

Abstract

Riboswitches that couple binding of ligands to conformational changes offer sensors and control elements for RNA synthetic biology and medical biotechnology. However, design of these riboswitches has required expert intuition or software specialized to transcription or translation outputs; design has been particularly challenging for applications in which the riboswitch output cannot be amplified by other molecular machinery. We present a fully automated design method called RiboLogic for such “stand-alone” riboswitches and test it via high-throughput experiments on 2875 molecules using RNA-MaP (RNA on a massively parallel array) technology. These molecules consistently modulate their affinity to the MS2 bacteriophage coat protein upon binding of flavin mononucleotide, tryptophan, theophylline, and microRNA miR-208a, achieving activation ratios of up to 20 and significantly better performance than control designs. By encompassing a wide diversity of stand-alone switches and highly quantitative data, the resulting ribologic-solves experimental data set provides a rich resource for further improvement of riboswitch models and design methods.
Keywords: riboswitch, RNA, molecular design, high-throughput measurements, thermodynamic model, computer-assisted design
Riboswitches use RNA conformational changes to transduce sensing of molecules in the cellular milieu into modulation of RNA transcription, ribosomal translation, pre-mRNA splicing, and RNA cleavage.1 The ability to perform de novo design of arbitrary riboswitches would have broad impacts in synthetic biology as well as for RNA diagnostics, therapeutics, and biomedical imaging. Supporting these efforts, there are a rapidly growing number of synthetic and natural RNA “aptamer” sequences that bind drugs, metabolites, proteins, and other biologically important molecules that expand the possible inputs for novel riboswitches, and powerful design rules and software to create riboswitches with transcription and translation outputs.2−4 Similarly, the possible outputs of riboswitches are being expanded to triggering of “light-up” fluorescence and toggling activities of CRISPR/Cas and other ribonucleoprotein complexes.5−9 These newer applications would benefit from riboswitch mechanisms that do not require external molecular machinery or energy dissipation but instead broadcast their output simply after reaching thermodynamic equilibrium. Such molecules would be more likely to retain their functions when moved into different RNA contexts or used in extracellular environments where energy or additional molecular machinery cannot be provided. Inspired by the concept of stand-alone executables in software engineering, we term such molecules “stand-alone” riboswitches.
Creating stand-alone riboswitches leads to a new design challenge. Natural and synthetic riboswitches achieve maximal activation ratios—defined as the ratio of observed output signal in the presence and absence of the input ligand—by toggling between states that are barely activated to states that are weakly activated, rather than to states that saturate the output.10 Biological control is then achieved by subsequent amplification steps such as ribosomal translation of many proteins per activation step.10,11 Effective stand-alone riboswitches, which forego such amplification machinery, require quantitative conversion between two distinct states, rather than changing the frequency of transient sampling of an active state. This design constraint necessitates a trade-off with good activation ratios.10 Testifying to the difficulty of this additional trade-off, development of light-up sensors has required significant trial-and-error; success has been achieved through screening of many constructs, the majority of which exhibit little to no switching, with median activation ratios close to 1 and best-case activation ratios of 10.5,6,9,10 Moreover, computational predictions of the success of light-up designs are poor (Figure S1), suggesting the need for richer datasets characterizing diverse RNAs. Further exemplifying their inherent design difficulty, stand-alone switches for CRISPR/Cas9 or other ribonucleoprotein complexes, which would enable reversible control of these complexes in therapeutic settings, have not been achieved.12−14
Here, we present a detailed computational and experimental study involving thousands of diverse molecules to test the fully automated design of stand-alone riboswitches. For computational design, we describe RiboLogic, an algorithm for designing sequences of RNA molecules that are predicted to change their secondary structure in response to interactions with other molecules. Unlike prior software that might be applied to stand-alone switch design,3,15−19 this package only requires the user to provide small aptamer segments to bind desired input molecules and the desired structures adopted in each state. For experimental characterization, we evaluate the switching of thousands of designed RNA molecules in vitro using repurposed Illumina sequencers, through the recently developed RNA-MaP (RNA on a massively parallel array) platform.20−23
RiboLogic designs stand-alone riboswitches based on a flexible set of user-specified constraints. The algorithm accounts for any number of folding conditions, as defined by the concentrations of ligands defined by the user. These ligands can be small molecules, proteins with known aptamers, or other RNA strands engaged through base-pairing interactions. For example, in some of our tests below, we used flavin mononucleotide (FMN) as an input ligand; FMN binds to a small aptamer sequence discovered by in vitro selection (Figure 1A,D).24 The user only needs to specify the sequence of this aptamer and the estimated dissociation constant of the aptamer-ligand complex under the experimental conditions, and RiboLogic will place this “input” segment within the design and optimize the surrounding sequence in each of the riboswitch states, simulating ligand binding to the aptamer (see Methods for details). In this example, the two states are RNA with no FMN present and with a concentration of 200 μM FMN (Figure 1B). For each of the target riboswitch states, the user can specify either a full desired secondary structure or, more simply, the substructure of an “output” segment that must be adopted or not adopted by the RNA in order to trigger or suppress an output, respectively. For example, in some of our tests below, we used binding of a fluorescently tagged MS2 viral coat protein to an MS2 RNA hairpin segment within the design as an output (Figure 1A,D); such interactions underlie most systems for CRISPR interference and activation and in situ RNA visualization but have not yet been used in standalone switch design.5−9,12−14 The user only needs to specify the sequence and “active” secondary structure of this output element, and RiboLogic places this sequence relative to the input aptamer element and optimize surrounding sequences during its design process. We note that unlike prior natural and synthetic riboswitches, we demand that the RNA’s MS2 output segment take on the desired hairpin secondary structure as its dominant structure in the ON state, rather than simply sampling this structure more frequently than in the OFF state. Such complete conversion of structure is needed for a stand-alone riboswitch that would work without further amplification.
Figure 1.

RiboLogic uses a graph representation and two scoring functions to design stand-alone riboswitches. (A) This energy diagram represents the thermodynamic model used, where the ligand-bound state is given an energetic bonus due to the chemical potential of the binding of the ligand. (B) A user specifies design constraints for a riboswitch of interest, e.g., the formation of the MS2 hairpin in the absence of a ligand and the nonformation of the hairpin in the absence of a ligand. (C) The sequence is initialized to all A’s except for known sequence constraints. (D) A graph representation is used to constrain the sequence space that is sampled by RiboLogic. In this example, the goal is to design a riboswitch whose formation of the MS2 RNA hairpin is modulated by the presence of the flavin mononucleotide (FMN) molecule. Bases connected by an arc are part of these secondary structure elements and are constrained to be complementary in sequence update. (E) Two scoring metrics are used to evaluate each design candidate. The base pair distance measures the number of base pairs that must be broken or formed to reach the target structure, while the base pair probability (bpp) score quantifies the probability of formation of each base pair in the target structure. (F) The scores change as expected during computational design, with the base pair distance decreasing and the base pair probability score increasing over optimization steps.
RiboLogic uses simulated annealing to sample the space of possible sequences to satisfy the given constraints (Figure 1D,E). At each step, the sequence is mutated either at a single base or by sliding the position of a functional element (e.g., the FMN aptamer or MS2 hairpin; colored nucleotides in Figure 1D). For each sequence that is sampled, the minimum free energy secondary structure is determined for each solution condition (e.g., without and with 200 μM FMN) and evaluated by two scores (Figure 1E,F). The first score is a base pair distance that measures the number of base pairs that must be broken or formed to obtain the target structure or substructures in each solution condition, summed over the different solution conditions. The second score is a base pair probability score that sums the probabilities of formation of all base pairs that should form in the target structure or substructures, providing a smoother quantitative measure of structure formation than the first base pair distance score. RiboLogic implements several additional strategies to narrow the sequence space being explored. Mutation of the sampled sequences leverages a dependency graph-based approach, which ensures that bases that are paired in any target structure are always complementary in sequence (e.g., N’s connected by blue lines in Figure 1D).25 In the case of designing riboswitches responsive to other input RNA molecules, the algorithm provides the option to automatically introduce the sequence complementary to the input in order to promote favorable interactions between the designed RNA and input RNA.
As test cases for our methods, we designed stand-alone riboswitches where the binding of a small molecule or oligonucleotide ligand modulates the formation of the MS2 RNA hairpin, which can then transduce outputs by recruiting machinery coupled to the MS2 bacteriophage coat protein. This is the first example of MS2-controlling riboswitches, which could have broad applications.26−28 We applied a quantitative, high-throughput array technology that enables fluorescence measurements over millions of individual RNA clusters generated on an Illumina array, which has been extensively tested using the MS2 system (Figure 2A,B).20,22,23 The formation of the MS2 RNA hairpin was detected by flowing fluorescently labeled MS2 protein at increasing concentrations to get a binding curve (Figure 2B,C). The dissociation constant Kd was fit over tens to hundreds of clusters for each design, yielding a distribution of Kd measurements for each state (Figure 2D). By taking the median of each distribution, we calculated a Kd as a quantitative measure of the binding of each design, and the ratio of these Kd values with and without input ligand (e.g., FMN) gives an activation ratio, which we use as our figure of merit for riboswitches. This activation ratio is equal to the ratio of fluorescence of the riboswitch with and without input ligand at low MS2 concentrations and is therefore the most relevant performance measure for stand-alone switches that need to work without output amplification.10 By carrying out fits of data from subnanomolar to many micromolar MS2 concentrations, we achieve high precision in these measurements. The resulting Kd values and activation ratios were strongly correlated across experimental replicates, confirming the high precision of the method (r2 = 0.94 for log Kd; errors in activation ratios well under 2-fold; see Figure S2).
Figure 2.

Functional tests of riboswitches using a high-throughput array. (A) Each cluster on the array initially contained a single species of ssDNA from a synthesized oligo pool. dsDNA was generated by Klenow extension with a biotinylated primer, and RNA was transcribed by RNA polymerase until being stalled at the streptavidin roadblock. (B) Fluorescently labeled MS2 protein was flowed in at varying concentrations to enable measurement of binding. (C) The array technology enables measurement of binding curves over tens or hundreds of replicate clusters for each design and solution condition. (D) The median over the distribution of fit Kd’s was used to estimate the activation ratio of switching. In this example of an ON switch, the activation ratio of 11 was measured over 172 independent clusters displaying the same switch.
We applied the algorithm to design switches responsive to three different small molecules–flavin mononucleotide (FMN), theophylline, and tryptophan. For stand-alone OFF switches, the MS2 hairpin should form when the ligand is absent and be disrupted when the ligand is added (Figure 3A). For ON switches, the MS2 hairpin should form only when the FMN is present and otherwise be disrupted (Figure 3B). By applying secondary structure constraints to the MS2 hairpin region in both the absence and presence of the ligand, we set up a two-state design problem. We were able to obtain a set of structurally diverse designs (Figures 3 and 4A), and we experimentally characterized thousands of these molecules with the RNA-MaP method.
Figure 3.

Top ligand-responsive riboswitch designs. (A) Predicted secondary structures for a top OFF switches show disruption of the MS2 hairpin (red) upon binding of FMN, theophylline, or tryptophan (blue). (B) Predicted secondary structures for top ON switches show formation of the MS2 hairpin (red) upon binding of FMN, theophylline, or tryptophan (blue).
Figure 4.

Design of ligand-responsive riboswitches. (A) Clustering of FMN switches based on the sum of base pair distances of predicted secondary structures reveals that RiboLogic designs with diverse structures achieve high activation ratios. (B) Distributions of experimentally measured activation ratios are shown for various types of designs, with medians shown as vertical lines. RiboLogic generally achieves significantly better activation ratios than baseline, as determined by a Wilcoxon rank-sum test (***p < 0.001). Baseline is the measured activation ratio for sequences made for other design problems. (C) In practice, several of the most promising designs would be experimentally screened to evaluate switch efficiency. To mimic this, we bootstrapped sets of ten designs and chose the design with the best activation ratio. The distributions of activation ratios for these best-of-ten designs were compared between RiboLogic and baseline. A best-of-ten strategy yields designs with significantly higher activation ratios than baseline.
We found that RiboLogic designs achieved activation ratios significantly better than unrelated designs made for other ligands, which were used as baseline comparisons (Figure 4B). For example, theophylline and tryptophan designs, which are expected not to respond to FMN-binding, were used as baseline measurements for comparison to FMN designs. For example, the median activation ratio for RiboLogic designs of FMN-responsive ON switches was 1.5 (Figure 4B, Table 1, Table S1). As the baseline comparison, the median activation ratios with respect to FMN for designs meant to be responsive to theophylline or tryptophan was 1.2. For each of the six switch design challenges (three ligands, ON vs OFF) the difference was significant (p < 10–10; Figure 4B, Table S2). In addition, RiboLogic designs also perform significantly better than no switching (activation ratio 1) in almost all design problems. We also provide a success rate by counting the number of designs that perform better than the median or 95th percentile of baseline designs (Table S3). Since other existing automated methods are not compatible with our design problem, we also compare our performance to previous rational design efforts of similar systems. Previous characterization of reversible riboswitches yielded a median activation ratio of 1.2.6,9,29
Table 1. Activation Ratios for RiboLogic Designs.
| design | maximum AR | median AR | best-of-ten median AR | count |
|---|---|---|---|---|
| FMN OFF | 9.74 | 0.987 | 2.57 | 1357 |
| FMN ON | 14.4 | 1.46 | 3.89 | 853 |
| theophylline OFF | 9.92 | 1.73 | 4.86 | 97 |
| theophylline ON | 15.4 | 0.991 | 3.44 | 99 |
| tryptophan OFF | 4.29 | 1.17 | 2.28 | 89 |
| tryptophan ON | 4.55 | 1.08 | 2.09 | 94 |
| miRNA OFF | 21.8 | 0.825 | 1.66 | 188 |
| miRNA ON | 20.0 | 1.17 | 2.84 | 98 |
For each of these six small-molecule-triggered challenges, the best activation ratio was over 4-fold, and extended up to 15-fold for the theophylline ON switch tests (Figure 4B). In addition, previous design efforts generally involve experimentally testing several designs and choosing the best one.3,6,9,30 Thus, we conducted a best-of-ten analysis, in which we randomly drew subsets of 10 designs and scored the best activation ratios. These best-of-ten trials showed clear separation of the activation ratios from baselines, and in the majority of cases gave activation ratios of 2.0 or greater (Figure 4C, Table S4). In addition, most designs exhibited Kd’s close to the affinity of the MS2 coat protein under the conditions in which they were supposed to be active (with ligand for ON switches; without ligand for OFF switches) (Figure S3). This bias likely reflects our design constraint that the stand-alone riboswitches should quantitatively convert to MS2-binding structures when activated rather than requiring subsequent molecular machinery to amplify their output. The stand-alone switch with the highest activation ratio of 15.4 achieved a Kd of 10 nM in the activated state, within experimental error of the intrinsic dissociation constant of the MS2 coat protein-RNA hairpin interaction (6 nM, measured in the same experiment). However, the activation ratios fell short of the thermodynamic optimum described by Wayment-Steele et al.(10) (Figure S4 and S5).
We further tested if RiboLogic could design stand-alone riboswitches that are responsive to RNA inputs instead of small molecule ligands. Specifically, we applied the algorithm to design 286 switches that modulate MS2 binding based on the presence of miR-208a, a 22-nt miRNA implicated in cardiac hypertrophy.31 This type of RNA-based system could be used in diagnostic devices or linked to downstream therapeutic events. Using RiboLogic, we were able to design both ON and OFF switches triggered by the miRNA strand (Figure 5A,B). We found that these designs generally took more iterations of optimization to satisfy the constraints as compared to the ligand-responsive switches (Figure S6), but diverse mechanisms were achieved (Figure 5C). Disappointingly, experimental evaluation did not show a significant difference between RiboLogic and baseline designs in terms of activation ratio. Nevertheless, the best-of-ten comparison showed significant differences and maximum activation ratios of 20 exceeded those of small molecule activated switches (Figure 5D,E, Table 1). These computational and experimental observations suggest that design for RNA-responsive switches may be intrinsically more difficult, despite the larger binding energy of the RNA compared to the small molecule ligands, perhaps due to a large number of competing binding modes where the input RNAs hybridize to alternative locations in the riboswitch design. At the same time, this automated procedure can still lead to excellent microRNA sensors, at the expense of characterizing more designs.
Figure 5.

Design of miRNA-responsive riboswitches. (A) This OFF switch is predicted to form the MS2 hairpin (red) only in the absence of the miRNA (blue). (B) This ON switch is predicted to form the MS2 hairpin (red) only in the presence of the miRNA (blue). (C) Clustering of miRNA switches based on the base pair distance between predicted secondary structures in the absence of the miRNA reveals that RiboLogic designs with diverse structures achieve high activation ratios. (D) The distribution of experimentally measured activation ratios are shown as scatter and violin plots, with medians shown as horizontal lines. Across all design problems, there is no significant difference between RiboLogic and baseline designs, as determined by a Wilcoxon rank-sum test. (E) We conducted a best-of-ten analysis by bootstrapping sets of ten designs and choosing the design with the best activation ratio. The distributions of activation ratios for these best-of-ten designs were compared between RiboLogic and baseline. This analysis results in designs with significantly higher activation ratios, but the distributions remain similar, with the exception of a few high performaning designs.
Across these design challenges, we found that stand-alone riboswitches with high activation ratios could take a variety of forms. Some high performing designs had the MS2 sequence nested between the two sides of the aptamer, while others had the MS2 outside, with only a short hairpin between the two halves of the ligand-binding internal loop (Figure 3; compare designs 2297 and 2343 to 512 and 2534). Some designs formed relatively simple secondary structures with long stems, while others formed more complex folds with three-way junctions (Figure 3; compare designs 512 and 2357 to 1555 and 2534). Several structures contain large single-stranded regions, while some have regions designed to bind the functional elements when they are inactive (Figure 3; compare design 2534 to 512). The size of our dataset enabled statistical analyses of these secondary structure features, highlighting several that were correlated with activation ratios (Figure S7). For example, the data showed that having more base pairs shared between states correlated with higher resulting activation ratios. Still, the correlations of any single feature with activation ratio, while statistically significant, were weak (r2 < 0.01). Machine learning models that take into account multiple features to predict design success will be interesting to develop and test prospectively.
A related insight into current design limitations is also enabled by the diversity and large number of our riboswitches. We note that the designs produced by RiboLogic have features that are distinct from designs created by human experts. For the small molecule sensitive riboswitches (Figure 3), the RiboLogic designs include numerous stems outside the aptamer segments that need to be broken or formed. These designs are not as “concise” as expert-designed riboswitches seen in the literature,5,19 although it should be noted that some natural riboswitches do involve ornate conformational rearrangements.32 For the miRNA-sensitive riboswitches (Figures 5), the binding of the input miRNA and the RiboLogic riboswitch is typically not through a completely contiguous, long RNA–RNA duplex, as is typically the case in, e.g., toehold riboswitches33,34 or DNA logical devices35,36 designed by human experts. Automated riboswitch design might improve if these features seen in human designs were rewarded or seeded into the RiboLogic design algorithm.
We hypothesized that errors in current RNA secondary structure energetic models might be limiting for RiboLogic stand-alone riboswitch designs. We carried out comparisons of Kd’s and activation ratios predicted by the ViennaRNA and NUPACK packages for small molecule and miRNA riboswitches, respectively. We saw poor correlations for both (r2 of 0.06 and 0.01 for small molecule and miRNA riboswitches, respectively; Figures S8 and 6). Several designs predicted to have poor activation ratios (near or lower than 1.0) in fact gave activation ratios near 10.0; and other designs predicted to have outstanding activation ratios (greater than 100.0) gave experimental activation ratios lower than 1.0 (Figure 6B). This experiment–theory correlation was better for small-molecule riboswitches compared to the miRNA riboswitches, consistent with the generally better activation ratios of the former, relative to baseline measurements (compare Figures 4B and 5D; Table S1). Future design efforts would benefit from more accurate computational models of RNA folding energetics. We present all data collected herein as the ribologic-solves dataset (Supplemental Data) to help guide and validate such improvements.
Figure 6.

Comparison of predicted and measured activation ratios. (A) For small molecule riboswitches, the predicted activation ratio is somewhat correlated with measured activation ratio. (B) For miRNA riboswitches, the correlation between prediction and experiment is poor.
Here, we have presented RiboLogic, an automated algorithm for designing stand-alone riboswitches that transduce input ligand binding into output effector binding without energy input or amplification by other molecular machines. We show that RiboLogic generates designs with diverse structural mechanisms and achieves activation ratios comparable to previous efforts in rational design of reversible riboswitches. In combination with improved thermodynamic models and high-throughput measurement techniques, we expect that this method and these data will enable improved automated design of switchable RNA elements for a wide variety of applications in biotechnology and medicine.
Methods
Design Algorithm
Overview
Given secondary structure constraints in multiple states defined by ligands or short RNA inputs, our method optimizes an RNA sequence using a simulated annealing algorithm. The starting sequence is arbitrarily set to all A’s, with the exception of known sequence constraints and updates to ensure complementarity in the target secondary structures. The length is specified by the user and is not changed during sequence optimization. In each step, a random mutation is made, and the new sequence is evaluated using a base pair distance and a base pair probability score. The sequence is updated on the basis of a Metropolis–Hastings acceptance criterion:
| 1 |
where ΔG is the difference in score between the updated and current sequences and Tdesign is the temperature parameter. This temperature parameter is decreased over the course of the optimization and can be tuned by the user. By default, it decreases linearly from 5 to 1 over the course of design. This process is repeated until a satisfactory sequence is found or the maximum number of iterations specified by the user is reached.
Constraints
Sequence constraints can include fixed bases at specified positions as well as substrings that are disallowed from the final sequence. Secondary structure constraints can be given for multiple user-specified states, as defined by varying concentrations of the input ligands. For small molecule and protein ligands, the aptamer sequence, secondary structure, and dissociation constant must be specified. For each state, secondary structure constraints can be applied to any part of the input sequence, including any RNA inputs, and bases can be specified to be unpaired, paired to any other base, or paired with a specific other base. Secondary structure elements’ positions can be left unspecified, and RiboLogic will optimize its position as well. To further ensure diversity, for the tests herein, we enforced two different global arrangements of the aptamer and MS2 hairpin elements—one with the two parts of the aptamer loop adjacent to each other and one with the MS2 sequence nested within the aptamer segments.
Sequence Update
Sequences are represented in a dependency graph structure as described by Flamm et al.(25) Briefly, each base is a node and each base pair in the constraints forms an edge between nodes. The graph is maintained such that nodes connected by an edge are always complementary. Each time a base is mutated, its entire connected component is mutated accordingly to ensure that all nodes connected to the selected base maintains complementarity. In addition, sequence constraints are incorporated into this graph, disallowing mutations that would force a constrained base to change. In the case of RNA inputs, our method provides the option to automatically introduce the complement of the input sequence into the design sequence in order to promote interactions between strands. This complementary segment can be altered in length, moved, or mutated as a sequence update step.
Scoring Functions
Two scoring functions are used: a primary score based on a single minimum free energy secondary structure, and a base pair probability-based secondary score that is used in the primary score’s place when the it is the same between two sequences. On the basis of the predicted minimum free energy structures in each state, a base pair distance to the target secondary structure is calculated. The base pair distance is the number of base pairs that must be broken or formed in order to get from one secondary structure to the other.37 If only a substructure is specified, this can include the breaking of base pairs formed with nucleotides outside of the subsequence specified. In addition, for small molecule riboswitches, if the energy of the ligand-bound conformation, with energetic bonus, is not lower than the ligand-free conformation, a penalty equal to the ΔG between the two states is applied to the base pair distance.
| 2 |
where ΔG–aptamer is the
free energy of the RNA alone in kcal/mol, [L] is
the concentration of the input ligand, KdL is the affinity of the input ligand, ΔG+aptamer is the free energy of the RNA constrained
to form the aptamer, R is the gas constant, T is the experimental temperature (37 °C = 310.15 K).
We consider only structures that form the desired aptamer, as opposed
to doing a minimum free energy calculation with an energetic bonus.
This allows the algorithm to guide the sequence toward those that
have a more favorable aptamer-forming conformation, even if it is
not the minimum free energy structure. We used a value of 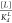 of 133 for FMN and 150 for theophylline
and tryptophan, based on initial Kd estimates for those input ligands (Figure S4) and experimental [L] = 200 μM,
2 mM, and 2.4 mM (FMN, theophylline, and tryptophan, respectively).
of 133 for FMN and 150 for theophylline
and tryptophan, based on initial Kd estimates for those input ligands (Figure S4) and experimental [L] = 200 μM,
2 mM, and 2.4 mM (FMN, theophylline, and tryptophan, respectively).
However, since the score in eq 2 is not highly sensitive to single mutations, a secondary base pair probability score is used when the base pair distance is unchanged between sequence updates. This measure of secondary structure formation over the full ensemble is defined by
| 3 |
where s is the index of the folding state, i and j are indices of the base position in the sequence, Xsij is an indicator variable representing whether base i and j should be paired in state s, and psij is the probability of base i and j forming in state s according to the partition function calculation. The value of the indicator variable is 1 if the base pair should be formed, −1 if it should not be formed, and 0 if it is unconstrained.
Folding of each sequence can be modeled using either ViennaRNA38 or NUPACK.39 NUPACK 3.0.5.39 was used for design involving more than one RNA, in order to properly model multistrand RNA folding, while ViennaRNA 2.1.938 was used for designs involving small molecule aptamers.
The score used for the Metropolis–Hastings criterion in eq 1 was:
By default, the sequence search terminates once the base pair distance reaches 0 or the number of steps reaches 10 000 steps. The software also provides the option to continue optimizing the sequence after the base pair distance reaches 0. Sequences were not filtered in any way before proceeding to experimental characterization.
Computation and Code Availability
All computation was performed on Intel Xeon Processors E5–2650. The code is available at https://github.com/wuami/RiboLogic.
Average computation time for the design of a ligand-induced riboswitch varied widely, both across runs and depending on the design problem (Figure S6). Every 1000 iterations took about 2 min on one core.
High-Throughput Array Experiments
The experimental methods have been described in detail previously.20,22 Briefly, DNA templates for designs were synthesized (CustomArray, Bothell, WA) and sequenced on Illumina MiSeq instruments, and RNA was transcribed directly on the sequencing chip in a repurposed Illumina Genome Analyzer II instrument. Fluorescently labeled MS2 protein was introduced at concentrations from 1.5 nM to 3 μM at room temperature. Incubation times varied from 0.8 to 1.5 h at the lowest concentrations to 10–20 min at the highest concentrations. Fluorescence images were collected and quantified to generate binding curves in buffer of 100 mM Tris-HCl pH 7.5, 80 mM KCl, 4 mM MgCl2, 0.1 mg/mL BSA, 1 mM DTT, 10 μg/mL yeast tRNA, 0.012% Tween20. These curves were measured in the absence and presence of the ligand of interest, with concentrations of 200 μM FMN, 2 mM theophylline, 4 mM tryptophan, and 100 nM miR-208a. These conditions were selected based on the Kd of each ligand. Each design was measured over an average of about 100 individual clusters on the flow cell. Median fit Kd values over all clusters for each design were used to compute the activation ratio. Designs were prepared and analyzed as part of the Eterna massive open laboratory experiments (rounds R95, R101, and R107).42
Designs for which Kd measurements were made over fewer than 10 clusters were excluded from our analysis to avoid poor quality measurements. For diversity analysis, Levenshtein distance was computed between each pair of sequences to obtain a distance matrix. Complete-linkage hierarchical clustering was performed to obtain a dendrogram with each design as a leaf (hclust in R). For statistical analysis, two-sided Wilcoxon rank sum test was used to determine if activation ratios between design types were significantly different. Predicted Kd’s were computed as described by Wayment-Steele et al.(10) Calculations were performed in R,40 with example scripts available at https://github.com/wuami/RiboLogic. The full dataset is available as Supplementary Data.
Chemical Mapping Experiments
One-dimensional chemical mapping measurements were performed as described previously.41 1M7 was used for FMN and tryptophan aptamers, while DMS was used for the theophylline aptamer.
Acknowledgments
We thank F. Portela, J. Anderson-Lee, E. Fisker, and R. Wellington-Oguri for discussions of these designs. This work was funded through a Burroughs-Wellcome Foundation Career Award (to RD), NIH Grant R01 GM100953 (to RD), NIH Grant R01 GM111990 and P50HG007735 (to WJG), Stanford School of Medicine Discovery Innovation Award (to RD), and a JIMB Seed Grant (to RD and WJG). MJW was supported by NSF Graduate Research Fellowship DGE-114747, NLM Biomedical Informatics Training Grant T15 LM007033, and NIH Ruth L. Kirschstein National Research Service Award F31GM125151. WJG acknowledges support as a Chan-Zuckerberg Investigator. Computational design was performed on the Stanford BioX3 cluster, supported by NIH Shared Instrumentation Grant S10 RR02664701.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acssynbio.9b00142.
Author Contributions
MW and RD conceived and planned the computational framework. MW implemented the computational framework. MW, JA, WG, and RD conceived and planned the experiments. JA and WK collected the data. MW and RD wrote the manuscript with input from all authors.
The authors declare no competing financial interest.
Supplementary Material
References
- Tucker B. J.; Breaker R. R. (2005) Riboswitches as Versatile Gene Control Elements. Curr. Opin. Struct. Biol. 15 (3), 342–348. 10.1016/j.sbi.2005.05.003. [DOI] [PubMed] [Google Scholar]
- Rodrigo G.; Landrain T. E.; Majer E.; Daròs J.-A.; Jaramillo A. (2013) Full Design Automation of Multi-State RNA Devices to Program Gene Expression Using Energy-Based Optimization. PLoS Comput. Biol. 9 (8), e1003172 10.1371/journal.pcbi.1003172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Espah Borujeni A.; Mishler D. M.; Wang J.; Huso W.; Salis H. M. (2016) Automated Physics-Based Design of Synthetic Riboswitches from Diverse RNA Aptamers. Nucleic Acids Res. 44 (1), 1–13. 10.1093/nar/gkv1289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ceres P.; Garst A. D.; Marcano-Vela J. G.; Batey R. T. (2013) Modularity of Select Riboswitch Expression Platforms Enables Facile Engineering of Novel Genetic Regulatory Devices. ACS Synth. Biol. 2, 463. 10.1021/sb4000096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kellenberger C. A.; Wilson S. C.; Sales-Lee J.; Hammond M. C. (2013) RNA-Based Fluorescent Biosensors for Live Cell Imaging of Second Messengers Cyclic Di-GMP and Cyclic AMP-GMP. J. Am. Chem. Soc. 135 (13), 4906–4909. 10.1021/ja311960g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kellenberger C. A.; Chen C.; Whiteley A. T.; Portnoy D. A.; Hammond M. C. (2015) RNA-Based Fluorescent Biosensors for Live Cell Imaging of Second Messenger Cyclic Di-AMP. J. Am. Chem. Soc. 137 (20), 6432–6435. 10.1021/jacs.5b00275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- You M.; Litke J. L.; Jaffrey S. R. (2015) Imaging Metabolite Dynamics in Living Cells Using a Spinach-Based Riboswitch. Proc. Natl. Acad. Sci. U. S. A. 112 (21), E2756–65. 10.1073/pnas.1504354112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paige J. S.; Nguyen-Duc T.; Song W.; Jaffrey S. R. (2012) Fluorescence Imaging of Cellular Metabolites with RNA. Science 335 (6073), 1194. 10.1126/science.1218298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Truong J.; Hsieh Y.-F.; Truong L.; Jia G.; Hammond M. C. (2018) Designing Fluorescent Biosensors Using Circular Permutations of Riboswitches. Methods 143, 102–109. 10.1016/j.ymeth.2018.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wayment-Steele H.; Wu M.; Gotrik M.; Das R. (2019) Evaluating Riboswitch Optimality. Methods Enzymol. 623, 417–450. 10.1016/bs.mie.2019.05.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Espah Borujeni A.; Salis H. M. (2016) Translation Initiation Is Controlled by RNA Folding Kinetics via a Ribosome Drafting Mechanism. J. Am. Chem. Soc. 138 (22), 7016–7023. 10.1021/jacs.6b01453. [DOI] [PubMed] [Google Scholar]
- Tang W.; Hu J. H.; Liu D. R. (2017) Aptazyme-Embedded Guide RNAs Enable Ligand-Responsive Genome Editing and Transcriptional Activation. Nat. Commun. 8, 15939. 10.1038/ncomms15939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y.; Zhan Y.; Chen Z.; He A.; Li J.; Wu H.; Liu L.; Zhuang C.; Lin J.; Guo X.; et al. (2016) Directing Cellular Information Flow via CRISPR Signal Conductors. Nat. Methods 13 (11), 938–944. 10.1038/nmeth.3994. [DOI] [PubMed] [Google Scholar]
- Ferry Q. R. V.; Lyutova R.; Fulga T. A. (2017) Rational Design of Inducible CRISPR Guide RNAs for de Novo Assembly of Transcriptional Programs. Nat. Commun. 8, 14633. 10.1038/ncomms14633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyngsø R. B.; Anderson J. W. J.; Sizikova E.; Badugu A.; Hyland T.; Hein J. (2012) Frnakenstein: Multiple Target Inverse RNA Folding. BMC Bioinf. 13 (1), 260. 10.1186/1471-2105-13-260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- zu Siederdissen C. H.; Hammer S.; Abfalter I.; Hofacker I. L.; Flamm C.; Stadler P. F. (2013) Computational Design of RNAs with Complex Energy Landscapes. Biopolymers 99 (12), 1124–1136. 10.1002/bip.22337. [DOI] [PubMed] [Google Scholar]
- Findeiß S.; Hammer S.; Wolfinger M. T.; Kühnl F.; Flamm C.; Hofacker I. L. (2018) In Silico Design of Ligand Triggered RNA Switches. Methods 143, 90. 10.1016/j.ymeth.2018.04.003. [DOI] [PubMed] [Google Scholar]
- Taneda A. (2015) Multi-Objective Optimization for RNA Design with Multiple Target Secondary Structures. BMC Bioinf. 16 (1), 280. 10.1186/s12859-015-0706-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodrigo G.; Jaramillo A. (2014) RiboMaker: Computational Design of Conformation-Based Riboregulation. Bioinformatics 30 (17), 2508–2510. 10.1093/bioinformatics/btu335. [DOI] [PubMed] [Google Scholar]
- Buenrostro J. D.; Araya C. L.; Chircus L. M.; Layton C. J.; Chang H. Y.; Snyder M. P.; Greenleaf W. J. (2014) Quantitative Analysis of RNA-Protein Interactions on a Massively Parallel Array Reveals Biophysical and Evolutionary Landscapes. Nat. Biotechnol. 32 (6), 562–568. 10.1038/nbt.2880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denny S. K.; Greenleaf W. J. (2018) Linking RNA Sequence, Structure, and Function on Massively Parallel High-Throughput Sequencers. Cold Spring Harbor Perspect. Biol. a032300 10.1101/cshperspect.a032300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denny S. K.; Bisaria N.; Yesselman J. D.; Das R.; Herschlag D.; Greenleaf W. J. (2018) High-Throughput Investigation of Diverse Junction Elements in RNA Tertiary Folding. Cell 174 (2), 377–390. 10.1016/j.cell.2018.05.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- She R.; Chakravarty A. K.; Layton C. J.; Chircus L. M.; Andreasson J. O. L.; Damaraju N.; McMahon P. L.; Buenrostro J. D.; Jarosz D. F.; Greenleaf W. J. (2017) Comprehensive and Quantitative Mapping of RNA-Protein Interactions across a Transcribed Eukaryotic Genome. Proc. Natl. Acad. Sci. U. S. A. 114 (14), 3619–3624. 10.1073/pnas.1618370114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burgstaller P.; Famulok M. (1994) Isolation of RNA Aptamers for Biological Cofactors by In Vitro Selection. Angew. Chem., Int. Ed. Engl. 33 (10), 1084–1087. 10.1002/anie.199410841. [DOI] [Google Scholar]
- Flamm C.; Hofacker I. L.; Maurer-Stroh S.; Stadler P. F.; Zehl M. (2001) Design of Multistable RNA Molecules. RNA 7, 254–265. 10.1017/S1355838201000863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zalatan J. G.; Lee M. E.; Almeida R.; Gilbert L. A.; Whitehead E. H.; La Russa M.; Tsai J. C.; Weissman J. S.; Dueber J. E.; Qi L. S.; et al. (2015) Engineering Complex Synthetic Transcriptional Programs with CRISPR RNA Scaffolds. Cell 160 (1–2), 339–350. 10.1016/j.cell.2014.11.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mali P.; Aach J.; Stranges P. B.; Esvelt K. M.; Moosburner M.; Kosuri S.; Yang L.; Church G. M. (2013) CAS9 Transcriptional Activators for Target Specificity Screening and Paired Nickases for Cooperative Genome Engineering. Nat. Biotechnol. 31 (9), 833–838. 10.1038/nbt.2675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konermann S.; Brigham M. D.; Trevino A. E.; Joung J.; Abudayyeh O. O.; Barcena C.; Hsu P. D.; Habib N.; Gootenberg J. S.; Nishimasu H.; et al. (2015) Genome-Scale Transcriptional Activation by an Engineered CRISPR-Cas9 Complex. Nature 517 (7536), 583–588. 10.1038/nature14136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X. C.; Wilson S. C.; Hammond M. C. (2016) Next-Generation RNA-Based Fluorescent Biosensors Enable Anaerobic Detection of Cyclic Di-GMP. Nucleic Acids Res. 44 (17), e139 10.1093/nar/gkw580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodrigo G.; Landrain T. E.; Jaramillo A. (2012) De Novo Automated Design of Small RNA Circuits for Engineering Synthetic Riboregulation in Living Cells. Proc. Natl. Acad. Sci. U. S. A. 109 (38), 15271–15276. 10.1073/pnas.1203831109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Callis T. E.; Pandya K.; Seok H. Y.; Tang R.-H.; Tatsuguchi M.; Huang Z.-P.; Chen J.-F.; Deng Z.; Gunn B.; Shumate J.; et al. (2009) MicroRNA-208a Is a Regulator of Cardiac Hypertrophy and Conduction in Mice. J. Clin. Invest. 119 (9), 2772–2786. 10.1172/JCI36154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yanofsky C. (2000) Transcription Attenuation: Once Viewed as a Novel Regulatory Strategy. J. Bacteriol. 182 (1), 1–8. 10.1128/JB.182.1.1-8.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin P.; Choi H. M. T.; Calvert C. R.; Pierce N. A. (2008) Programming Biomolecular Self-Assembly Pathways. Nature 451 (7176), 318–322. 10.1038/nature06451. [DOI] [PubMed] [Google Scholar]
- Green A. A.; Silver P. A.; Collins J. J.; Yin P. (2014) Toehold Switches: De-Novo-Designed Regulators of Gene Expression. Cell 159 (4), 925–939. 10.1016/j.cell.2014.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Penchovsky R.; Breaker R. R. (2005) Computational Design and Experimental Validation of Oligonucleotide-Sensing Allosteric Ribozymes. Nat. Biotechnol. 23 (11), 1424–1433. 10.1038/nbt1155. [DOI] [PubMed] [Google Scholar]
- Penchovsky R. (2012) Engineering Integrated Digital Circuits with Allosteric Ribozymes for Scaling up Molecular Computation and Diagnostics. ACS Synth. Biol. 1 (10), 471–482. 10.1021/sb300053s. [DOI] [PubMed] [Google Scholar]
- Ding Y.; Chan C. Y.; Lawrence C. E. (2006) Clustering of RNA Secondary Structures with Application to Messenger RNAs. J. Mol. Biol. 359 (3), 554–571. 10.1016/j.jmb.2006.01.056. [DOI] [PubMed] [Google Scholar]
- Andronescu M.; Fejes A. P.; Hutter F.; Hoos H. H.; Condon A. (2004) A New Algorithm for RNA Secondary Structure Design. J. Mol. Biol. 336 (3), 607–624. 10.1016/j.jmb.2003.12.041. [DOI] [PubMed] [Google Scholar]
- Zadeh J. N.; Steenberg C. D.; Bois J. S.; Wolfe B. R.; Pierce M. B.; Khan A. R.; Dirks R. M.; Pierce N. A. (2011) NUPACK: Analysis and Design of Nucleic Acid Systems. J. Comput. Chem. 32 (1), 170–173. 10.1002/jcc.21596. [DOI] [PubMed] [Google Scholar]
- Lee J.; Kladwang W.; Lee M.; Cantu D.; Azizyan M.; Kim H.; Limpaecher A.; Gaikwad S.; Yoon S.; Treuille A.; Das R.; EteRNA Participants (2014) RNA design rules from a massive open laboratory. Proc. Natl. Acad. Sci. U. S. A. 111 (6), 2122–2127. 10.1073/pnas.1313039111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- R Core Team (2018) R: A Language and Environment for Statistical Computing, Vienna, Austria.
- Kladwang W.; Mann T. H.; Becka A.; Tian S.; Kim H.; Yoon S.; Das R. (2014) Standardization of RNA Chemical Mapping Experiments. Biochemistry 53 (19), 3063–3065. 10.1021/bi5003426. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.