

Single-cell analysis of CD4 T cells reveals a population structure associated with inflammation and declined immunity in aging.
Abstract
Age-associated changes in CD4 T-cell functionality have been linked to chronic inflammation and decreased immunity. However, a detailed characterization of CD4 T cell phenotypes that could explain these dysregulated functional properties is lacking. We used single-cell RNA sequencing and multidimensional protein analyses to profile thousands of CD4 T cells obtained from young and old mice. We found that the landscape of CD4 T cell subsets differs markedly between young and old mice, such that three cell subsets—exhausted, cytotoxic, and activated regulatory T cells (aTregs)—appear rarely in young mice but gradually accumulate with age. Most unexpected were the extreme pro- and anti-inflammatory phenotypes of cytotoxic CD4 T cells and aTregs, respectively. These findings provide a comprehensive view of the dynamic reorganization of the CD4 T cell milieu with age and illuminate dominant subsets associated with chronic inflammation and immunity decline, suggesting new therapeutic avenues for age-related diseases.
INTRODUCTION
One of the key hallmarks of aging is the deterioration of the immune system, rendering the elderly more prone to infections, chronic inflammatory disorders, and vaccination failure (1). A marked change observed in aging relates to the composition and functionality of CD4 T cells, the main orchestrators of adaptive immune responses (1–3). In young rodents and humans, CD4 T cells comprise a high frequency of naïve cells, reflecting the ability of the immune system to encounter new antigens, respond to them effectively, and generate immune memory (4). With aging, the naïve subset shrinks along with the accumulation of highly differentiated memory cells, which often show dysregulated properties (5, 6). For example, previous studies performed on classical CD4 T cell subsets described reduced proliferation, lower secretion of interleukin-2 (IL-2) cytokine (7, 8), and accumulation of metabolic defects (9), along with higher production of pro-inflammatory mediators (10). Furthermore, regulatory T cells (Tregs) appear to accumulate with age and further contribute to reduced responsiveness of effector T cells (11, 12). These changes are assumed to result from age-related thymus involution, repeated antigen encounters, and intrinsic cellular senescence processes (4, 13). In addition, systemic low-grade chronic inflammation that develops with age, referred to as “inflammaging”, also appears to affect the phenotype and function of CD4 T cells (14, 15). Overall, aging is associated with a change in the structure of the CD4 T cell compartment, where accumulating dysfunctional subsets contribute to immune failure.
Only recently, studies performed at a single-cell resolution showed elevated cell-to-cell transcriptional variability in the activation program of CD4 T cells (16) and in leukocytes from lungs of old mice (17). In line with these findings, increased variation of chromatin modifications was recently observed in immune cells from elderly individuals (18). While these studies provided insights into the functional alterations occurring in aging, they primarily relied on previously described markers of CD4 T cell subsets or on small numbers of cells, which might obscure more complex cellular compositions. Therefore, an unsupervised, large-scale view on the organization of cell subsets within the CD4 T cell population in aging is needed.
Here, we applied 3′-based single-cell RNA sequencing (scRNA-seq) to characterize CD4 T cells from young and aged C57BL/6 mice. Transcriptomic profiles of 24,007 CD4 T cells from both age groups revealed the emergence of distinct activated regulatory, exhausted, and cytotoxic CD4 T cell subsets in aged mice, which were very rare in young mice, and accounted for about 30% of the CD4 T cell compartment of old mice. We further show that these subsets gradually accumulate with age and have distinct pro- and anti-inflammatory functional properties.
RESULTS
scRNA-seq reveals complex gene expression signature of CD4 T cells in aged mice
We generated scRNA-seq data from splenic CD4 T cells of young (2 to 3 months; n = 4) and old (22 to 24 months; n = 4) healthy mice, henceforth denoted young and old cells, respectively (Fig. 1A; fig. S1, A and B; and Materials and Methods). Cells were subjected to two rounds of CD4 enrichment followed by sorting for CD4+TCRb+CD8−CD19−CD11b−NK1.1− cells to achieve highly pure (>99%) CD4 T cells (Fig. 1B and fig. S1C). To assess the gross shift of CD4 T cells from naïve to memory phenotype in aging, we measured canonical surface markers using flow cytometry. As expected (19), we observed a significant reduction in the abundance of naïve cells (CD4+CD62L+CD44−, P = 0.0006) and an increase in the frequency of effector-memory cells (CD4+CD44+CD62L−) in the old versus the young splenic CD4 T cells (Fig. 1C). Next, we sequenced thousands of these cells using the 10x Genomics GemCode Chromium platform (20) to explore the transcriptomic differences between the age groups. After quality control steps, we obtained expression profiles of 13,186 and 10,821 pure CD4 T cells from young and old mice, respectively (figs. S1D and S2A, table S1, and Materials and Methods).
Fig. 1. Gene expression signatures of CD4 T cells in young and old mice.
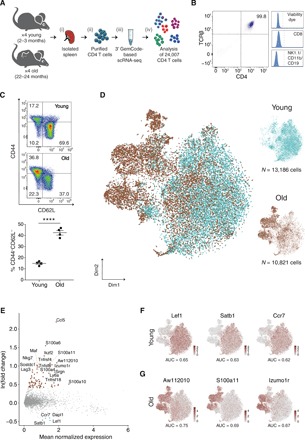
(A) Experimental flowchart: (i) Splenocytes were harvested from young (2 to 3 months, n = 4) and old (22 to 24 months, n = 4) mice; (ii) CD4 T cells were purified using magnetic separation and sorting; (iii) cells’ mRNAs were barcoded using 10x Genomics Chromium platform and sequenced; and (iv) data were computationally analyzed. (B) Representative flow cytometry plots showing highly pure CD4+TCRβ+ T cells after magnetic enrichment and sorting, discarding cells that were positive for CD8, CD19, CD11b, and/or NK1.1. These cells were used for the scRNA-seq experiments. (C) Analysis of the sorted young and old CD4 T cells stained for CD44 and CD62L surface markers. Top: Representative flow cytometry plots of cells from young and old mice. Bottom: Cells from old mice show a shift toward effector-memory identity. Data from two different experiments (n = 2 in each age group, per experiment). Each dot represents a mouse, bars represent mean ± SEM (unpaired t test, ****P < 10−4). (D) t-SNE projections of CD4 T cells including 13,186 and 10,821 cells from young (turquoise) and old (brown) mice, respectively. Each dot represents a single cell. (E) MA plot showing differentially expressed genes between age groups. Each dot represents a gene, with significantly up-regulated genes [ln(fold change) > 0.4, adjusted P < 10−3] in young and old mice colored turquoise and brown, respectively. (F and G) t-SNE projections with cells colored by the expression levels of age marker genes. Markers were selected as differentially expressed genes within an age group [ln(fold change) > 0.4] that best distinguish between age groups according to a receiver operating characteristic analysis [(F) AUC > 0.61, power > 0.23 and (G) AUC > 0.66, power > 0.33].
Next, we applied dimensionality reduction to their profiles. For this, we selected genes with variable expression and projected them on the first 20 principal components (PCs), followed by a t-distributed stochastic neighbor embedding (t-SNE; fig. S2, B and C, and Materials and Methods). Cells did not cluster topologically by depth of sequencing, experimental batch, or individual mouse (fig. S2, D and E). Instead, old cells grouped together in a distinctive manner, thus highlighting the impact of aging on the transcriptome of CD4 T cells (Fig. 1D). Differential expression analysis revealed that while genes up-regulated in young cells [ln(fold change) > 0.4, adjusted P < 10−3] were associated with a naïve phenotype [e.g., Lef1, Ccr7, and Dapl1 genes (16)], genes up-regulated in old cells presented a mix of inflammatory (e.g., S100a4, S100a11, and Ccl5 genes) and regulatory (e.g., Maf, Ikzf2, and Tnfrsf4 genes) signatures (21), as well as exhaustion markers [e.g., Lag3 and Tnfsf8 genes (22); Fig. 1E and Materials and Methods]. In contrast to the relatively homogeneous signature of young cells, the highly expressed inflammatory, regulatory, and exhaustion genes in old cells suggest an intricacy of the CD4 T cell compartment in old mice. Next, we searched for shared genes exclusively expressed in old or young cells by using receiver operating characteristic analysis. We focused on genes that were highly differentially expressed in each age group (Materials and Methods). Lef1, Satb1, and Ccr7 genes were the top three markers common to young cells [AUC (area under the curve) > 0.61 and power > 0.23], supporting the dominancy of naïve CD4 T cells in young age (Fig. 1F). The three top markers common to old cells were the genes Aw112010, S100a11, and Izumo1r (AUC > 0.66 and power > 0.33; Fig. 1G), which were recently reported to be up-regulated under chronic inflammatory conditions (23–25). These genes imply a strong relationship between the inflammatory state and the identity of CD4 T cells in aging and can be used as general markers for aged CD4 T cells.
CD4 T cells undergo extensive diversification with age, resulting in a new population structure
To classify CD4 T cell subsets in an unbiased manner, we clustered cells by their transcriptomic profiles and assessed the robustness of the clusters’ identity [Materials and Methods; (26)]. Combining all cells from young and old mice, we identified seven distinct and robust clusters (Fig. 2A and fig. S2F). To associate each cluster with a known CD4 T cell subset, we screened the most significantly up-regulated genes (combined P < 10−3) of each cluster and compared them to previously reported T cell subsets and to canonical markers (Fig. 2, B and C; fig. S3, A and B; table S2; and Materials and Methods). Of the seven distinct clusters, three were matching established subsets: a population of naïve T cells overexpressing Lef1, Sell, and Igfbp4 genes (denoted naïve); a population of resting regulatory T cells (rTregs), labeled on the basis of their classical expression of Foxp3 and Il2ra (encoding CD25 protein) genes, together with the expression of naïve-associated genes Lef1 and Sell (21); and effector-memory T cells (TEM) expressing the Igals1, S100a4, and Itgb1 genes (27). We also observed a population of naïve CD4 T cells (denoted Naïve_Isg15) expressing the Lef1, Sell, and Igfbp4 genes along with type I interferon (IFN) response genes (Ifit1, Ifit3, Isg15, and Stat1), which possibly indicate an intermediate state of T cell receptor–mediated activation or direct IFN signaling (28). The transcriptional signatures of the three remaining subsets have not been previously defined in the context of aging and include activated regulatory T cells (aTregs) overexpressing Foxp3, Cd81, Cd74, and Cst7 genes, together with aTregs-associated genes such as Tnfrsf4, Tnfrsf9, Tnfrsf18, and Ikzf2 (21, 29, 30); cells with an exhaustion signature (denoted exhausted, fig. S3B) overexpressing the Lag3, Tbc1d4, Sostdc1, and Tnfsf8 genes (22, 31); and cells overexpressing genes such as Eomes, Gzmk, and Ctla2a, which are commonly associated with CD8 T cells (denoted cytotoxic) and were previously described in the context of viral infection and cancer as CD4 cytotoxic T cells (32–34).
Fig. 2. CD4 T cell subset composition in aging.
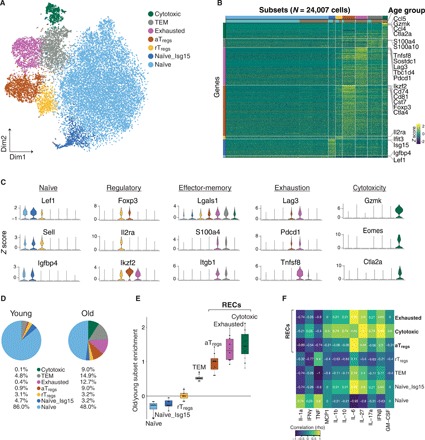
(A) t-SNE projection of all 24,007 CD4 T cells presenting seven different subsets, identified via shared nearest neighbor modularity optimization-based clustering algorithm, followed by merging of similar clusters. (B) Heatmap of gene expression z scores across cells. All CD4 T cells were grouped by subset and age (horizontal bars). Genes shown were up-regulated significantly [ln(fold change) > 0.4, combined P < 10−3] in at least one subset compared to all other cells. Genes were ordered by significance and associated with the subset with higher detection rates. (C) Violin plots showing the expression (z score) of selected canonical marker genes across all seven subsets. (D) Representative pie charts showing the percentage of cells belonging to each of the seven subsets in a young mouse and an old mouse. (E) Enrichment was computed as the log odds ratio between the frequency of each subset in old versus young mice, across all pairs. Naïve subsets were enriched in young mice [Naïve: log(median ratio) = −0.27, P = 0.03 and Naïve_Isg15: log(median ratio) = −0.23, P = 0.03]. rTregs subset was equally distributed [log(median ratio) = 0.02, P = 0.89]. Four subsets were enriched in every old mouse: TEM [log(median ratio) = 0.51, P = 0.03], aTregs [log(median ratio) = 1, P = 0.03], exhausted [log(median ratio) = 1.32, P = 0.03], and cytotoxic [log(median ratio) = 1.46, P = 0.03] subsets. Mann-Whitney U test. (F) A heatmap showing Spearman correlation coefficient (rho) between the frequency of each subset and cytokine concentration (in micrograms per milliliter) measured in serum of old mice. Because of the limited number of mice, we considered as meaningful only strong correlations (absolute Spearman correlation coefficient > 0.6).
Next, we compared the proportion of each subset in old versus young mice (Fig. 2, D and E, and fig. S3C). Whereas the two naïve subsets were significantly enriched in young mice [Naïve: log(median ratio) = −0.27, P = 0.03 and Naïve_Isg15: log(median ratio) = −0.23, P = 0.03], the rTregs subset had a similar abundance in both age groups [log(median ratio) = 0.02, P = 0.89], while the TEM subset was dominant in old mice [log(median ratio) = 0.51, P = 0.03]. Notably, the aTregs, exhausted, and cytotoxic subsets (collectively denoted RECs to represent these regulatory, exhausted, and cytotoxic subsets) were highly enriched in all aged mice [log(median ratio) = 1, P = 0.03; log(median ratio) = 1.32, P = 0.03; and log(median ratio) = 1.46, P = 0.03; respectively], accounting for ~30% of the CD4 T cells and were negligible in young mice (~1%).
Since the frequency of RECs substantially differed between old mice (Fig. 2E and fig. S3C), we sought to examine whether this pattern represents different stages of chronic inflammation. We thus measured the levels of serum cytokines in the old group and analyzed their correlation with the proportion of RECs (Spearman correlations; Fig. 2F and fig. S4A). Whereas the cytotoxic cells were highly correlated with IL-27 (r = 1) and IFNβ (r = 0.95), aTregs and exhausted cells were highly correlated with IL-6 (r = 1 and r = 0.95, respectively).
Overall, these results demonstrate that aging is marked by a complex landscape of CD4 T cells, with increased frequencies of subsets with effector (including TEM, exhausted, and cytotoxic cells; fig. S4B) and regulatory (aTregs) signatures. Notably, the frequency of these subsets was associated with elevated levels of inflammatory cytokines in the sera (primarily IL-27, IFNβ, and IL-6), suggesting a link between the process of inflammaging (35) and the fate of CD4 T cells in aging.
RECs are distinct CD4 T cell subsets exhibiting a gradual increase in frequency with age
To gain insight into the identity of RECs, we compared their expression profiles to the profiles of closely related, well-established subsets (table S3 and Materials and Methods). First, we compared the cytotoxic and exhausted cells to TEMs (Fig. 3, A and B): The cytotoxic cells exhibited higher levels of Nkg7, Eomes, Gzmk, and Runx3 genes, together with genes encoding inflammatory chemokines [e.g., Ccl3, Ccl4, and Ccl5 (36); Fig. 3A]; the exhausted cells expressed relatively high levels of coinhibitory genes [e.g., Cd200, Lag3, and Pdcd1 (22)], as well as genes related to cellular oxidative stress [e.g., Hif1a and Tbc1d4 (37); Fig. 3B]; the TEM subset expressed higher levels of memory-associated genes [e.g., Itgb7, Itgb1, and Il18r1; (27)] in both comparisons. Next, we compared the gene expression profiles of the two Tregs subsets (Fig. 3C). Notably, whereas the rTregs subset expressed higher levels of quiescence genes (e.g., Ms4a4b and Sell), aTregs subset expressed higher levels of coinhibitory (e.g., Pdcd1 and Lag3) and costimulatory genes [e.g., Tnfrsf9 and Tnfrsf4; (38)], activation genes [S100a11 and Il1r2 (IL1 decoy receptor); (39)], and the transcription factor (TF) Ikzf2 (30).
Fig. 3. The genes and proteins characterizing cytotoxic, exhausted, and aTregs subsets.
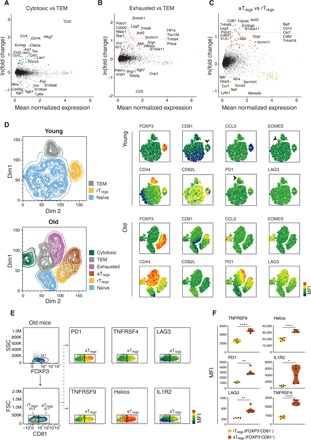
(A to C) MA plots showing differentially expressed genes between (A) cytotoxic and TEM cells, (B) exhausted and TEM cells, and (C) aTregs and rTregs cells. Only cells from old mice were considered in the analysis. Each dot represents a gene; genes that were up-regulated consistently across mice [ln(fold change) > 0.4] were colored by the corresponding subset. (D) Left: Representative t-SNE plots of CD4 T cells coming from young (top) and old (bottom) mice colored by subset identity. Right: Analysis was based on the expression of 10 marker proteins chosen according to the gene expression profiles. The mean fluorescence intensities (MFIs) of each marker are presented. (E) Representative flow cytometry plots gated on FOXP3+ cells showing the MFI of selected marker proteins that relate to Tregs activation, projected on CD81− (rTregs) and CD81+ (aTregs) populations. (F) Violin plots quantitatively showing the MFI of each marker in rTregs and aTregs. Each dot represents a mouse (n = 6, from two different experiments). Paired t test, *P < 0.05, **P < 0.01, and ****P < 10−4.
We further validated the subsets’ identities by measuring the levels of key surface and intracellular proteins (chosen on the basis of the gene expression profiles) in CD4 T cells from young (2 months) and old (20 months) mice, using multicolor flow cytometry. Clustering analysis revealed that whereas the young cells comprised mainly naïve cells and defined clusters of effector-memory and rTregs cells, the old cells included a prominent cytotoxic CD4 T cell subset, expressing high levels of the EOMES (Eomesodermin) TF and the CCL5 chemokine (Fig. 3D and fig. S5A). We also identified naïve, TEM, exhausted (based on the expression of CD62L, CD44, PD1, and LAG3), and the two Tregs subsets (all were FOXP3+ and differed by expression levels of CD81, PD1, and LAG3). Further analysis of the Tregs subsets in the old cells revealed that CD81 [a member of the tetraspanin family (40)] is a specific marker that differentiates between rTregs and aTregs. CD81 was coexpressed with PD1, TNFRSF4, TNFRSF9, Helios, IL1R2, and LAG3, which together affirmed the activated phenotype of aTregs [Fig. 3, E and F; (21)]. The Tregs marker CD25 was similarly expressed by aTregs and rTregs (fig. S5B).
We next sought to assess the dynamics of RECs over time and thus measured their relative abundance in spleens of healthy mice at 2, 6, 12, 16, and 24 months of age, using flow cytometry. The frequency of exhausted cells (defined as CD4+PD1+CD62L−FOXP3−EOMES−CCL5−) steadily increased from 6 months of age (r = 0.94, P = 1.5 × 10−12, Spearman correlation) and coincided with continuous decreased proportions of naïve cells (defined as CD4+CD62L+PD1−FOXP3−EOMES−CCL5−; r = −0.96, P = 1.7 × 10−14, Spearman correlation; Fig. 4A). Out of the regulatory cells (CD4+FOXP3+), the relative abundance of aTregs, (defined as CD81+) also increased with age, reaching a peak at 16 months of age and slightly declining at 24 months [r = 0.64, P = 1.81 × 10−5, Spearman correlation; Fig. 4B]. Cytotoxic cells (defined as CD4+EOMES+CCL5+) were observed in a later time point at 12 months, and their fraction sharply increased with age (r = 0.71, P = 4.0 × 10−8, Spearman correlation; Fig. 4C). To assess whether the high RECs frequencies in old mice reflected an increase in the absolute number of cells, or merely a decrease in the naïve compartment, we measured the absolute numbers of RECs per gram of spleen in young (2 months) and old (24 months) mice (fig. S5C). While a marked decline in total CD4 cells was observed in old mice (P = 0.0014, Fig. 4D), the absolute numbers of cytotoxic and aTregs cells significantly increased (P = 0.016 and P = 0.03, respectively; Fig. 4E). The absolute number of exhausted cells was also increased in old mice but did not significantly differ from young mice (P = 0.21). In general, the number of RECs was highly variable among individual mice, possibly representing the individual aging process.
Fig. 4. Dynamics of RECs across age and immune sites.
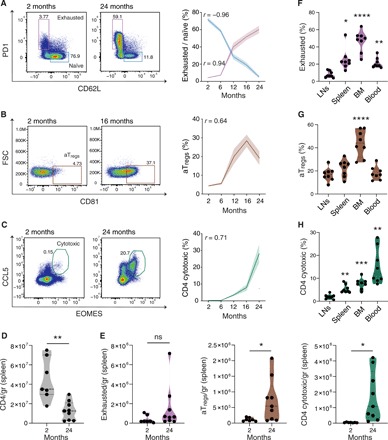
(A to C) Prevalence of RECs and naïve cells measured via flow cytometry (representative panels, left) in mice at the age of 2, 6, 12, 16, and 24 months (right). Color-shaded area of each graph represents ± SEM. Spearman correlation coefficients (r) are shown. (A) Exhausted cells (pink), defined as PD1+CD62L− cells, and naïve cells (blue), defined as CD62L+PD1− out of CD4+FOXP3−EOMES−CCL5−cells. (B) aTregs cells, defined as CD81+ cells out of CD4+FOXP3+ cells. (C) Cytotoxic cells, defined as EOMES+CCL5+ out of CD4+ cells. Gating was based on unstained samples and fluorescence minus one. Data include two (A) or three (B and C) different experiments, n = 5 to 15 per time point. (D and E) Absolute numbers of total CD4 (D) and RECs (E) cells out of total live cells in each spleen, calculated as specific cell count per gram of spleen in young (2 months, n = 7) and old (24 months, n = 9) mice. (F to H) Percentages of RECs in different immune sites: (F) exhausted cells, (G) aTregs cells, and (H) cytotoxic cells. LNs represent the mean cell frequencies of four LN sites. Data include three different experiments, n = 2 to 3 per experiment. P values computed via one-way analysis of variance (ANOVA) test with Tukey correction for multiple comparisons (*P < 0.05, **P < 0.01, and ****P < 10−4).
Since the cellular composition of the spleen is different from other immunological tissues and dynamically changes with aging (41), we also examined site-specific proportions of RECs. For this, we purified CD4 T cells from seven immune compartments of 16-month-old mice, including the spleen, bone marrow (BM), blood, and lymph nodes (LNs) from axillary, cervical, inguinal, and mesenteric sites, and analyzed the percentage of RECs in each compartment using flow cytometry (fig. S5D). All RECs showed the lowest proportion in the LNs. Whereas aTregs were highly abundant in BM, exhausted and cytotoxic cells were enriched also in blood and spleen (Fig. 4, F to H, and fig. S5, E and F). Together, these findings indicate that the RECs’ accumulation with time predominantly occurs in non-lymphatic immune compartments, as opposed to lymphatic sites, which exhibit a relatively preserved T cell composition.
RECs exhibit extreme regulatory and effector properties
As the regulation of CD4 T cell identity and function is largely coordinated by the combined activity of TFs (42), we examined the specific set of working regulons (namely, TFs and their target gene modules) that appear to be active within the identified subsets and specifically within the RECs. For this, we applied the SCENIC workflow on the transcriptional signatures of the old cells (43). We identified 27 high-confidence regulons that were active consistently across all four old mice (Fig. 5A and Materials and Methods). Clustering cells hierarchically by their active regulons revealed that cells were largely grouped by the subsets that we identified. The three regulons that were active in all subsets were governed by the TFs Bclaf1, Elf1, and Ets1, previously shown as key regulators of T cell homeostasis and activation [fig. S6A; (44, 45)]. The RECs exhibited an active Prdm1 regulon, suggesting a certain degree of exhaustion in these subsets (22). Among the RECs, the aTregs subset displayed elevated activity of the regulons corresponding to Foxp3, nuclear factor κB (NF-κB) TF family (Nfkb1, Nfkb2, Rel, and Relb), Irf4, and Maf, which were previously associated with activation phenotype of Tregs [Fig. 5B; (46, 47)]. To assess whether the two Tregs subsets differ in functionality, we sorted aTregs (CD25highCD81+) and rTregs (CD25highCD81−) from old (16 months) and young (2 months) mice (fig. S6B), respectively, and compared their suppressive activity on the proliferation of CD4 T cells, in vitro (Materials and Methods). aTregs exhibited a significantly higher suppressive activity than rTregs (Fig. 5C).
Fig. 5. The regulatory and functional properties of RECs.
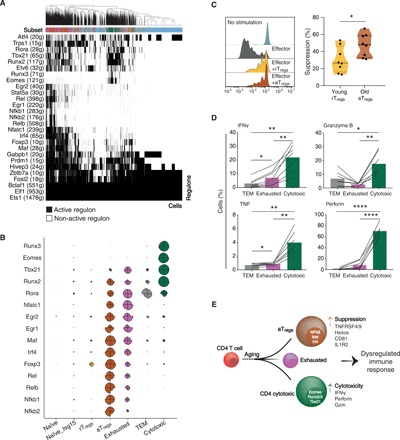
(A) A heatmap of old CD4 T cells showing 27 high-confidence regulons that were active consistently across all old mice. Active regulons per cell appear in black; the horizontal color bar indicates the subset associated with each cell. Numbers in parentheses represent the number of genes comprising the regulon. (B) Radared-balloon plot showing regulons’ activity per subset per mouse. Each circle corresponds to a single subset and is divided into four slices, one per mouse. Slice size reflects the fraction of subset cells with active regulon, normalized to the maximal fraction of that regulon across mice and subsets (fig. S6A shows other regulons). (C) Ex vivo suppression activity of sorted CD25highCD81− (yellow, cells derived from young mice), referred to as rTregs, or CD25highCD81+ (brown, cells derived from old mice), referred to as aTregs, after 72 hours of coculture with activated naïve CD4 T cells from young CD45.1 mice. The reduction in the proliferation of the activated CD4 T cells was measured via flow cytometry and calculated as percentage of suppression (Materials and Methods). Left: Representative flow cytometry plots showing reduced proliferation in the presence of aTregs. Right: Violin plot showing the suppression ability (%) of rTregs versus aTregs. Each dot represents cells pulled from three mice (n = 8, from two independent experiments). Unpaired t test (*P < 0.05). (D) The percentage of cells positive to pro-inflammatory and cytotoxic cytokines in TEM, exhausted, and cytotoxic subsets after 48 hours of activation, measured by flow cytometry. Lines connect measurements within the same mouse. Data from two independent experiments, n = 7 mice. Paired t test (*P < 0.05, **P < 0.01, and ****P < 10−4). (E) Schematic illustration of the accumulation of RECs with age, showing their key transcription factors and markers (within and to the right of each cell, respectively), which point to a dysregulated immune response.
The cytotoxic subset appeared to be dominantly regulated by TFs associated with cytotoxicity and T helper 1 (TH1) polarization, including Eomes, Runx2, Runx3, and Tbx21 regulons [Fig. 5B; (42, 48)]. In activated CD8 T cells, these TFs regulate the production of cytokines and lytic proteins, such as tumor necrosis factor (TNF), IFNγ, granzyme B (GzmB), and perforin (48). To evaluate the activity of these regulons in aged cytotoxic CD4 T cells, we used flow cytometry to measure subset-specific secretion of these molecules from activated CD4 T cells extracted from 16-month-old mice (Materials and Methods). Compared to exhausted and TEM subsets, the cytotoxic subset showed the highest percentage of cells that produced TNF, IFNγ, perforin, and GzmB, indicating their primary contribution to the cytotoxic and pro-inflammatory mediators that were released following stimulation (Fig. 5D and fig. S6C). We also observed higher frequency of cells producing IL-21 and IL-17a within cytotoxic cells (fig. S7A), probably due to the active Rora regulon [Fig. 5B; (49)]. Visualizing the expression of key TH1 and TH17 markers on the t-SNE map revealed primarily TH1—rather than TH17—polarization in the cytotoxic subset (fig. S7, B and C), which was validated by the high versus low percentage of IFNγ- and IL-17–secreting activated cytotoxic CD4 T cells, respectively (fig. S7D). Exhausted cells presented a high activity of Nfatc1 regulon, which is presumably involved in regulating the exhaustion process (50–53). Together, these results demonstrate that each of the three RECs uses defined sets of genes and TFs, which together promote an extreme immune phenotype that can hinder immunity in old age (Fig. 5E).
DISCUSSION
In the current study, we aimed to comprehensively describe how the CD4 T cell compartment is sculptured during the process of aging. Our scRNA-seq and flow cytometry data, together with the functional assays we performed, allowed in-depth characterization of CD4 T cell subsets in aging. We identified a gradual reorganization of the CD4 T cell compartment, where regulatory, exhausted, and cytotoxic phenotypes co-emerge with age.
Our initial analysis of the young and old mRNA profiles pointed to mixed signatures of pro- and anti-inflammatory gene expression in old CD4 T cells, compared with the naïve-associated signature in young cells. Among the top differentially expressed genes in old CD4 T cells, S100a11, Izumor1, and Aw112010 were found to be strong markers of old cells. Up-regulation of these genes in leukocytes was previously reported in the context of long-term inflammation (54). Furthermore, a recent study has shown that the Aw112010 gene is induced in acute and chronic inflammation and is essential for translation of inflammatory proteins (25). Whereas the mechanisms underlying the induction of these genes in old CD4 T cells is yet unclear, together with the increased levels of circulating cytokines and chemokines, they may serve a useful set of biomarkers for immune senescence.
The single-cell analysis we performed uncovered heterogeneity in the CD4 T cell compartment, which is composed of a number of distinct cell subsets in aging. Pro- and anti-inflammatory signatures were thus not common to all old CD4 T cells but were distributed primarily among four distinct subsets, expressing gene sets of TEM, aTregs, exhausted, and cytotoxic cells. Whereas effector-memory and exhaustion phenotypes were already implicated in aging, the aTregs and the cytotoxic CD4 T cell subsets were previously described in cancer and chronic inflammation but not in aging (30, 32). The gene signatures of the aTregs and the cytotoxic CD4 T cells indicate their enhanced regulatory and effector cytotoxic profiles, respectively, suggesting that these subsets play a key role in the deterioration of the immune system with age. Since these subsets accumulate at different stages of aging and are enriched in the BM, blood, and spleen compartments, it is plausible that their molecular phenotype and function represent two parallel processes in the timeline of aging.
The aTregs subset showed a gradual increased frequency with age, reaching ~50% of all FOXP3+ CD4 T cells in the BM. Compared with the rTregs, the aTregs expressed increased levels of activation genes such as Helios, IL1R2, TNFRSF9, and TNFRSF4, as well as their encoded proteins. These marker proteins were coexpressed with CD81, which we found to be a highly specific marker for aTregs in mice. In line with other recent reports, the aTregs were governed by TFs of the NF-kB TF family (Nfkb1, Nfkb2, Rel, and Relb), Irf4, and Maf, which were associated with their distinct functional characteristics (46, 47). Notably, old mice-derived aTregs had a greater suppressive effect than rTregs from young mice in an ex vivo suppression assay. The aTregs we identified are thus reminiscent of Tregs with enhanced suppression activity previously observed within tumors of patients with solid cancers (55). Further research is required to determine the environmental cues that promote the differentiation of aTregs with age, and whether they originate primarily from thymus-derived Tregs or from other cells such as the exhausted subset, which shows similar spatiotemporal and gene module patterns. It is intriguing that while presumably attenuating the process of chronic inflammation, these Tregs may represent a gradual process that substantially compromises immunity to self- and non–self-antigens and thus need to be specifically targeted therapeutically.
Whereas the sharpest increase in frequency of TEMs, exhausted cells, and aTregs occurred between 6 and 12 months of age, the cytotoxic CD4 T cells accumulated primarily after 12 months of age. Furthermore, we found that the gene modules driven by Eomes, Runx2, Runx3, and Tbx21 TFs, which are required for the cytotoxic CD8 lineage, are active in this cytotoxic subset, and that in vitro stimulation of these cells results in a significantly higher expression of pro-inflammatory and cytotoxic molecules, such as IFNγ, TNF, GzmB, and perforin, as compared to the TEM and exhausted subsets. Cytotoxic CD4 cells were previously observed and therapeutically used in mouse models of colitis (33) and cancer (56), and in human viral infection (32, 34). However, neither were they linked to aging nor did they reach the frequencies observed in our findings. While the differentiation pathways, antigen specificity, function, and accumulation of cytotoxic CD4 cells in aging are yet to be revealed, their correlation with increased levels of circulating inflammatory cytokines suggests that they may mark a stage in aging with a robust immune failure and chronic inflammation.
Overall, our data depict a complex and dynamic landscape of CD4 T cells in aging. The composition of the CD4 T cell compartment undergoes massive rearrangement characterized by a decline of naïve cells and co-emerging cytotoxic, exhausted, and aTregs subsets, which at least in part feed the process of chronic inflammation on one hand and declined immunity on the other hand. Notably, whereas this cellular reorganization occurred in all the mice we analyzed, there was a substantial variability in the frequency and dynamics of each subset among individual mice, which, in general, may reflect possible impacts of environmental factors, such as exposure to stress, pathogens, and a diverse microbiome (57). It is thus intriguing to speculate that the dynamics of RECs reflects the aging process in each individual in a manner that might render them prone to a myriad of age-related diseases (such as cancer and neurodegeneration), which commonly exhibit cell senescence, inflammation, and compromised repair mechanisms (5, 58–60). For example, both aTregs and exhausted cells have been directly implicated in the tumor microenvironment as cells that can suppress tumor immunity (29, 30), and thus a decline in T cell effector functions along with chronic inflammation and increased frequencies of RECs may play a key role in age-related onset of cancer. While further work is needed to dissect the mechanisms underlying the accumulation and function of each subset with age, our study opens avenues to the understanding of age-related immune failure and can be relevant for improving diagnostic and therapeutic strategies for its associated pathologies.
MATERIALS AND METHODS
Mice
Wild-type (WT) C57BL/6 and CD45.1 (B6.SJL-Ptprca Pepcb/BoyJ) mice were purchased from the Jackson Laboratory (Bar Harbor, ME) and were housed under specific pathogen–free conditions at the animal facility of Ben-Gurion University. WT C57BL/6 mice were kept in different age batches from 2 to 24 months. All mice were checked for any macroscopic abnormalities (according to the Jackson guide—“AGED C57BL/6J MICE FOR RESEARCH STUDIES”). Animals with skin lesions, organ-specific problems, or behavioral issues were discarded from the study. All surgical and experimental procedures were approved by the Institutional Animal Care and Use committee of Ben-Gurion University of the Negev, Israel.
Sample processing for scRNA-seq
Young (2 to 3 months) and old (22 to 24 months) mice were euthanized using isoflurane overdose. Then, spleens were harvested and mashed into a 70-μm cell strainer. Lysis of red blood cells was performed using 300 μl of Ammonium-Chloride-Potassium (ACK) buffer for 1 min (Lonza, Basel Switzerland). Then, CD4+ T cells were purified from total splenocytes using a magnetic microbeads negative selection kit (EasySep Mouse CD4+ T Cell Isolation Kit, STEMCELL Technologies), according to the manufacturer’s instructions. In all processing steps, wide-bore pipette tips were used, and centrifugation condition did not exceed 400g, to minimize physical damage to the cells caused by shearing forces.
FACS sorting
To enhance purity and discard dead cells, CD4 T cells were further purified using FACS. Cells were sorted as eFluor780-Live cells+ (Fixable Viability dye, eBioscience), PerCP/cy5.5-conjugated anti-CD8− (53-6.7; BioLegend), phycoerythrin (PE)–conjugated anti-CD19− (6D5; BioLegend), PE-conjugated anti-CD11b− (M170; BioLegend), PE-conjugated anti-NK1.1− (PK136; BioLegend), BV421-conjugated anti-CD4+ (GK1.5; BioLegend), and fluorescein isothiocyanate (FITC)–conjugated anti-TCRb+ (H57-597; BioLegend) using the FACSAria III instrument (BD Biosciences, San Jose, CA). To minimize physical stress to the cells, a 100-μm nozzle and 20-psi sheet fluid pressure were used. Cells were sorted into RPMI (Life Technologies)–enriched media with 10% fetal bovine serum, 10 mM Hepes, 1 mM sodium pyruvate, 10 mM nonessential amino acids, 1% penicillin-streptomycin-nystatin, and 50 μM 2-mercaptoethanol (2-ME). After sorting, CD4 T cells in all samples reached >99% purity. Before the loading on Chromium Controller Instrument (10x Genomics), cells were suspended in phosphate-buffered saline (PBS) supplemented with 0.04% bovine serum albumin, counted four times under a light microscope, and passed through a 40-μm strainer (for extended protocol, see “Cell preparation guide,” 10x Genomic protocol; https://support.10xgenomics.com/single-cell-gene-expression/sample-prep). To reach 2000 to 3000 recovered cells, ~5000 cells per sample were loaded to the 10x Genomics platform.
Tissue processing for flow cytometry and in vitro assays
Spleen
See the “Sample processing for scRNA-seq” section.
Lymph nodes
Mice were euthanized with isoflurane overdose, and lymph nodes were harvested from inguinal, mesenteric, cervical, and axillar areas. Then, lymph nodes were mashed into a 70-μm cell strainer, and cells were washed and counted.
Blood
Blood was collected into EDTA-coated tubes (MiniCollect, Greiner Bio-One) from euthanized mice using cardiac puncture. Red blood cells were then lysed using blood lysis buffer (BD Biosciences), and the remaining leukocytes were washed twice and counted.
Bone marrow
Mice were euthanized with isoflurane overdose. Femurs and tibias were collected. Cells from the BM were obtained by flushing the bones with injected sterile PBS. Red blood cells were removed using 500 μl of (ACK) lysis buffer for 1.5 min.
Sequencing library construction using the 10x Genomics Chromium platform
Single-cell suspension was loaded on a Chromium Controller Instrument (10x Genomics) to generate single-cell gel bead-in-emulsions (GEMs). Barcoding and complementary DNA (cDNA) libraries were prepared using the Single-Cell 3′ Library & Gel Bead Kit, according to the manufacturer’s instructions. GEM-reverse transcription (RT) was performed in a Mastercycler Nexus Thermal cycler (Eppendorf). After RT, GEMs were broken, and the cDNA was cleaned up with Dynabeads MyOne Silane Beads (Thermo Fisher Scientific) and amplified. Amplified cDNA product was cleaned up with the SPRIselect Reagent Kit (0.6× SPRI; Beckman Coulter). Indexed sequencing libraries were constructed using the Chromium Single-Cell 3′ Library Kit (10x Genomics) for enzymatic fragmentation, end-repair, A-tailing, adaptor ligation, post-ligation cleanup, sample index polymerase chain reaction (PCR), and PCR cleanup. Final libraries were quantified by Qubit, TapeStation, and quantitative PCR using NEBNext Library Quant Kit (BioLabs). Libraries were loaded at 2 to 2.2 pM on an Illumina NextSeq500 using the high-output 75-cycle kit with the following parameters: 26 bp for Read1, 56 bp for Read2, and 8 bp for I7 Index. Each library was sequenced twice using Illumina’s NextSeq 500 sequencing platform. Sequencing depth was 50,000 to 100,000 reads per cell.
RNA sequencing data processing
The output Illumina base call files from both sequencing runs were merged and converted to FASTQ files using Cell Ranger software v2.0.1, which included Illumina’s bcl2fastq v2.19.1.403. FASTQ files of each mouse sample were converted to count matrices using Cell Ranger software, based on transcriptome reference of Mus musculus (mm10 1.2.0). Barcodes (25,707) were identified as cells, according to Cell Ranger’s algorithm of calling cell barcodes. This algorithm takes as input the expected number of recovered cells, which was set to 3000 cells in our study. It then orders all barcodes by their unique molecular identifier (UMI) content and computes the number of UMIs at the 99th percentile. All barcodes whose total UMI count was at most 1/10 of this number were referred to as low-quality cells and discarded.
Quality control
Quality control and further analyses were done in R v3.4.2, using Seurat package v2.0.1 [fig. S1D; (26)]. To discard doublets, cells were ordered by their number of UMIs, and the top (N/1000) percentage of cells per sample were filtered out, where N is the total number of called cell barcodes identified per sample (20). To discard broken cells (lysed cells with retained mitochondrial RNA), cells with more than 5% UMIs mapped to mitochondrial DNA were filtered out (61). The numbers of cells per sample filtered out by the different criteria appear in table S1. Together, 24,007 cells were kept for further analysis after quality control. Within each cell, the expression level of each gene was normalized cell-wise, by using the NormalizeData function of Seurat. This function divides the expression level of a gene by the total expression of all genes in that cell, multiplies the quotient by a scale factor of 10,000, and log-transforms the product (base e) after the addition of a pseudocount of 1.
Dimensionality reduction
Dimensionality reduction was done using Seurat package in R. First, we selected variable genes by using the FindVariableGenes function. To identify variable genes across a range of expression values, this function first computes the mean expression and dispersion [i.e., log(variance/mean)] per gene. Then, it bins genes according to their mean expression and calculates z scores for dispersion within each bin. Last, it then uses the dispersion-based z scores to select variable genes from the different bins. Only genes with mean expression between 0.0125 and 2 and dispersion >0.9 were considered (fig. S2B), resulting in 2399 variable genes. These variable genes were then used as input to linear dimensionality reduction based on principal component analysis (PCA), executed via the RunPCA function. This function identifies the PCs that account for the largest variability in the data, in a decreasing order. We estimated the significance of PCs by using the JackStraw function. This function repeatedly permutes a subset of the genes and calculates the PC scores of each gene to assess the likelihood that the PC score of a gene was obtained by chance. Accordingly, significant PCs are those enriched for genes with low P values. To create the t-SNE projections, the first 20 PCs (all of which were significant, P < 7.2 × 10−18) were used via RunTSNE function by the elbow method (fig. S2C). Perplexity of the t-SNE projection was set to 10, 30 (default), 50, 100, and 150, and showed similar shapes of projections. Thus, default perplexity was used.
Differential gene expression analysis and discriminating markers between cells from young and old mice
Both analyses were carried via Seurat R package, using FindMarkers function. In the differential gene expression analysis, only genes whose differential expression was relatively high and significant [|ln(fold change)| > 0.4, adjusted P < 10−3] were considered as differentially expressed. In the discriminating markers analysis, age group markers were calculated using the FindMarkers function in Seurat, with test.use parameter set to “roc.” This function calculates per gene its differential expression (fold change and P value) between age groups. The function uses a standard AUC classifier to estimate the ability to correctly assign a cell to its age group based on the expression level of the putative marker gene.
Differential gene expression analysis between RECs and their closely related well-established subsets was done by using cells from old mice and was calculated via the FindMarkers function (using the Wilcoxon rank sum statistical test). Only highly and significantly expressed genes were counted as differentially expressed [|ln(fold change)| > 0.4, adjusted P < 10−3].
Clustering process, robustness analysis, and subsets’ identity association
We clustered cells that passed our quality control by applying the Seurat FindClusters function. This function calculates k-nearest neighbors according to the PCA and constructs a shared nearest neighbor graph. It then optimizes a modularity function (Louvain algorithm) to determine cell clusters. We used the first 20 PCs and calculated the k-nearest neighbors according to the expression profiles of variable genes in all cells. To cope with overclustering of large cell subsets, we assessed the confidence of cluster splits. First, using the BuildClusterTree function, we averaged the gene expression within each cluster to create a representative cluster signature, which we hierarchically ordered, resulting in a cluster tree. Second, we applied the AssessNodes function to each split within the tree. This function judges the confidence of the split by testing the assignment of cells to one cluster or the other, using out-of-bag (OOB) error of a random forest classifier. Split clusters that were indistinguishable from each other (OOB error > 0.05) were merged. To assess the robustness of the seven identified clusters, we tested whether results differ upon selection of different numbers of PCs and different values for the resolution parameter. We repeated the clustering process based on the first 10, 19, and 21 PCs. These led to similar clustering patterns on the t-SNE projection. We set the resolution parameter to several values [0.6, 0.8 (default), 1, and 1.2, as recommended by Seurat]. The different values resulted in varying numbers of clusters, yet clusters were then merged by the AssessNodes function to seven clusters with similar positions on the t-SNE projections. To test the sensitivity of the analysis to the dispersion threshold selected (0.9), we repeated the analyses upon setting it to 0.87 and 1.4, which resulted in 3231 and 277 variable genes, respectively. As can be seen (fig. S2F), the t-SNEs obtained for the different thresholds were similar to each other. Moreover, subsets were colocalized to similar, well-defined regions of the t-SNE, together supporting the robustness of the analysis to the selection of variable genes.
To identify subset-specific markers, we applied the FindConservedMarkers function in Seurat to each subset. For each age group, this function computes the differential expression (fold change and P value) of genes in the studied subset relative to all other cells. It then calculates a combined P value to identify genes whose differential expression is conserved between the age groups. Only highly and significantly expressed genes were counted as differentially expressed [|ln(fold change)| > 0.4 and combined P < 10−3]. To associate each cluster with a potential identity, we manually compared the top differentially expressed genes per subset to canonical markers of previously reported CD4 T cell subsets.
We also examined the similarities between the identified subsets to previously published datasets of T cells: a dataset of naïve and TEM subsets (16); a dataset of Tregs in resting and activated states (21), pertinent to our Tregs subsets; and a dataset of tumor-infiltrating lymphocytes (22), related to our cytotoxic and exhausted subsets (fig. S3A). First, we compared gene signatures of each pair of subsets using Jaccard index-based metric, implemented via the matchSCore package (62). Gene signatures were defined as the top 500 most up-regulated genes of each subset, relative to other subsets taken from the same study (a pseudocount of 1 was added to the expression level of each gene to allow fold change analysis). Second, Spearman correlation coefficients between the gene expression profiles of each pair of subsets were calculated. Expression profiles included only genes expressed above noise levels (mean normalized expression > 0.0125) in both subsets, resulting in at least 7400 genes per comparison.
Regulons
Regulons were identified in SCENIC v0.1.6 package in R (43). We used expression data pertaining to cells from old mice. Only genes with at least one raw count in at least one cell were analyzed throughout the pipeline. All gene raw counts were then log-transformed. Regulons were identified by SCENIC only if they were found to be active in more than 1% of all cells and correlated with at least one other regulon (|r| > 0.3). In downstream analysis, we focused on high-confidence regulons, which we defined as active in more than 10% of the cells in at least one of the subsets in all mice and including high-confidence relationships between the transcription factor and at least nine target genes.
Flow cytometry
For extracellular staining, cells were washed with FACS staining buffer (PBS supplemented with 2% fetal bovine serum and 1 mM EDTA) and incubated with Fc receptor blocker (TrueStain fcX; BioLegend) for 5 min at 4°C. To differentiate between live and dead cells, a viability staining step was done using the eFluor780-Fixable Viability Dye (eBioscience) following the manufacturer’s instructions. Cells were then incubated with primary antibodies for 25 min at 4°C and were washed twice with a FACS staining buffer. The following antibodies were used for membranal staining: PE-conjugated anti-CTLA4 (4C10-4B9; BioLegend), PE/cy7-conjugated anti-CD25 (3C7; BioLegend), AF700-conjugated anti-CD62L (Mel-14; BioLegend), BV605- or BV785-conjugated anti-PD1 (29F.1A12; BioLegend), APC-conjugated anti-CD81 (Eat-2; BioLegend), FITC-conjugated anti-CD8 (53-6.7; BioLegend), PerCP/cy5.5-conjugated anti-CD44 (IM7; BioLegend), AF700- or BV785-conjugated anti-CD4 (RM4-5; BioLegend), PE-conjugated anti-CD121b (4E2; BD Biosciences), BV421-conjugated anti-CD25 (PC61; BioLegend), BV605-conjugated anti-CD195 (C34-3448; BD Biosciences), BV785-conjugated anti-LAG3 (C9B7W; BioLegend), BV421-conjugated anti-CD4 (GK1.5; BioLegend), PerCP/cy5.5-conjugated anti-CD8 (53-6.7; BioLegend), PE-conjugated anti-CD137 (17B5; BioLegend), PE/cy7-conjugated anti-CD134 (OX-86; BioLegend), and PE-conjugated anti-CD178 (MFL3; eBioscience). After staining for membranal markers, intracellular labeling was performed: Cells were fixed and permeabilized using the FOXP3/Transcription Factor Staining Kit (eBioscience), blocked with Rat serum (1 μl per 100 μl of staining buffer), and stained with the following antibodies: BV605-conjugated anti-TNF (MP6-XT22; BioLegend), BV605-conjugated anti–IL-17a (TC11-18H10.1; BioLegend), FITC- or BV510-conjugated anti–IL-2 (JES6-5H4; BioLegend), BV421- or BV786-conjugated anti-IFNγ (XMG1.2; BioLegend), BV421-conjugated anti–IL-10 (JES5-16E3; BioLegend), APC-conjugated anti-GzmB (QA16A02; BioLegend), PE-conjugated anti-CCL5 (2E9/CCL5; BioLegend), PE/cy7-conjugated anti-EOMES (Dan11mag; eBioscience), AF488-conjugated anti-FOXP3 (150D; BioLegend), APC-conjugated anti–IL-21 (149204; R&D systems), PE/dazzle-conjugated anti–Helios (22F6; BioLegend), and APC-conjugated anti-Perforin (B-D48; BioLegend). All flow cytometry experiments were performed with the CytoFLEX instrument (Beckman Coulter). Data were analyzed with the FlowJo (v10.5.3) software. Gating strategies were set on the basis of fluorescence minus one, unstained samples, and unstimulated samples (when needed). All the samples in the experiment excluded dead cells, clumps, and debris.
Clustering analysis of flow cytometry data
Clustering analysis of flow cytometry data was done using FlowJo (v10.5.3). First, we excluded dead cells, doublets, and non-lymphocyte cells (based on viability staining and FSC/SSC parameters). CD4+ cells were used for further analysis. Data were sampled using the “down sampling” function to obtain 40,000 representative cells from each sample. Then, we applied t-SNE algorithm with the following parameters: Iteration = 1000, Perplexity = 40, and Learning rate (eta) = 2800. Mean fluorescence intensity projected on the t-SNE plots for each protein to infer the cluster’s identity.
Proliferation assay and cytokine measurements
CD4+ T cells were isolated from spleens of mice with a magnetic microbeads negative selection kit (EasySep Mouse CD4+ T Cell Isolation Kit; STEMCELL Technologies) and labeled with 5 μM carboxyfluorescein diacetate succinimidyl ester (CFSE; CellTrace Proliferation Kit, Invitrogen) for 5 min in PBS containing 5% fetal bovine serum, at room temperature. After incubation, cells were washed twice with PBS, resuspended in RPMI-enriched medium (composition stated above), and activated with anti-CD3/anti-CD28 beads (Dynabeads, Gibco) in 96-well (U shape) plates (80 × 105 cells per well). After 72 hours, cells were collected and analyzed by flow cytometry.
Cytokine measurements
Isolated CD4+ T cells were incubated with anti-CD3/anti-CD28 beads for 48 hours. Six hours before analysis of TNF, IL-10, IFNγ, GzmB, perforin, IL-17a, IL-21, IL-2, and CCL5, cells were treated with brefeldin A (GolgiPlug, BD Biosciences) and cell culture (1 μl/ml, containing ~106 cells). The cells producing the cytokines were identified with flow cytometry (see the “Flow cytometry” section). Cells were cultured at 37°C and 5% CO2.
Suppression assay
For in vitro suppression assay, naïve CD4+ T cells were isolated from spleens of young (2 months) CD45.1 mice using a naïve isolation kit (EasySep Mouse Naïve CD4+ T Cell Isolation Kit, STEMCELL Technologies), labeled with CFSE (CellTrace Proliferation Kit, Invitrogen), and used as responder cells (2 × 104 cells per well). Then, cells were cultured in 96-well plates with irradiated 2 × 104 APCs (as feeder cells) in the presence of sorted CD25highCD81− or CD25highCD81+ Tregs at 1:1, 1:2, and 1:4 responders:Tregs ratios. Cells were stimulated with anti-CD3 (1 μg/ml) for 72 hours. Proliferation (defined as all cells with CFSE dilution) of responder cells was analyzed to assess the suppression of Tregs cells. The percentage of suppression was determined as [100 − (% of proliferating cells with Tregs)/(% of proliferating cells without Tregs)].
Serum cytokine measurements
Mouse peripheral blood was extracted after right atrial puncture into a 2-ml Eppendorf tube. Then, blood tubes were incubated at room temperature for coagulation (15 min). After incubation, tubes undergo a centrifugation step (450g), and serum was collected. For cytokine measurement, we used LEGENDplex Mouse Inflammation Kit (BioLegend) following the manufacturer’s instruction. Data were acquired on a CytoFLEX instrument (Beckman Coulter) and analyzed using LEGENDplex analysis software (BioLegend).
Statistical analysis for flow cytometry experiments
Spearman correlation between the age of mice and the proportions of RECs and naïve cells in spleen (Fig. 4, A to C) was computed in R v3.4.2 using stats package v3.4.1. For statistical analysis, GraphPad Prism (version 7.0a) was used. Paired t test was used for comparisons between two groups from the same biological samples. For analysis of more than two groups, one-way ANOVA was used, corrected by Bonferroni correction for multiple comparisons.
Supplementary Material
Acknowledgments
We thank M. Sklarz and L. Levin from the Bioinformatics Core Facility in the National Institute for Biotechnology in the Negev for help in computational analysis; H. Keren-Shaul and the Advanced sequencing technologies (Sandbox) unit from the Weizmann Institute of Science for assistance with 10x Genomics Platform and helpful discussions; and T. Pecht and Y. Greenshpan from the National Institute for Biotechnology in the Negev and U. Hadad from the Ilse Katz Institute for Nanoscale Science and Technology in Ben-Gurion University of the Negev for technical support and helpful discussions. Funding: This work was supported by the Israel Science Foundation (no. 684/14) and the Litwin and Gural Foundations. Author contributions: Y.E., I.H., I.E.-M., N.F., V.C.-C., E.Y.-L., and A.M. designed the experiments; Y.E. performed the experimental analysis; I.H., A.V., and E.S. performed the computational analysis; I.E.-M., O.B., I.S., M.S., K.M., A.N., and E.E. provided technical support; Y.E., I.H., I.E.-M., N.F., E.Y.-L., and A.M. wrote the manuscript. All authors commented on and approved the manuscript. Competing interests: I.H., E.Y.-L., A.M., and Y.E. are inventors on a provisional patent application related to this work, filed by the National Institute of Biotechnology in the Negev, Ben-Gurion University (no. 62813235, 4 March 2019). The other authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and the Supplementary Materials. Additional data related to this paper can be obtained in Single Cell Portal (https://portals.broadinstitute.org/single_cell/study/SCP490/aging-promotes-reorganization-of-the-cd4-t-cell-landscape-toward-extreme-regulatory-and-effector-phenotypes) or may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/5/8/eaaw8330/DC1
Fig. S1. Purification of CD4 T cells from young and old mice.
Fig. S2. Dimensionality reduction of scRNA-seq data.
Fig. S3. Subset identity and abundance in mice used for scRNA-seq.
Fig. S4. RECs activation state and correlation with serum cytokines.
Fig. S5. RECs abundance with time and across different immunological sites.
Fig. S6. Gene regulatory networks and cytokine secretion in RECs.
Fig. S7. Characterization of T helper polarizations within RECs.
Table S1. The number of cells per mouse before and after quality control.
Table S2. Subset and age-group expression of genes.
Table S3. RECs comparison to closely related subsets.
REFERENCES AND NOTES
- 1.Goronzy J. J., Weyand C. M., Successful and maladaptive T cell aging. Immunity 46, 364–378 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Nikolich-Žugich J., Aging of the T cell compartment in mice and humans: from no naive expectations to foggy memories. J. Immunol. 193, 2622–2629 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Alpert A., Pickman Y., Leipold M., Rosenberg-Hasson Y., Ji X., Gaujoux R., Rabani H., Starosvetsky E., Kveler K., Schaffert S., Furman D., Caspi O., Rosenschein U., Khatri P., Dekker C. L., Maecker H. T., Davis M. M., Shen-Orr S. S., A clinically meaningful metric of immune age derived from high-dimensional longitudinal monitoring. Nat. Med. 25, 487–495 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nikolich-Žugich J., The twilight of immunity: Emerging concepts in aging of the immune system. Nat. Immunol. 19, 10–19 (2018). [DOI] [PubMed] [Google Scholar]
- 5.Akbar A. N., Henson S. M., Lanna A., Senescence of T lymphocytes: Implications for enhancing human immunity. Trends Immunol. 37, 866–876 (2016). [DOI] [PubMed] [Google Scholar]
- 6.Tsukamoto H., Clise-Dwyer K., Huston G. E., Duso D. K., Buck A. L., Johnson L. L., Haynes L., Swain S. L., Age-associated increase in lifespan of naive CD4 T cells contributes to T-cell homeostasis but facilitates development of functional defects. Proc. Natl. Acad. Sci. U.S.A. 106, 18333–18338 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Li G., Yu M., Lee W.-W., Tsang M., Krishnan E., Weyand C. M., Goronzy J. J., Decline in miR-181a expression with age impairs T cell receptor sensitivity by increasing DUSP6 activity. Nat. Med. 18, 1518–1524 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Haynes L., Eaton S. M., Burns E. M., Randall T. D., Swain S. L., CD4 T cell memory derived from young naive cells functions well into old age, but memory generated from aged naive cells functions poorly. Proc. Natl. Acad. Sci. U.S.A. 100, 15053–15058 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lanna A., Gomes D. C. O., Muller-Durovic B., McDonnell T., Escors D., Gilroy D. W., Lee J. H., Karin M., Akbar A. N., A sestrin-dependent Erk-Jnk-p38 MAPK activation complex inhibits immunity during aging. Nat. Immunol. 18, 354–363 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Harpaz I., Bhattacharya U., Elyahu Y., Strominger I., Monsonego A., Old mice accumulate activated effector CD4 T cells refractory to regulatory T cell-induced immunosuppression. Front. Immunol. 8, 283 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Han G.-M., Zhao B., Jeyaseelan S., Feng J.-M., Age-associated parallel increase of Foxp3+CD4+ regulatory and CD44+CD4+ memory T cells in SJL/J mice. Cell. Immunol. 258, 188–196 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Haynes L., Maue A. C., Effects of aging on T cell function. Curr. Opin. Immunol. 21, 414–417 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Carr E. J., Dooley J., Garcia-Perez J. E., Lagou V., Lee J. C., Wouters C., Meyts I., Goris A., Boeckxstaens G., Linterman M. A., Liston A., The cellular composition of the human immune system is shaped by age and cohabitation. Nat. Immunol. 17, 461–468 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tsukamoto H., Senju S., Matsumura K., Swain S. L., Nishimura Y., IL-6-mediated environmental conditioning of defective Th1 differentiation dampens antitumour immune responses in old age. Nat. Commun. 6, 6702 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Furman D., Chang J., Lartigue L., Bolen C. R., Haddad F., Gaudilliere B., Ganio E. A., Fragiadakis G. K., Spitzer M. H., Douchet I., Daburon S., Moreau J.-F., Nolan G. P., Blanco P., Déchanet-Merville J., Dekker C. L., Jojic V., Kuo C. J., Davis M. M., Faustin B., Expression of specific inflammasome gene modules stratifies older individuals into two extreme clinical and immunological states. Nat. Med. 23, 174–184 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Martinez-Jimenez C. P., Eling N., Chen H.-C., Vallejos C. A., Kolodziejczyk A. A., Connor F., Stojic L., Rayner T. F., Stubbington M. J. T., Teichmann S. A., de la Roche M., Marioni J. C., Odom D. T., Aging increases cell-to-cell transcriptional variability upon immune stimulation. Science 355, 1433–1436 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Angelidis I., Simon L. M., Fernandez I. E., Strunz M., Mayr C. H., Greiffo F. R., Tsitsiridis G., Ansari M., Graf E., Strom T.-M., Nagendran M., Desai T., Eickelberg O., Mann M., Theis F. J., Schiller H. B., An atlas of the aging lung mapped by single cell transcriptomics and deep tissue proteomics. Nat. Commun. 10, 963 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cheung P., Vallania F., Warsinske H. C., Donato M., Schaffert S., Chang S. E., Dvorak M., Dekker C. L., Davis M. M., Utz P. J., Khatri P., Kuo A. J., Single-cell chromatin modification profiling reveals increased epigenetic variations with aging. Cell 173, 1385–1397.e14 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Moro-García M. A., Alonso-Arias R., López-Larrea C., When aging reaches CD4+ T-cells: Phenotypic and functional changes. Front. Immunol. 4, 107 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zheng G. X. Y., Terry J. M., Belgrader P., Ryvkin P., Bent Z. W., Wilson R., Ziraldo S. B., Wheeler T. D., McDermott G. P., Zhu J., Gregory M. T., Shuga J., Montesclaros L., Underwood J. G., Masquelier D. A., Nishimura S. Y., Schnall-Levin M., Wyatt P. W., Hindson C. M., Bharadwaj R., Wong A., Ness K. D., Beppu L. W., Deeg H. J., McFarland C., Loeb K. R., Valente W. J., Ericson N. G., Stevens E. A., Radich J. P., Mikkelsen T. S., Hindson B. J., Bielas J. H., Massively parallel digital transcriptional profiling of single cells. Nat. Commun. 8, 14049 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zemmour D., Zilionis R., Kiner E., Klein A. M., Mathis D., Benoist C., Single-cell gene expression reveals a landscape of regulatory T cell phenotypes shaped by the TCR. Nat. Immunol. 19, 291–301 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chihara N., Madi A., Kondo T., Zhang H., Acharya N., Singer M., Nyman J., Marjanovic N. D., Kowalczyk M. S., Wang C., Kurtulus S., Law T., Etminan Y., Nevin J., Buckley C. D., Burkett P. R., Buenrostro J. D., Rozenblatt-Rosen O., Anderson A. C., Regev A., Kuchroo V. K., Induction and transcriptional regulation of the co-inhibitory gene module in T cells. Nature 558, 454–459 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Andrés Cerezo L., Šumová B., Prajzlerová K., Veigl D., Damgaard D., Nielsen C. H., Pavelka K., Vencovský J., Šenolt L., Calgizzarin (S100A11): A novel inflammatory mediator associated with disease activity of rheumatoid arthritis. Arthritis Res. Ther. 19, 79 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hibiya S., Tsuchiya K., Hayashi R., Fukushima K., Horita N., Watanabe S., Shirasaki T., Nishimura R., Kimura N., Nishimura T., Gotoh N., Oshima S., Okamoto R., Nakamura T., Watanabe M., Long-term inflammation transforms intestinal epithelial cells of colonic organoids. J. Crohns Colitis 11, 621–630 (2017). [DOI] [PubMed] [Google Scholar]
- 25.Jackson R., Kroehling L., Khitun A., Bailis W., Jarret A., York A. G., Khan O. M., Brewer J. R., Skadow M. H., Duizer C., Harman C. C. D., Chang L., Bielecki P., Solis A. G., Steach H. R., Slavoff S., Flavell R. A., The translation of non-canonical open reading frames controls mucosal immunity. Nature 564, 434–438 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Butler A., Hoffman P., Smibert P., Papalexi E., Satija R., Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat. Biotechnol. 36, 411–420 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Weng N.-p., Araki Y., Subedi K., The molecular basis of the memory T cell response: Differential gene expression and its epigenetic regulation. Nat. Rev. Immunol. 12, 306–315 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Szabo P. A., Levitin H. M., Miron M., Snyder M. E., Senda T., Yuan J., Cheng Y. L., Bush E. C., Dogra P., Thapa P., Farber D. L., Sims P. A., A single-cell reference map for human blood and tissue T cell activation reveals functional states in health and disease. bioRxiv, 555557 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zheng C., Zheng L., Yoo J.-K., Guo H., Zhang Y., Guo X., Kang B., Hu R., Huang J. Y., Zhang Q., Liu Z., Dong M., Hu X., Ouyang W., Peng J., Zhang Z., Landscape of infiltrating T cells in liver cancer revealed by single-cell sequencing. Cell 169, 1342–1356.e16 (2017). [DOI] [PubMed] [Google Scholar]
- 30.Guo X., Zhang Y., Zheng L., Zheng C., Song J., Zhang Q., Kang B., Liu Z., Jin L., Xing R., Gao R., Zhang L., Dong M., Hu X., Ren X., Kirchhoff D., Roider H. G., Yan T., Zhang Z., Global characterization of T cells in non-small-cell lung cancer by single-cell sequencing. Nat. Med. 24, 978–985 (2018). [DOI] [PubMed] [Google Scholar]
- 31.Wherry E. J., Kurachi M., Molecular and cellular insights into T cell exhaustion. Nat. Rev. Immunol. 15, 486–499 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Patil V. S., Madrigal A., Schmiedel B. J., Clarke J., O’Rourke P., de Silva A. D., Harris E., Peters B., Seumois G., Weiskopf D., Sette A., Vijayanand P., Precursors of human CD4+ cytotoxic T lymphocytes identified by single-cell transcriptome analysis. Sci. Immunol. 3, eaan8664 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mucida D., Husain M. M., Muroi S., van Wijk F., Shinnakasu R., Naoe Y., Reis B. S., Huang Y., Lambolez F., Docherty M., Attinger A., Shui J. W., Kim G., Lena C. J., Sakaguchi S., Miyamoto C., Wang P., Atarashi K., Park Y., Nakayama T., Honda K., Ellmeier W., Kronenberg M., Taniuchi I., Cheroutre H., Transcriptional reprogramming of mature CD4+ helper T cells generates distinct MHC class II-restricted cytotoxic T lymphocytes. Nat. Immunol. 14, 281–289 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Juno J. A., van Bockel D., Kent S. J., Kelleher A. D., Zaunders J. J., Munier C. M. L., Cytotoxic CD4 T cells—Friend or foe during viral infection? Front. Immunol. 8, 19 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Franceschi C., Campisi J., Chronic inflammation (inflammaging) and its potential contribution to age-associated diseases. J. Gerontol. A Biol. Sci. Med. Sci. 69 ( suppl. 1), S4–S9 (2014). [DOI] [PubMed] [Google Scholar]
- 36.Conti P., DiGioacchino M., MCP-1 and RANTES are mediators of acute and chronic inflammation. Allergy Asthma Proc. 22, 133–137 (2001). [DOI] [PubMed] [Google Scholar]
- 37.MacIver N. J., Michalek R. D., Rathmell J. C., Metabolic regulation of T lymphocytes. Annu. Rev. Immunol. 31, 259–283 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ward-Kavanagh L. K., Lin W. W., Šedý J. R., Ware C. F., The TNF receptor superfamily in co-stimulating and co-inhibitory responses. Immunity 44, 1005–1019 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wang D., Zhang S., Li L., Liu X., Mei K., Wang X., Structural insights into the assembly and activation of IL-1β with its receptors. Nat. Immunol. 11, 905–911 (2010). [DOI] [PubMed] [Google Scholar]
- 40.Hemler M. E., Tetraspanin functions and associated microdomains. Nat. Rev. Mol. Cell Biol. 6, 801–811 (2005). [DOI] [PubMed] [Google Scholar]
- 41.Shifrut E., Baruch K., Gal H., Ndifon W., Deczkowska A., Schwartz M., Friedman N., CD4+ T cell-receptor repertoire diversity is compromised in the spleen but not in the bone marrow of aged mice due to private and sporadic clonal expansions. Front. Immunol. 4, 379 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhu J., Yamane H., Paul W. E., Differentiation of effector CD4 T cell populations. Annu. Rev. Immunol. 28, 445–489 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Aibar S., González-Blas C. B., Moerman T., Huynh-Thu V. A., Imrichova H., Hulselmans G., Rambow F., Marine J.-C., Geurts P., Aerts J., van den Oord J., Atak Z. K., Wouters J., Aerts S., SCENIC: Single-cell regulatory network inference and clustering. Nat. Methods 14, 1083–1086 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.McPherson J. P., Sarras H., Lemmers B., Tamblyn L., Migon E., Matysiak-Zablocki E., Hakem A., Azami S. A., Cardoso R., Fish J., Sanchez O., Post M., Hakem R., Essential role for Bclaf1 in lung development and immune system function. Cell Death Differ. 16, 331–339 (2009). [DOI] [PubMed] [Google Scholar]
- 45.Muthusamy N., Barton K., Leiden J. M., Defective activation and survival of T cells lacking the Ets-1 transcription factor. Nature 377, 639–642 (1995). [DOI] [PubMed] [Google Scholar]
- 46.Cretney E., Xin A., Shi W., Minnich M., Masson F., Miasari M., Belz G. T., Smyth G. K., Busslinger M., Nutt S. L., Kallies A., The transcription factors Blimp-1 and IRF4 jointly control the differentiation and function of effector regulatory T cells. Nat. Immunol. 12, 304–311 (2011). [DOI] [PubMed] [Google Scholar]
- 47.Grinberg-Bleyer Y., Oh H., Desrichard A., Bhatt D. M., Caron R., Chan T. A., Schmid R. M., Hayden M. S., Klein U., Ghosh S., NF-κB c-Rel Is crucial for the regulatory T cell immune checkpoint in cancer. Cell 170, 1096–1108.e13 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Pearce E. L., Mullen A. C., Martins G. A., Krawczyk C. M., Hutchins A. S., Zediak V. P., Banica M., DiCioccio C., Gross D. A., Mao C.-a., Shen H., Cereb N., Yang S. Y., Lindsten T., Rossant J., Hunter C. A., Reiner S. L., Control of effector CD8+ T cell function by the transcription factor Eomesodermin. Science 302, 1041–1043 (2003). [DOI] [PubMed] [Google Scholar]
- 49.Yang X. O., Pappu B. P., Nurieva R., Akimzhanov A., Kang H. S., Chung Y., Ma L., Shah B., Panopoulos A. D., Schluns K. S., Watowich S. S., Tian Q., Jetten A. M., Dong C., T helper 17 lineage differentiation is programmed by orphan nuclear receptors RORα and RORγ. Immunity 28, 29–39 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bengsch B., Wherry E. J., The importance of cooperation: Partnerless NFAT induces T cell exhaustion. Immunity 42, 203–205 (2015). [DOI] [PubMed] [Google Scholar]
- 51.Martinez G. J., Pereira R. M., Äijö T., Kim E. Y., Marangoni F., Pipkin M. E., Togher S., Heissmeyer V., Zhang Y. C., Crotty S., Lamperti E. D., Ansel K. M., Mempel T. R., Lähdesmäki H., Hogan P. G., Rao A., The transcription factor NFAT promotes exhaustion of activated CD8+ T cells. Immunity 42, 265–278 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Oestreich K. J., Yoon H., Ahmed R., Boss J. M., NFATc1 regulates PD-1 expression upon T cell activation. J. Immunol. 181, 4832–4839 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ames R. Y., Ting L.-M., Gendlina I., Kim K., Macian F., The transcription factor NFAT1 participates in the induction of CD4+ T cell functional exhaustion during Plasmodium yoelii infection. Infect. Immun. 85, e00364-17 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Madeddu S., Woods T. A., Mukherjee P., Sturdevant D., Butchi N. B., Peterson K. E., Identification of glial activation markers by comparison of transcriptome changes between astrocytes and microglia following innate immune stimulation. PLOS ONE 10, e0127336 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Tanaka A., Sakaguchi S., Regulatory T cells in cancer immunotherapy. Cell Res. 27, 109–118 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Quezada S. A., Simpson T. R., Peggs K. S., Merghoub T., Vider J., Fan X., Blasberg R., Yagita H., Muranski P., Antony P. A., Restifo N. P., Allison J. P., Tumor-reactive CD4+ T cells develop cytotoxic activity and eradicate large established melanoma after transfer into lymphopenic hosts. J. Exp. Med. 207, 637–650 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Tao L., Reese T. A., Making mouse models that reflect human immune responses. Trends Immunol. 38, 181–193 (2017). [DOI] [PubMed] [Google Scholar]
- 58.Muñoz-Espín D., Serrano M., Cellular senescence: From physiology to pathology. Nat. Rev. Mol. Cell Biol. 15, 482–496 (2014). [DOI] [PubMed] [Google Scholar]
- 59.He S., Sharpless N. E., Senescence in health and disease. Cell 169, 1000–1011 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Jurk D., Wilson C., Passos J. F., Oakley F., Correia-Melo C., Greaves L., Saretzki G., Fox C., Lawless C., Anderson R., Hewitt G., Pender S. L. F., Fullard N., Nelson G., Mann J., van de Sluis B., Mann D. A., von Zglinicki T., Chronic inflammation induces telomere dysfunction and accelerates ageing in mice. Nat. Commun. 2, 4172 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ilicic T., Kim J. K., Kolodziejczyk A. A., Bagger F. O., McCarthy D. J., Marioni J. C., Teichmann S. A., Classification of low quality cells from single-cell RNA-seq data. Genome Biol. 17, 29 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Mereu E., Iacono G., Guillaumet-Adkins A., Moutinho C., Lunazzi G., Santos C. P., Miguel-Escalada I., Ferrer J., Real F. X., Gut I., Heyn H., matchSCore: Matching single-cell phenotypes across tools and experiments. bioRxiv, 314831 (2018). [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/5/8/eaaw8330/DC1
Fig. S1. Purification of CD4 T cells from young and old mice.
Fig. S2. Dimensionality reduction of scRNA-seq data.
Fig. S3. Subset identity and abundance in mice used for scRNA-seq.
Fig. S4. RECs activation state and correlation with serum cytokines.
Fig. S5. RECs abundance with time and across different immunological sites.
Fig. S6. Gene regulatory networks and cytokine secretion in RECs.
Fig. S7. Characterization of T helper polarizations within RECs.
Table S1. The number of cells per mouse before and after quality control.
Table S2. Subset and age-group expression of genes.
Table S3. RECs comparison to closely related subsets.