

Summary
Studies with gene‐deficient and gnotobiotic mice have identified many host and microbial factors that contribute to induced colitis, but information on whether specific factors determine susceptibility under more physiological conditions is lacking. Using wild‐type strains that differ in their IgA response but harbor a diverse gut microbiome, we found that the IgA‐high strain CBA/CaJ (CBA) is resistant to acute colitis induced with dextran sodium sulfate (DSS), unlike the IgA‐low strain C57BL/6 (B6). Resistance was associated with extensive IgA‐coating of fecal bacteria, lower fecal bacterial loads and greater abundance of barrier‐protective transcripts in colonic tissues under homeostatic conditions. Fecal microbial transplant (FT) experiments revealed that disease induction in B6 mice was associated with a cohort of bacteria that are not targeted by IgA. However, CBA mice continued to be resistant to colitis induction following FTs from B6 mice, indicating that they are able to contain such colitogenic members. In support of a role for bacterial exclusion in resistance, oral administration of immunoglobulins decreased DSS‐induced disease in B6 mice. In F1 mice derived separately with CBA and B6 dams and in F1 mice backcrossed to the two parental strains, resistance segregated with the IgA response of the pups and not with barrier‐associated transcripts or bacterial loads. Interestingly, B6 pups foster‐nursed on CBA dams continued to be susceptible in later life, whereas CBA pups foster‐nursed on B6 dams continued to be resistant. Together, the data indicate that a high‐IgA response in adult life can protect against colitis and compensate for IgA deficiency in early life.
Keywords: backcrosses, colitis, F1 mice, fecal microbial transplants, foster nursing
Abbreviations
- AMP
antimicrobial peptide
- APC‐Cy7
allophycocyanin‐Cychrome 7
- BGG
bovine γ‐globulin
- DSS
dextran sodium sulfate
- FITC
fluorescein isothiocyanate
- FT
fecal microbial transplant
- IL‐5
interleukin‐5
- MNZ
metronidazole
- NGAL
neutrophil gelatinase‐associated lipocalin
- PBS
phosphate‐buffered saline
- PE
phycoerythrin
- Tff
trefoil factor
- Tg
transgenic
- Th17
T helper type 17
- Treg
regulatory T
- VNAM
vancomycin, neomycin, ampicillin and metronidazole
- ZO
zona occludens
Introduction
A layer of simple epithelium separates systemic tissues from potential pathogens in the mammalian intestinal lumen. Under homeostatic conditions, a selectively permeable barrier, mucus, antimicrobial peptides (AMPs), and IgA‐mediated immune exclusion/containment of bacteria offer the first line of defense against bacterial invasion, allowing immune cells in the intestine to deal with the occasional invader without the generation of potent and tissue‐damaging inflammatory responses. However, intestinal homeostasis is an active and dynamic process and may be disrupted by damage to the barrier, poor wound healing, significant alterations in the resident microbial community and poor, deviant or uncontrolled immune responses.1, 2, 3 In humans, such disruptions can lead to inflammatory bowel diseases, such as Crohn's disease or ulcerative colitis. Treatment of mice with dextran sodium sulfate (DSS) has been used to study experimental colitis in mice, and a number of host and microbial factors have been reported to influence the induction and severity of such induced colitis.4, 5, 6, 7, 8, 9, 10, 11, 12, 13
Most of the studies have, however, relied on the use of gene‐deficient mice or on the colonization of germ‐free or limited flora mice with specific bacterial taxa. As IgA, received either passively by neonatal mice or made actively by adults, influences host–microbe homeostasis in the gut by several mechanisms including early bacterial colonization events, immune exclusion of pathogenic bacteria, containment of inflammation through effects on regulatory T (Treg) cell generation and strengthening of the intestinal barrier,10, 14, 15 we have tested whether IgA is necessary and sufficient to protect conventional wild‐type mice from DSS‐colitis, using mice that differ in IgA amount. We chose the CBA/CaJ (CBA, IgAhigh) and C57BL/6 (B6, IgAlow) strains for our study and found that the CBA strain was resistant to DSS‐colitis, unlike the B6 strain. Under homeostatic conditions, fecal pellets from CBA mice had lower bacterial loads, and a greater proportion of the bacteria was coated with IgA. Transcripts for some genes that contribute to barrier resilience were also more abundant in the colonic tissue of CBA mice. To understand how IgA mediates protection, we looked at disease induction and other parameters in mice receiving reciprocal fecal microbial transplants (FTs), in F1 and backcross progeny derived from crosses in which the proximal dam was either B6 or CBA, in foster‐nursed mice, and in mice given bovine γ‐globulin (BGG) orally to provide surrogate non‐specific bacterial coating. Our data indicate that active IgA made in adult life may be the prime determinant of resistance to DSS‐colitis.
Materials and methods
Reagents
Dextran sodium sulfate (36 000–50 000 MW) was purchased from MP Biomedicals (Illkirch, France); sodium butyrate, BGG, fluorescein isothiocyanate (FITC) ‐dextran (4000 MW, FITC : glucose = 1:250), bovine serum albumin (BSA) (essentially globulin‐free), vancomycin, neomycin, ampicillin and metronidazole were supplied by Sigma Aldrich (St. Louis, MI).
Mice
C57BL/6ByJ, CBA/CaJ, DBA/2J and BALB/cJ mice, from Jackson Laboratories (Bar Harbor, ME). were maintained in the Small Animal Facility of The National Institute of Immunology. The interleukin‐5 (IL‐5) transgenic (Tg) mouse strain was a kind gift of Prof. T.V. Rajan, University of Connecticut Medical School, Farmington. F1, ‘Parent × F1’ and ‘F1 × Parent’ crosses were carried out in‐house and the nomenclature used for them lists the female first; for example, CBAB6F1 are F1 mice generated with a CBA female and a B6 male and CBA×B6CBAF1 are backcross progeny of CBA females crossed with F1 males that had been generated with B6 dams. Mice of both sexes, aged 8–12 weeks, were used. All mouse protocols were carried out in accordance with Institutional Animal Ethics Committee guidelines.
Measurement of fecal bacterial loads, IgA and bacterial coating
For loads, bacterial DNA was extracted with the Qiagen DNA Stool Mini kit (Hilden, Germany), quantified (NanoDrop, Thermo Scientific, Waltham, MA) and expressed as μg yield of DNA/g fecal weight, as reported previously.16 Fecal IgA was measured in supernatants collected after homogenization of fecal pellets in phosphate‐buffered saline (PBS) and centrifugation at 16 000 g for 10 min as reported elsewhere.17 Appropriate dilutions of the supernatant were loaded on ELISA plates coated with goat anti‐mouse immunoglobulin, and goat anti‐mouse IgA‐horseradish peroxidase was used to detect the captured IgA (Southern Biotech, Birmingham, AL). IgA concentrations were read off a standard curve with an IgA myeloma standard that was run in parallel. For determination of IgA‐coated bacteria, fecal pellets were homogenized in filtered PBS, debris was removed by centrifugation at 800 g for 5 min, and bacteria were pelleted by centrifugation at 9200 g for 10 min. Pelleted bacteria were stained with biotinylated goat anti‐mouse IgA (Southern Biotech), followed by streptavidin‐allophycocyanin‐Cychrome 7 (APC‐Cy7; BD Biosciences, San Jose, CA) and the DNA‐binding dye Syto‐13 (Invitrogen, Eugene, OR) in saline. IgA staining was recorded on bacteria (low forward scatter/side scatter, Syto13+), as reported previously.18
DSS‐colitis
A total of 2·5% DSS was added to autoclaved drinking water and replaced every 72 hr. The scoring disease index used was as follows: 0 = normal fecal pellet, 1 = few formed pellets to semi‐solid stool, 2 = semi‐solid to fluid stool with or without blood, 3 = bloody stool, 4 = bloody fluid, 5 = dead on arrival. Colon lengths were measured from the base of the cecum to the end. Intestinal permeability was assayed by administering 8 mg FITC‐dextran orally to mice that had been starved overnight, and measuring fluorescence in serum separated from blood collected 4 hr later (CLARIOstar; BMG LABTECH, Ortenberg, Germany). Concentrations were read out from a standard curve of FITC‐dextran run in parallel. In some experiments, mice were treated orally with 200 mm sodium butyrate from day –2 of DSS treatment or with BGG or BSA (10 mg/mouse in 3·5% NaHCO3) from day –3 onwards.
Fecal microbial transplants
Fecal microbial transplants with unfractionated bacteria were as described previously19 or with IgA‐coated and IgA‐uncoated bacteria that were separated on streptavidin‐MACS columns (Miltenyi Biotec, Bergisch‐Gladbach, Germany) after staining fecal bacteria with biotinylated goat anti‐mouse IgA (Southern Biotech). Preparations were routinely > 85% pure. Three FTs were carried out with a gap of 2–3 days between transfers and mice were rested for 10 days before exposure to DSS.
Assay for neutrophil activity
Neutrophil gelatinase‐associated lipocalin (NGAL) and calprotectin amounts in fecal pellets were measured with a Lipocalin‐2/NGAL Picokine ELISA kit (Boster Bio, Pleasanton, CA) and Mouse CALP (Calprotectin) ELISA kit (Elabscience, Houston, TX) as recommended, and amounts were read off standard curves run in parellel.
Fluorescence‐activated cell sorting
Pacific Blue‐CD45·2 (104), FITC‐CD11b (M1/70), phycoerythrin (PE) ‐Siglec‐F (E50‐2440), BV‐421‐RORγt (Q31‐378), APC‐CD4 (RM4‐5) and APC‐Cy7‐B220 (RA3‐6B2) were obtained from BD. PE‐Cy7‐F4/80 (BM8) and FoxP3 (FJK‐16s), APC‐Gr‐1 (RB6‐8C5) and APC‐Cy7‐CD90·2 (53–2·1) were from eBiosciences (San Diego, CA) and APC‐Cy7‐CD45·2 (104) was supplied by BioLegend (Cambridge, UK). Data were acquired on facsverse (BD) and were analyzed with flojo software (TreeStar, Ashland, OR).
Staining of colonic lamina propria cells
Colons were slit longitudinally, washed with ice‐cold PBS, minced and incubated in 20 ml Ca2+/Mg2+‐free Hanks’ balanced salt solution containing 10% fetal bovine serum (Gibco, Grand Island, NY), 1 mm dithiothreitol, 2 mm EDTA and 25 mm HEPES (Sigma) for 20 min at 37°. Tissue pieces were washed, chopped and digested in 20 ml RPMI‐1640 (Biological Industries, Cromwell, CT) containing 5% fetal bovine serum, 300 U/ml Collagenase Type 4, 10 U/ml DNaseI (both from Worthington, Lakewood, NJ) and 0·5 mg/ml Dispase (Gibco) for 60 min at 37°. The cell suspension was cleared, and cells were pelleted and washed. On CD45·2+ B220− CD90·2− cells, granulocytes were identified as CD11b+ and further characterized as eosinophils (SiglecF+), neutrophils (Gr‐1+, F4/80−) and macrophages (Gr‐1−, F4/80+). Treg cells and T helper type 17 (Th17) cells were identified as CD4+ FoxP3/RORγt+, respectively. Cells were fixed and permeabilized using FoxP3/Transcription factor staining buffer set (eBiosciences).
RNA extraction
For colonic epithelial cells, washed pieces of colon were incubated twice in Ca2+/Mg2+‐free Hanks’ balanced salt solution containing 30 mm EDTA at 37° for 15 min and RNA extracted from pooled cells with TRIzol reagent (Invitrogen). For colonic cells, a 1·5‐cm length of tissue from the central part of longitudinally slit colons was washed with ice‐cold PBS, patted dry, chopped into small pieces, RNA extracted (Qiagen RNeasy Kit; Qiagen), and cDNA synthesized (Verso cDNA synthesis kit, Thermo Scientific) and amplified with Power SYBR Green PCR master mix (Applied Biosystems, Warrington, UK) and an ABI Prism 7000 cycler. For DSS‐treated tissues, RNA was passed over two or three columns. Data are shown as fold change of mRNA using the ΔΔ threshold cycle (Ct) method.
Quantification of relative abundance of bacterial groups
Bacterial DNA was amplified with primers for phylum/family/taxon‐specific 16S rDNA and expressed relative to all bacteria (with universal 16S primers) by the ΔΔCt method. For measuring bacterial translocation into mesenteric lymph nodes, whole genomic DNA from the tissue was extracted with HiYield™ Genomic DNA Mini Kit (Real Biotech, Taipei, Taiwan), as recommended. Bacterial loads in the tissue were then determined by polymerase chain reaction with universal 16S primers for detecting all bacteria and expressed relative to GAPDH to normalize for tissue amounts, using the ΔΔCt method.
Primers
The primers were obtained from Sigma: muc1, 5′‐TCGTCTATTTCCTTGCCCTG‐3′ and 5′‐ATTACCTGCCGAAACCTCCT‐3′; muc2, 5′‐CCCAGAAGGGACTGTGTATG‐3′ and 5′‐TTGTGTTCGCTCTTGGTCAG‐3′; muc3, 5′‐TGGTCAACTGCGAGAATGGA‐3′ and 5′‐TACGCTCTCCACCAGTTCCT‐3′; muc4, 5′‐GTCTCCCATCACGGTTCAGT‐3′ and 5′‐TGTCATTCCACACTCCCAGA‐3′; cldn1, 5′‐TCCTTGCTGAATCTGAACA‐3′ and 5′‐AGCCATCCACATCTTCTG‐3′; cldn2, 5′‐GTCATCGCCCATCAGAAGAT‐3′ and 5′‐ACTGTTGGACAGGGAACCAG‐3′; cldn5, 5′‐GCTCTCAGAGTCCGTTGACC‐3′ and 5′‐CTGCCCTTTCAGGTTAGCAG‐3′, jam‐a, 5′‐CACCTTCTCATCCAGTGGCATC‐3′ and 5′‐CTCCACAGCATCCATGTGTGC‐3′; occludin, 5′‐CACCTTCTCATCCAGTGGCATC‐3′ and 5′‐CTCCACAGCATCCATGTGTGC‐3′; tff3, 5′‐TCTGGCTAATGCTGTTGGTG‐3′ and 5′‐CTCCTGCAGAGGTTTGAAGC‐3′; zo1, 5′‐CCACCTCTGTCCAGCTCTTC‐3′, and 5′‐CACCGGAGTGATGGTTTTCT‐3′; Il‐1b, 5′‐ACCTTCCAGGATGAGGACATGAG‐3′ and 5′‐CATCCCATGAGTCACAGAGGATG‐3′; Ifn‐a, 5′‐CCTGAGAGGAAGAAACACACC‐3′ and 5′‐GGCTCTCCAGACTTCTGCTCTG‐3′; Il‐6, 5′‐CTCTGCAAGAGACTTCCATCCAGT‐3′ and 5′‐CGTGGTTGTCACCAGCATCA‐3′; Il‐10, 5′‐GGCCCAGAAATCAAGGAGCAT‐3′, and 5′‐GAGAAATCGATGACAGCGCCT‐3′; Il‐12p35, 5′‐TGATGACCCTGTGCCTTGGT‐3′ and 5′‐AGTGCTGCGTTGATGGCCT‐3′; Il‐17a, 5′‐TCCAGAAGGCCCTCAGACTA‐3′ and 5′‐CAGGATCTCTTGCTGGATG‐3′; Il‐18, 5′‐GCCGCCTCAAACCTTCCAA‐3′ and 5′‐TGGCAGCCATTGTTCCTGG‐3′; Il‐22, 5′‐ACCGCTGATGTGACAGGAGC‐3′ and 5′‐AGGTGGTGCCTTTCCTGACC‐3′; Il‐23a, 5′‐AATGTGCCCCGTATCCAGTGT‐3′ and 5′‐CCTTTGCAAGCAGAACTGGC‐3′; Ccl‐19, 5′‐TGCTGGTTCTCTGGACCTTCC‐3′ and 5′‐GCATCATTAGCACCCCCCA‐3′; Ccl‐20, 5′‐AGATGGCCGATGAAGCTTGT‐3′ and 5′‐TGGATCAGCGCACACAGATT‐3′; Ccl‐21, 5′‐ TGGACCCAAGGCAGTGATG‐3′ and 5′‐TGGCTGTACTTAAGGCAGCAGTC‐3′; Cxcl‐1, 5′‐GCTAAAAGGTGTCCCCAAGTAACG‐3′ and 5′‐GCTAAAAGGTGTCCCCAAGTAACG‐3′; Ifn‐g, 5′‐CAGCAACAGCAAGGCGAAA‐3′ and 5′‐ AGCTCATTGAATGCTTGGCG‐3′; Tnf‐a, 5′‐AATGGCCTCCCTCTCATCAG‐3′ and 5′‐GCTACGACGTGGGCTACAGG‐3′; ATF4, 5′‐ATGGCCGGCTATGGATGAT‐3′ and 5′‐CGAAGTCAAACTCTTTCAGATCCATT‐3′; spliced XBP‐1, 5′‐ACACGCTTGGGAATGGACAC‐3′ and 5′‐CCATGGGAAGATGTTCTGGG‐3′; BiP, 5′‐GAAAGGATGGTTAATGATGCTGAG‐3′ and 5′‐GTCTTCAATGTCCGCATCCTG‐3′; Edem‐1, 5′‐ATCCGAGTTCCAGAAGGCAGTT‐3′ and 5′‐GCTTCCCAGAACCCTTATCGT‐3′; CHOP, 5′‐CATACACCACCACACCTGAAAG‐3′ and 5′‐CCGTTTCCTAGTTCTTCC TTGC‐3′; Ire‐1b, 5′‐CCTGGGTCCTCTACCTGATG‐3′ and 5′‐AAGGAAATCTTCCCCACCAC‐3′; GLUT1, 5′‐TCATCCCAGCCCTGCTACAG‐3′ and 5′‐ACACTCTTGGCCCGGTTCT‐3′; Gapdh, 5′‐ATGGCCTTCCGTGTTCCTA‐3′ and 5′‐TGAAGTCGCAGGAGACAACCT‐3′.
Eubacteria (all groups), 5′‐ACTCCTACGGGAGGCAGCAG‐3′ and 5′‐ATTACCGCGGCTGCTGG‐3′; Actinobacteria, 5′‐CGCGGCCTATCAGCTTGTTG‐3′ and 5′‐ATTACCGCGGCTGCTGG‐3′; Bacteroidetes, 5′‐GGARCATGTGGTTTAATTCGATGAT‐3′ and 5′‐AGCTGACGACAACCATGCAG‐3′; Firmicutes, 5′‐GGAGYATGTGGTTTAATTCGAAGCA‐3′ and 5′‐AGCTGACGACAACCATGCAC‐3′; Bifidobacterium, 5′‐TCGCGTC(C/T) GGTGTGAAAG‐3′ and 5′‐CCACATCCAGC(A/G)TCCAC‐3′; Lactobacillus, 5′‐AGCAGTAGGGAATCTTCCA‐3′ and 5′‐CACCGCTACACATGGAG‐3′; Bacillus, 5′‐GCGGCGTGCCTAATACATGC‐3′ and 5′‐CTTCATCACTCACGCGGCGT‐3′; Bacteroides‐Prevotella‐Porphyromonas (BPP), 5′‐GGTGTCGGCTTAAGTGCCAT‐3′ and 5′‐CGGA(C/T)GTAAGGGCCGTGC‐3′; segmented filamentous bacteria, 5′‐GACGCTGAGGCATGAGAGCAT‐3′ and 5′‐GACGGCACGGATTGTTATTCA‐3′; Helicobacter, 5′‐CTTAACCATAGAACTGCATTTGAAACTAC‐3′ and 5′‐GGTCGCCTTCGCAATGAGTA‐3′; Enterobacteriaceae, 5′‐GTGCCAGCMGCCGCGGTAA‐3′ and 5′‐GCCTCAAGGGCACAACCTCCAAG‐3′; Betaproteobacteria, 5′‐TCACTGCTACACGYG‐3′ and 5′‐ACTCCTACGGGAGGCAGCAG‐3′; Gammaproteobacteria, 5′‐TCGTCAGCTCGTGTYGTGA‐3′ and 5′‐CGTAAGGGCCATGATG‐3′; Faecalibacterium prausnitzii, 5′‐AGATGGCCTCGCGTCCGA‐3′ and 5′‐CCGAAGACCTTCTTCCTCC‐3′; Peptostreptococcus productus, 5′‐AACTCCGGTGGTATCAGATG‐3′ and 5′‐GGGGCTTCTGAGTCAGGTA‐3′; Clostridium clostridioforme, 5′‐CCGCATGGCAGTGTGTGAAA‐3′ and 5′‐CTGCTGATAGAGCTTTACATA‐3′; Akkermansia, 5′‐CAGCACGTGAAGGTGGGGAC‐3′ and 5′‐CCTTGCGGTTGGCTTCAGAT‐3′; Sutterella, 5′‐CGCGAAAAACCTTACCTAGCC‐3′ and 5′‐GACGTGTGAGGCCCTAGCC‐3′; Acinetobacter, 5′‐TTTAAGCGAGGAGGAGG‐3′ and 5′‐ATTCTACCATCCTCTCCC‐3′; CRAMP, 5′‐CCGAGCTGTGGATGACTTCAA‐3′ and 5′‐CTGCCCCCATACACTGCTTCAC‐3′; Crs1, 5′‐GTCTCCTTTGGAGGCACAGA‐3′ and 5′‐GCTTGGGTGGTGATAGCAGT‐3′; MMP‐7, 5′‐TTTGATGGGCCAGGGAACACTCTA‐3′ and 5′ATGGGTGGCAGCAAACAGGAAGT‐3′; Cryptdin 1, 5′‐AAGAGACTAAAACTGAGGAGCAGC‐3′ and 5′‐CGCAGCAGAGCGTGTA‐3′; Cryptdin 4, 5′‐AAGAGACTAAAACTGAGGAGCAGC‐3′ and 5′‐CGGCGGGGGCAGCAGTA‐3′; Cryptdin 5, 5′‐AAGAGACTAAAACTGAGGAGCAGC‐3′ and 5′‐GCAGCAGAATACGAAAGT‐3′; Global alpha‐defensins, 5′‐GGTGATCATCAGACCCCAGCATCAGT‐3′ and 5′‐AAGAGACTAAAACTGAGGAGCAGC‐3′; Lysozyme, 5′‐GGATCAATTGCACTGCTCTG‐3′ and 5′‐CAGTTCCGAATATACTGGGAC‐3′;Reg‐III‐γ, 5′‐GTATGATGCAGATATGGCCTG‐3′ and 5′‐ATATTGGCCACTGTTACCAC‐3′; LC3, 5′‐AAGATCCCAGTGATTATAGAGCGA‐3′ and 5′‐ATTGCTGTCCCGAATGTCTC‐3′.
Statistical analyses
Data were analyzed by Student's t‐test and corrected for false discovery rate. Error bars indicate SEM.
Results
In CBA mice, resistance to DSS‐colitis correlates with extensive IgA‐coating of fecal bacteria and low fecal bacterial loads
In preliminary experiments, we measured fecal IgA amounts in a number of inbred mouse strains and found that DBA/2, BALB/c and CBA strains all have more fecal IgA than the B6 strain (Fig. 1a). When treated with DSS, IgAlow B6 mice showed severe colitis as read out by mortality, weight loss, disease activity, colon shortening and increased intestinal permeability (Fig. 1b–f), whereas the IgAhigh CBA strain was essentially disease‐free. These data are in keeping with previous reports of a higher IgA response in BALB/c mice than in B6 mice17 and of the relative resistance of DBA/2 and BALB/c mice to induced colitis.20, 21 Most fecal bacteria in CBA mice were coated with IgA (Fig. 1g, see Supplementary material, Fig. S1a) and this appears to contain bacterial numbers, as fecal bacterial loads were significantly lower in this strain (Fig. 1h). Basal expression of AMPs in colonic tissues from the two strains was similar, indicating that bacterial loads were not related to differences in AMP expression (see Supplementary material, Fig. S1c). Bacterial translocation into mesenteric lymph nodes was, however, relatively higher in DSS‐treated B6 mice (see Supplementary material, Fig. S1b). Next, we determined whether the relative representation of major phyla and/or their members differed in the two strains using primers that amplify the following: (i) Phylum Bacteroidetes (and included group BPP, i.e. Bacteroides, Prevotella, Porphyromonas), (ii) Phylum Firmicutes (and included members Bacillus, Clostridium, Faecalibacterium, Lactobacillus, Peptostreptococcus and segmented filamentous bacteria), (iii) Phylum Actinobacteria (and included member Bifidobacterium) (iv) Class α‐Proteobacteria, Class β‐Proteobacteria (and included member Sutterella) and Class γ‐Proteobacteria (and included members Acinetobacteria and Enterobacteriaceae). Primers for amplifying Helicobacter (from ε‐Proteobacteria) and Akkermansia (from Phylum Verrucomicrobia) were also used (Fig. 1i).
Figure 1.
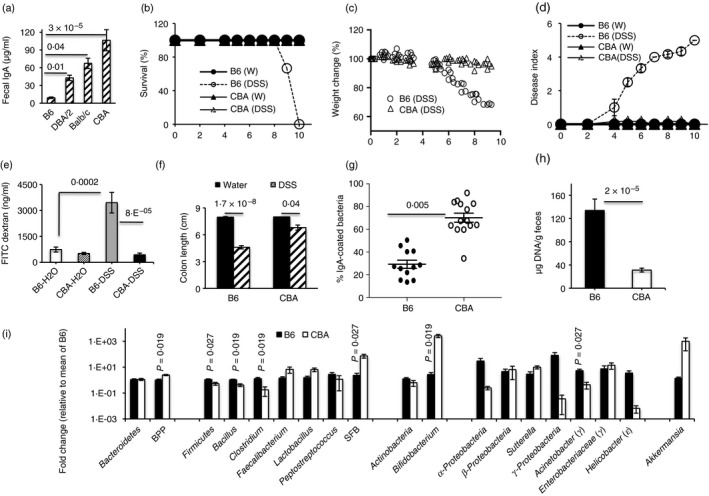
Resistance to dextran sodium sulfate (DSS)‐colitis correlates with IgA amounts and low fecal bacterial loads. Fecal IgA amounts in inbred mouse strains (a). Data were pooled from 20 mice for B6 and CBA, 10 for BALB/c and 5 for DBA/2. Survival, weight change and disease index over time in B6 and CBA given 2·5% DSS in drinking water (b–d). FITC‐dextran concentrations in serum of mice on day 5 of treatment with H2O or DSS (e). Colon length on day 7 of DSS treatment (f). Proportion of Syto13+ fecal bacteria that are coated with IgA in untreated mice (g). Fecal bacterial loads in untreated B6 and CBA mice (h). Relative representation of major bacterial phyla/taxa in the two strains (i). Data are representative of four experiments with five or six mice per group (b–d), two experiments (e), five experiments (f), three experiments (g), means of 19–21 mice (h) and 11 to 12 mice (i) per group.
Representation of Firmicutes, Bacillus, Clostridium and Acinetobacter was significantly higher, whereas that of BPP was lower in B6 fecal pellets. Representation of Helicobacter, α‐Proteobacteria and γ‐Proteobacteria was also higher, whereas that of Sutterella and Akkermansia was lower, in B6 pellets; however, the differences were not statistically significant.
CBA mice are resistant to DSS‐colitis in the presence of a barrier‐disruptive agent
Dextran sodium sulfate is known to decrease colonic mucus thickness,22 raising the possibility that a thin mucus layer may be more easily stripped off, allowing otherwise excluded bacteria access to epithelial cells. Further, mice with deficiencies in barrier genes have enhanced intestinal permeability and bacterial translocation into systemic tissues,23 and a deficit of IgA in early life has been shown to compromise intestinal barrier development.10 To determine whether the two strains differed in barrier integrity, we quantified transcripts of genes that encode mucins and barrier proteins in colonic tissues of untreated mice and found that CBA colonic tissues had higher abundance of transcripts for Muc1, Claudin 1, Occludin, Zona Occludens‐1 (ZO‐1), and trefoil factor 3 (Tff3) under homeostatic conditions (Fig. 2a). Further, when colitis was induced in the presence of a barrier‐disruptive dose (200 nm) of oral sodium butyrate,24 the CBA strain continued to be resistant, whereas the sensitive B6 strain showed more severe disease (Fig. 2b). Oral supplementation with a barrier‐protective dose (20 mm), on the other hand, protected B6 mice against colitis (see Supplementary material, Fig. S1d).
Figure 2.
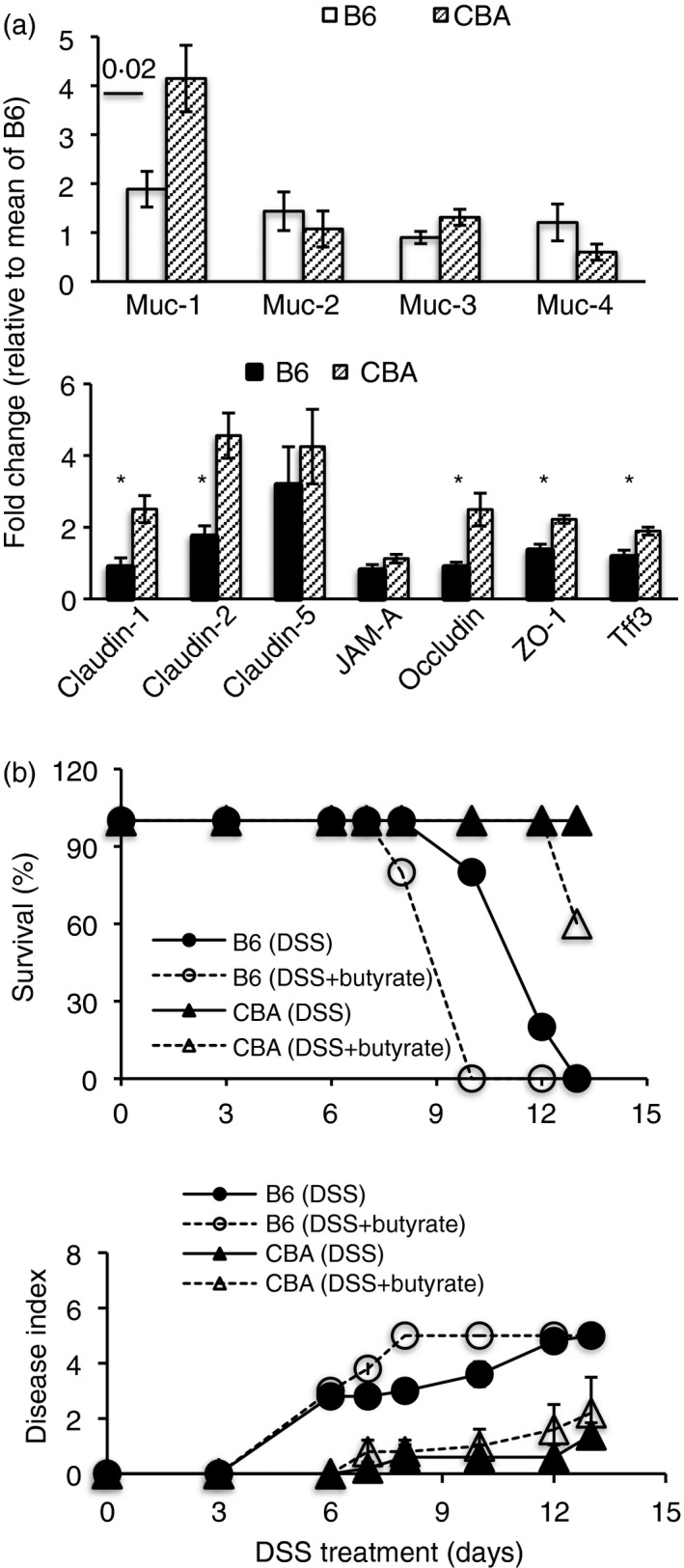
Resistance to dextran sodium sulfate (DSS)‐colitis correlates with barrier resilience. Relative expression of various genes in colonic tissues of untreated mice (a), *P ≤ 0·02. Colitis readouts in mice given 2·5% DSS in drinking water ± 200 mm sodium butyrate from day –2 to end of experiment (b). Data are from four mice per group (a) and five or six mice per group (b). Representative of two experiments.
DSS induces a higher inflammatory response in B6 colons
IgA has been shown to limit innate cell activation in the intestine of gnotobiotic mice25 and hence we determined whether innate cell accumulation or activity were higher in DSS‐treated B6 mice. We found higher granulocyte and macrophage numbers in B6 colons on day 3 of DSS treatment (Fig. 3a). However, neutrophil numbers were greatly elevated in B6 colons at all time‐points tested, and B6 fecal pellets showed greater neutrophil activity as measured by calprotectin and NGAL amounts (Fig. 3b,c). Eosinophil numbers were also higher in DSS‐treated B6 mice on day 5 and day 7 of DSS treatment (Fig. 3a); however, IL‐5 Tg mice, harboring elevated eosinophil frequencies, were marginally less sensitive to DSS‐colitis than B6 mice (see Supplementary material, Fig. S1e), indicating that eosinophils are probably not involved in disease, and may have a protective function, as reported.26 Analysis of Th17 and Treg cells, subsets that have been extensively linked to colitis, showed that both subsets were less numerous in CBA colons on day 3 of DSS treatment; however, the trend reversed, with numbers being significantly higher on day 7 (Fig. 3d) and it is possible that they may contribute to barrier repair, as reported earlier.27 Frequencies of these cells as a proportion of CD4+ cells in the colon, were, however, similar (Fig. 3e) and the higher numbers probably reflect longer colons and higher cell yields in DSS‐treated CBA mice. Disease activity in DSS‐treated B6 mice correlated with higher levels of transcripts (see Supplementary material, Fig. S2) for the inflammatory cytokines interferon‐γ, IL‐17, IL‐1β, tumor necrosis factor‐α and IL‐23, and for the chemokines CXCL1, CCL‐19 and CCL‐21, which are involved in recruitment of neutrophils, dendritic cells and activated T cells.12, 27, 28, 29, 30 Both interferon‐α and IL‐22 have been reported to have a role in epithelial repair/regeneration and we found lower induction of interferon‐α, but higher induction of IL‐22 in B6 colonic tissue. No significant induction of IL‐6, IL‐10, IL‐12 and IL‐18 was seen in either strain.
Figure 3.
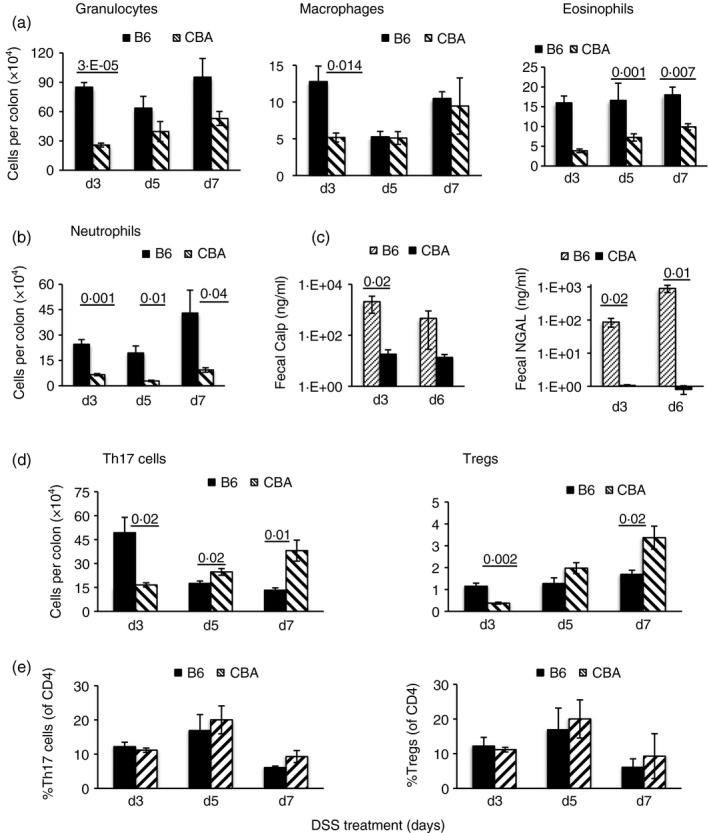
Immune cells in colon and neutrophil activity in fecal pellets of dextran sodium sulfate (DSS)‐treated mice. Numbers of indicated cells (mean ± SEM)/colon on days 3, 5 and 7 of DSS treatment and calprotectin (Calp) and neutrophil gelatinase‐associated lipocalin (NGAL) amounts in fecal pellets on day 3 and day 6 of DSS treatment (a–d). Frequencies of T helper type 17 (Th17) cells and regulatory T (Treg) cells as % of CD4+ cells (e). Data are from five mice per group and representative of two experiments.
Colitis in B6 mice is associated with IgA‐uncoated bacteria
So far, the data indicate that resistance to colitis correlates with extensive IgA‐coating of fecal bacteria, low bacterial loads, and a barrier that can resist damage and contain colonic inflammation. We used a number of approaches to determine which, if any, of these was particularly important in conferring resistance. To determine whether disease susceptibility correlated with the presence of IgA‐uncoated bacteria that are over‐represented in B6 mice (Fig. 1), we treated B6 mice with a cocktail of vancomycin, neomycin, ampicillin and metronidazole (VNAM) and then carried out FTs with homologous (B6) bacteria or equivalent loads of heterologous (CBA) bacteria. VNAM treatment protected against colitis, and susceptibility was restored following homologous (B6) FTs but not heterologous (CBA) FTs (Fig. 4a). Next, we separated B6 fecal bacteria into IgA‐coated and IgA‐uncoated fractions (see Supplementary material, Fig. S3a) and transferred them, load‐normalized, into VNAM‐treated B6 recipients. We found that only recipients of IgA‐uncoated bacteria were susceptible to DSS‐colitis (Fig. 4b). Surprisingly, CBA mice continued to be resistant to colitis induction following FTs with B6 flora (Fig. 4c) even though they were transplanted with the higher bacterial loads seen in the latter.
Figure 4.
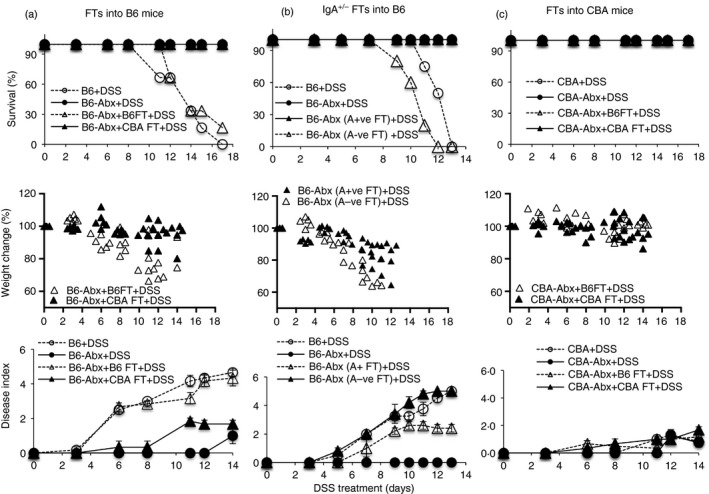
Dextran sodium sulfate (DSS‐colitis in B6 mice is associated with IgA‐uncoated bacteria. B6 mice treated with DSS without antibiotic treatment [B6 + DSS], after 4 weeks of antibiotic treatment [B6‐Abx+DSS], after antibiotic treatment and B6 fecal transplantation (FT) [B6‐Abx+B6FT+DSS], and after antibiotic treatment and CBA FTs [B6‐Abx+CBAFT+DSS] (a). B6 mice treated with DSS without antibiotic treatment [B6+DSS], after antibiotic treatment [B6‐Abx+DSS], after antibiotic treatment and FTs with homologous IgA‐coated bacteria [B6‐Abx (A+ve FT)+DSS], and after antibiotic treatment and FTs with homologous IgA‐uncoated bacteria [B6‐Abx (A‐ve FT)+DSS] (b). CBA mice treated with DSS without antibiotic treatment [CBA+DSS], after antibiotic treatment [CBA‐Abx+DSS], after antibiotic treatment and B6 FTs [CBA‐Abx+B6FT+DSS], and after antibiotic treatment and CBA FTs [CBA‐Abx+CBAFT+DSS] (c). Data are representative of four experiments (a) and two experiments (b, c) with fecal pellets pooled from seven or eight mice for each IgA+/− separation.
Treatment with VNAM decreased bacterial loads in B6 mice (Fig. 5a), and loads were high after B6 FTs (Fig. 5a, grey bar) but low after CBA FTs (Fig. 5a). Loads were also high after FT with IgA‐uncoated bacteria (Fig. 5a, black bar) and low after FT with IgA‐coated bacteria (Fig. 5a). VNAM treatment also led to an increase in the frequency of IgA‐coated bacteria in B6 mice (Fig. 5b) and to a major reduction in the Phyla Bacteroidetes, Firmicutes and Actinobacteria as well as various members within these phyla. Interestingly, γ‐Proteobacteria, β‐Proteobacteria and Sutterella, taxa that have been associated with dysbiosis or disease, were proportionately higher after VNAM treatment (Fig. 5c). The IgA‐uncoated fraction was also enriched for some taxa that have been associated with dysbiosis or disease. These included Phylum Actinobacteria (and member Bifidobacterium), Classes β‐Proteobacteria and γ‐Proteobacteria (and member Sutterella) from Phylum Proteobacteria and Bacillus from Phylum Firmicutes, although some differences were not statistically significant. (Fig. 5d). A previous report has indicated that ampicillin, but not metronidazole, eliminates a cohort of bacteria in B6 mice that is associated with low fecal IgA,19 and we found that ampicillin treatment decreased disease severity in B6 mice (see Supplementary material, Fig. S3b), whereas metronidazole had a minimal effect (see Supplementary material, Fig. S3c). Together, the data indicate that disease in B6 mice is associated with bacteria that are not targeted by IgA. However, if these include ‘colitogenic’ members, they are unable to cause disease in the CBA strain and it appears that their loads may determine induction of disease in B6 mice.
Figure 5.
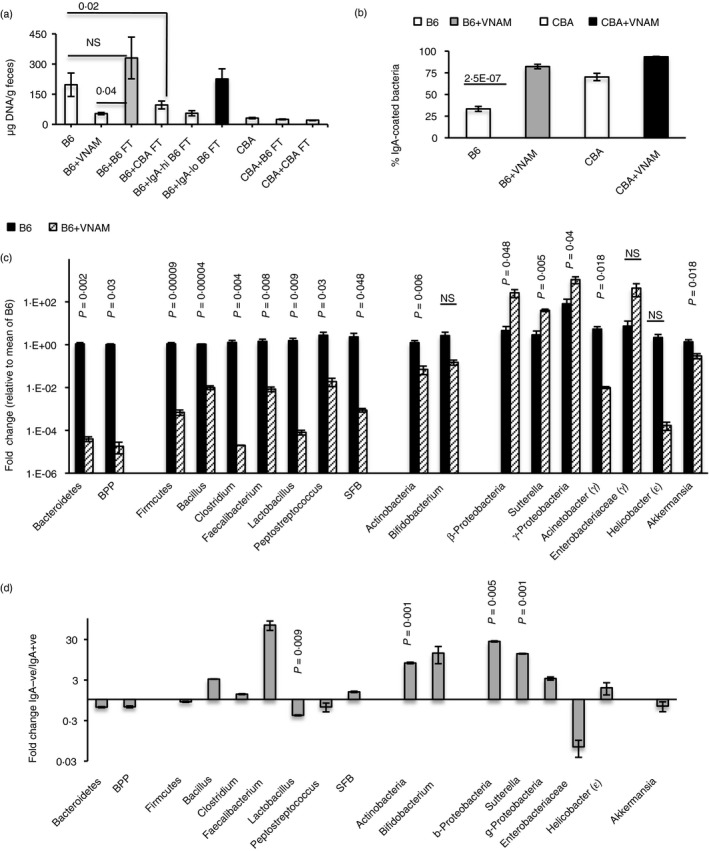
Effect of vancomycin, neomycin, ampicillin and metronidazole (VNAM) treatment on bacterial loads, IgA‐coating, bacterial members, and bacterial representation in IgA‐coated/uncoated fractions. (a) Fecal bacterial loads in untreated mice, and after VNAM treatment ± various fecal transplants (FTs) as indicated. Data are pooled from 6 to 20 mice. In the interests of clarity, P‐values for only three comparisons are shown. (b) IgA‐coating after VNAM treatment. Data are from ten mice per group. (c) Changes in bacterial taxon representation after VNAM treatment. Data are from 11 untreated mice and 8 treated mice. (d) Representation of bacterial members in the IgA‐uncoated fraction of B6 fecal bacteria compared with the IgA‐coated fraction. Data are representative of two experiments (a–c) and are from replicate polymerase chain reactions from two independent separations used for calculation of mean Ct value.
In F1 and ‘Parent × F1’ progeny, resistance segregates with the IgA response of the proximal dam
The combination‐antibiotic treatment required for FT experiments causes dramatic reductions and alterations in the resident microbiome, and can change subsequent colonization dynamics. In this context, and in extension of previous data,17 we found that cohousing the two strains for 3 months did not lead to any increase in fecal IgA amounts in the B6 strain or increase their resistance to DSS‐colitis (Fig. 6a,b), indicating that host–microbe homeostasis in adults is resilient to changes. Hence we carried out genetic experiments to further assess the contribution of IgA to colitis‐resistance. We made two sets of F1 mice, one with a CBA dam (CBAB6F1) and one with a B6 dam (B6CBAF1) to determine whether differences in passive IgA received by pups in early life impacted on their susceptibility to colitis induction in later life. We found that F1 mice generated with a CBA dam were more resistant to DSS‐colitis than those generated with a B6 dam (Fig. 6c). No major differences in microbiota representation were seen between the two F1 strains; (Fig. 7a). Next, we made two sets of ‘Parent × F1’ backcrosses and tested them for susceptibility to colitis. Resistant F1 males (CBAB6F1) were crossed with susceptible B6 dams to generate B6×CBAB6F1 progeny, and susceptible F1 males were crossed with resistant CBA dams to generate CBA×B6CBAF1 progeny. We found that resistance was associated with the IgA‐phenotype of the proximal dam (Fig. 7b). Hence, the CBA dam in the backcross imposed resistance, whereas the B6 dam in the backcross imposed susceptibility. The data indicate that maternal effects dominate in determining resistance to DSS‐colitis.
Figure 6.
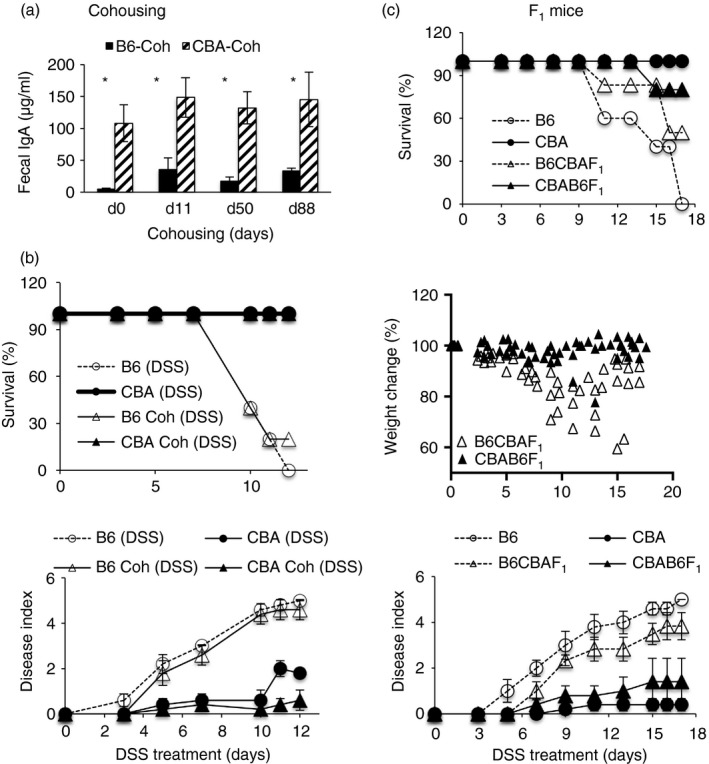
Dextran sodium sulfate (DSS)‐colitis in cohoused mice and F1 mice. Fecal IgA amounts over time (a) and DSS‐colitis readouts (b) in ten B6 and ten CBA mice housed separately or cohoused (Coh) for 3 months. *P < 0·01. DSS‐colitis in F1 progeny derived with a CBA dam and B6 sire (CBAB6F1) or a B6 dam and CBA sire (B6CBAF1) compared with colitis in the parental strains (c). Data are with eight or nine mice per group and representative of two experiments (a,b) and four experiments (c).
Figure 7.
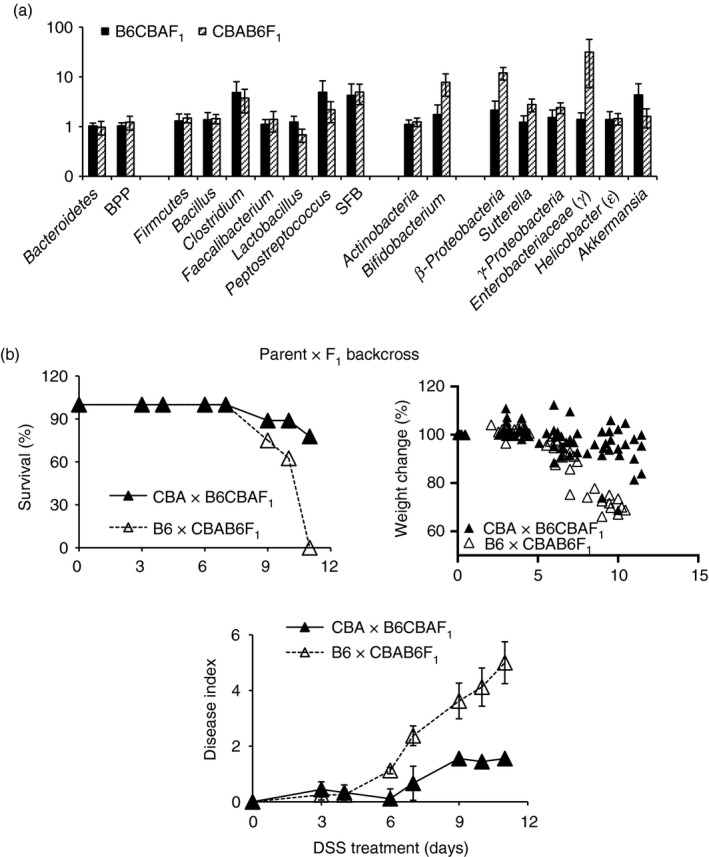
Flora in F1 mice and colitis in Parent × F1 backcrossed progeny. Relative representation of bacterial taxa in B6CBAF1 and CBAB6F1 mice (a). Dextran sodium sulfate (DSS)‐colitis in progeny of CBA×B6CBAF1 and B6×CBAB6F1 crosses (b). Data are from five or six mice per group (a) and eight or nine mice per group (b) and are representative of two experiments.
Passive IgA received by neonates is insufficient to protect against colitis in later life
As high‐IgA response of the proximal dam correlated with protection, we carried out foster‐nursing experiments to determine whether the quantity and/or quality of passive IgA received by pups was sufficient to protect them in later life. To this end, B6 and CBA pups derived from timed pregnancies were foster‐nursed on the reciprocal dams and treated with DSS at 8 weeks of age. We found that in adult foster‐nursed mice, fecal bacterial loads and IgA‐coating of fecal bacteria reverted to those found in adults of the original strain (Fig. 8a,b). Notably, B6 pups that had been foster‐nursed on CBA dams continued to be susceptible to colitis induction, whereas CBA pups that had been foster‐nursed on B6 dams continued to be resistant (Fig. 8c). Hence, maternally acquired IgA is not sufficient to protect adult mice if they make insufficient active IgA.
Figure 8.
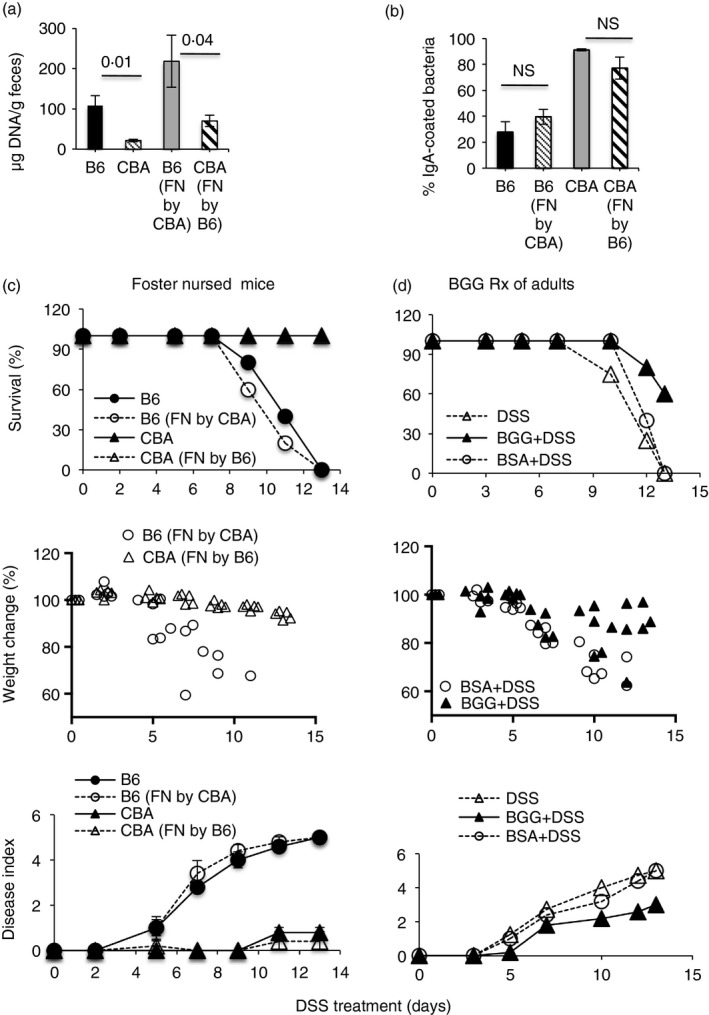
IgA received by neonates is not sufficient to protect adult B6 mice from dextran sodium sulfate (DSS)‐colitis. Proportion of fecal bacteria coated with IgA (a) fecal bacterial loads (b) and DSS‐colitis (c) in adult B6 and CBA mice that had been nursed by the birth dam (B6, CBA), or had been foster nursed [B6 (FN by CBA) and CBA (FN by B6)]. DSS‐colitis in adult B6 mice that received 10 mg bovine γ‐globulin (BGG) or bovine serum albumin (BSA) from day –3 to day +4 of DSS treatment (d). Data are representative of two experiments each with five mice/group.
To determine whether this insufficiency in adults could be corrected with oral supplementation of antibodies, we administered BGG orally to adult B6 mice to provide surrogate coating of bacteria and found that the treatment afforded some protection against colitis compared with mice treated with BSA (Fig. 8d). Bacterial loads were not different in mice treated with BGG or not (134·3 ± 19·3 and 147 ± 40·1 μg bacterial DNA/g feces, respectively, n = 10), indicating that the protection was likely mediated by immune exclusion of bacteria.
In ‘F1 × Parent’ progeny, resistance segregates with IgA response of the pups
In the F1 mice (Fig. 7a), the genetic landscape is similar in the two sets of pups and hence the role of IgA contributed by the proximal dam is clear. However, in the ‘Parent × F1’ backcross experiment (Fig. 7b), the genetic landscape of the resistant set (CBA×B6CBAF1) is ‘more CBA‐like’ with CBA elements being contributed by the parent as well as the F1 mice whereas that of the susceptible set (B6×CBAB6F1) is ‘more B6‐like’, with CBA elements being contributed only by the F1 mice. Together with the inability of foster nursing to protect adult B6 mice from DSS‐colitis, the data raise the possibility that CBA‐associated genetic components other than the effects of maternal IgA in early life may be involved in resistance. To address this, we made another set of backcrosses in which dams of the two F1 sets were crossed with males of the two parental strains, as ‘F1 × Parent’ crosses. We found that resistance was associated with inheritance of a greater genetic component from the resistant strain (Fig. 9) and not the IgA‐phenotype of the proximal dam. Hence, when susceptible B6CBAF1 dams were crossed with resistant CBA males to generate B6CBAF1×CBA mice (CBA elements contributed by both, but less maternal IgA), the progeny were resistant to DSS‐colitis, whereas when resistant CBAB6F1 dams were crossed with susceptible B6 males to generate CBAB6F1×B6 mice (CBA elements contributed only by the F1 mice, but more maternal IgA), the progeny were susceptible. We compared fecal IgA amounts, fecal bacterial loads, IgA‐coating and barrier‐gene transcripts in the progeny of all crosses and we found that resistance correlated with high fecal IgA amounts and high‐IgA coating of fecal bacteria and not with fecal bacterial loads or barrier transcripts in the F1 mice (Fig. 10a) and backcrossed mice (Fig. 10b). Although fecal IgA amounts in the resistant CBAB6F1 mice were lower than in the parent CBA strain they appear to be sufficient to coat an equivalent proportion of bacteria (63·08 ± 6·31 in the F1s against 70·19 ± 4·23 in the parent), indicating that a threshold amount of IgA may be required in vivo.
Figure 9.
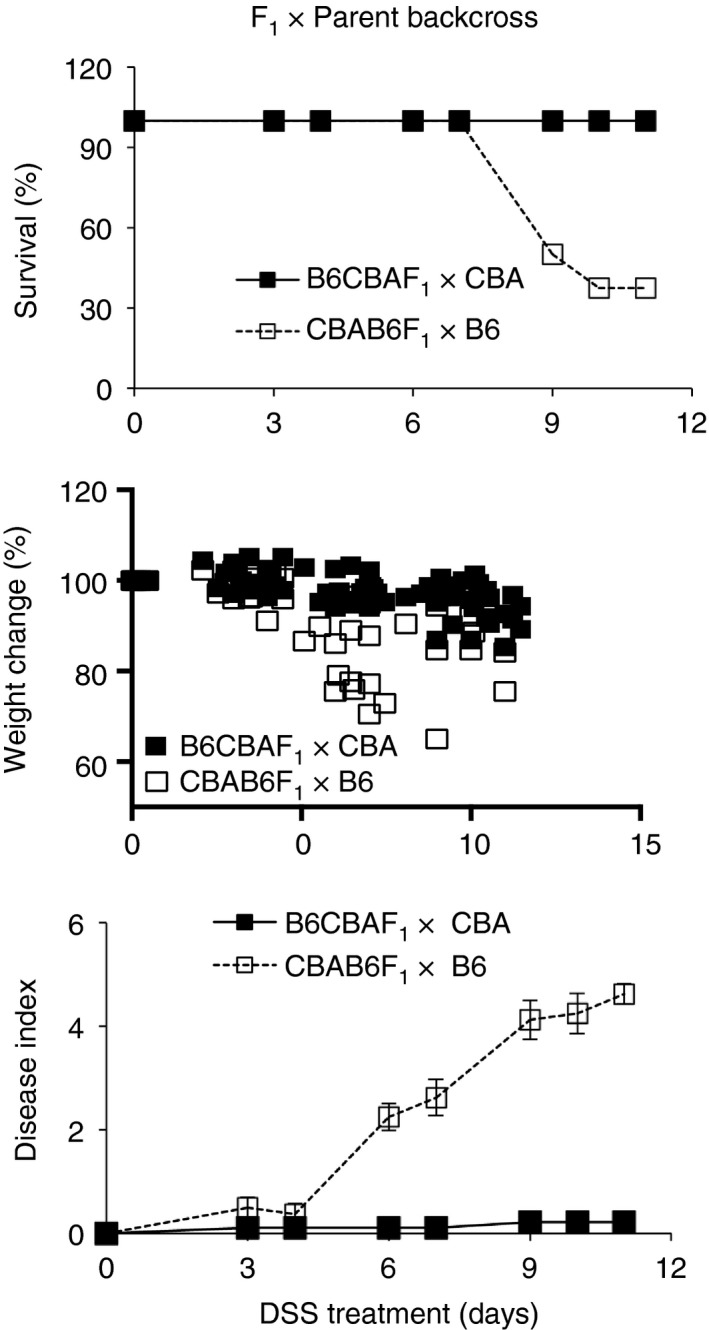
Response of F1 × Parent backcross progeny to colitis induction. Dextran sodium sulfate (DSS)‐colitis in progeny of B6CBAF1×CBA and CBAB6F1×B6 crosses. Data are with eight or nine mice per group and representative two experiments.
Figure 10.
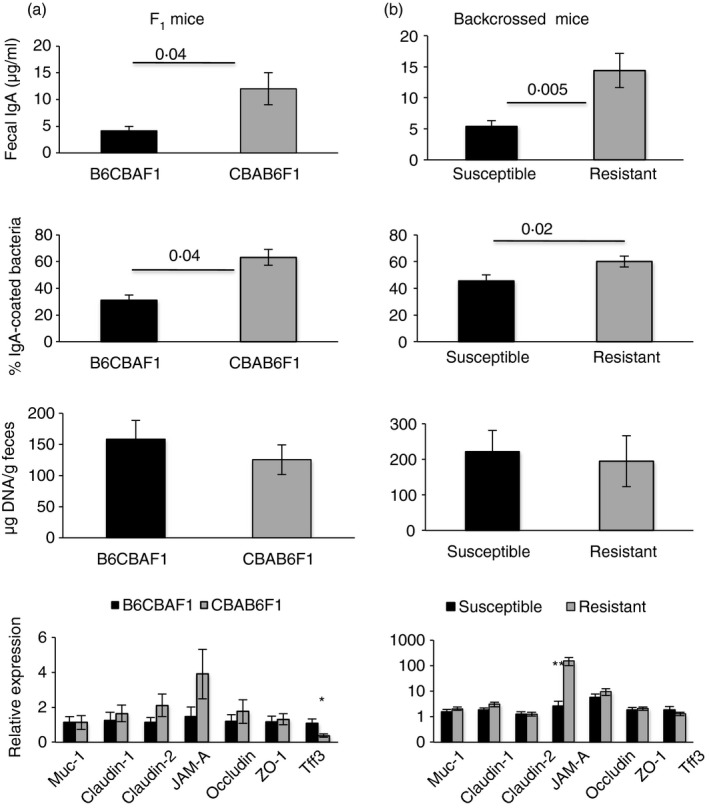
In genetic experiments, resistance to dextran sodium sulfate (DSS)‐colitis correlates with IgA response of progeny. Fecal IgA concentrations, proportion of IgA‐coated fecal bacteria, fecal bacterial loads and relative barrier‐gene transcript abundance (as labeled) in the F1 progeny (a, five mice per group) and in backcrossed progeny (b, 10–20 mice per group). For (b), the two susceptible groups and the two resistant groups were pooled for analysis. Data are representative of two experiments.
Discussion
Due to the extraordinary complexity and changing nature of the intestinal environment, investigators have often relied on gene‐deficient, germ‐free and gnotobiotic mice to understand the mechanistic processes involved in the induction of inflammatory bowel disease. These efforts have indicated, variously, a role for IgA, mucins, tight junction proteins, inflammatory cells, AMPs, cytokines, Treg cells, microbial diversity, specific bacterial groups, microbe‐derived metabolic products, dysregulated endoplasmic reticulum stress response, autophagy and poor wound repair, among others, as contributory factors.1, 2, 3, 14, 24, 31 Because IgA is a major player in the establishment and maintenance of a healthy intestinal microbiome, and modulates many of the other factors, we examined its role in determining disease induction in the wild‐type inbred strains B6 and CBA, which differ in the amount of IgA they make, but are otherwise healthy and harbor a diverse microbiome.
We found that resistance to DSS‐colitis in CBA mice correlated with a higher coating of fecal bacteria with IgA, lower fecal bacterial loads and resistance to disease even when DSS was administered in the presence of a barrier‐disruptive agent,24 a treatment that exacerbated disease in B6 mice. In support of a more resilient barrier, colonic tissues in CBA mice showed higher expression of transcripts for barrier‐protective genes such as Muc1, claudin 1, occludin, ZO‐1 and Tff3. Mice deficient in the gel‐forming mucin Muc2 are reported to have a relatively thin adherent mucus layer and increased susceptibility to DSS‐colitis,9 but Muc2 transcript amounts were similar in colonic tissue from the two strains.
The intestinal epithelium requires steady‐state maintenance of an unfolded protein response to support high‐level synthesis of various secreted molecules, transcytosis of IgA and continuous cell movement along the crypt–villus axis, and mice deficient in endoplasmic reticulum stress response components are more sensitive to DSS‐colitis.31, 32 In keeping with these studies, transcripts for some endoplasmic reticulum stress genes were less abundant in epithelial cells isolated from B6 mice (see Supplementary material, Fig. S4). It is, therefore, possible that intestinal permeability may be slightly higher in B6 mice under homeostatic conditions; nevertheless, B6 mice develop disease only after exposure to DSS, as do mice with modest barrier deficiencies.4, 11
Decreasing bacterial loads with VNAM treatment protected B6 mice, as has been reported earlier,33, 34 and most of the residual bacteria in IgA‐coated B6 mice were also resistant to FTs with CBA flora, which are mostly IgA‐coated. Together, these observations raised the possibility that disease could be associated with a ‘colitogenic’ cohort of bacteria that are not targeted by IgA, and are over‐represented in this strain. In support, disease in B6 mice was seen only in recipients of IgA‐uncoated homologous FTs. Our data differ from an earlier report identifying colitogenic members in the IgA‐coated fraction of fecal bacteria in patients with inflammatory bowel disease and in ASC–/– mice,8 but are in keeping with the heightened susceptibility of IgA‐low B6 mice to DSS‐colitis.19 The IgA‐uncoated fraction was enriched in γ‐Proteobacteria, segmented filamentous bacteria and Helicobacter identified with colitis in the first study, and with Sutterella identified in the second.
DSS treatment led to greater accumulation of neutrophils in colonic tissues of B6 mice, greater neutrophil transmigration as measured by fecal neutrophil activity, and greater abundance of some pro‐inflammatory cytokines and chemokines. The extent of intestinal inflammation depends on cytokine balances12, 27, 28, 29, 30 and our analysis indicates that disease in B6 mice is associated with a pro‐inflammatory landscape and elevated neutrophil recruitment whose transmigration may disrupt tight junctions and weaken the barrier further. Th17 and Treg frequencies were similar in the two strains, but their numbers were higher in CBA mice during DSS treatment. Th17 cells have been reported to promote inflammation as well as repair25, 35, 36, 37 and they may prevent overt disease by facilitating repair and controlling barrier breach.
Possible pathobionts present in B6 mice failed to induce disease in CBA mice. High IgA amounts in this strain may modulate immune exclusion in taxon‐specific and non‐specific ways; a large proportion of IgA induced by potent stimulators such as segmented filamentous bacteria is polyreactive,38 binding of IgA to commensals can occur independently of the Fab portion of the antibody molecule,39 the IgA hinge region is similar to a portion of mucin glycoproteins and allows for reversible anchoring of coated bacteria to mucus40 and favours ‘host‐supported’ biofilms,15 and free SC can bind to and inactivate IL‐8, preventing its neutrophil chemotactic potential.41 Our finding that administration of BGG to B6 mice afforded some protection against colitis, as indicated previously,42 without decreasing bacterial loads, suggests that immune exclusion can prevent colitis.
Fecal transplant experiments can lead to blooms of specific bacterial groups and they do not address the contribution of maternal effects on neonates. Maternal microbiota and IgA are known to modulate the expression of barrier genes, influence microbial colonization, regulate translocation of gut bacteria to systemic tissues in neonates, and influence susceptibility to colitis and intestinal inflammation in later life.7, 10, 43 Hence, we generated F1 mice with a CBA or B6 dam, allowing for segregation of pups receiving differing amounts of maternal IgA and maternally transmitted flora, and found that F1 mice of CBA dams were more resistant to DSS‐colitis than those of B6 dams. Further, when resistant F1 males were backcrossed to B6 dams, the progeny were susceptible, and when susceptible F1 males were backcrossed to CBA dams the progeny were resistant, together indicating a role for the proximal dam in determining resistance. In the F1 and backcross progeny, resistance correlated with IgA amounts and IgA‐coating of fecal bacteria, but not with bacterial loads, taxon abundance or barrier transcripts. Interestingly, early maternal effects were insufficient to protect mice in later life as foster‐nursed B6 adults were susceptible to colitis induction.
Our data indicate that resistance to DSS‐colitis correlates most consistently with active IgA made by adults, a critical amount of which may be required for maintenance of a healthy microbiome and immune exclusion. Data with gene‐deficient mice have identified multiple individual elements that determine susceptibility, but most of these are on the sensitive B6 background and single gene deficiencies that affect barrier integrity, for instance, may not have any deleterious consequences in the CBA strain. Together with our data and those of others indicating that decreasing the load of IgA‐uncoated bacteria or administering appropriate doses of short‐chain fatty acids or aryl hydrocarbon receptor‐ligands6, 44 protects B6 mice against colitis, it appears that other factors may play a protective role when IgA is limiting. A comprehensive microbiome sequencing analysis to determine the abundance of specific colitogenic members in the B6 gut, the critical numbers of these needed for disease induction, and differential immune responses of the two strains to these members will shed more light on the generation and maintenance of microbe‐immune homeostasis in the gut in health and disease.
Disclosures
The authors declare no conflict of interest.
Supporting information
Figure S1. (a) Representative flow cytometric identification of IgA‐coated fecal bacteria. (b) Relative abundance of bacterial DNA, relative to tissue DNA in mesenteric lymph nodes of B6 and CBA mice at indicated times after dextran sodium sulfate (DSS) treatment, mean of four or five mice. (c) Relative expression of Cathelin‐related antimicrobial peptide (CRAMP), Chemokine recognition site‐1 (Crs1), Matrix metalloproteinase‐1 (MMP‐7), Regenerating islet‐derived IIIγ (Reg‐IIIγ), Cryptdins (1, 4, 5), α‐defensins, lysozyme and Light chain‐3 (LC3) transcripts (left panel) in colonic tissue and fecal calprotectin amounts in fecal pellets (right panel) of untreated B6 and CBA mice. (d) Protective effect of low‐dose SCFA (20 nm) treatment during colitis induction in B6 mice. (e) DSS‐colitis in interleukin ‐5 (IL‐5) transgenic mice. Data are from five or six mice per group, representative of two experiments.
Figure S2. Relative abundance of transcripts for various cytokines and chemokines (as labeled) in colonic tissues of mice 5 days after initiation of dextran sodium sulfate treatment, mean ± SEM of five or six mice per group. Representative of two experiments
Figure S3. (a) Representative enrichment of IgA‐coated and IgA‐uncoated fecal bacteria after flow cytometric sorting. (b,c) Effect of Ampicillin (b) and metronidazole (c) treatment on induction of dextran sodium sulfate‐colitis in B6 mice. Five or six mice per group, representative of two experiments.
Figure S4. Relative expression of transcripts for endoplasmic reticulum stress genes in colonic tissues of untreated B6 and CBA mice. Five or six mice per group, representative of two experiments.
Acknowledgements
SG and SB performed experiments and helped in their design and analysis. SR, VB and AG designed and planned the experiments and analyzed and interpreted data. SG, VB and SR helped AG with writing the manuscript. This work was supported in part by grants from the Department of Biotechnology, Government of India (BT/PR‐14592/BRB/10/858/2010 to SR and No. BT/PR13699/BRB/10/1514/2016 to AG) and the Science and Engineering Research Board, Government of India (to AG and VB). The National Institute of Immunology is supported by the Department of Biotechnology, Government of India. We thank Dr. Nagarajan for expert assistance and help for breeding and maintenance of mouse strains. This manuscript is dedicated to the memory of Prof. John J. Cebra.
References
- 1. Kurashima Y, Goto Y, Kiyono H. Mucosal innate immune cells regulate both gut homeostasis and intestinal inflammation: HIGHLIGHTS. Eur J Immunol 2013; 43(12):3108–15. [DOI] [PubMed] [Google Scholar]
- 2. Peterson LW, Artis D. Intestinal epithelial cells: regulators of barrier function and immune homeostasis. Nat Rev Immunol 2014; 14(3):141–53. [DOI] [PubMed] [Google Scholar]
- 3. Wei M, Shinkura R, Doi Y, Maruya M, Fagarasan S, Honjo T. Mice carrying a knock‐in mutation of Aicda resulting in a defect in somatic hypermutation have impaired gut homeostasis and compromised mucosal defense. Nat Immunol 2011; 12(3):264–70. [DOI] [PubMed] [Google Scholar]
- 4. Choi W, Yeruva S, Turner JR. Contributions of intestinal epithelial barriers to health and disease. Exp Cell Res 2017; 358(1):71–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Kamada N, Seo S‐U, Chen GY, Núñez G. Role of the gut microbiota in immunity and inflammatory disease. Nat Rev Immunol 2013; 13(5):321–35. [DOI] [PubMed] [Google Scholar]
- 6. Medina‐Contreras O, Harusato A, Nishio H, Flannigan KL, Ngo V, Leoni G et al Cutting edge: IL‐36 receptor promotes resolution of intestinal damage. J Immunol 2016; 196(1):34–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Mirpuri J, Raetz M, Sturge CR, Wilhelm CL, Benson A, Savani RC et al Proteobacteria‐specific IgA regulates maturation of the intestinal microbiota. Gut Microbes 2014; 5(1):28–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Palm NW, de Zoete MR, Cullen TW, Barry NA, Stefanowski J, Hao L et al Immunoglobulin A coating identifies colitogenic bacteria in inflammatory bowel disease. Cell 2014; 158(5):1000–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Petersson J, Schreiber O, Hansson GC, Gendler SJ, Velcich A, Lundberg JO et al Importance and regulation of the colonic mucus barrier in a mouse model of colitis. Am J Physiol‐Gastrointest Liver Physiol 2011; 300(2):G327–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Rogier EW, Frantz AL, Bruno MEC, Wedlund L, Cohen DA, Stromberg AJ et al Secretory antibodies in breast milk promote long‐term intestinal homeostasis by regulating the gut microbiota and host gene expression. Proc Natl Acad Sci U S A 2014; 111(8):3074–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Su L, Shen L, Clayburgh DR, Nalle SC, Sullivan EA, Meddings JB et al Targeted epithelial tight junction dysfunction causes immune activation and contributes to development of experimental colitis. Gastroenterology 2009; 136(2):551–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Sugimoto K, Ogawa A, Mizoguchi E, Shimomura Y, Andoh A, Bhan AK et al IL‐22 ameliorates intestinal inflammation in a mouse model of ulcerative colitis. J Clin Invest 2008; 118(2):534–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Torow N, Hornef MW. The neonatal window of opportunity: setting the stage for life‐long host‐microbial interaction and immune homeostasis. J Immunol 2017; 198(2):557–63. [DOI] [PubMed] [Google Scholar]
- 14. Kawamoto S, Maruya M, Kato LM, Suda W, Atarashi K, Doi Y et al Foxp3+ T cells regulate immunoglobulin a selection and facilitate diversification of bacterial species responsible for immune homeostasis. Immunity 2014; 41(1):152–65. [DOI] [PubMed] [Google Scholar]
- 15. Mathias A, Corthésy B. N‐Glycans on secretory component: mediators of the interaction between secretory IgA and Gram‐positive commensals sustaining intestinal homeostasis. Gut Microbes 2011; 2(5):287–93. [DOI] [PubMed] [Google Scholar]
- 16. Li Y, Innocentin S, Withers DR, Roberts NA, Gallagher AR, Grigorieva EF et al Exogenous stimuli maintain intraepithelial lymphocytes via aryl hydrocarbon receptor activation. Cell 2011; 147(3):629–40. [DOI] [PubMed] [Google Scholar]
- 17. Fransen F, Zagato E, Mazzini E, Fosso B, Manzari C, El Aidy S et al BALB/c and C57BL/6 mice differ in polyreactive IgA abundance, which impacts the generation of antigen‐specific IgA and microbiota diversity. Immunity 2015; 43(3):527–40. [DOI] [PubMed] [Google Scholar]
- 18. Bunker JJ, Flynn TM, Koval JC, Shaw DG, Meisel M, McDonald BD et al Innate and adaptive humoral responses coat distinct commensal bacteria with immunoglobulin A. Immunity 2015; 43(3):541–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Moon C, Baldridge MT, Wallace MA, Burnham C‐AD, Virgin HW et al Vertically transmitted faecal IgA levels determine extra‐chromosomal phenotypic variation. Nature 2015; 521(7550):90–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Mähler M, Bristol IJ, Leiter EH, Workman AE, Birkenmeier EH, Elson CO et al Differential susceptibility of inbred mouse strains to dextran sulfate sodium‐induced colitis. Am J Physiol 1998; 274(3 Pt 1):G544–51. [DOI] [PubMed] [Google Scholar]
- 21. Melgar S, Karlsson A, Michaëlsson E. Acute colitis induced by dextran sulfate sodium progresses to chronicity in C57BL/6 but not in BALB/c mice: correlation between symptoms and inflammation. Am J Physiol Gastrointest Liver Physiol 2005; 288(6):G1328–38. [DOI] [PubMed] [Google Scholar]
- 22. Johansson MEV, Gustafsson JK, Sjöberg KE, Petersson J, Holm L, Sjövall H et al Bacteria penetrate the inner mucus layer before inflammation in the dextran sulfate colitis model. PLoS ONE 2010; 5(8):e12238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Martini E, Krug SM, Siegmund B, Neurath MF, Becker C. Mend your fences: the epithelial barrier and its relationship with mucosal immunity in inflammatory bowel disease. Cell Mol Gastroenterol Hepatol 2017; 4(1):33–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kaiko GE, Ryu SH, Koues OI, Collins PL, Solnica‐Krezel L, Pearce EJ et al The colonic crypt protects stem cells from microbiota‐derived metabolites. Cell 2016; 167(4):1137. [DOI] [PubMed] [Google Scholar]
- 25. Peterson DA, McNulty NP, Guruge JL, Gordon JI. IgA response to symbiotic bacteria as a mediator of gut homeostasis. Cell Host Microbe 2007; 2(5):328–39. [DOI] [PubMed] [Google Scholar]
- 26. Masterson JC, McNamee EN, Fillon SA, Hosford L, Harris R, Fernando SD et al Eosinophil‐mediated signalling attenuates inflammatory responses in experimental colitis. Gut 2015; 64(8):1236–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Song X, Dai D, He X, Zhu S, Yao Y, Gao H et al Growth factor FGF2 cooperates with interleukin‐17 to repair intestinal epithelial damage. Immunity 2015; 43(3):488–501. [DOI] [PubMed] [Google Scholar]
- 28. Wirtz S, Neurath MF. Illuminating the role of type I IFNs in colitis. J Clin Invest 2005; 115(3):586–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Bradford EM, Ryu SH, Singh AP, Lee G, Goretsky T, Sinh P et al Epithelial TNF receptor signaling promotes mucosal repair in inflammatory bowel disease. J Immunol 2017; 199(5):1886–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Pickert G, Neufert C, Leppkes M, Zheng Y, Wittkopf N, Warntjen M et al STAT3 links IL‐22 signaling in intestinal epithelial cells to mucosal wound healing. J Exp Med 2009; 206(7):1465–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Lopes F, Keita ÅV, Saxena A, Reyes JL, Mancini NL, Al Rajabi A et al ER‐stress mobilization of death‐associated protein kinase‐1‐dependent xenophagy counteracts mitochondria stress‐induced epithelial barrier dysfunction. J Biol Chem 2018; 293(9):3073–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Bertolotti A, Wang X, Novoa I, Jungreis R, Schlessinger K, Cho JH et al Increased sensitivity to dextran sodium sulfate colitis in IRE1β‐deficient mice. J Clin Invest 2001; 107(5):585–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Hu S, Peng L, Kwak Y‐T, Tekippe EM, Pasare C, Malter JS et al The DNA sensor AIM2 maintains intestinal homeostasis via regulation of epithelial antimicrobial host defense. Cell Rep 2015; 13(9):1922–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Tanaka Y, Ito S, Isobe K. Vancomycin‐sensitive bacteria trigger development of colitis‐associated colon cancer by attracting neutrophils. Sci Rep 2016; 6(1):23920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Aden K, Rehman A, Falk‐Paulsen M, Secher T, Kuiper J, Tran F et al Epithelial IL‐23R signaling licenses protective IL‐22 responses in intestinal inflammation. Cell Rep 2016; 16(8):2208–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Cao AT, Yao S, Gong B, Elson CO, Cong Y. Th17 cells upregulate polymeric Ig receptor and intestinal IgA and contribute to intestinal homeostasis. J Immunol 2012; 189(9):4666–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Izcue A, Hue S, Buonocore S, Arancibia‐Cárcamo CV, Ahern PP, Iwakura Y et al Interleukin‐23 restrains regulatory T cell activity to drive T cell‐dependent colitis. Immunity 2008; 28(4):559–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Talham GL, Jiang HQ, Bos NA, Cebra JJ. Segmented filamentous bacteria are potent stimuli of a physiologically normal state of the murine gut mucosal immune system. Infect Immun 1999; 67(4):1992–2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Royle L, Roos A, Harvey DJ, Wormald MR, van Gijlswijk‐Janssen D, Redwan E‐RM et al Secretory IgA N‐ and O‐glycans provide a link between the innate and adaptive immune systems. J Biol Chem 2003; 278(22):20140–53. [DOI] [PubMed] [Google Scholar]
- 40. Clamp JR. The relationship between secretory immunoglobulin A and mucus. Proc Biochem Soc Trans 1977; 5(5):1579–81. [DOI] [PubMed] [Google Scholar]
- 41. Marshall LJ, Perks B, Ferkol T, Shute JK. IL‐8 released constitutively by primary bronchial epithelial cells in culture forms an inactive complex with secretory component. J Immunol 2001; 167(5):2816–23. [DOI] [PubMed] [Google Scholar]
- 42. Bodammer P, Zirzow E, Klammt S, Maletzki C, Kerkhoff C. Alteration of DSS‐mediated immune cell redistribution in murine colitis by oral colostral immunoglobulin. BMC Immunol 2013; 14:10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Bergström A, Kristensen MB, Bahl MI, Metzdorff SB, Fink LN, Frøkiaer H et al Nature of bacterial colonization influences transcription of mucin genes in mice during the first week of life. BMC Res Notes 2012; 5:402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. D'Souza WN, Douangpanya J, Mu S, Jaeckel P, Zhang M, Maxwell JR et al Differing roles for short chain fatty acids and GPR43 agonism in the regulation of intestinal barrier function and immune responses. PLoS ONE 2017; 12(7):e0180190. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. (a) Representative flow cytometric identification of IgA‐coated fecal bacteria. (b) Relative abundance of bacterial DNA, relative to tissue DNA in mesenteric lymph nodes of B6 and CBA mice at indicated times after dextran sodium sulfate (DSS) treatment, mean of four or five mice. (c) Relative expression of Cathelin‐related antimicrobial peptide (CRAMP), Chemokine recognition site‐1 (Crs1), Matrix metalloproteinase‐1 (MMP‐7), Regenerating islet‐derived IIIγ (Reg‐IIIγ), Cryptdins (1, 4, 5), α‐defensins, lysozyme and Light chain‐3 (LC3) transcripts (left panel) in colonic tissue and fecal calprotectin amounts in fecal pellets (right panel) of untreated B6 and CBA mice. (d) Protective effect of low‐dose SCFA (20 nm) treatment during colitis induction in B6 mice. (e) DSS‐colitis in interleukin ‐5 (IL‐5) transgenic mice. Data are from five or six mice per group, representative of two experiments.
Figure S2. Relative abundance of transcripts for various cytokines and chemokines (as labeled) in colonic tissues of mice 5 days after initiation of dextran sodium sulfate treatment, mean ± SEM of five or six mice per group. Representative of two experiments
Figure S3. (a) Representative enrichment of IgA‐coated and IgA‐uncoated fecal bacteria after flow cytometric sorting. (b,c) Effect of Ampicillin (b) and metronidazole (c) treatment on induction of dextran sodium sulfate‐colitis in B6 mice. Five or six mice per group, representative of two experiments.
Figure S4. Relative expression of transcripts for endoplasmic reticulum stress genes in colonic tissues of untreated B6 and CBA mice. Five or six mice per group, representative of two experiments.