

Abstract
The T-type Ca2+ channels encoded by the CaV3 genes are well established electrogenic drivers for burst discharge. Here, using CaV3.3−/− mice we found that CaV3.3 channels trigger synaptic plasticity in reticular thalamic neurons. Burst discharge via CaV3.3 channels induced long-term potentiation at thalamoreticular inputs when coactivated with GluN2B-containing NMDA receptors, which are the dominant subtype at these synapses. Notably, oscillatory burst discharge of reticular neurons is typical for sleep-related rhythms, suggesting that sleep contributes to strengthening intrathalamic circuits.
Introduction
T-type Ca2+ channels generate low-threshold discharges essential for neuronal rhythmogenesis (Huguenard, 1996), and, additionally, are involved in a variety of physiological and pathological conditions, such as sensory transmission, neurotransmitter release, neuronal development and pain (Cueni et al., 2009). The three T-channel subtypes (CaV3.1–3) show different expression patterns and distinct biophysical and pharmacological properties (Talley et al., 1999; Perez-Reyes, 2003). Whether such diversity accounts for specific roles has not yet been fully addressed, mainly because the pharmacological tools available are not subtype-specific, and, additionally, exert nonselective actions on other Ca2+ channels (Perez-Reyes, 2003).
A role for T-channels in promoting synaptic plasticity has been suggested by several studies, e.g., in cortex and hippocampus (Thomas et al., 1998; Schmidt-Hieber et al., 2004; Nevian and Sakmann, 2006; Lanté et al., 2011). Here, we made use of a mutant mouse lacking the CaV3.3 gene (Astori et al., 2011) to demonstrate the involvement of CaV3.3 channels in a novel form of intrathalamic plasticity. In the nucleus reticularis thalami (nRt), a component of the thalamocortical system, CaV3.3 channels represent a potent source for dendritic [Ca2+]i elevations (Cueni et al., 2008; Astori et al., 2011). Moreover, NMDA receptors (NMDARs), another Ca2+ source and trigger for synaptic plasticity, are expressed at both thalamoreticular and corticoreticular synapses (Gentet and Ulrich, 2003, 2004), but their proplastic role has not been explored.
We first investigated NMDAR signaling at thalamic inputs by means of patch-clamp electrophysiology on acute slices ex vivo, and found that GluN2B-containing NMDARs (GluN2B-NMDARs) represent the major NMDAR subtype at both developing and mature synapses. GluN2B-NMDAR activation associated with oscillatory low-threshold spiking induced long-term potentiation of thalamoreticular synapses. Potentiation was lacking in CaV3.3−/− mice, in which burst discharge is largely suppressed. Notably, oscillatory bursting resembles physiological activity patterns typical for NREM sleep (Steriade, 2006), suggesting that CaV3.3 channels and GluN2B-NMDARs may contribute to strengthen intrathalamic circuits and regulate sleep waves.
Materials and Methods
Electrophysiological recordings and analyses.
All procedures were approved by the Veterinary Office of Canton de Vaud. Typically, acute horizontal brain slices (300 μm thick) were prepared as previously described (Cueni et al., 2008) from 3- to 4-week-old C57BL/6J and CaV3.3−/− mice of either sex. In a subset of experiments, 2- or 7- to 8-week-old animals were used. For comparative characterization of cortical versus thalamic inputs, thalamocortical slices (Agmon and Connors, 1991) were prepared. Animals were maintained under a 12 h light/dark schedule (lights on at 7:00 AM), and slices were prepared between 11AM and 12AM. The CaV3.3−/− mouse line, originally obtained from GlaxoSmithKline, was maintained in the institute's animal facility and genotyped as previously described (Astori et al., 2011).
Slices were constantly superfused with oxygenated artificial CFS (ACSF) containing (in mm): 125 NaCl, 25 NaHCO3, 2.5 KCl, 1.25 NaH2PO4, 1.2 MgCl2, 2 CaCl2, 25 glucose, 1.7 l(+)-ascorbic acid, 0.1 picrotoxin, 0.01 glycine. Recordings were performed at 30–32°C. Visually identified nRt neurons were whole-cell patched with borosilicate glass pipettes (TW150F-4, WPI). Typically, pipettes (3–5 MΩ) were filled with (in mm): 140 KMeSO4, 10 KCl, 10 HEPES, 0.1 EGTA, 4 Mg-ATP, 0.2 Na-GTP, 10 phosphocreatine (290–300 mOsm, pH 7.25). For some recordings, 0.5 mm N-(2,6-dimethylphenylcarbamoylmethyl)-triethylammonium-chloride (QX-314) or 2 mm 1,2-bis(o-aminophenoxy)ethane-N,N,N′,N′-tetraacetic acid (BAPTA) were included in the pipette. A liquid junction potential of −10 mV was taken into account. For NMDA-EPSCs, pipettes (3–4 MΩ) were filled with (in mm): 127 CsGluconate, 10 HEPES, 2 BAPTA, 6 MgCl2, 2 Mg-ATP, 0.2 Na-GTP, 10 phosphocreatine (290–300 mOsm, pH 7.25), 2.5 QX-314. A liquid junction potential of −8 mV was corrected for. In horizontal slices, EPSCs were evoked every 30 s by stimulation in the internal capsule with an ACSF-filled electrode. In thalamocortical slices, the electrode was placed in cortical layer 6 or in the striatum adjacent to the cortex. Paired-pulse ratio (PPR, 50 ms interval) was measured from average responses (≥4 sweeps) as peak2/peak1.
For NMDA/AMPA ratios and NMDAR pharmacology, EPSCs were first evoked at −70 mV. Subsequently, cells were depolarized to +40 mV, and DNQX (40 μm) was bath-applied to block AMPAR-mediated transmission. NMDA-EPSC amplitude was measured as mean of 1 ms around the absolute peak of the average of 4 sweeps. This value was divided by the average EPSC peak at −70 mV to evaluate NMDA/AMPA ratios. NMDAR-GluN2-directed blockers were bath-applied for >4 min.
For plasticity experiments, baseline EPSCs were recorded for 10 min at −70 mV. Subsequently, cells were hyperpolarized to −80/−90 mV in current-clamp with DC injections, to ensure complete T-channel recovery from inactivation. Induction protocols consisted of sinusoidal current injections for 3 or 6 min (1 Hz, 0.15–0.3 nA maximal amplitude) to elicit low-threshold bursting, accompanied by paired or unpaired synaptic stimulation, as stated. Given the variability of burst onset and duration, it was not possible to define a fixed timing for the presynaptic stimulation relative to postsynaptic discharge. For series with 3 min or 6 min paired oscillations, we measured post hoc the time interval (Δt) between the first action potential and stimulation onset at three time points (beginning, middle, and end of the oscillation) to obtain a mean Δt for each cell. Mean Δt values ranged between −73/+76 ms (on average 23 ± 10 ms, n = 14). No correlation was found between Δt values and extent of plasticity induced (Pearson's correlation coefficient: r = 0.24, p = 0.41). For unpaired induction protocols, stimulation was delivered at the trough of the current injection (Δt = −400/−450 ms). To test for tonic firing-induced plasticity, membrane potential was adjusted to ∼−60 mV, and 200-ms-long squared pulses of 0.5/0.6 nA were provided at 1 Hz with synaptic stimulation.
After induction, EPSC recording was resumed for 30 min. Change in synaptic efficacy was calculated by comparing average EPSCs during the last 10 min of recording with baseline average EPSCs. Series resistance (Rs) was monitored throughout recordings by brief voltage-pulses, and data were rejected for Rs changes >25%. Data were acquired through a Digidata1320 digitizer. Signals were amplified through a Multiclamp700B amplifier (Molecular Devices), sampled at 20 kHz and filtered at 10 kHz using Clampex10 (Molecular Devices). Clampfit10 (Molecular Devices) and Igor5 Pro (WaveMetrics) were used for data analysis.
Data are presented as mean ± SEM. Paired or unpaired Student's t test was used as appropriate with significance accepted for p < 0.05. For comparison between age groups, one-way ANOVA was used followed by post hoc Student's t test.
Chemicals.
All standard salts and chemicals were purchased from Sigma-Aldrich, except the following: KMeSO4 (ICN Biomedicals), l(+)-ascorbic acid (VWR Prolabo), picrotoxin and DNQX (Abcam), QX-314 and PPDA ((2S*,3R*)-1-(phenanthren-2-carbonyl)piperazine-2,3-dicarboxylic acid; Tocris Bioscience). NVP-AAM077 was provided by Novartis Pharma. CP101,606 was provided by Pfizer Pharmaceuticals.
Results
GluN2B-NMDAR-mediated transmission at thalamoreticular synapses
GABAergic nRt cells receive glutamatergic inputs from thalamocortical cells and from layer 6 pyramidal cells. We first verified in the thalamocortical slice preparation that these two classes of inputs can be separated based on different short-term plasticity (Fig. 1). When stimulated in layer 6, corticoreticular responses consisted of long-latency facilitating EPSCs of relatively small amplitude. In contrast, antidromic stimulation of myelinated thalamic axons elicited short-latency EPSCs with larger depressant amplitudes. These findings were consistent with previous literature (Golshani et al., 2001; Gentet and Ulrich, 2003, 2004), and allowed identification of thalamoreticular depressant EPSCs when stimulating the internal capsule in horizontal slices in subsequent recordings.
Figure 1.

Thalamoreticular versus corticoreticular inputs. A, Scheme of a thalamocortical slice with recording electrode in the nRt and stimulating electrode in cortical layer 6. B, Average EPSCs evoked by two stimuli (50 ms), showing either paired-pulse depression (black) or paired-pulse facilitation (gray). Inset, superimposition of the first peaks reveals different response latencies. C, Plot of latency values versus PPR, indicating that depressant responses (n = 9, black) display shorter latencies than facilitating responses (n = 8, gray; p = 0.01 between groups). Filled circles represent mean values. D, Depressant inputs (PPD) display significantly larger amplitudes than facilitating inputs (PPF). *p < 0.05.
We examined basic properties of NMDAR transmission at thalamoreticular synapses. In 3- to 4-week-old mice, a significant NMDAR component was present at resting potential, as revealed by bath-application of 100 μm dl-APV, which accelerated decay kinetics of EPSCs at −70 mV (decay slope: 223 ± 50 pA/ms vs 290 ± 60 pA/ms, n = 9, p < 0.01; Fig. 2A). NMDAR blockade affected neither peak amplitude nor PPR of EPSCs (Fig. 2B), indicating that presynaptic NMDARs are unlikely to modulate basal transmission. In addition, these findings ensured that the pharmacological profile of postsynaptic NMDARs could be assessed without interfering with glutamate release. The NMDA/AMPA ratio significantly decreased after the second postnatal week from 0.42 ± 0.06 (n = 9) to 0.27 ± 0.04 (n = 14), and remained stable afterward at least until 7–8 weeks (0.26 ± 0.03, n = 10, p < 0.05; Fig. 2C), indicating synaptic maturation, as found in other brain structures (Liu et al., 1996; Brill and Huguenard, 2008).
Figure 2.
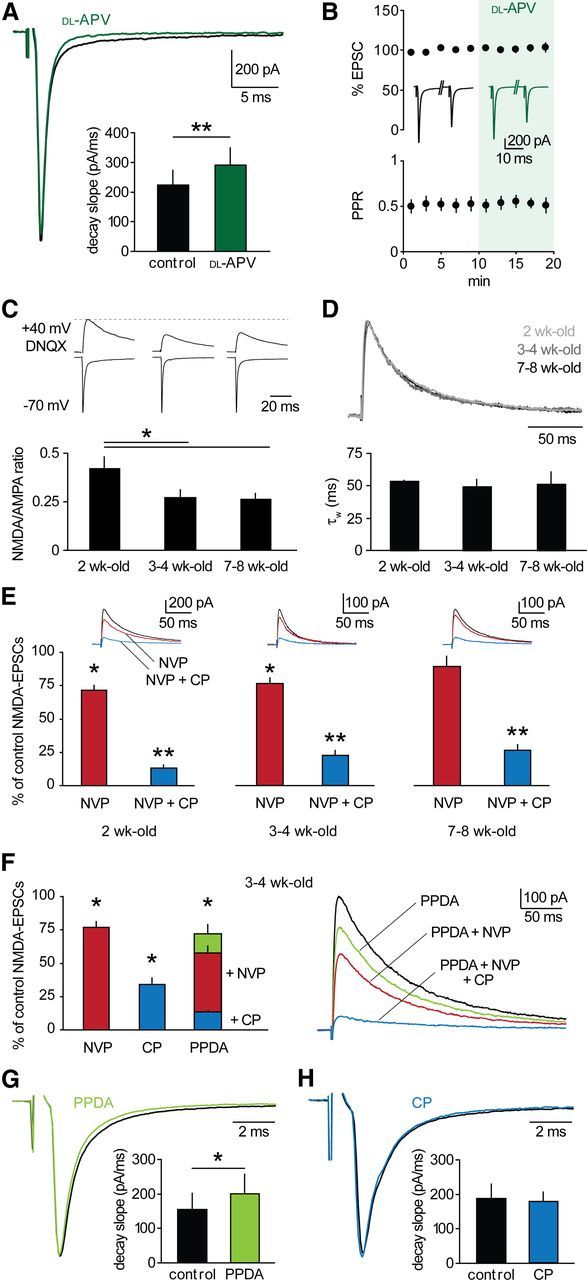
GluN2B-NMDARs dominate at thalamoreticular synapses. A, Thalamoreticular EPSCs in a cell voltage-clamped at −70 mV in control (black) and in the presence of 100 μm dl-APV (green). NMDAR blockade induced an increase in decay slope (bars; n = 9; p < 0.01). B, NMDAR blockade did not affect EPSC peak and PPR (n = 9, p > 0.05). Inserted traces are average EPSCs in control and in dl-APV. C, Top, examples of AMPAR- and NMDAR-mediated components at −70 mV and +40 mV, respectively. NMDA-EPSCs were isolated with 40 μm DNQX. Bottom, NMDA/AMPA ratio significantly decreased after 2 weeks (2-week-old, n = 9; 3- to 4-week-old, n = 14; 7- to 8-week-old, n = 10, *p < 0.05). D, Top, overlay of scaled NMDA-EPSCs at different developmental stages, coded in gray scale. No change in decay kinetics occurred, as indicated by comparable values of weighted τ (τW) of biexponential fit (bars: 2-week-old, n = 6; 3- to 4-week-old, n = 16; 7- to 8-week-old, n = 6). E, Pharmacological profile of NMDA-EPSCs (2-week-old, n = 6; 3- to 4-week-old, n = 8; 7- to 8-week-old, n = 6). Upper insets, example NMDA-EPSCs showing progressive reduction of control responses (black) after superfusion of NVP (red) and NVP+CP (blue). F, Left, mean effects of GluN2-specific blockers in 3- to 4-week-old mice (NVP, n = 8; CP, n = 8; PPDA, n = 7). Right, example NMDA-EPSCs showing progressive reduction upon blocker superfusion, as indicated. G, H, EPSCs in a cell voltage-clamped at −70 mV in control (black) and in the presence of 500 nm PPDA (G, green) or 10 μm CP (H, blue). GluN2C/D inhibition induced an increase in decay slope (bars; n = 8; p < 0.05), whereas GluN2B blockade had no effect (bars; n = 6; p > 0.05). *p < 0.05, **p < 0.01 drug versus control.
We next examined NMDAR subunit composition. GluN2 subunit expression is typically developmentally regulated such that GluN2B predominates at birth, while GluN2A expression starts in the first postnatal week and increases with maturation in most brain regions (Monyer et al., 1994). Surprisingly, decay times of NMDA-EPSCs remained constant at the three developmental stages examined (p > 0.05; Fig. 2D), suggesting no major changes in GluN2 content. Indeed, the pharmacological profile of NMDA-EPSCs was comparable at all stages (p > 0.05; Fig. 2E). The GluN2A-preferring antagonist NVP-AAM077 (NVP, 50 nm) (Auberson et al., 2002; Berberich et al., 2005) reduced NMDA-EPSCs in 2- and 3- to 4-week-old mice to a similar extent (2-week-old: 71.6 ± 3.6% of control, n = 6, p < 0.05 drug vs control; 3- to 4-week-old: 76.6 ± 4.1%, n = 8, p < 0.05). Subsequent application of the GluN2B-specific blocker CP101,606 (CP, 10 μm) (Mott et al., 1998) provoked a further reduction to 12.9 ± 2.2% and 22.7 ± 3.4%, respectively (p < 0.01 in both cases). Similarly, NVP had a minor effect compared with CP in 7- to 8-week-old mice (NVP: 86.5 ± 7.1%, NVP+CP: 24.8 ± 4.1%, n = 6, p = 0.36 and p < 0.01, respectively). In 3- to 4-week-old mice, we quantified the relative contribution of GluN2-subtypes to NMDAR transmission. When CP was applied in the absence of NVP, to avoid unspecific actions of NVP on GluN2B-NMDARs (Berberich et al., 2005; Longordo et al., 2009), NMDA-EPSCs were reduced to 34.1 ± 3.6% of control (3- to 4-week-old, n = 8, p = 0.07 compared with NVP+CP; Fig. 2F). Moreover, NMDA-EPSCs were sensitive to the GluN2C/D-preferring antagonist PPDA (500 nm; Harney et al., 2008), which reduced control responses to 71.6 ± 6.6% (n = 7, p < 0.05). Subsequent application of NVP and CP further reduced NMDA-EPSCs to 57.6 ± 4.8% (p < 0.05) and 13.3 ± 3.2% (p < 0.01), respectively (Fig. 2F). Notably, PPDA, similarly to dl-APV, slightly but significantly accelerated decay kinetics of EPSCs at −70 mV (decay slope: 155 ± 47 pA/ms vs 201 ± 56 pA/ms, n = 8, p < 0.05; Fig. 2G), whereas CP had no effect (185 ± 42 pA/ms vs 176 ± 26 pA/ms, n = 6, p > 0.05; Fig. 2H). Thus, in addition to GluN2B- and GluN2A-NMDARs, GluN2C/D-NMDARs are present at thalamoreticular synapses and are likely to be recruited during glutamatergic transmission at resting membrane potential.
In summary, our data suggest a complex composition of NMDARs at thalamoreticular synapses. Although a contribution of triheteromeric NMDARs (Rauner and Köhr, 2011) with undefined sensitivity to the antagonists used here cannot be excluded, our results indicate that GluN2B-containing NMDARs remain the predominant subtype throughout development and adulthood.
Burst-induced, NMDAR-dependent plasticity in nRt cells
We hypothesized that rhythmic discharge occurring during sleep waves, which relies on coincident activation of neurons in the thalamocortical system, could be conducive for intrathalamic plasticity. To mimic these rhythms in acute slices ex vivo, after recording baseline EPSCs, we elicited repetitive low-threshold discharge in hyperpolarized nRt cells by sinusoidal current injections in the δ frequency range (1 Hz; see Materials and Methods). Pairing synaptic inputs with burst discharge resulted in subsequent EPSC long-term potentiation (Fig. 3A,B). The duration of the pairing protocol (3 min vs 6 min) seemed to affect the time course of EPSC potentiation, but had no significant effect on its final extent (38 ± 9%, n = 7 vs 52 ± 18%, n = 7, p > 0.05 between groups). Lack of change in the PPR of baseline EPSCs versus potentiated EPSCs (Fig. 3G) suggested a postsynaptic locus of expression. To assess the requirement of synaptic activity for plasticity induction, we elicited burst discharge alone or with temporally unpaired synaptic inputs, both of which resulted in no potentiation (Fig. 3C,D). Next, we asked whether pairing-induced plasticity is NMDAR dependent. EPSC potentiation was prevented by CP (−0.7 ± 2.9%, n = 9, p > 0.05; Fig. 3E), but still inducible in the presence of NVP and PPDA (36.0 ± 9.3%, n = 10, p < 0.05; Fig. 3F), indicating an obligatory requirement for induction via GluN2B-NMDARs.
Figure 3.
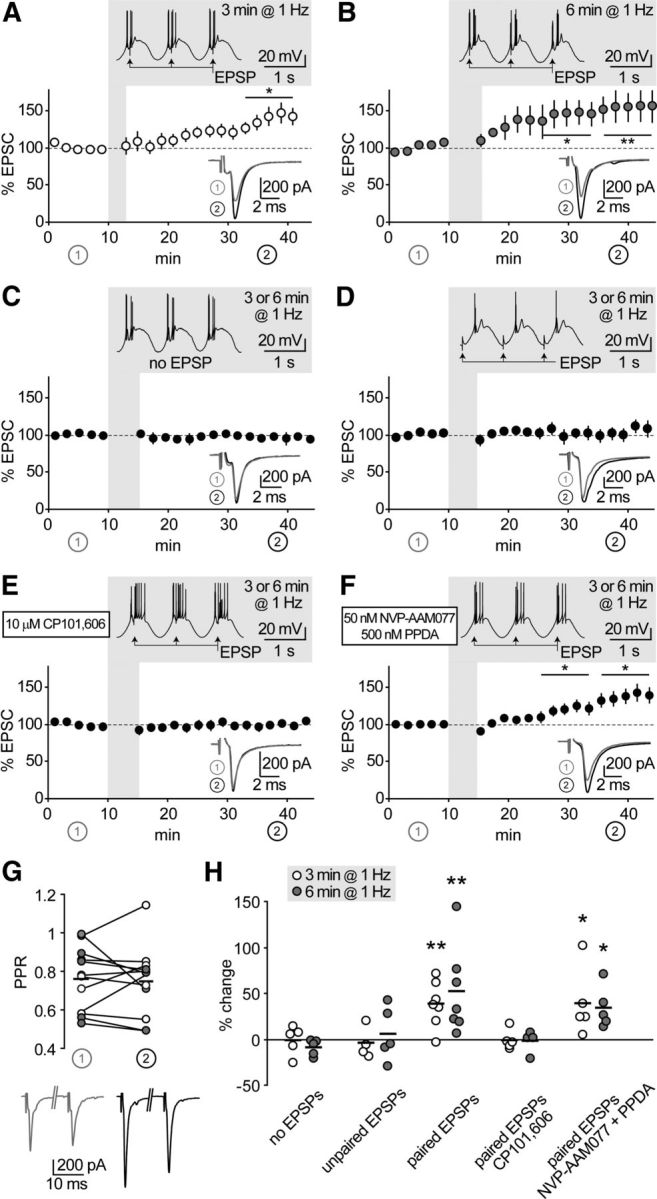
GluN2B-NMDARs mediate thalamoreticular plasticity. A, B, Time course of EPSCs at thalamoreticular synapses. Shadowed insets show pairing protocol applied after 10 min baseline. Low-threshold bursts were paired with synaptic stimulation (EPSP) for 3 min (A, n = 7) or 6 min (B, n = 7), which induced EPSC potentiation. Traces in the lower insets are average EPSCs evoked during baseline (1, gray) and during the last 10 min of recording (2, black). *p < 0.05, **p < 0.01 baseline versus end of recording. C–F, Same representation as in A and B. Synaptic potentiation was not induced when EPSPs were omitted (C, n = 10), or not paired with bursts (D, n = 9). Pairing-induced plasticity was prevented by GluN2B-NMDAR blockade with CP (E, n = 9), but not by inhibition of GluN2A and GluN2C/D with NVP and PPDA (F, n = 10). G, Potentiation was not accompanied by significant changes in PPR, as tested at the beginning of baseline (1) and at the end of the recording (2) in a subset of cells from A and B. H, Summary of data presented in A–F. Short horizontal lines represent means from series with 3 min (white circles) and 6 min (gray circles) oscillations. Asterisks represent significant difference from the corresponding “no EPSP” series (one-way ANOVA on log-transformed values, followed by post hoc Student's t test; *p < 0.05, **p < 0.01).
Requirement of CaV3.3 channels for burst-induced plasticity
Low-threshold bursts in nRt cells consist of large Ca2+ spikes crowned by Na+-dependent action potentials and arranged in cycles that are sustained by SK2-channel-mediated repolarization (Cueni et al., 2008). On the one hand, Ca2+ spikes alone could sufficiently depolarize nRt dendrites to relieve the Mg2+ block of NMDARs. On the other hand, the robust Ca2+ influx mediated by T-channels could be necessary to induce plasticity in synergy with GluN2B-NMDARs.
To test for the first hypothesis, we took two complementary approaches. First, we suppressed action potentials by blocking NaV channels with intracellular QX-314 (0.5 mm). This concentration was chosen to minimize unspecific blockade of T-channels (Talbot and Sayer, 1996), while ensuring sufficient NaV channel blockade at the subthreshold potentials reached during current injections (Fig. 4A, inset). Pairing-induced plasticity could still occur in this configuration (38 ± 13%, n = 7, p < 0.05; Fig. 4A), although the time course of the potentiation appeared to be slower compared with control. Thus, action potential discharge is not necessary for plasticity induction. Second, we modified the induction protocol to promote tonic discharge over low-threshold bursting: we applied depolarizing squared pulses to nRt cells held at −60 mV (see Materials and Methods), which resulted in robust suprathreshold firing with negligible T-channel contribution (Cueni et al., 2008; Astori et al., 2011). No change in synaptic efficacy was induced by pairing tonic firing with synaptic stimulation (6 ± 7%, n = 8, p > 0.05; Fig. 4B), indicating that repetitive action potential discharge is not sufficient to induce plasticity.
Figure 4.

CaV3.3 channels are required for thalamoreticular plasticity. A, Time course of EPSCs in nRt cells patched with a solution containing 0.5 mm QX-314 (n = 7). Shadowed insets show pairing protocols applied after 10 min baseline, with examples of average EPSCs during baseline (1) and at the end of recording (2). During induction, low-threshold bursts were largely preserved, while action potentials were blocked, which resulted in significant potentiation (n = 7). B, Same representation as in A. Sinusoidal current injections were replaced by squared current pulses applied to nRt cell held at −60 mV, to promote tonic firing over low-threshold bursting. No change in synaptic efficacy was induced (n = 8). C, D, Same representation as in A. Suppression of burst-induced Ca2+ with intracellular BAPTA (n = 6) and in CaV3.3−/− mice (n = 8) prevented potentiation. *p < 0.05 baseline versus end of recording.
To test for the second hypothesis, we included the Ca2+ chelator BAPTA (2 mm) in the intracellular solution, which prevented potentiation (8.7 ± 8.8%, n = 6, p > 0.05; Fig. 4C). Finally, we made use of CaV3.3−/− mice, in which nRt low-threshold bursting is largely suppressed (Astori et al., 2011). These mice displayed comparable thalamoreticular transmission to wild-type mice (NMDA/AMPA ratio: 0.31 ± 0.03, n = 13) and, more specifically, the same NMDAR pharmacological profile (NVP: 89.7 ± 9.6%, NVP+CP: 20.1 ± 4.2%, n = 7, p = 0.6 and p < 0.01, respectively). Although action potentials could still be elicited by sinusoidal oscillations, plasticity did not occur (7.2 ± 7.7%, n = 7, p > 0.05; Fig. 4D), indicating that CaV3.3 channel activation is a requirement for synaptic potentiation.
Discussion
Within thalamic regions, CaV3.3 channels are restricted to the nRt (Talley et al., 1999) and endow reticular neurons with oscillatory properties that are the cellular basis of sleep spindles (Astori et al., 2011). In the present study, we have identified a further role for CaV3.3 channels as triggers of synaptic plasticity. Repetitive bursting associated with synaptic simulation-induced long-term potentiation of thalamic inputs. Potentiation required GluN2B-NMDARs and could be induced even in the absence of Na+-action potentials, indicating that low-threshold spiking was sufficient to enable coincidence detection via NMDARs. In contrast, if postsynaptic depolarization was provided by Na+-dependent firing without T-channel contribution, or if low-threshold bursting was suppressed by CaV3.3 deletion, no potentiation occurred.
To repetitively coactivate CaV3.3 channels and NMDARs, we applied sinusoidal current injections to nRt cells conjointly with stimulation of thalamic inputs. This paradigm mimics some of the major electrical patterns occurring in vivo during physiological slow-wave sleep (Crunelli et al., 2006; Steriade, 2006). In this stage of NREM sleep, the thalamocortical system displays oscillatory electrical activity in the δ frequency range (0.5–4 Hz) generated by synchronous neuronal discharge. During NREM sleep, nRt cells are hyperpolarized, and low-threshold bursting becomes the dominant discharge mode, whereas tonic discharge is detected during waking and REM (Fuentealba and Steriade, 2005). Notably, sustained tonic discharge failed to induce plasticity. The modest Ca2+ influx generated by high-voltage-gated Ca2+ channels in nRt cells during action potentials (Cueni et al., 2008; Crandall et al., 2010) is probably insufficient to trigger plasticity, despite the contribution of NMDAR-mediated Ca2+ signaling. An alternative explanation is that a precise timing between presynaptic and postsynaptic signals could be required to generate spike-timing-dependent plasticity, which is an interesting subject for future investigations. Altogether, our data provide first evidence that thalamic centers are capable of long-term synaptic plasticity and suggest a mechanism by which intrathalamic synapses could boost sleep rhythms.
The persistence of GluN2B-NMDARs throughout development and adulthood is a remarkable feature of thalamoreticular synapses. The lack of a developmental switch in favor of GluN2A-NMDARs has been documented in only few other glutamatergic synapses, e.g., in the amygdala (Lopez de Armentia and Sah, 2003). Notably, thalamocortical neurons have already been reported to retain an immature phenotype by showing high expression of the differentiation-promoting transcription factor LEF1 (Wisniewska et al., 2010) and of HCN4 channels (Wenzel et al., 1997; Kanyshkova et al., 2009) that promote oscillatory discharge. We also found a component of NMDAR currents that was recruited at resting potential and sensitive to the GluN2C/D-preferring blocker PPDA. This component is likely to be mediated by GluN2C-NMDARs, that are expressed in thalamic regions (Monyer et al., 1994; Wenzel et al., 1997), and our data provide functional evidence for their presence at thalamoreticular synaptic sites. The unusual composition of NMDARs in nRt might have interesting functional implications. GluN2 composition is highly susceptible to sensory experience, environmental enrichment and learning (Kopp et al., 2007). Additionally, in different brain areas, GluN2 subunit trafficking has been shown to be sensitive to sleep deprivation (Kopp et al., 2007; Longordo et al., 2009) and to be modulated by arousal and sleep-promoting agents, such as orexins and adenosine (Borgland et al., 2006; Deng et al., 2011). Whether there are factors that modify intrathalamic NMDARs or whether there are protective mechanisms that retain the juvenile phenotype is an intriguing question arising from this work that may shed novel light on thalamic function and its unusual CaV3.3-gated plasticity.
Footnotes
This work was supported by the Swiss National Science Foundation (Ambizione grant to S.A. and nr. 129810 to A.L.) and by the Synapsis Foundation. We thank all laboratory members for critical reading of the manuscript and Dr. Georg Köhr for helpful comments and discussion. We are grateful to Dr. Ceri Davis for acting as a GlaxoSmithKline referent.
The authors declare no competing financial interests.
References
- Agmon A, Connors BW. Thalamocortical responses of mouse somatosensory (barrel) cortex in vitro. Neuroscience. 1991;41:365–379. doi: 10.1016/0306-4522(91)90333-j. [DOI] [PubMed] [Google Scholar]
- Astori S, Wimmer RD, Prosser HM, Corti C, Corsi M, Liaudet N, Volterra A, Franken P, Adelman JP, Lüthi A. The CaV3.3 calcium channel is the major sleep spindle pacemaker in thalamus. Proc Natl Acad Sci U S A. 2011;108:13823–13828. doi: 10.1073/pnas.1105115108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Auberson YP, Allgeier H, Bischoff S, Lingenhoehl K, Moretti R, Schmutz M. 5-Phosphonomethylquinoxalinediones as competitive NMDA receptor antagonists with a preference for the human 1A/2A, rather than 1A/2B receptor composition. Bioorg Med Chem Lett. 2002;12:1099–1102. doi: 10.1016/s0960-894x(02)00074-4. [DOI] [PubMed] [Google Scholar]
- Berberich S, Punnakkal P, Jensen V, Pawlak V, Seeburg PH, Hvalby Ø, Köhr G. Lack of NMDA receptor subtype selectivity for hippocampal long-term potentiation. J Neurosci. 2005;25:6907–6910. doi: 10.1523/JNEUROSCI.1905-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borgland SL, Taha SA, Sarti F, Fields HL, Bonci A. Orexin A in the VTA is critical for the induction of synaptic plasticity and behavioral sensitization to cocaine. Neuron. 2006;49:589–601. doi: 10.1016/j.neuron.2006.01.016. [DOI] [PubMed] [Google Scholar]
- Brill J, Huguenard JR. Sequential changes in AMPA receptor targeting in the developing neocortical excitatory circuit. J Neurosci. 2008;28:13918–13928. doi: 10.1523/JNEUROSCI.3229-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crandall SR, Govindaiah G, Cox CL. Low-threshold Ca2+ current amplifies distal dendritic signaling in thalamic reticular neurons. J Neurosci. 2010;30:15419–15429. doi: 10.1523/JNEUROSCI.3636-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crunelli V, Cope DW, Hughes SW. Thalamic T-type Ca2+ channels and NREM sleep. Cell Calcium. 2006;40:175–190. doi: 10.1016/j.ceca.2006.04.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cueni L, Canepari M, Luján R, Emmenegger Y, Watanabe M, Bond CT, Franken P, Adelman JP, Lüthi A. T-type Ca2+ channels, SK2 channels and SERCAs gate sleep-related oscillations in thalamic dendrites. Nat Neurosci. 2008;11:683–692. doi: 10.1038/nn.2124. [DOI] [PubMed] [Google Scholar]
- Cueni L, Canepari M, Adelman JP, Lüthi A. Ca2+ signaling by T-type Ca2+ channels in neurons. Pflugers Arch. 2009;457:1161–1172. doi: 10.1007/s00424-008-0582-6. [DOI] [PubMed] [Google Scholar]
- Deng Q, Terunuma M, Fellin T, Moss SJ, Haydon PG. Astrocytic activation of A1 receptors regulates the surface expression of NMDA receptors through a Src kinase dependent pathway. Glia. 2011;59:1084–1093. doi: 10.1002/glia.21181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuentealba P, Steriade M. The reticular nucleus revisited: intrinsic and network properties of a thalamic pacemaker. Prog Neurobiol. 2005;75:125–141. doi: 10.1016/j.pneurobio.2005.01.002. [DOI] [PubMed] [Google Scholar]
- Gentet LJ, Ulrich D. Strong, reliable and precise synaptic connections between thalamic relay cells and neurones of the nucleus reticularis in juvenile rats. J Physiol. 2003;546:801–811. doi: 10.1113/jphysiol.2002.032730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gentet LJ, Ulrich D. Electrophysiological characterization of synaptic connections between layer VI cortical cells and neurons of the nucleus reticularis thalami in juvenile rats. Eur J Neurosci. 2004;19:625–633. doi: 10.1111/j.1460-9568.2004.03168.x. [DOI] [PubMed] [Google Scholar]
- Golshani P, Liu XB, Jones EG. Differences in quantal amplitude reflect GluR4− subunit number at corticothalamic synapses on two populations of thalamic neurons. Proc Natl Acad Sci U S A. 2001;98:4172–4177. doi: 10.1073/pnas.061013698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harney SC, Jane DE, Anwyl R. Extrasynaptic NR2D-containing NMDARs are recruited to the synapse during LTP of NMDAR-EPSCs. J Neurosci. 2008;28:11685–11694. doi: 10.1523/JNEUROSCI.3035-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huguenard JR. Low-threshold calcium currents in central nervous system neurons. Annu Rev Physiol. 1996;58:329–348. doi: 10.1146/annurev.ph.58.030196.001553. [DOI] [PubMed] [Google Scholar]
- Kanyshkova T, Pawlowski M, Meuth P, Dubé C, Bender RA, Brewster AL, Baumann A, Baram TZ, Pape HC, Budde T. Postnatal expression pattern of HCN channel isoforms in thalamic neurons: relationship to maturation of thalamocortical oscillations. J Neurosci. 2009;29:8847–8857. doi: 10.1523/JNEUROSCI.0689-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kopp C, Longordo F, Lüthi A. Experience-dependent changes in NMDA receptor composition at mature central synapses. Neuropharmacology. 2007;53:1–9. doi: 10.1016/j.neuropharm.2007.03.014. [DOI] [PubMed] [Google Scholar]
- Lanté F, Toledo-Salas JC, Ondrejcak T, Rowan MJ, Ulrich D. Removal of synaptic Ca2+-permeable AMPA receptors during sleep. J Neurosci. 2011;31:3953–3961. doi: 10.1523/JNEUROSCI.3210-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu YB, Lio PA, Pasternak JF, Trommer BL. Developmental changes in membrane properties and postsynaptic currents of granule cells in rat dentate gyrus. J Neurophysiol. 1996;76:1074–1088. doi: 10.1152/jn.1996.76.2.1074. [DOI] [PubMed] [Google Scholar]
- Longordo F, Kopp C, Mishina M, Luján R, Lüthi A. NR2A at CA1 synapses is obligatory for the susceptibility of hippocampal plasticity to sleep loss. J Neurosci. 2009;29:9026–9041. doi: 10.1523/JNEUROSCI.1215-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez de Armentia M, Sah P. Development and subunit composition of synaptic NMDA receptors in the amygdala: NR2B synapses in the adult central amygdala. J Neurosci. 2003;23:6876–6883. doi: 10.1523/JNEUROSCI.23-17-06876.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monyer H, Burnashev N, Laurie DJ, Sakmann B, Seeburg PH. Developmental and regional expression in the rat brain and functional properties of four NMDA receptors. Neuron. 1994;12:529–540. doi: 10.1016/0896-6273(94)90210-0. [DOI] [PubMed] [Google Scholar]
- Mott DD, Doherty JJ, Zhang S, Washburn MS, Fendley MJ, Lyuboslavsky P, Traynelis SF, Dingledine R. Phenylethanolamines inhibit NMDA receptors by enhancing proton inhibition. Nat Neurosci. 1998;1:659–667. doi: 10.1038/3661. [DOI] [PubMed] [Google Scholar]
- Nevian T, Sakmann B. Spine Ca2+ signaling in spike-timing-dependent plasticity. J Neurosci. 2006;26:11001–11013. doi: 10.1523/JNEUROSCI.1749-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez-Reyes E. Molecular physiology of low-voltage-activated T-type calcium channels. Physiol Rev. 2003;83:117–161. doi: 10.1152/physrev.00018.2002. [DOI] [PubMed] [Google Scholar]
- Rauner C, Köhr G. Triheteromeric NR1/NR2A/NR2B receptors constitute the major N-methyl-d-aspartate receptor population in adult hippocampal synapses. J Biol Chem. 2011;286:7558–7566. doi: 10.1074/jbc.M110.182600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt-Hieber C, Jonas P, Bischofberger J. Enhanced synaptic plasticity in newly generated granule cells of the adult hippocampus. Nature. 2004;429:184–187. doi: 10.1038/nature02553. [DOI] [PubMed] [Google Scholar]
- Steriade M. Grouping of brain rhythms in corticothalamic systems. Neuroscience. 2006;137:1087–1106. doi: 10.1016/j.neuroscience.2005.10.029. [DOI] [PubMed] [Google Scholar]
- Talbot MJ, Sayer RJ. Intracellular QX-314 inhibits calcium currents in hippocampal CA1 pyramidal neurons. J Neurophysiol. 1996;76:2120–2124. doi: 10.1152/jn.1996.76.3.2120. [DOI] [PubMed] [Google Scholar]
- Talley EM, Cribbs LL, Lee JH, Daud A, Perez-Reyes E, Bayliss DA. Differential distribution of three members of a gene family encoding low voltage-activated (T-type) calcium channels. J Neurosci. 1999;19:1895–1911. doi: 10.1523/JNEUROSCI.19-06-01895.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas MJ, Watabe AM, Moody TD, Makhinson M, O'Dell TJ. Postsynaptic complex spike bursting enables the induction of LTP by theta frequency synaptic stimulation. J Neurosci. 1998;18:7118–7126. doi: 10.1523/JNEUROSCI.18-18-07118.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wenzel A, Fritschy JM, Mohler H, Benke D. NMDA receptor heterogeneity during postnatal development of the rat brain: differential expression of the NR2A, NR2B, and NR2C subunit proteins. J Neurochem. 1997;68:469–478. doi: 10.1046/j.1471-4159.1997.68020469.x. [DOI] [PubMed] [Google Scholar]
- Wisniewska MB, Misztal K, Michowski W, Szczot M, Purta E, Lesniak W, Klejman ME, Dabrowski M, Filipkowski RK, Nagalski A, Mozrzymas JW, Kuznicki J. LEF1/β-catenin complex regulates transcription of the CaV3.1 calcium channel gene (Cacna1g) in thalamic neurons of the adult brain. J Neurosci. 2010;30:4957–4969. doi: 10.1523/JNEUROSCI.1425-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]