

Abstract
Introduction:
Expression of fusion oncoproteins generated by recurrent chromosomal translocations represents a major tumorigenic mechanism characteristic of multiple cancers, including one-third of all sarcomas. Oncogenic fusion genes provide novel targets for therapeutic intervention. The PAX3-FOXO1 oncoprotein in alveolar rhabdomyosarcoma (ARMS) is presented as a paradigm to examine therapeutic strategies for targeting sarcoma-associated fusion genes.
Areas covered:
This review discusses the role of PAX3-FOXO1 in ARMS tumors. In addition to evaluating various approaches to molecularly target PAX3-FOXO1 itself, this review highlights therapeutically attractive downstream genes activated by PAX3-FOXO1.
Expert opinion:
Oncogenic fusion proteins represent desirable therapeutic targets because their expression is specific to tumor cells, but these fusions generally characterize rare malignancies. Full development and testing of potential drugs targeted to these fusions are complicated by the small numbers of patients in these disease categories. Although efforts to develop targeted therapies against fusion proteins should continue, molecular targets that are applicable to a broader tumor landscape should be pursued. A shift of the traditional paradigm to view therapeutic intervention as target-specific rather than tumor-specific will help to circumvent the challenges posed by rare tumors and maximize the possibility of developing successful new treatments for patients with these rare translocation-associated sarcomas.
Keywords: fusion gene, PAX3-FOXO1, rhabdomyosarcoma, sarcoma, therapeutic target, translocation
1. Introduction
Sarcomas are a rare, heterogeneous array of mesenchymal tumors that encompass more than 50 subtypes 1–4 Tumors can arise from bone, cartilage, or connective tissues and present virtually anywhere in the body 1,5. While they account for only 1% of all cancers, sarcomas are more prevalent in children than adults and represent approximately 13% of malignancies affecting patients less than 20 years of age 1, 3.
Sarcomas have traditionally been divided into two major categories: 1) tumors with nonspecific genetic lesions and complex karyotypes; and 2) tumors with simple genetic alterations and nearly diploid karyotypes 3, 5. Tumors classified in the second category often arise de novo and harbor chromosomal translocations 5. Although non-random translocations are generally rare in solid tumors, they are associated with about one-third of all sarcomas 6. Indeed, recurrent translocations have been identified in 19 sarcoma subtypes 2. The majority of these non-random chromosomal translocations generate chimeric transcription factors, which aberrantly transactivate target gene expression 2, 5. In Ewing’s sarcoma, for example, the common t(11;22)(q24;q12) translocation creates the EWSR1-FLI1 fusion transcription factor 1. A different mechanism is demonstrated in dermatofibrosarcoma protuberans (DFSP) where a COL1A1-PDGFB fusion constitutively drives PDGFB expression from COL1A1 regulatory elements 1,2, 5.
Studies of recurrent chromosomal translocations and their associated fusion genes have contributed much to sarcoma research from both basic biology and clinical perspectives. Gene fusions have not only furthered understanding of sarcomagenesis, but have improved diagnosis, as the presence of defined fusion genes can be detected by RT-PCR and FISH approaches 7 Moreover, these specific fusion genes provide additional targets for therapeutic intervention in sarcoma. While advances in targeted therapy have been made in a few categories - for example, PDGFR inhibition by the tyrosine kinase inhibitor imatinib has been demonstrated as an effective therapy for DFSP 1, 8 - useful therapies targeting chimeric transcription factors remain largely undeveloped.
This review will focus on PAX3-FOXO1, the gene product resulting from the chromosomal translocation t(2;13)(q35;q14), in alveolar rhabdomyosarcoma (ARMS). PAX3-FOXO1 in ARMS is highlighted as an excellent paradigm to target sarcoma-associated fusion genes based on the following: 1) development of skeletal muscle, which is the lineage related to ARMS tumors, has been extensively described 9; 2) wild-type PAX3, PAX7 10–14, and FOXO1 15–17 as well as resulting chimeric products 18–21 have been well characterized; and 3) model systems—comprising both cell culture 22–26 and whole animal, including a conditional knock-in mouse model of PAX3-FOXO1-induced ARMS 27–30—have been developed. Through examination of PAX3-FOXO1, this review will discuss a conceptual framework of therapeutic strategies that applies not only to this ARMS-specific gene fusion, but also to oncogenic transcription factor chimeras of other sarcoma subtypes.
2. Rhabdomyosarcoma (RMS) Family of Tumors
RMS is a heterogeneous family of pediatric soft tissue tumors associated with the skeletal muscle lineage 31. Although rare in adults, RMS is the most common pediatric soft tissue sarcoma, accounting for approximately 50% of all soft tissue sarcomas in children and adolescents 32, 33. With an annual incidence of 4.5 cases per million children in the United States, which corresponds to roughly 350 new cases per year, RMS represents about 3–4% of all childhood malignancies 33–35.
Alveolar rhabdomyosarcoma (ARMS) and embryonal rhabdomyosarcoma (ERMS) constitute the two major histopathologic subtypes of RMS. These two variants are not only histologically distinguishable, they are also associated with clinically distinct phenotypes 32, 36. ARMS accounts for 20–30% of RMS, affects children as well as adolescents and young adults, and tends to occur in the extremities and trunk 32, 33, 36. In contrast, ERMS represents 70–80% of all RMS cases 32, 33. Typically presenting in patients less than 10 years of age, ERMS predominantly occurs in the head and neck and genitourinary tract 36. ARMS is clinically more aggressive than ERMS and is associated with an unfavorable prognosis, which is partially attributable to its propensity for early dissemination, poor response to therapy, and frequent relapses following therapy 21, 31, 32, 37, 38. The 5-year overall survival for ARMS is ~50% compared to ~75% for ERMS 34
2.1. Molecular Genetics of ARMS
The considerable clinical and pathologic dissimilarities between ARMS and ERMS reflect genetic differences between these RMS subtypes. In contrast to 11p15.5 allelic loss and point mutations that occur frequently in ERMS 32, 33, 39–44, recurrent chromosomal translocations characterize 70–80% of ARMS tumors 33, 45. Two specific translocations are unique to ARMS tumors: the majority of ARMS cases harbor the common translocation, t(2;13)(q35;q14), whereas a smaller subset of ARMS harbor a variant translocation, t(1; 13)(p36;q14) 7, 33, 45. In these translocations, PAX3 is the gene rearranged on chromosome 2 46, and PAX7 is the gene rearranged on chromosome 1 19 (Figure 1). PAX3 and PAX7 encode highly homologous members of the paired box family of transcription factors. Structurally, both proteins contain N-terminal DNA binding domains, which are comprised of paired box and homeobox motifs, and C-terminal transcriptional activation domains 11, 32. The gene located at the chromosome 13 locus in these translocations is FOXO1, encoding a member of the O subfamily of forkhead box transcription factors 18, 19, 32. FOXO1 contains a forkhead DNA binding domain at its N-terminus and a transcriptional activation domain at its C-terminus 6.
Figure 1.
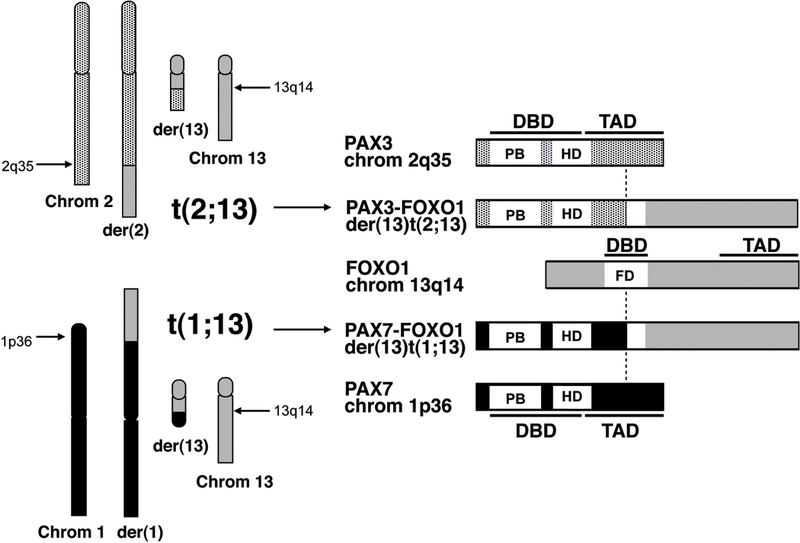
Schematic depiction of t(2;13)(q35;q14) and t(1;13)(p36;q14) chromosomal translocations and resultant PAX3/7-FOXO1 chimeric fusion products. The vertical dashed line denotes the fusion point. DBD: DNA binding domain; FD: Forkhead domain; HD: Homeobox domain; PB: Paired box; TAD: Transcriptional activation domain.
The 2;13 and 1;13 translocations break within the seventh intron of PAX3 or PAX7 and within the first intron of FOXO1 32. Chimeric genes are thereby generated and encode chimeric proteins consisting of the PAX3 or PAX7 N-terminal DNA binding domain fused to the FOXO1 C-terminal transactivation domain 20, 32 (Figure 1). PAX3/PAX7 and FOXO1 coding sequences are fused in-frame, creating functional—albeit aberrant—transcription factors. PAX-FOXO1 fusion proteins are discussed in greater detail below.
Molecular pathology studies of the chimeric products reveal that ~60% of ARMS tumors are PAX3-FOXO1-positive, ~20% are PAX7-FOXO1-positive, and ~20% are fusion-negative 45,47 These studies thus confirm that there is a subset of histologically defined ARMS tumors that are negative for the hallmark translocations generating PAX3-FOXO1 or PAX7-FOXO1 33. In rare cases, alternative translocations, such as t(2;2)(p23;q35) and t(2;8)(q35;q13), result in fusion of PAX3 to nuclear receptor coactivator genes NCOA1 and NCOA2, respectively 48. Most cases in this PAX3-FOXO1 and PAX7-FOXO1-negative subset show no detectable rearrangements involving PAX3, PAX7, or FOXO1, providing evidence for bona fide fusion-negative ARMS cases 49. Interestingly, fusion-negative ARMS demonstrates genetic changes characteristic of ERMS, which is consistent with the similar expression patterns 50 and clinical outcomes 51 of fusion-negative ARMS and ERMS cases.
2.2. PAX-FOXO1 Oncogenicity
The PAX-FOXO1 fusion products have altered expression, subcellular localization, and function, compared to wild-type PAX3, PAX7, or FOXO1. Both PAX-FOXO1 fusion proteins are expressed at higher levels than their wild-type PAX counterparts; PAX7-FOXO1 overexpression results from gene amplification while PAX3-FOXO1 overexpression occurs via copy number-independent enhanced transcription 52. In contrast to the wild-type FOXO1 protein that can shuttle between the nucleus and cytoplasm, the PAX3- or PAX7-FOXO1 protein is localized exclusively in the nucleus. Finally, these fusion proteins activate transcription of target genes 10–100 fold more potently than wild-type PAX3 and PAX7 32, 53, 54
Numerous studies have demonstrated the oncogenic capacity of the PAX3/PAX7-FOXO1 fusion protein. In chicken embryo fibroblasts and murine NIH 3T3 fibroblasts, ectopic expression of PAX3-FOXO1, but not wild-type PAX3, resulted in transformation as evidenced by focus formation and anchorage-independent growth in soft agar 55–57 Based on these early studies, PAX3-FOXO1 appears to function as a dominant-acting oncogene 36. This fusion likely contributes to tumorigenesis through several mechanisms 58. The finding that an engineered PAX3-KRAB repressor suppressed the oncogenicity of Rh30 ARMS cells in vitro and in vivo, supports the hypothesis that PAX3-FOXO1’s aberrant transcriptional activity lies at the heart of its oncogenic potential 59.
Despite early reports demonstrating the transformative capacity of the PAX3-FOXO1 fusion 55–57, additional studies revealed that PAX3-FOXO1 is generally not sufficient for complete oncogenic transformation 33, 58, 60. Ectopic expression of PAX3-FOXO1 alone failed to result in transformation of human myoblasts or murine mesenchymal stem cells 25, 61. In fact, high expression levels of PAX3-FOXO1, comparable to endogenous fusion expression levels in human ARMS tumor cells, were found to be anti-proliferative in immortalized murine cell lines62.
In collaboration with added genetic lesions, PAX3-FOXO1 expression is capable of transforming human and murine cells to recapitulate ARMS tumors 33, 58. For example, immortalized human myoblasts expressing PAX3-FOXO1 were transformed upon introduction of MYCN 61. Similarly, p53 inactivation was required in PAX3-FOXO1-expressing murine mesenchymal stem cells to elicit ARMS-like tumor formation when injected into immunocompromised mice 25. Human skeletal muscle myoblasts stably expressing PAX3-FOXO1 can produce ARMS-like tumors after addition of TERT and MYCN and loss of CDKN2A 26.In studies of a mouse model using a conditional PAX3-FOXO1 knock-in allele, ARMS formed at low frequency, but addition of conditional Trp53 or Cdkn2a inactivation increased ARMS tumor incidence, providing further evidence that the PAX3-FOXO1 fusion requires accompanying genetic lesions for ARMS pathogenesis 27
Although PAX3-FOXO1 and PAX7-FOXO1 are virtually indistinguishable in structure 31,PAX3-FOXO1 expression portends an especially unfavorable outcome relative to PAX7-FOXO1 63. In recent analyses of a cohort of fusion-positive RMS, PAX7-FOXO1 fusion status demonstrated a statistically significant association with improved overall survival (p=0.0012) 63. The poorer prognosis associated with PAX3-FOXO1-positive tumors coupled with the ARMS- specific nature of PAX3-FOXO1 expression make this oncogenic chimera a very appealing therapeutic target. It is worth noting that, to date, most functional studies have concentrated on PAX3-FOXO1, though many findings can be extended in conception to include PAX7-FOXO1. This review will focus on molecular therapeutic strategies to abrogate oncogenic activity driven by the PAX3-FOXO1 fusion in ARMS.
3. Targeting PAX3-FOXO1
3.1. Regulating PAX3-FOXO1 Expression
3.1.1. RNA Interference and Antisense Technologies
While PAX3-FOXO1 expression is typically not sufficient for full oncogenic transformation 33, 58, 60, the fusion protein plays a necessary and fundamental role in ARMS tumorigenesis 64. Indeed, reduced cellular proliferation, decreased motility and invasion, and increased myogenic differentiation were observed upon PAX3-FOXO1 depletion 64 These phenotypic effects can be attributed to PAX3-FOXO1, as depletion of the oncogenic chimera was achieved by siRNA specifically targeting the PAX3-FOXO1 fusion 64 Furthermore, in another study that selectively decreased expression of PAX3-FOXO1 using shRNA directed against the PAX3-FOXO1 fusion point, PAX3-FOXO1-expressing human myoblast-derived tumor cells and ARMS cells displayed significantly reduced proliferation rates and transformation capabilities coupled with elevated myogenic differentiation relative to cells transduced with control shRNA 61.
PAX3-FOXO1 may also be required for cell survival, though findings are not as conclusive as those described above. Antisense oligonucleotide- or siRNA-mediated depletion of PAX3-FOXO1 induced apoptosis, suggesting that the fusion protein is essential in cell viability 65, 66. The caveat is that these approaches 65, 66 were directed toward 5’ PAX3 sequences and thereby targeted wild-type PAX3 as well as the fusion, thus compromising a definitive interpretation of PAX3-FOXO1 as anti-apoptotic.
The anti-tumor effects of PAX3-FOXO1 depletion provide proof of principle for therapeutic strategies designed to abrogate PAX3-FOXO1 expression. Although additional technological advances are required, siRNA/shRNA approaches targeting the oncogenic PAX3-FOXO1 fusion may become a viable method for therapy. Despite existing limitations that impede full clinical translation of antisense therapeutics 67, antisense oligonucleotide-mediated depletion of PAX3-FOXO1 is a potential therapeutic alternative. In a related study, antisense oligodeoxynucleotide treatment against EWSR1-FLI1 in Ewing’s sarcoma induced tumor regression 68. Moreover, EWSR1-FLI1-targeting antisense oligonucleotides loaded onto nanosphere-chitosan resulted in efficient and tumor-specific delivery of the antisense oligonucleotides 69.
3.1.2. Other Translational or Post-Translational Mechanisms
Preliminary studies of RMS cell lines in vitro identified camptothecin as a selective chemotherapeutic agent in ARMS 70. Camptothecin is a topoisomerase I inhibitor, and its derivatives topotecan and irinotecan have both been evaluated for their utility in RMS in Phase II clinical trials, though neither significantly improved survival 32. Interestingly, the sensitivity of ARMS cells to camptothecin appeared to depend not on topoisomerase I, but on the transcriptional activity of PAX3-FOXO1, as ectopic expression of PAX3-FOXO1 in ERMS cells increases sensitivity to campothecin 70. Further studies revealed that camptothecin reduces PAX3-FOXO1 transactivation by decreasing its protein expression. Camptothecin-mediated downregulation of fusion protein levels was not attributable to AKT dephosphorylation, p53 function, or reduced PAX3-FOXO1 mRNA expression. Collectively, these data suggest that camptothecin may enhance degradation of the PAX3-FOXO1 fusion protein, and camptothecin is thus postulated to modulate PAX3-FOXO1’s ubiquitination status 70. Ubiquitylation of PAX3-FOXO1 has been demonstrated previously 71, and additional studies of camptothecin may provide a paradigm for therapeutic strategies to stimulate proteasomal degradation of oncogenic fusion protein.
3.2. Regulating the Phosphorylation Status of PAX3-FOXO1
3.2.1. The C-Terminal FOXO1 Portion
As a member of the FOXO transcription factor family, wild-type FOXO1 is regulated by a variety of posttranslational modifications, including deacetylation, ubiquitination, and phosphorylation 15. The FOXO1 protein shuttles between the nucleus and cytoplasm, with its subcellular localization regulated by the canonical PI3K/AKT signaling pathway 15, 16, 31 (Figure 2). Phosphorylation of FOXO1 confers cytoplasmic sequestration, and dephosphorylation allows nuclear translocation 16 (Figure 2). While multiple serine/threonine kinases, such as members of the AGC protein kinase family 72, CDK1 73, CDK2, CK1, and DYRK1 have been reported to phosphorylate FOXO1 at various sites, AKT is regarded as the primary kinase involved in phosphorylation-dependent modulation of FOXO1 subcellular localization and consequent transcriptional activity 15–17 FOXO1 harbors three evolutionarily conserved AKT phosphorylation sites located at threonine 24, serine 256, and serine 319 17 Upon activation, AKT translocates to the nucleus and directly phosphorylates FOXO1 74–76. AKT-directed phosphorylation at these residues appears to have no direct consequences on FOXO1 function, but rather facilitates 14–3-3 protein docking and binding, leading to cytoplasmic accumulation of this complex 17 (Figure 2). Thus, phosphorylation by AKT indirectly inactivates FOXO1 transcriptional function.
Figure 2.
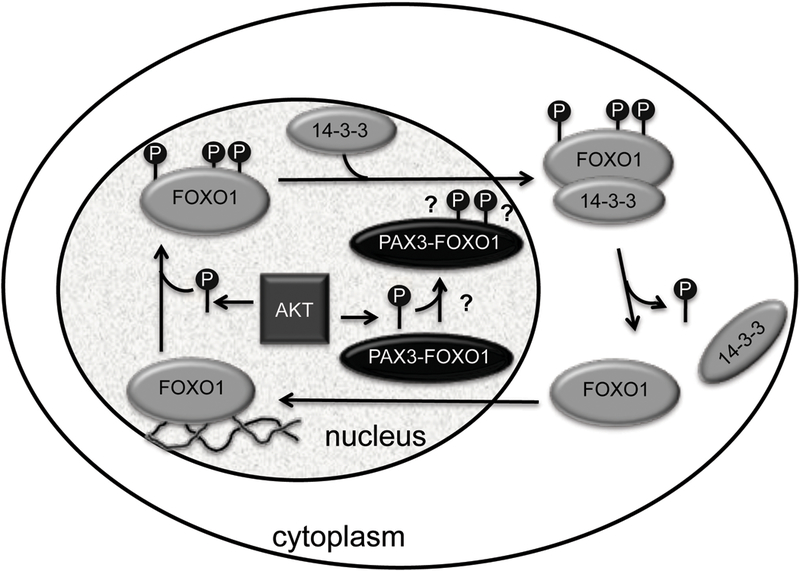
Phosphorylation-mediated regulation of FOXO1, but not PAX3-FOXO1, subcellular localization. Wild-type FOXO1 contains three evolutionarily conserved AKT phosphorylation sites (P). AKT-driven phosphorylation at these FOXO1 residues promotes 14–3-3 protein docking and binding, resulting in inactivation of FOXO1 transcriptional activity via cytoplasmic sequestration. PAX3-FOXO1 is resistant to AKT-mediated regulation by phosphorylation as evidenced by its constant nuclear localization.
PAX3-FOXO1 retains two of the three consensus AKT phosphorylation residues found in wild-type FOXO1 32. Using a FOXO1 mutant in which threonine 24 was replaced by alanine but serines 256 and 319 were unaltered, studies demonstrated that the presence of these two AKT phosphorylation sites matching those preserved in the PAX3-FOXO1 chimera are sufficient for AKT-mediated cytoplasmic sequestration and inhibition of FOXO1 transcriptional activity 77 A promising prediction was derived: if serine 256 and serine 319 in the FOXO1 portion of PAX3- FOXO1 were phosphorylated by AKT, then PAX3-FOXO1 should be relocalized to the cytoplasm, thereby quelling its ability to aberrantly transactivate target gene expression. In HEK293T and NIH 3T3 cells, transfection of PAX3-FOXO1 or a form with the two serines mutated, with or without constitutively active AKT, showed no effect on PAX3-FOXO1 nuclear localization or transcriptional activity 77. Possible explanations are that PAX3-FOXO1 adopts a conformation that precludes phosphorylation of relevant FOXO1 residues or that nuclear localization is controlled by N-terminal PAX3 domains.
A recent study using murine cell lines derived from conditional PAX3-FOXO1 knock-in mice and cultured in low serum provided evidence that PAX3-FOXO1 can be phosphorylated by hyperactivated AKT in this setting 78. Phosphorylation rendered PAX3-FOXO1 transcriptionally inactive but induced no change in nuclear localization of the fusion protein 78. The persistent nuclear presence of PAX3-FOXO1 suggests that, even if PAX3-FOXO1 undergoes phosphorylation, it is not conducive to 14–3-3 protein binding and resultant cytoplasmic sequestration.
While interesting, there is no available data to indicate that such AKT hyperactivation—and resulting PAX3-FOXO1 inactivation—can be induced in human ARMS tumors. IGF2 is overexpressed in human ARMS cells 31, and high phospho-AKT levels were observed in human ARMS tumors and cell lines, indicating the presence of endogenous AKT activation 79. Thus, human ARMS tumors retain PAX3-FOXO1 transcriptional activity despite the presence of activated AKT. Regardless of the mechanism conferring fusion protein resistance to AKT-mediated regulation in human ARMS, therapeutic approaches would likely be ineffective if they aimed at controlling PAX3-FOXO1 subcellular localization or transcriptional activity by modulating phosphorylation of the FOXO1 portion.
3.2.2. The N-Terminal PAX3 Portion
Compared to phosphorylation of FOXO1, kinases, residues, and functional consequences associated with PAX3 phosphorylation are poorly understood. Additionally, because wild-type PAX3 is exclusively nuclear 80, 81, phosphorylation status of the PAX3 region of PAX3-FOXO1 cannot be exploited using the same conceptual subcellular relocalization framework that applied to the fusion’s FOXO1 portion. In vitro and in vivo studies of murine primary myoblasts revealed serine 201, serine 205, and serine 209 as the only sites of phosphorylation in wild-type PAX3, and all three serine residues are retained in the oncogenic PAX3-FOXO1 fusion 82. CK2 and GSK3P have been identified as the kinases that phosphorylate wild-type PAX3 and PAX3- FOXO1 at serines 205 and 201, respectively 82, 83. Recently, CK2 was also found to be responsible for serine 209 phosphorylation 84
Wild-type PAX3 and PAX3-FOXO1 demonstrate distinct patterns of phosphorylation throughout early myogenic differentiation, leading to proposal of separate models for wild-type PAX3 versus PAX3-FOXO1 fusion phosphorylation 82. It appears that wild-type PAX3 undergoes GSK3β-mediated phosphorylation at serine 201 only after phosphorylation at serine 205 by CK2. Serine 201 phosphorylation subsequently promotes serine 205 dephosphorylation, as evidenced by the absence of PAX3 species exhibiting simultaneous phosphorylation at serines 201 and 205. Phosphorylation at serine 201 persists as phosphorylation at serine 209 becomes detectable 82.
Experiments in mouse primary myoblasts and human ARMS cell lines indicate that the oncogenic PAX3-FOXO1 fusion is phosphorylated by CK2 at serine 205 followed by GSK3β-driven phosphorylation at serine 201. In contrast to wild-type PAX3, however, these coincident phosphorylations are then maintained throughout early differentiation, and phosphorylation at serine 209 is never detected in PAX3-FOXO1 82. The precise functional consequences of each of these phosphorylation events have not yet been elucidated, though the notion that an altered phosphorylation status of the 5’ PAX3 portion of PAX3-FOXO1 relative to wild-type PAX3 contributes to ARMS is provocative. Mutational analyses of PAX3-FOXO1 engineered to mimic wild-type PAX3 phosphorylation patterns and vice versa as well as identification and characterization of additional putative phosphorylation sites, kinases, and phosphatases involved in PAX3 phosphorylation are needed and may inform a new avenue of therapeutic development for ARMS tumors.
Studies of a small-molecule inhibitor have already provided evidence that inhibiting phosphorylation of the PAX3 region of PAX3-FOXO1 can have anti-tumorigenic effects in ARMS cell lines and xenograft models 85. PKC412, an inhibitor of multiple kinases such as PKC, FGFR, AKT, FLT3, CDK1, and c-Kit, is a staurosporine derivative that suppresses ARMS proliferation and induces caspase 3-dependent apoptosis in vitro and reduced proliferation, increased apoptosis, and inhibited tumor growth in vivo 85. PKC412 treatment decreases DNA binding of PAX3-FOXO1, thereby abrogating its transcriptional activity in a phosphorylation-dependent mechanism without affecting its nuclear localization 85. Unlike the previously described studies identifying three PAX3 serine residues as phosphorylation sites 82, investigators here detected six potential phosphorylation residues at serines 187, 193, 197, 201, 205, and 209. It should be noted that simultaneous mutation of all six serines to phosphorylation-mimicking aspartate residues was required to overcome PKC412 inhibition but was not sufficient to rescue complete transcriptional activity of PAX3-FOXO1 85, suggesting that PKC412 inhibits fusion protein transactivation potential by mechanisms beyond these six phosphorylation events. Despite concerns of potential toxicity from off-target effects, this study demonstrates that small- molecule-mediated modulation of PAX3-FOXO1 post-transcriptional modifications, namely phosphorylation, is a promising approach for ARMS treatment.
3.3. Recognizing PAX3-FOXO1 as a Tumor Antigen
In pediatric sarcomas, translocation fusion products have long been regarded as potential tumor antigens 86, 87 In particular, it was hypothesized that tumor-specific peptides spanning the translocation breakpoint are proteolytically processed, bound to major histocompatibility complex (MHC) class I molecules, and presented on the surface of the tumor cell 86, 87 Displayed peptides could thereby target tumor cells for recognition and killing by CD8+ cytotoxic T cell lymphocytes (CTL) 86, 87 In a pilot study, apheresis fractions of monocytes and dendritic cells were pulsed with synthetic peptides comprising the PAX3-FOXO1 fusion breakpoint region sequence, and peptide pulsed vaccines were administered with interleukin-2 (IL-2) to ARMS patients 88. Vaccination, however, failed to affect clinical outcome 88.
In a subsequent study of immunotherapy in ARMS, dendritic cells were pulsed with a specific PAX3-FOXO1 fusion protein breakpoint peptide identified to bind HLA-B7 MHC class I molecules 89. These dendritic cells were then used to generate a lymphocyte-derived human CTL line capable of lysing PAX3-FOXO1 and HLA-B7-expressing ARMS tumor cells, but not PAX3-FOXO1 fusion-negative ERMS cells 89. Although this neoantigen is specific for only tumor cells expressing the oncogenic fusion protein and thus allows for highly targeted immunotherapy, HLA-B7 is expressed in less than 25% of the population, suggesting that the majority of ARMS patients is unlikely to benefit from this therapy 89. Other MHC class I molecules, including HLA-A1, HLA-A2, and HLA-A3, have been evaluated, but none were found to present neoantigens corresponding to the PAX3-FOXO1 fusion breakpoint region 90.
The most promising data comes from a more recent pilot study of consolidative immunotherapy 91. ARMS patients in remission following multimodal therapy were vaccinated with dendritic cells pulsed with PAX3-FOXO1 fusion protein breakpoint peptides in combination with autologous lymphocyte infusions with or without IL-2. This consolidation therapy regimen was well tolerated, and ARMS patients who received immunotherapy demonstrated significantly improved survival compared to those who did not 91. Results should be interpreted with caution, however, as patients with rapidly progressive disease were excluded from immunotherapy in this study. Therefore, it is likely that the observed increase in survival rate is at least partially attributable to patient selection 91. Nevertheless, immunotherapy represents a plausible strategy to antagonize the PAX3-FOXO1 oncoprotein. Investigations to optimize cancer immunotherapeutic efficacy, including alternative methods for enhancement of antigen immunogenicity and induction of dendritic cell maturation, are underway 91.
4. Targeting Downstream Factors of PAX3-FOXO1
Given the uniqueness of PAX3-FOXO1 to ARMS tumors, the oncogenic fusion itself is a very desirable therapeutic target. Nearly two decades after its initial characterization 18, 46, however, the chimeric transcription factor remains a difficult pharmacological target. Thus, it has become necessary to conceive of alternative treatment approaches.
Much progress has been made in recent years to generate a gene expression profile of ARMS that is distinct from that of ERMS. Multiple downstream genes activated by PAX3- FOXO1 have been identified from this ARMS expression profile, and provide another important source of potential targets for therapeutic intervention 4, 30, 66, 92–100. While this discussion is not all-inclusive, the presentation below and in Table 1 highlights the most therapeutically promising genes downstream of the PAX3-FOXO1 oncoprotein. Our selections are strongly based not only on molecular targets that promote ARMS tumorigenesis and metastasis, but also on those that contribute to tumor development, maintenance, and progression in other cancer categories.
Table 1.
Therapeutic targets implicated in ARMS and additional cancer categories.
| Target | Agent | Alteration in RMS | Alteration/Implication in Other Tumor Types |
|---|---|---|---|
| CNR1/CB1 | AM251; HU210; Delta(9)-tetrahydro-cannabinol169, 170 | Overexpression; Induced by PAX3-FOXO1 169, 170 | Invasion in breast cancer; migration in kidney 171, 172 |
| CPT1A | Etomoxir 173 | Direct PAX3-FOXO1 transcriptional target 174 | Proliferation and motility in lung 175 and prostate cancer 175, 176 |
| FGFR4 | PD173074; AZD4547; AZ12908010 109,112,123, 124 | Overexpression in RMS; Direct PAX3-FOXO1 transcriptional target; Association with PAX3-FOXO1-positive ARMS and reduced overall survival; Activating mutations found in 7.5% of RMS tumors (mostly ERMS) 95, 105–109, 111 | Overexpression in several malignancies (e.g., breast, gynecologic, lung, liver, pituitary, prostate, and pancreas) 116–122, 177 |
| IGF1R | Cixutumumab; Figitumumab; Teprotumumab 1, 2 | Overexpression; Association with aggressive behavior and reduced failure-free survival in ARMS 2, 178 | Overexpression in numerous human cancers (e.g., breast, colorectal, melanoma, liver, prostate, and sarcoma) 179, 180 |
| MET | Crizotinib; Tivantinib; OA-5D5; DN30; K252; SU11274; PHA665752; PF2341066; XL880; MK2461; MP470; SGX523; JNJ38877605 1, 2, 128 | Association with migration, invasion, and metastasis; Correlation of high MET expression with advanced stage, worse outcome, ARMS histology, and PAX3- FOXO1 expression; Direct PAX3-FOXO1 transcriptional target 142–147, 149, 150 | Overexpression in almost all carcinoma types (e.g., breast, colorectal, hepatocellular, oral squamous cell, ovarian, pancreatic, prostatic, renal cell, and thyroid) 128, 131–141 |
| MTOR (mTOR) | Everolimus; Ridaforolimus; Sirolimus; Temsirolimus 1, 2 | Strong association between activation of mTOR signaling components and poor failure-free or overall survival 181, 182 | Deregulation in several cancers (e.g., hamartoma syndromes, lymphomas, breast, and melanoma) 183–187 |
| MYCN | PNA-M7CW165 | Amplification, most frequently in ARMS; Correlation between high MYCN expression and PAX3/7-FOXO1 -positive ARMS tumors and poorer clinical outcome; Direct PAX3-FOXO1 transcriptional target 111,148, 161–166 | Aberrant expression of MYCN as a result of MYCN amplification detected in several malignancies (e.g., neuroblastoma, glioblastoma, medulloblastoma, retinoblastoma, anaplastic large cell lymphoma, and small cell lung carcinoma) 154, 159 |
| PDGFR | Imatinib; Olaratumab; Sorafenib; Dasatinib; Sunitinib; Axitinib; Pazopanib 1, 2 | Overexpression of PDGFR-A in ARMS and ERMS; Association between PDGFR expression and decreased survival 178, 188 | Aberrant expression or overexpression in a variety of cancers (e.g., glioma, breast, ovarian, prostate, and lung) 189–192 |
| PI3K | GSK1059615; BEZ235 2 | Putative overexpression/gain-of-function mutations based on downstream activation of AKT-mTOR axis 79, 181 | PIK3CA mutations in several malignancies (e.g., breast, colon, endometrial, glioblastoma, ovarian); PIK3CA amplification in many other tumors (e.g., head and neck, squamous cell lung carcinoma, cervical, gastric, and esophageal) 193 |
| VEGFR | Bevacizumab; Brivanib; Cediranib; Sorafenib; Sunitinib; Axitinib; Pazopanib 1, 2 | Higher VEGFR expression in ARMS versus ERMS implicated in metastatic phenotype of ARMS 194 | Aberrant expression or overexpression in vasculature of numerous solid tumors (e.g., breast, ovarian, colorectal, and lung) 195, 196 |
ARMS: Alveolar rhabdomyosarcoma; CNR1/CB1: Cannabinoid receptor 1; CPT1A: Carnitine palmitoyltransferase 1A; ERMS: Embryonal rhabdomyosarcoma; FGFR4: Fibroblast growth receptor 4; IGF1R: Insulin-like growth factor 1 receptor; MTOR: Mammalian target of rapamycin; PDGFR: Platelet-derived growth factor receptor; PI3K: Phosphatidylinositol 3-kinase; PIK3CA: Phosphatidylinositol-4,5-bisphosphate 3-kinase, catalytic subunit alpha; PNA: Peptide nucleic acid; RMS: Rhabdomyosarcoma; VEGFR: Vascular endothelial growth factor receptor
4.1. FGFR4
Fibroblast growth factor receptor 4 (FGFR4) encodes a member of the FGFR family of receptor tyrosine kinases (RTK) that is necessary during normal myogenic differentiation and injury-induced muscle regeneration, but not in mature, differentiated skeletal muscle 101–104. In RMS, FGFR4 was identified to be overexpressed at the mRNA and protein levels 105–108. Analysis of primary RMS tumors revealed a strong correlation of high FGFR4 expression with PAX3-FOXO1-positive ARMS 37, 107, advanced clinical stage, and lower overall survival 109. In functional studies using Rh30 cells, FGFR4 depletion mediated by inducible shRNA targeting FGFR4 inhibited proliferation in vitro and reduced proliferation and lung metastasis in vivo 109, further suggesting that FGFR4 is oncogenic in RMS. In addition, activating mutations in the tyrosine kinase domain of FGFR4 were found in 7.5% of RMS tumors (mostly ERMS) 95, and transduction of murine RMS772 cells with two of these FGFR4 mutants resulted in elevated proliferation, invasion, and metastatic capacity relative to wild-type FGFR4-transduced cells in vitro and in vivo 109. Recent findings in primary mouse myoblasts demonstrated that ectopic expression of a constitutively active FGFR4 mutant, but not wild-type FGFR4, is sufficient to contribute to ARMS tumorigenesis 110.
Subsequent work demonstrated that FGFR4 is a direct transcriptional target of PAX3-FOXO1 111, elucidating a second, mutation-independent mechanism for FGFR4 activation. This additional mechanism of overexpression is consistent with the observation that FGFR4 expression is higher in ARMS tumors than in PAX3-FOXO1-lacking ERMS tumors 109,112. Using ChlP-seq, two PAX3-FOXO1 binding sites were identified downstream of FGFR4, and functional examination revealed that one of the binding sites is a bona fide PAX3-FOXO1-dependent enhancer 111. These data solidified the role of FGFR4 as an important oncoprotein in RMS—a mutant protein in a subset of ERMS and an overexpressed wild-type protein in most fusion-positive ARMS.
As a kinase, FGFR4 is inherently more amenable to pharmacologic inhibition than the PAX3-FOXO1 transcription factor. In a very promising finding in vitro, FGFR4 mutant-expressing RMS772 cells exhibited enhanced sensitivity to FGFR inhibitor PD173074, as apoptotic cell death was higher in RMS772 cells transduced with FGFR4 mutants compared to those expressing wild-type FGFR4 109. PD173074 also attenuated cell proliferation of ARMS and ERMS cell lines overexpressing wild-type FGFR; therefore, FGFR mutations are not required for pharmacologic efficacy of PD173074 in vitro 112. In the in vivo setting, however, the small-molecule inhibitor was found to have a narrow therapeutic window and high toxicity, eliminating PD173074 as a viable option in ARMS therapy 112. Nonetheless, FGFR4 is clearly a very attractive candidate for targeted therapy in RMS and especially in ARMS.
Given that resources are limited, it is most prudent and efficient to develop targeted therapies with applicability to multiple cancer categories. Evidence not only indicated that ARMS tumors are likely addicted to the FGFR4 oncogene 109, 112–115, but FGFR4 has also been proposed as an oncogene in other tumor types, such as cancers of the liver, pituitary, lung, breast, and prostate 116–122. Thus, FGFR4 represents a prime target for pharmacologic intervention, particularly in the adjuvant setting. Other small-molecule inhibitors targeting FGFRs, including AZD4547 and AZ12908010, have already been developed and are undergoing preclinical and clinical evaluation 123, 124 Moreover, neutralizing or high-affinity monoclonal antibodies against FGFR4 have been generated, and strategies to target FGF19—the ligand of FGFR4—are under investigation 125–127.
4.2. MET
Like FGFR4, MET encodes a RTK proto-oncogene. Upon activation by hepatocyte growth factor (HGF, also referred to as scatter factor) binding, MET promotes cellular proliferation, motility, invasion, and survival 128, 129. Collectively, the cellular responses evoked by MET have been referred to as “invasive growth,” 130–132 a program that, if dysregulated, can have profound tumorigenic and metastatic consequences.
Amplification of MET, gain-of-function mutations, and transcriptional upregulation are mechanisms leading to MET overexpression and/or activation, which has been reported in a myriad of human primary tumors 128. Amplification-driven MET overexpression and constitutive kinase activation have been observed in medulloblastomas, esophageal and gastric carcinomas, and colorectal cancer 133–138. Additionally, MET amplification was found in non-small cell lung cancers with acquired resistance to erlotinib or gefitinib, two epidermal growth factor receptor inhibitors 139, 140. Activating mutations in MET were detected in pediatric hepatocellular carcinoma, head and neck squamous cell carcinoma, papillary renal cancer, gastric cancer, and melanoma 128, 131, 141. Remarkably, nearly all carcinoma types, such as breast, colorectal, hepatocellular, oral squamous cell, ovarian, pancreatic, prostatic, renal cell, and thyroid exhibit elevated MET expression resulting from transcriptional upregulation 128, 132, 141. In glioblastoma, osteosarcoma, breast carcinoma, and RMS 128, expression of HGF has been proposed to aberrantly activate MET through an autocrine loop 33, 128, 142.
Expression of MET in RMS is associated with migration, invasion, and metastasis 142–144 Thus, it is not surprising that high MET expression levels correlated with advanced stage, worse outcome, and ARMS histology, specifically PAX3-FOXO1 expression 145, 146. Although other studies found MET expressed in ERMS as well 147, 148, high MET expression is consistently observed in ARMS 143, 145, 146, 149, 150, whereas its expression levels are more variable in ERMS 147 These findings are in agreement with reports that MET is a PAX3-FOXO1 target gene that plays an essential role in mediating the fusion protein’s oncogenicity 5, 147, 149. Importantly, both ARMS and ERMS tumors demonstrated oncogene addiction to MET, as MET depletion abrogated proliferation, invasiveness, survival, and anchorage-independent growth in vitro and arrested tumor growth in vivo 147 Therefore, both RMS subtypes—in addition to the multitude of cancer types described above—could potentially benefit from MET-directed therapies.
Many therapeutic agents targeting MET have been developed (Table 1) and are at stages ranging from preclinical evaluation to Phase II clinical trials 128, 151. The sheer variety of compounds and strategies designed to antagonize MET holds much promise: HGF antagonists to block interaction of MET with its ligand, HGF and MET neutralizing antibodies to interfere with HGF-MET binding and to downregulate MET, MET decoys to sequester HGF and obstruct receptor dimerization, and small-molecule inhibitors to impair catalytic activity 128. MET inhibitors have been discussed comprehensively in a recent review 128. As clinical trials of MET inhibitors progress in other tumor types, evaluation of their safety and efficacy in RMS is clearly warranted and anticipated.
4.3. MYCN
Unlike the RTKs FGFR4 and MET, MYCN belongs to a transcription factor family of proto-oncogenes that includes MYC and MYCL 152, 153. MYCN is a basic helix-loop-helix/leucine zipper transcription factor that is expressed predominantly in neuronal tissues during embryogenesis 153, 154 Following heterodimerization with MAX, MYCN activates transcription of target genes, such as TERT, ODC, MDM2, and IGF1R 155. Recent reports that the related MYC protein is a global gene expression amplifier 156, 157, however, may foreshadow a comparable, more universal model of gene expression regulation for MYCN.
Normal MYCN expression is virtually undetectable in mature, post-embryonic tissues 153, 154, 158. Aberrant expression of MYCN as a result of MYCN amplification, however, has been detected in several malignancies, including neuroblastoma, glioblastoma, medulloblastoma, retinoblastoma, anaplastic large cell lymphoma, and small cell lung carcinoma 154, 159. MYCN amplification is perhaps best known in neuroblastoma, in which it clearly correlates with poor outcome 154, 160.
Several studies also demonstrated MYCN amplification in RMS tumors 148, 161–166. MYCN gene amplification occurs predominantly in ARMS 161–163 though recent studies revealed a low frequency of MYCN amplification in ERMS 148, 164, 165. In the latest investigation, MYCN amplification was present in 25% of ARMS patient samples versus 6% of ERMS 165. High MYCN expression levels significantly correlated with PAX3/7-FOXO1 -positive ARMS tumors and poorer clinical outcome for ARMS patients 165. Association of high MYCN expression with fusion gene positivity is consistent with previous findings that PAX3-FOXO1 increases MYCN mRNA expression 26, 148, suggesting that MYCN is a direct transcriptional target of the PAX3-FOXO1 oncoprotein. Moreover, ChIP-seq analysis revealed a PAX3-FOXO1 binding site downstream of the MYCN transcription start site, providing further evidence that PAX3-FOXO1 directly activates MYCN transcription 111.
As a transcription factor lacking enzymatic activity, perturbation of MYCN function is challenging. Numerous studies of the related MYC protein focused on disrupting MYC-MAX dimerization 152, which is conceptually a viable strategy. In vitro data provided proof of principle, but in vivo functionality of this approach remains to be evaluated 152. Preclinical studies of MYCN antigene therapy, however, are promising 165. In both ARMS and ERMS cell lines in vitro, treatment with an antigene peptide nucleic acid (PNA-MYCN) oligonucleotide that specifically inhibited MYCN mRNA expression abrogated proliferation and induced apoptosis 165.Antitumor activity of PNA-MYCN was especially marked and intriguing in ARMS; not only was PNA-MYCN effectiveness validated in vivo using a murine xenograft model, but also a novel interaction between MYCN and PAX3-FOXO1 was illuminated. Decreased MYCN levels resulted in reduced PAX3-FOXO1 expression, while MYCN overexpression led to elevated PAX3-FOXO1 levels 165. This novel positive feedback mechanism has profound potential for therapeutic exploitation in ARMS, as anti-MYCN therapy will directly inhibit MYCN, thereby indirectly suppressing PAX3-FOXO1 as well. Like killing two birds with one stone, one agent could kill two oncogenes. Thus, in refractory ARMS tumors, MYCN-directed therapy may provide a surrogate strategy for PAX3-FOXO1 inhibition 167.
5. Conclusion
Although recurrent chromosomal translocations are uncommon in solid tumors, they are characteristic of approximately one-third of all sarcomas 6. Studies of non-random chromosomal translocations and their resultant fusion genes have expanded our understanding of sarcoma biology, facilitated diagnosis and prognosis, and underscored the value of developing therapies to target oncogenic fusion genes. Using PAX3-FOXO1 in ARMS as a paradigm, ample evidence advocates two broad approaches for the treatment of translocation-associated sarcomas: 1) targeted therapies directed against the oncogenic chimera itself, and 2) therapeutic strategies targeting downstream genes activated by the fusion oncoprotein.
6. Expert Opinion
The uniqueness of PAX3-FOXO1 expression to ARMS is a double-edged sword—on one hand, it is advantageous for PAX3-FOXO1-directed therapies, conferring cytotoxicity specifically to cancer cells. On the other hand, however, PAX3-FOXO1 is exclusively expressed in ARMS 1, 7, a rare pediatric tumor. The same scenario is mirrored in other translocation-associated sarcomas, including ASPSCR1-TFE3 in alveolar soft part sarcoma and FUS-DDIT3 in myxoid liposarcoma 1.
Although targeting specific oncogenic chimeras is a viable therapeutic approach, the rare tumor context in which these fusion genes are expressed presents considerable challenges. Currently, despite more than 800 new anticancer drugs estimated to be in clinical development for adult tumors, the biopharmaceutical industry does not conduct preclinical research and development for rare cancers 168. From an economic perspective, the rationale of pharmaceutical companies is uncomplicated: drugs indicated for a rare malignancy have a smaller market and will therefore garner less profit compared to a more prevalent tumor. Drug research and development for rare cancers, such as ARMS and other translocation-associated sarcomas, is thus relegated to the academic sector funded by federal, foundation, and private grants. However, even if the pharmaceutical industry were to extend studies to include rare malignancies, patient enrollment in clinical trials would remain a major impediment to drug evaluation. Common cancers benefit by virtue of a greater patient pool, whereas resources for uncommon tumors are inherently limited, with accrual taking years rather than months. To compensate for the small patient population, clinical trials examining treatment for rare malignancies must recruit patients from numerous sites, which is the impetus for creation of cooperative clinical oncology trials groups such as the Children’s Oncology Group.
While efforts to develop targeted therapies against fusion proteins should not be abandoned, one must also be cognizant of molecular targets that are applicable to a broader tumor landscape. This review has highlighted FGFR4, MET, and MYCN as examples of such targets, but numerous other oncogenes, including IGF1R, PDGFR, VEGFR, PI3K, and MTOR (Table 1), are relevant to both rare, translocation-associated sarcomas 2 and to more common cancers. Such oncogenes may be found not only as genes downstream of the fusion oncoproteins but also potentially related to genetic events that cooperate with the fusion oncoproteins. By focusing on more generalizable molecular targets, the paradigm shifts to regard therapies not as tumor-specific, but as target-specific. With a higher likelihood for investment and a larger patient population from which to enroll for clinical trials, this approach provides the potential for including more rare malignancies in trials of mainstream targeted cancer therapeutics.
7. Article Highlights.
Although recurrent chromosomal translocations are typically rare in solid tumors, they are associated with approximately one-third of all sarcomas.
The majority of recurrent chromosomal translocations in sarcomas generate oncogenic chimeric transcription factors, which aberrantly transactivate target gene expression.
Using the PAX3-FOXO1 fusion oncoprotein in ARMS as an example, evidence is discussed that constructs a conceptual framework of therapeutic strategies to target not only the fusion product itself, but also the downstream products mediating fusion protein oncogenicity.
Most fusion genes are expressed in rare tumors, creating considerable challenges for drug development and clinical evaluation. Therefore, an important additional approach is to exploit downstream targets in these translocation-associated sarcomas that are also applicable to more common cancer categories.
Acknowledgments
* This work was supported by the Intramural Research Program of the National Cancer Institute
Abbreviations
- ARMS
Alveolar rhabdomyosarcoma
- CDK1/2
Cyclin-dependent kinase 1/2
- ChIP
Chromatin immunoprecipitation
- CK1/2
Casein kinase 1/2
- CNR1/CB1
Cannabinoid receptor 1
- COL1A1-PDGFB
Collagen type I alpha 1-Platelet-derived growth factor beta
- CPT1A
Carnitine palmitoyltransferase 1A
- CTL
Cytotoxic T cell lymphocytes
- DFSP
Dermatofibrosarcoma protuberans
- EGFR
Epidermal growth factor receptor
- ERMS
Embryonal rhabdomyosarcoma
- EWSR1-FLI1
Ewing’s sarcoma breakpoint region 1-Friend leukemia virus integration 1
- FGFR
Fibroblast growth factor receptor
- FISH
fluorescence in situ hybridization
- HGF
Hepatocyte growth factor
- HLA
Human leukocyte antigen
- IGF2
Insulin-like growth factor 2
- IGF1R
Insulin-like growth factor 1 receptor
- IL-2
Interleukin-2
- MHC
Major histocompatibility complex
- MTOR
Mammalian target of rapamycin
- NCOA1/2
Nuclear receptor coactivator 1
- ODC
Ornithine decarboxylase
- PDGFR
Platelet-derived growth factor receptor
- PI3K
Phosphatidylinositol 3-kinase
- PIK3CA
Phosphatidylinositol-4,5-bisphosphate 3-kinase, catalytic subunit alpha
- PKC
Protein kinase C
- PNA
Peptide nucleic acid
- RMS
Rhabdomyosarcoma
- RTK
Receptor tyrosine kinase
- RT-PCR
Reverse transcription polymerase chain reaction
- TERT
Telomerase reverse transcriptase
- TK
Tyrosine kinase
- VEGFR
Vascular endothelial growth factor receptor
Bibliography
* of importance
** of considerable importance
- 1.Anderson JL, Denny CT, Tap WD, Federman N. Pediatric Sarcomas: Translating Molecular Pathogenesis of Disease to Novel Therapeutic Possibilities. Pediatric research 2012. April 30 * This recent review provides a comprehensive description of translocation-associated sarcomas and the current status of targeted agents under clinical evaluation. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Demicco EG, Maki RG, Lev DC, Lazar AJ. New therapeutic targets in soft tissue sarcoma. Advances in anatomic pathology 2012. May;19(3):170–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Teicher BA. Searching for molecular targets in sarcoma. Biochemical pharmacology 2012. July 1;84(1):1–10. [DOI] [PubMed] [Google Scholar]
- 4.Barretina J, Taylor BS, Banerji S, Ramos AH, Lagos-Quintana M, Decarolis PL, et al. Subtype-specific genomic alterations define new targets for soft-tissue sarcoma therapy. Nature genetics 2010. August;42(8):715–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Taylor BS, Barretina J, Maki RG, Antonescu CR, Singer S, Ladanyi M. Advances in sarcoma genomics and new therapeutic targets. Nature reviews Cancer 2011. August;11(8):541–57.* This review discusses the sarcoma genome and identification of therapeutic targets from an integrated genomics persepctive. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Xia SJ, Barr FG. Chromosome translocations in sarcomas and the emergence of oncogenic transcription factors. Eur J Cancer 2005. November;41(16):2513–27. [DOI] [PubMed] [Google Scholar]
- 7.Slater O, Shipley J. Clinical relevance of molecular genetics to paediatric sarcomas. Journal of clinical pathology 2007. November;60(11): 1187–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.McArthur GA, Demetri GD, van Oosterom A, Heinrich MC, Debiec-Rychter M, Corless CL, et al. Molecular and clinical analysis of locally advanced dermatofibrosarcoma protuberans treated with imatinib: Imatinib Target Exploration Consortium Study B2225. Journal of clinical oncology : official journal of the American Society of Clinical Oncology 2005. February 1;23(4):866–73. [DOI] [PubMed] [Google Scholar]
- 9.Mok GF, Sweetman D. Many routes to the same destination: lessons from skeletal muscle development. Reproduction 2011. March;141(3):301–12. [DOI] [PubMed] [Google Scholar]
- 10.Relaix F, Rocancourt D, Mansouri A, Buckingham M. A Pax3/Pax7-dependent population of skeletal muscle progenitor cells. Nature 2005. June 16;435(7044):948–53. [DOI] [PubMed] [Google Scholar]
- 11.Tremblay P, Gruss P. Pax: genes for mice and men. Pharmacology & therapeutics 1994;61(1–2):205–26. [DOI] [PubMed] [Google Scholar]
- 12.Li CG, Eccles MR. PAX Genes in Cancer; Friends or Foes? Front Genet 2012;3:6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kuang S, Charge SB, Seale P, Huh M, Rudnicki MA. Distinct roles for Pax7 and Pax3 in adult regenerative myogenesis. J Cell Biol 2006. January 2;172(1): 103–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Relaix F, Montarras D, Zaffran S, Gayraud-Morel B, Rocancourt D, Tajbakhsh S, et al. Pax3 and Pax7 have distinct and overlapping functions in adult muscle progenitor cells. J Cell Biol 2006. January 2;172(1):91–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhang Y, Gan B, Liu D, Paik JH. FoxO family members in cancer. Cancer biology & therapy 2011. August 15;12(4):253–9. [DOI] [PubMed] [Google Scholar]
- 16.Zhang X, Tang N, Hadden TJ, Rishi AK. Akt, FoxO and regulation of apoptosis. Biochimica et biophysica acta 2011. November;1813(11):1978–86. [DOI] [PubMed] [Google Scholar]
- 17.Tzivion G, Dobson M, Ramakrishnan G. FoxO transcription factors; Regulation by AKT and 14–3-3 proteins. Biochimica et biophysica acta 2011. November;1813(11):1938–45. [DOI] [PubMed] [Google Scholar]
- 18.Galili N, Davis RJ, Fredericks WJ, Mukhopadhyay S, Rauscher FJ 3rd, Emanuel BS, et al. Fusion of a fork head domain gene to PAX3 in the solid tumour alveolar rhabdomyosarcoma. Nature genetics 1993. November;5(3):230–5. ** This paper was the first to describe the PAX3-FOXO1 fusion in ARMS tumors. [DOI] [PubMed] [Google Scholar]
- 19.Davis RJ, D’Cruz CM, Lovell MA, Biegel JA, Barr FG. Fusion of PAX7 to FKHR by the variant t(1; 13)(p36;q14) translocation in alveolar rhabdomyosarcoma. Cancer Res 1994. June 1;54(11):2869–72. **This paper was the first to report a variant chromosomal translocation resulting in PAX7-FOXO1 fusion gene expression in ARMS. [PubMed] [Google Scholar]
- 20.Davis RJ, Bennicelli JL, Macina RA, Nycum LM, Biegel JA, Barr FG. Structural characterization of the FKHR gene and its rearrangement in alveolar rhabdomyosarcoma. Human molecular genetics 1995. December;4(12):2355–62. [DOI] [PubMed] [Google Scholar]
- 21.Xia SJ, Rajput P, Strzelecki DM, Barr FG. Analysis of genetic events that modulate the oncogenic and growth suppressive activities of the PAX3-FKHR fusion oncoprotein. Lab Invest 2007. April;87(4):318–25. [DOI] [PubMed] [Google Scholar]
- 22.Avirneni-Vadlamudi U, Galindo KA, Endicott TR, Paulson V, Cameron S, Galindo RL. Drosophila and mammalian models uncover a role for the myoblast fusion gene TANC1 in rhabdomyosarcoma. J Clin Invest 2012. January 3;122(1):403–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Linardic CM, Downie DL, Qualman S, Bentley RC, Counter CM. Genetic modeling of human rhabdomyosarcoma. Cancer Res 2005. June 1;65(11):4490–5. [DOI] [PubMed] [Google Scholar]
- 24.Linardic CM, Naini S, Herndon JE 2nd, Kesserwan C, Qualman SJ, Counter CM. The PAX3-FKHR fusion gene of rhabdomyosarcoma cooperates with loss of p16INK4A to promote bypass of cellular senescence. Cancer Res 2007. July 15;67(14):6691–9. [DOI] [PubMed] [Google Scholar]
- 25.Ren YX, Finckenstein FG, Abdueva DA, Shahbazian V, Chung B, Weinberg KI, et al. Mouse mesenchymal stem cells expressing PAX-FKHR form alveolar rhabdomyosarcomas by cooperating with secondary mutations. Cancer Res 2008. August 15;68(16):6587–97. [DOI] [PubMed] [Google Scholar]
- 26.Naini S, Etheridge KT, Adam SJ, Qualman SJ, Bentley RC, Counter CM, et al. Defining the cooperative genetic changes that temporally drive alveolar rhabdomyosarcoma. Cancer Res 2008. December 1;68(23):9583–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Keller C, Arenkiel BR, Coffin CM, El-Bardeesy N, DePinho RA, Capecchi MR. Alveolar rhabdomyosarcomas in conditional Pax3:Fkhr mice: cooperativity of Ink4a/ARF and Trp53 loss of function. Genes Dev 2004. November 1;18(21):2614–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Keller C, Hansen MS, Coffin CM, Capecchi MR. Pax3:Fkhr interferes with embryonic Pax3 and Pax7 function: implications for alveolar rhabdomyosarcoma cell of origin. Genes Dev 2004. November 1;18(21):2608–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Keller C, Capecchi MR. New genetic tactics to model alveolar rhabdomyosarcoma in the mouse. Cancer Res 2005. September 1;65(17):7530–2. [DOI] [PubMed] [Google Scholar]
- 30.Nishijo K, Chen QR, Zhang L, McCleish AT, Rodriguez A, Cho MJ, et al. Credentialing a preclinical mouse model of alveolar rhabdomyosarcoma. Cancer Res 2009. April 1;69(7):2902–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Barr FG. Gene fusions involving PAX and FOX family members in alveolar rhabdomyosarcoma. Oncogene 2001. September 10;20(40):5736–46. [DOI] [PubMed] [Google Scholar]
- 32.Barr FG, Womer R. Rhabdomyosarcoma In: Orkin SH FD, Look AT, Lux SE, Ginsburg D, Nathan DG, ed. Oncology of Infancy and Childhood. Philadelphia: Saunders; 2009:743–828. [Google Scholar]
- 33.De Giovanni C, Landuzzi L, Nicoletti G, Lollini PL, Nanni P. Molecular and cellular biology of rhabdomyosarcoma. Future Oncol 2009. November;5(9):1449–75. [DOI] [PubMed] [Google Scholar]
- 34.Ognjanovic S, Linabery AM, Charbonneau B, Ross JA. Trends in childhood rhabdomyosarcoma incidence and survival in the United States, 1975–2005. Cancer 2009. September 15;115(18):4218–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Huh WW, Skapek SX. Childhood rhabdomyosarcoma: new insight on biology and treatment. Curr Oncol Rep 2010. November;12(6):402–10. [DOI] [PubMed] [Google Scholar]
- 36.Xia SJ, Pressey JG, Barr FG. Molecular pathogenesis of rhabdomyosarcoma. Cancer biology & therapy 2002. Mar-Apr;1(2):97–104. [DOI] [PubMed] [Google Scholar]
- 37.Wachtel M, Runge T, Leuschner I, Stegmaier S, Koscielniak E, Treuner J, et al. Subtype and prognostic classification of rhabdomyosarcoma by immunohistochemistry. Journal of clinical oncology : official journal of the American Society of Clinical Oncology 2006. February 10;24(5):816–22. [DOI] [PubMed] [Google Scholar]
- 38.Mercado GE, Barr FG. Fusions involving PAX and FOX genes in the molecular pathogenesis of alveolar rhabdomyosarcoma: recent advances. Current molecular medicine 2007. February;7(1):47–61. [DOI] [PubMed] [Google Scholar]
- 39.Scrable HJ, Witte DP, Lampkin BC, Cavenee WK. Chromosomal localization of the human rhabdomyosarcoma locus by mitotic recombination mapping. Nature 1987. October 15–21;329(6140):645–7. [DOI] [PubMed] [Google Scholar]
- 40.Scrable H, Cavenee W, Ghavimi F, Lovell M, Morgan K, Sapienza C. A model for embryonal rhabdomyosarcoma tumorigenesis that involves genome imprinting. Proc Natl Acad Sci U S A 1989. October;86(19):7480–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Besnard-Guerin C, Newsham I, Winqvist R, Cavenee WK. A common region of loss of heterozygosity in Wilms’ tumor and embryonal rhabdomyosarcoma distal to the D11S988 locus on chromosome 11p15.5. Human genetics 1996. February;97(2):163–70. [DOI] [PubMed] [Google Scholar]
- 42.Visser M, Sijmons C, Bras J, Arceci RJ, Godfried M, Valentijn LJ, et al. Allelotype of pediatric rhabdomyosarcoma. Oncogene 1997. September; 15(11): 1309–14. [DOI] [PubMed] [Google Scholar]
- 43.Feinberg AP. Imprinting of a genomic domain of 11p15 and loss of imprinting in cancer: an introduction. Cancer Res 1999. April 1;59(7 Suppl):1743s–6s. [PubMed] [Google Scholar]
- 44.Smith AC, Choufani S, Ferreira JC, Weksberg R. Growth regulation, imprinted genes, and chromosome 11p15.5. Pediatric research 2007. May;61(5 Pt 2):43R–7R. [DOI] [PubMed] [Google Scholar]
- 45.Sorensen PH, Lynch JC, Qualman SJ, Tirabosco R, Lim JF, Maurer HM, et al. PAX3-FKHR and PAX7-FKHR gene fusions are prognostic indicators in alveolar rhabdomyosarcoma: a report from the children’s oncology group. Journal of clinical oncology : official journal of the American Society of Clinical Oncology 2002. June 1;20(11):2672–9. [DOI] [PubMed] [Google Scholar]
- 46.Barr FG, Galili N, Holick J, Biegel JA, Rovera G, Emanuel BS. Rearrangement of the PAX3 paired box gene in the paediatric solid tumour alveolar rhabdomyosarcoma. Nature genetics 1993. February;3(2):113–7. [DOI] [PubMed] [Google Scholar]
- 47.Barr FG, Smith LM, Lynch JC, Strzelecki D, Parham DM, Qualman SJ, et al. Examination of gene fusion status in archival samples of alveolar rhabdomyosarcoma entered on the Intergroup Rhabdomyosarcoma Study-III trial: a report from the Children’s Oncology Group. The Journal of molecular diagnostics : JMD 2006. May;8(2):202–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sumegi J, Streblow R, Frayer RW, Dal Cin P, Rosenberg A, Meloni-Ehrig A, et al. Recurrent t(2;2) and t(2;8) translocations in rhabdomyosarcoma without the canonical PAX-FOXO1 fuse PAX3 to members of the nuclear receptor transcriptional coactivator family. Genes, chromosomes & cancer 2010. March;49(3):224–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Barr FG, Qualman SJ, Macris MH, Melnyk N, Lawlor ER, Strzelecki DM, et al. Genetic heterogeneity in the alveolar rhabdomyosarcoma subset without typical gene fusions. Cancer Res 2002. August 15;62(16):4704–10. [PubMed] [Google Scholar]
- 50.Davicioni E, Anderson MJ, Finckenstein FG, Lynch JC, Qualman SJ, Shimada H, et al. Molecular classification of rhabdomyosarcoma--genotypic and phenotypic determinants of diagnosis: a report from the Children’s Oncology Group. Am J Pathol 2009. February;174(2):550–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Williamson D, Missiaglia E, de Reynies A, Pierron G, Thuille B, Palenzuela G, et al. Fusion gene-negative alveolar rhabdomyosarcoma is clinically and molecularly indistinguishable from embryonal rhabdomyosarcoma. Journal of clinical oncology : official journal of the American Society of Clinical Oncology 2010. May 1;28(13):2151–8. [DOI] [PubMed] [Google Scholar]
- 52.Davis RJ, Barr FG. Fusion genes resulting from alternative chromosomal translocations are overexpressed by gene-specific mechanisms in alveolar rhabdomyosarcoma. Proc Natl Acad Sci U S A 1997. July 22;94(15):8047–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bennicelli JL, Edwards RH, Barr FG. Mechanism for transcriptional gain of function resulting from chromosomal translocation in alveolar rhabdomyosarcoma. Proc Natl Acad Sci U S A 1996. May 28;93(11):5455–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Bennicelli JL, Advani S, Schafer BW, Barr FG. PAX3 and PAX7 exhibit conserved cis-acting transcription repression domains and utilize a common gain of function mechanism in alveolar rhabdomyosarcoma. Oncogene 1999. July 29;18(30):4348–56. [DOI] [PubMed] [Google Scholar]
- 55.Scheidler S, Fredericks WJ, Rauscher FJ 3rd, Barr FG, Vogt PK. The hybrid PAX3-FKHR fusion protein of alveolar rhabdomyosarcoma transforms fibroblasts in culture. Proc Natl Acad Sci U S A 1996. September 3;93(18):9805–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lam PY, Sublett JE, Hollenbach AD, Roussel MF. The oncogenic potential of the Pax3-FKHR fusion protein requires the Pax3 homeodomain recognition helix but not the Pax3 paired-box DNA binding domain. Mol Cell Biol 1999. January;19(1):594–601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Cao Y, Wang C. The COOH-terminal transactivation domain plays a key role in regulating the in vitro and in vivo function of Pax3 homeodomain. The Journal of biological chemistry 2000. March 31;275(13):9854–62. [DOI] [PubMed] [Google Scholar]
- 58.Linardic CM. PAX3-FOXO1 fusion gene in rhabdomyosarcoma. Cancer Lett 2008. October 18;270(1): 10–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Fredericks WJ, Ayyanathan K, Herlyn M, Friedman JR, Rauscher FJ, 3rd. An engineered PAX3-KRAB transcriptional repressor inhibits the malignant phenotype of alveolar rhabdomyosarcoma cells harboring the endogenous PAX3-FKHR oncogene. Mol Cell Biol 2000;20(14):5019–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Hettmer S, Wagers AJ. Muscling in: Uncovering the origins of rhabdomyosarcoma. Nat Med 2010. February;16(2):171–3. [DOI] [PubMed] [Google Scholar]
- 61.Xia SJ, Holder DD, Pawel BR, Zhang C, Barr FG. High expression of the PAX3-FKHR oncoprotein is required to promote tumorigenesis of human myoblasts. Am J Pathol 2009. December;175(6):2600–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Xia SJ, Barr FG. Analysis of the transforming and growth suppressive activities of the PAX3-FKHR oncoprotein. Oncogene 2004. September 9;23(41):6864–71. [DOI] [PubMed] [Google Scholar]
- 63.Duan F, Smith LM, Gustafson DM, Zhang C, Dunlevy MJ, Gastier-Foster JM, et al. Genomic and clinical analysis of fusion gene amplification in rhabdomyosarcoma: a report from the Children’s Oncology Group. Genes, chromosomes & cancer 2012. July;51(7):662–74. * This study describes prognostic differences between PAX3-FOXO1 and PAX7-FOXO1-positive ARMS, consistent with the initial report in reference 45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kikuchi K, Tsuchiya K, Otabe O, Gotoh T, Tamura S, Katsumi Y, et al. Effects of PAX3-FKHR on malignant phenotypes in alveolar rhabdomyosarcoma. Biochem Biophys Res Commun 2008. January 18;365(3):568–74. [DOI] [PubMed] [Google Scholar]
- 65.Bernasconi M, Remppis A, Fredericks WJ, Rauscher FJ, 3rd, Schafer BW. Induction of apoptosis in rhabdomyosarcoma cells through down-regulation of PAX proteins. Proc Natl Acad Sci U S A 1996. November 12;93(23):13164–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Ebauer M, Wachtel M, Niggli FK, Schafer BW. Comparative expression profiling identifies an in vivo target gene signature with TFAP2B as a mediator of the survival function of PAX3/FKHR. Oncogene 2007. November 8;26(51):7267–81. [DOI] [PubMed] [Google Scholar]
- 67.Mansoor M, Melendez AJ. Advances in antisense oligonucleotide development for target identification, validation, and as novel therapeutics. Gene regulation and systems biology 2008;2:275–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Asami S, Chin M, Shichino H, Yoshida Y, Nemoto N, Mugishima H, et al. Treatment of Ewing’s sarcoma using an antisense oligodeoxynucleotide to regulate the cell cycle. Biol Pharm Bull 2008. March;31(3):391–4. [DOI] [PubMed] [Google Scholar]
- 69.Elhamess H, Bertrand JR, Maccario J, Maksimenko A, Malvy C. Antitumor vectorized oligonucleotides in a model of ewing sarcoma: unexpected role of nanoparticles. Oligonucleotides 2009. September;19(3):255–64. [DOI] [PubMed] [Google Scholar]
- 70.Zeng FY, Cui J, Liu L, Chen T. PAX3-FKHR sensitizes human alveolar rhabdomyosarcoma cells to camptothecin-mediated growth inhibition and apoptosis. Cancer Lett 2009. November 1;284(2):157–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Roeb W, Boyer A, Cavenee WK, Arden KC. Guilt by association: PAX3-FOXO1 regulates gene expression through selective destabilization of the EGR1 transcription factor. Cell Cycle 2008. April 1;7(7):837–41. [DOI] [PubMed] [Google Scholar]
- 72.Pearce LR, Komander D, Alessi DR. The nuts and bolts of AGC protein kinases. Nature reviews Molecular cell biology 2010. January;11(1):9–22. [DOI] [PubMed] [Google Scholar]
- 73.Yuan Z, Becker EB, Merlo P, Yamada T, DiBacco S, Konishi Y, et al. Activation of FOXO1 by Cdk1 in cycling cells and postmitotic neurons. Science 2008. March 21;319(5870):1665–8. [DOI] [PubMed] [Google Scholar]
- 74.Greer EL, Brunet A. FOXO transcription factors at the interface between longevity and tumor suppression. Oncogene 2005. November 14;24(50):7410–25. [DOI] [PubMed] [Google Scholar]
- 75.Dejana E, Taddei A, Randi AM. Foxs and Ets in the transcriptional regulation of endothelial cell differentiation and angiogenesis. Biochimica et biophysica acta 2007. June;1775(2):298–312. [DOI] [PubMed] [Google Scholar]
- 76.Cabodi S, Morello V, Masi A, Cicchi R, Broggio C, Distefano P, et al. Convergence of integrins and EGF receptor signaling via PI3K/Akt/FoxO pathway in early gene Egr-1 expression. Journal of cellular physiology 2009. February;218(2):294–303. [DOI] [PubMed] [Google Scholar]
- 77.del Peso L, Gonzalez VM, Hernandez R, Barr FG, Nunez G. Regulation of the forkhead transcription factor FKHR, but not the PAX3-FKHR fusion protein, by the serine/threonine kinase Akt. Oncogene 1999; 18(51):7328–33. * This study demonstrates resistance of PAX3-FOXO1 to AKT-driven regulation. [DOI] [PubMed] [Google Scholar]
- 78.Jothi M, Nishijo K, Keller C, Mal AK. AKT and PAX3-FKHR cooperation enforces myogenic differentiation blockade in alveolar rhabdomyosarcoma cell. Cell Cycle 2012. March 1; 11(5). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Cen L, Hsieh FC, Lin HJ, Chen CS, Qualman SJ, Lin J. PDK-1/AKT pathway as a novel therapeutic target in rhabdomyosarcoma cells using OSU-03012 compound. Br J Cancer 2007. September 17;97(6):785–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Corry GN, Hendzel MJ, Underhill DA. Subnuclear localization and mobility are key indicators of PAX3 dysfunction in Waardenburg syndrome. Human molecular genetics 2008. June 15; 17(12): 1825–37. [DOI] [PubMed] [Google Scholar]
- 81.Corry GN, Raghuram N, Missiaen KK, Hu N, Hendzel MJ, Underhill DA. The PAX3 paired domain and homeodomain function as a single binding module in vivo to regulate subnuclear localization and mobility by a mechanism that requires base-specific recognition. Journal of molecular biology 2010. September 10;402(1):178–93. [DOI] [PubMed] [Google Scholar]
- 82.Dietz KN, Miller PJ, Iyengar AS, Loupe JM, Hollenbach AD. Identification of serines 201 and 209 as sites of Pax3 phosphorylation and the altered phosphorylation status of Pax3-FOXO1 during early myogenic differentiation. Int J Biochem Cell Biol 2011. June;43(6):936–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Dietz KN, Miller PJ, Hollenbach AD. Phosphorylation of serine 205 by the protein kinase CK2 persists on Pax3-FOXO1, but not Pax3, throughout early myogenic differentiation. Biochemistry 2009. December 15;48(49):11786–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Iyengar AS, Loupe JM, Miller PJ, Hollenbach AD. Identification of CK2 as the kinase that phosphorylates Pax3 at Ser209 in early myogenic differentiation. Biochem Biophys Res Commun 2012. November 9;428(1):24–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Amstutz R, Wachtel M, Troxler H, Kleinert P, Ebauer M, Haneke T, et al. Phosphorylation regulates transcriptional activity of PAX3/FKHR and reveals novel therapeutic possibilities. Cancer Res 2008. May 15;68(10):3767–76. [DOI] [PubMed] [Google Scholar]
- 86.Goletz TJ, Mackall CL, Berzofsky JA, Helman LJ. Molecular alterations in pediatric sarcomas: potential targets for immunotherapy. Sarcoma 1998;2(2):77–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Mackall C, Berzofsky J, Helman LJ. Targeting tumor specific translocations in sarcomas in pediatric patients for immunotherapy. Clin Orthop 2000(373):25–31. [DOI] [PubMed] [Google Scholar]
- 88.Dagher R, Long LM, Read EJ, Leitman SF, Carter CS, Tsokos M, et al. Pilot trial of tumor-specific peptide vaccination and continuous infusion interleukin-2 in patients with recurrent Ewing sarcoma and alveolar rhabdomyosarcoma: An inter-institute NIH study. Med Pediatr Oncol 2002;38(3):158–64. [DOI] [PubMed] [Google Scholar]
- 89.van den Broeke LT, Pendleton CD, Mackall C, Helman LJ, Berzofsky JA. Identification and epitope enhancement of a PAX-FKHR fusion protein breakpoint epitope in alveolar rhabdomyosarcoma cells created by a tumorigenic chromosomal translocation inducing CTL capable of lysing human tumors. Cancer Res 2006. February 1;66(3): 1818–23. [DOI] [PubMed] [Google Scholar]
- 90.Rodeberg DA, Nuss RA, Heppelmann CJ, Celis E. Lack of effective T-lymphocyte response to the PAX3/FKHR translocation area in alveolar rhabdomyosarcoma. Cancer immunology, immunotherapy : CII 2005. June;54(6):526–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Mackall CL, Rhee EH, Read EJ, Khuu HM, Leitman SF, Bernstein D, et al. A pilot study of consolidative immunotherapy in patients with high-risk pediatric sarcomas. Clin Cancer Res 2008. August 1;14(15):4850–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Barber TD, Barber MC, Tomescu O, Barr FG, Ruben S, Friedman TB. Identification of target genes regulated by PAX3 and PAX3-FKHR in embryogenesis and alveolar rhabdomyosarcoma. Genomics 2002. March;79(3):278–84. [DOI] [PubMed] [Google Scholar]
- 93.Begum S, Emami N, Cheung A, Wilkins O, Der S, Hamel PA. Cell-type-specific regulation of distinct sets of gene targets by Pax3 and Pax3/FKHR. Oncogene 2005. March 10;24(11):1860–72. [DOI] [PubMed] [Google Scholar]
- 94.Bai Y, Li J, Fang B, Edwards A, Zhang G, Bui M, et al. Phosphoproteomics identifies driver tyrosine kinases in sarcoma cell lines and tumors. Cancer Res 2012. May 15;72(10):2501–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Shukla N, Ameur N, Yilmaz I, Nafa K, Lau CY, Marchetti A, et al. Oncogene mutation profiling of pediatric solid tumors reveals significant subsets of embryonal rhabdomyosarcoma and neuroblastoma with mutated genes in growth signaling pathways. Clin Cancer Res 2012. February 1;18(3):748–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Lae M, Ahn EH, Mercado GE, Chuai S, Edgar M, Pawel BR, et al. Global gene expression profiling of PAX-FKHR fusion-positive alveolar and PAX-FKHR fusion-negative embryonal rhabdomyosarcomas. J Pathol 2007. June;212(2):143–51. [DOI] [PubMed] [Google Scholar]
- 97.Khan J, Bittner ML, Saal LH, Teichmann U, Azorsa DO, Gooden GC, et al. cDNA microarrays detect activation of a myogenic transcription program by the PAX3-FKHR fusion oncogene. Proc Natl Acad Sci U S A 1999. November 9;96(23):13264–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Missiaglia E, Selfe J, Hamdi M, Williamson D, Schaaf G, Fang C, et al. Genomic imbalances in rhabdomyosarcoma cell lines affect expression of genes frequently altered in primary tumors: an approach to identify candidate genes involved in tumor development. Genes, chromosomes & cancer 2009. June;48(6):455–67. [DOI] [PubMed] [Google Scholar]
- 99.Beck AH, West RB, van de Rijn M. Gene expression profiling for the investigation of soft tissue sarcoma pathogenesis and the identification of diagnostic, prognostic, and predictive biomarkers. Virchows Arch 2010. February;456(2):141–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Nielsen TO, West RB. Translating gene expression into clinical care: sarcomas as a paradigm. Journal of clinical oncology : official journal of the American Society of Clinical Oncology 2010. April 1;28(10):1796–805. [DOI] [PubMed] [Google Scholar]
- 101.Marics I, Padilla F, Guillemot JF, Scaal M, Marcelle C. FGFR4 signaling is a necessary step in limb muscle differentiation. Development 2002. October;129(19):4559–69. [DOI] [PubMed] [Google Scholar]
- 102.Zhao P, Hoffman EP. Embryonic myogenesis pathways in muscle regeneration. Dev Dyn 2004. February;229(2):380–92. [DOI] [PubMed] [Google Scholar]
- 103.Zhao P, Caretti G, Mitchell S, McKeehan WL, Boskey AL, Pachman LM, et al. Fgfr4 is required for effective muscle regeneration in vivo. Delineation of a MyoD-Tead2-Fgfr4 transcriptional pathway. The Journal of biological chemistry 2006. January 6;281(1):429–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Lagha M, Kormish JD, Rocancourt D, Manceau M, Epstein JA, Zaret KS, et al. Pax3 regulation of FGF signaling affects the progression of embryonic progenitor cells into the myogenic program. Genes Dev 2008. July 1;22(13): 1828–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Khan J, Wei JS, Ringner M, Saal LH, Ladanyi M, Westermann F, et al. Classification and diagnostic prediction of cancers using gene expression profiling and artificial neural networks. Nat Med 2001. June;7(6):673–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Baird K, Davis S, Antonescu CR, Harper UL, Walker RL, Chen Y, et al. Gene expression profiling of human sarcomas: insights into sarcoma biology. Cancer Res 2005. October 15;65(20):9226–35. [DOI] [PubMed] [Google Scholar]
- 107.Davicioni E, Finckenstein FG, Shahbazian V, Buckley JD, Triche TJ, Anderson MJ. Identification of a PAX-FKHR gene expression signature that defines molecular classes and determines the prognosis of alveolar rhabdomyosarcomas. Cancer Res 2006. July 15;66(14):6936–46. ** This study describes a PAX3/7-FOXO1-specific gene expression profile derived from primary RMS tumors and cell lines. [DOI] [PubMed] [Google Scholar]
- 108.Yu SJ, Zheng L, Ladanyi M, Asa SL, Ezzat S. Sp1-mediated transcriptional control of fibroblast growth factor receptor 4 in sarcomas of skeletal muscle lineage. Clin Cancer Res 2004. October 1;10(19):6750–8. [DOI] [PubMed] [Google Scholar]
- 109.Taylor JGt, Cheuk AT, Tsang PS, Chung JY, Song YK, Desai K, et al. Identification of FGFR4-activating mutations in human rhabdomyosarcomas that promote metastasis in xenotransplanted models. J Clin Invest 2009. November;119(11):3395–407. ** This study was the first to demonstrate FGFR4 tyrosine kinase domain mutations in RMS tumors, representing the first reported RTK mutations in RMS. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Marshall AD, van der Ent MA, Grosveld GC. PAX3-FOXO1 and FGFR4 in alveolar rhabdomyosarcoma. Molecular carcinogenesis 2012. October;51(10):807–15. [DOI] [PubMed] [Google Scholar]
- 111.Cao L, Yu Y, Bilke S, Walker RL, Mayeenuddin LH, Azorsa DO, et al. Genome-wide identification of PAX3-FKHR binding sites in rhabdomyosarcoma reveals candidate target genes important for development and cancer. Cancer Res 2010. August 15;70(16):6497–508. ** This paper identifies therapeutic targets in ARMS by whole genome mapping of PAX3-FOXO1 binding sites. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Crose LE, Etheridge KT, Chen C, Belyea B, Talbot LJ, Bentley RC, et al. FGFR4 blockade exerts distinct antitumorigenic effects in human embryonal versus alveolar rhabdomyosarcoma. Clin Cancer Res 2012. July 15;18(14):3780–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Weinstein IB. Cancer. Addiction to oncogenes--the Achilles heal of cancer . Science 2002. July 5;297(5578):63–4. [DOI] [PubMed] [Google Scholar]
- 114.Weinstein IB, Joe A. Oncogene addiction. Cancer Res 2008. May 1;68(9):3077–80; discussion 80. [DOI] [PubMed] [Google Scholar]
- 115.Luo J, Solimini NL, Elledge SJ. Principles of cancer therapy: oncogene and non-oncogene addiction. Cell 2009. March 6;136(5):823–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.French DM, Lin BC, Wang M, Adams C, Shek T, Hotzel K, et al. Targeting FGFR4 inhibits hepatocellular carcinoma in preclinical mouse models. PLoS One 2012;7(5):e36713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Ezzat S, Zheng L, Zhu XF, Wu GE, Asa SL. Targeted expression of a human pituitary tumor-derived isoform of FGF receptor-4 recapitulates pituitary tumorigenesis. J Clin Invest 2002. January;109(1):69–78. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 118.Ding L, Getz G, Wheeler DA, Mardis ER, McLellan MD, Cibulskis K, et al. Somatic mutations affect key pathways in lung adenocarcinoma. Nature 2008. October 23;455(7216):1069–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Roidl A, Foo P, Wong W, Mann C, Bechtold S, Berger HJ, et al. The FGFR4 Y367C mutant is a dominant oncogene in MDA-MB453 breast cancer cells. Oncogene 2010. March 11;29(10): 1543–52. [DOI] [PubMed] [Google Scholar]
- 120.Tateno T, Asa SL, Zheng L, Mayr T, Ullrich A, Ezzat S. The FGFR4-G388R polymorphism promotes mitochondrial STAT3 serine phosphorylation to facilitate pituitary growth hormone cell tumorigenesis. PLoS Genet 2011. December;7(12):e1002400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Jain VK, Turner NC. Challenges and opportunities in the targeting of fibroblast growth factor receptors in breast cancer. Breast Cancer Res 2012. June 19;14(3):208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Sahadevan K, Darby S, Leung HY, Mathers ME, Robson CN, Gnanapragasam VJ. Selective over-expression of fibroblast growth factor receptors 1 and 4 in clinical prostate cancer. J Pathol 2007. September;213(1):82–90. [DOI] [PubMed] [Google Scholar]
- 123.Gavine PR, Mooney L, Kilgour E, Thomas AP, Al-Kadhimi K, Beck S, et al. AZD4547: an orally bioavailable, potent, and selective inhibitor of the fibroblast growth factor receptor tyrosine kinase family. Cancer Res 2012. April 15;72(8):2045–56. [DOI] [PubMed] [Google Scholar]
- 124.Chell V, Balmanno K, Little AS, Wilson M, Andrews S, Blockley L, et al. Tumour cell responses to new fibroblast growth factor receptor tyrosine kinase inhibitors and identification of a gatekeeper mutation in FGFR3 as a mechanism of acquired resistance. Oncogene 2012. August 6. [DOI] [PubMed] [Google Scholar]
- 125.Chen C, Patel S, Corisdeo S, Liu X, Micolochick H, Xue J, et al. Generation and characterization of a panel of monoclonal antibodies specific for human fibroblast growth factor receptor 4 (FGFR4). Hybridoma (Larchmt) 2005. June;24(3):152–9. [DOI] [PubMed] [Google Scholar]
- 126.Desnoyers LR, Pai R, Ferrando RE, Hotzel K, Le T, Ross J, et al. Targeting FGF19 inhibits tumor growth in colon cancer xenograft and FGF19 transgenic hepatocellular carcinoma models. Oncogene 2008. January 3;27(1):85–97. [DOI] [PubMed] [Google Scholar]
- 127.Miura S, Mitsuhashi N, Shimizu H, Kimura F, Yoshidome H, Otsuka M, et al. Fibroblast growth factor 19 expression correlates with tumor progression and poorer prognosis of hepatocellular carcinoma. BMC Cancer 2012;12:56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Comoglio PM, Giordano S, Trusolino L. Drug development of MET inhibitors: targeting oncogene addiction and expedience. Nature reviews Drug discovery 2008. June;7(6):504–16. [DOI] [PubMed] [Google Scholar]
- 129.Corso S, Comoglio PM, Giordano S. Cancer therapy: can the challenge be MET? Trends in molecular medicine 2005. June;11(6):284–92. [DOI] [PubMed] [Google Scholar]
- 130.Trusolino L, Comoglio PM. Scatter-factor and semaphorin receptors: cell signalling for invasive growth. Nature reviews Cancer 2002. April;2(4):289–300. [DOI] [PubMed] [Google Scholar]
- 131.Stella GM, Benvenuti S, Comoglio PM. Targeting the MET oncogene in cancer and metastases. Expert opinion on investigational drugs 2010. November;19(11):1381–94. [DOI] [PubMed] [Google Scholar]
- 132.Migliore C, Giordano S. Molecular cancer therapy: can our expectation be MET? Eur J Cancer 2008. March;44(5):641–51. [DOI] [PubMed] [Google Scholar]
- 133.Tong CY, Hui AB, Yin XL, Pang JC, Zhu XL, Poon WS, et al. Detection of oncogene amplifications in medulloblastomas by comparative genomic hybridization and array-based comparative genomic hybridization. Journal of neurosurgery 2004. February;100(2 Suppl Pediatrics):187–93. [DOI] [PubMed] [Google Scholar]
- 134.Rajcevic U, Juvan R, Gazvoda B, Repse S, Komel R. Assessment of differential expression of oncogenes in gastric adenocarcinoma by fluorescent multiplex RT-PCR assay. Pflugers Archiv : European journal of physiology 2001;442(6 Suppl 1):R190–2. [DOI] [PubMed] [Google Scholar]
- 135.Hara T, Ooi A, Kobayashi M, Mai M, Yanagihara K, Nakanishi I. Amplification of c-myc, K-sam, and c-met in gastric cancers: detection by fluorescence in situ hybridization. Lab Invest 1998. September;78(9):1143–53. [PubMed] [Google Scholar]
- 136.Seruca R, Suijkerbuijk RF, Gartner F, Criado B, Veiga I, Olde-Weghuis D, et al. Increasing levels of MYC and MET co-amplification during tumor progression of a case of gastric cancer. Cancer genetics and cytogenetics 1995. July 15;82(2):140–5. [DOI] [PubMed] [Google Scholar]
- 137.Miller CT, Lin L, Casper AM, Lim J, Thomas DG, Orringer MB, et al. Genomic amplification of MET with boundaries within fragile site FRA7G and upregulation of MET pathways in esophageal adenocarcinoma. Oncogene 2006. January 19;25(3):409–18. [DOI] [PubMed] [Google Scholar]
- 138.Umeki K, Shiota G, Kawasaki H. Clinical significance of c-met oncogene alterations in human colorectal cancer. Oncology 1999;56(4):314–21. [DOI] [PubMed] [Google Scholar]
- 139.Bean J, Brennan C, Shih JY, Riely G, Viale A, Wang L, et al. MET amplification occurs with or without T790M mutations in EGFR mutant lung tumors with acquired resistance to gefitinib or erlotinib. Proc Natl Acad Sci U S A 2007. December 26;104(52):20932–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Turke AB, Zejnullahu K, Wu YL, Song Y, Dias-Santagata D, Lifshits E, et al. Preexistence and clonal selection of MET amplification in EGFR mutant NSCLC. Cancer Cell 2010. January 19;17(1):77–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Danilkovitch-Miagkova A, Zbar B. Dysregulation of Met receptor tyrosine kinase activity in invasive tumors. J Clin Invest 2002. April;109(7):863–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Rees H, Williamson D, Papanastasiou A, Jina N, Nabarro S, Shipley J, et al. The MET receptor tyrosine kinase contributes to invasive tumour growth in rhabdomyosarcomas. Growth Factors 2006. September;24(3):197–208. [DOI] [PubMed] [Google Scholar]
- 143.Ferracini R, Olivero M, Di Renzo MF, Martano M, De Giovanni C, Nanni P, et al. Retrogenic expression of the MET proto-oncogene correlates with the invasive phenotype of human rhabdomyosarcomas. Oncogene 1996. April 18;12(8):1697–705. [PubMed] [Google Scholar]
- 144.Lukasiewicz E, Miekus K, Kijowski J, Drabik G, Wilusz M, Bobis-Wozowicz S, et al. Inhibition of rhabdomyosarcoma’s metastatic behavior through downregulation of MET receptor signaling. Folia histochemica et cytobiologica / Polish Academy of Sciences, Polish Histochemical and Cytochemical Society 2009. January;47(3):485–9. [DOI] [PubMed] [Google Scholar]
- 145.Chen Y, Takita J, Mizuguchi M, Tanaka K, Ida K, Koh K, et al. Mutation and expression analyses of the MET and CDKN2A genes in rhabdomyosarcoma with emphasis on MET overexpression. Genes, chromosomes & cancer 2007. April;46(4):348–58. [DOI] [PubMed] [Google Scholar]
- 146.Diomedi-Camassei F, McDowell HP, De Ioris MA, Uccini S, Altavista P, Raschella G, et al. Clinical significance of CXC chemokine receptor-4 and c-Met in childhood rhabdomyosarcoma. Clin Cancer Res 2008. July 1;14(13):4119–27. [DOI] [PubMed] [Google Scholar]
- 147.Taulli R, Scuoppo C, Bersani F, Accornero P, Forni PE, Miretti S, et al. Validation of met as a therapeutic target in alveolar and embryonal rhabdomyosarcoma. Cancer Res 2006. May 1;66(9):4742–9. ** This paper confirms MET as a direct PAX3-FOXO1 target and provides evidence of oncogene addiction to MET in both ARMS and ERMS tumors. [DOI] [PubMed] [Google Scholar]
- 148.Mercado GE, Xia SJ, Zhang C, Ahn EH, Gustafson DM, Lae M, et al. Identification of PAX3-FKHR-regulated genes differentially expressed between alveolar and embryonal rhabdomyosarcoma: focus on MYCN as a biologically relevant target. Genes, chromosomes & cancer 2008. June;47(6):510–20. [DOI] [PubMed] [Google Scholar]
- 149.Ginsberg JP, Davis RJ, Bennicelli JL, Nauta LE, Barr FG. Up-regulation of MET but not neural cell adhesion molecule expression by the PAX3-FKHR fusion protein in alveolar rhabdomyosarcoma. Cancer Res 1998. August 15;58(16):3542–6. [PubMed] [Google Scholar]
- 150.Epstein JA, Shapiro DN, Cheng J, Lam PY, Maas RL. Pax3 modulates expression of the c-Met receptor during limb muscle development. Proc Natl Acad Sci U S A 1996. April 30;93(9):4213–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Crose LE, Linardic CM. Receptor tyrosine kinases as therapeutic targets in rhabdomyosarcoma. Sarcoma 2011;2011:756982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Dang CV. MYC on the path to cancer. Cell 2012. March 30;149(1):22–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Lu X, Pearson A, Lunec J. The MYCN oncoprotein as a drug development target. Cancer Lett 2003. July 18;197(1–2): 125–30. [DOI] [PubMed] [Google Scholar]
- 154.Morgenstern DA, Anderson J. MYCN deregulation as a potential target for novel therapies in rhabdomyosarcoma. Expert Rev Anticancer Ther 2006. February;6(2):217–24. [DOI] [PubMed] [Google Scholar]
- 155.Pession A, Tonelli R. The MYCN oncogene as a specific and selective drug target for peripheral and central nervous system tumors. Curr Cancer Drug Targets 2005. June;5(4):273–83. [DOI] [PubMed] [Google Scholar]
- 156.Lin CY, Loven J, Rahl PB, Paranal RM, Burge CB, Bradner JE, et al. Transcriptional amplification in tumor cells with elevated c-Myc. Cell 2012. September 28;151(1):56–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Nie Z, Hu G, Wei G, Cui K, Yamane A, Resch W, et al. c-Myc is a universal amplifier of expressed genes in lymphocytes and embryonic stem cells. Cell 2012. September 28;151(1):68–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Thomas WD, Raif A, Hansford L, Marshall G. N-myc transcription molecule and oncoprotein. Int J Biochem Cell Biol 2004. May;36(5):771–5. [DOI] [PubMed] [Google Scholar]
- 159.Schwab M MYCN in neuronal tumours. Cancer Lett 2004. February 20;204(2):179–87. [DOI] [PubMed] [Google Scholar]
- 160.Schwab M, Westermann F, Hero B, Berthold F. Neuroblastoma: biology and molecular and chromosomal pathology. Lancet Oncol 2003. August;4(8):472–80. [DOI] [PubMed] [Google Scholar]
- 161.Driman D, Thorner PS, Greenberg ML, Chilton-MacNeill S, Squire J. MYCN gene amplification in rhabdomyosarcoma. Cancer 1994. April 15;73(8):2231–7. [DOI] [PubMed] [Google Scholar]
- 162.Hachitanda Y, Toyoshima S, Akazawa K, Tsuneyoshi M. N-myc gene amplification in rhabdomyosarcoma detected by fluorescence in situ hybridization: its correlation with histologic features. Mod Pathol 1998. December;11(12):1222–7. [PubMed] [Google Scholar]
- 163.Toffolatti L, Frascella E, Ninfo V, Gambini C, Forni M, Carli M, et al. MYCN expression in human rhabdomyosarcoma cell lines and tumour samples. J Pathol 2002. April;196(4):450–8. [DOI] [PubMed] [Google Scholar]
- 164.Williamson D, Lu YJ, Gordon T, Sciot R, Kelsey A, Fisher C, et al. Relationship between MYCN copy number and expression in rhabdomyosarcomas and correlation with adverse prognosis in the alveolar subtype. Journal of clinical oncology : official journal of the American Society of Clinical Oncology 2005. February 1;23(4):880–8. [DOI] [PubMed] [Google Scholar]
- 165.Tonelli R, McIntyre A, Camerin C, Walters ZS, Di Leo K, Selfe J, et al. Antitumor Activity of Sustained N-Myc Reduction in Rhabdomyosarcomas and Transcriptional Block by Antigene Therapy. Clin Cancer Res 2011. November 7 ** This paper demonstrates antitumor activity of MYCN silencing and novel positive feedback mechanisms between MYCN and PAX3-FOXO1. [DOI] [PubMed] [Google Scholar]
- 166.Barr FG, Duan F, Smith LM, Gustafson D, Pitts M, Hammond S, et al. Genomic and clinical analyses of 2p24 and 12q13-q14 amplification in alveolar rhabdomyosarcoma: a report from the Children’s Oncology Group. Genes, chromosomes & cancer 2009. August;48(8):661–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Barr FG. New Treatments for Rhabdomyosarcoma: the Importance of Target Practice. Clin Cancer Res 2012. February 1;18(3):595–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Norris RE, Adamson PC. Challenges and opportunities in childhood cancer drug development. Nature reviews Cancer 2012. November;12(11):776–82. [DOI] [PubMed] [Google Scholar]
- 169.Oesch S, Walter D, Wachtel M, Pretre K, Salazar M, Guzman M, et al. Cannabinoid receptor 1 is a potential drug target for treatment of translocation-positive rhabdomyosarcoma. Mol Cancer Ther 2009. July;8(7):1838–45. [DOI] [PubMed] [Google Scholar]
- 170.Marshall AD, Lagutina I, Grosveld GC. PAX3-FOXO1 Induces Cannabinoid Receptor 1 to Enhance Cell Invasion and Metastasis. Cancer Res 2011. December 15;71(24):7471–80. [DOI] [PubMed] [Google Scholar]
- 171.Sarnataro D, Pisanti S, Santoro A, Gazzerro P, Malfitano AM, Laezza C, et al. The cannabinoid CB1 receptor antagonist rimonabant (SR141716) inhibits human breast cancer cell proliferation through a lipid raft-mediated mechanism. Molecular pharmacology 2006. October;70(4): 1298–306. [DOI] [PubMed] [Google Scholar]
- 172.Song ZH, Zhong M. CB1 cannabinoid receptor-mediated cell migration. The Journal of pharmacology and experimental therapeutics 2000. July;294(1):204–9. [PubMed] [Google Scholar]
- 173.Hernlund E, Ihrlund LS, Khan O, Ates YO, Linder S, Panaretakis T, et al. Potentiation of chemotherapeutic drugs by energy metabolism inhibitors 2-deoxyglucose and etomoxir. Int J Cancer 2008. July 15;123(2):476–83. [DOI] [PubMed] [Google Scholar]
- 174.Liu L, Wang YD, Wu J, Cui J, Chen T. Carnitine palmitoyltransferase 1A (CPT1A): a transcriptional target of PAX3-FKHR and mediates PAX3-FKHR-dependent motility in alveolar rhabdomyosarcoma cells. BMC Cancer 2012. April 25; 12(1): 154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Zaugg K, Yao Y, Reilly PT, Kannan K, Kiarash R, Mason J, et al. Carnitine palmitoyltransferase 1C promotes cell survival and tumor growth under conditions of metabolic stress. Genes Dev 2011. May 15;25(10):1041–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Lin H, Lu JP, Laflamme P, Qiao S, Shayegan B, Bryskin I, et al. Inter-related in vitro effects of androgens, fatty acids and oxidative stress in prostate cancer: a mechanistic model supporting prevention strategies. Int J Oncol 2010. October;37(4):761–6. [DOI] [PubMed] [Google Scholar]
- 177.Sugiyama N, Varjosalo M, Meller P, Lohi J, Hyytiainen M, Kilpinen S, et al. Fibroblast growth factor receptor 4 regulates tumor invasion by coupling fibroblast growth factor signaling to extracellular matrix degradation. Cancer Res 2010. October 15;70(20):7851–61. [DOI] [PubMed] [Google Scholar]
- 178.Blandford MC, Barr FG, Lynch JC, Randall RL, Qualman SJ, Keller C. Rhabdomyosarcomas utilize developmental, myogenic growth factors for disease advantage: a report from the Children’s Oncology Group. Pediatr Blood Cancer 2006. March;46(3):329–38. [DOI] [PubMed] [Google Scholar]
- 179.Chu E The IGF-1R pathway as a therapeutic target. Oncology (Williston Park) 2011. May;25(6):538–9, 43. [PubMed] [Google Scholar]
- 180.Rikhof B, de Jong S, Suurmeijer AJ, Meijer C, van der Graaf WT. The insulin-like growth factor system and sarcomas. J Pathol 2009. March;217(4):469–82. [DOI] [PubMed] [Google Scholar]
- 181.Petricoin EF 3rd, Espina V, Araujo RP, Midura B, Yeung C, Wan X, et al. Phosphoprotein pathway mapping: Akt/mammalian target of rapamycin activation is negatively associated with childhood rhabdomyosarcoma survival. Cancer Res 2007. April 1;67(7):3431–40. [DOI] [PubMed] [Google Scholar]
- 182.Wan X, Helman LJ. The biology behind mTOR inhibition in sarcoma. Oncologist 2007. August;12(8): 1007–18. [DOI] [PubMed] [Google Scholar]
- 183.Janus A, Robak T, Smolewski P. The mammalian target of the rapamycin (mTOR) kinase pathway: its role in tumourigenesis and targeted antitumour therapy. Cellular & molecular biology letters 2005;10(3):479–98. [PubMed] [Google Scholar]
- 184.Guertin DA, Sabatini DM. Defining the role of mTOR in cancer. Cancer Cell 2007. July;12(1):9–22. [DOI] [PubMed] [Google Scholar]
- 185.Lane HA, Breuleux M. Optimal targeting of the mTORC1 kinase in human cancer. Current opinion in cell biology 2009. April;21(2):219–29. [DOI] [PubMed] [Google Scholar]
- 186.Garcia-Garcia C, Ibrahim YH, Serra V, Calvo MT, Guzman M, Grueso J, et al. Dual mTORC1/2 and HER2 blockade results in antitumor activity in preclinical models of breast cancer resistant to anti-HER2 therapy. Clin Cancer Res 2012. May 1;18(9):2603–12. [DOI] [PubMed] [Google Scholar]
- 187.Romeo Y, Moreau J, Zindy PJ, Saba-El-Leil M, Lavoie G, Dandachi F, et al. RSK regulates activated BRAF signalling to mTORC1 and promotes melanoma growth. Oncogene 2012. July 16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Taniguchi E, Nishijo K, McCleish AT, Michalek JE, Grayson MH, Infante AJ, et al. PDGFR-A is a therapeutic target in alveolar rhabdomyosarcoma. Oncogene 2008. November 20;27(51):6550–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Wang H, Yin Y, Li W, Zhao X, Yu Y, Zhu J, et al. Over-expression of PDGFR-beta promotes PDGF-induced proliferation, migration, and angiogenesis of EPCs through PI3K/Akt signaling pathway. PLoS One 2012;7(2):e30503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Uehara H, Kim SJ, Karashima T, Shepherd DL, Fan D, Tsan R, et al. Effects of blocking platelet-derived growth factor-receptor signaling in a mouse model of experimental prostate cancer bone metastases. Journal of the National Cancer Institute 2003. March 19;95(6):458–70. [DOI] [PubMed] [Google Scholar]
- 191.Antoniades HN, Galanopoulos T, Neville-Golden J, O’Hara CJ. Malignant epithelial cells in primary human lung carcinomas coexpress in vivo platelet-derived growth factor (PDGF) and PDGF receptor mRNAs and their protein products. Proc Natl Acad Sci U S A 1992. May 1;89(9):3942–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Henriksen R, Funa K, Wilander E, Backstrom T, Ridderheim M, Oberg K. Expression and prognostic significance of platelet-derived growth factor and its receptors in epithelial ovarian neoplasms. Cancer Res 1993. October 1;53(19):4550–4. [PubMed] [Google Scholar]
- 193.Chen CY, Cheng KC, Chang AY, Lin YT, Hseu YC, Wang HM. 10-Shogaol, an Antioxidant from Zingiber officinale for Skin Cell Proliferation and Migration Enhancer. International journal of molecular sciences 2012;13(2):1762–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Onisto M, Slongo ML, Gregnanin L, Gastaldi T, Carli M, Rosolen A. Expression and activity of vascular endothelial growth factor and metalloproteinases in alveolar and embryonal rhabdomyosarcoma cell lines. Int J Oncol 2005. September;27(3):791–8. [PubMed] [Google Scholar]
- 195.Spannuth WA, Nick AM, Jennings NB, Armaiz-Pena GN, Mangala LS, Danes CG, et al. Functional significance of VEGFR-2 on ovarian cancer cells. Int J Cancer 2009. March 1; 124(5): 1045–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Smith NR, Baker D, James NH, Ratcliffe K, Jenkins M, Ashton SE, et al. Vascular endothelial growth factor receptors VEGFR-2 and VEGFR-3 are localized primarily to the vasculature in human primary solid cancers. Clin Cancer Res 2010. July 15;16(14):3548–61. [DOI] [PubMed] [Google Scholar]