

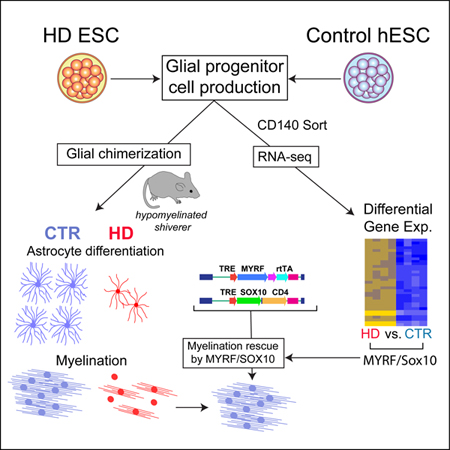
Abstract
Huntington’s disease (HD) is characterized by hypomyelination as well as neuronal loss. To assess the basis for myelin loss in HD, we generated bipotential glial progenitor cells (GPCs) from human embryonic stem cells (hESCs), derived from huntingtin (mHTT)-mutant embryos or normal controls, and performed RNAseq to assess mHTT-dependent changes in gene expression. In hGPCs derived from 3 mHTT hESC lines, transcription factors associated with glial differentiation and myelin synthesis were sharply down-regulated relative to normal hESC GPCs; NKX2.2, OLIG2, SOX10, MYRF and their downstream targets were all suppressed. Accordingly, when mHTT hGPCs were transplanted into hypomyelinated shiverer mice, the resultant glial chimeras were hypomyelinated; this defect could be rescued by forced expression of SOX10 and MYRF by mHTT hGPCs. The mHTT hGPCs also manifested impaired astrocytic differentiation, and developed abnormal fiber architecture. White matter involution in HD is thus a product of the cell autonomous, mHTT-dependent suppression of glial differentiation.
Graphical Abstract
eTOC
The authors show that glial progenitor cells derived from human ES cells expressing mutant Huntingtin (mHTT) are delayed and defective in their maturation; their suppressed transcription of differentiation-associated genes leads to both astrocytic dysfunction and myelin deficiency in vivo. Glial pathology may thus contribute to disease phenotype in Huntington’s disease.
Introduction
Huntington’s disease (HD) is a fatal, autosomal-dominant neurodegenerative disorder characterized by progressive behavioral, cognitive, and motor dysfunction. HD is caused by a CAG trinucleotide repeat in the first exon of the huntingtin (HTT) gene, encoding a polyglutamine expansion. Its age of onset and severity are proportional to the length of this repeat expansion, with CAG lengths over 35 invariably leading to clinical disease. While HD neuropathology is characterized by the intracellular aggregation of mutant HTT (mHTT) and progressive loss of striatal medium spiny neurons, MRI studies have revealed that HD is also characterized by the early appearance of demyelination and white matter loss, which can appear before symptoms arise (Tabrizi et al., 2012). Similarly, studies in mouse models of HD have revealed early dysmyelination (Teo et al., 2016), attended by a deficit in the critical myelinogenic gene MYRF (Huang et al., 2015; Jin et al., 2015). Together, these observations suggest that HD is associated with white matter loss, which may reflect the concurrent dysfunction of myelinogenic oligodendrocytes.
Yet despite these data associating early dysmyelination with HD, and parallel studies indicating that glial replacement may ameliorate symptoms in HD transgenic mice (Benraiss et al., 2016), neither the cellular nor molecular bases for glial pathology in human HD have been well-explored. To address this issue, we asked if the gene expression patterns of mHTT-expressing human glial progenitor cells (hGPCs) might reflect cell-autonomous molecular pathology, and if so whether that might predict the white matter disease of HD. We first generated bipotential oligodendrocyte-astrocyte hGPCs from human embryonic stem cells (hESCs), derived from either huntingtin mutant embryos or their sibling controls. (Of note, GPCs are variably referred to as oligodendrocyte progenitor cells, NG2 cells, polydendrocytes and glial restricted precursors in the literature. We will use the terminology GPC, which we believe the most empirically accurate, in this report.) We then used fluorescence-activated cell sorting (FACS) to isolate these cells based on their expression of the GPC-selective CD140a (Sim et al., 2011; Wang et al., 2013), followed by whole transcriptome RNA sequence analysis (RNA-seq) to assess mHTT-dependent changes in their gene expression. We found that in hGPCs produced from hESCs derived from three different HD embryos, a coherent set of key transcription factors associated with both astroglial and oligodendroglial differentiation, as well as with downstream myelin biosynthesis, was significantly down-regulated relative to controls as a function of mHTT expression. Accordingly, when HD hESC-derived GPCs were transplanted into neonatal myelin-deficient and immunodeficient shiverer mice (MBPshi/shi), the resultant glial chimeras myelinated more slowly and less completely than did littermate controls transplanted with GPCs derived from normal control hESCs. In addition, chimeras established with HD hGPCs manifested a marked delay and disruption in astrocytic morphogenesis relative to mice chimerized with normal sibling GPCs. Together, these data suggest that white matter failure and hypomyelination in human HD, rather than being secondary to neuronal loss, might instead be the result of a cell-autonomous defect in the terminal glial differentiation of mHTT-expressing GPCs, the occurrence of which may be central to the pathogenesis and neurological manifestations of HD.
RESULTS
mHTT GPCs down-regulate transcriptional determinants of glial lineage progression
To address the role of glial transcriptional abnormalities in the pathogenesis of HD, we first assessed differential gene expression by bipotential GPCs derived from huntingtin mutant hESCs. To that end, we generated and purified GPCs from three distinct lines of hESCs derived from mHTT-expressing blastocysts (GENEA17, 18 and 20; GENEA Biocells), and from two control lines (GENEA02 and 19) (Bradley et al., 2011). GPCs were produced from human ESCs using our previously described methods (Wang et al., 2013), followed by CD140a-based FACS to isolate the resulting GPC fraction (>99% CD140a+) (Sim et al., 2011). Importantly, one of the controls (GENEA19; 18 CAG) was a sibling to one of the mHTT-expressing lines (GENEA20, 48 CAG); these lines, donated by the same parents, were fraternal female twins.
mHTT and control hGPCs were harvested as stably expanding GPCs, after average propagation times of 190 ± 16 and 174 ± 14 days, respectively. Flow cytometry revealed that 54 ± 12% of normal (GENEA02 and 19; n=12 culture runs) and 44 ± 13% of huntingtin mutant cells (GENEA17, 18 and 20; n=16) expressed CD140a at these time-points (means ± SD). The CD140a fraction of each culture was then isolated to near-purity by FACS, and RNA-seq performed uisng an Illumina HiSeq 2500 sequencer, which showed profound transcriptional dysregulation in the GPCs derived from the three HD lines, relative to the pooled control hESC GPCs. Principle component analysis (PCA) showed clear segregation of the mHTT-expressing and control hGPCs (Figure 1A). As a group, using a 2-fold change (FC) cut-off and 1% FDR, 239 genes were up- and 530 genes were down-regulated in the mHTT hGPCs relative to their controls (Figure 1B). To further refine the resultant list of differentially-expressed genes, we then compared the differential expression of GENEA20 (mHTT)-derived hGPCs to their sibling GENEA19-derived controls, and added that sibling comparison to our overall comparison; this acted as an additional filter, and yielded a tighter differentially-expressed gene list, comprised of 64 up- and 365 down-regulated genes in hGPCs derived from all HD-derived hGPC cell lines relative to their pooled control hGPCs (Figures 1B–1C).
Figure 1. HD hESC-derived hGPCs display profound mHTT-dependent changes in gene expression.

A, Principal component analysis (PCA) based on expression of approximately 26,000 transcripts. The expression data are shown as transcripts per million (TPM), post-normalization to account for variance (Risso et al., 2014). The PCA plot shows the distinct transcriptome-wide expression signature of HD-derived hGPCs. B, Venn diagram shows intersections of lists of differentially expressed genes (DEGs) (green, down-regulated; red, up-regulated; fold change, FC > 2.0, FDR 1%), obtained by comparing hGPCs derived from 3 different HD patients to pooled control hGPCs from 2 donors. The list of DEGs shared by the 3 HD patients was then filtered by intersecting with those DEGs (FC > 2.0, FDR 1%) found in patient HD20 (GENEA20-derived) vs. its normal sibling CTR19 (GENEA19); this filtration step further increased the specificity of mHTT-associated DEGs. The gray-highlighted intersections together comprise the entire set of genes differentially expressed by all HD lines relative to their pooled controls. C, Expression heat-map based on transcripts per million (TPM) values for 429 DEGs highlighted in B, showing clustering of hGPCs by disease status. Dendrogram shows hierarchical clustering based on Euclidean distance calculated from log2-TPM values from the three HD-ESCs lines (HD-17, HD-18, HD-20) and the two matched control lines (CTR19, CTR02).
D, Network representation of functional annotations (Gene Ontology: Biological Process and Cellular Component, Bonferroni-corrected p<0.01) for the 429 intersection DEGs highlighted in B. Genes are round nodes with border colors representing their direction of dysregulation (green, down-regulated; red, up-regulated). Rounded rectangle nodes represent annotation terms. Nodes are sized by degree and colored by closely interconnected modules (M1 though M3) identified by community detection. For each module, 3 of the top annotations by significance and fold-enrichment are listed. Selected gene nodes are labeled, and include genes encoding key hGPC lineage transcription factors and stage-regulated proteins. E, Expression heat-map of 63 conserved DEGs identified in M1 (purple in D), annotations related to glial cell differentiation and myelination. F, Expression heat-map of 56 conserved DEGs identified in M2 (lilac in D), annotations related to axon guidance and axonogenesis. G, Expression heat-map of 68 conserved DEGs identified in M3 (yellow in D), annotations related to regulation of synapse structure and synaptic signaling.
All DE results are 1% FDR and FC >2; GO annotation results are Bonferroni-corrected to p<0.01.
Using this gene set, we performed functional analysis with annotation from the Gene Ontology (GO), by which we identified 50 significantly associated GO annotation terms (Bonferroni-corrected p<0.01, among terms in the Biological Process and Cellular Component GO domains), that represented 187 of the 429 differentially regulated genes (Figures S1 and S2). By network analysis, these annotation terms together with their associated genes were further grouped into three functionally related modules, each of which was characterized by its most significant annotation terms (Figure 1D). The three modules represented genes and functions related to (1) glial cell differentiation and myelination; (2) axon guidance and axonogenesis; and (3) regulation of synapse structure and synaptic signaling (Figure 1D). The first and second modules were closely interconnected, and contained an array of critical oligodendrocyte lineage transcription factors, including SOX10, SIRT2, MYRF, NKX2–2, TCF7L2, OLIG1 and OLIG2, as well as stage-regulated and myelin-associated proteins, which included TF, MBP, MAG, OMG, UGT8, and FA2H; all of these were significantly down-regulated in HD hGPCs. The third module contained genes concerned with the regulation of components of synaptic transmission, most notably SYNDIG1, BCAN, NETO1, and SNPH, as well as genes encoding the glutamate receptor signaling proteins GRIA2, GRIA4, GRID1, GRID2, and GRIK4, and the potassium channels encoded by KCND2, KCNJ9, KCNQ1, and KCNS3; all of these were significantly down-regulated (Figures 1E–G). Together, these HD-dysregulated genes and their associated functions suggest an HD-dependent suppression in the differentiation of hGPCs into mature oligodendroglia.
mHTT hGPCs down-regulate transcriptional determinants of myelinogenesis
As revealed by our differential expression analysis, a key set of transcription factors associated with both oligodendroglial differentiation and myelin biosynthesis were significantly and substantially down-regulated as a function of mHTT expression. These included the early oligodendroglial regulators NKX2.2, OLIG2 and SOX10, each of which was sharply down-regulated in mHTT-expressing hGPCs (Figure 1E). Moreover, downstream of the mHTT-suppressed oligodendroglial lineage transcription factors, the mHTT GPCs expressed sharply reduced levels of MYRF, the myelin-regulatory factor. MYRF coordinately activates a number of genes necessary for myelin formation (Bujalka et al., 2013), and its production has been noted to be deficient in mouse mHTT-transgenic oligodendrocytes (Huang et al., 2015). Among our human ESC-derived hGPCs, the MYRF-regulated myelinogenic transcripts MBP, MAG, OMG, PLP1 and MOG were all significantly down-regulated (Figure 1E). Moreover, when we directly compared the expression pattern of hGPCs derived from the sibling pair (GENEA20 for mHTT, and GENEA19 for normal HTT), which have minimal background genetic variation between them, we again noted the differential down-regulation in mHTT GPCs of those genes associated with myelinogenesis. These included MYRF (−4.04 fold lower in mHTT hGPCs; log2 scale), MAG (−6.78), MBP (−5.14), MOG (−10.35), OMG (−5.15), and PLP1 (−2.22), indicating a broad down-regulation of myelinogenesis-associated transcripts in HD GPCs. Importantly, when we compared the RNA expression patterns of hGPCs derived from three different mHTT hESC lines, GENEA17, 18 and 20, whose HTT genes have 40, 46 and 48 CAG repeats respectively, we noted that longer CAG repeat lengths correlated strongly with the progressive down-regulation of these same differentiation and myelinogenesis-associated genes (Figure 2). Importantly, there was a high degree of overlap between those genes and ontologies found to be increasingly dysregulated with longer CAG repeat length in hGPCs, with those genes and ontologies increasingly dysregulated with CAG repeat length in HD transgenic mice (Langfelder et al., 2016) (Figure S3).
Figure 2. Increasing CAG lengths correlate with diminished oligodendroglial gene expression.

Correlation analysis between the differential expression fold-change (FC), and the number of CAG repeats, indicates a strong inverse relationship between glial gene expression and the CAG repeat number. A, Expression heat map based on transcripts per million (TPM) values calculated from raw counts of 429 differentially expressed genes (DEGs) (1% FDR, FC>2.0) found in the intersection of DEGs by comparisons of hGPCs derived from each of the three different HD patients against pooled control hGPCs from two different donors. Row side colors show the Pearson’s R correlation coefficient between fold change of that gene in each HD-derived hGPC line against pooled controls and the corresponding CAG repeat number in that HD line (HD17 = 40x CAG, HD18 = 46x CAG, and HD20 = 48x CAG). Selected genes encoding transcription factors and stage-regulated proteins involved in glial differentiation and myelination are listed. B, Combined scatterplot with linear fit lines, obtained by regression of fold-changes of each of the 429 DEGs shown in heat map in A, against the CAG repeat number in the corresponding hGPC line. C, Histogram showing the distribution of Pearson’s R coefficients for correlation between fold changes of DEGs in 3 HD lines, to corresponding CAG length. For 255 of the 429 genes (|Pearson’s R| > 0.75), the correlation analysis indicated that the absolute magnitude of the fold-change increased with CAG repeat number; 228 of these genes displayed an inverse correlation of gene expression level to the CAG repeat number, with longer repeats associated with diminished glial gene expression.
To validate these RNA-seq-based expression data, we then used RT-qPCR with TaqMan Low Density Arrays (TLDA) to compare the expression levels of these differentiation-associated genes between mHTT and control hGPCs. The majority of those genes identified in RNA-seq analysis as differentially dysregulated in the mHTT GPCs were confirmed as such (Figure S4). These genes included the key oligodendroglial lineage transcription factors MYRF, SOX10, OLIG2, as well as their downstream myelinogenesis-associated targets, including PLP1, MOG and MBP. Based on the down-regulation of this broad set of myelination-associated genes, we predicted a significant disruption in both myelin biogenesis and maintenance by mHTT hGPCs.
mHTT-associated differentiation arrest suppressed potassium channel expression
Among the functionally-related genes most differentially dysregulated by mHtt expression were those encoding ion channels and transporters, in particular the potassium channels. This large group of genes includes 117 known members in the human genome (Pruitt et al., 2006), of which 93 were detectably expressed by hGPCs (raw counts > 5 in at least 3 samples across the dataset). Among these, 25 of the 93 identified K+ channel and transporter genes were dysregulated in the HD GPCs, relative to their pooled hESC GPC controls, using a FC>2.0 cutoff and 5% FDR threshold; 23 of these remained significantly dysregulated even at a 1% FDR (Figure S5). These genes included a number of inwardly rectifying K+ channels, the coordinate suppression of which suggested a basis for the disrupted potassium buffering of the HD brain (Tong et al., 2014). To further refine and prioritize within this large set of coordinately dysregulated K+ channel genes, we included the GENEA20 vs GENEA19 sibling pair comparison as an additional filtration. By this most stringent analysis, 4 genes - KCND2, KCNJ9, KCNQ1, and KCNS3 - remained strongly and significantly down-regulated, both in all HD hGPC lines relative to pooled controls, and within the sibling set of mHTT and normal hESC-derived GPCs. Together, the dysregulated expression of these K+ channel genes are of special significance given their role in maintaining stable interstitial K+ levels, and in determining action potential thresholds. As such, the mHTT-associated suppression of the hGPC K+ channels, which among other roles mediate the glial reuptake of synaptic K+, may causally contribute to the neuronal hyper-excitability observed among striatal neurons in HD (Benraiss et al., 2016; Shin et al., 2005; Tong et al., 2014).
In light of the concurrent dysregulation of glial differentiation as well as K+ channel expression, and the dependence of the latter upon the former, we asked whether a common upstream regulator might exist that is dysregulated itself as a function of mHTT expression. Using Ingenuity Pathway Analysis (IPA; https://www.qiagenbioinformatics.com/products/ingenuity-pathway-analysis), we found that TCF7L2 was predicted as a positive regulator of a broad variety of glial differentiation-associated genes, including several that have been reported to regulate K+ channel gene expression – for instance, the SOX10-modulated KCNB1 (Liu et al., 2015), which was down-regulated in hGPCs derived from all three of our tested HD lines. Among these glial differentiation-associated genes were a number whose expression was markedly deficient in mHTT glia relative to their controls (Figure S4). On that basis, we queried our RNA-seq datasets for both TCF7L2 and TCF7L2-regulated transcripts, and found that TCF7L2 was indeed differentially down-regulated in HD relative to normal hGPCs, while TCF7L2-regulated genes were concomitantly down-regulated (Figure S4). Since TCF7L2 has been strongly implicated in glial differentiation, and oligodendroglial differentiation in particular, these results further emphasized the cell intrinsic nature of the glial differentiation block in HD.
HD hGPCs exhibited impaired myelinogenesis in vivo
Since mHTT hGPCs appeared deficient in their acquisition of gene expression patterns typifying oligodendrocyte maturation and myelinogenesis, we asked if hypomyelinated mice engrafted with HD GPCs were deficient in myelination competence, relative to those engrafted with GPCs from a normal sibling. To this end, mHTT-expressing and control GPCs, respectively derived from the sibling female GENEA20 and GENEA19 lines in matched cultures, were transplanted neonatally into immunodeficient shiverer mice, using our described multisite injection protocol with bilateral hemispheric injections. This protocol yields a stereotypic pattern and time course of donor-derived myelination in host brains, when using normal pluripotent stem cell-derived or tissue-derived hGPCs (Wang et al., 2013; Windrem et al., 2008). In this case, while non-isogenic – no truly isogenic lines for normal and mutant huntingtin have yet been reported – the use of sibling lines for this experiment minimized genetic variation to the extent possible. Using these paired lines and this in vivo model, the oligodendrocytic differentiation and myelination patterns of engrafted mice were assessed at 8, 13 and 18 weeks of age (n=3–5/mice/time point, totaling 12 HD hGPC- and 10 control hGPC-engrafted mice). The brains of these mice were cryo-sectioned, immunolabeled for both oligodendroglial and myelin antigens, and confocal imaged, so as to compare the differentiation and myelination efficiency of HD and control-derived hESC hGPCs in vivo.
We found that the appearance of both oligodendroglial phenotypic markers and indices of myelin protein production occurred significantly earlier in the control hGPC-engrafted mice, relative to the HD hGPC-engrafted animals. Whereas the expression of axonally-engaged myelin basic protein was apparent using control hGPCs by 8 weeks after neonatal graft, mice engrafted with HD hGPCs manifested no evident MBP immunolabeling by that time point (Figures 3A–B). By 12–13 weeks of age, a point by which mice engrafted with control hGPCs exhibited robust myelin production, only scattered islands of MBP expressed by immature oligodendroglia were noted in matched recipients of HD GPCs (Figures 3C–D). The relatively delayed myelination of HD GPC-engrafted white matter persisted for at least 4 months; whereas by 18 weeks control GPC-engrafted mice exhibited dense callosal and capsular myelination, confluent regions of MBP-defined myelination were only just arising in the mHTT-engrafted brains (Figures 3E–F). Accordingly, the fractions of human donor cells that differentiated as transferrin+ oligodendrocytes (Figures 3H–I) and their derivatives, MBP+ myelinating oligodendrocytes (Figures 3J–K), was significantly higher in mice engrafted with GENEA19 control GPCs than with GENEA20 mHTT GPCs. Similarly, myelin luminance, as assessed on MBP-immunostained sections, was significantly higher at both time-points in control GPC-engrafted corpus callosa than in their mHTT GPC-engrafted counterparts (Figures 3L). Nonetheless, neither the density nor distribution of engraftment by human GPCs differed significantly between control and HD-derived cells (Figures 3G, 3M–N), indicating that the myelination defect in HD hGPC-engrafted brains was due to an mHTT-associated impediment in donor cell oligodendrocytic differentiation and myelin production, rather than in differential engraftment.
Figure 3. Myelination was impaired in mice chimerized with mHTT-expressing human GPCs.

Human glial chimeric mice were established by neonatal injection of hGPCs into shiverer x rag2 hosts, which were sacrificed at 8, 13 and 18 weeks. A-B Whereas myelin basic protein (MBP) expression by control hGPCs (GENEA19) was evident by 8 weeks after neonatal graft (A), mice engrafted with HD-derived, mHTT-expressing hGPCs (GENEA20) manifested little or no MBP immunolabeling by that point (B). C-D, By 13 weeks, by which time mice engrafted with control GPCs exhibited robust myelin production (C), only scattered islands of MBP expression were noted in matched recipients of HD-derived GPCs (D). E-F, Control GPC-derived myelination was increasingly robust by 18 weeks (E), relative to mHTT GPC chimeric mice (F). G-I, The density of engrafted human GPCs did not differ between control and mHTT GPCs at any timepoint assessed (G), but the fraction of these GPCs that differentiated as transferrin (TF)+ oligodendrocytes was significantly lower among mHTT-expressing GPCs (H), resulting in fewer TF-defined oligodendrocytes in chimeras engrafted with mHTT GPCs (I). J-L, Among donor-derived oligodendrocytes, the proportion that became myelinogenic, as defined by MBP co-expression of human TF and MBP, was significantly lower in mHTT- than control GPC-engrafted chimeric brains (J). Similarly, the fraction of all donor cells that developed MBP expression was significantly higher in mice engrafted with control compared to HD-derived GPCs (K). Accordingly, myelin luminance, as assessed on MBP-immunostained sections, was significantly higher in control-engrafted corpus callosa than in corresponding mHTT GPC-engrafted white matter (L). Neither the density (G) nor the distribution of engrafted human GPCs (M-N, dot maps) differed significantly between control and HD-derived GPCs, indicating that the myelination defect in mHTT GPC-engrafted brains was due to impaired oligodendroglial differentiation and myelinogenesis, rather than to differential engraftment.
Scale: 50 μm. Values are represented as mean ± SEM. **p<0.01 and ***p<0.001 by two-way ANOVA with Bonferroni post-hoc tests.
The mHTT-associated delay in myelination had significant consequences in the rate and efficiency of axonal myelination. When callosal myelination was analyzed by high resolution confocal imaging of individual callosal axons, it was evident that axonal ensheathment was impaired in mHTT hGPC-engrafted brains (Figures 4A–F). At both the 13- and 18-week time-points, the mHTT hGPC chimeric brains exhibited fewer myelinated axons (Figure 4G); a greater proportion of those axons that myelinated did so incompletely along the length of visualized axons, while fewer axons were ensheathed per MBP+ human oligodendrocyte identified (Figure 4H). Together, these data indicate that shiverer mice rendered chimeric for mHTT-expressing hGPCs failed to myelinate as quickly or as well as those engrafted with normal hESC hGPCs, yielding relatively hypomyelinated animals with deficient axonal ensheathment. Thus, the mHtt-associated differentiation block suggested by the expression profiles of mHTT hGPCs appears to be reflected by their relative deficiency in oligodendrocytic differentiation competence, leading to hypomyelination in vivo.
Figure 4. mHTT GPC-engrafted brains exhibited diminished and delayed axonal myelination.

A-F, Confocal images of hGPC-engrafted shiverer corpus callosum show the greater MBP expression and higher proportion of ensheathed axons in mice engrafted with GENEA19 control hGPCs (A-C), compared to mice engrafted with GENEA20-derived mHTT-expressing hGPCs (D-F). A’ and B’, Confocal z-stacks with orthogonal views of donor-derived MBP+ oligodendrocytes. C’, Higher magnification of C, showing MBP immunoreactivity surrounding ensheathed axons. G-H, The proportion of MBP-ensheathed NF+ host axons overall (G), and per MBP+ donor-derived oligodendrocyte (H).
Scale: A-F, 20 μm; A’,B’,C’, 5 μm. Values represented as mean ± SEM. **p<0.01 and ***p<0.001 by 2-way ANOVA with Bonferroni post-hoc tests.
Myelin gene expression and myelinogenesis in vivo could be rescued by SOX10 and MYRF
In light of the primacy of SOX10 and MYRF in regulating myelin synthesis (Bujalka et al., 2013; Emery et al., 2009; Lopez-Anido et al., 2015), and their role as terminal effectors of myelin gene expression, our data suggested that the transcriptional activation of SOX10 and MYRF might be sufficient to rescue the myelination defect of HD. On that basis, we next asked whether the forced expression of SOX10 and MYRF in mHTT-expressing hGPCs rescued the expression of MAG, MBP and other critical genes involved in myelin biosynthesis. To this end, we induced expression of SOX10 and MYRF in both mHTT and normal control hESC-derived hGPCs (GENEA20 and GENEA19 respectively), via plasmid transfection using a bicistronic plasmid in which both genes were placed under the control of the constitutive EF1α promoter. We then compared their expression of downstream myelinogenic genes, including MAG, MBP, MOG, PDGFRA, PLP1, TF, and LINGO1 in SOX10-MYRF and control plasmid-transfected cells, using qPCR. We found that overexpression of SOX10-MYRF indeed rescued the expression of most myelin-associated genes in the transfected mHTT hGPCs (Table S1; also Figure S6).
On the basis of these data, we next asked whether SOX10 and MYRF over-expression were sufficient to rescue downstream oligodendrocyte differentiation and myelinogenesis. To this end, we developed a doxycycline-regulated dual vector lentiviral transduction strategy, that allowed the DOX-triggered, interdependent over-expression of SOX10 and MYRF, with concurrent expression of CD4 to permit FACS-based immunoisolation of SOX10-MYRF-transduced hGPCs (see Figure 5A). We first assessed the effects of SOX10 and MYRF over-expression in mHTT-expressing hGPCs, by transducing matched sets of 180 DIV Genea20-derived hGPCs with doxycycline-regulated lentiviral SOX10/MYRF, and then exposing some cultures to doxycycline, while leaving matched control cultures untreated. We confirmed that in cells raised in the absence of DOX, SOX10 and MYRF expression were no different than that of untransduced Genea20-derived hGPCs. After an additional week in vitro, the cells were then immunostained for the oligodendrocytic sulfatide recognized by O4, which is expressed by lineage-restricted, largely post-mitotic human oligodendrocytes. Without DOX, the mHTT GPCs were maintained as such, and expressed no detectable O4. In contrast, those mHTT GPCs raised in DOX, with up-regulated SOX10 and MYRF expression, exhibited a sharp and significant increment in oligodendrocyte differentiation, with >15% expressing O4 immunoreactivity (Figures 5B–D).
Figure 5. SOX10 and MYRF rescued oligodendrocyte differentiation and myelinogenesis by mHTT GPCs.

A, A doxycycline-regulated dual vector lentiviral (LV) transduction strategy allowed the DOX-triggered, inter-dependent over-expression of SOX10 and MYRF, with concurrent expression of CD4 to permit FACS-based immunoisolation of SOX10-MYRF-transduced hGPCs. B-D. The effects of SOX10 and MYRF over-expression in mHTT-expressing hGPCs were assessed by transducing matched sets of 180 DIV Genea20-derived hGPCs with doxycycline-regulated lentiviral SOX10/MYRF, and exposing some cultures to doxycycline, while leaving matched control cultures untreated. After an additional week in vitro, the cells were immunostained using mAb O4, which recognizes oligodendrocytic sulfatide. B, Without DOX, the mHTT GPCs were stably maintained and expressed no detectable O4. C, In contrast, those mHTT GPCs raised in DOX, with up-regulated SOX10 and MYRF expression, exhibited a sharp and significant increment in oligodendrocyte differentiation (D).
E, This schematic outlines the experimental design used to assess the in vivo myelinogenic competence of HD-derived hGPCs, with and without rescue of SOX10 and MYRF expression. All cells were exposed transiently to doxycycline in vitro, so as to initiate CD4 expression and permit FACS isolation before transplant into neonatal immunodeficient shiverer mice. When nine weeks of age, the engrafted mice were either given doxycycline for another 4 weeks to initiate SOX10 and MYRF expression (+DOX), ornot so treated (-DOX, controls). F, Shiverer mice engrafted neonatally with hGPCs derived from normal HTT-expressing hESCs (Genea 19) developed abundant myelin basic protein (MBP) expression and oligodendrocytic morphologies by 13 weeks in vivo. G, In contrast, mice engrafted with mHTT-expressing hGPCs produced from HD hESCs (Genea 20, or G20) developed little detectable MBP by that point. H-I, At 9 weeks of age, some Genea 20 mHTT hGPC-engrafted mice were given oral doxycycline to trigger SOX10 and MYRF expression, while matched controls were not given dox. H, The DOX(+) mice, in whose donor-derived hGPCs SOX10 and MYRF were induced, exhibited significant numbers of MBP+ myelinating oligodendrocytes in the engrafted white matter. I-J, By that same time point, no donor cells in the DOX(−) control mice had developed MBP expression, despite analogous donor cell engraftment (K). In the DOX(+) mice engrafted with SOX10/MYRF-transduced Genea20 GPCs, the donor-derived oligodendrocytes induced the robust formation of nodes of Ranvier, evidenced by the clustering of ßIV-spectrin flanked by Caspr protein that typifies nodal architecture, and which is otherwise absent in untreated shiverer brain (Figures 5L–M).
Scale: B-C, F-I, 50 μm. L, 1 μm; M, 0.5 μm. Means ± SEM. ***p<0.001, t-tests.
Since the induction of SOX10 and MYRF expression appeared sufficient to rescue oligodendrocyte differentiation from mHTT hGPCs in vitro, we next asked if SOX10 and MYRF expression were similarly sufficient to rescue myelinogenesis in vivo. To this end, we transduced Genea 20-derived HD hGPCs with doxycycline-regulated lentiviral SOX10/MYRF as above, using our vector system by which concurrent SOX10 and MYRF expression were reported by CD4 expression, sorted the cells on CD4, and then transplanted the SOX10/MYRF-transduced mHTT hGPCs into neonatal shiverer mice. At 9 weeks of age, some of the transplanted mice were given doxycycline (orally, introduced into their water ad lib) so as to trigger SOX10 and MYRF expression, while others were not given dox, thereby serving as matched controls (Figure 5E). At 13 weeks of age - a time point by which normal hGPCs typically initiate myelination, while untreated mHTT hGPCs have not yet done so (Figures 5F–G) - the mice were all sacrificed and their brains sectioned and immunostained for myelin basic protein (MBP). We found that the DOX(+) mice, in whose donor-derived hGPCs SOX10 and MYRF were induced, exhibited significant numbers of MBP+ myelinating oligodendrocytes in the host’s engrafted white matter. Quantitatively, the DOX(+) mice engrafted with SOX10/MYRF-transduced, DOX-regulated Genea20- GPCs exhibited robust myelinogenesis: 28.6 ± 0.8% (n=3 mice; mean ± SEM) of donor cells expressed MBP by 13 weeks, while no donor cells in identically-engrafted DOX(−) mice (n=6 mice) developed detectable MBP expression (p<0.0001). By way of comparison, 18.1 ± 2.1% (n=5) of normal Genea 19-derived GPCs developed MBP expression by that same time point, indicating that the SOX10/MYRF-transduced HD GPCs were at least as efficient as normal GPCs in MBP-defined myelinogenesis in vivo.
In the DOX(+) mice engrafted with SOX10/MYRF-transduced Genea20 GPCs, the resultant oligodendrocytes proved sufficient to induce the formation of nodes of Ranvier by resident shiverer axons, which exhibited the typical clustering of ßIV-spectrin flanked by Caspr that characterizes nodal architecture (Figures 5L–M). In contrast, by that same time point, no donor cells whatsoever in the DOX(−) control mice had yet developed MBP expression (Figures 5H–I, 5J), nor were clearly-defined nodes observed, despite analogous donor cell engraftment (Figure 5K). These data indicated that the forced expression of SOX10 and MYRF was sufficient to rescue both oligodendrocyte differentiation and myelination by mHTT-expressing glial progenitor cells.
mHTT impairs human astroglial differentiation in vivo
Since hGPCs give rise to astrocytes as well as oligodendrocytes, the mHTT-associated defect in oligodendroglial lineage progression, along with our RNA expression data indicating a transcriptional impediment to glial differentiation upstream of the astrocyte-oligodendrocyte fate choice, together suggested an analogous impediment to astrocytic differentiation. On that basis, we next asked if mice neonatally injected with mHTT-expressing hGPCs (GENEA20-derived) exhibited any differences in astrocytic differentiation in vivo, relative to mice injected with normal HTT sibling control hGPCs (GENEA19). To that end, we used the same mice examined earlier for the effect of HD genotype on myelinogenesis, to assess its effect on the maturation of glial fibrillary acidic protein (GFAP)-defined white matter astrocytes. We immunostained the control and HD hGPC-engrafted shiverer brains at 8, 13 and 18 weeks after neonatal graft, using a species-specific anti-human GFAP antibody.
We found that astrocytic maturation from engrafted hGPCs was markedly deficient in the HD (GENEA20) hGPC-engrafted brains assessed (n=12 total, across the 3 time-points), relative to their control (GENEA19) hGPC-engrafted counterparts (n=10). Focusing on the most rapidly and heavily engrafted white matter compartments of the corpus callosum and internal capsules, we found that GFAP-defined astrocytic differentiation by HD hGPCs was significantly diminished relative to that of control GPCs, and remained so through the 18-week observation point (Figures 6A–F). To validate this observation quantitatively, we scored those brains sacrificed at both 13 weeks and 18 weeks. At 13 weeks, the control hGPC-engrafted mice showed appreciable GFAP+ astrocytic maturation, such that 5.9 ± 0.5% of human donor cells in the corpus callosum expressed GFAP (n=4 mice; included 170 GFAP+/2669 total scored donor cells); in contrast, only 3.3 ± 0.3% of human cells were GFAP+ in mHTT GPC-engrafted callosa (n=5 mice; 60 GFAP+/2153 scored donor cells) (p=0.026) (Figure 6I). By 18 weeks, the mHTT-dependent suppression of astrocytic maturation remained pronounced; by that point, 8.5 ± 1.0% of control-derived cells had developed a GFAP+ astrocytic phenotype (n=3 mice; 209 GFAP+/2452 scored donor cells), while only 4.9 ± 0.8% of mHTT-expressing human donor cells did so (n=4 mice; 147 GFAP+/3522 scored donor cells) (p<0.005) (Figure 6I). Together, these data indicate that astrocytic differentiation by mHTT-expressing hESC GPCs is significantly delayed relative to normal hESC GPCs (F=16.31 [1,16 d.f.], 2 way-ANOVA; p=0.0009 overall). As a result, one might expect that the developmental circuit integration as well as the adult function of astrocytes might be impaired in HD.
Figure 6. Astrocytic differentiation is delayed in mHTT glial progenitor cells.

Astrocytic differentiation was significantly delayed in mHTT glial chimeras. Mice neonatally transplanted with normal HTT GENEA19-derived GPCs began to develop significant donor-derived GFAP+ astrocytes by 8 weeks (A), robustly so by 13 weeks (B), with dense astrocytic colonization of the callosal white matter by 18 weeks (C). In contrast, mHTT-expressing hGPCs derived from GENEA20 sibling hESCs developed astrocytic phenotype more slowly, with little evident GFAP expression at 8 and 13 weeks (D and E), and only modest GFAP+ astrocytic maturation at 18 weeks (F). G-H, The mature astrocytic morphologies of mHTT-expressing and control astrocytes differed, in that mHTT astrocytes typically failed to manifest the degree of radial symmetry of their control-derived counterparts. I, The proportion of GFAP-expressing cells among all donor cells was consistently lower in mHTT hGPC-engrafted than control-engrafted mice. J-K, Sholl analysis of cells traced in NeuroLucida in 3D and shown flattened (O-P), revealed that normal donor astrocytes exhibited greater fiber complexity (J), more primary processes (K), yet shorter average and maximal fiber lengths (L-M), than mHTT-expressing astroglia. N-P, Fan-in radial analysis of volume occupancy (Dang et al., 2014) revealed that mHTT astrocytes had significantly more regions unoccupied by glial processes than did control astrocytes (N; illustrations in O and P), indicating their discontiguous domain structure.
Means ± SEM. *p<0.05; **p<0.01; ***p<0.001 by: I, 2-way ANOVA with Bonferroni’s post-hoc tests; J, comparison of non-linear regressions, p<0.0001; K-N, unpaired t-tests comparing per-mouse average values across all cells scored (n=4 control, 7 mHTT mice). Scale: A-F, 50 μm; G-H, O-P, 25 μm.
mHTT GPC white matter astrocytes developed abnormal fiber distributions and domains
In light of the diminished and delayed astrocytic differentiation noted in the mHTT hGPC-engrafted mice, we next asked whether the morphologies developed by those HD astrocytes that did mature were normal, or whether their mature architectures ultimately differed from those of their more rapidly-developing control GPC-derived counterparts. Gross assessment revealed that the mature astrocytic morphologies of mHTT-expressing and control astrocytes differed, in that the mHTT-expressing, HD-derived astrocytes typically failed to manifest the degree of radial symmetry of their control-derived counterparts (Figures 6G–H). To investigate this observation, we used Sholl analysis to assess the complexity of individual astroglial morphologies; Sholl analysis is based on the number of intersections of cellular processes with concentric circles placed at sequentially more distant radii (Sholl, 1953). By imaging anti-human GFAP-immunostained cells in z-stacks of 150 μm sections and reconstructing these in Neurolucida (MBF Biosciences), we compared the fiber architectures of donor-derived astrocytes in the white matter of mice engrafted with two different lines of mHTT hESC hGPCs (GENEA18 and 20), to those engrafted with hGPCs derived from two control hESC lines (C27 iPSCs and GENEA19 hESCs, the latter sibling to GENEA20). Sholl analysis revealed that the fiber complexity of the mHTT-expressing astrocytes was substantially diminished relative to astrocytes derived from their sibling control GPCs (Figure S7). This effect was particularly evident in the comparison of mHTT astrocytes derived from GENEA20 hESCs to normal astrocytes derived from their matched GENEA19 siblings (Figures 6J–P). The human astrocytes in the mHTT GPC-engrafted chimeras differed significantly from those in the normal GPC-engrafted mice, with less fiber network complexity (Figure 6J), characterized by fewer yet longer processes (Figures 6K–M). When the 3-dimensional Neurolucida tracings (Figures 6O–P) were additionally assessed by Fan-in radial analysis to assess the extent to which the fiber domain of each cell occupied its immediate volumetric environment (Dang et al., 2014), we found that mHTT astrocytes exhibited significantly more regions that were unoccupied by glial processes than did control-derived astrocytes (Figures 6N–6P), indicative of a discontiguous and incomplete domain structure.
To better undersatnd the transcriptional concomitants to these HD-asociated morphological abnormalities in astrocyte morphology, we next assessed the gene expression patterns of HD vs. control-derived astrocytes. To do so, we generated CD140a-defined GPCs as per our standard protocol, and then directed these towards astrocytic differentiation by transitioning the cells to serum-containing media supplemented with 20 ng/ml BMP4. The cells were then sorted on the basis of CD44, which among brain cells is differentially expressed by astrocytes and their committed precursor cells (Cai et al., 2012; Liu et al., 2004). RNAseq was then performed on the extracted RNA of HD and control-derived CD44-defined astrocytes, which were confirmedd as such by their virtually uniform expression of GFAP. This analysis revealed significant differences in gene expression by mHTT-expressing astroglia, relative to control-derived CD44+ astroglia (Figures 7A–C). Network analysis revealed the differential expression of four discrete modules, which included functional ontologies referable to: 1) synaptic, post-synaptic and receptor-associated genes; 2) endosomal transcripts; 3) desmosomal and cell-cell junction genes; and 4) extracellular matrix components (Figures 7D and 7E–H). Of these, the largest set of differentially-expressed genes were those referable to synaptic and receptor modulation; these included a number of genes that regulate fiber outgrowth and motility, including MYL7 and MYKL2, the myosin light chain-7 and myosin light chain kinase-2, which were both sharply down-regulated in mHTT-expressing astrocytes relative to controls (Figure 7E). Importantly, the glial myosins and their kinases are involved not only in glial fiber elaboration, but also in astroglial calcium signaling (Cotrina et al., 1998). Their deficient expression in HD astroglia might then contribute to the abnormal morphological development of HD astrocytes (Khakh et al., 2017; Octeau et al., 2018), while predicting aberrant signaling within the glial syncytium of the HD brain (Jiang et al., 2016). Together, these data serve to emphasize that HD is associated with deficient astrocytic differentiation and functional development, as well as with impaired oligodendrocytic maturation and myelination.
Figure 7. HD hESC-derived CD44+ astroglia exhibit mHTT-dependent changes in gene expression.

A, Principal component analysis performed as in Figure 1 but using CD44-sorted astroglia and their precursors validates the segregated expression signatures of HD-derived and normal cells. B, The Venn diagram highlights the intersection of lists of differentially expressed genes (DEGs) (green, down-regulated; red, up-regulated; FDR 5%), obtained by comparing astroglia derived from 3 HD patients against pooled control cells, using the same cell lines and analytic pipeline as in Figure 1. The list of DEGs shared by the 3 HD patients was filtered by those genes differentially expressed by patient HD20 (GENEA20) relative to its sibling donor CTR19 (GENEA19). C, The heatmap based on log2-transformed TPM values calculated from raw counts of the 114 DEGs highlighted in B, shows clustering by disease status. D, Network representation of functional annotations (Gene Ontology: Cellular Component, FDR-corrected p<0.1) for the 114 intersection DEGs highlighted in B. Genes are designated as round nodes (green, down-regulated; red, up-regulated); rounded rectangular nodes represent annotation terms. Nodes are sized by degree and grouped as interconnected modules (M1 though M4) identified by community detection. For each colored module, three of the top significant annotations are listed and labeled in the network. E, Expression heatmap of 14 conserved DEGs identified in M1 (yellow in D), annotations related to post-synaptic and receptor complex components. F, Heatmap of 9 conserved DEGs identified in M2 (grey in D), annotated to perinuclear and early endosome components. G, Heatmap of 11 conserved DEGs identified in M3 (blue in D), annotations related to plasma membrane, cell-cell junction, and desmosomal components. H, Heatmap of 8 DEGs identified in M4 (orange in D); annotations related to extracellular matrix components.
DISCUSSION
These experiments suggest that white matter failure in Huntington’s disease is a product of an mHTT-dependent block in differentiation by affected glial progenitor cells, such that mRNAs encoding a group of critical glial lineage transcription factors are differentially down-regulated in mHTT-expressing GPCs. The mHTT-associated inhibition of oligodendroglial differentiation in particular, as manifested by the down-regulated expression of NKX2.2, OLIG2 and SOX10, is accompanied by the diminished expression of the SOX10-regulated myelin regulatory factor, MYRF. This results in the suppression of myelination, which requires the MYRF-dependent transcription of critical mRNAs associated with myelin biogenesis, such as MAG and MBP (Bujalka et al., 2013; Emery et al., 2009). Interestingly, the down-regulation of MYRF has been similarly noted in the mature oligodendrocytes of HD transgenic mice expressing especially long CAG repeats (150Q and 250Q) (Jin et al., 2015). Our data reveal that in humans, the mHTT-associated block in glial differentiation occurs at an earlier stage than previously appreciated, and is apparent in bipotential GPCs that generate astrocytes as well as oligodendrocytes. As such, we found that mutant HTT significantly impedes the development of both glial lineages in HD, and importantly, that this developmental arrest occurs in human GPCs expressing CAG repeat expansion lengths of 40–48Q, which typify human HD.
These expression data, implicating the mHTT-dependent suppression of NKX2.2, OLIG2 and SOX10 in the white matter deficiency of HD, suggested that efforts to over-express or otherwise activate the transcription of SOX10 and MYRF might be sufficient to relieve the myelination defect of this disease. We found this to be the case, in that forced expression of SOX10 and MYRF in mHTT-expressing hGPCs rescued the expression of critical genes involved in myelin biogenesis, and restored myelination by HD-derived glia in vivo. As such, the targeted activation or upregulation of SOX10 and MYRF might serve as a means of restoring the myelination competence of mHTT-expressing oligodendrocytes in HD.
Besides the defects in oligodendrocyte maturation and myelination associated with mHTT, we noted that astrocytic differentiation was also impaired, as might have been expected given the dysregulation of glial transcription as early as the NKX2.2 and OLIG2 stages, proximal to the astrocyte-oligodendrocyte fate choice. Such defective astrocytic maturation of HD hGPCs suggests that the Huntington disease phenotype might have a significant developmental component, in that any delay in astrocytic differentiation by mHTT-expressing GPCs might impair developmental synaptogenesis and circuit formation, each of which depend upon astrocytic guidance (Clarke and Barres, 2013; Ullian et al., 2001). In addition, any such disease-dependent delay in astrocytic maturation might be expected to contribute to the delayed - and ultimately deficient - myelination of HD, given the metabolic dependence of oligodendrocytes upon local astrocytes (Amaral et al., 2013). It remains to be seen whether the rescue of astrocytic maturation by HD-derived GPCs might relieve these effects on synaptic development and organization; if so, we may predict that astrocytic replacement might be sufficient to rescue the synaptic pathology of HD, in the manner that the rescue of oligodendrocytic differentiation appears sufficient to relieve the myelination defect of HD.
Besides their contributions to neural network formation and synaptic architecture, both GPCs and astrocytes are intimately involved with maintaining adult interstitial ion homeostasis, and with the regulation of neuronal excitability. We were thus intrigued to note that the arrested terminal differentiation of mHTT-expressing hGPCs was associated with the widespread suppression of several families of glial potassium channels. These included the inwardly rectifying K+ channels of the KCNJ family, including KCNJ8 and KCNJ9 among others. This mHTT-associated suppression of inwardly rectifying K+ channels, which are responsible for potassium import into cells, might contribute to the hyper-excitability of HD neurons, by inhibiting the glial reuptake of synaptically-released K+ (Shin et al., 2005). In that regard, Khakh and colleagues have reported a deficit in astrocytic expression of the inwardly-rectifying channel Kir4.1 (KCNJ10) in mouse models of HD (Tong et al., 2014), which might similarly reflect the effect of disrupted glial maturation on potassium channel expression and glial K+ uptake. In the mHTT-expressing human GPC, arrested at a stage before terminal astrocytic maturation, it appears that a large set of K+ channel transcripts are coordinately suppressed, suggesting the inhibition of a shared upstream activator of K+ channel gene expression. While the upstream regulators of these potassium channel genes have not yet been identified, it is reasonable to posit that the mHTT-dependent suppression of terminal glial differentiation might lead to a failure in the development of glial potassium homeostatic mechanisms that would otherwise regulate and protect neuronal activity.
Together, these observations suggest that any disruption in astrocytic maturation by HD hGPCs might be expected to significantly influence both the development and adult performance of neural networks in HD. Importantly, a corollary of these findings is that the replacement of mHTT-expressing GPCs by their wild-type or genetically-corrected counterparts might be sufficient to restore functional astrocytes and oligodendroglia to affected HD brain. This possibility was first suggested by the ability of neonatally-delivered wild-type hGPCs to out-compete diseased hGPCs in models of congenital hypomyelination (Windrem et al., 2008), and we have similarly noted that neonatal glial replacement is sufficient to correct deficient potassium homeostasis in HD transgenic mice as well (Benraiss et al., 2016). Whether such competitive dominance of healthy over diseased cells may occur in adult HD remains to be established, but should this prove feasible, such a strategy of glial replacement might prove a realistic therapeutic avenue for disease amelioration in HD.
STAR METHODS AND KEY RESOURCES
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to, and will be fulfilled by, the lead and corresponding author, Steve Goldman (steven_goldman@urmc.rochester.edu or goldman@sund.ku.dk).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Production of glial progenitor cells (GPCs) from human embryonic stem cells (hESCs)
Glial progenitor cells were generated from human embryonic stem cells (ESCs) using our previously described protocol (Wang et al., 2013; Windrem et al., 2017), which is outlined in great methodological detail in the supplemental experimental procedures of Wang et al., 2013. Cells were harvested between 160–240, by which time the majority typically expressed the bipotential glial progenitor cell marker CD140a, while the remainder were composed predominantly of A2B5+/CD140a− immature astrocytes. No SSEA4 expressing cells were detectable. Human ES cells were obtained from GENEA, Inc. (Sydney, Australia), as lines GENEA02 and 19 (normal HTT: 15/18 CAG) and GENEA 17, 18 and 20 (mutant HTT: 40/12, 46/17 and 48/17 CAG, respectively) (Bradley et al., 2011). GENEA02 and 17 are male, and GENEA18, 19, and 20 are female. Of note, GENEA 19 and 20 were donated and derived as a pair of female siblings, one normal and one with HD. The C27 control line is male.
Hosts
Homozygous shiverer mice (The Jackson Laboratory, Bar Harbor, ME) were crossed with homozygous rag2-null immunodeficient mice (Shinkai et al., 1992) on the C3h background (Taconic, Germantown, NY, USA) to generate shi/shi x rag2−/− myelin-deficient, immunodeficient mice (Windrem et al., 2008). Mice were maintained in a temperature and humidity-controlled housing (64–79°F; 30–70% humidity), in a pathogen-free colony room on a 12:12 hr light cycle. They were fed ad lib Mod LabDiet 5P00 with 0.025% trimethoprim/0.124%sulfamethoxyzole (www.testdiet.com) and autoclaved acid water (pH 2.5–3.0).
Suspensions of single-cells or small clusters of hESC-derived GPCs were spun down to 100,000 cells/μl. Neonates were anesthetized by cooling, and transplanted bilaterally in the corpus callosum with a total of 200,000 cells, as described (Windrem et al., 2004); see Method Details section for transplant procedure. At 8, 12–13 or 18 weeks of age, the transplanted mice were anesthetized with pentobarbital, then perfusion fixed with cold HBSS followed by 4% paraformaldehyde. Brains were removed and post-fixed for 2 hrs in cold paraformaldehyde.
All procedures were approved by the University of Rochester’s Committee on Animal Resources (UCAR), under protocol 2004–129.
METHOD DETAILS
Cell preparation for transplantation
Prior to injection, flow cytometry was performed to confirm CD140a predominance in each culture. The suspended cell clusters were then collected from the well, spun down, and resuspended in a small volume of Ca2+/Mg2+-free HBSS. The resuspended clusters were transferred to a 100 mm cell culture dish, then cut with a no. 11 surgical scalpel to obtain pieces 100–200 μm in diameter. These fragments were then collected, spun down, washed with Ca2+/Mg2+-free HBSS, and resuspended to an approximate concentration of 105 cells/μl in Ca2+/Mg2+-free HBSS.
Transplantation
Shiverer x Rag2-null neonatal mice were transplanted on postnatal day 1 or 2. Half of the litter was removed from the dam and placed in a humidified warming chamber. For this we used a sterilized plastic box, lined with sterile gauze dampened with Hanks balanced salt solution, and warmed on a heating block. The pups to be injected were then wiped with Povidone-Iodine and wrapped in sterile gauze to prevent direct contact with ice, then cryo-anesthetized for 2 to 6 minutes, depending on size. The pups were then removed from ice and cleaned with an alcohol prep pad, then laid in a customized neonatal mouse holder made of baked molded clay. The pups were injected directly through the skin and skull osteoid into both the rostral (AP + 1.0 mm; ML ± 1.0 mm, ventral 1.0 mm) and caudal (AP −1.0, ML ± 1.0 mm, ventral 0.9 mm) corpus callosum. Following injections, pups were cleaned with alcohol prep pads and returned to the warming chamber for recovery. Upon recovery, the first half of the litter was returned to the dam, and the second half put in the humidified chamber. Pups were weaned between 21 and 28 days, then group housed.
Immunolabeling of tissue sections
Brains were cryopreserved, embedded in OCT (Tissue-Tek OCT, Sakura Finetek, Torrance, CA) and sectioned at 20 μm, either sagittally or coronally, on a cryostat. Human cells were identified with mouse anti-human nuclei, clone 235–1 at 1:800 (MAB1281; EMD Millipore, Billerica, MA). Oligodendrocytes were labeled with myelin basic protein with rat anti-MBP at 1:25 (Ab7349; Abcam, Cambridge, MA), astrocytes with anti-human-specific GFAP (SMI 21 at 1:1000, Covance, Princeton, NJ), and axons with mouse anti-neurofilament at 1:5000 (SMI-311) or 1:1000 (SMI-312; Covance, Princeton, NJ). Alexa Fluor secondary antibodies, goat anti-mouse and anti-rat 488, 568, 594, and 647 were used at 1:400 (Life Technologies, Carlsbad, CA).
Antibodies and dilutions used
See Resource Table.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse monoclonal anti-human nuclei, 1:800 | Millipore | Cat #MAB1281; RRID:AB_94090 |
| Mouse monoclonal anti-human nuclei, conjugated with Alexa 488. 1:200 | Millipore | Cat #MAB1281A4 |
| Rat monoclonal anti-myelin basic protein, 1:25 | Abcam | Cat #ab7349; RRID: AB_305869 |
| Rabbit polyclonal anti-transferrin, 1:800 | Abcam | Cat #ab9538; RRID: AB_307325 |
| Mouse monoclonal anti-human GFAP, SMI 21R, 1:600 | Covance Research Products Inc | Cat #SMI-21R-500; RRID: AB_509979 |
| Rabbit polyclonal anti-olig2, 1:500 | Neuromics | Cat #RA25017, 25081 |
| Mouse anti-neurofilament, smi-311, 1:5000 | Covance | Cat #SMI-311R-100, RRID:AB_509991 |
| Mouse anti-neurofilament, smi-312 1:1000 | Covance | Cat #SMI-312R-100, RRID:AB_509993) |
| Goat anti-mouse IgG (H+L) Alexa Fluor 647, 1:400 | ThermoFisher Scientific | Cat #A-21235; RRID: AB_2535804 |
| Goat anti-mouse IgG1 Alexa Fluor 568, 1:400 | ThermoFisher Scientific | Cat #A-21124; RRID: AB_2535766 |
| Goat anti-mouse IgG1 Alexa Fluor 488, 1:400 | ThermoFisher Scientific | Cat #A-21121; RRID: AB_2535764 |
| Goat anti-Rabbit IgG (H+L) Alexa Fluor 568, 1:400 | ThermoFisher Scientific | Cat #A-11036; RRID: AB_2534094 |
| Goat anti-Rabbit IgG (H+L) Alexa Fluor 488, 1:400 | ThermoFisher Scientific | Cat #A-11034; RRID: AB_2576217 |
| Goat anti-Rat IgG (H+L) Alexa Fluor 568 | ThermoFisher Scientific | Cat #A-11077; RRID: AB_2534121 |
| Goat anti-Rat IgG (H+L) Alexa Fluor 488 | ThermoFisher Scientific | Cat #A-11006; RRID: AB_2534074 |
| Alexa Fluor 488-SSEA4 | Invitrogen | Cat #A14810 |
| APC-conjugated mouse IgG1, Isotype Control | Miltenyi Biotec | Cat #130-092-214 |
| APC-mouse IgM, Isotype Control | Miltenyi Biotec | Cat #130-093-176 |
| APC-conjugated mAb A2B5 | Miltenyi Biotec | Cat #130-093-582 |
| APC-conjugated anti-CD44 | Miltenyi Biotec | Cat #130-095-177 |
| APC-conjugated anti-CD133/1 | Miltenyi Biotec | Cat #130-090-826 |
| PE-conjugated anti-CD140a | BD Pharmingen | Cat #556002 |
| Anti-olig2 | R&D Systems | Cat #AF2418 |
| PE-conjugated mouse IgG2a, Isotype Control | BD Pharmingen | Cat #555574 |
| Anti-PDGFRα | Cell Signaling Tech. | Cat #5241S |
| Chemicals, Peptides, and Recombinant Proteins | ||
| bFGF | Sigma | Cat #F0291 |
| Biotin | Sigma | Cat #B4639 |
| dibutyryl cAMP | Sigma | Cat #D0260 |
| Heparin | Fisher | Cat #NC9484621 |
| IGF-1 | R&D Systems | Cat #291-G1-050 |
| Laminin | Corning | Cat #354232 |
| NT3 | R&D Systems | Cat #267-N3-025 |
| PDGFaa | R&D Systems | Cat #221-AA-50 |
| Purmorphamine | Calbiochem | Cat #80603-730 |
| Retinoic acid | Sigma | Cat #R2625 |
| T3 | Sigma | Cat #T5516-1MG |
| Critical Commercial Assays | ||
| Custom TaqMan Array Card | Applied Biosystems | N/A |
| Ovation PicoSL WTA System V2 | NuGEN | Cat#3312 |
| RNeasy mini kit | QIAGEN | Cat#74104 |
| Taqman Universal master mix | Applied Biosystems | Cat #4304437 |
| TruSeq RNA library Prep Kit V2 | Illumina | Cat#RS-122-2001 |
| Deposited Data | ||
| Raw RNA-seq data | GEO datasets: https://www.ncbi.nlm.nih.gov/gds/ | GEO accession number: GSE105041 |
| Processed RNA-seq data (count matrix) and R scripts for data analysis | This paper | GitHub repository: https://github.com/cbtncph/HD-Glial-Differentiation-Block-Goldman-Lab-2017 |
| Lab-based interactive differential expression database | This paper | https://ctngoldmanlab.genialis.com |
| Human reference genome NCBI build 38, GRCh38 | Genome Reference Consortium | https://www.ncbi.nlm.nih.gov/grc/human |
| Mendeley dataset, with links | This paper | doi:10.17632/5gmpkdwcz4.1 |
| Experimental Models: Cell Lines | ||
| C27 iPSCs | Dr. Lorenz Studer, SKI | N/A |
| GENEA17 hESCs | Genea Biocells | http://geneabiocells.com/services/shelf-products/human-embryonic-stem-cells/ |
| GENEA18 hESCs | Genea Biocells | http://geneabiocells.com/ |
| GENEA19 hESCs | Genea Biocells | http://geneabiocells.com/ |
| GENEA20 hESCs | Genea Biocells | http://geneabiocells.com/ |
| GENEA02 hESCs | Genea Biocells | http://geneabiocells.com/ |
| Experimental Models: Organisms/Strains | ||
| Mouse: C3Fe.SWV-Mbpshi/J | Jackson Laboratory | Cat #001428 |
| Mouse: C3H.129S6(B6)-Rag2tm1FwaN12 | Taconic | Cat #000602-M |
| Experimental Models: Housing | ||
| Temperature | 64-79F | |
| Relative Humidity Range: | 30-70% | |
| Light-cycle | 12/12 | |
| Water | Autoclaved acid water (pH 2.5–3.0) in sterile bottles | |
| Cages and bedding | irradiated | |
| Diet | Mod LabDiet® 5P00 w/0.025%Trimethoprim/0.124%Sulfameth-5TK5 | |
| Colony room | Pathogen free | |
| Bacterial and Virus Strains | ||
| NA | ||
| Software and Algorithms | ||
| Photoshop CS6 | Adobe | |
| Illustrator CS6 | Adobe | |
| StereoInvestigator v11 | MBF Bioscience | |
| Neurolucida 360 v2 | MBF Bioscience | |
| Neurolucida Explorer v11 | MBF Bioscience | |
| Leica Metamorph AF v2 | Leica Biosystems | |
| Leica Application Suite X | Leica Biosystems | |
| FlowJo | TreeStar Inc. | N/A |
| Trimmomatic (version 0.32) | Bolger et al., 2014 | http://www.usadellab.org/cms/?page=trimmomatic |
| Subread (version 1.5.1) | Liao et al., 2013 | http://subread.sourceforge.net/ |
| featureCounts (version 1.5.1) | Liao et al., 2013 | http://subread.sourceforge.net/ |
| R | R Core Team, 2016 | https://www.R-project.org/ |
| RUVSeq (version 1.6.2) | Risso et al., 2014 | http://www.bioconductor.org/packages/release/bioc/html/RUVSeq.html |
| edgeR (version 3.14.0) | Robinson et al., 2010 | http://www.bioconductor.org/packages/release/bioc/html/edgeR.html |
| DESeq2 (version 1.12.4) | Love et al., 2014 | http://www.bioconductor.org/packages/release/bioc/html/DESeq2.html |
| ToppCluster | Kaimal et al., 2010 | https://toppcluster.cchmc.org/ |
| Gephi (version 0.9.1) | Bastian et al., 2009 | https://gephi.org/ |
| Ingenuity Pathway Analysis | QIAGEN | https://www.qiagenbioinformatics.com/products/ingenuity-pathway-analysis/ |
| ExpressionSuite Software (version 1.1) | Applied Biosystems | https://www.thermofisher.com/dk/en/home/technical-resources/software-downloads/expressionsuite-software.html |
| Other | ||
| Agilent Bioanalyzer | Agilent | N/A |
| BD FACS Aria IIIU | BD Biosciences | N/A |
| HiSeq 2500 | Illumina Inc. | N/A |
| Nanodrop 1000 spectrophotometer | Nanodrop | N/A |
| Olympus IX71 Inverted Microscope | Olympus | N/A |
| QuantStudio 12K Flex Real-Time PCR | Applied Biosystems | N/A |
| Cryostat | Hacker Instruments | Model OTF |
| Cryostat | Leica Biosystems | Cat #CM3050S |
| Vibratome | Vibratome | 1000 Plus |
| Disposable microtome blades | C.L. Sturkey | Cat #DT315G50 |
| PTFE coated stainless steel blades | Ted Pella Inc | Cat #121-6 |
| Surgipath X-tra precleaned micro slides | Leica Biosystems | Cat #38002002 |
| DMi8 | Leica Biosystems | |
| DM6000B | Leica Biosystems | |
| DFC 360 FX camera | Leica Biosystems | |
| BX51 | Olympus | |
| DP30BW camera | Olympus | |
| Orca-R2 Digital CCD Camera | Hamamatsu | Cat #C10600-10B |
| MAC 5000 | Ludl Electronic Prods. | Cat #73005001 |
| Focus DR Linear encoder | Ludl Electronic Prods. | Cat #99A420 |
| STG 4”x3” Stepper | Ludl Electronic Prods. | Cat #99S100LE2MBF |
RNA-seq
hGPCs assessed for gene expression were first sorted by fluorescence-activated cell sorting on the basis of the cell surface marker CD140a (BD Pharmingen), as described (Sim et al., 2011), using a FACS Aria IIIu (Becton-Dickinson) (Figure S2). The mRNA was isolated by polyA-selection protocol from FACS-sorted PDGFRa-positive glial progenitor cell (GPC) lines produced from human embryonic stem (ES) cells derived from 3 Huntington’s Disease (HD) patients (designated to HD lines 17 [N = 5 independent cell set preparations], 18 [N = 5], and 20 [N = 6]) and 2 healthy controls (designated to CTR lines 02 [N = 6], and 19 [N = 6], sibling of HD20). Sequencing libraries were prepared with the Illumina TruSeq RNA v2 kit and sequenced on an Illumina HiSeq 2500 sequencer, yielding approximately 45 million 100-bp single-end reads per sample for all cell lines except for control line CTR02, which was sequenced to similar depth but in 125-bp paired-end read mode. The sequencing reads were then pre-processed by trimming off adapter and low-quality sequences using Trimmomatic (Bolger et al., 2014). The quality of reads before and after pre-processing was assessed with FastQC (Andrews, 2010). The pre-processed reads were then aligned to the RefSeq NCBI reference human genome version GRCh38 (Pruitt et al., 2006), with Subread read aligner (Liao et al., 2013b). Raw gene counts were obtained from BAM alignment files with featureCounts (Liao et al., 2013a).
For bioinformatic analysis, see Quantification and Statistical Analysis section below
SOX10/MYRF rescue of myelinogenic gene expression
Both mHTT and normal sibling hESC-derived hGPCs were transfected with plasmids expressing either SOX10 and MYRF in tandem, in a bicistronic vector under the regulatory control of EF1α (pTANK-EF1α-Sox10-P2A-Myrf–T2A-EGFP-WPRE), or a control plasmid expressing only EGFP (pTANK-EF1α-EGFP-WPRE). Transfection was performed using Nucleofector (Lonza, Germany), using the CA205 transfection program in P3 buffer following the manufacturer’s protocol. Cells were collected 72 hrs after transfection for RT-qPCR of potential SOX10 and MYRF target genes. RNA was extracted using the Qiagen RNeasy Micro Kit (Qiagen, Germany). The first-strand cDNA was synthetized using TaqMan Reverse Transcription Reagents (Applied Biosystems). 5 ng of RNA input was used for each reaction; these were performed using FastStart Universal SybrGreen Mastermix (Roche Diagnostics, Germany), on a real-time PCR instrument (CFX Connect Real-Time System thermocycler; Bio-Rad). Samples from G19- and G20-derived hGPCs were each assayed in triplicate for each target gene assayed (primers available in Table S2). Melting-curve analysis was performed after each PCR to confirm the specificity of the reaction, and to identify peaks of interest in all samples. Results were normalized to the expression level of 18S from the same sample.
SOX10/MYRF rescue of myelination
For this set of experiments., SOX10 and MYRF transcripts were cloned in two separate lentiviral vectors: pTANK-TRE-MYRF-CAG-rtTA3G-WPRE and pTANK-TRE-Sox10-P2A-DC4-WPRE. In this Tet-On system, the cell surface expression of the selectable marker CD4 requires the expression from both viruses, thus ensuring co-expression of the MYRF and SOX10 transgenes. Virus particles pseudotyped with vesicular stomatitis virus G glycoprotein were produced, concentrated by ultracentrifugation, and titrated on 293HEK cells. G20 hGPC cultures were infected at 1.0 MOI in glial media. Cells were washed with HBSS and maintained in glial media supplemented with 1 μg/ml doxycycline (Millipore-Sigma St. Louis, MO) for 4 days. hGPCs were then selected for membrane expression of CD4 using MACS (Miltenyi, Germany) as described (Windrem et al., 2008).
Rescue of oligodendrocytic differentiation in vitro
MACS isolated CD4+ cells were allowed to attach overnight in glial media (Wang et al., 2013); doxycycline was maintained in overexpression conditions throughout the differentiation induction. The next day, cells were washed once in HBSS and transitioned to differentiation media (Neurobasal medium (Gibco), 1x N2 (Thermo Fisher), 1x B27 (Thermo Fisher), 1x GlutaMAX (Thermo Fisher), 20 ng/ml BDNF (R&D Systems), 0.2 mM L-Ascorbic Acid (Sigma), 60 ng/ml T3 (Sigma), 0.2 mM dibutyrl cyclicAMP (Sigma), 100 ng/ml biotin (Sigma), 1x insulin-transferrin-selenium (ThermoFisher), 10 ng/ml NT3 (R&D), and 100 ng/ml IGF1 (R&D). Media were changed every other day for 2 weeks before fixation. Oligodendrocyte differentiation was quantified via O4 immunostaining.
Rescue of oligodendrocytic differentiation in vitro
Cells were prepared for transplantation and then injected into the corpus callosa of neonatal shiverer mice, at 2 sites unilaterally. Starting at 9 weeks of age, half of the transplanted mice were administered either doxycycline (2 mg/ml with 5% sucrose in water (Chow et al., 2012) or normal drinking water in their water bottles, for 5 weeks. Both control and experimental mice were then sacrificed at 13 weeks of age and prepared for immunolabeled for myelin basic protein and human nuclear antigens, then imaged by confocal to assess axonal ensheathment by MBP-expressing oligodendrocytes as described.
QUANTIFICATION AND STATISTICAL ANALYSIS
Imaging and quantitative histology
To map the distribution of human nuclei, sections were imaged on a Nikon Instruments Ni-E equipped with Nikon Digital Sight Camera DS-Fi1, and counts scored in Nikon NIS Elements v4.5. For photographing the distribution of myelin at low power, whole brain sections were imaged on a Leica LMD 6500. Higher power confocal images of myelin ensheathment were obtained using a Nikon C2+ confocal, and images were acquired with a 100x objective using 0.2 μm steps. Imaging for cell type-specific markers was performed on an Olympus BX51 using a Hammamatsu camera driven by Stereo Investigator software (MBF, Williston, VT). Higher magnification confocal stacks of astrocytes subjected to Sholl analysis were obtained using a Leica SP8 confocal (see below).
Cell counting
Quantification of donor cell density in the corpus callosum was based on counts of 1 mm lateral from midline. Randomly initiated, uniformly sampled coronal sections of the brains were labeled for human nuclei, DAPI and other phenotype-specific markers (Olig2, hGFAP, TF and MBP). For Olig2 and hGFAP quantification, the regions of interest of each section were imaged using an Olympus BX51 equipped with a Hamamatsu camera, at 40x. Z-stacks were obtained with a step size of 1µm. For TF and MBP quantification, the regions of interest were imaged using a Nikon Ni-E Eclipse microscope equipped with a DS-Fi1 camera, at 20X. Z-stacks were obtained with a step size of 0.7–1 µm. Immunolabeled cells were counted using high intensity projection of the z-stacked images on three evenly-spaced coronal sections from each mouse, in Nikon NIS Elements v.4.5.
Astrocyte morphometrics
Shiverer x rag2-null mice were sacrificed at 18 weeks of age and their white matter astrocyte morphologies assessed. 150 μm thick coronal slices were taken by Vibratome at Bregma −1.0 mm from control (GENEA19) or HD (GENEA20) hGPC-engrafted mice, incubated in mouse anti-hGFAP for 1 week at 4°C, then 4 hrs in Alexa 568 goat anti-mouse antisera. The slices were mounted on slides and imaged at 100x by confocal (Leica SP8). The images were traced using Neurolucida 360 (MicroBrightfield, Inc.); all tracings were done by experimenters blinded as to the treatment condition.
Individual astrocytes were selected from the middle of the corpus callosum at mid-depth so as to capture cells and their processes in their entirety. Cells were analyzed by Neurolucida with Sholl analysis, as 3 cells/slice and 3 slices/brain, taken at 500, 1000, and 1500 μm lateral of the midline. A total of 14 neonatally-engrafted brains (GENEA18, n=21 cells/3 brains; GENEA19, 32 cells/4 brains; GENEA20, 42 cells/7 brains) were assessed, yielding 63 traced mHTT astrocytes (GENEA18- and 20-derived), and 32 control (GENEA19) astrocytes. For Sholl analysis, concentric shells placed at successively increasing diameters of 5 μm were centered on the cell body, and the number of intersections between cell processes and shells counted (Sholl, 1953). For the assessment and quantitative description of astrocytic fiber 3D architecture, Fan-in analysis (MBF Biosciences) was used as previously described for studies of dendritic topology (Dang et al., 2014).
Myelin luminance analysis
To measure forebrain myelination, we used luminance analysis based on measurement of myelin basic protein (MBP) immunofluorescence. Evenly-spaced and uniformly sampled coronal sections were stained for MBP as described, and images taken at 10x using a Nikon Ni-E and Nikon DS-Fi1 camera. The corpus callosum was selected as region of interest, and mean intensity values were obtained using NIS Elements v.4.5.
Statistical analysis of histological data
All analyses were done with Prism® v.7 (GraphPad Software) using two-way ANOVA and post hoc Bonferroni t tests. Statistical significance was considered as P-values less than 0.05. Significances were represented as *p<0.05, **p<0.01 and ***p<0.001. Graphs and figures were made and assembled with Prism 7, and all data are shown as mean ± standard error of the mean (SEM).
Bioinformatics
After examining principal component and hierarchical clustering plots generated with native R functions (R Core Team, 2016) (https://www.R-project.org), one mis-clustered outlier sample was removed from analysis in line HD17 (GENEA17), as were 2 outliers in lines HD20 (GENEA20) and CTR19 (GENEA19). After eliminating lowly expressed transcripts leaving those with a count of at least 5 reads in more than 3 samples, the count data were normalized using RUVSeq (Risso et al., 2014). The R Bioconductor package (Gentleman et al., 2004) was used to account for variance. As described in the RUVSeq documentation, normalization was accomplished in the following three-step procedure: 1) negative in silico control genes were determined by first-pass differential expression analysis by edgeR (Robinson et al., 2010) and DESeq2 (Love et al., 2014) R Bioconductor packages, including those genes with FDR-adjusted p values >0.75, as calculated by both methods; 2) the negative in silico control genes were then used in the RUVg function of RUVSeq to calculate variance factors; and 3) the second-pass differential expression analysis (1% FDR and log2 fold-change >1) was performed to determine disease-dysregulated genes, using the original raw counts, and adjusting for RUVg-calculated variance factors by multi-factor GLM models implemented in both the edgeR and DESeq2 packages.
This three-step analysis, with filtering out low- and non-expressed transcripts, was used to compare each HD-derived hGPC cell line to the pooled CTR-derived hGPCs, as well as for the sibling pair comparison of HD20 vs. HD19. In all comparisons, one RUVg-calculated variance factor was used. The intersection of the resulting four lists of differentially expressed genes was taken as the conserved representative list of HD-dysregulated genes. To obtain average fold changes and p values for dysregulated genes in all three HD-derived GPC lines, a differential expression comparison of pooled HD to pooled CTR lines was performed by the same workflow with the same number of variance factors.
For all comparisons of differential expression, only the significant results that agreed between edgeR and DESeq2 were used in downstream analysis. Fold-changes and FDR-adjusted p values reported in the Results were calculated by edgeR. Functional annotation of the conserved set of HD-dysregulated genes was performed using ToppCluster (Kaimal et al., 2010) and Ingenuity Pathway Analysis (IPA) (Qiagen; https://www.qiagenbioinformatics.com/products/ingenuity-pathway-analysis) (Krämer et al., 2013).
Network visualization and analysis
The ToppCluster annotation tool was used for its ability to represent term to gene associations as a network (Kaimal et al., 2010). The annotation results were exported with ToppCluster’s Network Generator as a list of term to gene associations representing network edges. For all subsequent network visualizations and analyses, the term to gene association networks were imported into Gephi graph visualization software (http://gephi.org) (Jacomy et al., 2014). Basic node centrality measures and node degrees were calculated and the networks were arranged with Force Atlas layout using default parameters. Closely interconnected node modules were determined with the built-in community detection algorithm (Blondel et al., 2008), using a randomization and resolution parameter of 1.3 for CD140a-derived, and 2.0 for CD44-derived annotation networks, so as to optimize both the grouping and number of communities.
TaqMan RT-qPCR arrays for gene expression validation
Extracted total RNA was amplified using ribo-SPIA based whole transcriptome based amplification (NuGen). The expression of cell type markers and pathway-specific genes was assessed by real-time polymerase chain reaction (RT-PCR) using a 48-gene Taqman low-density array (TLDA) (Applied Biosystems). The relative abundance of transcript expression was calculated by ΔΔCt analysis, and the expression data normalized to the mean of 18S and GAPDH as endogenous controls. The difference of expression in HD and control GPCs was assessed by paired t-test followed by multiple testing correction by Benjamini-Hochberg (BH) procedure (Benjamini and Hochberg, 1995). Analysis of TLDA data was performed in ExpressionSuite Software version 1.1 supplied by Applied Biosciences.
DATA AND SOFTWARE AVAILABILITY
All raw RNA-seq data have been deposited to GEO, accession number GSE105041. The complete reproducible workflow, including R scripts and count matrix, was deposited to https://github.com/cbtncph/HD-Glial-Differentiation-Block-Goldman-Lab-2017.
All differential expression data have been uploaded to a publicly-accessible, interactive lab-based website, https://ctngoldmanlab.genialis.com, within which further evaluation and interrogation of our differentially-expressed gene sets may be performed by interested users. All data have also been uploaded to Mendeley Data (https://data.mendeley.com), at doi:10.17632/5gmpkdwcz4.1.
Supplementary Material
Highlights.
Mutant HTT-expressing hESCs exhibit a relative block in glial lineage progression
Differentiation-associated genes are down-regulated in mHTT glial progenitor cells
Human glial chimeras established with mHTT glial progenitors are hypomyelinated
mHTT human glial chimeras manifest disrupted astrocytic differentiation
Acknowledgements:
Supported by CHDI, NIMH, NINDS, the Dr. Miriam and Sheldon G. Adelson Medical Research Foundation, the Lundbeck Foundation and the Novo Nordisk Foundation. We thank Thomas Vogt, Dan Felsenfeld and Ignacio Munoz-Sanjuan of CHDI for helpful comments, Lorenz Studer (Sloan-Kettering Institute) for the C27 hiPSC cell line, and Matt Rasband (Baylor) for anti-Caspr antibody. All genomic data have been deposited to NCBI GEO, accession number GSE105041.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Interests: Drs. Goldman and Windrem hold a patent on human glial chimeric mice, US 7,524,491, from which they receive no financial remuneration. Drs. Goldman and Osipovitch have a patent application pending covering genes differentially expressed by glial cells in Huntington’s disease, and modulators thereof.
Citations
- Amaral AI, Meisingset TW, Kotter MR, and Sonnewald U (2013). Metabolic aspects of neuron-oligodendrocyte-astrocyte interactions. Front Endocrinol 4, 54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrews S (2010). FastQC: a quality control tool for high throughput sequence data
- Benjamini Y, and Hochberg Y (1995). Controlling the false discovery rate: a practical and powerful approach to multiple testing. Journal of the royal statistical society Series B (Methodological), 289–300.
- Benraiss A, Wang S, Herrlinger S, Li X, Chandler-Militello D, Mauceri J, Burm H, Toner M, Osipovitch M, Xu Q, et al. (2016). Human glia can both induce and rescue aspects of phenotype in Huntington Disease. Nature communications 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blondel V, Guillaume J-L, Lambiotte R, and Lefebvre E (2008). Fast unfolding of communities in large networks. J Statistical Mechanics doi: 10.1088/1742-5468/2008/10/P10008. [DOI] [Google Scholar]
- Bolger AM, Lohse M, and Usadel B (2014). Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradley CK, Scott HA, Chami O, Peura TT, Dumevska B, Schmidt U, and Stojanov T (2011). Derivation of Huntington’s disease-affected human embryonic stem cell lines. Stem Cells Dev 20, 495–502. [DOI] [PubMed] [Google Scholar]
- Bujalka H, Koenning M, Jackson S, Perreau VM, Pope B, Hay CM, Mitew S, Hill AF, Lu QR, Wegner M, et al. (2013). MYRF is a membrane-associated transcription factor that autoproteolytically cleaves to directly activate myelin genes. PLoS biology 11, e1001625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai N, Kurachi M, Shibasaki K, Okano-Uchida T, and Ishizaki Y (2012). CD44-positive cells are candidates for astrocyte precursor cells in developing mouse cerebellum. Cerebellum 11, 181–193. [DOI] [PubMed] [Google Scholar]
- Chow JD, Price JT, Bills MM, Simpson ER, and Boon WC (2012). A doxycycline-inducible, tissue-specific aromatase-expressing transgenic mouse. Transgenic Res 21, 415–428. [DOI] [PubMed] [Google Scholar]
- Clarke LE, and Barres BA (2013). Glia keep synapse distribution under wraps. Cell 154, 267–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cotrina ML, Lin JH, and Nedergaard M (1998). Cytoskeletal assembly and ATP release regulate astrocytic calcium signaling. J Neurosci 18, 8794–8804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dang V, Medina B, Das D, Moghadam S, Martin KJ, Lin B, Naik P, Patel D, Nosheny R, Wesson Ashford J, et al. (2014). Formoterol, a long-acting beta2 adrenergic agonist, improves cognitive function and promotes dendritic complexity in a mouse model of Down syndrome. Biol Psychiatry 75, 179–188. [DOI] [PubMed] [Google Scholar]
- Emery B, Agalliu D, Cahoy JD, Watkins TA, Dugas JC, Mulinyawe SB, Ibrahim A, Ligon KL, Rowitch DH, and Barres BA (2009). Myelin gene regulatory factor is a critical transcriptional regulator required for CNS myelination. Cell 138, 172–185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gentleman RC, Carey VJ, Bates DM, Bolstad B, Dettling M, Dudoit S, Ellis B, Gautier L, Ge Y, and Gentry J (2004). Bioconductor: open software development for computational biology and bioinformatics. Genome biology 5, R80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang B, Wei W, Wang G, Gaertig MA, Feng Y, Wang W, Li XJ, and Li S (2015). Mutant huntingtin downregulates myelin regulatory factor-mediated myelin gene expression and affects mature oligodendrocytes. Neuron 85, 1212–1226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacomy M, Venturini T, Heymann S, and Bastian M (2014). ForceAtlas2, a continuous graph layout algorithm for handy network visualization designed for the Gephi software. PloS one 9, e98679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang R, Diaz-Castro B, Looger LL, and Khakh BS (2016). Dysfunctional Calcium and Glutamate Signaling in Striatal Astrocytes from Huntington’s Disease Model Mice. J Neurosci 36, 3453–3470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin J, Peng Q, Hou Z, Jiang M, Wang X, Langseth AJ, Tao M, Barker PB, Mori S, Bergles DE, et al. (2015). Early white matter abnormalities, progressive brain pathology and motor deficits in a novel knock-in mouse model of Huntington’s disease. Human molecular genetics 24, 2508–2527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaimal V, Bardes EE, Tabar SC, Jegga AG, and Aronow BJ (2010). ToppCluster: a multiple gene list feature analyzer for comparative enrichment clustering and network-based dissection of biological systems. Nucleic Acids Res 38, W96–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khakh BS, Beaumont V, Cachope R, Munoz-Sanjuan I, Goldman SA, and Grantyn R (2017). Unravelling and Exploiting Astrocyte Dysfunction in Huntington’s Disease. Trends Neurosci 40, 422–437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krämer A, Green J, Pollard J Jr, and Tugendreich S (2013). Causal analysis approaches in ingenuity pathway analysis. Bioinformatics 30, 523–530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langfelder P, Cantle JP, Chatzopoulou D, Wang N, Gao F, Al-Ramahi I, Lu XH, Ramos EM, El-Zein K, Zhao Y, et al. (2016). Integrated genomics and proteomics define huntingtin CAG length-dependent networks in mice. Nature neuroscience 19, 623–633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao Y, Smyth GK, and Shi W (2013a). featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30, 923–930. [DOI] [PubMed] [Google Scholar]
- Liao Y, Smyth GK, and Shi W (2013b). The Subread aligner: fast, accurate and scalable read mapping by seed-and-vote. Nucleic acids research 41, e108–e108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Magri L, Zhang F, Marsh NO, Albrecht S, Huynh JL, Kaur J, Kuhlmann T, Zhang W, Slesinger PA, et al. (2015). Chromatin landscape defined by repressive histone methylation during oligodendrocyte differentiation. J Neurosci 35, 352–365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Han SS, Wu Y, Tuohy TM, Xue H, Cai J, Back SA, Sherman LS, Fischer I, and Rao MS (2004). CD44 expression identifies astrocyte-restricted precursor cells. Developmental biology 276, 31–46. [DOI] [PubMed] [Google Scholar]
- Lopez-Anido C, Sun G, Koenning M, Srinivasan R, Hung HA, Emery B, Keles S, and Svaren J (2015). Differential Sox10 genomic occupancy in myelinating glia. Glia 63, 1897–1914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love MI, Huber W, and Anders S (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome biology 15, 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Octeau JC, Chai H, Jiang R, Bonanno SL, Martin KC, and Khakh BS (2018). An Optical Neuron-Astrocyte Proximity Assay at Synaptic Distance Scales. Neuron 98, 49–66 e49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pruitt KD, Tatusova T, and Maglott DR (2006). NCBI reference sequences (RefSeq): a curated non-redundant sequence database of genomes, transcripts and proteins. Nucleic acids research 35, D61–D65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- R Core Team (2016). R: A Language and Environment for Statistical Computing (R Foundation for Statistical Computing; ). [Google Scholar]
- Risso D, Ngai J, Speed TP, and Dudoit S (2014). Normalization of RNA-seq data using factor analysis of control genes or samples. Nature biotechnology 32, 896–902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson MD, McCarthy DJ, and Smyth GK (2010). edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26, 139–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin JY, Fang ZH, Yu ZX, Wang CE, Li SH, and Li XJ (2005). Expression of mutant huntingtin in glial cells contributes to neuronal excitotoxicity. J Cell Biol 171, 1001–1012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shinkai Y, Rathbun G, lam K, Oltz E, Stewart V, Mendelsohn M, Charron J, Datta M, Young F, Stall A, et al. (1992). RAG2-deficient mice lack mature lymphocytes owing to inability to initiate V(D)J rearrangement. Cell 68, 855–867. [DOI] [PubMed] [Google Scholar]
- Sholl DA (1953). Dendritic organization in the neurons of the visual and motor cortices of the cat. J Anat 87, 387–406. [PMC free article] [PubMed] [Google Scholar]
- Sim FJ, McClain CR, Schanz SJ, Protack TL, Windrem MS, and Goldman SA (2011). CD140a identifies a population of highly myelinogenic, migration-competent and efficiently engrafting human oligodendrocyte progenitor cells. Nature biotechnology 29, 934–941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tabrizi SJ, Reilmann R, Roos RA, Durr A, Leavitt B, Owen G, Jones R, Johnson H, Craufurd D, Hicks SL, et al. (2012). Potential endpoints for clinical trials in premanifest and early Huntington’s disease in the TRACK-HD study: analysis of 24 month observational data. The Lancet Neurology 11, 42–53. [DOI] [PubMed] [Google Scholar]
- Teo RT, Hong X, Yu-Taeger L, Huang Y, Tan LJ, Xie Y, To XV, Guo L, Rajendran R, Novati A, et al. (2016). Structural and molecular myelination deficits occur prior to neuronal loss in the YAC128 and BACHD models of Huntington disease. Human molecular genetics 25, 2621–2632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tong X, Ao Y, Faas GC, Nwaobi SE, Xu J, Haustein MD, Anderson MA, Mody I, Olsen ML, Sofroniew MV, et al. (2014). Astrocyte Kir4.1 ion channel deficits contribute to neuronal dysfunction in Huntington’s disease model mice. Nature neuroscience 17, 694–703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ullian EM, Sapperstein SK, Christopherson KS, and Barres BA (2001). Control of synapse number by glia. Science (New York, NY) 291, 657–661. [DOI] [PubMed] [Google Scholar]
- Wang S, Bates J, Li X, Schanz S, Chandler-Militello D, Levine C, Maherali N, Studer L, Hochedlinger K, Windrem M, et al. (2013). Human iPSC-derived oligodendrocyte progenitor cells can myelinate and rescue a mouse model of congenital hypomyelination. Cell Stem Cell 12, 252–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Windrem MS, Nunes MC, Rashbaum WK, Schwartz TH, Goodman RA, McKhann G, Roy NS, and Goldman SA (2004). Fetal and adult human oligodendrocyte progenitor cell isolates myelinate the congenitally dysmyelinated brain. Nature medicine 10, 93–97. [DOI] [PubMed] [Google Scholar]
- Windrem MS, Osipovitch M, Liu Z, Bates J, Chandler-Militello D, Zou L, Munir J, Schanz S, McCoy K, Miller RH, et al. (2017). Human iPSC glial mouse chimeras reveal glial contributions to schizophrenia. Cell Stem Cell 21, 195–208 e196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Windrem MS, Schanz SJ, Guo M, Tian GF, Washco V, Stanwood N, Rasband M, Roy NS, Nedergaard M, Havton LA, et al. (2008). Neonatal chimerization with human glial progenitor cells can both remyelinate and rescue the otherwise lethally hypomyelinated shiverer mouse. Cell Stem Cell 2, 553–565. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All raw RNA-seq data have been deposited to GEO, accession number GSE105041. The complete reproducible workflow, including R scripts and count matrix, was deposited to https://github.com/cbtncph/HD-Glial-Differentiation-Block-Goldman-Lab-2017.
All differential expression data have been uploaded to a publicly-accessible, interactive lab-based website, https://ctngoldmanlab.genialis.com, within which further evaluation and interrogation of our differentially-expressed gene sets may be performed by interested users. All data have also been uploaded to Mendeley Data (https://data.mendeley.com), at doi:10.17632/5gmpkdwcz4.1.