

Abstract
Objective
The role of hepatocyte ATP binding cassette transporter A1 (Abca1) in trafficking hepatic free cholesterol (FC) into plasma versus bile for reverse cholesterol transport (RCT) is poorly understood. We hypothesized that hepatocyte Abca1 recycles plasma HDL cholesterol (HDL-C) taken up by the liver back into plasma, maintaining the plasma HDL-C pool and decreasing HDL-mediated RCT into feces.
Approach and Results
Chow-fed hepatocyte-specific Abca1 knockout (HSKO) and control mice were injected with human HDL radiolabeled with 125I-tyramine cellobiose (125I-TC;protein) and 3H-cholesteryl oleate (3H-CO). 125I-TC and 3H-CO plasma decay, plasma HDL 3H-CO selective clearance (i.e., 3H-125I fractional catabolic rate), liver radiolabel uptake, and fecal 3H-sterol were significantly greater in HSKO versus control mice, supporting increased plasma HDL RCT. Twenty-four hours after 3H-CO-HDL injection, HSKO mice had reduced total hepatic 3H-FC (i.e., 3H-CO hydrolyzed to 3H-FC in liver) resecretion into plasma, demonstrating Abca1 recycled HDL-derived hepatic 3H-FC back into plasma. Despite similar liver LDL receptor (LDLr) expression between genotypes, HSKO mice treated with LDLr-targeting versus control antisense oligonucleotide had slower plasma 3H-CO-HDL decay, reduced selective 3H-CO clearance, and decreased fecal 3H-sterol excretion that were indistinguishable from control mice. Increased RCT in HSKO mice was selective for 3H-CO-HDL, since macrophage RCT was similar between genotypes.
Conclusions
Hepatocyte Abca1 deletion unmasks a novel and selective FC trafficking pathway that requires LDLr expression, accelerating plasma HDL-selective CE uptake by the liver and promoting HDL RCT into feces, consequently reducing HDL-derived hepatic FC recycling into plasma.
Keywords: Abca1, High Density Lipoproteins, Cholesterol, Lipoproteins/Kinetics, Liver Metabolism
Subject terms: Lipids and Cholesterol, Metabolism
Graphical Abstract
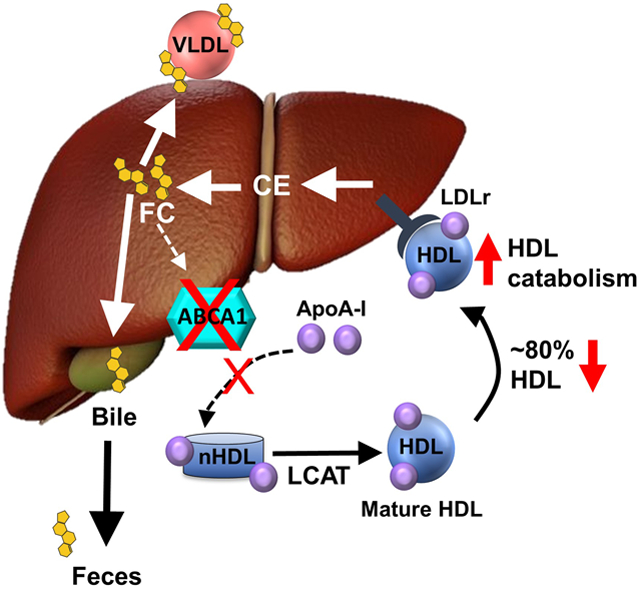
Introduction
Coronary heart disease (CHD) remains the leading cause of death worldwide. Statins, which reduce circulating plasma low density lipoprotein (LDL) concentrations, have lowered mortality attributable to CHD 1, yet CHD risk remains in statin-treated individuals 2. Epidemiologic studies have documented a strong inverse association between plasma high density lipoprotein cholesterol (HDL-C) concentrations and CHD, suggesting that HDL is an anti-atherogenic lipoprotein 3-5. However, treatments to raise plasma HDL concentrations and reduce CHD risk have had limited success 6-8. HDL’s atheroprotective role is attributed to its participation in reverse cholesterol transport (RCT), where HDL mobilizes extrahepatic cholesterol, particularly from arterial macrophage foam cells, and transports it to the liver for bile secretion and fecal excretion 9. HDL cholesterol efflux capacity measures the ability of plasma HDL (i.e., apoB lipoprotein deleted plasma or serum) to accept cellular free cholesterol, the first step in the biogenesis of HDL and RCT. HDL cholesterol efflux capacity better predicts CHD risk than plasma HDL-C concentrations 10, 11.
HDL biogenesis is initiated via ATP binding cassette transporter A1 (ABCA1), a cellular plasma membrane cholesterol and phospholipid efflux protein that assembles effluxed cellular lipids with apolipoproteins, such as apolipoprotein A-I and E, forming nascent HDL particles 12-14. Although ABCA1 is expressed throughout the body, hepatocyte ABCA1 expression is largely responsible for maintaining plasma HDL-C levels 14. Mutations that inactivate ABCA1 lead to Tangier disease, a disorder characterized by near-absence of plasma HDL, increased extra-hepatic tissue cholesterol accumulation, decreased plasma LDL concentrations, and increased plasma TG levels 15-17. Studies using hepatocyte-specific Abca1 knockout (HSKO) mice have shown that the Tangier disease plasma lipid and lipoprotein phenotype can be explained almost entirely by loss of hepatocyte Abca1 activity 18, except for ~20–30% of plasma HDL generated by the intestine and adipose tissue 19, 20. This marked reduction of plasma HDL-C is due to defective assembly of nascent HDL by hepatocytes and increased plasma clearance of HDL protein and cholesteryl ester (CE) 14, 18, 21, 22. Although our knowledge of hepatocyte Abca1 in plasma lipoprotein formation and catabolism is growing, the role of Abca1 in trafficking HDL-C taken up by the liver into the RCT pathway is less clear.
Recently, Yamamoto et al. 23 demonstrated that mice fed probucol, an Abca1 inhibitor, had increased plasma HDL RCT into feces, despite ~80% lower plasma HDL-C. This suggests that without hepatic Abca1 activity, plasma HDL-C taken up by the liver is preferentially trafficked into the RCT pathway for biliary secretion and fecal excretion, not resecreted into plasma. However, probucol has pleotropic effects and is not a specific hepatocyte Abca1 inhibitor 24, 25. Furthermore, direct evidence that Abca1 recycles plasma HDL-derived hepatic FC is lacking, indicating significant gaps in knowledge about hepatocyte Abca1 in HDL-C RCT.
The current study was designed to investigate the role of hepatocyte Abca1 in plasma HDL RCT and recycling of hepatic cholesterol into plasma. We hypothesized that hepatocyte Abca1 recycles a significant amount of plasma HDL-C, taken up by the liver, back into plasma, maintaining the plasma HDL-C pool and decreasing HDL-mediated RCT into feces. Our results demonstrate that hepatocyte Abca1 is pivotal in resecreting plasma-derived HDL-C back into plasma versus trafficking into RCT excretion pathways and reveal a novel role for the hepatic LDL receptor (LDLr) in stimulating plasma HDL selective CE uptake and trafficking of sterol into RCT when hepatocyte Abca1 is absent. Our data also suggest that hepatocyte Abca1 inhibition or haploinsufficiency due to coding variants may promote HDL RCT in parallel with a significant reduction in plasma HDL-C.
Materials and Methods
(please see the Major Resources Table in the Supplemental Material for reagent details)
Animals
For each experiment, chow-fed age-matched 12- to 24-week-old male and female Abca1flox/flox or C57BL/6J (The Jackson Laboratory; Stock # 000664) mice (controls) and hepatocyte-specific Abca1 knockout (HSKO) mice were used. HSKO mice were generated by crossing Abca1flox/flox mice 14 (backcrossed >99% into the C57BL/6 background) with albumin Cre recombinase transgenic mice (The Jackson Laboratory; Stock # 003574). All mice were maintained in a specific pathogen-free environment on a 12:12 h light:dark cycle (dark cycle, 6 p.m. to 6 a.m.) and allowed free access to standard chow diet (Purina – LabDiet; Prolab RMH 3000) and water. Cage bedding was Bed-o’Cobs (1/8”) from Andersons Lab Bedding and mice were provided with EnviroPak for enrichment (W.F. Fisher & Son, Inc.). Mouse studies were performed in facilities approved by the American Association for Accreditation of Laboratory Animal Care using a protocol approved by the Institutional Animal Care and Use Committee at Wake Forest School of Medicine.
Lipoprotein Preparation and Radiolabeling
Pooled human plasma was obtained from the American Red Cross and mouse plasma was obtained via cardiac puncture from C57BL6/J mice. HDL (d=1.063–1.21 g/mL) was isolated from plasma by sequential density ultracentrifugation 26.
HDL was radiolabeled with 3H-cholesteryl oleate (3H-CO) (Perkin Elmer) using human lipoprotein-deficient serum (LPDS) as a source of cholesteryl ester transfer protein 27. HDL protein was radiolabeled using 125I-tyramine cellobiose (125I-TC) (Perkin Elmer) according to methods previously described 28, 29.
HDL Turnover Studies
Control and HSKO mice were anesthetized with isoflurane and intravenously (retroorbital) injected with 125I-TC (0.1–0.3×106 cpm; 50 cpm/ng HDL protein) and 3H-CO-HDL (0.5–1×106 dpm; 200 dpm/ng HDL CE) tracers. Blood samples were taken 2 minutes, 30 minutes, 1, 3, 5, 8, 24, and 48 hours after tracer injection and experiments terminated at 24 or 48 hours. Plasma samples were directly quantified for 125I-TC protein radiolabel using a gamma counter, or lipid extracted 30 to quantify 3H radiolabel using a liquid scintillation counter. Plasma HDL turnover curves were generated by plotting the percentage of the two-minute radiolabel remaining in plasma at each time point. The total amount of plasma radiolabel at each time point was determined by multiplying the 125I cpm/ml or 3H dpm/ml plasma by total plasma volume, estimated as 3.5% of body weight. Fractional catabolic rates (FCR) were calculated by using SigmaPlot software to fit curves to a double exponential decay curve with 4 parameters (y=ae–bx+ce-dx), as previously described 31.
Liver, kidney, and feces were collected at completion of each turnover study. 125I-TC tissue uptake was measured by quantifying 125I radiolabel in 50–100 mg of tissue using a gamma counter. To quantify 3H tissue uptake and fecal excretion, tissue and feces were lipid extracted and 3H was quantified by liquid scintillation counting 32, 33. Selective HDL 3H-CO removal from plasma was calculated as 3H FCR-125I FCR 34. To determine the role of liver LDLr in HDL turnover, control and HSKO mice were randomly assigned to received intraperitoneal injections of either LDLr targeting antisense oligonucleotides (ASO) (Ionis Pharmaceuticals, Inc; ION 713852) or control ASO (ION 740133) at 5 mg/kg/week for 4 weeks. HDL turnover studies were then performed over 48 hours as described above.
Hepatic Recycling of Plasma HDL 3H-CO
Control and HSKO mice were intravenously injected with 3H-CO radiolabeled (2–4×106 dpm) HDL and blood samples were taken at 2 minutes, 1, 3, 5, 8, and 24 hours as described above. Plasma was isolated from blood samples and FC and individual CE fatty acyl species were separated by HPLC as described previously 35 with minor modifications. Briefly, plasma was lipid extracted 30 and the extract was filtered (0.22 μm), dried under N2, reconstituted in 10 μL of tetrahydrofuran-acetonitrile 80:20 (v:v), and injected onto a reverse phase HPLC column (250 × 4.6mm, Ultrasphere 5μm ODS column, Mac-Mod Analytical Inc.). FC and CEs were eluted from the column with a mobile phase of acetonitrile:isopropanol 50:50 (v:v) and fractions were collected every minute for 30 minutes (flow rate=2 mL/min). Each fraction was transferred to a scintillation vial, the solvent evaporated under N2, and a scintillation cocktail was added before 3H quantification by liquid scintillation counting 35. Radioactivity in FC and CE fractions at each time point of the turnover study was normalized to percentage of the two-minute plasma total radioactivity.
To determine 3H radiolabel distribution among plasma lipoproteins, terminal plasma isolated from blood taken 24 hours after 3H-CO-HDL tracer injection was fractionated by fast protein liquid chromatography (FPLC) using a Superose 6 column; fractions were collected every minute (0.4 mL/min). Radiolabel in the FPLC fractions was quantified by liquid scintillation counting and fractions corresponding to VLDL, LDL, and HDL, based on elution position, were pooled, lipid extracted, and fractionated into FC and CE fatty acyl species by reverse phase HPLC analysis as described above.
In Vivo Macrophage RCT
Macrophage RCT assays were conducted as described previously 33, 36. J774 macrophages were radiolabeled with 3H-cholesterol and cholesterol-loaded with acetylated LDL. Five hundred μL of cell suspension containing ~1×107 cells/mL and ~4×106 dpm/mL was injected into the peritoneal cavity of recipient mice. Plasma samples were collected at 3, 6, 24, 48, 72, and 96 hours. Feces were collected continuously from 0–48 hours and 48–96 hours. At the termination of the study, liver was harvested and 3H tracer in plasma, liver, and feces was quantified after lipid extraction and scintillation counting 32, 33. One hundred μL of pooled plasma from terminal blood draws were fractionated by FPLC to determine 3H radiolabel distribution among lipoproteins.
Plasma Lipid and Lipoprotein Analysis
Plasma was collected by tail bleeding from mice fasted for 4 hours following 4 weeks of ASO treatment. Total plasma cholesterol (TPC) concentrations were determined by enzymatic assay 37. Cholesterol distribution among lipoproteins was quantified after fractionation of plasma by FPLC size-exclusion chromatography 36.
Quantitative RT-PCR
Total RNA was extracted from snap-frozen liver tissue using TRI-Reagent (Molecular Research Center, Inc). Target gene mRNA abundance was quantified by quantitative real-time PCR using Luna® Universal One-Step RT-qPCR Kit (NEB, E3005L). GAPDH mRNA abundance was used to correct the target gene expression data. Relative quantification was calculated using the ΔΔ comparative threshold formula 38. Primers used in this study were: GAPDH; TGTGTCCGTCGTGGATCTGA (forward), CCTGCTTCACCACCTTCTTGAT (reverse); ABCA1: CGTTTCCGGGAAGTGTCCTA (forward), GCTAGAGATGACAAGGAGGATGGA (reverse); LDLR: AGGCTGTGGGCTCCATAG (forward), TGCGGTCCAGGGTCATCT (reverse); SR-BI: TCCCCATGAACTGTTCTGTGAA (forward), TGCCCGATGCCCTTGA (reverse).
Immunoblotting
Frozen liver (50–100 mg) was homogenized using a polytron-aggregate tissue homogenizer in RIPA buffer containing protease inhibitor cocktail (Roche 05892791001). Proteins were fractioned by 4–20% Criterion™ TGX™ precast gels (Bio-Rad; 5671094) and transferred to a PVDF membrane. Membranes were blocked with 5% nonfat dry milk in Tris-buffered saline + 1% tween 20 (TBST), incubated with primary antibody at 4°C overnight, washed three times with TBST, and then incubated with secondary antibody for 1 h at room temperature. Blots were incubated with SuperSignal West Pico chemiluminescence substrate (Pierce) and visualized with a Fujifilm LAS-3000 camera. Primary antibodies used for immunoblots included the following: Abca1 (custom mademade 39; 1:1000), SR-BI (Abcam; ab217318;0.187 ug/mL), LDLr (gift from Dr. Joachim Herz; 1:1000), and GAPDH was used as a loading control (Santa Cruz; 32233;0.02 ug/mL). Secondary antibodies used for immunoblots were HRP-conjugated anti-mouse (1:10,000) and anti-rabbit (GE Healthcare; 1:10,000). Band intensities were quantified using Image Studio Lite (LI-COR Biosciences).
Statistics
All data are presented as mean ± standard error of the mean (SEM). Data were tested for normality (Kolmogorov and Smirnov test) and equal variance (Bartlett or Brown-Forsythe test) using GraphPad prism 7 software. Statistical analyses were performed using an unpaired two-tailed Student’s t-test, one-way ANOVA with Tukey’s post hoc analysis, or repeated measures ANOVA. For the ASO studies in which the number of mice per group was too few to test for normality, the non-parametric Kruskal-Wallis test was used.
Results
Hepatocyte-specific Abca1 deletion accelerates HDL 125I-TC tracer plasma decay and tissue uptake
A major goal of this study was to determine how hepatocyte Abca1 affects plasma HDL-C catabolism and RCT. We previously reported increased plasma clearance and kidney uptake of 125I-TC radiolabeled mouse HDL in HSKO versus control mice; however, liver uptake of the mouse 125I-TC HDL tracer was similar for both genotypes 14. Because mouse HDL particles are monodispersed, whereas human HDL particles are polydispersed 40, exhibiting distinct size subfractions, we used human HDL tracer for our turnover studies to capture the complexity of HDL-C catabolism that likely impacts RCT. Figure 1 summarizes plasma decay and tissue accumulation of 125I-TC human HDL tracer for two independent 24- or 48-hour studies. Plasma decay of human HDL 125I-TC protein was 2–3 fold greater in HSKO recipient mice than control mice (Fig. 1A, B). 125I-TC human HDL tracer accumulation by liver (Fig. 1C) and kidney (Fig. 1D) was also significantly greater in HSKO compared to control mice. The apparent decrease in liver 125I-TC accumulation at 48 versus 24 hours (Fig. 1C) was likely due to cellular release and urinary excretion of 125I-TC (or free 125I) that occurs during prolonged turnover studies 41. When analyzed separately, 125I-TC-HDL FCRs were similar for male and female recipient mice, indicating no sex-related differences in HDL protein turnover (data not shown).
Figure 1.

In vivo catabolism of 125I-TC radiolabeled human HDL. 125I-TC radiolabeled human HDL was injected intravenously into chow-fed control (n=6) and HSKO (n=6) mice in two separate turnover studies lasting 24 or 48 hours. Periodic blood samples were taken over 24 or 48 hours to analyze plasma decay (A), plasma FCR (B), liver (C), and kidney accumulation (D) of the 125I-TC tracer. Data are mean ± SEM. Control turnover curves in panel A are nearly identical and SEM in nearly all points falls within the symbol. **p < 0.01; ***p < 0.001.
Hepatocyte-specific Abca1 deletion accelerates HDL cholesteryl oleate plasma decay, selective CE clearance, and RCT
Next, we explored the role of hepatocyte Abca1 in plasma HDL-derived CE RCT. Once in the liver, HDL 3H-CO is hydrolyzed to 3H-FC, which can be 1) resecreted into plasma in very low density lipoprotein (VLDL) or HDL particles, 2) esterified by acyl:CoA cholesterol acyltransferase 2, forming CE that can be stored in lipid droplets, 3) secreted into bile as 3H-FC, or 4) converted to bile acid and secreted into bile. The latter two pathways increase RCT. We radiolabeled isolated human HDL particles with 3H-CO and injected them into control and HSKO mice to measure radiolabel plasma clearance, CE selective clearance (i.e., 3H-125I FCR), liver radiolabel accumulation, and fecal radiolabel excretion. Plasma decay of HDL 3H-CO was markedly faster in HSKO versus control recipient mice in two turnover studies (Fig. 2A). Die-away curves for the two studies over the first 24 hours were remarkably similar. Plasma FCR (Fig. 2B) for the HDL 3H-CO was 5–7-fold greater in HSKO recipient mice compared with controls in both studies. Plasma HDL 3H-CO selective clearance, calculated as plasma HDL 3H-CO minus 125I-TC HDL FCR (from Figure 1) was 10- to 20-fold greater in HSKO mice (Fig. 2C). Liver accumulation of the HDL 3H-CO tracer in HSKO mice (Fig. 2D) was 2-fold greater at 24 hours and 1.9-fold greater at 48 hours than in control mice, showing more rapid removal of tracer from plasma in HSKO mice. Seventy percent of the liver 3H radiolabel at the 24-hour time point was FC for both genotypes of mice (Supplemental Figure I), suggesting efficient and equivalent intrahepatic hydrolysis of plasma HDL-derived 3H-CO to 3H-FC. Plasma HDL 3H-radiolabel RCT was similar between genotypes in the 24-hour study, but 2-fold higher in HSKO recipient mice in the 48-hour study (Fig. 2E), indicating that plasma HDL RCT is increased in the absence of hepatocyte Abca1 expression 48 hours after 3H-CO HDL tracer injection.
Figure 2.

In vivo catabolism and RCT of plasma HDL CO. 3H-CO radiolabeled human HDL was injected intravenously in control (n=6) and HSKO (n=6) mice with 125I-TC HDL (see Figure 1). Periodic blood samples were taken over 24 or 48 hours in two separate experiments to analyze plasma decay (A), plasma FCR (B), plasma HDL 3H-CO selective clearance, using data from Figure 1B for 125I-TC FCR (C), liver 3H-radiolabel accumulation (D), and fecal 3H-radiolabel excretion (E). Data are mean ± SEM. Control turnover curves in panel A are nearly identical and SEM in nearly all points falls within the symbol. **p < 0.01; ***p < 0.001.
Hepatocyte Abca1 recycles plasma HDL-derived cholesterol taken up by the liver into plasma
To determine whether hepatocyte Abca1 recycles plasma HDL-derived cholesterol taken up by the liver into plasma, we injected 3H-CO-radiolabeled human HDL into control and HSKO recipient mice, collected blood samples over 24 hours, and measured appearance of 3H-FC and individual 3H-CE fatty acyl species in plasma. Since in vitro incubations of HDL tracer with plasma demonstrated that 3H-CO remained associated with the HDL fraction and that there was minimal 3H-CO hydrolysis to 3H-FC over 24 hours (Supplemental Figure IIA and B, respectively), plasma appearance of 3H-FC and 3H-CE fatty acyl species can only result from hepatic uptake of plasma HDL 3H-CO, hydrolysis of 3H-CO to 3H-FC (see Supplemental Figure I), and resecretion into plasma as 3H-FC or as 3H-CE fatty acyl species, other than CO, generated by liver acyl CoA:cholesterol acyltransferase 2 (ACAT2)- or plasma lecithin:cholesterol acyl transferase (LCAT)-mediated esterification. HPLC separation of plasma lipid extracts (Fig. 3) showed that 3H-CO radiolabel decreased more rapidly in HSKO recipient mice than control mice, in agreement with 3H-CO die-away data in Figure 2. There was little detectable 3H radiolabel in plasma other than CO before 24 hours (Fig. 3A-E). However, at the 24-hour time point, considerable 3H radiolabel was in FC and CE fatty acyl species besides CO (Fig. 3F), such as cholesteryl docosahexaenoate (C22:6 n-3), cholesteryl arachidonate (C20:4), and cholesteryl linoleate (C18:2); less 3H radiolabel was found in all fractions in HSKO compared to control plasma samples. This occurred despite more 3H radiolabel in HSKO livers 24 hours after HDL tracer injection (Fig. 2D), suggesting markedly decreased resecretion of plasma HDL-derived hepatic 3H cholesterol in HSKO mice.
Figure 3.

Resecretion of HDL-C into plasma. 3H-CO-radiolabeled HDL was injected intravenously into control (n=5) and HSKO (n=5) mice. Periodic blood samples (A-F) were taken over 24 hours to trace disappearance of cholesterol oleate (C18:1) and reappearance of free cholesterol (FC), cholesteryl docoasahexanoate (C22:6), cholesteryl arachidonate (C20:4), and cholesteryl linoleate (C18:2) in plasma by reverse phase HPLC. Each data point is the mean ± SEM.
At 24 hours, overall recycling of HDL-C taken up by the liver was decreased in HSKO mice (Fig. 4A). We fractionated the 24-hour terminal plasma samples by FPLC size exclusion chromatography to determine radiolabel distribution of FC and CE fatty acyl species among VLDL, LDL and HDL. HSKO plasma had an increased proportion of 3H radiolabel distributed in VLDL and less HDL, with a small increase in LDL, compared with control plasma (Fig. 4B, C). There was a 5-fold increase in VLDL 3H-FC in HSKO plasma compared to control (Fig. 4D), no difference in LDL 3H-FC (Fig. 4E), and an 80% reduction in HDL 3H-FC (Fig. 4F). Minimal 3H radiolabel was found in VLDL CE species (Fig. 4D). The LDL fraction contained 3H-CE fatty acyl species similarly distributed in HSKO and control plasma (Fig 4E). 3H-CE fatty acyl species also appeared in the HDL fraction where the amount of radiolabel was less in HSKO versus control plasma (Fig. 4F). These combined results provide direct support for the concept that Abca1 recycles hepatic FC derived from plasma HDL CO uptake back into plasma.
Figure 4.

Plasma lipoprotein 3H radiolabel distribution 24 hours after injection of 3H-CO radiolabeled human HDL. Total 3H-CO HDL radiolabel removed by the liver and recycled into plasma at 24 hours was calculated as the sum of radiolabel in fractions 0-20 from Figure 3F (i.e., 3H-FC + 3H-CE radioactivity exclusive of 3H-C18:1) (A). Plasma isolated from 24-hour terminal blood samples was fractionated by FPLC and percentage radiolabel distribution in each lipoprotein fraction was plotted (B). 3H-radiolabel percentage distribution in panel B, calculated as area under the curve from FPLC profiles (C). VLDL, LDL, and HDL FPLC fractions (i.e., panel B) were pooled, lipid extracted, FC and CE fatty acyl species fractionated by HPLC, and radiolabeled 3H-FC and 3H-CE fatty acyl species in VLDL (D), LDL (E), and HDL (F) quantified by liquid scintillation counting. Data are mean ± SEM; n=5/genotype. *p < 0.05; **p < 0.01; ***p < 0.001.
Plasma HDL-3H-CO hypercatabolism and increased RCT in HSKO mice depend on hepatic LDLr expression
We hypothesized that rapid plasma removal of human HDL 3H-CO in HSKO recipient mice was due to increased expression of hepatic SR-BI, which mediates CE-selective uptake from HDL 42. However, we observed significantly decreased hepatic SR-BI protein expression in HSKO compared to control livers (Fig. 5A-B), whereas hepatic SR-BI mRNA expression was similar between the two genotypes (Fig. 5C), making it unlikely that SR-BI expression was directly responsible for HDL-CE hypercatabolism in HSKO mice. In a previous study, hepatic LDLr expression was increased nearly 2-fold in HSKO versus control mice 18 and doubling hepatic LDLr expression decreases plasma HDL-C in chow-fed mice 43. We hypothesized that increased LDLr expression in HSKO mice led to HDL hypercatabolism. However, in contrast to our previous study 18, we observed similar hepatic LDLr protein expression (Fig. 5A-B) and mRNA abundance (Fig. 5C) in HSKO vs. control liver.
Figure 5.

Hepatic Abca1, LDLr, and SR-BI protein expression and gene expression. Whole liver lysates from control (n=6) and HSKO (n=6) were immunoblotted for Abca1, LDLr,SR-BI, and GAPDH, as loading control (A). Immunoblots in panel A for LDLr and SR-BI were quantified by calculating fold change of the protein/GAPDH ratio relative to control livers (B). Abca1, LDLr, and SR-BI gene expression was analyzed by real-time PCR and the mRNA/GAPDH ratio relative to control livers was quantified (C). Data are mean ± SEM; n=6/genotype from recipient mice used for the 24-hour turnover study in figures 1 and 2. *p < 0.05; ***p < 0.001.
Increased LDLr expression is associated with reduced plasma HDL-C concentrations in mice 43, 44. Although hepatic LDLr expression was similar for HSKO and control mice, hepatic LDLr surface expression or recycling may be increased in the absence of hepatocyte Abca1, leading to increased catabolism of plasma HDL. To investigate whether liver LDLr is involved in plasma HDL hypercatabolism in HSKO mice, we treated control and HSKO mice with a control ASO or LDLr-targeting ASO for 4 weeks. LDLr ASO treatment significantly increased plasma cholesterol concentrations in both strains relative to control ASO (Supplemental Figure IIIA). Most of the plasma cholesterol increase with LDLr ASO treatment was in the LDL fraction, as anticipated, but HDL-C was also considerably increased in LDLr ASO-treated HSKO mice versus control ASO-treated HSKO mice (Supplemental Figure IIIB). LDLr-targeting ASO treatment eliminated hepatic LDLr protein (Supplemental Figure IIIC-D) and reduced gene expression by ~75% (Supplemental Figure IIIE) relative to control ASO in both genotypes of mice. On the other hand, hepatic SR-BI protein and gene expression were unaffected by LDLr ASO treatment (Supplemental Figure IIIC-E). LDLr ASO treatment of HSKO mice normalized plasma HDL 125I-TC die-way curves (Fig 6A) and FCR (Fig. 6B) to those of control mice treated with control or LDLr-targeting ASO. These results suggest that HDL protein (i.e., 125I-TC) hypercatabolism in HSKO recipient mice depends on hepatic LDLr expression.
Figure 6.

Effect of LDLr ASO treatment on in vivo catabolism of 125I-TC radiolabeled human HDL. 125I-TC radiolabeled HDL was injected intravenously into control and HSKO mice treated with a control (Cntl) or LDLr-targeting ASO. Periodic blood samples were taken over 48 hours to analyze plasma decay (A) and FCR (B). Data are mean ± SEM. Groups with different letters are statistically different (p<0.05), n=3/group.
Next, we examined plasma HDL-CE metabolism in mice treated with control or LDLr ASO. Human HDL 3H-CO plasma decay (Fig. 7A) and FCR (Fig. 7B) in HSKO recipient mice treated with LDLr ASO was normalized to those of control recipient mice treated with either control or LDLr ASO. Plasma selective HDL 3H-CO removal in HSKO mice was also diminished to control levels with LDLr ASO treatment (Fig. 7C). Hepatic accumulation of 3H-radiolabel was higher in HSKO than control mice. Hepatic 3H radiolabel was similar in HSKO mice treated with LDLr ASO or control ASO (Fig. 7D). HSKO versus control mice treated with control ASO had significantly more fecal 3H-sterol excretion (Fig. 7E), similar to earlier results for untreated mice (Fig. 2E). Treatment of HSKO mice with LDLr ASO reduced fecal 3H-sterol excretion to levels indistinguishable from HSKO or control mice treated with control ASO. Similar trends occurred with fecal cholesterol and bile acid 3H-radiolabel (Fig. 7E).
Figure 7.

Effect of LDLr ASO treatment on in vivo catabolism of 3H-CO radiolabeled human HDL. 3H-CO-radiolabeled HDL was injected intravenously in control and HSKO mice treated with a control or LDLr targeting ASO. Periodic blood samples were taken over 48 hours to analyze plasma decay (A), plasma FCR (B), and plasma HDL 3H-CO selective clearance, using data from Figure 7B for 125I-TC FCR (C). Tissues were then harvested to quantify liver accumulation (D) and fecal excretion (E) of the 3H-tracer as bile acid, cholesterol or total sterol (bile acids + cholesterol). Data are mean ± SEM. Groups with different letters are statistically different (p<0.05), n=3/group. Data in panel B were analyzed with the Kruskal-Wallis non-parametric ANOVA (p=0.0001), followed by Dunn’s multiple comparisons test.
Human HDL particles are polydispersed, with multiple size subfractions, whereas mouse HDL particles are monodispersed 40. To determine whether the more rapid turnover of plasma HDL tracer in HSKO depended on use of human HDL tracer particles, we isolated plasma HDL from C57Bl/6 donor mice, radiolabeled HDL with 125I-TC or 3H-CO, and repeated the plasma turnover and RCT studies. Mouse HDL 125I-TC displayed more rapid plasma decay and FCR in HSKO versus control recipient mice (Supplemental Figure IV), similar to results with human HDL 125I-TC tracer (Figure 1); LDLr ASO treatment of recipient mice had minimal influence on HDL 125I-TC catabolism (Supplemental Figure IV). Qualitatively similar results were obtained for plasma HDL 3H-CO decay, FCR, HDL 3H-CO selective clearance from plasma, hepatic accumulation, and fecal 3H radiolabel excretion (Supplemental Figure V; panels A-E) as with human HDL 3H-CO tracer (Figure 2) and LDLr ASO treatment of HSKO recipient mice normalized plasma decay, selective CE clearance, and RCT (Supplemental Figure V). These results support the conclusion that the more rapid plasma HDL catabolism, increased selective HDL CE plasma clearance, and greater RCT in HSKO mice was independent of the source of HDL tracer.
Macrophage reverse cholesterol transport is unaffected by hepatocyte Abca1 deletion
We previously reported that macrophage RCT is not compromised in hyperlipidemic HSKO mice (i.e., HSKO mice in the LDLrKO background fed an atherogenic diet) compared to controls 36. To determine whether macrophage RCT is intact in normolipidemic HSKO mice, chow-fed control and HSKO mice were injected in the peritoneal cavity with 3H-cholesterol-loaded J774 macrophages and appearance of 3H radiolabel in plasma, liver, and feces measured. HSKO plasma had markedly less 3H radiolabel appearance than control plasma during the 96-hour RCT experiment (Fig. 8A). In control plasma, 3H distribution was associated with HDL and to a lesser extent, LDL (Fig. 8B), similar to cholesterol mass distribution (Supplemental Figure IIIB), whereas 3H radiolabel in HSKO plasma was extremely low in HDL and LDL fractions. However, 3H radiolabel in liver (Fig. 8C) or feces (Fig. 8D) were similar between genotypes.
Figure 8.

Macrophage reverse cholesterol transport. Control (n=6) and HSKO (n=6) mice were injected intraperitoneally with 3H-cholesterol-loaded J774 macrophages. Blood samples were taken over 96 hours to monitor plasma appearance of 3H-radiolabel (A). 48-hour plasma samples were fractionated by FPLC to determine 3H-radiolabeled lipoprotein distribution (B). At 96 hours, mice were sacrificed and liver 3H radiolabel uptake was measured (C). Feces were collected from 0-48 hours and 48-96h and 3H-radiolabeled cholesterol (Chol) and bile acid (BA) excretion were quantified (D). Total sterol 3H-radiolabel = Chol + BA radiolabel. Data are mean ± SEM.
Discussion
Hepatocyte Abca1 plays a key role in production and catabolism of all three major plasma lipoprotein classes (VLDL, LDL, and HDL) that contribute to cardiometabolic risk 18, 22. In addition, chow-fed HSKO mice closely phenocopy lipid and lipoprotein alterations in Tangier disease subjects, suggesting HSKO mice are ideal for investigating in vivo metabolic pathways impacted by diminished hepatocyte Abca1 expression. Despite our current understanding of hepatocyte Abca1 in plasma lipoprotein metabolism, we do not understand its involvement in hepatic FC trafficking into plasma versus bile and feces for RCT. Our current study addresses this gap in knowledge and contributes three novel findings. First, HSKO mice showed increased selective HDL-CE removal from plasma and preferential trafficking of plasma-derived HDL-C into feces, increasing HDL RCT, compared to control mice. Second, absence of hepatocyte Abca1 expression diminishes overall recycling of plasma HDL-C, taken up by the liver, back into the plasma compartment, but increases the portion of resecreted FC in VLDL particles at the expense of HDL particles, thereby affecting quantity and compartmentalization of resecreted hepatic FC. These results suggest that hepatocyte Abca1 is an important gatekeeper for regulating hepatic FC flux between the plasma compartment and bile. Finally, increased selective HDL-CE removal from plasma and trafficking into the RCT pathway in HSKO mice depends on hepatic LDLr expression, identifying a novel role for the LDLr in plasma HDL RCT. Thus, our data suggest that hepatocyte Abca1 inhibition or haploinsufficiency due to coding variants may promote HDL-C RCT despite a concomitant reduction in plasma HDL.
Specific genetic deletion of hepatocyte Abca1 results in diminished nascent HDL particle assembly 22 and increased HDL protein and CE catabolism 14, 21, resulting in reduced plasma HDL-C concentrations in chow-fed HSKO versus control mice. However, trafficking of HDL 3H-CO tracer through the RCT pathway has not been investigated in HSKO mice. Sterol balance studies in whole body Abca1 knockout mice have suggested either no effect 45 or increased fecal sterol excretion due to reduced intestinal cholesterol absorption 46. However, the role of hepatocyte Abca1 in hepatic cholesterol trafficking is confounded in whole body Abca1 knockout mice because of Abca1’s widespread and variable tissue and cell expression 47. Yamamoto et al 23 showed increased fecal excretion of plasma HDL-derived cholesterol in mice fed probucol, an Abca1 inhibitor. Our study is the first to use a specific genetic deletion of hepatocyte Abca1 and supports their conclusion. We also observed increased selective HDL 3H-CO removal from plasma in HSKO versus control mice, in agreement with two turnover studies using probucol inhibition in mice 23, 48. Decreased plasma HDL pool size is unlikely to explain the more rapid plasma decay of HDL 3H-CO in HSKO recipient mice, since low plasma HDL in apoA-I knockout mice did not affect plasma removal of HDL 3H-FC or 3H-CE 49. Further, normalization of the HDL pool in a Tangier disease subject by infusion of human HDL did not normalize rapid plasma decay of HDL 50. Our results demonstrate a novel role for hepatocyte Abca1 in regulating plasma HDL-C trafficking into the RCT pathway for fecal excretion, and suggest that loss or reduction of hepatocyte Abca1 activity might enhance net removal of cholesterol from the body.
The concept that hepatic Abca1 facilitates HDL-derived hepatic cholesterol resecretion into plasma, diverting it from RCT, was suggested by Yamamoto et al 23; however, in vivo support was lacking. Our study demonstrates that HSKO mice had reduced resecretion of plasma HDL-derived cholesterol, taken up by the liver, back into plasma and altered compartmentalization of cholesterol with a relative increase in VLDL FC secretion and a decrease in HDL FC and CE secretion (Fig. 4). To quantify hepatic resecretion of internalized plasma HDL-CE, two criteria must be met. First, HDL 3H-CO tracer must not undergo significant hydrolysis in plasma during the turnover study and second, 3H-CO must be significantly hydrolyzed after hepatic uptake to release 3H-FC for resecretion. Our control data demonstrate that these criteria were met (Supplemental Fig. I and II). Our previous study showed increased secretion of larger, TG-enriched VLDL particles in HSKO mice, hepatocytes from HSKO mice, and hepatoma cells with silenced Abca1 18, 51, 52, compatible with increased VLDL 3H-FC appearance in HSKO plasma 24 hours after HDL 3H-CO injection, relative to controls. This finding also agrees with results demonstrating that Abca1-stimulated FC efflux from hepatoma cells and primary hepatocytes decreases the pool of hepatocyte FC available for VLDL secretion 53. These data support an emerging concept that Abca1 is critical in affecting hepatic lipid (TG and FC) trafficking into several pathways, including biliary secretion for RCT, VLDL secretion, and HDL particle assembly.
We previously showed that HSKO mice in a mixed (80% C57Bl/6– 20% 129/SvEv) genetic background had significantly higher selective hepatic uptake of HDL CE than control mice, presumably via SR-BI, although hepatic SR-BI protein expression was similar between the two genotypes 21. However, selective plasma removal of HDL 3H-CO in HSKO versus control mice in our current study seemed too high to be explained by SR-BI expression alone. For example, hepatic-specific SR-BI transgenic mice, exhibiting 12-fold overexpression of mouse SR-BI, had a 2-fold increase in selective HDL CE removal from plasma 54, less than the 4-fold increase in plasma HDL-CE selective removal in HSKO mice, relative to control (Fig. 2C). Furthermore, we observed a significant decrease, not increase, in hepatic SR-BI protein expression in HSKO mice compared to controls across several cohorts. We verified that SR-BI protein expression, but not mRNA abundance, was significantly decreased in another cohort of HSKO relative to control mice (n=5/group; data not shown; 48-hour macrophage RCT study). The difference in liver SR-BI expression compared to our past study 21 may relate to our use of HSKO and control mice that were >99% in the C57Bl/6 background 18. These combined results suggested another explanation besides SR-BI expression for the more rapid selective removal of plasma HDL 3H-CO in HSKO mice.
In our previous study, liver LDLr expression was increased ~2-fold in HSKO mice versus controls, resulting in increased plasma 125I-LDL turnover 18. This magnitude of LDLr overexpression is associated with significantly reduced plasma HDL-C concentrations in other mouse models 43, 44. We tested the hypothesis that higher LDLr expression in HSKO liver increased plasma HDL 3H-CO removal, compared to control mice, but found no difference in hepatic LDLr expression. In other unpublished studies, we have observed significantly increased hepatic LDLr protein expression and mRNA abundance in HSKO versus control mice after a 24-hour fast. Recipient mice in the current study were not fasted before sacrifice after 24- or 48-hour turnover studies. Whether the physiologic extremes of fasting and fasting-refeeding result in a unique regulation of hepatic LDLr expression in HSKO mice requires further investigation.
Despite similar hepatic LDLr expression in HSKO and control mice in this study, hepatic LDLr silencing normalized rapid removal of HDL 3H-CO from plasma, selective plasma HDL 3H-CO removal, and RCT in HSKO mice to those of control mice (Fig. 7). Similar, but less striking, trends were observed with control mice treated with LDLr ASO versus control ASO, in general agreement with results of Rinninger et al 55, who demonstrated that plasma HDL CE selective removal and hepatic selective uptake of HDL CE were nearly eliminated in LDLr knockout mice versus wild-type controls. HSKO mice treated with LDLr ASO had similar hepatic 3H radiolabel accumulation as HSKO mice treated with control ASO, but the former had decreased fecal 3H radiolabel (Fig. 7D-E), suggesting that hepatic LDLr may also play a role in intrahepatic FC trafficking into bile and feces, resulting in diminished fecal 3H radiolabel when the LDLr is silenced. We speculate that there may be greater hepatocyte LDLr surface expression or faster endocytic recycling back to the plasma membrane in HSKO mice to explain the dependence of increased HDL catabolism on hepatic LDLr expression.
One perplexing observation is why macrophage RCT is not decreased when hepatocyte Abca1 is absent (Fig. 8). We previously demonstrated that macrophage RCT was not compromised in atherogenic diet-fed, hyperlipidemic HSKO/LDLr double knockout mice versus controls (i.e., LDLrKO), suggesting hepatic Abca1 deletion has no impact on macrophage RCT 36. Our current study shows this outcome was unrelated to hyperlipidemic background or atherogenic diet feeding. Our results agree with those of Yamamoto et al 23, who showed that macrophage RCT was not stimulated in probucol-fed mice although plasma HDL CE RCT was increased versus controls. One possible explanation is that not all radiolabeled cholesterol in macrophages used for RCT studies is esterified and RCT for macrophage 3H-FC may be more efficient than 3H-CE, which must first be hydrolyzed to 3H-FC before it can be effluxed from macrophages. A small, dynamic HDL pool, such as pre-β1 HDL 56, may efficiently remove excess macrophage 3H-FC, which is quantitatively insignificant relative to the mass of cholesterol in plasma, and rapidly transport it to the liver for excretion without a detectable increase in plasma HDL-C. Another potential explanation is that red blood cells become quantitatively more important for macrophage RCT in low plasma HDL situations, such as HSKO and apoA-I knockout mice 57. Finally, although much attention has been focused on macrophage Abca1 in RCT, our study clearly documents a critical role for hepatocyte Abca1 in hepatic FC trafficking and plasma HDL RCT.
Supplementary Material
Highlights.
Plasma HDL CE clearance is increased in the absence of hepatic Abca1, contributing to reduced plasma HDL cholesterol concentrations
Increased plasma HDL CE clearance in mice lacking hepatocyte Abca1 requires hepatic LDL receptor expression
In the absence of hepatocyte Abca1, less plasma HDL-derived hepatic FC is recycled back into plasma as nascent HDL and a relatively greater proportion of HDL-derived hepatic FC is secreted from liver in VLDL particles
Reverse cholesterol transport of plasma HDL cholesterol into feces is increased in mice lacking hepatocyte Abca1
Acknowledgments:
The authors gratefully acknowledge Abraham K. Gebre for technical assistance and Dr. Joachim Herz, University of Texas, Southwestern, for providing anti-mouse LDLr antiserum. We also acknowledge the editorial assistance of Karen Klein, MA, in the Wake Forest Clinical and Translational Science Institute (UL1 TR001420; PI: McClain).
Sources of funding: This project was supported by funding from the National Institutes of Health R01 HL119962 (to J.S.P.) and T32 HL091797 (A.C.B.)
Non-standard Abbreviations and Acronyms:
- Abca1
ATP binding cassette transport A1
- FC
Free cholesterol
- HDL
High density lipoprotein
- HDL-C
High density lipoprotein cholesterol
- RCT
Reverse cholesterol transport
- HSKO
Hepatocyte-specific Abca1 knockout
- CO
Cholesteryl oleate
- CE
Cholesteryl ester
- VLDL
Very low density lipoprotein
- SR-BI
Scavenger receptor class B type 1
- LDLr
Low density lipoprotein receptor
- CHD
Coronary heart disease
- LDL
Low density lipoprotein
- ASO
Antisense oligonucleotide
- TC
Tyramine cellobiose
- LPDS
Lipoprotein-deficient serum
- FPLC
Fast protein liquid chromatography
- HPLC
High performance liquid chromatography
- TBST
Tris-buffered saline + 1% tween 20
- TPC
Total plasma cholesterol
- FCR
Fractional catabolic rate
Footnotes
Disclosures: Drs. Mullick and Lee are employees of Ionis Pharmaceuticals, Inc. All other authors have no conflicts of interest.
References
- 1.Singh IM, Shishehbor MH, Ansell BJ. High-density lipoprotein as a therapeutic target: A systematic review. JAMA. 2007;298:786–798 [DOI] [PubMed] [Google Scholar]
- 2.Baigent C, Keech A, Kearney PM, Blackwell L, Buck G, Pollicino C, Kirby A, Sourjina T, Peto R, Collins R, Simes R, Cholesterol Treatment Trialists C. Efficacy and safety of cholesterol-lowering treatment: Prospective meta-analysis of data from 90,056 participants in 14 randomised trials of statins. Lancet. 2005;366:1267–1278 [DOI] [PubMed] [Google Scholar]
- 3.Miller GJ, Miller NE. Plasma-high-density-lipoprotein concentration and development of ischaemic heart-disease. Lancet. 1975;1:16–19 [DOI] [PubMed] [Google Scholar]
- 4.Castelli WP, Doyle JT, Gordon T, Hames CG, Hjortland MC, Hulley SB, Kagan A, Zukel WJ. Hdl cholesterol and other lipids in coronary heart disease. The cooperative lipoprotein phenotyping study. Circulation. 1977;55:767–772 [DOI] [PubMed] [Google Scholar]
- 5.Miller NE, Thelle DS, Forde OH, Mjos OD. The Tromso heart-study. High-density lipoprotein and coronary heart-disease: A prospective case-control study. Lancet. 1977;1:965–968 [DOI] [PubMed] [Google Scholar]
- 6.Barter PJ, Caulfield M, Eriksson M, Grundy SM, Kastelein JJ, Komajda M, Lopez-Sendon J, Mosca L, Tardif JC, Waters DD, Shear CL, Revkin JH, Buhr KA, Fisher MR, Tall AR, Brewer B, Investigators I. Effects of torcetrapib in patients at high risk for coronary events. N. Engl. J. Med 2007;357:2109–2122 [DOI] [PubMed] [Google Scholar]
- 7.Schwartz GG, Olsson AG, Abt M, Ballantyne CM, Barter PJ, Brumm J, Chaitman BR, Holme IM, Kallend D, Leiter LA, Leitersdorf E, McMurray JJ, Mundl H, Nicholls SJ, Shah PK, Tardif JC, Wright RS, dal OI. Effects of dalcetrapib in patients with a recent acute coronary syndrome. N. Engl. J. Med 2012;367:2089–2099 [DOI] [PubMed] [Google Scholar]
- 8.Investigators A-H, Boden WE, Probstfield JL, Anderson T, Chaitman BR, Desvignes-Nickens P, Koprowicz K, McBride R, Teo K, Weintraub W. Niacin in patients with low hdl cholesterol levels receiving intensive statin therapy. N. Engl. J. Med 2011;365:2255–2267 [DOI] [PubMed] [Google Scholar]
- 9.Lewis GF, Rader DJ. New insights into the regulation of hdl metabolism and reverse cholesterol transport. Circulation Res. 2005;96:1221–1232 [DOI] [PubMed] [Google Scholar]
- 10.Khera AV, Cuchel M, de la Llera-Moya M, Rodrigues A, Burke MF, Jafri K, French BC, Phillips JA, Mucksavage ML, Wilensky RL, Mohler ER, Rothblat GH, Rader DJ. Cholesterol efflux capacity, high-density lipoprotein function, and atherosclerosis. N. Engl. J. Med 2011;364:127–135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rohatgi A, Khera A, Berry JD, Givens EG, Ayers CR, Wedin KE, Neeland IJ, Yuhanna IS, Rader DR, de Lemos JA, Shaul PW. Hdl cholesterol efflux capacity and incident cardiovascular events. N. Engl. J. Med 2014;371:2383–2393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Francis GA. The complexity of hdl. Biochim Biophys Acta. 2010;1801:1286–1293 [DOI] [PubMed] [Google Scholar]
- 13.Oram JF, Heinecke JW. ATP-binding cassette transporter a1: A cell cholesterol exporter that protects against cardiovascular disease. Physiol. Rev. 2005;85:1343–1372 [DOI] [PubMed] [Google Scholar]
- 14.Timmins JM, Lee JY, Boudyguina E, Kluckman KD, Brunham LR, Mulya A, Gebre AK, Coutinho JM, Colvin PL, Smith TL, Hayden MR, Maeda N, Parks JS. Targeted inactivation of hepatic abca1 causes profound hypoalphalipoproteinemia and kidney hypercatabolism of apoa-i. J. Clin. Invest 2005;115:1333–1342 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bodzioch M, Orso E, Klucken J, Langmann T, Bottcher A, Diederich W, Drobnik W, Barlage S, Buchler C, Porsch-Ozcurumez M, Kaminski WE, Hahmann HW, Oette K, Rothe G, Aslanidis C, Lackner KJ, Schmitz G. The gene encoding atp-binding cassette transporter 1 is mutated in tangier disease. Nat. Genet 1999;22:347–351 [DOI] [PubMed] [Google Scholar]
- 16.Rust S, Rosier M, Funke H, Real J, Amoura Z, Piette JC, Deleuze JF, Brewer HB, Duverger N, Denefle P, Assmann G. Tangier disease is caused by mutations in the gene encoding atp-binding cassette transporter 1. Nat. Genet 1999;22:352–355 [DOI] [PubMed] [Google Scholar]
- 17.Brooks-Wilson A, Marcil M, Clee SM, Zhang LH, Roomp K, van Dam M, Yu L, Brewer C, Collins JA, Molhuizen HO, Loubser O, Ouelette BF, Fichter K, Ashbourne-Excoffon KJ, Sensen CW, Scherer S, Mott S, Denis M, Martindale D, Frohlich J, Morgan K, Koop B, Pimstone S, Kastelein JJ, Genest J, Jr., Hayden MR. Mutations in abc1 in tangier disease and familial high-density lipoprotein deficiency. Nat. Genet 1999;22:336–345 [DOI] [PubMed] [Google Scholar]
- 18.Chung S, Timmins JM, Duong M, Degirolamo C, Rong S, Sawyer JK, Singaraja RR, Hayden MR, Maeda N, Rudel LL, Shelness GS, Parks JS. Targeted deletion of hepatocyte abca1 leads to very low density lipoprotein triglyceride overproduction and low density lipoprotein hypercatabolism. J. Biol. Chem 2010;285:12197–12209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Brunham LR, Kruit JK, Iqbal J, Fievet C, Timmins JM, Pape TD, Coburn BA, Bissada N, Staels B, Groen AK, Hussain MM, Parks JS, Kuipers F, Hayden MR. Intestinal abca1 directly contributes to hdl biogenesis in vivo. J. Clin. Invest 2006;116:1052–1062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chung S, Sawyer JK, Gebre AK, Maeda N, Parks JS. Adipose tissue atp binding cassette transporter a1 contributes to high-density lipoprotein biogenesis in vivo. Circulation. 2011;124:1663–1672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Singaraja RR, Stahmer B, Brundert M, Merkel M, Heeren J, Bissada N, Kang M, Timmins JM, Ramakrishnan R, Parks JS, Hayden MR, Rinninger F. Hepatic atp-binding cassette transporter a1 is a key molecule in high-density lipoprotein cholesteryl ester metabolism in mice. Arterioscler. Thromb. Vasc. Biol 2006;26:1821–1827 [DOI] [PubMed] [Google Scholar]
- 22.Liu M, Chung S, Shelness GS, Parks JS. Hepatic abca1 and vldl triglyceride production. Biochim. Biophys. Acta 2012;1821:770–777 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yamamoto S, Tanigawa H, Li X, Komaru Y, Billheimer JT, Rader DJ. Pharmacologic suppression of hepatic atp-binding cassette transporter 1 activity in mice reduces high-density lipoprotein cholesterol levels but promotes reverse cholesterol transport. Circulation. 2011;124:1382–1390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yamashita S, Masuda D, Matsuzawa Y. Did we abandon probucol too soon? Curr. Opin. Lipidol 2015;26:304–316 [DOI] [PubMed] [Google Scholar]
- 25.Yamashita S, Matsuzawa Y. Where are we with probucol: A new life for an old drug? Atherosclerosis. 2009;207:16–23 [DOI] [PubMed] [Google Scholar]
- 26.Havel RJ, Eder HA, Bragdon JH. The distribution and chemical composition of ultracentrifugally separated lipoproteins in human serum. J Clin Invest. 1955;34:1345–1353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Terpstra AH, Nicolosi RJ, Herbert PN. In vitro incorporation of radiolabeled cholesteryl esters into high and low density lipoproteins. J. Lipid Res. 1989;30:1663–1671 [PubMed] [Google Scholar]
- 28.Pittman RC, Carew TE, Glass CK, Green SR, Taylor CA Jr., Attie AD. A radioiodinated, intracellularly trapped ligand for determining the sites of plasma protein degradation in vivo. Biochem. J. 1983;212:791–800 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lee JY, Lanningham-Foster L, Boudyguina EY, Smith TL, Young ER, Colvin PL, Thomas MJ, Parks JS. Prebeta high density lipoprotein has two metabolic fates in human apolipoprotein a-i transgenic mice. J Lipid Res. 2004;45:716–728 [DOI] [PubMed] [Google Scholar]
- 30.Bligh EG, Dyer WJ. A rapid method of total lipid extraction and purification. Can J Biochem Physiol. 1959;37:911–917 [DOI] [PubMed] [Google Scholar]
- 31.Parks JS, Rudel LL. Metabolism of the serum amyloid a proteins (ssa) in high-density lipoproteins and chylomicrons of nonhuman primates (vervet monkey). Am J Pathol. 1983;112:243–249 [PMC free article] [PubMed] [Google Scholar]
- 32.Folch J, Lees M, Sloane Stanley GH. A simple method for the isolation and purification of total lipides from animal tissues. J. Biol. Chem 1957;226:497–509 [PubMed] [Google Scholar]
- 33.Temel RE, Sawyer JK, Yu L, Lord C, Degirolamo C, McDaniel A, Marshall S, Wang N, Shah R, Rudel LL, Brown JM. Biliary sterol secretion is not required for macrophage reverse cholesterol transport. Cell Metab. 2010;12:96–102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Varban ML, Rinninger F, Wang N, Fairchild-Huntress V, Dunmore JH, Fang Q, Gosselin ML, Dixon KL, Deeds JD, Acton SL, Tall AR, Huszar D. Targeted mutation reveals a central role for sr-bi in hepatic selective uptake of high density lipoprotein cholesterol. Proc Natl Acad Sci U S A. 1998;95:4619–4624 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Thomas MS, Rudel LL. Intravascular metabolism of lipoprotein cholesteryl esters in african green monkeys: Differential fate of doubly labeled cholesteryl oleate. J. Lipid Res. 1987;28:572–581 [PubMed] [Google Scholar]
- 36.Bi X, Zhu X, Duong M, Boudyguina EY, Wilson MD, Gebre AK, Parks JS. Liver abca1 deletion in ldlrko mice does not impair macrophage reverse cholesterol transport or exacerbate atherogenesis. Arterioscler. Thromb. Vasc. Biol 2013;33:2288–2296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Allain CC, Poon LS, Chan CS, Richmond W, Fu PC. Enzymatic determination of total serum cholesterol. Clinical chemistry. 1974;20:470–475 [PubMed] [Google Scholar]
- 38.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative pcr and the 2(-delta delta c(t)) method. Methods. 2001;25:402–408 [DOI] [PubMed] [Google Scholar]
- 39.Lee JY, Timmins JM, Mulya A, Smith TL, Zhu Y, Rubin EM, Chisholm JW, Colvin PL, Parks JS. Hdls in apoa-i transgenic abca1 knockout mice are remodeled normally in plasma but are hypercatabolized by the kidney. J. Lipid Res 2005;46:2233–2245 [DOI] [PubMed] [Google Scholar]
- 40.Blanche PJ, Gong EL, Forte TM, Nichols AV. Characterization of human high-density lipoproteins by gradient gel electrophoresis. Biochim Biophys Acta. 1981;665:408–419 [DOI] [PubMed] [Google Scholar]
- 41.Huggins KW, Burleson ER, Sawyer JK, Kelly K, Rudel LL, Parks JS. Determination of the tissue sites responsible for the catabolism of large high density lipoprotein in the african green monkey. J. Lipid Res 2000;41:384–394 [PubMed] [Google Scholar]
- 42.Acton S, Rigotti A, Landschulz KT, Xu S, Hobbs HH, Krieger M. Identification of scavenger receptor sr-bi as a high density lipoprotein receptor. Science. 1996;271:518–520 [DOI] [PubMed] [Google Scholar]
- 43.Knouff C, Malloy S, Wilder J, Altenburg MK, Maeda N. Doubling expression of the low density lipoprotein receptor by truncation of the 3’-untranslated region sequence ameliorates type iii hyperlipoproteinemia in mice expressing the human apoe2 isoform. J. Biol. Chem 2001;276:3856–3862 [DOI] [PubMed] [Google Scholar]
- 44.Rashid S, Curtis DE, Garuti R, Anderson NN, Bashmakov Y, Ho YK, Hammer RE, Moon YA, Horton JD. Decreased plasma cholesterol and hypersensitivity to statins in mice lacking pcsk9. Proc. Natl. Acad. Sci. U. S. A 2005;102:5374–5379 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Groen AK, Bloks VW, Bandsma RH, Ottenhoff R, Chimini G, Kuipers F. Hepatobiliary cholesterol transport is not impaired in abca1-null mice lacking hdl. J. Clin. Invest 2001;108:843–850 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Drobnik W, Lindenthal B, Lieser B, Ritter M, Christiansen Weber T, Liebisch G, Giesa U, Igel M, Borsukova H, Buchler C, Fung-Leung WP, Von Bergmann K, Schmitz G. Atp-binding cassette transporter a1 (abca1) affects total body sterol metabolism. Gastroenterology. 2001;120:1203–1211 [DOI] [PubMed] [Google Scholar]
- 47.Wellington CL, Walker EK, Suarez A, Kwok A, Bissada N, Singaraja R, Yang YZ, Zhang LH, James E, Wilson JE, Francone O, McManus BM, Hayden MR. Abca1 mrna and protein distribution patterns predict multiple different roles and levels of regulation. Lab. Invest 2002;82:273–283 [DOI] [PubMed] [Google Scholar]
- 48.Rinninger F, Wang N, Ramakrishnan R, Jiang XC, Tall AR. Probucol enhances selective uptake of hdl-associated cholesteryl esters in vitro by a scavenger receptor b-i-dependent mechanism. Arterioscler. Thromb. Vasc. Biol 1999;19:1325–1332 [DOI] [PubMed] [Google Scholar]
- 49.Ji Y, Wang N, Ramakrishnan R, Sehayek E, Huszar D, Breslow JL, Tall AR. Hepatic scavenger receptor bi promotes rapid clearance of high density lipoprotein free cholesterol and its transport into bile. J. Biol. Chem 1999;274:33398–33402 [DOI] [PubMed] [Google Scholar]
- 50.Schaefer EJ, Blum CB, Levy RI, Jenkins LL, Alaupovic P, Foster DM, Brewer HB, Jr. Metabolism of high-density lipoprotein apolipoproteins in tangier disease. N. Engl. J. Med 1978;299:905–910 [DOI] [PubMed] [Google Scholar]
- 51.Chung S, Gebre AK, Seo J, Shelness GS, Parks JS. A novel role for abca1-generated large pre-beta migrating nascent hdl in the regulation of hepatic vldl triglyceride secretion. J. Lipid Res 2010;51:729–742 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Liu M, Chung S, Shelness GS, Parks JS. Hepatic abca1 deficiency is associated with delayed apolipoprotein b secretory trafficking and augmented vldl triglyceride secretion. Biochim. Biophys. Acta 2017;1862:1035–1043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sahoo D, Trischuk TC, Chan T, Drover VA, Ho S, Chimini G, Agellon LB, Agnihotri R, Francis GA, Lehner R. Abca1-dependent lipid efflux to apolipoprotein a-i mediates hdl particle formation and decreases vldl secretion from murine hepatocytes. J. Lipid Res 2004;45:1122–1131 [DOI] [PubMed] [Google Scholar]
- 54.Wang N, Arai T, Ji Y, Rinninger F, Tall AR. Liver-specific overexpression of scavenger receptor bi decreases levels of very low density lipoprotein apob, low density lipoprotein apob, and high density lipoprotein in transgenic mice. J. Biol. Chem 1998;273:32920–32926 [DOI] [PubMed] [Google Scholar]
- 55.Rinninger F, Heine M, Singaraja R, Hayden M, Brundert M, Ramakrishnan R, Heeren J. High density lipoprotein metabolism in low density lipoprotein receptor-deficient mice. J. Lipid Res 2014;55:1914–1924 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.de la Llera-Moya M, Drazul-Schrader D, Asztalos BF, Cuchel M, Rader DJ, Rothblat GH. The ability to promote efflux via abca1 determines the capacity of serum specimens with similar high-density lipoprotein cholesterol to remove cholesterol from macrophages. Arterioscler. Thromb. Vasc. Biol 2010;30:796–801 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hung KT, Berisha SZ, Ritchey BM, Santore J, Smith JD. Red blood cells play a role in reverse cholesterol transport. Arterioscler. Thromb. Vasc. Biol 2012;32:1460–1465 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.