

Abstract
Drugs targeting the orthosteric, primary binding site of G-protein coupled receptors are the most common therapeutics. Allosteric binding sites, elsewhere on the receptors, are less well-defined, and so less exploited clinically. We report the crystal structure of the prototypic beta-2 adrenergic receptor in complex with an orthosteric agonist and Compound-6FA, a positive allosteric modulator of this receptor. It binds on the receptor’s inner surface in a pocket created by intracellular loop 2 and transmembrane segments 3 and 4, stabilizing the loop in an alpha helical conformation required to engage the G-protein. Structural comparison explains the selectivity of the compound for beta-2 over the beta-1 adrenergic receptor. Diversity in location, mechanism, and selectivity of allosteric ligands provides potential to expand the range of receptor drugs.
One Sentence Summary:
Crystal Structure elucidates mechanism of action and specificity of a positive allosteric modulator of the β2AR.
G-protein-coupled receptors (GPCRs) regulate virtually all physiological processes in humans and are the subjects of intense drug discovery efforts(1, 2). Recent structures of GPCRs bound to allosteric modulators have revealed that receptor surfaces are decorated with diverse cavities and crevices that may serve as allosteric modulatory sites(3). This substantiates the notion that GPCRs are structurally plastic and can be modulated by a variety of allosteric ligands through distinct mechanisms(4–9). The majority of these structures have been solved with negative allosteric modulators (NAMs), which stabilize receptors in their inactive-states(3). To date only a single structure of an active GPCR bound to a small molecule positive allosteric modulator (PAM) has been reported, namely the M2 muscarinic acetylcholine receptor with LY2119620(10). Thus, mechanisms of PAMs and their potential binding sites remain largely unexplored.
The β2-adrenergic receptor (β2AR), a prototypic class A GPCR, plays essential roles in cardiovascular and respiratory physiology, and is the therapeutic target of clinical drugs such as beta-blockers and beta-agonists(11). We previously reported a cell-permeable NAM of the β2AR, Compound-15 (Cmpd-15), which binds to a cytoplasmic pocket within the central helical core of the receptor and prevents structural rearrangements necessary for receptor activation(12, 13). We recently identified Compound-6 (Cmpd-6), a positive allosteric modulator of the β2AR, through affinity-based screening with DNA-encoded small molecule libraries against purified agonist-bound β2AR(14). Cmpd-6 [(R)-N-(4-amino-1-(4-(tert-butyl)phenyl)-4-oxobutan-2-yl)-5-(N-isopropyl-N-methylsulfamoyl)-2-((4-methoxyphenyl)-thio)benzamide] exhibits robust positive cooperativity with orthosteric agonists and transducers (β-arrestins and G-proteins), and potentiates agonist-induced cAMP production and β-arrestin recruitment to the β2AR(14). Furthermore, Cmpd-6 is negatively cooperative with Cmpd-15 (Fig. S1A) and reduces antagonist binding to the β2AR (Fig. S1B). To both visualize the interaction of Cmpd-6 with the β2AR, and to understand the mechanism behind its positive allostery, we sought to obtain a crystal structure of the active receptor in complex with Cmpd-6.
The active-state of the β2AR is conformationally unstable and the addition of a G-protein or a G-protein mimetic is required for capturing this state by crystallography(15–18). We used nanobody 6B9 (Nb6B9) to stabilize the active-state of β2AR, as Cmpd-6 does not interfere with the ability of Nb6B9 to bind to agonist-occupied β2AR and enhance agonist affinity (Fig. S1, C and D). To facilitate crystallogenesis, we used a previously described amino-terminal T4 lysozyme-β2AR (T4L-β2AR) fusion protein(19). Cmpd-6 maintains its ability to enhance agonist binding of the T4L-β2AR (Fig. S1E).
We initially crystallized the T4L-β2AR bound to the agonist BI-167107, and Nb6B9 with 30 μM of Cmpd-6 (solubility limit) in the crystallization solution. However, we were not able to identify electron density representing Cmpd-6. This was attributed to Cmpd-6’s micromolar affinity and poor solubility. To improve solubility, we synthesized a derivative of Cmpd-6 (Cmpd-6FA) having a free carboxylic acid group (acetic acid) at its terminal amide site previously used for conjugating its DNA tag (Fig. 1A). Cmpd-6FA has approximately 6-fold higher solubility in aqueous solutions and recapitulates the pharmacological properties of the parent compound in in vitro assays with purified β2AR (Fig. 1B). To assess the direct molecular interaction between Cmpd-6FA and wild-type β2AR, we performed isothermal titration calorimetry (ITC) in the presence of BI-167107. The interaction is exothermic and enthalpically favorable with a stoichiometry of 1 and dissociation constant (KD) of 1.2 μM (Fig. 1C). When tested using β2AR bound to the antagonist carazolol, ITC detected no measurable physical interaction between the receptor and Cmpd-6FA, consistent with its role as a PAM (Fig. S2). Like the parent Cmpd-6, Cmpd-6FA displays positive cooperativity with the stimulatory G-protein (Gs) and β-arrestin1 (βarr) (Fig. S3)(14).
Fig. 1. Structure of the active-state T4L-β2AR in complex with the orthosteric agonist BI-167107, nanobody 6B9, and Compound-6FA.
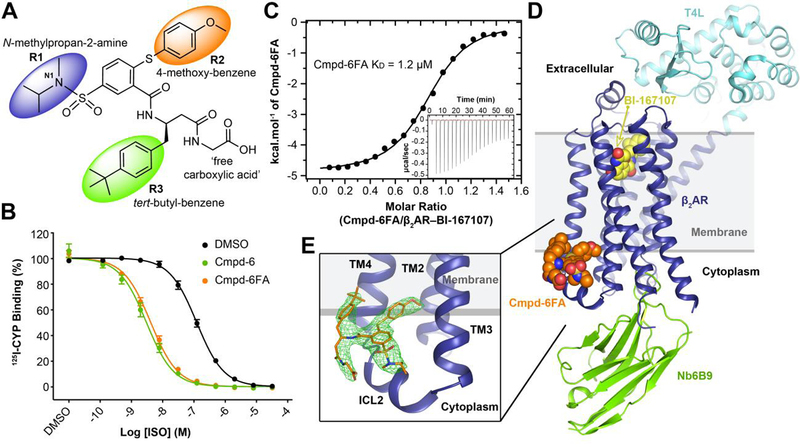
A, The chemical structure of Compound-6FA (Cmpd-6FA). B, Isoproterenol (ISO) competition binding with 125I-cyanopindolol (CYP) to the β2AR reconstituted in nanodiscs in the presence of vehicle (0.32 % dimethylsulfoxide; DMSO), Cmpd-6, or Cmpd-6FA at 32 μM. Values were normalized to percentages of the maximal 125I-CYP binding level obtained from a one-site competition binding-Log IC50 curve fit. Binding curves were generated by GraphPad Prism. Points on curves represent mean ± SEM obtained from five independent experiments performed in duplicate. C, Analysis of Cmpd-6FA interaction with the BI-167107-bound β2AR by isothermal titration calorimetry (ITC). Representative thermogram (insert) and binding isotherm, of three independent experiments, with the best titration curve fit are shown. Summary of thermodynamic parameters obtained by ITC: binding affinity (Kd = 1.2 ± 0.1 µM), stoichiometry (N = 0.9 ± 0.1 sites), enthalpy (ΔH = 5.0 ± 1.2 kcal/mol), and entropy (ΔS =13 ± 2.0 cal/mol/deg). D, Side-view of T4L-β2AR bound to the orthosteric agonist BI-167107, nanobody 6B9 (Nb6B9), and Cmpd-6FA. The gray box indicates the membrane layer as defined by the OPM database. e, Close-up view of Cmpd-6FA binding site. Covering Cmpd-6FA is 2Fo – Fc electron density contoured at 1.0 σ (green mesh).
We obtained crystals of the active T4L-β2AR bound to BI-167107 and Nb6B9 in a crystallization condition saturated with 3 mM of Cmpd-6FA. The complex structure was resolved to 3.2 Å by the molecular replacement method using the T4L-β2AR–Nb6B9 structure(17) as the search model (Table S1). Clear electron density was observed for Cmpd-6FA, and was localized at the interface between the cytoplasm and membrane (Fig. 1, D and E, and Fig. S4). However, the simulated annealing omit map was relatively weak and a contour level of 2σ was required to see the shape of Cmpd-6FA (Fig. S4A). We therefor generated a polder map, which excludes bulk solvent around the ligand (see Methods) and provides better density for compound (Fig. S4C). The Cmpd-6FA we used in crystallographic work is a mixture of two enantiomers with N1 as chiral center. An S configuration at nitrogen N1 was docked in our model based on the density, but due to the limitation of the crystal resolution, we cannot exclude the possibility that the R enantiomer is the correct one. The newly identified allosteric binding site is formed by the transmembrane helices (TMs) 2, 3, 4, and intracellular loop (ICL) 2 (Fig. 1E). Notably this site was not previously predicted to be a druggable allosteric site for the β2AR(20); therefore, its identification not only improves our understanding of the druggable molecular landscape on the β2AR but also expands our concept of what surface features can constitute an allosteric site.
The majority of contacts at the binding interface between Cmpd-6FA and the β2AR are mediated by hydrophobic and van der Waals interactions, which is consistent with the hydrophobic nature of the compound (Fig. S5). Cmpd-6FA can be subdivided into four chemical groups: core scaffold, N-methylpropan-2-amine (R1), 4-methoxy-benzene (R2), and tert-butylbenzene (R3) (Fig. 1A). The R1, R2, and R3 chemical moieties interact with three surface features of the β2AR – the cytoplasmic, membrane-proximal, and membrane-embedded surfaces (Fig. 2A). The core scaffold spans both the membrane-proximal and cytoplasmic surfaces, and provides a central point for the R1, R2, and R3 groups to extend into their respective binding surface features.
Fig. 2. Intermolecular interaction between the β2AR and Cmpd-6FA.
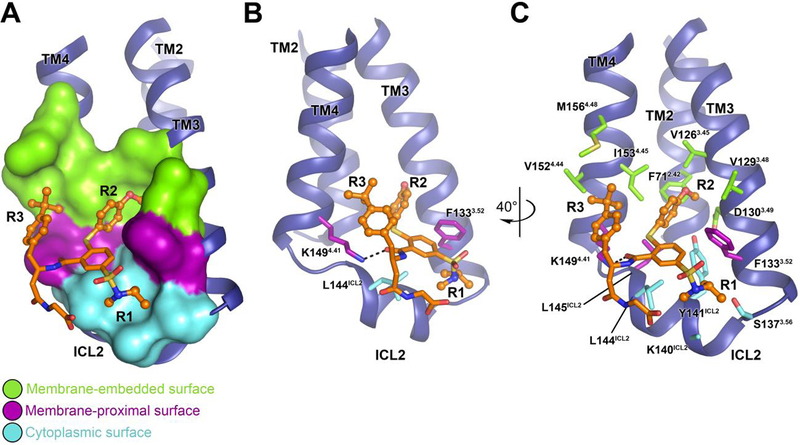
A, Overview of the Cmpd-6FA binding site. The molecular surface of the β2AR bound to Cmpd-6FA is subdivided into three surface regions: membrane embedded (green), membrane-proximal (purple), and cytoplasmic (cyan). B, C, Detailed interactions of the β2AR bound to Cmpd-6FA. B, The side-chains of those residues that interact with the core scaffold of Cmpd-6FA are shown. The hydrogen bond is indicated by a black dashed line. C, Rotated view showing all β2AR residues that contribute at the β2AR–Cmpd-6FA interface.
The core scaffold of Cmpd-6FA forms a hydrogen bond with Lys1494.41 (the only hydrogen bond between the compound and β2AR), and van der Waals contacts with Phe1333.52 and Leu144ICL2 (Fig. 2B). The acetate group of Cmpd-6FA, which was chemically added to enhance the compound solubility, makes minimal contacts with the β2AR and points towards solvent in the crystal structure. The R1, R2, and R3 moieties provide the majority of interactions with the protein surface (Fig. 2C).
To understand the structural basis of PAM activity, we compared our structure of the active β2AR–Cmpd-6FA occupied by the agonist BI-167107 to the structure of the inactive β2AR occupied by the antagonist carazolol (21). A superposition of these models reveals structural changes associated with receptor activation consistent with previous crystallographic studies of the β2AR(16, 18), such as the reorganization of ICL2 from a random coil to a two-turn α-helix (Fig. 3A). Tyr141ICL2 is positioned to both form a hydrogen bond with Asp1303.49 of the conserved (DRY) motif in TM 3 and stabilize the α-helical conformation of ICL2 within the active structure. Conversely, Tyr141ICL2 is situated nearly 12 Å away from the aspartate residue in the inactive structure. As a result of these structural changes, particularly in ICL2, the topography of the Cmpd-6FA binding site is particular to the active-state of the β2AR (Fig. 3B). The α-helical conformation of the ICL2 favors G protein binding because Phe139ICL2 adopts a suitable orientation to interact with a hydrophobic pocket on Gαs (Fig. 3, A and C).
Fig. 3. Mechanism of allosteric activation of the β2AR by Cmpd-6FA.
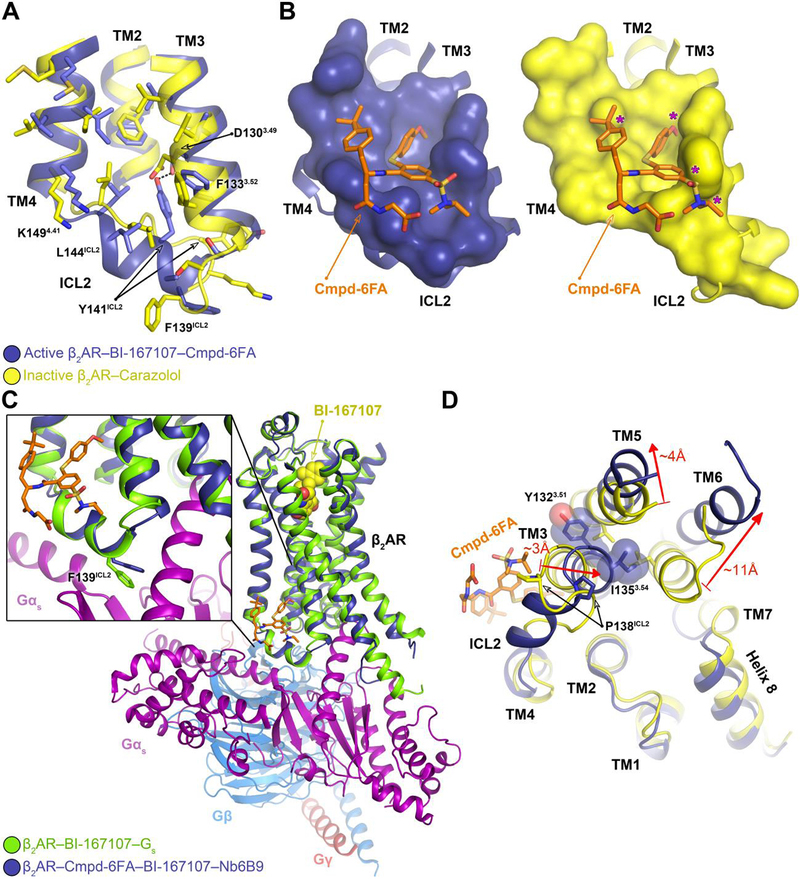
A, Superposition of the inactive β2AR bound to the antagonist carazolol (PDB code: 2RH1) and the active β2AR bound to the agonist BI-167107, Cmpd-6FA, and Nb6B9. Close-up view of the Cmpd-6FA binding site is shown. The residues of the inactive (yellow) and active (blue) β2AR are depicted and the hydrogen bond formed between Asp1303.49 and Tyr141ICL2 in the active-state is indicated by a black dashed line. B, Topography of Cmpd-6FA binding surface on the active β2AR (left, blue) and the corresponding surface of the inactive β2AR (right, yellow) with Cmpd-6FA (orange sticks) docked on top. Molecular surfaces are of only those residues involved in interaction with Cmpd-6FA. Steric clash between Cmpd-6FA and the surface of inactive β2AR is represented by a purple asterisk. C, Overlay of the β2AR bound to BI-167107, Nb6B9, and Cmpd-6FA with the β2AR–Gs complex (PDB code: 3SN6). The inset shows the position of Phe139ICL2 relative to the α-subunit of Gs. D, Superposition of the active β2AR bound to the agonist BI-167107, Nb6B9, and Cmpd-6FA (blue) with the inactive β2AR bound to carazolol (yellow) (PDB code: 2RH1) as viewed from the cytoplasm. For clarity, Nb6B9 and the orthosteric ligands are omitted. The arrows indicate shifts in the intracellular ends of the TMs 3, 5, and 6 upon activation and their relative distances.
The stabilization of the ICL2 α-helix by Cmpd-6FA may explain its ability to enhance agonist affinity (Fig. 1B). The formation of the two-turn α-helix in ICL2 requires an inward displacement of Pro138ICL2. This displacement results in a ~3Å inward movement of TM3, which in turn dictates an outward movement of TM5 and TM6 to prevent steric clashes between Tyr1323.51 and Ile1353.54 and residues on TM5 and TM6 (Fig. 3D). This outward movement of TM5 and TM6 is a hallmark of GPCR activation. In this way, Cmpd-6FA binding increases the population of receptors adopting active conformations, which have higher affinity for agonists (16–18, 22). Conversely, the structural rearrangements that are required when ICL2 forms an α-helix would be disfavored when the receptor is bound to an orthosteric inverse agonist, explaining the observation that Cmpd-6FA does not bind to carazolol-bound β2AR (Fig. S2). Further, Cmpd-6 retards agonist dissociation (Fig. S1F) and exerts its “agonist trapping” effect(23) not through direct steric occlusion but rather through stabilizing the active-state of the receptor with a closed hormone-binding site. This mode of action differs from the mechanism previously reported for the PAM that targets the M2 muscarinic acetylcholine receptor, whereby the allosteric modulator binds within the extracellular vestibule to directly prevent agonist dissociation(10).
To determine if Cmpd-6FA stabilizes a distinct conformation in the β2AR, we superimposed the β2AR–BI-167107–Nb6B9-Cmpd-6FA structure with the previously reported β2AR–BI-167107–Nb6B9 structure(17). Although the superposition showed the overall conformation of the receptors and agonist interactions to be nearly identical (r.m.s.d 0.37 Å, Fig. S6A), there are local conformational differences at the Cmpd-6FA binding site (Fig. S6B). In particular, the side-chains of Phe1333.52 and Tyr141ICL2 of the β2AR bound to Cmpd-6FA are pivoted to provide optimal shape complementarity with the allosteric ligand.
To validate our crystal structure, we performed a structure-activity relationship study using a set of Cmpd-6 derivatives. Fig. S7 summarizes the allosteric effects of these analogs on agonist binding affinity for the β2AR. The reduced function of the analogs agrees well with the Cmpd-6FA interactions observed in the crystal structure.
Allosteric sites may not face the same evolutionary pressure as do orthosteric sites, and thus are more divergent across subtypes within a receptor family(24–26). Therefore, allosteric sites may provide a greater source of specificity for targeting GPCRs. Cmpd-6 is much more efficacious in enhancing agonist binding and receptor-mediated signaling for the human β2AR, compared to the closely related β1AR subtype (Fig. 4). Sequence alignment of the β2AR and β1AR reveals that only 7 out of the 14 amino acid residues that form the Cmpd-6FA allosteric site are conserved (Fig. 4A). To investigate the roles of the 7 different residues, we constructed a series of β1AR mutants matching these residues to their counterparts in the β2AR. Subsequently, we performed [125I]CYP competition binding assays with these mutants in the presence or absence of Cmpd-6. Consistent with previous studies, Cmpd-6 enhances the agonist ISO binding affinity of the β2AR by 31-fold (Fig. 4B), and has only marginal effect toward the β1AR (2.8-fold increase, Fig. 4C). Interestingly, when all of the 7 residues of the β1AR were mutated to match their counterparts in the β2AR, agonist affinity was enhanced by 13-fold in the presence of Cmpd-6 (Fig. 4D). We speculated that the mutations at Leu1583.52 and Arg1744.41 may be the major determinants of the observed gain-of-function with the β1AR, as their β2AR counterparts play significant roles in binding to the core scaffold of Cmpd-6FA binding (Fig. 2, B and C, and Fig. S8, A and B). In fact, the single mutations of Leu1583.52 to phenylalanine, and Arg1744.41 to lysine resulted in a 10 and 7.3-fold increase in agonist affinity, respectively, in the presence of Cmpd-6 (Fig. S9, A and B), and the β1AR containing both of the Leu1583.52Phe and Arg1744.41Lys mutations conferred a 20-fold shift (Fig. 4E). It is interesting that the Arg1744.41Lys mutation has such a dramatic effect given that they are both basic amino acids. However, the structure reveals that the bulkier side-chain of an arginine would not fit into the space between ICL2 and Cmpd-6FA (Fig. S8C). Taken together, these gain-of-function studies are in accordance with the crystal structure of β2AR–Cmpd-6FA and strongly support the proposed allosteric site and binding mode of Cmpd-6FA.
Fig. 4. Gain-of-function of Cmpd-6 on the human β1AR mutants as compared to wild-type.
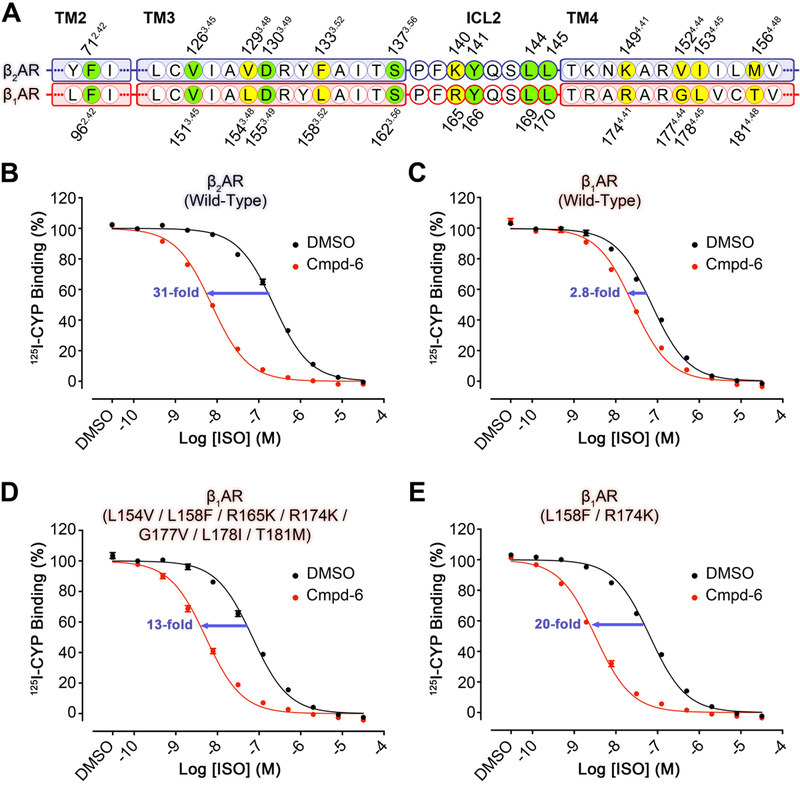
A, Partial protein sequence alignment of the human β2AR (blue highlight) and β1AR (red highlight) that includes only the relevant locations that engage Cmpd-6FA. Those residues that interact with Cmpd-6FA and are conserved are highlighted in green. Residues that are not conserved, but also interact with Cmpd-6FA are highlighted in yellow. Residue numbers for the β2AR and β1AR are shown above and below the alignment, respectively. B-E, Cmpd-6 positive allosteric effect on agonist binding of B, wild-type β2AR C, wild-type β1AR D, L154V / L158F / R165K / R174K / G177V / L178I / T181M β1AR mutant and E, L158F / R174K β1AR mutant. 125I-CYP vs. ISO competition binding was done using membrane preparations from HEK293 cells expressing the indicated receptor in the absence or presence of Cmpd-6 at 25 μM. Values were normalized to percentages of the maximal 125I-CYP binding level obtained from a one-site competition binding-Log IC50 curve fit (GraphPad Prism) of the vehicle (0.25% dimethylsulfoxide; DMSO) control data. Points on curves represent mean ± SEM obtained from four independent experiments performed in duplicate.
Recently, the crystal structure of the free fatty acid receptor GPR40 in complex with its partial agonist MK-8666 and allosteric agonist AP8 was reported(27). Although the complex represents the inactive-state of the receptor, it reveals the binding site of AP8, which is located outside the 7-transmembrane helical bundle at a lipid-facing region formed by TMs 3, 4, and 5 (Fig. S10A). An overlay of this structure and our β2AR structure bound to Cmpd-6FA shows that AP8 partially extends into the membrane-embedded surface and overlaps with the R2 moiety of Cmpd-6FA (Fig. S10B). More recently, mutagenesis and chimeric receptor studies suggested that the binding site for DETQ, a PAM for the dopamine D1 receptor, is located near ICL2, although no structural data were presented (28). Thus, this general locale in Class A GPCRs can exert positive allostery, and may be exploited for therapeutic purposes.
In summary, we identified a binding pocket on the β2AR that engages Cmpd-6FA, which is positioned on the surface of the helical bundle at the interface of the cytoplasm and membrane. Through structure-activity relationship studies and gain-of-function mutagenesis on the closely related β1AR subtype, we validated the binding site of Cmpd-6FA. Comparing the structure of the β2AR bound to Cmpd-6FA with the active and inactive receptors provided a mechanistic understanding of the positive allosteric modulatory effects of Cmpd-6FA. These structural comparisons suggest that Cmpd-6FA enhances both orthosteric agonist binding and transducer coupling by stabilizing the active-state of the β2AR.
Supplementary Material
Acknowledgments:
Funding: Supported by the National Institute of Health grants NS028471 and GM106990 (B.K.K.), HL16037 (R.J.L.), HL016037-45S1 (A.W.K.), and T32HL007101 (A.M.), Beijing Advanced Innovation Center for Structural Biology, School of Medicine, Tsinghua University (X-Y.L.) and the Mathers Foundation (B.K.K.).
Footnotes
Competing interests: Authors declare no competing interests.
Data and materials availability: Atomic coordinates and structure factors have been deposited in the Protein Data Bank (PDB) under accession code 6N48. All other data is available in the main text or Supplementary Material.
References and Notes:
- 1.Pierce KL, Premont RT, Lefkowitz RJ, Seven-transmembrane receptors. Nat Rev Mol Cell Biol 3, 639–650 (2002). [DOI] [PubMed] [Google Scholar]
- 2.Hauser AS, Attwood MM, Rask-Andersen M, Schioth HB, Gloriam DE, Trends in GPCR drug discovery: new agents, targets and indications. Nat Rev Drug Discov 16, 829–842 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Thal DM, Glukhova A, Sexton PM, Christopoulos A, Structural insights into G-protein-coupled receptor allostery. Nature 559, 45–53 (2018). [DOI] [PubMed] [Google Scholar]
- 4.Wacker D, Stevens RC, Roth BL, How Ligands Illuminate GPCR Molecular Pharmacology. Cell 170, 414–427 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Staus DP et al. , Allosteric nanobodies reveal the dynamic range and diverse mechanisms of G-protein-coupled receptor activation. Nature 535, 448–452 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Manglik A et al. , Structural Insights into the Dynamic Process of beta2-Adrenergic Receptor Signaling. Cell 161, 1101–1111 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ye L, Van Eps N, Zimmer M, Ernst OP, Prosser RS, Activation of the A2A adenosine G-protein-coupled receptor by conformational selection. Nature 533, 265–268 (2016). [DOI] [PubMed] [Google Scholar]
- 8.Van Eps N et al. , Conformational equilibria of light-activated rhodopsin in nanodiscs. Proc Natl Acad Sci U S A 114, E3268–E3275 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dror RO et al. , Structural basis for modulation of a G-protein-coupled receptor by allosteric drugs. Nature 503, 295–299 (2013). [DOI] [PubMed] [Google Scholar]
- 10.Kruse AC et al. , Activation and allosteric modulation of a muscarinic acetylcholine receptor. Nature 504, 101–106 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Goodman LS, Brunton LL, Chabner B, Knollmann B. r. C., Goodman & Gilman’s pharmacological basis of therapeutics (McGraw-Hill, New York, ed. 12th, 2011), pp. 2084 p. [Google Scholar]
- 12.Ahn S et al. , Allosteric “beta-blocker” isolated from a DNA-encoded small molecule library. Proc Natl Acad Sci U S A 114, 1708–1713 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Liu X et al. , Mechanism of intracellular allosteric beta2AR antagonist revealed by X-ray crystal structure. Nature 548, 480–484 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ahn S et al. , Small-Molecule Positive Allosteric Modulators of the beta2-Adrenoceptor Isolated from DNA-Encoded Libraries. Mol Pharmacol 94, 850–861 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rosenbaum DM et al. , Structure and function of an irreversible agonist-beta(2) adrenoceptor complex. Nature 469, 236–240 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rasmussen SG et al. , Structure of a nanobody-stabilized active state of the beta(2) adrenoceptor. Nature 469, 175–180 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ring AM et al. , Adrenaline-activated structure of beta2-adrenoceptor stabilized by an engineered nanobody. Nature 502, 575–579 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rasmussen SG et al. , Crystal structure of the beta2 adrenergic receptor-Gs protein complex. Nature 477, 549–555 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zou Y, Weis WI, Kobilka BK, N-terminal T4 lysozyme fusion facilitates crystallization of a G protein coupled receptor. PLoS One 7, e46039 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ivetac A, McCammon JA, Mapping the druggable allosteric space of G-protein coupled receptors: a fragment-based molecular dynamics approach. Chem Biol Drug Des 76, 201–217 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cherezov V et al. , High-resolution crystal structure of an engineered human beta2-adrenergic G protein-coupled receptor. Science 318, 1258–1265 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Huang W et al. , Structural insights into micro-opioid receptor activation. Nature 524, 315–321 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.DeVree BT et al. , Allosteric coupling from G protein to the agonist-binding pocket in GPCRs. Nature 535, 182–186 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Christopoulos A, Advances in G protein-coupled receptor allostery: from function to structure. Mol Pharmacol 86, 463–478 (2014). [DOI] [PubMed] [Google Scholar]
- 25.Wootten D, Christopoulos A, Sexton PM, Emerging paradigms in GPCR allostery: implications for drug discovery. Nat Rev Drug Discov 12, 630–644 (2013). [DOI] [PubMed] [Google Scholar]
- 26.Langmead CJ, Determining allosteric modulator mechanism of action: integration of radioligand binding and functional assay data. Methods Mol Biol 746, 195–209 (2011). [DOI] [PubMed] [Google Scholar]
- 27.Lu J et al. , Structural basis for the cooperative allosteric activation of the free fatty acid receptor GPR40. Nat Struct Mol Biol 24, 570–577 (2017). [DOI] [PubMed] [Google Scholar]
- 28.Wang X et al. , Intracellular Binding Site for a Positive Allosteric Modulator of the Dopamine D1 Receptor. Mol Pharmacol 94, 1232–1245 (2018). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.