion. In line with this notion, POLH or POLK knockouts, two closely related Y-family members show selective sensitivity for UV and MMS, respectively (74,192,193). In line, expression of Y-family TLS polymerases POLI and POLK correlated inversely with the mutation load in invasive breast cancers, suggesting that the absence of one TLS polymerase leads to increased point mutagenesis. This mutagenic phenotype likely relates to the fact that the backup TLS polymerases are more error-prone in tolerating a specific lesion. Missing TLS polymerases are likely to be back-upped by other members that further increases the error-rate and consequently mutation-rate (106). In line with this hypothesis, both Polh and Polk knockout mouse models show increased mutagenesis under specific conditions, suggesting that each TLS polymerase has its preferred lesion(s). Polh deficiency leads skin UV-induced mutagenesis in the skin, Polk deficiency leads to an increase of spontaneous mutations during ageing in an organ-specific manner, respectively (174,194).
In order to provide a comprehensible overview on the effect of TLS on mutagenesis, we focus here on UV-induced mutagenesis. Exposure of DNA to UV leads to the formation of CPDs and 6-4PP (195). Upon UV-B exposure, these CPDs and 6-4PP are produced in a declining frequency: TT CPD > TC CPD > TC 6-4PP > CT CPD > CC CPD > TT 6-4PP (196). The UV signature is composed of primarily of TC > TT or CC > TT transitions caused by CPDs (197). Daylight UV and more specifically UV-B exposure leads to preferential formation of CPDs at 5-methylcytosine bases, though this preference for 5-methylcytosine bases seems to be absent upon UV-C irradiation (198,199). Replication across the U in CPDs can explain the TC > TT and CC > TT UV mutagenic signature. TLS has been involved in UV mutagenesis, as POLH-deficiency leads to increased mutagenesis and XP-V (168,169). As mentioned previously, POLH can synthesize DNA across TT CPDs in an fairly error-free manner (200). The increase of mutagenesis in XP-V seems to be due to POLI activity, as Polh;Poli double mutant mice have a lower mutation rate upon UV exposure, compared to Polh defective mice (174,175).
In contrast to POLH, POLQ was found to act error-prone on UV-induced lesions even in the presence of POLH, as Polq-deficient cells show a level of UV-induced mutagenesis decreased similar to the non-treated control (30). These data are in line with the notion that specific TLS polymerases bypass different types of DNA damage. In order to study selectively CPDs or 6-4PP, CPDs or 6-4PP specific photolyases are often used to repair either CPDs or 6-4PP (201). Whether an impairment in the activity of a given TLS polymerase will be pro- or anti-mutagenic appears to depend on the type of the DNA polymerase and lesion (64). The authors exposed MEFs express (6–4) PP and CPD specific photolyases to UV light and show that knockdown of Polh or Rev1 leads to an increased number of mutations in the 6-4PP lyase setting, but a decrease of mutations when the CPD specific lyase is expressed.
Whereas loss of individual TLS polymerases generally increases mutagenesis, impaired regulation of TLS decreases UV-induced mutagenesis. Regarding UV-damage induced mutagenesis, both REV1 and PCNA K164 were found to stimulate mutagenesis (64,73,202). Thus, if there is inefficient recruitment due to a defective TLS regulator, there are fewer mutations, at least in the case of UV irradiation. Whether the same holds for the plethora of endogenous lesions remains to be determined further. However, the loss of an individual non-regulatory TLS polymerase increases mutagenesis, likely caused by an alternative TLS polymerase more error-prone at a given lesion.
Homology-directed DNA damage tolerance in genome rearrangements
Both template switching and fork reversal prevent point mutations by avoiding the use of the damaged template; however, they are associated with the extension of inverted repeats and other genome rearrangements (70). Homology-directed DDT has been shown to be involved in genome rearrangements induced by template switching during pausing at inverted repeats, as shown in Schizosaccharo pompe and S. cerevisiae (203–205). The genome rearrangements fuelled by inverted repeats can lead to acentric and dicentric chromosomes. Also in the evolution of the human genome, homology-directed DDT induced genome rearrangements seems to have a large contribution (206).
In conclusion, DDT can have both pro- and anti-mutagenic activities. Both homology-directed modes of DDT prevent point mutations, though they likely come wish the risk of increased genome rearrangements. The error-proneness of TLS depends on the specific lesion and the type of TLS polymerase replicating this lesion. It remains unknown whether there are mechanisms that recruit specific TLS polymerases to specific lesions.
THE ROLE OF DDT IN AGEING AND STEM CELLS
Stem cells depend on their own genome stability to both maintain an adequate stem cell pool and differentiate to enable long-term tissue homeostasis. The majority of work on genome stability in stem cells and its effect on ageing has been focussed on hematopoietic stem cells (HSC). Long-term HSC (LT-HSC) are mainly quiescent and thought to differentiate into short-term HSC (ST-HSC) (207). ST-HSC further differentiate into multipotent progenitors (MPP) 2, 3 and 4, which give rise to the majority of blood cells to warrant hematopoiesis (207–210).
Increased genome instability leads to a decrease in HSC potential and this progresses with age, suggesting that DNA damage accelerates HSC ageing (211). Similarly, different mouse models have shown that increased replication stress leads to premature ageing of HSCs (125,212–214). DDT impaired PcnaK164R/K164R mice show premature ageing of HSCs and skewing of differentiation toward the myeloid/erythroid lineage and increased DNA damage within the LSKs (213). Both myeloid/erythroid progenitor skewing and increased DNA damage in HSCs are signs of increased ageing. Myeloid/erythroid skewing of HSC differentiation is known to increase upon ageing and is thought to be at the basis of lymphoid decline in aged mice (213,215,216). In line with the role of DDT in HSCs, TLS regulator and Y-family polymerase Rev1 mutant mice show a reduction of LSK cells in 5-month-old mice. The Rev1 defect can be increased through introduction of the Xpc nucleic excision repair defect (214). As many lesions are not repaired, the Rev1;Xpc double defect leads to increased replication stress, anemia and bone marrow failure. Knockout mice lacking regulators of TLS such as Paf15 a PCNA associated factor suffer from severe HSC defects (217). Another regulator of PCNA, Sprtn hypomorphic variant mice displayed a progeroid phenotype. The hematopoietic progenitor compartment was not investigated, though likely to be affected as well. Next to the genes involved in TLS, mutation of genes involved in fork reversal may also have HSC defects. Mouse models of translocase Smarcal1 also displayed decreased number of HSC upon induction of replication of stem and progenitors (218). Strikingly, Smarcal1 defective cells are more sensitive to fork stalling by 5-FU, but not double strand break inducing γ-irradiation, indicating that Smarcal1 deficiency affects DDT but not double strand break repair. In Brca1/Fancs hypo-morphic mice, HSCs are marginalized and the mice show reduced bone marrow cellularity (219). In line, a fully defective Brca1/Fancs bone marrow specific deletion using a bone marrow specific Cre leads to bone marrow failure and hematologic malignancies (220). Furthermore, Brca2/Fancd1 contributes to HSC function, as a hypo-morphic homozygous Brca2/Fancd1 allele also shows an HSC repopulating defect and a decreased number of cells in the bone marrow (132). Next to HSCs, other stem cells also appear sensitive to DDT defects. For example, germ stem cells were completely absent in PncaK164R/K164R mice, in both male and female mice (221).
In summary, increased DNA damage and replication stress due to DDT defects can accelerate ageing as shown in the hematopoietic system. It will be interesting to assess the relevance of DDT in determining the ageing of other tissues and their stem cells. Although in the liver DNA damage has been shown to contribute to ageing, independent of mutagenesis (222). The role of mutations in HSC and ageing is less clear, many DDT defective mouse models will have both increased DNA damage and mutagenesis, it remains unclear what the contribution of mutations to ageing is.
DDT AS TARGET FOR CANCER THERAPY
Paradoxically, while DNA needs to be kept in pristine condition, both DNA itself and the DNA damage response are prime targets for anti-cancer therapeutics. Many chemotherapeutic agents act through damaging the DNA or impeding replication (1,223). Defective DDT often leads to increased sensitivity to chemotherapeutically induced replication stalling DNA lesions. In line, increased expression levels of POLH have been found to correlate with decreased survival in non-small cell lung cancer treated with platinating agents (224). As many cancers have somatic mutations in DDT genes, targeting defects in the DDT system is an attractive way to optimize the treatment of cancers (225). One example already present in the clinic is PARP inhibitors in BRCA1/2-deficient cancers, though the mechanism of sensitivity to PARP inhibitors has been hypothesized to be due to defective homologous recombination (226). However, PARP inhibitor resistance mechanisms involve a rescue of replication fork stability, suggesting that one homology-directed DDT mechanism that is defective in Brca1/2-deficient cells plays a role in determining the sensitivity to PARP inhibitors (227). Furthermore, PCNA K164 ubiquitination inhibition is also synthetic lethal with Brca1, in combination with UV or cisplatin treatment (228). Another potential candidate for synthetic lethality with DDT defects leading to increased replication stress could be ATM inhibitors, as tumours with mutated DDT genes should show increased replication stress and ATM inhibitors are synthetic lethal with increased replication stress (229). Furthermore, synthetic lethality with Rad18 or Polk can be induced by inhibitor of WEE1- and oncogene-induced stress (230). Individual TLS DNA polymerases may be attractive anti-cancer targets, because they have a bigger active site than replicative polymerases and therefore could be targeted specifically. Screens for TLS polymerase inhibitors for POLK have identified new lead compounds, which await further development (231). As PCNA is a key regulator of DDT, inhibitors of PCNA function are being developed. T2AA is a PCNA ubiquitination inhibitor isolated from fungi (232). Next to T2AA, short peptides inhibiting PCNA interactions revealed efficacy in mouse tumour models (233–235). These PCNA inhibitors affected the AlkB homologue 2 PCNA-interacting motif (APIM), PIP motif or Y211 phosphorylation, and thereby prevent proper response to DNA damage. Furthermore, targeting ubiquitin-binding motif of REV1 that binds PCNA also sensitizes tumours to DNA damaging agents in vitro (236). However, mouse models have shown that targeting PCNA K164 for chemo-sensitization strategies is not a good approach, as the side effects of PCNA inhibition combined with platinum treatment become severe, mainly due to bone marrow failure. However, like for high dose chemotherapy, bone marrow failure may be compensated or prohibited by autologous HSC transplantation (213,225). Though, PCNA K164 targeting could be feasible in a setting where there is a tumour-specific mutation in a gene that is synthetic lethal with PCNA K164 inhibition. Phenotypes of mice treated with drugs that target PCNA K164 specifically are likely a lot less sensitive to platinating agents, as compared to a systemic PCNA K164R mutation (213,225).
Inhibition of PCNA deubiquitinase USP1 functioning also leads to cellular sensitization to chemotherapeutics. Targeting USP1 can be accomplished through targeting the USP1 UAF1 complex formation, which is needed for the activity of USP1. The activity of USP1 on FANCD2-Ub and PCNA-Ub has successfully been targeted leading to increased sensitivity to cisplatin in vitro (237).
In summary, DDT should be further exploited for synthetic lethal anti-cancer strategies and tumour-specific vulnerabilities to further optimize cancer therapy. In the future, personalized cancer medicine that targets tumour-specific vulnerabilities with adequate drugs will be achieved.
CONCLUSION
DDT is a key component of the DNA damage response network and hence has a central role in genome maintenance, through preventing replication stress and secondary DNA damage. Defects in the DDT system are associated with specific diseases. Disease phenotypes generally include increased ageing, cancer and neurological symptoms. Besides being causal in some cancers, DDT defects can lead to decreased genome stability, which at the same time opens a therapeutic window that renders primarily the tumour vulnerable to targeted therapies. Further research is required to unravel the choice and regulation of specific DDT pathways. These insights will be instrumental in treating DDT-related pathologies.
ACKNOWLEDGEMENTS
The authors thank Aldo Spanjaard, Daniel de Groot, Paul van den Berk and Ronak Shah for providing feedback on the manuscript.
FUNDING
Netherlands Organization for Scientific Research [ZonMW Top 91213018 to H.J.]; Dutch Cancer Society [KWF NKI-2012-5243, KWF NKI-2016-10032, KWF NKI-2016-10796]. Funding for open access charge: Dutch Cancer Society [KWF NKI-2016-10032].
Conflict of interest statement. None declared.
REFERENCES
- 1. Hoeijmakers J.H. DNA damage, aging, and cancer. N. Engl. J. Med. 2009; 361:1475–1485. [DOI] [PubMed] [Google Scholar]
- 2. Lindahl T., Barnes D.E.. Repair of endogenous DNA damage. Cold Spring Harb. Symp. Quant. Biol. 2000; 65:127–133. [DOI] [PubMed] [Google Scholar]
- 3. De Bont R., van Larebeke N.. Endogenous DNA damage in humans: a review of quantitative data. Mutagenesis. 2004; 19:169–185. [DOI] [PubMed] [Google Scholar]
- 4. Ciccia A., Elledge S.J.. The DNA damage response: making it safe to play with knives. Mol. Cell. 2010; 40:179–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Roberts J.D., Thomas D.C., Kunkel T.A.. Exonucleolytic proofreading of leading and lagging strand DNA replication errors. Proc. Natl. Acad. Sci. U.S.A. 1991; 88:3465–3469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Pursell Z.F., Isoz I., Lundstrom E.B., Johansson E., Kunkel T.A.. Yeast DNA polymerase epsilon participates in leading-strand DNA replication. Science. 2007; 317:127–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Burgers P.M.J., Gordenin D., Kunkel T.A.. Who is leading the replication fork, Pol epsilon or Pol delta?. Mol. Cell. 2016; 61:492–493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Lange S.S., Takata K., Wood R.D.. DNA polymerases and cancer. Nat. Rev. Cancer. 2011; 11:96–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Lazzaro F., Novarina D., Amara F., Watt D.L., Stone J.E., Costanzo V., Burgers P.M., Kunkel T.A., Plevani P., Muzi-Falconi M.. RNase H and postreplication repair protect cells from ribonucleotides incorporated in DNA. Mol. Cell. 2012; 45:99–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Schiavone D., Guilbaud G., Murat P., Papadopoulou C., Sarkies P., Prioleau M.N., Balasubramanian S., Sale J.E.. Determinants of G quadruplex-induced epigenetic instability in REV1-deficient cells. EMBO J. 2014; 33:2507–2520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Stingele J., Schwarz M.S., Bloemeke N., Wolf P.G., Jentsch S.. A DNA-dependent protease involved in DNA-protein crosslink repair. Cell. 2014; 158:327–338. [DOI] [PubMed] [Google Scholar]
- 12. Hamperl S., Bocek M.J., Saldivar J.C., Swigut T., Cimprich K.A.. Transcription-Replication conflict orientation modulates R-Loop levels and activates distinct DNA damage responses. Cell. 2017; 170:774–786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Glover T.W., Wilson T.E., Arlt M.F.. Fragile sites in cancer: more than meets the eye. Nat. Rev. Cancer. 2017; 17:489–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Friedberg E.C. Suffering in silence: the tolerance of DNA damage. Nat. Rev. Mol. Cell Biol. 2005; 6:943–953. [DOI] [PubMed] [Google Scholar]
- 15. Ulrich H.D. Deubiquitinating PCNA: a downside to DNA damage tolerance. Nat. Cell Biol. 2006; 8:303–305. [DOI] [PubMed] [Google Scholar]
- 16. Moldovan G.L., Pfander B., Jentsch S.. PCNA, the maestro of the replication fork. Cell. 2007; 129:665–679. [DOI] [PubMed] [Google Scholar]
- 17. Davies A.A., Huttner D., Daigaku Y., Chen S., Ulrich H.D.. Activation of ubiquitin-dependent DNA damage bypass is mediated by replication protein a. Mol. Cell. 2008; 29:625–636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Bianchi J., Rudd S.G., Jozwiakowski S.K., Bailey L.J., Soura V., Taylor E., Stevanovic I., Green A.J., Stracker T.H., Lindsay H.D. et al.. PrimPol bypasses UV photoproducts during eukaryotic chromosomal DNA replication. Mol. Cell. 2013; 52:566–573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Mouron S., Rodriguez-Acebes S., Martinez-Jimenez M.I., Garcia-Gomez S., Chocron S., Blanco L., Mendez J.. Repriming of DNA synthesis at stalled replication forks by human PrimPol. Nat. Struct. Mol. Biol. 2013; 20:1383–1389. [DOI] [PubMed] [Google Scholar]
- 20. Garcia-Gomez S., Reyes A., Martinez-Jimenez M.I., Chocron E.S., Mouron S., Terrados G., Powell C., Salido E., Mendez J., Holt I.J. et al.. PrimPol, an archaic primase/polymerase operating in human cells. Mol. Cell. 2013; 52:541–553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Branzei D., Szakal B.. DNA damage tolerance by recombination: Molecular pathways and DNA structures. DNA Repair (Amst.). 2016; 44:68–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Quinet A., Lemacon D., Vindigni A.. Replication fork reversal: Players and guardians. Mol. Cell. 2017; 68:830–833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Buoninfante O.A. Physiological and pharmacological relevance of DNA damage tolerance. 2018; The Netherlands: Universiteit van Amsterdam. [Google Scholar]
- 24. Sale J.E. Translesion DNA synthesis and mutagenesis in eukaryotes. Cold Spring Harbor Perspect. Biol. 2013; 5:a012708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Yang W., Gao Y.. Translesion and repair DNA polymerases: Diverse structure and mechanism. Annu Rev. Biochem. 2018; 87:239–261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Nair D.T., Johnson R.E., Prakash L., Prakash S., Aggarwal A.K.. Rev1 employs a novel mechanism of DNA synthesis using a protein template. Science. 2005; 309:2219–2222. [DOI] [PubMed] [Google Scholar]
- 27. Takata K.I., Reh S., Yousefzadeh M.J., Zelazowski M.J., Bhetawal S., Trono D., Lowery M.G., Sandoval M., Takata Y., Lu Y. et al.. Analysis of DNA polymerase ν function in meiotic recombination, immunoglobulin class-switching, and DNA damage tolerance. PLoS Genet. 2017; 13:e1006818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. van Schendel R., Roerink S.F., Portegijs V., van den Heuvel S., Tijsterman M.. Polymerase Θ is a key driver of genome evolution and of CRISPR/Cas9-mediated mutagenesis. Nat. Commun. 2015; 6:7394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Roerink S.F., van Schendel R., Tijsterman M.. Polymerase theta-mediated end joining of replication-associated DNA breaks in C. elegans. Genome Res. 2014; 24:954–962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Yoon J.H., McArthur M.J., Park J., Basu D., Wakamiya M., Prakash L., Prakash S.. Error-Prone replication through UV lesions by DNA polymerase θ protects against skin cancers. Cell. 2019; 176:1295–1309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Choe K.N., Moldovan G.L.. Forging ahead through darkness: PCNA, still the principal conductor at the replication fork. Mol. Cell. 2017; 65:380–392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Kannouche P.L., Wing J., Lehmann A.R.. Interaction of human DNA polymerase eta with monoubiquitinated PCNA: a possible mechanism for the polymerase switch in response to DNA damage. Mol. Cell. 2004; 14:491–500. [DOI] [PubMed] [Google Scholar]
- 33. Friedberg E.C., Lehmann A.R., Fuchs R.P.. Trading places: how do DNA polymerases switch during translesion DNA synthesis?. Mol. Cell. 2005; 18:499–505. [DOI] [PubMed] [Google Scholar]
- 34. Bienko M., Green C.M., Crosetto N., Rudolf F., Zapart G., Coull B., Kannouche P., Wider G., Peter M., Lehmann A.R. et al.. Ubiquitin-binding domains in Y-family polymerases regulate translesion synthesis. Science. 2005; 310:1821–1824. [DOI] [PubMed] [Google Scholar]
- 35. Hoege C., Pfander B., Moldovan G.L., Pyrowolakis G., Jentsch S.. RAD6-dependent DNA repair is linked to modification of PCNA by ubiquitin and SUMO. Nature. 2002; 419:135–141. [DOI] [PubMed] [Google Scholar]
- 36. Watanabe K., Tateishi S., Kawasuji M., Tsurimoto T., Inoue H., Yamaizumi M.. Rad18 guides poleta to replication stalling sites through physical interaction and PCNA monoubiquitination. EMBO J. 2004; 23:3886–3896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Zhang S., Chea J., Meng X., Zhou Y., Lee E.Y., Lee M.Y.. PCNA is ubiquitinated by RNF8. Cell Cycle. 2008; 7:3399–3404. [DOI] [PubMed] [Google Scholar]
- 38. Terai K., Abbas T., Jazaeri A.A., Dutta A.. CRL4(Cdt2) E3 ubiquitin ligase monoubiquitinates PCNA to promote translesion DNA synthesis. Mol. Cell. 2010; 37:143–149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Huang M., Zhou B., Gong J., Xing L., Ma X., Wang F., Wu W., Shen H., Sun C., Zhu X. et al.. RNA-splicing factor SART3 regulates translesion DNA synthesis. Nucleic Acids Res. 2018; 46:4560–4574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Povlsen L.K., Beli P., Wagner S.A., Poulsen S.L., Sylvestersen K.B., Poulsen J.W., Nielsen M.L., Bekker-Jensen S., Mailand N., Choudhary C.. Systems-wide analysis of ubiquitylation dynamics reveals a key role for PAF15 ubiquitylation in DNA-damage bypass. Nat. Cell Biol. 2012; 14:1089–1098. [DOI] [PubMed] [Google Scholar]
- 41. De Biasio A., de Opakua A.I., Mortuza G.B., Molina R., Cordeiro T.N., Castillo F., Villate M., Merino N., Delgado S., Gil-Carton D. et al.. Structure of p15(PAF)-PCNA complex and implications for clamp sliding during DNA replication and repair. Nat. Commun. 2015; 6:6439. [DOI] [PubMed] [Google Scholar]
- 42. De March M., Barrera-Vilarmau S., Crespan E., Mentegari E., Merino N., Gonzalez-Magana A., Romano-Moreno M., Maga G., Crehuet R., Onesti S. et al.. p15PAF binding to PCNA modulates the DNA sliding surface. Nucleic Acids Res. 2018; 46:9816–9828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Choe K.N., Nicolae C.M., Constantin D., Imamura Kawasawa Y., Delgado-Diaz M.R., De S., Freire R., Smits V.A., Moldovan G.L.. HUWE1 interacts with PCNA to alleviate replication stress. EMBO Rep. 2016; 17:874–886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Porro A., Berti M., Pizzolato J., Bologna S., Kaden S., Saxer A., Ma Y., Nagasawa K., Sartori A.A., Jiricny J.. FAN1 interaction with ubiquitylated PCNA alleviates replication stress and preserves genomic integrity independently of BRCA2. Nat. Commun. 2017; 8:1073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Tian F., Sharma S., Zou J., Lin S.Y., Wang B., Rezvani K., Wang H., Parvin J.D., Ludwig T., Canman C.E. et al.. BRCA1 promotes the ubiquitination of PCNA and recruitment of translesion polymerases in response to replication blockade. Proc. Natl. Acad. Sci. U.S.A. 2013; 110:13558–13563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Han J., Liu T., Huen M.S., Hu L., Chen Z., Huang J.. SIVA1 directs the E3 ubiquitin ligase RAD18 for PCNA monoubiquitination. J. Cell Biol. 2014; 205:811–827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Centore R.C., Yazinski S.A., Tse A., Zou L.. Spartan/C1orf124, a reader of PCNA ubiquitylation and a regulator of UV-induced DNA damage response. Mol. Cell. 2012; 46:625–635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Juhasz S., Balogh D., Hajdu I., Burkovics P., Villamil M.A., Zhuang Z., Haracska L.. Characterization of human Spartan/C1orf124, an ubiquitin-PCNA interacting regulator of DNA damage tolerance. Nucleic Acids Res. 2012; 40:10795–10808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Machida Y., Kim M.S., Machida Y.J.. Spartan/C1orf124 is important to prevent UV-induced mutagenesis. Cell Cycle. 2012; 11:3395–3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Ghosal G., Leung J.W., Nair B.C., Fong K.W., Chen J.. Proliferating cell nuclear antigen (PCNA)-binding protein C1orf124 is a regulator of translesion synthesis. J. Biol. Chem. 2012; 287:34225–34233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Kim M.S., Machida Y., Vashisht A.A., Wohlschlegel J.A., Pang Y.P., Machida Y.J.. Regulation of error-prone translesion synthesis by Spartan/C1orf124. Nucleic Acids Res. 2013; 41:1661–1668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Toth A., Hegedus L., Juhasz S., Haracska L., Burkovics P.. The DNA-binding box of human SPARTAN contributes to the targeting of Poleta to DNA damage sites. DNA Repair (Amst.). 2017; 49:33–42. [DOI] [PubMed] [Google Scholar]
- 53. Yanagihara H., Kobayashi J., Tateishi S., Kato A., Matsuura S., Tauchi H., Yamada K., Takezawa J., Sugasawa K., Masutani C. et al.. NBS1 recruits RAD18 via a RAD6-like domain and regulates Pol eta-dependent translesion DNA synthesis. Mol. Cell. 2011; 43:788–797. [DOI] [PubMed] [Google Scholar]
- 54. Huang T.T., Nijman S.M., Mirchandani K.D., Galardy P.J., Cohn M.A., Haas W., Gygi S.P., Ploegh H.L., Bernards R., D’Andrea A.D.. Regulation of monoubiquitinated PCNA by DUB autocleavage. Nat. Cell Biol. 2006; 8:339–347. [DOI] [PubMed] [Google Scholar]
- 55. Niimi A., Brown S., Sabbioneda S., Kannouche P.L., Scott A., Yasui A., Green C.M., Lehmann A.R.. Regulation of proliferating cell nuclear antigen ubiquitination in mammalian cells. Proc. Natl. Acad. Sci. U.S.A. 2008; 105:16125–16130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Lee K.Y., Yang K., Cohn M.A., Sikdar N., D’Andrea A.D., Myung K.. Human ELG1 regulates the level of ubiquitinated proliferating cell nuclear antigen (PCNA) through Its interactions with PCNA and USP1. J. Biol. Chem. 2010; 285:10362–10369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Lee K.Y., Fu H., Aladjem M.I., Myung K.. ATAD5 regulates the lifespan of DNA replication factories by modulating PCNA level on the chromatin. J. Cell Biol. 2013; 200:31–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Park J.M., Yang S.W., Yu K.R., Ka S.H., Lee S.W., Seol J.H., Jeon Y.J., Chung C.H.. Modification of PCNA by ISG15 plays a crucial role in termination of error-prone translesion DNA synthesis. Mol. Cell. 2014; 54:626–638. [DOI] [PubMed] [Google Scholar]
- 59. Guo C., Fischhaber P.L., Luk-Paszyc M.J., Masuda Y., Zhou J., Kamiya K., Kisker C., Friedberg E.C.. Mouse Rev1 protein interacts with multiple DNA polymerases involved in translesion DNA synthesis. EMBO J. 2003; 22:6621–6630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Ohashi E., Murakumo Y., Kanjo N., Akagi J., Masutani C., Hanaoka F., Ohmori H.. Interaction of hREV1 with three human Y-family DNA polymerases. Genes Cells. 2004; 9:523–531. [DOI] [PubMed] [Google Scholar]
- 61. Tissier A., Kannouche P., Reck M.P., Lehmann A.R., Fuchs R.P., Cordonnier A.. Co-localization in replication foci and interaction of human Y-family members, DNA polymerase pol eta and REVl protein. DNA Repair (Amst.). 2004; 3:1503–1514. [DOI] [PubMed] [Google Scholar]
- 62. Jansen J.G., Tsaalbi-Shtylik A., Langerak P., Calléja F., Meijers C.M., Jacobs H., de Wind N.. The BRCT domain of mammalian Rev1 is involved in regulating DNA translesion synthesis. Nucleic Acids Res. 2005; 33:356–365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Kim H., Yang K., Dejsuphong D., D’Andrea A.D.. Regulation of Rev1 by the Fanconi anemia core complex. Nat. Struct. Mol. Biol. 2012; 19:164–170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Yoon J.H., Park J., Conde J., Wakamiya M., Prakash L., Prakash S.. Rev1 promotes replication through UV lesions in conjunction with DNA polymerases eta, iota, and kappa but not DNA polymerase zeta. Genes Dev. 2015; 29:2588–2602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Guo C., Sonoda E., Tang T.S., Parker J.L., Bielen A.B., Takeda S., Ulrich H.D., Friedberg E.C.. REV1 protein interacts with PCNA: significance of the REV1 BRCT domain in vitro and in vivo. Mol. Cell. 2006; 23:265–271. [DOI] [PubMed] [Google Scholar]
- 66. Edmunds C.E., Simpson L.J., Sale J.E.. PCNA ubiquitination and REV1 define temporally distinct mechanisms for controlling translesion synthesis in the avian cell line DT40. Mol. Cell. 2008; 30:519–529. [DOI] [PubMed] [Google Scholar]
- 67. Wang Z., Huang M., Ma X., Li H., Tang T., Guo C.. REV1 promotes PCNA monoubiquitylation through interacting with ubiquitylated RAD18. J. Cell Sci. 2016; 129:1223–1233. [DOI] [PubMed] [Google Scholar]
- 68. McGlynn P., Lloyd R.G.. Recombinational repair and restart of damaged replication forks. Nat. Rev. Mol. Cell Biol. 2002; 3:859–870. [DOI] [PubMed] [Google Scholar]
- 69. Vujanovic M., Krietsch J., Raso M.C., Terraneo N., Zellweger R., Schmid J.A., Taglialatela A., Huang J.W., Holland C.L., Zwicky K. et al.. Replication fork slowing and reversal upon DNA damage require PCNA polyubiquitination and ZRANB3 DNA translocase activity. Mol. Cell. 2017; 67:882–890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Chiu R.K., Brun J., Ramaekers C., Theys J., Weng L., Lambin P., Gray D.A., Wouters B.G.. Lysine 63-polyubiquitination guards against translesion synthesis-induced mutations. PLos Genet. 2006; 2:e116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Krijger P.H., Lee K.Y., Wit N., van den Berk P.C., Wu X., Roest H.P., Maas A., Ding H., Hoeijmakers J.H., Myung K. et al.. HLTF and SHPRH are not essential for PCNA polyubiquitination, survival and somatic hypermutation: existence of an alternative E3 ligase. DNA Repair (Amst.). 2011; 10:438–444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Neelsen K.J., Lopes M.. Replication fork reversal in eukaryotes: from dead end to dynamic response. Nat. Rev. Mol. Cell Biol. 2015; 16:207–220. [DOI] [PubMed] [Google Scholar]
- 73. Stelter P., Ulrich H.D.. Control of spontaneous and damage-induced mutagenesis by SUMO and ubiquitin conjugation. Nature. 2003; 425:188–191. [DOI] [PubMed] [Google Scholar]
- 74. Wit N., Buoninfante O.A., van den Berk P.C., Jansen J.G., Hogenbirk M.A., de Wind N., Jacobs H.. Roles of PCNA ubiquitination and TLS polymerases kappa and eta in the bypass of methyl methanesulfonate-induced DNA damage. Nucleic Acids Res. 2015; 43:282–294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Ciccia A., Nimonkar A.V., Hu Y., Hajdu I., Achar Y.J., Izhar L., Petit S.A., Adamson B., Yoon J.C., Kowalczykowski S.C. et al.. Polyubiquitinated PCNA recruits the ZRANB3 translocase to maintain genomic integrity after replication stress. Mol. Cell. 2012; 47:396–409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Yuan J., Ghosal G., Chen J.. The HARP-like domain-containing protein AH2/ZRANB3 binds to PCNA and participates in cellular response to replication stress. Mol. Cell. 2012; 47:410–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Bansbach C.E., Betous R., Lovejoy C.A., Glick G.G., Cortez D.. The annealing helicase SMARCAL1 maintains genome integrity at stalled replication forks. Genes Dev. 2009; 23:2405–2414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Yusufzai T., Kadonaga J.T.. HARP is an ATP-driven annealing helicase. Science. 2008; 322:748–750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Yusufzai T., Kong X., Yokomori K., Kadonaga J.T.. The annealing helicase HARP is recruited to DNA repair sites via an interaction with RPA. Genes Dev. 2009; 23:2400–2404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Poole L.A., Cortez D.. Functions of SMARCAL1, ZRANB3, and HLTF in maintaining genome stability. Crit. Rev. Biochem. Mol. Biol. 2017; 52:696–714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Couch F.B., Bansbach C.E., Driscoll R., Luzwick J.W., Glick G.G., Betous R., Carroll C.M., Jung S.Y., Qin J., Cimprich K.A. et al.. ATR phosphorylates SMARCAL1 to prevent replication fork collapse. Genes Dev. 2013; 27:1610–1623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Kolinjivadi A.M., Sannino V., De Antoni A., Zadorozhny K., Kilkenny M., Techer H., Baldi G., Shen R., Ciccia A., Pellegrini L. et al.. Smarcal1-Mediated fork reversal triggers Mre11-Dependent degradation of nascent DNA in the absence of Brca2 and stable Rad51 nucleofilaments. Mol. Cell. 2017; 67:867–881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Mijic S., Zellweger R., Chappidi N., Berti M., Jacobs K., Mutreja K., Ursich S., Ray Chaudhuri A., Nussenzweig A., Janscak P. et al.. Replication fork reversal triggers fork degradation in BRCA2-defective cells. Nat. Commun. 2017; 8:859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Lemacon D., Jackson J., Quinet A., Brickner J.R., Li S., Yazinski S., You Z., Ira G., Zou L., Mosammaparast N. et al.. MRE11 and EXO1 nucleases degrade reversed forks and elicit MUS81-dependent fork rescue in BRCA2-deficient cells. Nat. Commun. 2017; 8:860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Schlacher K., Christ N., Siaud N., Egashira A., Wu H., Jasin M.. Double-strand break repair-independent role for BRCA2 in blocking stalled replication fork degradation by MRE11. Cell. 2011; 145:529–542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. D’Amours D., Jackson S.P.. The Mre11 complex: at the crossroads of dna repair and checkpoint signalling. Nat. Rev. Mol. Cell Biol. 2002; 3:317–327. [DOI] [PubMed] [Google Scholar]
- 87. Thangavel S., Berti M., Levikova M., Pinto C., Gomathinayagam S., Vujanovic M., Zellweger R., Moore H., Lee E.H., Hendrickson E.A. et al.. DNA2 drives processing and restart of reversed replication forks in human cells. J. Cell Biol. 2015; 208:545–562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Ray Chaudhuri A., Hashimoto Y., Herrador R., Neelsen K.J., Fachinetti D., Bermejo R., Cocito A., Costanzo V., Lopes M.. Topoisomerase I poisoning results in PARP-mediated replication fork reversal. Nat. Struct. Mol. Biol. 2012; 19:417–423. [DOI] [PubMed] [Google Scholar]
- 89. Gari K., Decaillet C., Stasiak A.Z., Stasiak A., Constantinou A.. The Fanconi anemia protein FANCM can promote branch migration of Holliday junctions and replication forks. Mol. Cell. 2008; 29:141–148. [DOI] [PubMed] [Google Scholar]
- 90. Gari K., Decaillet C., Delannoy M., Wu L., Constantinou A.. Remodeling of DNA replication structures by the branch point translocase FANCM. Proc. Natl. Acad. Sci. U.S.A. 2008; 105:16107–16112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Sun W., Nandi S., Osman F., Ahn J.S., Jakovleska J., Lorenz A., Whitby M.C.. The FANCM ortholog Fml1 promotes recombination at stalled replication forks and limits crossing over during DNA double-strand break repair. Mol. Cell. 2008; 32:118–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Giannattasio M., Zwicky K., Follonier C., Foiani M., Lopes M., Branzei D.. Visualization of recombination-mediated damage bypass by template switching. Nat. Struct. Mol. Biol. 2014; 21:884–892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Menolfi D., Delamarre A., Lengronne A., Pasero P., Branzei D.. Essential roles of the Smc5/6 complex in replication through natural pausing sites and endogenous DNA damage tolerance. Mol. Cell. 2015; 60:835–846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Garcia-Rodriguez N., Wong R.P., Ulrich H.D.. The helicase Pif1 functions in the template switching pathway of DNA damage bypass. Nucleic Acids Res. 2018; 46:8347–8356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Branzei D., Vanoli F., Foiani M.. SUMOylation regulates Rad18-mediated template switch. Nature. 2008; 456:915–920. [DOI] [PubMed] [Google Scholar]
- 96. Gali H., Juhasz S., Morocz M., Hajdu I., Fatyol K., Szukacsov V., Burkovics P., Haracska L.. Role of SUMO modification of human PCNA at stalled replication fork. Nucleic Acids Res. 2012; 40:6049–6059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Halas A., Krawczyk M., Sledziewska-Gojska E.. PCNA SUMOylation protects against PCNA polyubiquitination-mediated, Rad59-dependent, spontaneous, intrachromosomal gene conversion. Mutat. Res. 2016; 791–792:10–18. [DOI] [PubMed] [Google Scholar]
- 98. Muzi-Falconi M., Giannattasio M., Foiani M., Plevani P.. The DNA polymerase alpha-primase complex: multiple functions and interactions. Sci. World J. 2003; 3:21–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Lopes M., Foiani M., Sogo J.M.. Multiple mechanisms control chromosome integrity after replication fork uncoupling and restart at irreparable UV lesions. Mol. Cell. 2006; 21:15–27. [DOI] [PubMed] [Google Scholar]
- 100. Elvers I., Johansson F., Groth P., Erixon K., Helleday T.. UV stalled replication forks restart by re-priming in human fibroblasts. Nucleic Acids Res. 2011; 39:7049–7057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Wan L., Lou J., Xia Y., Su B., Liu T., Cui J., Sun Y., Lou H., Huang J.. hPrimpol1/CCDC111 is a human DNA primase-polymerase required for the maintenance of genome integrity. EMBO Rep. 2013; 14:1104–1112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Keen B.A., Jozwiakowski S.K., Bailey L.J., Bianchi J., Doherty A.J.. Molecular dissection of the domain architecture and catalytic activities of human PrimPol. Nucleic Acids Res. 2014; 42:5830–5845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Martinez-Jimenez M.I., Calvo P.A., Garcia-Gomez S., Guerra-Gonzalez S., Blanco L.. The Zn-finger domain of human PrimPol is required to stabilize the initiating nucleotide during DNA priming. Nucleic Acids Res. 2018; 46:4138–4151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Guilliam T.A., Brissett N.C., Ehlinger A., Keen B.A., Kolesar P., Taylor E.M., Bailey L.J., Lindsay H.D., Chazin W.J., Doherty A.J.. Molecular basis for PrimPol recruitment to replication forks by RPA. Nature Communications. 2017; 8:15222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Martinez-Jimenez M.I., Lahera A., Blanco L.. Human PrimPol activity is enhanced by RPA. Sci. Rep. 2017; 7:783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Pilzecker B., Buoninfante O.A., Pritchard C., Blomberg O.S., Huijbers I.J., van den Berk P.C., Jacobs H.. PrimPol prevents APOBEC/AID family mediated DNA mutagenesis. Nucleic Acids Res. 2016; 44:4734–4744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Tsaalbi-Shtylik A., Moser J., Mullenders L.H., Jansen J.G., de Wind N.. Persistently stalled replication forks inhibit nucleotide excision repair in trans by sequestering Replication protein A. Nucleic Acids Res. 2014; 42:4406–4413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Schiavone D., Jozwiakowski S.K., Romanello M., Guilbaud G., Guilliam T.A., Bailey L.J., Sale J.E., Doherty A.J.. PrimPol is required for replicative tolerance of G quadruplexes in vertebrate cells. Mol. Cell. 2016; 61:161–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Papadopoulou C., Guilbaud G., Schiavone D., Sale J.E.. Nucleotide pool depletion induces G-Quadruplex-Dependent perturbation of gene expression. Cell Rep. 2015; 13:2491–2503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Pilzecker B., Jacobs H.. Mutating for good: DNA damage responses during somatic hypermutation. Front. Immunol. 2019; 10:438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Kottemann M.C., Smogorzewska A.. Fanconi anaemia and the repair of Watson and Crick DNA crosslinks. Nature. 2013; 493:356–363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Deans A.J., West S.C.. DNA interstrand crosslink repair and cancer. Nat. Rev. Cancer. 2011; 11:467–480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Raschle M., Knipscheer P., Enoiu M., Angelov T., Sun J., Griffith J.D., Ellenberger T.E., Scharer O.D., Walter J.C.. Mechanism of replication-coupled DNA interstrand crosslink repair. Cell. 2008; 134:969–980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Knipscheer P., Raschle M., Smogorzewska A., Enoiu M., Ho T.V., Scharer O.D., Elledge S.J., Walter J.C.. The Fanconi anemia pathway promotes replication-dependent DNA interstrand cross-link repair. Science. 2009; 326:1698–1701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Budzowska M., Graham T.G., Sobeck A., Waga S., Walter J.C.. Regulation of the Rev1-pol zeta complex during bypass of a DNA interstrand cross-link. EMBO J. 2015; 34:1971–1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Semlow D.R., Zhang J., Budzowska M., Drohat A.C., Walter J.C.. Replication-Dependent unhooking of DNA interstrand Cross-links by the NEIL3 Glycosylase. Cell. 2016; 167:498–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Huang J., Liu S., Bellani M.A., Thazhathveetil A.K., Ling C., de Winter J.P., Wang Y., Wang W., Seidman M.M.. The DNA translocase FANCM/MHF promotes replication traverse of DNA interstrand crosslinks. Mol. Cell. 2013; 52:434–446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Rohleder F., Huang J., Xue Y., Kuper J., Round A., Seidman M., Wang W., Kisker C.. FANCM interacts with PCNA to promote replication traverse of DNA interstrand crosslinks. Nucleic Acids Res. 2016; 44:3219–3232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Kuchenbaecker K.B., Ramus S.J., Tyrer J., Lee A., Shen H.C., Beesley J., Lawrenson K., McGuffog L., Healey S., Lee J.M. et al.. Identification of six new susceptibility loci for invasive epithelial ovarian cancer. Nat. Genet. 2015; 47:164–171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Bell D.W., Sikdar N., Lee K.Y., Price J.C., Chatterjee R., Park H.D., Fox J., Ishiai M., Rudd M.L., Pollock L.M. et al.. Predisposition to cancer caused by genetic and functional defects of mammalian Atad5. PLos Genet. 2011; 7:e1002245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Kubota T., Myung K., Donaldson A.D.. Is PCNA unloading the central function of the Elg1/ATAD5 replication factor C-like complex?. Cell Cycle. 2013; 12:2570–2579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Zeman M.K., Cimprich K.A.. Causes and consequences of replication stress. Nat. Cell Biol. 2014; 16:2–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. O’Driscoll M., Ruiz-Perez V.L., Woods C.G., Jeggo P.A., Goodship J.A.. A splicing mutation affecting expression of ataxia-telangiectasia and Rad3-related protein (ATR) results in Seckel syndrome. Nat. Genet. 2003; 33:497–501. [DOI] [PubMed] [Google Scholar]
- 124. Tanaka A., Weinel S., Nagy N., O’Driscoll M., Lai-Cheong J.E., Kulp-Shorten C.L., Knable A., Carpenter G., Fisher S.A., Hiragun M. et al.. Germline mutation in ATR in autosomal- dominant oropharyngeal cancer syndrome. Am. J. Hum. Genet. 2012; 90:511–517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Murga M., Bunting S., Montana M.F., Soria R., Mulero F., Canamero M., Lee Y., McKinnon P.J., Nussenzweig A., Fernandez-Capetillo O.. A mouse model of ATR-Seckel shows embryonic replicative stress and accelerated aging. Nat. Genet. 2009; 41:891–898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Roy R., Chun J., Powell S.N.. BRCA1 and BRCA2: different roles in a common pathway of genome protection. Nat. Rev. Cancer. 2011; 12:68–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Gayther S.A., Warren W., Mazoyer S., Russell P.A., Harrington P.A., Chiano M., Seal S., Hamoudi R., van Rensburg E.J., Dunning A.M. et al.. Germline mutations of the BRCA1 gene in breast and ovarian cancer families provide evidence for a genotype-phenotype correlation. Nat. Genet. 1995; 11:428–433. [DOI] [PubMed] [Google Scholar]
- 128. Sawyer S.L., Tian L., Kahkonen M., Schwartzentruber J., Kircher M. University of Washington Centre for Mendelian, G. Consortium, F.C. . University of Washington Centre for Mendelian, G. Consortium, F.C. Majewski J., Dyment D.A., Innes A.M. et al.. Biallelic mutations in BRCA1 cause a new Fanconi anemia subtype. Cancer Discover. 2015; 5:135–142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Evers B., Jonkers J.. Mouse models of BRCA1 and BRCA2 deficiency: past lessons, current understanding and future prospects. Oncogene. 2006; 25:5885–5897. [DOI] [PubMed] [Google Scholar]
- 130. Wooster R., Bignell G., Lancaster J., Swift S., Seal S., Mangion J., Collins N., Gregory S., Gumbs C., Micklem G.. Identification of the breast cancer susceptibility gene BRCA2. Nature. 1995; 378:789–792. [DOI] [PubMed] [Google Scholar]
- 131. Howlett N.G., Taniguchi T., Olson S., Cox B., Waisfisz Q., De Die-Smulders C., Persky N., Grompe M., Joenje H., Pals G. et al.. Biallelic inactivation of BRCA2 in Fanconi anemia. Science. 2002; 297:606–609. [DOI] [PubMed] [Google Scholar]
- 132. Navarro S., Meza N.W., Quintana-Bustamante O., Casado J.A., Jacome A., McAllister K., Puerto S., Surralles J., Segovia J.C., Bueren J.A.. Hematopoietic dysfunction in a mouse model for Fanconi anemia group D1. Mol. Ther. 2006; 14:525–535. [DOI] [PubMed] [Google Scholar]
- 133. Pawlowska E., Szczepanska J., Blasiak J.. DNA2-An important player in DNA damage response or just another DNA maintenance protein?. Int. J. Mol. Sci. 2017; 18:E1562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Ronchi D., Di Fonzo A., Lin W., Bordoni A., Liu C., Fassone E., Pagliarani S., Rizzuti M., Zheng L., Filosto M. et al.. Mutations in DNA2 link progressive myopathy to mitochondrial DNA instability. Am. J. Hum. Genet. 2013; 92:293–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135. Shaheen R., Faqeih E., Ansari S., Abdel-Salam G., Al-Hassnan Z.N., Al-Shidi T., Alomar R., Sogaty S., Alkuraya F.S.. Genomic analysis of primordial dwarfism reveals novel disease genes. Genome Res. 2014; 24:291–299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Lin W., Sampathi S., Dai H., Liu C., Zhou M., Hu J., Huang Q., Campbell J., Shin-Ya K., Zheng L. et al.. Mammalian DNA2 helicase/nuclease cleaves G-quadruplex DNA and is required for telomere integrity. EMBO J. 2013; 32:1425–1439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137. Turner G., Gedeon A., Mulley J.. X-linked mental retardation with heterozygous expression and macrocephaly: pericentromeric gene localization. Am. J. Med. Genet. 1994; 51:575–580. [DOI] [PubMed] [Google Scholar]
- 138. Moortgat S., Berland S., Aukrust I., Maystadt I., Baker L., Benoit V., Caro-Llopis A., Cooper N.S., Debray F.G., Faivre L. et al.. HUWE1 variants cause dominant X-linked intellectual disability: a clinical study of 21 patients. Eur. J. Hum. Genet. 2018; 26:64–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139. Myant K.B., Cammareri P., Hodder M.C., Wills J., Von Kriegsheim A., Gyorffy B., Rashid M., Polo S., Maspero E., Vaughan L. et al.. HUWE1 is a critical colonic tumour suppressor gene that prevents MYC signalling, DNA damage accumulation and tumour initiation. EMBO Mol. Med. 2017; 9:181–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140. de Groot R.E., Ganji R.S., Bernatik O., Lloyd-Lewis B., Seipel K., Sedova K., Zdrahal Z., Dhople V.M., Dale T.C., Korswagen H.C. et al.. Huwe1-mediated ubiquitylation of dishevelled defines a negative feedback loop in the Wnt signaling pathway. Sci. Signal. 2014; 7:ra26. [DOI] [PubMed] [Google Scholar]
- 141. Zhao X., Heng J.I., Guardavaccaro D., Jiang R., Pagano M., Guillemot F., Iavarone A., Lasorella A.. The HECT-domain ubiquitin ligase Huwe1 controls neural differentiation and proliferation by destabilizing the N-Myc oncoprotein. Nat. Cell Biol. 2008; 10:643–653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142. Zhao F., Wu J., Xue A., Su Y., Wang X., Lu X., Zhou Z., Qu J., Zhou X.. Exome sequencing reveals CCDC111 mutation associated with high myopia. Hum. Genet. 2013; 132:913–921. [DOI] [PubMed] [Google Scholar]
- 143. Keen B.A., Bailey L.J., Jozwiakowski S.K., Doherty A.J.. Human PrimPol mutation associated with high myopia has a DNA replication defect. Nucleic Acids Res. 2014; 42:12102–12111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144. Baumann P., West S.C.. Role of the human RAD51 protein in homologous recombination and double-stranded-break repair. Trends Biochem. Sci. 1998; 23:247–251. [DOI] [PubMed] [Google Scholar]
- 145. Orr N., Lemnrau A., Cooke R., Fletcher O., Tomczyk K., Jones M., Johnson N., Lord C.J., Mitsopoulos C., Zvelebil M. et al.. Genome-wide association study identifies a common variant in RAD51B associated with male breast cancer risk. Nat. Genet. 2012; 44:1182–1184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146. Depienne C., Bouteiller D., Méneret A., Billot S., Groppa S., Klebe S., Charbonnier-Beaupel F., Corvol J.C., Saraiva J.P., Brueggemann N. et al.. RAD51 haploinsufficiency causes congenital mirror movements in humans. Am. J. Hum. Genet. 2012; 90:301–307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147. Ameziane N., May P., Haitjema A., van de Vrugt H.J., van Rossum-Fikkert S.E., Ristic D., Williams G.J., Balk J., Rockx D., Li H. et al.. A novel Fanconi anaemia subtype associated with a dominant-negative mutation in RAD51. Nat. Commun. 2015; 6:8829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148. Tsuzuki T., Fujii Y., Sakumi K., Tominaga Y., Nakao K., Sekiguchi M., Matsushiro A., Yoshimura Y., Morita T.. Targeted disruption of the Rad51 gene leads to lethality in embryonic mice. Proc. Natl. Acad. Sci. U.S.A. 1996; 93:6236–6240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149. Tomas-Roca L., Tsaalbi-Shtylik A., Jansen J.G., Singh M.K., Epstein J.A., Altunoglu U., Verzijl H., Soria L., van Beusekom E., Roscioli T. et al.. De novo mutations in PLXND1 and REV3L cause Mobius syndrome. Nat. Commun. 2015; 6:7199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150. Verzijl H.T., van der Zwaag B., Cruysberg J.R., Padberg G.W.. Mobius syndrome redefined: a syndrome of rhombencephalic maldevelopment. Neurology. 2003; 61:327–333. [DOI] [PubMed] [Google Scholar]
- 151. Bluteau D., Masliah-Planchon J., Clairmont C., Rousseau A., Ceccaldi R., d’Enghien C.D., Bluteau O., Cuccuini W., Gachet S., de Latour R.P. et al.. Biallelic inactivation of REV7 is associated with Fanconi anemia. J. Clin. Invest. 2017; 127:1117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152. Boersma V., Moatti N., Segura-Bayona S., Peuscher M.H., van der Torre J., Wevers B.A., Orthwein A., Durocher D., Jacobs J.J.L.. MAD2L2 controls DNA repair at telomeres and DNA breaks by inhibiting 5′ end resection. Nature. 2015; 521:537–540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153. Xu G., Chapman J.R., Brandsma I., Yuan J., Mistrik M., Bouwman P., Bartkova J., Gogola E., Warmerdam D., Barazas M. et al.. REV7 counteracts DNA double-strand break resection and affects PARP inhibition. Nature. 2015; 521:541–544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154. Boerkoel C.F., Takashima H., John J., Yan J., Stankiewicz P., Rosenbarker L., Andre J.L., Bogdanovic R., Burguet A., Cockfield S. et al.. Mutant chromatin remodeling protein SMARCAL1 causes Schimke immuno-osseous dysplasia. Nat. Genet. 2002; 30:215–220. [DOI] [PubMed] [Google Scholar]
- 155. Morimoto M., Lewis D.B., Lucke T., Boerkoel C.F.. Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJH, Stephens K, Amemiya A. Schimke Immunoosseous Dysplasia. 1993; Seattle: University of Washington. [Google Scholar]
- 156. Baradaran-Heravi A., Cho K.S., Tolhuis B., Sanyal M., Morozova O., Morimoto M., Elizondo L.I., Bridgewater D., Lubieniecka J., Beirnes K. et al.. Penetrance of biallelic SMARCAL1 mutations is associated with environmental and genetic disturbances of gene expression. Hum. Mol. Genet. 2012; 21:2572–2587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157. Betous R., Mason A.C., Rambo R.P., Bansbach C.E., Badu-Nkansah A., Sirbu B.M., Eichman B.F., Cortez D.. SMARCAL1 catalyzes fork regression and Holliday junction migration to maintain genome stability during DNA replication. Genes Dev. 2012; 26:151–162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158. Vaz B., Popovic M., Newman J.A., Fielden J., Aitkenhead H., Halder S., Singh A.N., Vendrell I., Fischer R., Torrecilla I. et al.. Metalloprotease SPRTN/DVC1 orchestrates replication-coupled DNA-Protein crosslink repair. Mol. Cell. 2016; 64:704–719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159. Stingele J., Bellelli R., Alte F., Hewitt G., Sarek G., Maslen S.L., Tsutakawa S.E., Borg A., Kjaer S., Tainer J.A. et al.. Mechanism and regulation of DNA-Protein crosslink repair by the DNA-Dependent metalloprotease SPRTN. Mol. Cell. 2016; 64:688–703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160. Lopez-Mosqueda J., Maddi K., Prgomet S., Kalayil S., Marinovic-Terzic I., Terzic J., Dikic I.. SPRTN is a mammalian DNA-binding metalloprotease that resolves DNA-protein crosslinks. eLife. 2016; 5:e21491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161. Lessel D., Vaz B., Halder S., Lockhart P.J., Marinovic-Terzic I., Lopez-Mosqueda J., Philipp M., Sim J.C., Smith K.R., Oehler J. et al.. Mutations in SPRTN cause early onset hepatocellular carcinoma, genomic instability and progeroid features. Nat. Genet. 2014; 46:1239–1244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162. Maskey R.S., Kim M.S., Baker D.J., Childs B., Malureanu L.A., Jeganathan K.B., Machida Y., van Deursen J.M., Machida Y.J.. Spartan deficiency causes genomic instability and progeroid phenotypes. Nat. Commun. 2014; 5:5744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163. Maskey R.S., Flatten K.S., Sieben C.J., Peterson K.L., Baker D.J., Nam H.J., Kim M.S., Smyrk T.C., Kojima Y., Machida Y. et al.. Spartan deficiency causes accumulation of Topoisomerase 1 cleavage complexes and tumorigenesis. Nucleic Acids Res. 2017; 45:4564–4576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164. Cleaver J.E. Defective repair replication of DNA in xeroderma pigmentosum. Nature. 1968; 218:652–656. [DOI] [PubMed] [Google Scholar]
- 165. Weeda G., van Ham R.C., Vermeulen W., Bootsma D., van der Eb A.J., Hoeijmakers J.H.. A presumed DNA helicase encoded by ERCC-3 is involved in the human repair disorders xeroderma pigmentosum and Cockayne's syndrome. Cell. 1990; 62:777–791. [DOI] [PubMed] [Google Scholar]
- 166. Kraemer K.H., DiGiovanna J.J.. Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJH, Stephens K, Amemiya A. Xeroderma Pigmentosum. 1993; Seattle: GeneReviews((R)). [PubMed] [Google Scholar]
- 167. Masutani C., Kusumoto R., Yamada A., Dohmae N., Yokoi M., Yuasa M., Araki M., Iwai S., Takio K., Hanaoka F.. The XPV (xeroderma pigmentosum variant) gene encodes human DNA polymerase eta. Nature. 1999; 399:700–704. [DOI] [PubMed] [Google Scholar]
- 168. Wang Y.C., Maher V.M., Mitchell D.L., McCormick J.J.. Evidence from mutation spectra that the UV hypermutability of xeroderma pigmentosum variant cells reflects abnormal, error-prone replication on a template containing photoproducts. Mol. Cell Biol. 1993; 13:4276–4283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169. Busuttil R.A., Lin Q., Stambrook P.J., Kucherlapati R., Vijg J.. Mutation frequencies and spectra in DNA polymerase eta-deficient mice. Cancer Res. 2008; 68:2081–2084. [DOI] [PubMed] [Google Scholar]
- 170. Johnson R.E., Prakash S., Prakash L.. Efficient bypass of a thymine-thymine dimer by yeast DNA polymerase, Poleta. Science. 1999; 283:1001–1004. [DOI] [PubMed] [Google Scholar]
- 171. McCulloch S.D., Kokoska R.J., Masutani C., Iwai S., Hanaoka F., Kunkel T.A.. Preferential cis-syn thymine dimer bypass by DNA polymerase eta occurs with biased fidelity. Nature. 2004; 428:97–100. [DOI] [PubMed] [Google Scholar]
- 172. Hendel A., Ziv O., Gueranger Q., Geacintov N., Livneh Z.. Reduced efficiency and increased mutagenicity of translesion DNA synthesis across a TT cyclobutane pyrimidine dimer, but not a TT 6-4 photoproduct, in human cells lacking DNA polymerase eta. DNA Repair (Amst.). 2008; 7:1636–1646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173. Lin Q., Clark A.B., McCulloch S.D., Yuan T., Bronson R.T., Kunkel T.A., Kucherlapati R.. Increased susceptibility to UV-induced skin carcinogenesis in polymerase eta-deficient mice. Cancer Res. 2006; 66:87–94. [DOI] [PubMed] [Google Scholar]
- 174. Dumstorf C.A., Clark A.B., Lin Q., Kissling G.E., Yuan T., Kucherlapati R., McGregor W.G., Kunkel T.A.. Participation of mouse DNA polymerase iota in strand-biased mutagenic bypass of UV photoproducts and suppression of skin cancer. Proc. Natl. Acad. Sci. U.S.A. 2006; 103:18083–18088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175. Kanao R., Yokoi M., Ohkumo T., Sakurai Y., Dotsu K., Kura S., Nakatsu Y., Tsuzuki T., Masutani C., Hanaoka F.. UV-induced mutations in epidermal cells of mice defective in DNA polymerase eta and/or iota. DNA Repair (Amst). 2015; 29:139–146. [DOI] [PubMed] [Google Scholar]
- 176. Nascimento R.M., Otto P.A., de Brouwer A.P., Vianna-Morgante A.M.. UBE2A, which encodes a ubiquitin-conjugating enzyme, is mutated in a novel X-linked mental retardation syndrome. Am. J. Hum. Genet. 2006; 79:549–555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177. Roest H.P., Baarends W.M., de Wit J., van Klaveren J.W., Wassenaar E., Hoogerbrugge J.W., van Cappellen W.A., Hoeijmakers J.H., Grootegoed J.A.. The ubiquitin-conjugating DNA repair enzyme HR6A is a maternal factor essential for early embryonic development in mice. Mol. Cell Biol. 2004; 24:5485–5495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178. Bruinsma C.F., Savelberg S.M., Kool M.J., Jolfaei M.A., Van Woerden G.M., Baarends W.M., Elgersma Y.. An essential role for UBE2A/HR6A in learning and memory and mGLUR-dependent long-term depression. Hum. Mol. Genet. 2016; 25:1–8. [DOI] [PubMed] [Google Scholar]
- 179. Hu Y., Wen W., Yu J.G., Qu S.Q., Wang S.S., Liu J., Li B.S., Luo Y.. Genetic association of UBE2B variants with susceptibility to male infertility in a Northeast Chinese population. Genet. Mol. Res. 2012; 11:4226–4234. [DOI] [PubMed] [Google Scholar]
- 180. Roest H.P., van Klaveren J., de Wit J., van Gurp C.G., Koken M.H., Vermey M., van Roijen J.H., Hoogerbrugge J.W., Vreeburg J.T., Baarends W.M. et al.. Inactivation of the HR6B ubiquitin-conjugating DNA repair enzyme in mice causes male sterility associated with chromatin modification. Cell. 1996; 86:799–810. [DOI] [PubMed] [Google Scholar]
- 181. Baarends W.M., Wassenaar E., Hoogerbrugge J.W., van Cappellen G., Roest H.P., Vreeburg J., Ooms M., Hoeijmakers J.H., Grootegoed J.A.. Loss of HR6B ubiquitin-conjugating activity results in damaged synaptonemal complex structure and increased crossing-over frequency during the male meiotic prophase. Mol. Cell Biol. 2003; 23:1151–1162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182. Gray M.D., Shen J.C., Kamath-Loeb A.S., Blank A., Sopher B.L., Martin G.M., Oshima J., Loeb L.A.. The Werner syndrome protein is a DNA helicase. Nat. Genet. 1997; 17:100–103. [DOI] [PubMed] [Google Scholar]
- 183. Chu W.K., Hickson I.D.. RecQ helicases: multifunctional genome caretakers. Nat. Rev. Cancer. 2009; 9:644–654. [DOI] [PubMed] [Google Scholar]
- 184. Oshima J., Martin G.M., Hisama F.M.. Adam M. P., Ardinger H. H., Pagon R. A., Wallace S. E., Bean L. J. H., Stephens K., Amemiya A.. Werner Syndrome. 1993; Seattle: GeneReviews((R)). [PubMed] [Google Scholar]
- 185. Yu C.E., Oshima J., Fu Y.H., Wijsman E.M., Hisama F., Alisch R., Matthews S., Nakura J., Miki T., Ouais S. et al.. Positional cloning of the Werner's syndrome gene. Science. 1996; 272:258–262. [DOI] [PubMed] [Google Scholar]
- 186. Mckusick V.A. MEDICAL genetics 1962. J. Chronic Dis. 1963; 16:457–634. [DOI] [PubMed] [Google Scholar]
- 187. Lebel M., Leder P.. A deletion within the murine Werner syndrome helicase induces sensitivity to inhibitors of topoisomerase and loss of cellular proliferative capacity. Proc. Natl. Acad. Sci. U.S.A. 1998; 95:13097–13102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188. Chang S., Multani A.S., Cabrera N.G., Naylor M.L., Laud P., Lombard D., Pathak S., Guarente L., DePinho R.A.. Essential role of limiting telomeres in the pathogenesis of Werner syndrome. Nat. Genet. 2004; 36:877–882. [DOI] [PubMed] [Google Scholar]
- 189. Gomes N.M., Ryder O.A., Houck M.L., Charter S.J., Walker W., Forsyth N.R., Austad S.N., Venditti C., Pagel M., Shay J.W. et al.. Comparative biology of mammalian telomeres: hypotheses on ancestral states and the roles of telomeres in longevity determination. Aging Cell. 2011; 10:761–768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190. Crabbe L., Verdun R.E., Haggblom C.I., Karlseder J.. Defective telomere lagging strand synthesis in cells lacking WRN helicase activity. Science. 2004; 306:1951–1953. [DOI] [PubMed] [Google Scholar]
- 191. Chang D.J., Cimprich K.A.. DNA damage tolerance: when it's OK to make mistakes. Nat. Chem. Biol. 2009; 5:82–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192. Hendel A., Krijger P.H., Diamant N., Goren Z., Langerak P., Kim J., Reissner T., Lee K.Y., Geacintov N.E., Carell T. et al.. PCNA ubiquitination is important, but not essential for translesion DNA synthesis in mammalian cells. PLoS Genet. 2011; 7:e1002262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193. Krijger P.H., van den Berk P.C., Wit N., Langerak P., Jansen J.G., Reynaud C.A., de Wind N., Jacobs H.. PCNA ubiquitination-independent activation of polymerase η during somatic hypermutation and DNA damage tolerance. DNA Repair (Amst.). 2011; 10:1051–1059. [DOI] [PubMed] [Google Scholar]
- 194. Stancel J.N., McDaniel L.D., Velasco S., Richardson J., Guo C., Friedberg E.C.. Polk mutant mice have a spontaneous mutator phenotype. DNA Repair (Amst.). 2009; 8:1355–1362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195. Pfeifer G.P., You Y.H., Besaratinia A.. Mutations induced by ultraviolet light. Mutat. Res. 2005; 571:19–31. [DOI] [PubMed] [Google Scholar]
- 196. Mouret S., Baudouin C., Charveron M., Favier A., Cadet J., Douki T.. Cyclobutane pyrimidine dimers are predominant DNA lesions in whole human skin exposed to UVA radiation. Proc. Natl. Acad. Sci. U.S.A. 2006; 103:13765–13770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197. You Y.H., Lee D.H., Yoon J.H., Nakajima S., Yasui A., Pfeifer G.P.. Cyclobutane pyrimidine dimers are responsible for the vast majority of mutations induced by UVB irradiation in mammalian cells. J. Biol. Chem. 2001; 276:44688–44694. [DOI] [PubMed] [Google Scholar]
- 198. Tommasi S., Denissenko M.F., Pfeifer G.P.. Sunlight induces pyrimidine dimers preferentially at 5-methylcytosine bases. Cancer Res. 1997; 57:4727–4730. [PubMed] [Google Scholar]
- 199. Ikehata H., Ono T.. The mechanisms of UV mutagenesis. J. Radiat. Res. 2011; 52:115–125. [DOI] [PubMed] [Google Scholar]
- 200. Johnson R.E., Haracska L., Prakash S., Prakash L.. Role of DNA polymerase eta in the bypass of a (6-4) TT photoproduct. Mol. Cell Biol. 2001; 21:3558–3563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201. Liu Z., Wang L., Zhong D.. Dynamics and mechanisms of DNA repair by photolyase. Phys. Chem. Chem. Phys. 2015; 17:11933–11949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202. Lawrence C.W., Krauss B.R., Christensen R.B.. New mutations affecting induced mutagenesis in yeast. Mutat. Res. 1985; 150:211–216. [DOI] [PubMed] [Google Scholar]
- 203. Lambert S., Watson A., Sheedy D.M., Martin B., Carr A.M.. Gross chromosomal rearrangements and elevated recombination at an inducible site-specific replication fork barrier. Cell. 2005; 121:689–702. [DOI] [PubMed] [Google Scholar]
- 204. Paek A.L., Kaochar S., Jones H., Elezaby A., Shanks L., Weinert T.. Fusion of nearby inverted repeats by a replication-based mechanism leads to formation of dicentric and acentric chromosomes that cause genome instability in budding yeast. Genes Dev. 2009; 23:2861–2875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205. Lambert S., Mizuno K., Blaisonneau J., Martineau S., Chanet R., Freon K., Murray J.M., Carr A.M., Baldacci G.. Homologous recombination restarts blocked replication forks at the expense of genome rearrangements by template exchange. Mol. Cell. 2010; 39:346–359. [DOI] [PubMed] [Google Scholar]
- 206. Loytynoja A., Goldman N.. Short template switch events explain mutation clusters in the human genome. Genome Res. 2017; 27:1039–1049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207. Wilson A., Laurenti E., Oser G., van der Wath R.C., Blanco-Bose W., Jaworski M., Offner S., Dunant C.F., Eshkind L., Bockamp E. et al.. Hematopoietic stem cells reversibly switch from dormancy to self-renewal during homeostasis and repair. Cell. 2008; 135:1118–1129. [DOI] [PubMed] [Google Scholar]
- 208. Morrison S.J., Uchida N., Weissman I.L.. The biology of hematopoietic stem cells. Annu. Rev. Cell Dev. Biol. 1995; 11:35–71. [DOI] [PubMed] [Google Scholar]
- 209. Cabezas-Wallscheid N., Klimmeck D., Hansson J., Lipka D.B., Reyes A., Wang Q., Weichenhan D., Lier A., von Paleske L., Renders S. et al.. Identification of regulatory networks in HSCs and their immediate progeny via integrated proteome, transcriptome, and DNA methylome analysis. Cell Stem Cell. 2014; 15:507–522. [DOI] [PubMed] [Google Scholar]
- 210. Pietras E.M., Reynaud D., Kang Y.A., Carlin D., Calero-Nieto F.J., Leavitt A.D., Stuart J.M., Gottgens B., Passegue E.. Functionally distinct subsets of lineage-biased multipotent progenitors control blood production in normal and regenerative conditions. Cell Stem Cell. 2015; 17:35–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211. Rossi D.J., Bryder D., Seita J., Nussenzweig A., Hoeijmakers J., Weissman I.L.. Deficiencies in DNA damage repair limit the function of haematopoietic stem cells with age. Nature. 2007; 447:725–729. [DOI] [PubMed] [Google Scholar]
- 212. Flach J., Bakker S.T., Mohrin M., Conroy P.C., Pietras E.M., Reynaud D., Alvarez S., Diolaiti M.E., Ugarte F., Forsberg E.C. et al.. Replication stress is a potent driver of functional decline in ageing haematopoietic stem cells. Nature. 2014; 512:198–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213. Pilzecker B., Buoninfante O.A., van den Berk P., Lancini C., Song J.Y., Citterio E., Jacobs H.. DNA damage tolerance in hematopoietic stem and progenitor cells in mice. Proc. Natl. Acad. Sci. U.S.A. 2017; 114:E6875–E6883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214. Martin-Pardillos A., Tsaalbi-Shtylik A., Chen S., Lazare S., van Os R.P., Dethmers-Ausema A., Fakouri N.B., Bosshard M., Aprigliano R., van Loon B. et al.. Genomic and functional integrity of the hematopoietic system requires tolerance of oxidative DNA lesions. Blood. 2017; 130:1523–1534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215. Young K., Borikar S., Bell R., Kuffler L., Philip V., Trowbridge J.J.. Progressive alterations in multipotent hematopoietic progenitors underlie lymphoid cell loss in aging. J. Exp. Med. 2016; 213:2259–2267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216. Leins H., Mulaw M., Eiwen K., Sakk V., Liang Y., Denkinger M., Geiger H., Schirmbeck R.. Aged murine hematopoietic stem cells drive aging-associated immune remodeling. Blood. 2018; 132:565–576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217. Amrani Y.M., Gill J., Matevossian A., Alonzo E.S., Yang C., Shieh J.H., Moore M.A., Park C.Y., Sant’Angelo D.B., Denzin L.K.. The Paf oncogene is essential for hematopoietic stem cell function and development. J. Exp. Med. 2011; 208:1757–1765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218. Puccetti M.V., Fischer M.A., Arrate M.P., Boyd K.L., Duszynski R.J., Betous R., Cortez D., Eischen C.M.. Defective replication stress response inhibits lymphomagenesis and impairs lymphocyte reconstitution. Oncogene. 2017; 36:2553–2564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219. Mgbemena V.E., Signer R.A.J., Wijayatunge R., Laxson T., Morrison S.J., Ross T.S.. Distinct Brca1 mutations differentially reduce hematopoietic stem cell function. Cell Rep. 2017; 18:947–960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220. Vasanthakumar A., Arnovitz S., Marquez R., Lepore J., Rafidi G., Asom A., Weatherly M., Davis E.M., Neistadt B., Duszynski R. et al.. Brca1 deficiency causes bone marrow failure and spontaneous hematologic malignancies in mice. Blood. 2016; 127:310–313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221. Langerak P., Nygren A.O., Krijger P.H., van den Berk P.C., Jacobs H.. A/T mutagenesis in hypermutated immunoglobulin genes strongly depends on PCNAK164 modification. J. Exp. Med. 2007; 204:1989–1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222. White R.R., Milholland B., de Bruin A., Curran S., Laberge R.M., van Steeg H., Campisi J., Maslov A.Y., Vijg J.. Controlled induction of DNA double-strand breaks in the mouse liver induces features of tissue ageing. Nat. Commun. 2015; 6:6790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223. Brown J.S., O’Carrigan B., Jackson S.P., Yap T.A.. Targeting DNA repair in cancer: Beyond PARP inhibitors. Cancer Discovery. 2017; 7:20–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224. Ceppi P., Novello S., Cambieri A., Longo M., Monica V., Lo Iacono M., Giaj-Levra M., Saviozzi S., Volante M., Papotti M. et al.. Polymerase eta mRNA expression predicts survival of non-small cell lung cancer patients treated with platinum-based chemotherapy. Clin. Cancer Res. 2009; 15:1039–1045. [DOI] [PubMed] [Google Scholar]
- 225. Buoninfante O.A., Pilzecker B., Aslam M.A., Zavrakidis I., van der Wiel R., van de Ven M., van den Berk P.C.M., Jacobs H.. Precision cancer therapy: profiting from tumor specific defects in the DNA damage tolerance system. Oncotarget. 2018; 9:18832–18843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226. Lord C.J., Ashworth A.. Mechanisms of resistance to therapies targeting BRCA-mutant cancers. Nat. Med. 2013; 19:1381–1388. [DOI] [PubMed] [Google Scholar]
- 227. Ray Chaudhuri A., Callen E., Ding X., Gogola E., Duarte A.A., Lee J.E., Wong N., Lafarga V., Calvo J.A., Panzarino N.J. et al.. Replication fork stability confers chemoresistance in BRCA-deficient cells. Nature. 2016; 535:382–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228. Villafañez F., García I.A., Carbajosa S., Pansa M.F., Mansilla S., Llorens M.C., Angiolini V., Guantay L., Jacobs H., Madauss K.P. et al.. AKT inhibition impairs PCNA ubiquitylation and triggers synthetic lethality in homologous recombination-deficient cells submitted to replication stress. Oncogene. 2019; 38:4310–4324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229. Toledo L.I., Murga M., Zur R., Soria R., Rodriguez A., Martinez S., Oyarzabal J., Pastor J., Bischoff J.R., Fernandez-Capetillo O.. A cell-based screen identifies ATR inhibitors with synthetic lethal properties for cancer-associated mutations. Nat. Struct. Mol. Biol. 2011; 18:721–727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230. Yang Y., Gao Y., Mutter-Rottmayer L., Zlatanou A., Durando M., Ding W., Wyatt D., Ramsden D., Tanoue Y., Tateishi S. et al.. DNA repair factor RAD18 and DNA polymerase Polkappa confer tolerance of oncogenic DNA replication stress. J. Cell Biol. 2017; 216:3097–3115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231. Yamanaka K., Dorjsuren D., Eoff R.L., Egli M., Maloney D.J., Jadhav A., Simeonov A., Lloyd R.S.. A comprehensive strategy to discover inhibitors of the translesion synthesis DNA polymerase kappa. PLoS One. 2012; 7:e45032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232. Inoue A., Kikuchi S., Hishiki A., Shao Y., Heath R., Evison B.J., Actis M., Canman C.E., Hashimoto H., Fujii N.. A small molecule inhibitor of monoubiquitinated Proliferating Cell Nuclear Antigen (PCNA) inhibits repair of interstrand DNA cross-link, enhances DNA double strand break, and sensitizes cancer cells to cisplatin. J. Biol. Chem. 2014; 289:7109–7120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233. Zhao H., Lo Y.H., Ma L., Waltz S.E., Gray J.K., Hung M.C., Wang S.C.. Targeting tyrosine phosphorylation of PCNA inhibits prostate cancer growth. Mol. Cancer Ther. 2011; 10:29–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234. Muller R., Misund K., Holien T., Bachke S., Gilljam K.M., Vatsveen T.K., Ro T.B., Bellacchio E., Sundan A., Otterlei M.. Targeting proliferating cell nuclear antigen and its protein interactions induces apoptosis in multiple myeloma cells. PLoS One. 2013; 8:e70430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235. Smith S.J., Gu L., Phipps E.A., Dobrolecki L.E., Mabrey K.S., Gulley P., Dillehay K.L., Dong Z., Fields G.B., Chen Y.R. et al.. A Peptide mimicking a region in proliferating cell nuclear antigen specific to key protein interactions is cytotoxic to breast cancer. Mol. Pharmacol. 2015; 87:263–276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236. Vanarotti M., Evison B.J., Actis M.L., Inoue A., McDonald E.T., Shao Y., Heath R.J., Fujii N.. Small-molecules that bind to the ubiquitin-binding motif of REV1 inhibit REV1 interaction with K164-monoubiquitinated PCNA and suppress DNA damage tolerance. Bioorg. Med. Chem. 2018; 26:2345–2353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237. Liang Q., Dexheimer T.S., Zhang P., Rosenthal A.S., Villamil M.A., You C., Zhang Q., Chen J., Ott C.A., Sun H. et al.. A selective USP1-UAF1 inhibitor links deubiquitination to DNA damage responses. Nat. Chem. Biol. 2014; 10:298–304. [DOI] [PMC free article] [PubMed] [Google Scholar]