

Summary
The microtubule (MT) network is an essential regulator of insulin secretion from pancreatic β-cells, which is central to blood-sugar homeostasis. We find that glucose metabolism induces insulin secretion but also increases formation of Golgi-derived microtubules (GDMTs), notably with the same biphasic kinetics as insulin exocytosis. Furthermore, GDMT nucleation is controlled by a glucose signal-transduction pathway through cAMP and its effector EPAC2. Preventing new GDMT nucleation dramatically affects the pipeline of insulin production, storage and release. There is an overall reduction of β-cell insulin content, and remaining insulin becomes retained within the Golgi, likely because of stalling of insulin-granule budding. While not preventing glucose-induced insulin exocytosis, the diminished granule availability substantially blunts the amount secreted. Constant dynamic maintenance of the GDMT network is therefore critical for normal β-cell physiology. Our study demonstrates that the biogenesis of post-Golgi carriers, particularly large secretory granules, requires ongoing nucleation/replenishment of the GDMT network.
Graphical Abstract

eTOC blurb:
Trogden et al. show that upon stimulation of pancreatic β-cells by glucose, the nucleation of Golgi-derived microtubules (GDMTs) is stimulated through the secretion-regulating cAMP/EPAC2 pathway. Glucose-dependent GDMTs are critical for replenishment of the insulin granule pool, proper secretion levels, and long-term β-cell functionality.
Introduction
Pancreatic β-cells secrete insulin to control glucose homeostasis in the body [1]. The failure of this process results in diabetes mellitus, a condition when peripheral tissues are unable to take up blood glucose [2], disorganizing energy metabolism and causing a variety of critical complications [3].
Glucose signaling is the chief determinant of insulin secretion from β-cells [4], while a combination of nutritional, hormonal, and neuronal inputs modulate the magnitude of response [1, 5, 6]. It is known that high glucose can promote cAMP production an important potentiator of insulin exocytosis, through several pathways including EPAC2 [7–12].
Besides the nature and strength of the stimuli, the dosage of insulin secretion is determined by the number of readily releasable secretory granules, stored in the β-cell cytoplasm in anticipation of a high-glucose signal [13, 14]. Although only a small fraction of granules are released at each stimulation, the granule pool has to be replenished in order to maintain readiness for another secretion stimulus and ensure long-term β-cell health [15]. This is accomplished by increasing insulin biogenesis as soon as the high glucose signal is received [16, 17]. The biogenesis process includes the translation of the insulin precursor, preproinsulin, its processing into proinsulin in the ER [18], and transition through the Golgi apparatus and the TGN into nascent secretory vesicles where it undergoes cleavage into the mature insulin polypeptide [19]. It is known from other cell types that MTs, cytoskeletal polymers that serve as tracks for molecular motors to drive secretory transport [20–24], provide a scaffold for vesicle budding from the TGN [25]. While smaller post-Golgi carriers are able to form without MTs [24, 26], the sheer size of insulin granules suggests that they required additional assistance for their budding, prompting our hypothesis that MTs are critical for insulin granule biogenesis at the TGN.
MTs in β-cells form a uniquely dense, interlocking network [27, 28]. Transport of insulin granules along these non-directional tracks (“random walk”) [28–30] serves to regulate availability of granules for secretion [27]. At the cell periphery, such random transport serves to move excessive granules away from the secretion sites and thus provides one method of fine-tuning glucose stimulated insulin secretion (GSIS). In high glucose, MTs are partially destabilized, allowing for more granules to be secreted [27]. This MT function is important in functional β-cells, where a large number of granules is stored awaiting the secretion stimulus.
Here, in addition to this short-term function of MTs in GSIS regulation, we investigate a long-term MT function in insulin biogenesis. Interestingly, a high glucose signal in β-cells promotes both insulin biogenesis and MT network remodeling. While in most vertebrate cells MTs are formed (nucleated) from the centrosome-based MT-organizing center (MTOC), in β-cells most MTs form at the cytosolic surface of the Golgi membrane [27] (Golgi-derived MTs, or GDMTs). We have previously observed that GDMT nucleation is stimulated by a high glucose stimulus [27], prompting our hypothesis that GDMTs are a part of the machinery whereby glucose-dependent pathways coordinate insulin-granule production, storage, and release. Being formed directly at the insulin-packaging organelle, the Golgi, GDMTs are perfectly positioned for this function.
To date, however, neither the regulation nor function of GDMTs in β-cells has been studied. Here, we explore how the increase in GDMTs is regulated and what role these MTs play in β-cells. Using both the secretion-competent MIN6 immortalized β-cell line and intact pancreatic islets, we show that the increase in GDMT nucleation upon glucose stimulation is regulated by EPAC2 downstream of cAMP signaling, and that GDMTs play a critical, multi-faceted role in β-cells, acting in insulin biogenesis and the secretion/storage balance of insulin granules.
Together, our previous and current data indicate that MTs, on one hand, are necessary for insulin-granule production, and, on the other hand, precisely balance storage vs. secretion of insulin granules [27]. Thus, MTs are critical for maintaining β-cell health and for the reestablishment of β-cell homeostasis after each round of insulin secretion.
Results
High glucose induces biphasic GDMT nucleation
We have observed previously that β-cell GDMT nucleation increases during high-glucose stimulation [27]. To characterize the time course of this change, we employed a MT regrowth assay that measures the capacity of MT-nucleating structures to produce new MTs. To achieve this, MIN6 β-cells and mouse pancreatic islets were treated with nocodazole, a compound that causes complete MT depolymerization and fragmentation of the Golgi (compare Figure S1A and S1B). After nocodazole washout and a brief MT-regrowth period, the cells or islets were fixed and immunostained to visualize newly formed MTs (Figures 1A–F and 1H–K, Video S1). The advantage of this assay is the detection of new MTs without the background of the preexisting MT network. We used a MT +TIP maker (either EB1 or EB3) to visualize newly formed MTs, which coats short MTs during regrowth (Figure S1C–H).
Figure 1. Rapid bi-phasic response of β-cell GDMT nucleation to glucose.
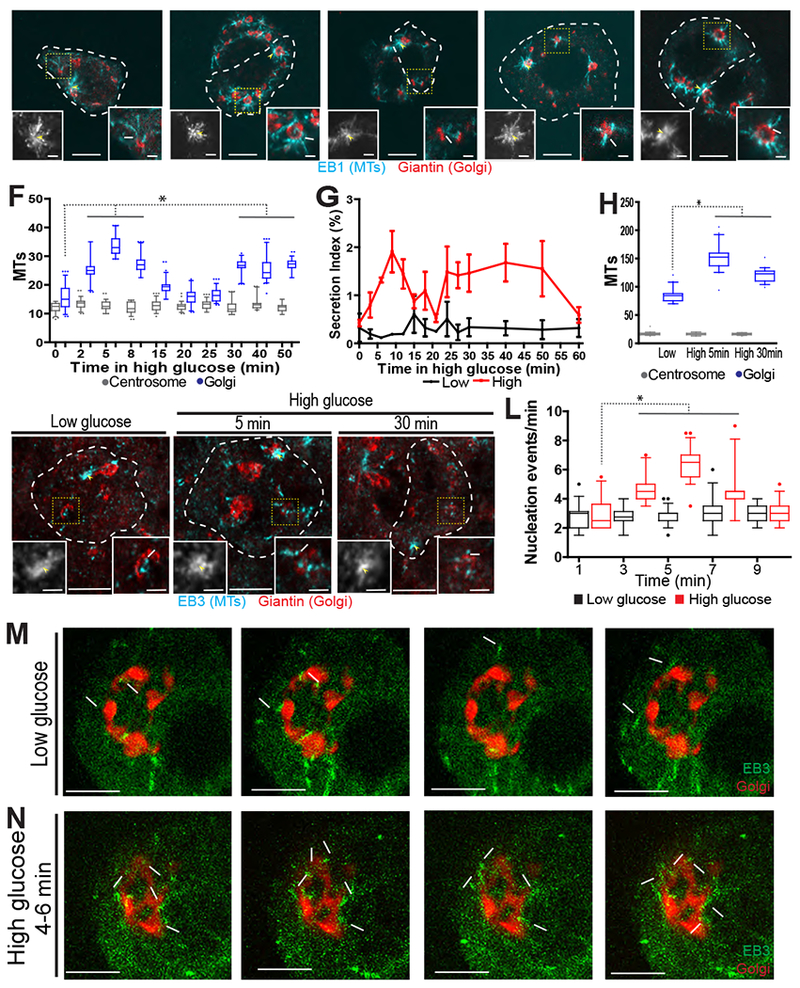
A-E) MIN6 cells following nocodazole washout. Cell outline, a white dotted line. Golgi (Giantin, red) and emerging MTs (EB1, cyan). Centrosome (left inset, yellow arrowhead) and GDMT nucleation (right inset, white arrow). F) GDMT (blue), but not centrosomal (grey) MT nucleation in MIN6 increases in two waves over time in high glucose. Number of new MTs (EB1) is shown as a 5-95% box plot. p < 0.001, one-way ANOVA and Tukey’s multiple comparison test; n = 56-81 cells. G) Secretion index (insulin released over cellular content) in MIN6 cells over time for low glucose (black line) and high glucose (red line). Error bars, standard deviation. H) GDMT (blue) and centrosomal (grey) nucleation (EB3) in islet β-cells. Shown as a 5-95% box plot. (p < 0.001, one-way ANOVA and Tukey’s multiple comparison test; n = 40 cells from 4 islets for each condition.) I-K) A single β-cell (outlined in a white dotted line) in an islet following nocodazole washout. Golgi (Giantin, red) and emerging MTs (EB3, cyan). Centrosome (left inset, yellow arrowhead) and an example of GDMT nucleation (right inset, white arrow). Low glucose (I), high glucose 5 minutes (J) and high glucose 30 minutes (K). L) GDMT nucleation events per minute in low (black) and high (red) glucose. p < 0.001, one-way ANOVA and Tukey’s multiple comparison test; n = 17-36 cells. M-N) Three stills from a time-lapse of MIN6 cells expressing EB3-mEmerald (+TIPs) and TGN-RFP (Golgi). Time, seconds. GDMTs (arrows) and centrosome (asterisk). Last panel is a maximum intensity projection over time (overlay). Low glucose (M), high glucose for 4-6 minutes (N). All images are maximum intensity projections of three confocal z-planes. See also Figures S1 and S4 and Videos S1 and S2.
Regrowth assays in MIN6 cells show that, in response to a high-glucose stimulus, GDMT nucleation increases in two waves (5 and 30 minutes after stimulation), which mirror the biphasic GSIS curve (Figure 1G), while centrosomal nucleation remains constant (Figure 1A–F). Furthermore, regrowth assays in intact mouse pancreatic islets show a similar trend (Figure 1H–K), indicating that this phenomenon is maintained in the presence of natural cell-cell communication.
In addition to measuring MT-nucleation potential of the Golgi membrane, we have validated GDMT formation as a part of physiological steady-state MT dynamics by live-cell imaging of MIN6 cells expressing fluorescently-tagged Golgi- and MT +TIP-markers (Video S2). To precisely calculate MTs originating from the Golgi, +TIP comets that emerged at the Golgi were manually quantified in 3-D stacks by both open (Figure 1L–N) and blinded analysis (Figure S1I). +TIP comets preexisting in time and/or approaching the Golgi from above and below were excluded from the analysis. Importantly, steady-state GDMT nucleation follows a similar rapid rise within five minutes in high glucose (Figure 1L–N), confirming that glucose signaling efficiently promotes GDMT nucleation.
Increased GDMT nucleation proceeds Golgi expansion
There are two potential explanations for the glucose-dependent GDMT nucleation increase. First, the density of specific nucleation events/sites per Golgi membrane unit could be increased by a specific mechanism [31]. Second, the known phenomenon of Golgi size expansion in response to high glucose [32] could lead to a proportional increase in nucleation site number. To distinguish between these possibilities, we analyzed Golgi expansion dynamics (Figure 2). We found that the first peak in GDMT nucleation precedes Golgi expansion (Figure 2), and thus is not a result of increased Golgi membrane area. This observation prompted our hypothesis that the first wave of GDMT nucleation might be involved in early insulin transport events at the Golgi. Accordingly, in this study we concentrate predominantly on this first nucleation wave, which occurs as a rapid response to glucose stimulation.
Figure 2. Golgi expands following the first phase of GSIS in MIN6 cells.
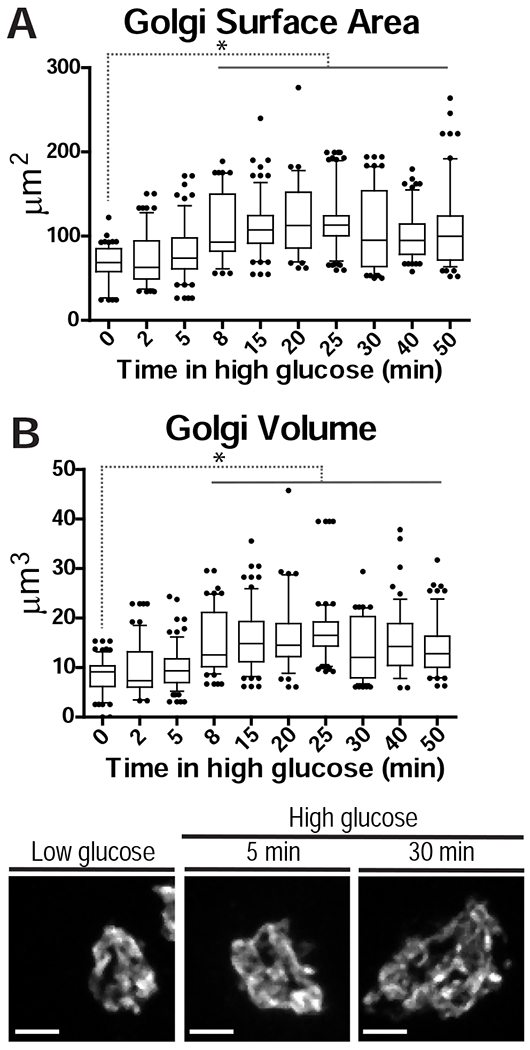
A-B) Golgi surface area (A) and volume (B) during high glucose treatment. p < 0.001, one-way ANOVA and Tukey’s multiple comparison test; n = 56-81 cells. C-E) Golgi (Giantin) at indicated high-glucose treatment times. All images are maximum intensity projections of whole-cell confocal z-stacks.
Regulation of GDMT nucleation by cAMP
The similarity of the GDMT and GSIS curves over time (Figure 1 F–G) suggested that a pathway involved in insulin secretion may also regulate GDMT nucleation. Thus, we performed a screen of small molecules that block or mimic different steps in the insulin secretion pathway (Figure S2A and S2B). Upon entrance into the β-cell, glucose is phosphorylated by glucokinase and processed in the glycolytic pathway and TCA cycle [5], producing intermediate metabolites and ATP, which triggers a calcium influx and, subsequently, insulin exocytosis [6]. Additionally, high glucose promotes cAMP production [7–9], which can also potentiate insulin release [33]. We found that glucokinase activation increased GDMT nucleation in low glucose, indicating that GDMT nucleation is tied to glucose metabolism (Figure S2C). A number of other downstream targets, including pyruvate, glycolysis, and GTP were not involved (Figure S2C).
We further tested the involvement of the cAMP and calcium signaling pathways, which are known to affect each other’s function [34, 35]. A cAMP analog, 8-br-cAMP, caused an increase in basal GDMT nucleation to high-glucose levels, as detected by MT regrowth assays in either MIN6 cells (Figure 3C–D and 3I) or intact islets (Figure 3 J–L) and by steady-state MT nucleation assays in MIN6 cells (Figure 3M–O, Video S3). Similar effects on GDMT nucleation were observed when intracellular calcium levels were changed by a number of means (Figure S2D and S3). However, the GDMT increase caused by 8-br-cAMP was not attenuated by calcium chelators EGTA (Figure 3E–F and 3I) or BAPTA-AM (Figure S3O–P and S3W), indicating that this effect is not the result of cAMP-induced release of intracellular calcium stores [36]. Additionally, the adenylyl cyclase activator Forskolin (Figure 3G–I) and the fast cell-permeable cAMP analog 8-br-cAMP-AM (Figure3I and S3K–L), showed similar results. Taken together, these results indicate that while calcium may play a role in GDMT nucleation in β-cells, cAMP is the predominant regulator.
Figure 3. cAMP regulates GDMT nucleation in β-cells.
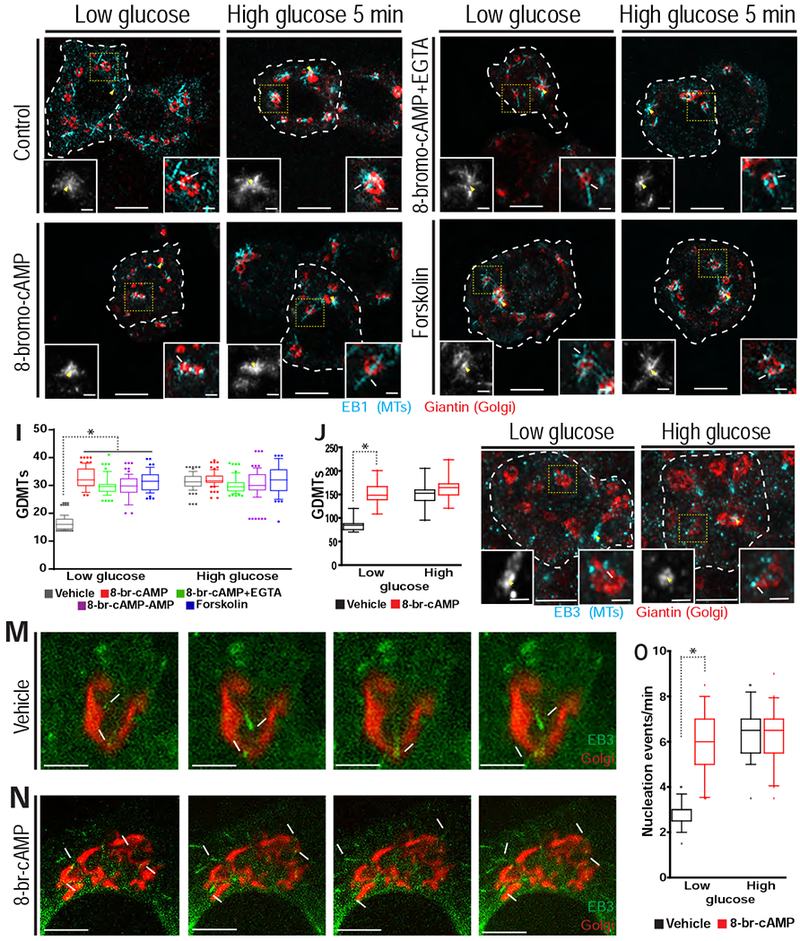
A-H) MIN6 cells (white dotted line) following nocodazole washout. A-B) DMSO control. C-D) 8-br-cAMP. E-F) 8-br-cAMP and EGTA. G-H) Forskolin. Golgi (Giantin, red) and emerging MTs (EB1, cyan). Centrosome (left inset, yellow arrowhead) and GDMT nucleation (right inset, white arrow). I) GDMT nucleation for low (left) and high glucose (right). p < 0.001, one-way ANOVA and Tukey’s multiple comparison test; n = 49-86 cells. J) GDMT nucleation in β-cells of intact islets in DMSO control (black) and 8-br-cAMP (red) in low and high glucose. p < 0.001, one-way ANOVA and Tukey’s multiple comparison test; n = 40 cells from 4 islets for each condition. K-L) A single β-cell (white dotted line) in an islet following nocodazole washout. 8-br-cAMP in low glucose (K) or high glucose 5 minutes (L). Golgi (Giantin, red) and emerging MTs (EB3, cyan). Centrosome (left inset, yellow arrowhead) and an example of GDMT nucleation (right inset, white arrow). M-N) Stills from a time lapse of MIN6 cells expressing EB3-mEmerald (+TIPs) and TGN-RFP (Golgi), during high glucose treatment combined with vehicle control (M) or 8-br-cAMP (N). Time, seconds. GDMTs (arrows) and centrosome (asterisk). Last panel is a maximum intensity projection over time (overlay). O) Nucleation events per minute for DMSO (black) and 8-br-cAMP (red). p < 0.001, one-way ANOVA and Tukey’s multiple comparison test; n = 36-41 cells. All images are maximum intensity projections of three confocal z-planes. See also Figures S2, S3 and S4 and Video S3.
EPAC2, not PKA, mediates the roles of cAMP in GDMT nucleation
cAMP has two downstream effectors, protein kinase A (PKA) and EPAC, a Rap1 guanine exchange factor (GEF). MIN6 cells express only the EPAC2 paralog at detectable levels, not the more ubiquitously expressed EPAC1 (Figure 4O top panel, compare to retinal pigment epithelial cells, RPE1 in Figure S5P). EPAC activator (8-CPT-2Me-cAMP) treatment resulted in increased GDMT formation in MT regrowth assays (Figure 4A–D, 4M–N and 4P–Q)) and steady-state assays (Figure 5A–B, 5G Video S4), mimicking the five-minute response to high glucose in control cells. The effect remained in a co-treatment with the EPAC activator and EGTA (Figure S3U–V and Figure 4M), indicating that it was not caused by calcium-induced calcium release from the ER downstream of EPAC2 [36].
Figure 4. EPAC2 regulates GDMT nucleation downstream of cAMP in β-cells.

A-L) MIN6 cells (white dotted line) following nocodazole washout. A-B) DMSO control. C-D) EPAC activator (8-pCPT-2O-Me-cAMP). E-F) EPAC inhibitor HJC0197. G-H) EPAC inhibitor ESI-09. I-J) 8-br-cAMP and EIS-09. K-L) KCl and ESI-09. Golgi (Giantin, red) and emerging MTs (EB1, cyan). Centrosome (left inset, yellow arrowhead) and GDMT nucleation (right inset, white arrow). M) GDMT nucleation in each condition for low (left) and high glucose (right). p < 0.001, one-way ANOVA and Tukey’s multiple comparison test; n = 51-86 cells. N) GDMT nucleation in DMSO (grey), EPAC activator (red) and ESI-09 (blue) in low and high glucose in β-cells of intact islets. 5-95% boxplots, p < 0.001, one-way ANOVA and Tukey’s multiple comparison test; n = 40 cells from 4 islets for each condition. O) Top: western blots for EPAC1 and EPAC2 in MIN6 cells. GAPDH, loading control. Bottom: western blots from MIN6 cells transduced with shRNA as indicated probed for EPAC2 and GAPDH (loading control). P-S) A single β-cell (white dotted line) in an islet following nocodazole washout. EPAC activator (P-O). ESI-09 (R-S). Low glucose (P, R). High glucose 5 minutes (Q, S). Golgi (Giantin, red) and emerging MTs (EB3, cyan). Centrosome (left inset, yellow arrowhead) and an example of GDMT nucleation (right inset, white arrow). T-W) MIN6 cells (white dotted line) following nocodazole washout. T-U) Scrambled shRNA. V-W) EPAC2 shRNA#1. Golgi (Giantin, red) and emerging MTs (EB1, cyan). Centrosome (lower, left inset, yellow arrowhead), GFP signal (upper right inset) and GDMT nucleation (lower, right inset, white arrow). All images are maximum intensity projections of three confocal z-planes. See also Figures S3, S4 and S5.
Figure 5. EPAC2 is the major homolog regulating GDMT nucleation.

A-D) Stills from a time-lapse (time, seconds) of MIN6 cells expressing EB3-mEmerald (+TIPs) and TGN-RFP (Golgi) in low glucose (A-B) and high glucose for 4-6 minutes (C-D). Vehicle control (A and C), EPAC activator (B) or HJC0197 (D). GDMTs (arrows) and centrosome (asterisk). Last panel is a maximum intensity projection over time (overlay). E-F) MIN6 cell immunostained for EPAC2 (cyan) and Golgi (GCC185, red). Low glucose (E) or high glucose 5 minutes (F). Insets (yellow dotted line) of the Golgi region (bottom panels). G) Nucleation events per minute for DMSO (grey), EPAC activator (red) and HJC0197 (blue) treated cells in low and high glucose. 5-95% boxplots, p < 0.001, one-way ANOVA and Tukey’s multiple comparison test; n = 36-40 cells. HI) MIN6 cell immunostained for EPAC2 (cyan) and MTs (red). DMSO (Vehicle, H) or nocodazole (I). A-D are maximum intensity projections of three confocal z-planes and E-F and H-I are maximum intensity projections of whole-cell confocal z-stacks. See also Videos S4 and S5.
Consistently, two different EPAC inhibitors (HJC0197 and ESI-09) blocked the increase in GDMT nucleation in high glucose (Figure 4E–H, 4M–N, 4R–S and Figure 5C–D, 5E, Video S5). shRNA knockdown of EPAC2 (Figure 4O bottom panel and Figure S5A–C) had a similar effect (Figure 4M, 4T–W and Figure S5D–E). Importantly, EPAC inhibition blocked GDMT induction by 8-br-cAMP, indicating that EPAC2 functions downstream of cAMP in this pathway (Figure 4I–J and 4M). Furthermore, EPAC2 inhibition blocked GDMT induction by KCl/calcium influx, indicating that the cAMP pathway takes precedence over the calcium pathway in regulating GDMT nucleation (Figure 4K–M).
We next determined the localization of EPAC2 in MIN6 cells. It is known that EPAC1 can localize to MTs [37–39]. EPAC2 in β-cells, however, is localized to the cell periphery in low glucose (Figure 5E). In high glucose, EPAC2 re-localizes to short stretches located throughout the cytoplasm (Figure 5F) and often in the Golgi area (Figure 5F, lower insets), placing EPAC2 at a vital location to regulate GDMTs. These stretches seen in high glucose are localized at or near MTs (Figure 5H) and disappear when MTs are depolymerized by nocodazole (Figure 5I), suggesting that EPAC2 can interact with MTs in β-cells. These localizations disappear upon EPAC2 depletion by two shRNA, indicating the specificity of the immunostaining (Figure S5A–C).
We have also evaluated the role of the other cAMP effector, PKA, by utilizing an inhibitor, PKI. In regrowth assays, PKA inhibition caused a modest decrease in high-glucose induced GDMT nucleation (Figure S5F–I and 5L), but it did not affect either the 8-br-cAMP-induced GDMT increase (Figure S5J–L) or steady-state GDMT nucleation levels (Figure S5M–O). This indicates that PKA is likely not involved in the cAMP-dependent GDMT regulation, at least during the first wave of glucose response.
Together, these data indicate that the rapid rise in glucose-dependent GDMT nucleation in β-cells is regulated through EPAC2 activation downstream of cAMP. Importantly, no detectable change was observed in centrosomal MT nucleation levels during any of the evaluated treatments (Figure S4), indicating that glucose and the cAMP/EPAC pathway regulate MT nucleation machinery at the Golgi only.
GDMTs affect insulin levels, but not secretory response to glucose
Next, we addressed the functional role of GDMTs in β-cells. We utilized two previously established dominant-negative approaches that specifically decrease GDMT levels, but not centrosomal MTs [31]. The first approach targets the scaffolding protein AKAP450 (AKAP9, AKAP-350, cG-NAP), which recruits the MT nucleation machinery to the Golgi [40]. Dominant-negative AKAP450 (DN-AKAP) contains the N-terminal Golgi-binding region (Figure S6A) and displaces endogenous AKAP450 from the Golgi, inhibiting GDMT formation [41]. The second approach targets Cdk5Rap2, an activator of the major MT-nucleation template, γ-TuRC [42]. Dominant negative Cdk5Rap2 is the γ-TuRC activating region (γ-TuNA) with a point mutation (F75A) (Figure S6A), which specifically suppresses formation of non-centrosomal MTs [42], such as GDMTs [31]. Both constructs are co-transcribed with GFP.
Firstly, we demonstrated that expression of either dominant-negative construct attenuated GDMT but not centrosomal MT nucleation in β-cells, as described for other cell types [31,40, 42]. These constructs decrease basal GDMT nucleation by half and block high-glucose dependent GDMT stimulation (Figure S6B, S6D, S6F and S6H). Also, the MT network density is decreased (Figure S6C, S6E and S6G).
We next tested whether these constructs affected insulin biogenesis and secretion. Immunostaining of insulin in control (GFP) cells showed that in low glucose insulin granules were localized throughout the cell with a small concentration in the Golgi region (Figure 6A and 6G–I). After five minutes in high glucose, insulin localization was similar with a slight, though not statistically significant, increase in Golgi insulin (Figure 6B and 6G–I). Strikingly, expression of either dominant-negative construct led to a decrease in cellular insulin levels (Figure 6C–F and 6G; see also Figure 6K for ELISA); the remaining insulin was mostly concentrated at the Golgi, leaving the rest of the cell almost devoid of insulin granules (Figure 6C–F, 6H and 6I). Glucose stimulation caused a slight increase in the insulin amount in the Golgi area, similar to controls (Figure 6C–F and 6H), suggesting that the absence of GDMTs did not influence ER-to-Golgi protein translocation.
Figure 6. Reduction of GDMTs reduces insulin content in β-cells.

A-F) MIN6 cells immunostained for insulin, cell outline (white dotted line) and Golgi outline (yellow dotted line) with whole cell (inset) staining of GFP (cyan) and Golgi (GM130, magenta). Cells expressing: AB) GFP. C-D) DN-AKAP co-expressed with GFP. E-F) γ-TuNA F75A co-expressed with GFP. G-H) Quantification of integrated density of insulin fluorescent signal in the whole cell (G) or within the Golgi region (H). p < 0.001, one-way ANOVA and Tukey’s multiple comparison test; n = 41-54 cells. I) Percent of insulin intensity in the Golgi region p < 0.001, one-way ANOVA and Tukey’s multiple comparison test; n = 41-54 cells. J) Insulin release (static GSIS assay). K) Total insulin content (ng/ml) in cells used for GSIS assay. All images are maximum intensity projections of whole-cell confocal stacks. See also Figure S6.
The observed phenotypes could result from deficient insulin-granule biogenesis, or from over-secretion of insulin. As expected for degranulated cells, cells expressing the dominant-negative constructs secreted significantly less insulin than controls (Figure 6J). However, these cells were still able to respond to a glucose stimulus: while the amount of secreted insulin was low, the fold increase of secretion caused by glucose triggering was similar to control (Figure 6J). This is in contrast to cells where all MT were disassembled by nocodazole resulting in a dramatic GSIS increase (Figure 6J), in agreement with our previous work [27]. This result indicates that new, glucose-stimulated GDMTs are not critical to insulin release, which may be the role of pre-existing MTs in the cytoplasm.
GDMTs are required for insulin exit from the Golgi
Given that the secretion response is not affected by the depletion of GDMTs, the low amount of insulin combined with the sequestration in the Golgi might indicate a defect in insulin granule biogenesis at the Golgi. To test this hypothesis, we performed a degranulation recovery assay. Following facilitated secretion caused by prolonged high glucose and KCl treatment, control cells became depleted of insulin granules (degranulated). Insulin content in these cells was decreased, with remaining insulin strongly concentrated in the Golgi region (Figure 7A–B and 7K–M). After two hours of recovery in high glucose, insulin content has returned to pre-treated levels (Figure 7C and 7K–M). After degranulation of cells expressing dominant-negative constructs, insulin was also concentrated in the Golgi region. However, these cells were unable to reform the cytosolic granules after two hours of recovery in high glucose, indicating that the exit of insulin from the Golgi and granule biogenesis were impaired in the absence of GDMTs (Figure 7D–I and 7K–M). Importantly, the ability of β-cells to recover their granule pool after degranulation was also impaired by the inhibition of EPAC2 (Figure 7J–M). These data support our conclusion that cAMP/EPAC2-dependent control of GDMT nucleation is essential for efficient refurnishing of the insulin granule pool in high glucose.
Figure 7. Reduction of GDMTs prevents β-cells from degranulation recovery.

A-I) MIN6 cells (white line) immunostained for insulin, Golgi outline (yellow dotted line) with whole cell (inset) staining of GFP (cyan) and Golgi (GM130, purple). A-C) Cells expressing GFP. D-F) Cells expressing DN-AKAP with GFP. G-I) Cells expressing γ-TuNA F75A with GFP. A, D, G) Cells in high glucose for 24 hours. B, E, H) Cells after 24-hour degranulation (high glucose/KCl). C, F, I) Cells after 2-hour recovery from degranulation. J) MIN6 cell (white line) immunostained for insulin and Golgi (yellow line and inset, magenta), treated with EPAC inhibitor (ESI-09) during recovery from degranulation. K-M) Measurement of integrated density of insulin fluorescent signal in the whole cell (K) or Golgi region (L). n=41-43 cells. M) Percent of integrated density of fluorescent signal in the Golgi region. n=41- 43. All images are maximum intensity projections of whole-cell confocal z-stacks. See also Figure S6.
Discussion
In this study we show that GDMTs in pancreatic β-cells are temporally regulated by glucose signaling and are highly functionally significant in insulin-granule biogenesis.
The site of origin of new MTs is critical for MT network configuration, because the majority of MT minus ends remain attached at their original location. Consistent with our earlier findings, we show that MTs in β-cells are predominantly Golgi-derived, and that the minor centrosomal array is not regulated by glucose [27]. This indicates that the centrosome in functional β-cells is silenced as an MTOC, similar to mature neurons [43, 44] and epithelia [45], and does not participate in regulation of insulin trafficking and/or secretion.
One important functional implication of MTs originating at the Golgi is supporting the β-cell-specific MT network, which is convoluted and resembles a dense mesh [27, 28]. The large Golgi surface in β-cells provides a much less centered site of origin than a centrosomal MTOC, resulting in MT minus-end scattering and contributing to the non-directional MT network. This network serves as tracks for the “random walk” of insulin granule movement, which follows constrained diffusion laws [27, 30]. We have shown previously that because of this configuration, MTs can exert restrictive tuning to GSIS, with many granules trapped in MT cages or being withdrawn from the cell edge [27]. Importantly, glucose causes a dual effect on the MT network: destabilization/depletion of preexisting MTs [27], and a boost of new MT nucleation ([27] and this study). The combination of these two processes result in a slightly sparser MT network as compared to low glucose. We have proposed that the rheostat-like balance between MT destabilization and GDMT nucleation serves for precise tuning of insulin release upon each stimulus. In this model, new, glucose-triggered GDMTs work to restore the dense MT network after glucose stimulation and to restrict insulin secretion in the short term (secretion restriction). It is also possible that new GDMTs provide a minor subset of direct MT tracks for releasable granule movement (secretion support) [46].
Importantly, this study reveals another important function of GDMTs in β-cells, which strongly supports insulin secretion in the long term. Specifically, we find that GDMTs are required for the biogenesis of new insulin granules. When GDMT formation is blocked, insulin accumulates in the Golgi, indicating that new GDMTs are not necessary for ER-to-Golgi transport of proinsulin, which depends on MTs [47] and thus is likely supported by a pre-existing MT sub-population.
We propose that new MTs nucleating at the Golgi in high glucose provide a scaffold for nascent insulin granule budding, similar to what has been reported for TGN carriers [25]. Interestingly, the number of new GDMTs exceeds the rate of nascent granule budding, as approximated from the known rates of insulin synthesis, combined with the number of insulin molecules in one granule. This rate in murine islets at the very peak of synthesis is estimated as five granules per minute [4, 48]. The number of granules budding off the Golgi in MIN6 cells is likely lower because, being a cell culture model, these cells are less efficient in insulin production than islet β-cells. Thus, the number of six to seven new MTs per minute that we detect in MIN6 cells should provide a sufficient scaffold for new insulin granule budding.
We reason that insulin granule budding, unlike small Golgi carrier formation [24], requires GDMTs because of the large size of insulin granules, which might need extra assistance to enter the highly crowded cytoplasm of β-cells. Together with the continued release of granules present in the cytosol, attenuated granule biogenesis leads to β-cell degranulation and severely affects the β-cell capacity for insulin secretion. Thus, defects in GDMTs function, as linked to insulin processing and vesicle storage, may be a fundamental deficiency in pathologies such as type 2 diabetes. Overall, our findings indicate that MT biology has a more complicated effect on insulin secretion, acting negatively in the short term at the cell periphery where insulin secretion occurs, and acting positively in the long term via insulin granule biogenesis at the Golgi.
Besides functional significance, we have made an important step in our understanding of the metabolic regulation of GDMT nucleation. So far, there is no complete explanation for the biphasic kinetics of GDMT nucleation, which is analogous to the lack of understanding of the biphasic nature of insulin secretion, still cryptic even after decades of exploration. We propose the biphasic nature of GDMT nucleation and its temporal alignment with insulin secretion as a reason to believe that GDMT formation may be regulated by the same signaling pathways as GSIS. We found that the glucose-dependent cAMP/EPAC2 signaling axis is necessary and sufficient for the early, rapid GDMT nucleation trigger in β cells. Interestingly, EPAC2-knockout mice have decreased granule exocytosis specifically during the first phase, indicating that EPAC2 plays an important role in this phase [11]; GDMT nucleation may be a part of this effect. Calcium influx, in addition, is also capable of stimulating GDMT nucleation, but it is neither necessary nor sufficient for this effect. It is possible that calcium facilitates GDMT formation by influencing cAMP signaling, since these two pathways are interdependent [34, 35].
Our finding that EPAC2 regulates GDMT nucleation is exciting in the light of previous observations that the more ubiquitous EPAC paralog, EPAC1, is capable of affecting MT stability and growth. While MT nucleation in cells relies on the γ-TuRC complex as a template, additional MT-binding proteins have been shown to increase MT nucleation rates. Examples include the MT polymerization enhancer XMAP215, which facilitates the first round of tubulin polymerization at γ-TuRC (templated nucleation) [49], MT-rescue promoter CLASP, which stabilizes the emerging nascent MTs [50], etc. A similar effect could be facilitated by EPAC, either directly, considering a reported binding of EPAC1 to MTs in vitro [37, 38] or indirectly, through an interaction of EPAC2 with the MT stabilizers LC1 or LC2 [39, 51]. We therefore hypothesize that the activation of EPAC2 by cAMP facilitates either templated nucleation or stabilization of nascent GDMTs. In addition, PKA might be involved in additional tuning of GDMT nucleation because PKA inhibition did decrease GDMT formation in the most sensitive assay (MT regrowth). On another note, the steady-state MT nucleation wave declined somewhat quicker than GDMT regrowth, which occurs in the excess of depolymerized tubulin, suggesting that regulation of the availability of tubulin dimers may also play a role in refining GDMT nucleation rates.
STAR★METHODS
LEAD CONTACT AND MATERIALS AVAILABILITY
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Irina Kaverina (irina.kaverina@Vanderbilt.Edu). Plasmids generated in this study are available upon request from the lead contact.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Animals
Species: Mice Mus musculus purchased from Charles River Inc. (Wilmington, MA).
Genotype: Male wild type CD-1 (ICR)
Age/developmental stage: less than 2-3 months old
Maintenance and care: Followed Vanderbilt Institutional Animal Care and Use Committee (IACUC) approved protocols.
Experimental Model and Subject Details for mice used in the study:
Health/immune status: healthy
Whether subjects were involved in previous procedures: not involved
Whether the subject is drug or test naïve: naive
Husbandry conditions of experimental animals: male, non-breeding
Housing conditions of experimental animals: 5/cage, according to Vanderbilt IACUC
Cell Lines
Culture conditions for in vitro systems:
MIN6 cells between passage 40-60 were utilized (obtained from J. Miyazaki [52]). Cells were maintained in 25 mM glucose Dulbecco’s modified eagle medium (DMEM) (Life Technologies, Frederick, MD) supplemented with 10% fetal bovine serum (FBS), 0.001% β-mercaptoethanol, 100 U/ml penicillin, and 0.1 mg/ml streptomycin in 5% CO2 at 3710. MIN6 cells were transfected using Nucleofection (Amaxa).
RPE1-hTert cells were maintained in DMEM/F12 with 10% FBS and grown at 371°C with 5% CO2.
Authentication of cell lines used:
MIN6 were authenticated by periodically glucose stimulated insulin release response test (see Figure 1 and 6).
Authenticated RPE1-hTert cells were recently purchased from the ATCC (Cat#: CRL-4000). Both cell lines were periodically tested for mycoplasma.
METHOD DETAILS
Experimental Design
Replication:
All experiments were replicated three times, with the exception of the islet regrowths (Figure 1L, Figure 3J, and Figure 4N) and insulin staining (Figure 6 and 7), which were repeated twice, and initial screen of insulin pathways (Figure S2C) which was performed once.
Strategy for randomization and/or stratification:
In imaging data, random fields were recorded across each sample. All recorded data points were used in the analyses. In all other types of data, each measurement was accounted for in the analysis.
Sample Size Estimation:
Sample size was determined by power analysis which allowed determination of the sample size required to detect an effect of a given size with a given degree of confidence. For MIN6 cell regrowth experiments: 10 images per condition were taken, containing a variable number of cells. At least 50 cells per condition were quantified. For Islet regrowth experiments: Four islets per treatment were imaged and 10 cells from each islet analyzed. Cells were chosen by insulin staining which indicated they were β-cells. For live cell nucleation: 8 movies per condition were taken, one per chamber in two separate 4-chamber dishes, containing a variable number of cells.
Blinding at any stage of the study:
Imaging for all experiments was typically not performed blind. Blind analysis of three of the movies was performed by another person to confirm (Figure S1I).
Statistical method of computation:
For all experiments, each point in the graph denotes one cell. Statistics were calculated by one-way ANOVA with Tukey’s multiple comparisons test. GraphPad Prism was used for statistical analyses and graphical representations. Significance was defined at p ≤ .05.
Inclusion and exclusion criteria of any data or subjects: No data was excluded after analysis.
Islet isolation
Mouse pancreatic islets used in this study were hand-picked following in situ collagenase perfusion and digestion. Islets were allowed to recover in Roswell Park Memorial Institute (RPMI) 1640 Medium (Life Technologies, Frederick, MD) supplemented with 10% fetal bovine serum (FBS), 100 U/ml penicillin, and 0.1 mg/ml streptomycin in 5% CO2 at 37°C for three hours then treated with 5 μM nocodazole in the same media for 12 hours.
DNA constructs
Dominant-negative AKAP450 (amino acids 159-463 [54]) and dominant-negative Cdk5Rap2 (amino acids 51-100, F75A, a kind gift from R. Qi, Hong Kong University of Science and Technology) [42] were inserted into the pCIG vector that expresses GFP and a gene of interest as separate proteins, the dominant negative Cdk5Rap2 GFP had an additional NLS signal. Constructs used for live cell imaging: Emerald-EB3 (kind gift from M. Davidson; Addgene plasmid # 54076), and TGN-RFP (a kind gift from E. Rodriguez-Boulan, Cornell University, New York) [53].
Reagents and antibodies
Primary antibodies for immunofluorescence were: mouse anti-EB1 (1:200), rat anti-EB3 (1:200), rabbit anti-Giantin (1:1000), mouse anti-GM130 (1:300), mouse anti-E-cadherin (1:500), rabbit EPAC2 (1:200), guinea pig anti-GCC185 (1:300), guinea pig anti-Insulin (1:1,000), rabbit anti-β-tubulin (1:1000) and anti-GFP FITC (1:1,000), Alexa488-, Alexa568-, and Alexa647-conjugated highly cross-absorbed secondary antibodies were from Invitrogen. Coverslips were mounted in Vectashield Mounting Medium. Nocodazole was prepared at 16.6 mM in DMSO (cell culture grade). A concentration of 5 μM was used to depolymerize MTs and added to the cells 4-14 hours. Cells were treated with indicated drugs for two hours unless otherwise indicated, drugs used were: Verapamil (10 μM), MPA (50 μg/ml, 16 hours), GKA50 (1 μM), Diazoxide (100 μM), Metformin (2.5 μg/ml), Pyruvate (1.4 or 10 mM), EGTA (5μM), KCl (25.2 μM, 1 minute), 8-br-cAMP (50 μM, 3 hours), 8-br-cAMP-AM (50μM, 30 minutes), lonomycin (100 μM, 2 minutes), PKI (1 μM), Forskolin (1 mM, 3 minutes), 8-pCPT-2O-Me-cAMP (50 μM, 30 minutes), HJC0197 (25 μM, 30 minutes), ESI-09 (20 μM, 30 minutes) and BAPTA-AM (50 μM, 3 minutes).
EPAC2 shRNAs
shRNA lentiviral GFP constructs were obtained from Origene. Virus was made using LHEK239T cells. Supernatant was ultracentrifuged at 25,0000 rpm for 90 minutes to concentrate the virus. Virus was then added to MIN6 cells at 5X concentration and incubated for two days with virus and an additional two days in fresh media before cell sorting.
Western blotting
Cell lysates from MIN6 and RPE were lysed using 1% CHAPs buffer on ice for 5 minutes. For EPAC2 knockdown, cells were first sorted for GFP expression. Samples were then spun down at 15K rpm for 15 minutes. Samples were loaded in sample buffer on a 10% acrylamide gel and run. A wet transfer was performed. Blots were blocked in 5% milk in TBST. Primary antibodies used were rabbit EPAC1 and EPAC2 (1:1000) and mouse GAPDH (1:5000). 680- and 800-Licor dyes were used as secondaries. Blots were imaged on a Licor system.
Nocodazole Regrowth Assays
Cells were cultured on coverslips (No. 1.5, 12 mm circular) coated in 10 μg/μl fibronectin for 24 h before experiments. Cells were incubated with 5 μM nocodazole 4-14 hrs. at 37°C with 5% CO2 to completely disassemble MTs. Cells are then switched out of their high glucose media into low glucose (2.8 mM) KRB (110 mM NaCl, 5 mM KCl, 1 mM MgSO4, 1 mM KH2PO4, 1 mM HEPES, 2.5 mM CaCl2 and 1 mg/ml BSA) for two hour incubations, then switched to high glucose (20 mM) KRB for the times indicated. These incubations contained 5 μM nocodazole and any drugs indicated in the experiment. To remove nocodazole, cells were placed on ice and washed five times with ice-cold KRB with low or high glucose. The cells were then shifted to 37°C for about a minute to allow for a burst of mic rotubule growth and immediately fixed in −20°C methanol. For mutant constructs, regrowth cell s were fixed in −20°C MeOH + 1% PFA to preserve GFP signal. This was followed by blocking in 1% bovine serum albumin (Fisher) and 5% donor horse serum (Atlanta Biologicals) in PBS.
Whole-islet MT regrowth assays followed a similar protocol. After 12-hour nocodazole treatment, islets were switched to low glucose KRB for two hours like MIN6 cells. Some islets were then switched to high glucose KRB for five or 30 minutes. Islets were then placed on ice and washed in either low or high glucose KRB five times to remove the nocodazole. Islets were then placed in a 37°C water bath for three minutes to allow for regrowth and immediately fixed in −20°C methanol then post-fixed in 4% paraformald ehyde for immunofluorescence.
Immunofluorescence microscopy of fixed samples
Fixed samples were imaged using a laser scanning confocal microscope Nikon A1r based on a TiE Motorized Inverted Microscope using a 100X lens, NA 1.49, run by NIS Elements C software. Cells were imaged in 2μm slices through the whole cell. Regrowth images in MIN6 and intact islets were depicted as three-slice maximum intensity projections to better show GDMT nucleation in these thick samples (Figure1A–E and I–K, Figure 3A–H and K–L, Figure 4A–L and P–W, Figure S1C–H, Figure S3 A–V, Figure S5 D–I, Figure S6 B,D,F). Golgi (Figure 2C–E), EPAC2 (Figure 5 E–F and H–I and Figure S5 A–C), Insulin (Figure 6 A–F and Figure 7 A–J), and Tubulin (Figure S1 A–B, Figure S6 C, E, G) staining was depicted as whole-cell maximum intensity projections.
Live cell imaging
Cells were cultured on 4-chamber MatTek dishes coated with 10 μg/μl fibronectin and transfected 24 h before experiment. For live-cell imaging of steady state MT dynamics, cells were transfected with Emerald-EB3 and TGN-RFP and imaged using a Nikon TiE inverted microscope equipped with 488- and 568-nm lasers, a Yokogawa CSU-X1 spinning disk head, a PLAN APO VC 100x NA1.4 oil lens, intermediate magnification 1.5X, and CMOS camera (Photometrics Prime 95B), controlled by Nikon Elements software. Image stacks with 0.3 μm between slices were recorded covering most of the Golgi (over 3-4 μm) for 2 min with a frame taken every 5 secs. These cells were imaged for 2 minutes in low glucose, then imaged again in high glucose for 2 minutes. Images of single time points were depicted as three-slice maximum intensity projections to better show individual nucleation events (Figure 1M–N, Figure 3M–N, Figure 5 A–D and Figure S5M–N).
Static GSIS assays
MIN6 cells for GSIS assays over time were plated in a 12 well dish and allowed to recover overnight. The next day, they were switched to low glucose KRB for two one-hour periods. Cells were then placed in low (half of the wells) or high (other half) glucose KRB which was removed and replaced with fresh KRB every three minutes for 50 minutes. Removed KRB was spun down to remove any cells or other debris. Static GSIS assay were carried out as previously described [27] using the ALPCO kit. GSIS assays for dominant negative mutants were performed in the same manner, with a few exceptions: Cells were obtained following flow cytometry to enrich for GFP-positive cells, performed at the Vanderbilt Flow Cytometry Shared Resource. The next day, cells were switched to low glucose KRB for two one-hour periods, with or without 5 μM nocodazole. KRB was collected after 15 minutes in fresh low or high glucose KRB then spun down to remove any cells or other debris.
Image Processing
In order to make small structures visible, adjustments were made to brightness, contrast and gamma settings of all fluorescent images presented here.
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistics
All quantitative data were collected from experiments performed in at least duplicate, usually triplicate. One-way ANOVA with Tukey’s multiple comparisons test was appropriate because the data were normally distributed (as determined by the D’Agostino & Pearson omnibus normality test) and we were comparing multiple groups. Box and whisker plots represent the 5-95 percentile of observed events.
Quantification of insulin localization
MIN6 cells on fibronectin-coated coverslips were pretreated in low-glucose KRB (see regrowth assays section above). Cells were then treated with high glucose KRB for five minutes or remained in low glucose. For degranulation assays, coverslips were placed in 25 mM glucose DMEM media either with or without 30 mM KCl for 24 hours. Half the KCl-treated coverslips were washed five times in media allowed to recover for two hours. For degranulation recovery upon EPAC2 inhibition, coverslips were treated with 20 μM ESI-09 for the last 30 minutes of the 24 hours and during the two-hour recovery period. Cells were fixed in 4% paraformaldehyde and immunostained with a Golgi marker, insulin, and GFP. To quantify insulin in the entire cells, a mask was drawn around the GFP and insulin in ImageJ using the multiply operation of the image calculator function. Insulin was then thresholded and the integrated density was measured using the analyze particles function as anything 25μm2 or larger to exclude any background staining. To quantify insulin in the Golgi, a mask was drawn around the Golgi marker (GM130) instead and quantified the same as the whole cell insulin.
Quantification of MT numbers
For regrowth assay in MIN6 cells, the MTs growing from the centrosome and Golgi were quantified manually using Imaris software (Bitplane). This software allows for quantification in 3D (see Video S1). GDMTs were counted as EB1 or EB3 comets originating at a Golgi mass. Centrosomal MTs were counted as EB1 or EB3 comets originating at an aster. For islet regrowth assays, quantification was done using ImageJ. Cell boundaries were delineated using E-cadherin staining and β-cells were identified using insulin staining. EB3 comets were counted manually when either coming from the Golgi or an aster in the cell.
For live cell imaging of nucleation, image stacks through 3-4μm of the Golgi taken in a two-minute movie were used. This allowed imaging of most, but usually not all of the Golgi in the cell, leading to an underestimation of GDMTs. To track the newly formed EB3 comets, the ImageJ plug-in MtrackJ was used [55]. Quantification was done in 3D. To ensure quantification of only newly nucleated MTs at the Golgi, tracks that start in the first time frame and tracks that appeared in the top or bottom slice of a z-stack were excluded.
Data and Code Availability
The data supporting the findings of this study are available from the Lead Contact on reasonable request.
Supplementary Material
Video S1. 3D Representation of a MIN6 Regrowth Cell. Related to Figure 1. A MIN6 cell immunostained for EB1 (cyan) and Giantin (red). Same cell as seen in Figure 1A.
Video S2. Live cell nucleation in control cells. Related to Figure 1. Maximum intensity projection of whole-cell spinning disk confocal stacks in a DMSO treated MIN6 cell expressing TGN-RFP (Golgi) and EB3-mEmerald (MT ends) in low glucose (left) and high glucose (right). Asterisk represents the centrosome location and arrows follow GDMTs from nucleation. Imaged for about two minutes at five second intervals.
Video S3. Live cell nucleation in 8-br-cAMP. Related to Figure 3. Maximum intensity projection of whole-cell spinning disk confocal stacks in an 8-br-cAMP treated MIN6 cell expressing TGN-RFP (Golgi) and EB3-mEmerald (MT ends) in low glucose (left) and high glucose (right). Asterisk represents the centrosome location and arrows follow GDMTs from nucleation. Imaged for about two minutes at five second intervals.
Video S4. Live cell nucleation with activated EPAC. Related to Figure 5. Maximum intensity projection of whole-cell spinning disk confocal stacks in an EPAC activator treated MIN6 cell expressing TGN-RFP (Golgi) and EB3-mEmerald (MT ends) in low glucose (left) and high glucose (right). Asterisk represents the centrosome location and arrows follow GDMTs from nucleation. Imaged for about two minutes at five second intervals.
Video S5. Live cell nucleation with inhibited EPAC. Related to Figure 5. Maximum intensity projection of whole-cell spinning disk confocal stacks in an EPAC inhibitor treated MIN6 cell expressing TGN-RFP (Golgi) and EB3-mEmerald (MT ends) in low glucose (left) and high glucose (right). Asterisk represents the centrosome location and arrows follow GDMTs from nucleation. Imaged for about two minutes at five second intervals.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse anti-EB1 | BD Transduction | Cat#: 610535 |
| Rat anti-EB3 | Absea | Cat#: 010314H04 |
| Rabbit anti-Giantin | Abcam | Cat#: ab24586 |
| Mouse anti-GM130 | BD Transduction | Cat#:610823 |
| Rabbit EPAC1 | Abcam | Cat#: ab124162 |
| Rabbit EPAC2 | Abcam | Cat#: ab124189 |
| Mouse GAPDH | Santa Cruz | Cat#: sc-32233 |
| Mouse anti-E cadherin | BD Transduction | Cat#: 610181 |
| Guinea Pig anti-GCC185 | [44] | N/A |
| Guinea Pig anti-Insulin | Agilent Technologies | Cat#: A0564 |
| Rabbit anti-β-tubulin | Abcam | Cat#: ab18251 |
| Anti-GFP FITC | Abcam | Cat#: ab6662 |
| Chemicals | ||
| Vectashield Mounting Medium | Vector Labs | Cat#: H-1000 |
| Nocodazole | Sigma-Aldrich | Cat#: M1404 |
| Verapamil hydrochloride | MP Biomedicals | Cat#: 0219554501 |
| MPA | Sigma-Aldrich | Cat#: M5255 |
| GKA50 | Sigma-Aldrich | Cat#: SML0849 |
| Diazoxide | Tocris Bioscience | Cat#: 0934 |
| Metformin | Sigma-Aldrich | Cat#: PHR1084 |
| Sodium Pyruvate | ThermoFisher | Cat#: 11360070 |
| KCl | EM Science | Cat#: PX1405-1 |
| 8-br-cAMP | Sigma-Aldrich | Cat#: B7880 |
| 8-br-cAMP-AM | Biolog | Cat#: B 020 |
| Ionomycin | Sigma-Aldrich | Cat#: I3909 |
| PKI | Sigma-Aldrich | Cat#: P9115 |
| Forskolin | Sigma-Aldrich | Cat#: F6886 |
| 8-pCPT-2O-Me-cAMP | Sigma-Aldrich | Cat#: C8988 |
| HJC0197 | Cayman Chemical | Cat#: 19092 |
| ESI-09 | Sigma-Aldrich | Cat#: SML0814 |
| BAPTA-AM | Invitrogen | Cat#: B1205 |
| D (+)-glucose | Acros | Cat# 41095-0010 |
| NaHCO3 | HyClone | Cat# SH30173.04 |
| EGTA | Sigma-Aldrich | Cat# E3889 |
| NaCl | Sigma-Aldrich | Cat# S9625 |
| MgSO4 | Sigma-Aldrich | Cat# M7506 |
| KH2PO4 | ThermoFisher | Cat# BP363 |
| HEPES | Sigma-Aldrich | Cat# H4034 |
| CaCl2 | Sigma-Aldrich | Cat# C1016 |
| BSA | ThermoFisher | Cat# BP1605 |
| Critical Commercial Assays | ||
| Mouse Ultrasensitive Insulin ELISA | Alpco | 80-INSMSU-E10 |
| Experimental Models: Cell Lines | ||
| MIN6 cells | [52] | N/A |
| hTERT RPE-1 cells | ATCC | Cat#: CRL-4000 |
| Experimental Models: Organisms/Strains | ||
| CD-1 (ICR) Mice | Charles River Inc. | Stain code:022 |
| Oligonucleotides | ||
| Cdk5Rap2 51-100 Fwd | Sigma-Aldrich | GGCTCGAGGCCGCGAATTCCACAGT |
| Cdk5Rap2 51-100 Rev | Sigma-Aldrich | GGATGCCACCCCGGGATCCTC |
| shRNA | ||
| EPAC2 Mouse shRNA #1 | Origene | TL509420A CGACAAGGAAGACTTCAATCGGATTCTGA |
| EPAC2 Mouse shRNA #2 | Origene | TL50942C CCATTACCACGCACAGCCTTCTCAAGGTA |
| Recombinant DNA | ||
| pCMV Cdk5Rap2 51-100 F75A | [42] | N/A |
| pCIG | McMahon Plasmid Database | 2165 |
| Emerald-EB3 | Addgene | Plasmid# 54076 |
| TGN-RFP | [53] | N/A |
| Dominant negative AKAP450 | [41] | N/A |
| Software and Algorithms | ||
| Graphpad Prism | Graphpad | https://www.graphpad.com |
| Image J | Image J | https://imagej.nih.gov/ij/ |
| Adobe illustrator/photoshop | Adobe | http://www.adobe.com |
| Imaris | Bitplane | http://www.bitplane.com |
Highlights:
Glucose triggers GDMT nucleation in β-cells, which mirrors biphasic insulin release
Glucose-dependent GDMT nucleation is regulated by EPAC2 downstream of cAMP
GDMTs are necessary for insulin granule biogenesis at the TGN
Glucose-dependent GDMT nucleation maintains the secretion/storage insulin balance
Acknowledgements
This work was supported by a Nation Institutes of Health (NIH) grants T32 DK07061 and 1F32DK117529 (to K.P.T.), R35-GM127098 (to I.K.), DK65949 (to G.G.) and R01-DK106228 (to I.K. and G.G.). We utilized the Vanderbilt Cell Imaging Shared Resource (funded by NIH grants CA68485, DK20593, DK58404, DK59637, EY08126, NIH SEI10, 1sS10, 1S10OD018075, and S10OD012324), Flow Cytometry experiments were performed in the VMC Flow Cytometry Shared Resource. The VMC Flow Cytometry Shared Resource is supported by the Vanderbilt Ingram Cancer Center (P30 CA68485) and the Vanderbilt Digestive Disease Research Center (DK058404) and the core(s) of the Vanderbilt Diabetes Research and Training Center (funded by NIH grant DK020593). We thank Brian S. Domin and Hamida Ahmed for technical help. The authors also thank Drs. Stephen Rogers, Alvin Powers and David Jacobson for suggestions and critical reading of the manuscript.
Abbreviations
- GSIS
Glucose stimulated insulin release
- MT
Microtubule
- GDMTs
Golgi-derived microtubules
- MTOC
Microtubule Organizing Center
- DN-AKAP
Dominant negative AKAP450
- γ-TuNA
Gamma Tubulin Nucleation Activating Region
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Interests
The authors declare no competing interests.
References
- 1.Roder PV, Wu B, Liu Y, and Han W (2016). Pancreatic regulation of glucose homeostasis. Exp. Mol. Med 48, e219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Saisho Y (2015). beta-cell dysfunction: Its critical role in prevention and management of type 2 diabetes. World J. Diabetes 6, 109–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Puchalska P, and Crawford PA (2017). Multi-dimensional Roles of Ketone Bodies in Fuel Metabolism, Signaling, and Therapeutics. Cell Metab 25, 262–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fu Z, Gilbert ER, and Liu D (2013). Regulation of insulin synthesis and secretion nd pancreatic Beta-cell dysfunction in diabetes. Curr. Diabetes Rev 9, 25–53. [PMC free article] [PubMed] [Google Scholar]
- 5.Matschinsky F, Liang Y, Kesavan P, Wang L, Froguel P, Velho G, Cohen D, Permutt MA, Tanizawa Y, Jetton TL, et al. (1993). Glucokinase as pancreatic beta cell glucose sensor and diabetes gene. J. Clin. Invest 92, 2092–2098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rorsman P, and Ashcroft FM (2018). Pancreatic beta-Cell Electrical Activity and Insulin Secretion: Of Mice and Men. Physiol. Rev 98, 117–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tengholm A (2012). Cyclic AMP dynamics in the pancreatic beta-cell. Ups. J. Med. Sci 117, 355–369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Drucker DJ, Philippe J, Mojsov S, Chick WL, and Habener JF (1987). Glucagon-like peptide I stimulates insulin gene expression and increases cyclic AMP levels in a rat islet cell line. Proc. Nat. Acad. Sci. U. S. A 84, 3434–3438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ramos LS, Zippin JH, Kamenetsky M, Buck J, and Levin LR (2008). Glucose and GLP-1 stimulate cAMP production via distinct adenylyl cyclases in INS-1E insulinoma cells. J. Gen. Physiol 132, 329–338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Renstrom E, Eliasson L, and Rorsman P (1997). Protein kinase A-dependent and -independent stimulation of exocytosis by cAMP in mouse pancreatic B-cells. J. Physiol 502 (Pt 1), 105–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Shibasaki T, Takahashi H, Miki T, Sunaga Y, Matsumura K, Yamanaka M, Zhang C, Tamamoto A, Satoh T, Miyazaki J, et al. (2007). Essential role of Epac2/Rap1 signaling in regulation of insulin granule dynamics by cAMP. Proc. Nat. Acad. Sci. U. S. A 104, 19333–19338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ozaki N, Shibasaki T, Kashima Y, Miki T, Takahashi K, Ueno H, Sunaga Y, Yano H, Matsuura Y, Iwanaga T, et al. (2000). cAMP-GEFII is a direct target of cAMP in regulated exocytosis. Nat. Cell Biol 2, 805–811. [DOI] [PubMed] [Google Scholar]
- 13.Dean PM (1973). Ultrastructural morphometry of the pancreatic -cell. Diabetologia 9, 115–119. [DOI] [PubMed] [Google Scholar]
- 14.Olofsson CS, Gopel SO, Barg S, Galvanovskis J, Ma X, Salehi A, Rorsman P, and Eliasson L (2002). Fast insulin secretion reflects exocytosis of docked granules in mouse pancreatic B-cells. Pflugers Arch 444, 43–51. [DOI] [PubMed] [Google Scholar]
- 15.Rorsman P, and Renstrom E (2003). Insulin granule dynamics in pancreatic beta cells. Diabetologia 46, 1029–1045. [DOI] [PubMed] [Google Scholar]
- 16.Itoh N, Sei T, Nose K, and Okamoto H (1978). Glucose stimulation of the proinsulin synthesis in isolated pancreatic islets without increasing amount of proinsulin mRNA. FEBS Lett 93, 343–347. [DOI] [PubMed] [Google Scholar]
- 17.Itoh N, and Okamoto H (1980). Translational control of proinsulin synthesis by glucose. Nature 283, 100–102. [DOI] [PubMed] [Google Scholar]
- 18.Dodson G, and Steiner D (1998). The role of assembly in insulin’s biosynthesis. Curr. Opin. Struct. Biol 8, 189–194. [DOI] [PubMed] [Google Scholar]
- 19.Orci L, Halban P, Perrelet A, Amherdt M, Ravazzola M, and Anderson RG (1994). pH-independent and -dependent cleavage of proinsulin in the same secretory vesicle. J. Cell Biol 126, 1149–1156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wacker I, Kaether C, Kromer A, Migala A, Almers W, and Gerdes HH (1997). Microtubule-dependent transport of secretory vesicles visualized in real time with a GFP-tagged secretory protein. J Cell Sci 110 ( Pt 13), 1453–1463. [DOI] [PubMed] [Google Scholar]
- 21.Hirschberg K, Miller CM, Ellenberg J, Presley JF, Siggia ED, Phair RD, and Lippincott-Schwartz J (1998). Kinetic analysis of secretory protein traffic and characterization of golgi to plasma membrane transport intermediates in living cells. J. Cell Biol 143, 1485–1503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kreitzer G, Marmorstein A, Okamoto P, Vallee R, and Rodriguez-Boulan E (2000). Kinesin and dynamin are required for post-Golgi transport of a plasma-membrane protein. Nat. Cell Biol 2, 125–127. [DOI] [PubMed] [Google Scholar]
- 23.Toomre D, Keller P, White J, Olivo JC, and Simons K (1999). Dual-color visualization of trans-Golgi network to plasma membrane traffic along microtubules in living cells. J Cell Sci 112 (Pt 1), 21–33. [DOI] [PubMed] [Google Scholar]
- 24.Miller PM, Folkmann AW, Maia AR, Efimova N, Efimov A, and Kaverina I (2009). Golgi-derived CLASP-dependent microtubules control Golgi organization and polarized trafficking in motile cells. Nat Cell Biol 11, 1069–1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Polishchuk EV, Di Pentima A, Luini A, and Polishchuk RS (2003). Mechanism of constitutive export from the golgi: bulk flow via the formation, protrusion, and en bloc cleavage of large trans-golgi network tubular domains. Mol. Biol. Cell 14, 4470–4485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Presley JF, Cole NB, Schroer TA, Hirschberg K, Zaal KJ, and Lippincott-Schwartz J (1997). ER-to-Golgi transport visualized in living cells. Nature 389, 81–85. [DOI] [PubMed] [Google Scholar]
- 27.Zhu X, Hu R, Brissova M, Stein RW, Powers AC, Gu G, and Kaverina I (2015). Microtubules Negatively Regulate Insulin Secretion in Pancreatic beta Cells. Dev. Cell 34, 656–668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Varadi A, Tsuboi T, Johnson-Cadwell LI, Allan VJ, and Rutter GA (2003). Kinesin I and cytoplasmic dynein orchestrate glucose-stimulated insulin-containing vesicle movements in clonal MIN6 beta-cells. Biochem. Biophys. Res. Commun 311, 272–282. [DOI] [PubMed] [Google Scholar]
- 29.Heaslip AT, Nelson SR, Lombardo AT, Beck Previs S, Armstrong J, and Warshaw DM (2014). Cytoskeletal dependence of insulin granule movement dynamics in INS-1 beta-cells in response to glucose. PloS One 9, e109082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tabei SM, Burov S, Kim HY, Kuznetsov A, Huynh T, Jureller J, Philipson LH, Dinner AR, and Scherer NF (2013). Intracellular transport of insulin granules is a subordinated random walk. Proc. Nat. Acad. Sci. U. S. A 110, 4911–4916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sanders A, Chang K, Zhu X, Thoppil RJ, Holmes WR, and Kaverina I (2017). Nonrandom gamma-TuNA-dependent spatial pattern of microtubule nucleation at the Golgi. Mol. Biol. Cell 28, 3181–3192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Batts AA, Gaal SA, and Tipton DL Jr. (1959). Changes in the Golgi apparatus of the islets of Langerhans in the rat following glucose and insulin administration. Endocrinology 64, 503–512. [DOI] [PubMed] [Google Scholar]
- 33.Seino S, Takahashi H, Fujimoto W, and Shibasaki T (2009). Roles of cAMP signalling in insulin granule exocytosis. Diabetes Obes. Metab 11 Suppl 4, 180–188. [DOI] [PubMed] [Google Scholar]
- 34.Landa LR Jr., Harbeck M, Kaihara K, Chepurny O, Kitiphongspattana K, Graf O, Nikolaev VO, Lohse MJ, Holz GG, and Roe MW (2005). Interplay of Ca2+ and cAMP signaling in the insulin-secreting MIN6 beta-cell line. J. Biol. Chem 280, 31294–31302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hofer AM (2012). Interactions between calcium and cAMP signaling. Curr. Med. Chem 19, 5768–5773. [DOI] [PubMed] [Google Scholar]
- 36.Kang G, Chepurny OG, Malester B, Rindler MJ, Rehmann H, Bos JL, Schwede F, Coetzee WA, and Holz GG (2006). cAMP sensor Epac as a determinant of ATP-sensitive potassium channel activity in human pancreatic beta cells and rat INS-1 cells. J. Physiol 573, 595–609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mei FC, and Cheng X (2005). Interplay between exchange protein directly activated by cAMP (Epac) and microtubule cytoskeleton. Mol. Biosyst 1, 325–331. [DOI] [PubMed] [Google Scholar]
- 38.Sehrawat S, Cullere X, Patel S, Italiano J Jr., and Mayadas TN (2008). Role of Epac1, an exchange factor for Rap GTPases, in endothelial microtubule dynamics and barrier function. Mol. Biol. Cell 19, 1261–1270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Borland G, Gupta M, Magiera MM, Rundell CJ, Fuld S, and Yarwood SJ (2006). Microtubule-associated protein 1B-light chain 1 enhances activation of Rap1 by exchange protein activated by cyclic AMP but not intracellular targeting. Mol. Pharmacol 69, 374–384. [DOI] [PubMed] [Google Scholar]
- 40.Rivero S, Cardenas J, Bornens M, and Rios RM (2009). Microtubule nucleation at the cis-side of the Golgi apparatus requires AKAP450 and GM130. EMBO J 28, 1016–1028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hurtado L, Caballero C, Gavilan MP, Cardenas J, Bornens M, and Rios RM (2011). Disconnecting the Golgi ribbon from the centrosome prevents directional cell migration and ciliogenesis. J. Cell Biol 193, 917–933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Choi YK, Liu P, Sze SK, Dai C, and Qi RZ (2010). CDK5RAP2 stimulates microtubule nucleation by the gamma-tubulin ring complex. J. Cell Biol 191, 1089–1095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Stiess M, Maghelli N, Kapitein LC, Gomis-Ruth S, Wilsch-Brauninger M, Hoogenraad CC, Tolic-Norrelykke IM, and Bradke F (2010). Axon extension occurs independently of centrosomal microtubule nucleation. Science 327, 704–707. [DOI] [PubMed] [Google Scholar]
- 44.Nguyen MM, Stone MC, and Rolls MM (2011). Microtubules are organized independently of the centrosome in Drosophila neurons. Neural Dev 6, 38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Feldman JL, and Priess JR (2012). A role for the centrosome and PAR-3 in the hand-off of MTOC function during epithelial polarization. Curr. Biol. CB 22, 575–582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hoboth P, Muller A, Ivanova A, Mziaut H, Dehghany J, Sonmez A, Lachnit M, Meyer-Hermann M, Kalaidzidis Y, and Solimena M (2015). Aged insulin granules display reduced microtubule-dependent mobility and are disposed within actin-positive multigranular bodies. Proc. Nat. Acad. Sci. U. S. A 112, E667–676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Malaisse-Lagae F, Amherdt M, Ravazzola M, Sener A, Hutton JC, Orci L, and Malaisse WJ (1979). Role of microtubules in the synthesis, conversion, and release of (pro)insulin. A biochemical and radioautographic study in rat islets. J. Clin. Invest 63, 1284–1296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Steiner DF, Park SY, Stoy J, Philipson LH, and Bell GI (2009). A brief perspective on insulin production. Diabetes Obes. Metab 11 Suppl 4, 189–196. [DOI] [PubMed] [Google Scholar]
- 49.Wieczorek M, Bechstedt S, Chaaban S, and Brouhard GJ (2015). Microtubule-associated proteins control the kinetics of microtubule nucleation. Nat. Cell Biol 17, 907–916. [DOI] [PubMed] [Google Scholar]
- 50.Efimov A, Kharitonov A, Efimova N, Loncarek J, Miller PM, Andreyeva N, Gleeson P, Galjart N, Maia AR, McLeod IX, et al. (2007). Asymmetric CLASP-dependent nucleation of noncentrosomal microtubules at the trans-Golgi network. Dev. Cell 12, 917–930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Gupta M, and Yarwood SJ (2005). MAP1A light chain 2 interacts with exchange protein activated by cyclic AMP 1 (EPAC1) to enhance Rap1 GTPase activity and cell adhesion. J. Biol. Chem 280, 8109–8116. [DOI] [PubMed] [Google Scholar]
- 52.Miyazaki J, Araki K, Yamato E, Ikegami H, Asano T, Shibasaki Y, Oka Y, and Yamamura K (1990). Establishment of a pancreatic beta cell line that retains glucose-inducible insulin secretion: special reference to expression of glucose transporter isoforms. Endocrinology 127, 126–132. [DOI] [PubMed] [Google Scholar]
- 53.Deora AA, Diaz F, Schreiner R, and Rodriguez-Boulan E (2007). Efficient electroporation of DNA and protein into confluent and differentiated epithelial cells in culture. Traffic 8, 1304–1312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Maia AR, Zhu X, Miller P, Gu G, Maiato H, and Kaverina I (2013). Modulation of Golgi-associated microtubule nucleation throughout the cell cycle. Cytoskeleton (Hoboken) 70, 32–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Meijering E, Dzyubachyk O, and Smal I (2012). Methods for cell and particle tracking. Methods Enzymol 504, 183–200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Video S1. 3D Representation of a MIN6 Regrowth Cell. Related to Figure 1. A MIN6 cell immunostained for EB1 (cyan) and Giantin (red). Same cell as seen in Figure 1A.
Video S2. Live cell nucleation in control cells. Related to Figure 1. Maximum intensity projection of whole-cell spinning disk confocal stacks in a DMSO treated MIN6 cell expressing TGN-RFP (Golgi) and EB3-mEmerald (MT ends) in low glucose (left) and high glucose (right). Asterisk represents the centrosome location and arrows follow GDMTs from nucleation. Imaged for about two minutes at five second intervals.
Video S3. Live cell nucleation in 8-br-cAMP. Related to Figure 3. Maximum intensity projection of whole-cell spinning disk confocal stacks in an 8-br-cAMP treated MIN6 cell expressing TGN-RFP (Golgi) and EB3-mEmerald (MT ends) in low glucose (left) and high glucose (right). Asterisk represents the centrosome location and arrows follow GDMTs from nucleation. Imaged for about two minutes at five second intervals.
Video S4. Live cell nucleation with activated EPAC. Related to Figure 5. Maximum intensity projection of whole-cell spinning disk confocal stacks in an EPAC activator treated MIN6 cell expressing TGN-RFP (Golgi) and EB3-mEmerald (MT ends) in low glucose (left) and high glucose (right). Asterisk represents the centrosome location and arrows follow GDMTs from nucleation. Imaged for about two minutes at five second intervals.
Video S5. Live cell nucleation with inhibited EPAC. Related to Figure 5. Maximum intensity projection of whole-cell spinning disk confocal stacks in an EPAC inhibitor treated MIN6 cell expressing TGN-RFP (Golgi) and EB3-mEmerald (MT ends) in low glucose (left) and high glucose (right). Asterisk represents the centrosome location and arrows follow GDMTs from nucleation. Imaged for about two minutes at five second intervals.
Data Availability Statement
The data supporting the findings of this study are available from the Lead Contact on reasonable request.