

Summary
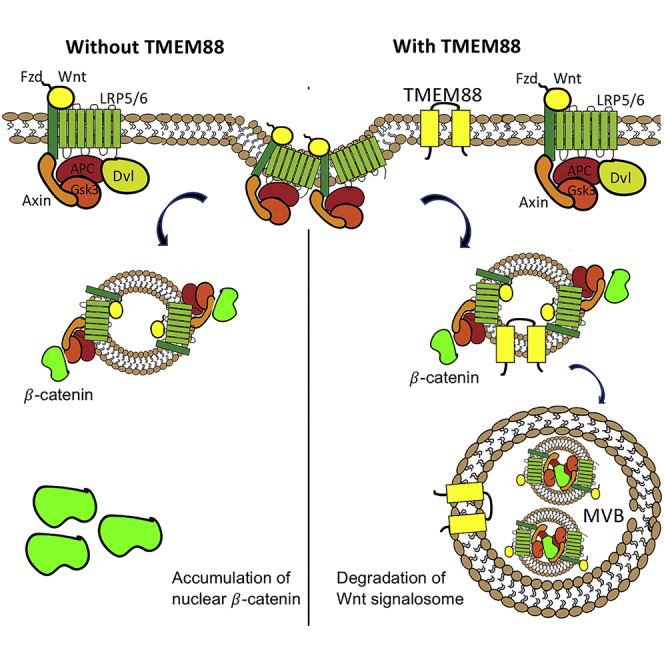
Wnt/β-catenin signaling is regulated in a bimodal fashion during cardiogenesis. Signaling is initially required to promote generation of precardiac mesoderm, but subsequently must be repressed for cardiac progenitor specification. TMEM88 was discovered recently as a negative regulator during the later phase of cardiac progenitor specification, but how TMEM88 functions was unknown. Based on a C-terminal PDZ-binding motif, TMEM88 was proposed to act by targeting the PDZ domain of Dishevelled, the positive Wnt signaling mediator. However, we discovered that TMEM88 acts downstream of the β-catenin destruction complex and can inhibit Wnt signaling independent of Dishevelled. TMEM88 requires the PDZ-binding motif for trafficking from Golgi to the plasma membrane and is also found in the multivesicular body (MVB) associated with the endocytosed Wnt signalosome. Expression of Tmem88 promotes association of the Wnt signalosome including β-catenin to the MVB, leading to reduced accumulation of nuclear β-catenin and repression of Wnt signaling.
Subject Areas: Molecular Mechanism of Behavior, Cell Biology, Stem Cells Research
Graphical Abstract
Highlights
-
•
Human ESCs with a targeted TMEM88 knockout are impaired for cardiac specification
-
•
TMEM88 does not require Dishevelled to inhibit Wnt signaling
-
•
TMEM88 is trafficked from Golgi to plasma membrane and then to the MVB
-
•
Expression of TMEM88 promotes association of the signalosome to the MVB
Molecular Mechanism of Behavior; Cell Biology; Stem Cells Research
Introduction
Canonical Wnt signaling regulates many aspects of embryonic development, and deregulation of the major effector protein β-catenin is associated with many diseases including cancer (Clevers and Nusse, 2012). Stage-specific activation and inhibition of Wnt signaling is critical for specification of cardiac progenitor fate during embryonic development (Naito et al., 2006, Ueno et al., 2007). Wnt signaling is negatively regulated at multiple levels, including extracellular ligand sequestration (Dkk, Sfrps, Cer, etc.), nuclear antagonists (Groucho, CtBP, NLK, etc.), and an intracellular “destruction complex” that targets the degradation of β-catenin, dependent on glycogen synthase kinase (GSK)-3β, Axin, and adenomatous polyposis coli (APC) (Clevers and Nusse, 2012, MacDonald et al., 2009, Nusse and Clevers, 2017). The destruction complex is recruited to the activated Wnt receptor complex (the Wnt signalosome) including LRP5/6 and Frizzled, followed by accumulation of cytosolic β-catenin that is able to traffic to the nucleus and stimulate transcription of target genes. Several models have been proposed regarding what happens next to the destruction complex. One study indicated that the destruction complex is removed from the receptor complex and can be recycled (Kim et al., 2013). Other studies demonstrated that the destruction complex becomes internalized into intraluminal vesicles (ILVs), which are subsequently sequestered into a multivesicular body (MVB) (Dobrowolski et al., 2012, Taelman et al., 2010, Vinyoles et al., 2014). Following Wnt signalosome internalization, the destruction complex becomes “locked down” in the MVB. This mechanism is thought to maintain a restricted low level of Wnt signaling after an initial acute inhibition of Wnt signaling by recruitment of the destruction complex to the membrane. The fate of proteins sequestered in the MVB is thought to be degradation, because the MVB eventually fuses with the lysosome (Dobrowolski et al., 2012, Metcalfe and Bienz, 2011, Piper and Katzmann, 2007).
The role of endocytosis in Wnt signaling has also been studied extensively. Internalization of LRP6 is important both for Wnt-mediated activation and Dkk-mediated inhibition of Wnt signaling, although the two processes use different endocytic routes, mediated by caveolin or clathrin, respectively (Yamamoto et al., 2006, Yamamoto et al., 2008). Caveolin is required for signalosome internalization upon Wnt ligand stimulation (Vinyoles et al., 2014). Inhibiting endocytosis blocks Wnt signaling activated by inhibition of GSK3 (Gagliardi et al., 2014, Saito-Diaz et al., 2018). The relationship and coordination between activation of Wnt signaling by sequestering the destruction complex to the membrane and subsequent activation of Wnt signaling by internalization of the destruction complex to the MVB is complex, and understanding regarding how this is regulated is limited.
TMEM88 is a putative transmembrane protein containing at the C terminus a tripeptide VWV sequence that can function to bind PDZ domains, including the PDZ domain in the positive regulatory protein of Wnt signaling, Dishevelled (DVL) (Lee et al., 2010). We showed previously that zebrafish Tmem88a functions downstream of the transcription factors Gata5/6 as an inhibitor of Wnt signaling during cardiac progenitor specification (Novikov and Evans, 2013). The loss of Tmem88a activity could be rescued by timed induction of Dkk1 expression, clearly supporting a normal function for Tmem88a in the suppression of Wnt activity (Novikov and Evans, 2013). TMEM88 was likewise identified as a Wnt regulator during in vitro human cardiomyocyte differentiation (Palpant et al., 2013). Several overexpression in vitro studies with cancer cells suggested an association between TMEM88 and DVL based on immunoprecipitation data, which led to the suggestion that TMEM88 might block Wnt signaling by physical sequestration of DVL (Ge et al., 2018, Lee et al., 2010, Zhang et al., 2015). However, this model has not been tested with respect to cardiogenesis.
Here, we confirmed using knockout (KO) human embryonic stem cells (hESCs) that human TMEM88 regulates cardiomyocyte specification, associated with suppression of Wnt signaling. Surprisingly, we found that TMEM88 is not dependent on DVL for this function. Using pathway inhibitors, we map the function of TMEM88 downstream of the destruction complex and upstream of β-catenin. Re-localization of the Wnt signalosome into the MVB was previously linked to activation of Wnt signaling (Ploper et al., 2015, Vinyoles et al., 2014). We found that expression of TMEM88 promotes sequestration of the Wnt signalosome including LRP6 and β-catenin to the MVB, as a previously unrecognized regulatory mechanism for repression of Wnt signaling.
Results
TMEM88 Knockout Cells Are Inefficient in Cardiomyocyte Differentiation
Previous studies in zebrafish embryos and hESCs showed that TMEM88 is expressed during the cardiac progenitor specification stage and that knockdown causes defects in cardiomyocyte development (Novikov and Evans, 2013, Palpant et al., 2013). To rigorously investigate the role of TMEM88 during cardiomyocyte development, we used CRISPR/Cas9 to target TMEM88 in hESCs to generate putative null alleles. Genomic sequencing confirmed the presence of compound heterozygous frameshift deletions of 5 and 7 bp within the first exon of the TMEM88 locus, predicted to generate early stop codons in both alleles of the mutant clone (Figure 1A). Using a standard directed differentiation assay (Figure 1B), TMEM88 protein was readily detected in lysates from wild-type cells as two isoforms at day 5 of differentiation, but was absent in lysates derived from the KO line (Figure 1C). The two isoforms of TMEM88 have been reported in various cell lines (Zhang et al., 2015); the smaller has a distinct C terminus lacking the PDZ-binding domain. Regardless, the western blot data confirmed the absence of both isoforms in cells derived from the mutant clone, demonstrating null alleles. Using directed differentiation to probe the impact of the TMEM88 mutation on cardiogenesis is complicated by the fact that the protocols for generating cardiomyocytes use exogenously added Wnt inhibitors, which might compensate for the loss of TMEM88. However, using conditions of limited Wnt repression, with addition of the small molecule Wnt inhibitor XAV939 for only 24 h from day 3 to 4 of differentiation, the generation of cardiac troponin T-positive cardiomyocytes was significantly compromised in TMEM88 KO cells (Figure 1D). Extending the duration of culture with XAV939 to day 5 largely rescued this defect, which confirms that the loss of TMEM88 can be compensated by a Wnt inhibitor (Figure 1D). Expression of mesoderm markers such as Brachyury, MIXL1, and EOMES was not impacted in TMEM88 KO cells using XAV939 treatment from day 3 to 4 of differentiation, indicating that cardiogenesis in TMEM88 KO is compromised at the later cardiac progenitor specification stage (Figure 1E).
Figure 1.

Knockout of TMEM88 Decreases Cardiomyocyte Differentiation In Vitro, which Is Rescued by an Extended Treatment with an Exogenous Wnt Inhibitor
(A) Schematics of guide RNA (gRNA)-binding site (red box) on the first exon of TMEM88. Genomic sequencing of the TMEM88 KO clone 22 showed compound heterozygous alleles of 5-bp deletion and 7-bp deletion, shown below the wild-type (WT) sequence. The red boxed area is the gRNA target. The PDZ-binding domain motif, Val-Trp-Val, is located at the C terminus followed by a stop codon.
(B) Schematic diagram of monolayer-directed differentiation of cardiomyocytes. See Methods for details.
(C) Western blot data show the absence of TMEM88 in TMEM88 KO cells in day 5 differentiating cardiomyocyte progenitor cells. WT embryonic stem cell (ESC) was used as a negative control.
(D) Percentage of cardiac troponin T-positive (cTnT+) cells by flow cytometry at day 14 of differentiation. XAV939 was added from day 3 to 4 for the samples labeled “XAV1d.” XAV939 was added from day 3 to 5 for the samples labeled “XAV2d.” Graphs show mean ± SEM. ∗∗p < 0.01; ∗∗∗p < 0.001.
(E) Expression of Brachyury, MIXL1, and EOMES in WT versus TMEM88 KO samples by qRT-PCR during cardiac differentiation. The results are displayed as normalized to WT ESC control or day1 control. Transcript levels in paired samples were not statistically different.
TMEM88 Is Associated with the Plasma Membrane, Golgi, and Multivesicular Bodies
Immunofluorescence assays confirmed that TMEM88 is expressed in hESC-derived cardiomyocyte progenitor cells marked by co-expression with the transcription factor NKX2.5 (Figure 2A), consistent with previous reports (Novikov and Evans, 2013, Palpant et al., 2013). NKX2.5 is exclusively nuclear, whereas TMEM88 could be detected at several distinct cellular locations (Figure 2A). TMEM88 staining specific to wild-type cardiac progenitors included the plasma membrane and a perinuclear region resembling the Golgi complex (Figure 2A). The Golgi localization was confirmed by co-staining for the Golgi marker Giantin, which showed a clear overlap with TMEM88 (Figure S1A). In some cells, only the Golgi staining was observed, suggesting that TMEM88 traffics from the Golgi to the plasma membrane. In addition to expression in the Golgi, we routinely found TMEM88 in the perinuclear regions that do not co-localize with Giantin (Figure S1A, white arrow), consistent with perinuclear MVBs. Immunoelectron microscopy coupled with gold-conjugated anti-TMEM88 antibody was used to more rigorously investigate the localization of TMEM88 in cardiac progenitors. TMEM88 was detected in MVB membranes (Figure 2B), as well as MVB vesicles and both Golgi and plasma membranes in day 7 cardiac progenitor cells (Figures 2B and S1B, black arrows). Perinuclear EEA1 is an established marker for MVB, best visualized when cells are permeabilized with digitonin before fixation, to remove cytosolic EEA1 (Vinyoles et al., 2014). Perinuclear digitonin-resistant EEA1+ structures are therefore used as a proxy for MVB (Vinyoles et al., 2014). Indeed, TMEM88 is present in EEA1+ digitonin-resistant bodies (Figure 2C). Western blot experiments confirmed that TMEM88 is highly enriched in a digitonin-resistant lysate compared with soluble fractions, co-segregating with the membrane marker NaK-ATPase (Figure 2D). In summary, TMEM88 is expressed in cardiac progenitor cells and found in the Golgi, plasma membrane, endocytic vesicles and MVB.
Figure 2.

TMEM88 Is Present in the Golgi, Plasma Membrane, and Digitonin-Resistant EEA1+ MVBs in Cardiac Progenitor Cells
(A) Immunofluorescence of TMEM88 and NKX2.5 in H1 embryonic stem cell (ESC)-derived day 7 cardiomyocyte progenitor cells. NKX2.5 is restricted to the nucleus, shown by Hoechst 33342 staining in the right panel. White arrow and arrowhead show localization of TMEM88 to the plasma membrane and MVB, respectively.
(B) Immunoelectron microscopy demonstrating presence of endogenous TMEM88 in MVB (arrows) in H1 ESC-derived day 7 cardiomyocyte progenitor cells. Arrows indicate gold-conjugated anti-TMEM88 antibody staining.
(C) Immunofluorescence of TMEM88 and an endosome marker, EEA1, in H1 ESC-derived day 7 differentiating cardiac progenitors. Cells were permeabilized with RB buffer supplemented with digitonin (25 μg/mL), fixed, and analyzed by immunofluorescence with the indicated antibody (Vinyoles et al., 2014).
(D) Western blots of digitonin-soluble fraction (DSF) and digitonin-resistant fraction (DRF) of day 5 ESC-derived differentiating cardiac progenitor cells using the CHIR99021/IWP2 protocol described in Methods. Membrane marker Na/K ATPase was used as a loading control for the DRF.
To monitor TMEM88 trafficking, we generated an N-terminal fusion of human TMEM88 with GFP (GFP-TMEM88) and evaluated the subcellular localization when expressed in cultured human embryonic kidney (HEK) 293T cells that lack endogenous TMEM88. GFP-TMEM88 was found associated with the plasma membrane (Figure 3A). Presence at the cell surface was confirmed by live imaging using total internal reflection fluorescence microscopy, demonstrating membrane-associated puncta with dynamic movements and internalization (Figure 3B and Video S1). Transfected cells subsequently treated with digitonin and co-stained with the MVB marker EEA1 showed the presence of GFP-TMEM88 or the non-tagged TMEM88 in digitonin-resistant EEA1+ MVB (Figure 3C, white arrows). GFP-TMEM88 and non-tagged TMEM88 were also found in the Golgi, suggesting that TMEM88 protein is being trafficked (Figure S2A). As in the cardiac progenitors, often the perinuclear GFP-TMEM88 did not co-stain with Giantin in 293T cells (Figure S2B, white arrow). Thus, TMEM88 expressed in 293T cells has cellular localizations consistent with endogenous TMEM88 in cardiomyocyte progenitor cells.
Figure 3.

Ectopically Expressed GFP-TMEM88 Is Found in the Golgi, Plasma Membrane, and Digitonin-Resistant EEA1+ MVBs in HEK293T Cells
(A) Live imaging of 293T cells transfected with GFP-TMEM88. Nucleus is stained with Hoechst 33342.
(B) A panel taken from total internal reflection fluorescence live imaging of 293T cells stably expressing GFP-TMEM88 (see also Video S1).
(C) 293T cells were transfected with GFP-TMEM88 or non-tagged TMEM88. Cells were permeabilized with RB buffer supplemented with digitonin (65 μg/mL), fixed, and analyzed by immunofluorescence with EEA1 antibody. White arrows indicate TMEM88 localized in the digitonin-resistant EEA1+ MVB.
(D) Live imaging of 293T cells transfected with GFP-TMEM88ΔC and stained with ER-tracker red dye in serum-free media. Nucleus is stained with Hoechst 33342.
The GFP+ puncta demonstrate membrane-localized GFP-TMEM88. The pattern is very dynamic, suggesting that GFP-TMEM88 is moving onto and then off of the plasma membrane (endocytosis).
The TMEM88 PDZ-Binding Motif Is Required for Trafficking to the Plasma Membrane
Previous studies showed that the VWV motif present at the carboxyl terminus of TMEM88 can bind to the PDZ domain of DVL, a positive regulator of Wnt signaling (Lee et al., 2010, Yu et al., 2015, Zhang et al., 2015), suggesting sequestration of DVL as a possible mechanism for Wnt signaling inhibition. To determine whether the PDZ-binding motif impacts the function of TMEM88, it was deleted from the fusion protein by replacing the sequences encoding the last three amino acids (VWV) with a translation stop codon (GFP-TMEM88ΔC). This mutant protein failed to localize to the plasma membrane or endosomal compartments, and instead was found exclusively in the endoplasmic reticulum (ER), co-localized with the ER dye, ER-tracker Red BODIPY TR (Figure 3D). This result suggests that a key function for the PDZ-binding motif is for interaction with Golgi-associated proteins containing a PDZ domain for trafficking to the plasma membrane (Romero et al., 2011).
TMEM88 Inhibits Wnt Signaling by Acting Downstream of the Destruction Complex and Upstream of Nuclear β-Catenin
To map where TMEM88 functions in the Wnt signaling pathway, a plasmid expressing GFP-TMEM88 was co-transfected in 293T cells with the Wnt-dependent T-cell factor reporter plasmid (TOP)-flash reporter (Veeman et al., 2003) and the Wnt pathway was activated at different levels. GFP-TMEM88 inhibited expression of Wnt3a-induced luciferase from the TOP-flash reporter, whereas the PDZ-binding mutant GFP-TMEM88ΔC failed to block reporter activity, even trending toward further activation (Figure 4A). As the mutant is blocked in the ER, this indicates that TMEM88 functions after trafficking to the membrane. To determine directly if TMEM88 functions by disabling DVL, the reporter assays were carried out in 293T cells that lack all DVL activity, carrying null mutations in each of the three DVL paralogs (Gammons et al., 2016, Jiang et al., 2015). This DVL triple knockout (TKO) cell line does not respond to Wnt ligands; however, blocking GSK3 with CHIR99021 (CHIR) can still activate Wnt signaling (Jiang et al., 2015). Strikingly, GFP-TMEM88 (but not GFP-TMEM88ΔC) fully inhibits TOP-flash activity induced by CHIR in DVL TKO cells (Figure 4B), indicating that DVL is dispensable for the repressive function of TMEM88. We also confirmed that GFP-TMEM88 (but not the mutant) also inhibits Wnt signaling activated by CHIR in wild-type 293T cells (Figure 4C). However, GFP-TMEM88 failed to inhibit TOP-flash reporter activity in the HCT116 cell line, which expresses a mutant form of β-catenin that is resistant to interaction with the destruction complex (Ilyas et al., 1997, Li et al., 2012, Saito-Diaz et al., 2018) and therefore fails to be degraded, accumulates in the nucleus, and activates the reporter even in the absence of Wnt induction (Figure 4D). We note that for these luciferase reporter experiments, we used 3 μM CHIR to inhibit GSK-3β, which is a relatively high concentration that might activate other kinases. However, titration experiments confirmed that 3 μM is necessary and sufficient for robust activation of the β-catenin-dependent TOP-flash reporter (Figure S3) and CHIR in the 3–6 μM range has been widely used in published cell-based Wnt reporter luciferase assays (Davidson et al., 2012, Kumar et al., 2016, Li et al., 2018). Taken together, these results show that GFP-TMEM88 must be trafficked out of the ER, but inhibits Wnt/β-catenin signaling downstream of the β-catenin destruction complex and upstream of activated β-catenin.
Figure 4.

GFP-TMEM88 Inhibits Wnt/β-Catenin Signaling Downstream of β-Catenin Destruction Complex and Upstream of β-Catenin
(A) TOP-flash luciferase reporter activity normalized to Renilla luciferase levels in control or Wnt3a-treated HEK293T cells transfected with plasmid vectors expressing GFP-TMEM88, GFP-TMEM88ΔC, or equimolar amount of GFP (control). Error bars, SEM. N = 4.
(B) TOP-flash luciferase reporter activity normalized to Renilla luciferase levels in CHIR-treated Dishevelled triple knockout (DVL TKO) HEK293T cells transfected with plasmid vectors expressing GFP-TMEM88, GFP-TMEM88ΔC, or equimolar amount of GFP (control). Error bars, SEM. N = 3.
(C) TOP-flash luciferase reporter activity normalized to Renilla luciferase levels in CHIR99021-treated HEK293T cells transfected with plasmid vectors expressing GFP-TMEM88, GFPTMEM88ΔC, or equimolar amount of GFP (control). Error bars, SEM. N = 3.
(D) TOP-flash/mutant TCF-binding sites (FOP)-flash luciferase reporter activity normalized to Renilla luciferase levels in HCT116 cells transfected with plasmid vectors expressing GFP-TMEM88, GFP-TMEM88ΔC, or equimolar amount of GFP (control). Error bars, SEM. N = 3.
All relative luciferase unit values were normalized to the negative control (control GFP vector-transfected cells treated with vehicle). A control vector/FOP-flash-transfected HCT116 was used as a negative control for normalization in (D). Graphs show mean ± SEM. N.S., non-significant; ∗p < 0.05, ∗∗∗p < 0.001.
The retention of TMEM88ΔC in the ER complicates analysis of whether TMEM88 normally functions at the plasma membrane. Therefore, we tagged the mutant protein with a membrane-bound form of GFP containing the palmitoylation signal MGSVSS from GAP-43 (Matsuda and Cepko, 2007, Okada et al., 1999) and evaluated if this could rescue function (mTMEM88ΔC). The tag was successful at moving TMEM88ΔC out of the ER and to the plasma membrane (Figure 5A). However, membrane localization of mGFP-TMEM88ΔC did not confer on the protein the ability to inhibit CHIR-activated reporter activity. Furthermore, the analogous membrane-localized wild-type version of GFP-TMEM88 (mGFP-TMEM88) lost the ability to block CHIR-activated reporter activity (Figure 5B). Therefore, although it may be important for TMEM88 to be trafficked to the membrane, the data are consistent with function not at the plasma membrane level, but following endocytic release from the membrane, for example, during Wnt signalosome trafficking.
Figure 5.

Membrane Localization of TMEM88 Is Not Sufficient for Its Function, and TMEM88 Inhibits Wnt Signaling Activated by Wnt Signalosome Internalization
(A) Live imaging of 293T cells transfected with membrane-targeted mGFP-TMEM88 or mGFP-TMEM88ΔC. Hoescht 33342 was used to stain the nucleus.
(B) TOP-flash luciferase reporter activity normalized to Renilla luciferase levels in CHIR99021-treated HEK293T cells transfected with plasmid vectors expressing mGFP-TMEM88, mGFP-TMEM88ΔC, or equimolar amount of mGFP (control). Error bars, SEM. N = 3.
(C) 293T cells transfected with plasmid vectors expressing CA-LRP6-GFP and GSK3-RFP were fixed and stained with Hoescht 33342.
(D) 293T cells transfected with a plasmid vector expressing CA-LRP6-GFP were permeabilized with RB buffer containing 65 μg/mL digitonin, fixed, and permeabilized with 0.2% Triton X-100 and stained with anti-EEA1 antibodies.
(E) TOP-flash/FOP-flash luciferase reporter activity normalized to Renilla luciferase levels in CA-LRP6-GFP-transfected 293T cells co-transfected with plasmid vectors expressing GFP-TMEM88, GFP-TMEM88ΔC, or equimolar amount of GFP (Control). pCS2 vector was used as a negative control for CA-LRP6-GFP.
(F) TOP-flash/FOP-flash luciferase reporter activity normalized to Renilla luciferase levels in CA-LRP6-GFP-transfected DVL TKO 293T cells co-transfected with plasmid vectors expressing GFP-TMEM88, GFP-TMEM88ΔC, or equimolar amount of GFP (control). pCS2 vector was used as a negative control for CA-LRP6-GFP.
All values were normalized to the negative control (control vector-transfected cells treated with vehicle). Graphs show mean ± SEM. N.S., non-significant; ∗p < 0.05.
TMEM88 Inhibits Signaling Activated by Wnt Signalosome Internalization
It is well established that endocytosis is required for the activation of Wnt signaling (Blitzer and Nusse, 2006, Brunt and Scholpp, 2018, Gagliardi et al., 2008). However, the relationship between endocytosis of the Wnt signalosome and function of the cytoplasmic β-catenin destruction complex is not entirely clear. One study (Gagliardi et al., 2014) showed that endocytosis inhibitors prevent Wnt signaling activated by blocking GSK-3β. Furthermore, CHIR has no effect on cytoplasmic β-catenin levels when endocytosis is inhibited by a temperature shift to 4°C (Saito-Diaz et al., 2018). These studies place the functional role of endocytosis for Wnt signaling downstream of the β-catenin destruction complex. Given that our results also place TMEM88 downstream of the β-catenin destruction complex, we tested whether TMEM88 regulates endocytic trafficking of the Wnt signalosome.
Following Wnt ligand activation, the internalized Wnt signalosome, composed of the Wnt receptor complex and the destruction complex, can be found in cytoplasmic vesicles (Bilic et al., 2007) and is sequestered into digitonin-resistant EEA1+ bodies (MVB) (Vinyoles et al., 2014). Wnt ligand activation can be modeled by expression of a constitutively active isoform of the co-receptor LRP6 that lacks the extracellular domain (CA-LRP6) (Bilic et al., 2007, Tamai et al., 2004, Vinyoles et al., 2014). We confirmed that CA-LRP6-GFP fusion protein is internalized in a ligand-independent manner and localizes to cytoplasmic puncta and the MVB. When co-expressed, both CA-LRP6-GFP and GSK3-RFP are co-sequestered into cytoplasmic puncta (Figure 5C), an indication of Wnt signalosome vesicle endocytosis (Bilic et al., 2007, Yamamoto et al., 2008), and to the MVB (Figure 5D), indicated by perinuclear digitonin-resistant EEA1+ structures (Taelman et al., 2010, Vinyoles et al., 2014). To investigate whether TMEM88 inhibits Wnt signaling by regulating Wnt signalosome trafficking, GFP-TMEM88 (or control GFP-TMEM88ΔC) was co-expressed with CA-LRP6-GFP and the TOP-flash reporter. GFP-TMEM88 (but not the mutant) inhibits TOP-flash activity induced by CA-LRP6-GFP, supporting a model in which TMEM88 regulates the internalized Wnt signalosome (Figure 5E). GFP-TMEM88 also inhibits TOP-flash activity induced by CA-LRP6-GFP in DVL TKO 293T cells, which is consistent with inhibition of Wnt signaling by GFP-TMEM88 being independent of DVL (Figure 5F). As expected, the mutant construct with the C-terminal VWV motif deleted, GFP-TMEM88ΔC, also fails to inhibit Wnt signaling activated by CA-LRP6-GFP in the DVL TKO cells (Figure 5F).
This model system was then used to investigate the impact of TMEM88 on localization of the Wnt signalosome. Expression of GFP-TMEM88 shifts CA-LRP6-mPlum localization from endocytic Wnt signalosome vesicles (puncta) to the MVB (Figures 6A and 6C). The cellular localization of CA-LRP6-mPlum depended on the location of GFP-TMEM88 in both 293T and DVL TKO 293T cells (Figures 6B and 6D). Thus, in cells with GFP-TMEM88 found primarily in the Golgi, based on co-localization with Giantin, CA-LRP6-GFP is found mostly in cytoplasmic puncta (Figures S4A and S4C). In contrast, cells with GFP-TMEM88 localized in the EEA1+ digitonin-resistant bodies show primarily co-localization of CA-LRP6-mPlum in the MVB (Figures S4B and S4C). These data indicate that TMEM88 promotes re-localization of internalized Wnt signalosome to the MVB to inhibit Wnt signaling.
Figure 6.

GFP-TMEM88 Localizing in EEA1+ Digitonin-Resistant Body (MVB) Enhances the MVB Co-localization of CA-LRP6-mPlum
(A) 293T cells were transfected with plasmid vectors expressing GFP-TMEM88 or GFP and CA-LRP6-mPlum; 24 h after transfection, the cells were fixed and the nucleus was stained with DAPI.
(B) Quantification of (A). The localization of CA-LRP6-mPlum was scored as either being present in cytoplasmic vesicles or in perinuclear MVB. ∗∗∗Chi squared p value: 1.14 × 10−13. For GFP, N = 46. For GFP-TMEM88, N = 53.
(C) 293T DVL TKO cells were transfected with plasmid vectors expressing GFP-TMEM88 or GFP and CA-LRP6-mPlum; 48 h after transfection, the cells were stained with DAPI and live imaged.
(D) Quantification of (C). The localization of CA-LRP6-mPlum was scored as either being present in cytoplasmic vesicles or in perinuclear MVB. ∗∗∗Chi squared p value: 4.39 × 10−9. For GFP, N = 40. For GFP-TMEM88, N = 60.
If this model were correct, TMEM88 might be expected to inhibit Wnt signaling by promoting co-localization of β-catenin to the MVB. Indeed, compared with control cells expressing GFP, cells expressing GFP-TMEM88 show a shifted co-localization of β-catenin predominantly to the MVB, indicated by digitonin-resistant structures stained with VPS4, another marker for MVB, as well as reduced nuclear β-catenin level in GFP-TMEM88-expressing cells compared with a control cell (GFP-expressing cells) (Figures 7A–7C). Reduced nuclear β-catenin level in cells expressing GFP-TMEM88 was also observed upon CHIR99021 treatment (Figure S5). Consistent with this model, TMEM88 and β-catenin show co-localization in cardiomyocyte progenitor cells within digitonin-resistant VPS4-positive structures (Figure 7D). A shift in β-catenin to the MVB associated with reduced nuclear β-catenin levels was also measured in DVL TKO cells expressing GFP-TMEM88 compared with control cells (expressing GFP), further supporting the hypothesis that TMEM88 function does not require DVL (Figures 8A–8C). These data indicate that TMEM88 enhances re-localization of β-catenin to MVB to inhibit Wnt signaling.
Figure 7.

GFP-TMEM88 Shifts the Accumulation of β-Catenin to Perinuclear MVB
(A) 293T cells were transfected with plasmids expressing GFP-TMEM88 or GFP and permeabilized with 65 μg/mL digitonin, fixed, and analyzed by immunofluorescence with the indicated antibodies. White arrowhead indicates β-catenin in MVB.
(B) 293T cells were transfected with plasmid vectors expressing GFP-TMEM88 or GFP and immunostained with β-catenin antibody; 24 h after transfection, the cells were fixed and the nucleus was stained with DAPI. The localization of β-catenin to the MVB was scored in a double-blinded manner. ∗∗∗Chi-squared p value: 5.01567 × 10−9. For GFP, N = 50. For GFP-TMEM88, N = 50.
(C) Quantification of nuclear β-catenin of Figure 7A. For GFP, N = 39. For GFP-TMEM88, N = 39. Graphs show mean ± SEM. ∗∗∗p < 0.001.
(D) Immunofluorescence of TMEM88 and β-catenin in H1 ESC-derived day 7 differentiating cardiac progenitor cells. Cells were permeabilized with RB buffer supplemented with digitonin (25 μg/mL), fixed, and analyzed by immunofluorescence with the indicated antibodies.
Figure 8.

GFP-TMEM88 Shifts the Accumulation of β-Catenin to Perinuclear MVB Even in the Absence of DVL
(A) DVL TKO 293T cells were transfected with plasmid vectors expressing GFP-TMEM88 or GFP; 24 h after transfection, the cells were fixed, immunostained with β-catenin antibody, and nuclei stained with DAPI.
(B) Quantification of localization of β-catenin of Figure 8A. ∗∗∗Chi-squared p value: 7.63 × 10−11. For GFP, N = 46. For GFP-TMEM88, N = 50.
(C) Quantification of nuclear β-catenin of Figure 8A. For GFP, N = 46. For GFP-TMEM88, N = 50. Graphs show mean ± SEM. ∗∗p < 0.01.
Discussion
TMEM88 was first identified in silico as a PDZ-domain-binding protein (Lee et al., 2010), based on the presence of a VWV motif that was known to interact with DVL. This led naturally to the investigation of its ability to modulate Wnt signaling, which was confirmed in reporter assays (Lee et al., 2010), of potential significance given the association of TMEM88 expression patterns described in various human neoplasias (Ma et al., 2017, Yu et al., 2015, Zhang et al., 2015). However, whether the impact of TMEM88 on Wnt signaling is actually mediated through interaction with DVL had not been previously tested. Our results using a DVL-null (triple mutant) cell line clearly indicate that TMEM88 can repress Wnt signaling in the absence of DVL, suggesting other means of regulation. We find that the VWV motif is required to repress Wnt signaling, but for trafficking out of the ER rather than interaction with DVL. There is precedence for PDZ domain proteins functioning in this context, for example, GOPC is a PDZ domain protein required for proper trafficking of the FZD receptor to the membrane surface (Yao et al., 2001). We cannot rule out that when DVL is expressed, TMEM88 interacts or antagonizes DVL function. In fact, we found the kinetics of signalosome localization to the MVB to be slower in the DVL TKO cells (localization measured at 48 h rather than 24 h post-transfection). We only observe that DVL is not required for TMEM88 to function in this pathway.
Instead, our data support a previously unexplored role for TMEM88, in sequestering the endocytosed Wnt signalosome, including cytoplasmic β-catenin, into the MVB, thereby limiting levels of nuclear accumulation. The impact on β-catenin during Wnt signalosome sequestration into MVB is not well understood. MVB is formed when the limiting membrane invaginates inward and buds into its own lumen to form ILVs. These ILVs get degraded when the MVB fuses with the lysosome, whereas proteins that remain on the limiting membrane of the MVB get recycled (Piper and Katzmann, 2007). β-Catenin being stabilized in MVB seemed unlikely based on the membrane topology of MVB (Piper and Katzmann, 2007). However, in their initial study showing that GSK3 is recruited into the MVB following Wnt activation, De Robertis and colleagues discovered that β-catenin is actually required for GSK sequestration, and mostly the phosphorylated, and therefore inactive, form is present in MVB. β-Catenin mutated at the GSK3 target sites (and therefore non-phosphorylated) accumulated in cytoplasmic particles that also sequester GSK3, although the identity of these cytoplasmic particles was not defined (Taelman et al., 2010). This counterintuitive role of β-catenin in MVB led to a model with β-catenin taking on a previously unknown function in sequestering GSK3 in the MVB, which may be independent of its transcriptional role in Wnt signaling (Taelman et al., 2010). A more recent study confirmed that only Ser27-phosphorylated β-catenin is detected in the MVB. In this case it is a stable form in this compartment due to the absence of β-TrCP1, the ubiquitin ligase responsible for modifying and eventually degrading phosphorylated β-catenin (Vinyoles et al., 2014). Our data show that the presence of TMEM88 in the MVB is associated with both accumulation of β-catenin in the MVB and the repression of Wnt signaling.
Our study confirmed that TMEM88 is important for inhibiting Wnt signaling during cardiac progenitor specification, and that this is associated with recruiting the Wnt signalosome to the MVB. Simply trafficking GFP-TMEM88 to the plasma membrane was not sufficient to inhibit Wnt signaling, and in fact, addition of a plasma membrane-tagging motif blocks the ability to inhibit Wnt signaling. This suggests that TMEM88 needs to be internalized and thereby promote internalization and trafficking of the Wnt signalosome from plasma membrane to the MVB. This mechanism appears distinct from Notch-mediated repression of Wnt signaling. In that case, membrane-bound Notch1 represses β-catenin signaling via Numb-mediated endosomal sorting, which leads to lysosomal degradation. However, cleavage of neither the Notch receptor nor the destruction complex is required for repression, which is fully dependent on Numb (Kwon et al., 2011). Previous studies have shown that inhibiting the lysosome increases Wnt signaling by enhancing the formation of endosomal ILVs, which sequester the Wnt signalosome (Dobrowolski et al., 2012), and that Wnt signaling is antagonized by exosomal release of β-catenin (Chairoungdua et al., 2010). Therefore, in principle the fate of the Wnt signalosome sequestered in the MVB by TMEM88 could be degradation by the lysosomal pathway or exosomal release. Our study indicates that sequestration in the MVB of a signalosome that retains β-catenin is yet another mechanism to control canonical Wnt signaling.
Limitations of the Study
TMEM88 is clearly important for regulating cardiogenesis, and our study further supports its function in the Wnt signaling pathway for specification of human cardiac progenitors. However, the bulk of our mechanistic studies implicating TMEM88 in promoting sequestration of β-catenin in the MVB were carried out for technical reasons in non-cardiac cell lines. Therefore, it is possible that TMEM88 has additional cardiac cell-specific functions that we have not revealed. The surprising finding that TMEM88 does not need to function through DVL must also be considered in context, because Wnt signaling normally functions optimally with DVL activity. Therefore, we cannot rule out whether TMEM88 normally can interact with or, in some manner, regulate DVL activity. We have used the small molecule CHIR99021 to inhibit GSK-3β and thereby activate the Wnt pathway, but with concentrations that might interfere with other kinases. Finally, we do not know why expression of TMEM88 recruits the signalosome into the MVB. This presumably results from higher-order protein complexes that assemble at the membrane or in the endocytosed vesicles and will be the focus of subsequent proteomic studies.
Methods
All methods can be found in the accompanying Transparent Methods supplemental file.
Acknowledgments
The authors are grateful to Dr. Danwei Huangfu (MSKCC) for the parental iCrispr H1 hESCs used to generate the TMEM88 mutant line and Dr. Stephane Angers (U. Toronto) for providing the DVL triple-KO cell line. We thank Drs. Anthony Brown (WCM), Anant Menon (WCM), and Marco Seandel (WCM) for helpful discussions. The WCM Optical Microscopy Core provided outstanding imaging support. These studies were supported by a grant from the NHLBI, United States (R35 HL135778 to T.E.).
Author Contributions
H.L. and T.E. conceived the study, designed experiments, and analyzed data. They also wrote, edited, and approved the final manuscript. H.L. performed all the experiments.
Declaration of Interests
The authors declare no competing interests.
Published: September 27, 2019
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.isci.2019.07.039.
Supplemental Information
References
- Bilic J., Huang Y.L., Davidson G., Zimmermann T., Cruciat C.M., Bienz M., Niehrs C. Wnt induces LRP6 signalosomes and promotes dishevelled-dependent LRP6 phosphorylation. Science. 2007;316:1619–1622. doi: 10.1126/science.1137065. [DOI] [PubMed] [Google Scholar]
- Blitzer J.T., Nusse R. A critical role for endocytosis in Wnt signaling. BMC Cell Biol. 2006;7:28. doi: 10.1186/1471-2121-7-28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunt L., Scholpp S. The function of endocytosis in Wnt signaling. Cell Mol. Life Sci. 2018;75:785–795. doi: 10.1007/s00018-017-2654-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chairoungdua A., Smith D.L., Pochard P., Hull M., Caplan M.J. Exosome release of beta-catenin: a novel mechanism that antagonizes Wnt signaling. J. Cell Biol. 2010;190:1079–1091. doi: 10.1083/jcb.201002049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clevers H., Nusse R. Wnt/beta-catenin signaling and disease. Cell. 2012;149:1192–1205. doi: 10.1016/j.cell.2012.05.012. [DOI] [PubMed] [Google Scholar]
- Davidson K.C., Adams A.M., Goodson J.M., McDonald C.E., Potter J.C., Berndt J.D., Biechele T.L., Taylor R.J., Moon R.T. Wnt/beta-catenin signaling promotes differentiation, not self-renewal, of human embryonic stem cells and is repressed by Oct4. Proc. Natl. Acad. Sci. U S A. 2012;109:4485–4490. doi: 10.1073/pnas.1118777109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobrowolski R., Vick P., Ploper D., Gumper I., Snitkin H., Sabatini D.D., De Robertis E.M. Presenilin deficiency or lysosomal inhibition enhances Wnt signaling through relocalization of GSK3 to the late-endosomal compartment. Cell Rep. 2012;2:1316–1328. doi: 10.1016/j.celrep.2012.09.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gagliardi M., Hernandez A., McGough I.J., Vincent J.P. Inhibitors of endocytosis prevent Wnt/Wingless signalling by reducing the level of basal beta-catenin/Armadillo. J. Cell Sci. 2014;127:4918–4926. doi: 10.1242/jcs.155424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gagliardi M., Piddini E., Vincent J.P. Endocytosis: a positive or a negative influence on Wnt signalling? Traffic. 2008;9:1–9. doi: 10.1111/j.1600-0854.2007.00662.x. [DOI] [PubMed] [Google Scholar]
- Gammons M.V., Rutherford T.J., Steinhart Z., Angers S., Bienz M. Essential role of the Dishevelled DEP domain in a Wnt-dependent human-cell-based complementation assay. J. Cell Sci. 2016;129:3892–3902. doi: 10.1242/jcs.195685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ge Y.X., Wang C.H., Hu F.Y., Pan L.X., Min J., Niu K.Y., Zhang L., Li J., Xu T. New advances of TMEM88 in cancer initiation and progression, with special emphasis on Wnt signaling pathway. J. Cell. Physiol. 2018;233:79–87. doi: 10.1002/jcp.25853. [DOI] [PubMed] [Google Scholar]
- Ilyas M., Tomlinson I.P., Rowan A., Pignatelli M., Bodmer W.F. Beta-catenin mutations in cell lines established from human colorectal cancers. Proc. Natl. Acad. Sci. U S A. 1997;94:10330–10334. doi: 10.1073/pnas.94.19.10330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang X., Charlat O., Zamponi R., Yang Y., Cong F. Dishevelled promotes Wnt receptor degradation through recruitment of ZNRF3/RNF43 E3 ubiquitin ligases. Mol. Cell. 2015;58:522–533. doi: 10.1016/j.molcel.2015.03.015. [DOI] [PubMed] [Google Scholar]
- Kim S.E., Huang H., Zhao M., Zhang X., Zhang A., Semonov M.V., MacDonald B.T., Zhang X., Garcia Abreu J., Peng L. Wnt stabilization of beta-catenin reveals principles for morphogen receptor-scaffold assemblies. Science. 2013;340:867–870. doi: 10.1126/science.1232389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar A., Chalamalasetty R.B., Kennedy M.W., Thomas S., Inala S.N., Garriock R.J., Yamaguchi T.P. Zfp703 is a Wnt/beta-catenin feedback suppressor targeting the beta-catenin/Tcf1 complex. Mol. Cell. Biol. 2016;36:1793–1802. doi: 10.1128/MCB.01010-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwon C., Cheng P., King I.N., Andersen P., Shenje L., Nigam V., Srivastava D. Notch post-translationally regulates beta-catenin protein in stem and progenitor cells. Nat. Cell Biol. 2011;13:1244–1251. doi: 10.1038/ncb2313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee H.J., Finkelstein D., Li X., Wu D., Shi D.L., Zheng J.J. Identification of transmembrane protein 88 (TMEM88) as a dishevelled-binding protein. J. Biol. Chem. 2010;285:41549–41556. doi: 10.1074/jbc.M110.193383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li V.S., Ng S.S., Boersema P.J., Low T.Y., Karthaus W.R., Gerlach J.P., Mohammed S., Heck A.J., Maurice M.M., Mahmoudi T. Wnt signaling through inhibition of beta-catenin degradation in an intact Axin1 complex. Cell. 2012;149:1245–1256. doi: 10.1016/j.cell.2012.05.002. [DOI] [PubMed] [Google Scholar]
- Li Y., Liu Y., Liu B., Wang J., Wei S., Qi Z., Wang S., Fu W., Chen Y.G. A growth factor-free culture system underscores the coordination between Wnt and BMP signaling in Lgr5(+) intestinal stem cell maintenance. Cell Discov. 2018;4:49. doi: 10.1038/s41421-018-0051-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma R., Feng N., Yu X., Lin H., Zhang X., Shi O., Zhang H., Zhang S., Li L., Zheng M. Promoter methylation of Wnt/beta-Catenin signal inhibitor TMEM88 is associated with unfavorable prognosis of non-small cell lung cancer. Cancer Biol. Med. 2017;14:377–386. doi: 10.20892/j.issn.2095-3941.2017.0061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacDonald B.T., Tamai K., He X. Wnt/beta-catenin signaling: components, mechanisms, and diseases. Dev. Cell. 2009;17:9–26. doi: 10.1016/j.devcel.2009.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuda T., Cepko C.L. Controlled expression of transgenes introduced by in vivo electroporation. Proc. Natl. Acad. Sci. U S A. 2007;104:1027–1032. doi: 10.1073/pnas.0610155104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metcalfe C., Bienz M. Inhibition of GSK3 by Wnt signalling–two contrasting models. J. Cell Sci. 2011;124:3537–3544. doi: 10.1242/jcs.091991. [DOI] [PubMed] [Google Scholar]
- Naito A.T., Shiojima I., Akazawa H., Hidaka K., Morisaki T., Kikuchi A., Komuro I. Developmental stage-specific biphasic roles of Wnt/beta-catenin signaling in cardiomyogenesis and hematopoiesis. Proc. Natl. Acad. Sci. U S A. 2006;103:19812–19817. doi: 10.1073/pnas.0605768103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novikov N., Evans T. Tmem88a mediates GATA-dependent specification of cardiomyocyte progenitors by restricting WNT signaling. Development. 2013;140:3787–3798. doi: 10.1242/dev.093567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nusse R., Clevers H. Wnt/beta-catenin signaling, disease, and emerging therapeutic modalities. Cell. 2017;169:985–999. doi: 10.1016/j.cell.2017.05.016. [DOI] [PubMed] [Google Scholar]
- Okada A., Lansford R., Weimann J.M., Fraser S.E., McConnell S.K. Imaging cells in the developing nervous system with retrovirus expressing modified green fluorescent protein. Exp. Neurol. 1999;156:394–406. doi: 10.1006/exnr.1999.7033. [DOI] [PubMed] [Google Scholar]
- Palpant N.J., Pabon L., Rabinowitz J.S., Hadland B.K., Stoick-Cooper C.L., Paige S.L., Bernstein I.D., Moon R.T., Murry C.E. Transmembrane protein 88: a Wnt regulatory protein that specifies cardiomyocyte development. Development. 2013;140:3799–3808. doi: 10.1242/dev.094789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piper R.C., Katzmann D.J. Biogenesis and function of multivesicular bodies. Annu. Rev. Cell Dev. Biol. 2007;23:519–547. doi: 10.1146/annurev.cellbio.23.090506.123319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ploper D., Taelman V.F., Robert L., Perez B.S., Titz B., Chen H.W., Graeber T.G., von Euw E., Ribas A., De Robertis E.M. MITF drives endolysosomal biogenesis and potentiates Wnt signaling in melanoma cells. Proc. Natl. Acad. Sci. U S A. 2015;112:E420–E429. doi: 10.1073/pnas.1424576112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romero G., von Zastrow M., Friedman P.A. Role of PDZ proteins in regulating trafficking, signaling, and function of GPCRs: means, motif, and opportunity. Adv. Pharmacol. 2011;62:279–314. doi: 10.1016/B978-0-12-385952-5.00003-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saito-Diaz K., Benchabane H., Tiwari A., Tian A., Li B., Thompson J.J., Hyde A.S., Sawyer L.M., Jodoin J.N., Santos E. APC inhibits ligand-independent Wnt signaling by the clathrin endocytic pathway. Dev. Cell. 2018;44:566–581.e8. doi: 10.1016/j.devcel.2018.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taelman V.F., Dobrowolski R., Plouhinec J.L., Fuentealba L.C., Vorwald P.P., Gumper I., Sabatini D.D., De Robertis E.M. Wnt signaling requires sequestration of glycogen synthase kinase 3 inside multivesicular endosomes. Cell. 2010;143:1136–1148. doi: 10.1016/j.cell.2010.11.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamai K., Zeng X., Liu C., Zhang X., Harada Y., Chang Z., He X. A mechanism for Wnt coreceptor activation. Mol. Cell. 2004;13:149–156. doi: 10.1016/s1097-2765(03)00484-2. [DOI] [PubMed] [Google Scholar]
- Ueno S., Weidinger G., Osugi T., Kohn A.D., Golob J.L., Pabon L., Reinecke H., Moon R.T., Murry C.E. Biphasic role for Wnt/beta-catenin signaling in cardiac specification in zebrafish and embryonic stem cells. Proc. Natl. Acad. Sci. U S A. 2007;104:9685–9690. doi: 10.1073/pnas.0702859104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veeman M.T., Slusarski D.C., Kaykas A., Louie S.H., Moon R.T. Zebrafish prickle, a modulator of noncanonical Wnt/Fz signaling, regulates gastrulation movements. Curr. Biol. 2003;13:680–685. doi: 10.1016/s0960-9822(03)00240-9. [DOI] [PubMed] [Google Scholar]
- Vinyoles M., Del Valle-Perez B., Curto J., Vinas-Castells R., Alba-Castellon L., Garcia de Herreros A., Dunach M. Multivesicular GSK3 sequestration upon Wnt signaling is controlled by p120-catenin/cadherin interaction with LRP5/6. Mol. Cell. 2014;53:444–457. doi: 10.1016/j.molcel.2013.12.010. [DOI] [PubMed] [Google Scholar]
- Yamamoto H., Komekado H., Kikuchi A. Caveolin is necessary for Wnt-3a-dependent internalization of LRP6 and accumulation of beta-catenin. Dev. Cell. 2006;11:213–223. doi: 10.1016/j.devcel.2006.07.003. [DOI] [PubMed] [Google Scholar]
- Yamamoto H., Sakane H., Yamamoto H., Michiue T., Kikuchi A. Wnt3a and Dkk1 regulate distinct internalization pathways of LRP6 to tune the activation of beta-catenin signaling. Dev. Cell. 2008;15:37–48. doi: 10.1016/j.devcel.2008.04.015. [DOI] [PubMed] [Google Scholar]
- Yao R., Maeda T., Takada S., Noda T. Identification of a PDZ domain containing Golgi protein, GOPC, as an interaction partner of frizzled. Biochem. Biophys. Res. Commun. 2001;286:771–778. doi: 10.1006/bbrc.2001.5430. [DOI] [PubMed] [Google Scholar]
- Yu X., Zhang X., Zhang Y., Jiang G., Mao X., Jin F. Cytosolic TMEM88 promotes triple-negative breast cancer by interacting with Dvl. Oncotarget. 2015;6:25034–25045. doi: 10.18632/oncotarget.4379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X., Yu X., Jiang G., Miao Y., Wang L., Zhang Y., Liu Y., Fan C., Lin X., Dong Q. Cytosolic TMEM88 promotes invasion and metastasis in lung cancer cells by binding DVLS. Cancer Res. 2015;75:4527–4537. doi: 10.1158/0008-5472.CAN-14-3828. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The GFP+ puncta demonstrate membrane-localized GFP-TMEM88. The pattern is very dynamic, suggesting that GFP-TMEM88 is moving onto and then off of the plasma membrane (endocytosis).