

Abstract
Voltage-gated sodium channel Nav1.7 is a central player in human pain. Mutations in Nav1.7 produce several pain syndromes, including inherited erythromelalgia (IEM), a disorder in which gain-of-function mutations render dorsal root ganglia (DRG) neurons hyperexcitable. Although patients with IEM suffer from episodes of intense burning pain triggered by warmth, the effects of increased temperature on DRG neurons expressing mutant Nav1.7 channels have not been well documented. Here, using structural modeling, voltage-clamp, current-clamp, and multielectrode array recordings, we have studied a newly identified Nav1.7 mutation, Ala1632Gly, from a multigeneration family with IEM. Structural modeling suggests that Ala1632 is a molecular hinge and that the Ala1632Gly mutation may affect channel gating. Voltage-clamp recordings revealed that the Nav1.7-A1632G mutation hyperpolarizes activation and depolarizes fast-inactivation, both gain-of-function attributes at the channel level. Whole-cell current-clamp recordings demonstrated increased spontaneous firing, lower current threshold, and enhanced evoked firing in rat DRG neurons expressing Nav1.7-A1632G mutant channels. Multielectrode array recordings further revealed that intact rat DRG neurons expressing Nav1.7-A1632G mutant channels are more active than those expressing Nav1.7 WT channels. We also showed that physiologically relevant thermal stimuli markedly increase the mean firing frequencies and the number of active rat DRG neurons expressing Nav1.7-A1632G mutant channels, whereas the same thermal stimuli only increase these parameters slightly in rat DRG neurons expressing Nav1.7 WT channels. The response of DRG neurons expressing Nav1.7-A1632G mutant channels upon increase in temperature suggests a cellular basis for warmth-triggered pain in IEM.
SIGNIFICANCE STATEMENT Inherited erythromelalgia (IEM), a severe pain syndrome characterized by episodes of intense burning pain triggered by warmth, is caused by mutations in sodium channel Nav1.7, which are preferentially expressed in sensory and sympathetic neurons. More than 20 gain-of-function Nav1.7 mutations have been identified from IEM patients, but the question of how warmth triggers episodes of pain in IEM has not been well addressed. Combining multielectrode array, voltage-clamp, and current-clamp recordings, we assessed a newly identified IEM mutation (Nav1.7-A1632G) from a multigeneration family. Our data demonstrate gain-of-function attributes at the channel level and differential effects of physiologically relevant thermal stimuli on the excitability of DRG neurons expressing mutant and WT Nav1.7 channels, suggesting a cellular mechanism for warmth-triggered pain episodes in IEM patients.
Keywords: chronic pain, man on fire syndrome, sensory neurons, temperature responses, thermosensation, voltage-gated sodium channel
Introduction
The Nav1.7 channel, encoded by the gene SCN9A is a member of the voltage-gated sodium channel family (Nav1.1-Nav1.9) that is preferentially expressed in sensory neurons of dorsal root ganglia (DRG) and sympathetic ganglia neurons (Dib-Hajj et al., 2013). Nav1.7 is activated at relatively hyperpolarized potentials (below the threshold for action potential generation), and it amplifies stimuli within this subthreshold domain, setting the gain in DRG neurons (Cummins et al., 1998; Waxman, 2006). Nav1.7 has been regarded as a central player in human pain signaling, as loss-of-function Nav1.7 mutations cause congenital indifference to pain (Cox et al., 2006), and gain-of-function Nav1.7 mutations produce several painful disorders, including inherited erythromelalgia (IEM) (Yang et al., 2004; Dib-Hajj et al., 2005), paroxysmal extreme pain disorder (Fertleman et al., 2006), and small fiber neuropathy (Faber et al., 2012).
IEM, sometimes called the “man-on-fire” syndrome, is characterized by episodes of intense burning pain in the distal extremities, which are triggered by warmth. Thus far, IEM mutations of Nav1.7 have been functionally assessed in several ways: (1) voltage-clamp recording permits an evaluation of the effects of the mutations on channel function (Cummins et al., 2004; Estacion et al., 2013; Yang et al., 2013a); and (2) at a higher integrative level, current-clamp recording and dynamic clamp permit an assessment of the effects of the mutant channels on excitability of neurons carrying the mutant channels (Dib-Hajj et al., 2005; Rush et al., 2006; Han et al., 2012; Vasylyev et al., 2014). Current-clamp and dynamic clamp assessment, however, involve patch-clamp recording, which entails perfusion of the cells under study. Although more than a dozen Nav1.7 IEM mutations have been described in patients displaying the characteristic warmth-evoked pain (Dib-Hajj et al., 2013), the question of how warmth triggers episodes of pain in IEM has not been well addressed. Multielectrode arrays (MEAs), on the other hand, provide a noninvasive, high-throughput, extracellular recording approach, allowing study of changes in excitability of a population of DRG neurons with intact cell membranes, and permitting assessment of the effect of changes in temperature on excitability (Spira and Hai, 2013). Combining MEA with voltage- and current-clamp recording, in the present study, we assessed a newly identified Nav1.7 mutation (Ala1632Gly) from a multigeneration family with IEM. Our data demonstrate pathogenic features of this mutation and demonstrate differential effects of physiologically relevant thermal stimuli on the excitability of DRG neurons expressing mutant and WT Nav1.7 channels.
Materials and Methods
Exon screening.
The proband was sequenced by GeneDx. Sequencing of the proband's 8 family members (7 affected; 1 unaffected, the father of the proband) was performed at the Howard Hughes Medical Institute/Keck Biotechnology Center at Yale University. Briefly, genomic DNA was purified from venous blood of the proband and her family members. Coding exons were amplified and compared with the reference Nav1.7 cDNA (Klugbauer et al., 1995) to identify sequence variation, as described previously (Dib-Hajj et al., 2005). Sequences were analyzed using BLAST (National Library of Medicine) and Lasergene (DNAStar).
Structural modeling.
Structural modeling was performed as described previously (Yang et al., 2012, 2013a). Briefly, sequence alignment was achieved using ClustalW2 and manually refined (Yarov-Yarovoy et al., 2012). Initial models of the four transmembrane domains of hNaV1.7 channel were created using GPCR-ITASSER (Roy et al., 2011; Zhang et al., 2015) and aligned to bacterial sodium channel (Protein Data Bank ID code 3RVY) (Payandeh et al., 2011) in a clockwise order viewed from the extracellular side (Dudley et al., 2000; Li et al., 2001). Four domain complex structural models were refined by Fragment-Guided Molecular Dynamics simulation (Zhang et al., 2011). Models were presented using PyMol (Schrödinger). Multistate modeling was performed using methods described previously (Yang et al., 2013b; Huang et al., 2014a) based on molecular dynamics simulations of NavAb (Amaral et al., 2012). Modeling was performed with SWISS-MODEL and further energy-minimized, analyzed, and visualized in Molecular Operating Environment (Chemical Computing Group). Ligand and receptor interaction maps were generated using Molecular Operating Environment.
Plasmid preparation and HEK293 cell transfection.
TTX-resistant human NaV1.7 wild-type (WT) channel (hNav1.7r) was created based on the hNav1.7 (mRNA: NM_002977.3; protein: NP_002968.1 of NCBI database) as a pcDNA3mod-pEGFP-2A construct (Ryan and Drew, 1994; Atkins et al., 2007; Luke et al., 2008), as we have described previously for another channel, Nav1.9 (Huang et al., 2014b). Briefly, using Mega mutagenesis protocol, a sequence encoding EGFP was cloned upstream of the Nav1.7 ATG with a “stopGo” 33 amino acid 2A linker, such that the GFP-2A adaptor and the Nav1.7 channel proteins are produced as independent proteins from the same mRNA. The Ala1632GLy mutation was created on the hNaV1.7r background using Quick Change XL Site-Directed Mutagenesis Kit (Stratagene). Mutant or WT Nav1.7 channels were transfected into HEK293 cells together with human β1 and β2 subunit (Lossin et al., 2002) using Lipofectamine reagent (Invitrogen). HEK293 cells were maintained in 1:1 DMEM/F-12 supplemented with 10% FBS (Hyclone) in a humidified 5% CO2 incubator at 37°C. HEK293 cells were seeded onto poly-L-lysine-coated glass coverslips (BD Biosciences) in a 24 well plate.
Voltage-clamp recording in HEK cells.
Whole-cell voltage-clamp recordings were obtained from HEK293 cells 24–36 h after transfection using an EPC-10 amplifier and the PatchMaster program (version 53; HEKA Elektronik) at room temperature. The extracellular solution contained the following (in mm): 140 NaCl, 3 KCl, 1 MgCl2, 1 CaCl2, 20 dextrose, and 10 HEPES, pH 7.3 with NaOH (320 mOsm adjusted with dextrose). The pipette solution contained the following (in mm): 140 Cs-fluoride, 10 NaCl, 1 EGTA, 10 HEPES, 20 dextrose, pH 7.3 with CsOH (310 mOsm adjusted with dextrose). Patch pipettes had a resistance of 1–2 mΩ when filled with pipette solution. Series resistance and prediction compensation (80%–90%) was applied to reduce voltage errors. The recorded currents were digitized at a rate of 50 kHz after passing through a low-pass Bessel filter setting of 10 kHz. Recording was initiated after a 5 min equilibration period after achieving whole-cell configuration.
Data were analyzed using Fitmaster (HEKA Elektronik) and OriginPro (Microcal Software) software. To generate activation curves, cells were held at −120 mV and stepped to potentials of −80 to 45 mV in 5 mV increments for 100 ms. Peak inward currents were automatically extracted by Origin and fitted with a Boltzmann function (BoltzIV) to determine the voltage at half-activation (V1/2), activation curve slope at half-activation (Z), and reversal potential (ENa) for each recording. Conductance was calculated as G = I/(Vm − ENa) and normalized by the maximum conductance value and fit with Boltzmann equation. To generate steady-state fast-inactivation curves, cells were stepped to inactivating potentials from −130 to −20 mV for 500 ms followed by a 40 ms step to 0 mV. Peak inward currents obtained from steady-state fast-inactivation were normalized by maximum current amplitude and fitted with a Boltzmann equation (Cummins et al., 2004; Dib-Hajj et al., 2005). Steady-state slow-inactivation was assessed by prepulsing the cells with a 30 s stimulus at a range of voltages from −130 to 30 mV in 10 mV increments, followed by a 100 ms step to −120 mV to remove fast-inactivation and then a 50 ms depolarizing step to 0 mV to elicit a test response, which reflects the remaining currents available for activation. Peak inward currents obtained from steady-state slow-inactivation were normalized by the maximum current amplitude and fit with a Boltzmann equation (Huang et al., 2014a).
Isolation and transfection of primary sensory neurons.
Animal studies followed a protocol approved by the Veterans Administration Connecticut Healthcare System Institutional Animal Care and Use Committee. DRGs from 4- to 6-week-old female and male Sprague Dawley rats were harvested and dissociated as described previously (Dib-Hajj et al., 2009; Huang et al., 2014a, b). Briefly, DRG neurons were dissociated with a 20 min incubation in 1.5 mg/ml Collagenase A (Roche) and 0.6 mm EDTA, followed by a 18 min incubation in 1.5 mg/ml Collagenase D (Roche), 0.6 mm EDTA, and 30 U/ml papain (Worthington Biochemical). DRGs were then centrifuged and triturated in 0.5 ml of DRG media containing 1.5 mg/ml BSA (low endotoxin) and 1.5 mg/ml trypsin inhibitor (Sigma). After trituration, 2 ml of DRG media was added to the cell suspension, which was filtered with 70 μm nylon mesh cell strainer (BD Biosciences). The mesh was washed twice with 2 ml of DRG media. The cells were then transfected with WT or mutant human Nav1.7 constructs with the stopGo 2A tag that provides green fluorescence (abbreviated as hNav1.7 hereafter) using a Nucleofector IIS (Lonza) and Amaxa Basic Neuron SCN Nucleofector Kit (VSPI-1003). Briefly, the cell suspension was centrifuged (100 × g for 3 min), and the cell pellet was resuspended in 20 μl of Nucleofector solution, mixed with 2 μg of hNav1.7 WT or Ala1632Gly construct, and transfected using Nucleofector IIS and protocol SCN-BNP 6. Our established protocol reproducibly yielded a ∼40%-60% transfection rate for WT and mutant channels (Dib-Hajj et al., 2009). After transfection, cells were allowed to recover in calcium-free DMEM, fed with DRG media supplemented with nerve growth factor (50 ng/ml) and glial cell line-derived neurotrophic factor (50 ng/ml), and maintained at 37°C in a 95% air/5% CO2 (v/v) incubator for 40–55 h before current-clamp recording. Live/death assays of transfected neurons were performed using a Nuclear-ID Blue/Red cell Viability Kit (Enzo Life Sciences) according to manufacturer's instruction.
Current-clamp recordings.
The pipette solution contained the following (in mm): 140 KCl, 0.5 EGTA, 5 HEPES, 3 Mg-ATP, 10 dextrose, pH 7.30 with KOH (adjusted to 310 mOsm with sucrose), and the bath solution contained the following (in mm): 140 NaCl, 3 KCl, 2 MgCl2, 2 CaCl2, 10 HEPES, 10 dextrose, pH 7.30 with NaOH (adjusted to 320 mOsm with sucrose). Whole-cell configuration was obtained in voltage-clamp mode before proceeding to the current-clamp recording mode. DRG neurons (20–30 μm) with green fluorescence were selected for recording. There was no significant difference in neuron diameter between WT and mutant neurons (p = 0.661). Small DRG neurons with stable (<10% variation) resting membrane potentials (RMPs) more negative than −40 mV and action potentials with overshoots >40 mV and amplitudes >85 mV were included in analysis. Input resistance was determined by the slope of a linear fit to hyperpolarizing responses to current steps from −5 pA to −40 pA in −5 pA increments. Threshold was determined by the first action potential elicited by a series of depolarizing current injections (200 ms) that increased in 5 pA increments. Action potential frequency was determined by quantifying the number of action potentials elicited in response to depolarizing current injections (500 ms). Finally, the expression level of endogenous Nav1.8 channels was examined by holding neurons at −50 mV (Cummins and Waxman, 1997). Neurons that expressed small Nav1.8 currents (<1 nA) or unable to generate all-or-none action potentials in response to 200 ms current stimulus were excluded from analysis (Huang et al., 2014a). Data were analyzed using Fitmaster (HEKA Elektronik) and Origin (Microcal Software) software. Current-clamp recordings to assess temperature sensitivity of DRG neurons were performed at room temperature, 33 ± 0.5°C and 40 ± 0.5°C, and analyzed using custom-written routines in MATLAB (The MathWorks).
MEA recording.
DRG neurons from Sprague Dawley rats (4–6 weeks old) were isolated and transfected as described above. Transfected neurons were allowed to recover for 5 min at 37°C in 0.5 ml of Ca2+-free DMEM. The cell suspension was then diluted with DRG media, and 30–60 μl volume of medium containing neurons was seeded on 12 well MEA plates (Axion Biosystems) coated with poly-d-lysine (50 μg/ml)/laminin (10 μg/ml). The cells were then incubated at 37°C in 5% CO2 for 50 min. DRG media (1.5 ml per well), supplemented with 50 ng/ml each of mouse NGF (Alomone Labs) and GDNF (Peprotec), was added to the cells. Cells were maintained at 37°C in a 5% CO2 incubator.
Neurons were studied using an MEA system after 3 d in culture in their normal culture medium. During recording, an ECmini unit provides environmental control, maintaining carbon dioxide concentrations around MEA cultures with a low flow of premixed gas to prevent experimental variability. Action potential activity in these neurons was assessed using a multiwell MEA system (Maestro, Axion Biosystems). A 12 well recording plate was used, consisting a total of 768 electrodes. Within each well, 64 low-noise individual embedded microelectrodes with integrated ground electrodes, capable of monitoring the firing of individual neurons simultaneously, form a recording grid across a 2 × 2 mm area. The diameter of the electrodes was 30 μm with 200 μm center-to-center spacing between electrodes. For each experiment, three wells (with ∼192 available electrodes for recording) were used to assess neurons expressing WT Nav1.7 channels or Nav1.7-A1632G mutant channels.
MEA is a population-based analysis; thus, we implemented an experimental design to minimize potential confounding effects of transfection and handling of DRG neurons transfected with either WT or the Ala1632Gly mutant channels; seeding density, number of viable cells, and transfection rate were comparable between groups. Multiple independent transfections were used to generate the MEA data, minimizing potential effects of variable transfection efficiency in any one transfection experiment. To minimize variations in seeding density and number of viable cells: (1) DRG tissue from several animals were pooled, and dissociated neurons were equally divided into two groups for transfections with WT and Ala1632Gly; (2) the transfection of WT and Ala1632Gly constructs and the plating of the transfected neurons onto the MEA plate were performed at the same time using the same procedure, ensuring minimal variation for transfection efficiency and seeding procedure; and (3) neurons transfected with either WT or Ala1632Gly constructs were randomly placed in different wells distributed over the same MEA plate (and blinded to the investigator) to further minimize the variations.
A spike detection criterion of >6 SDs above background signals was used to separate monophasic or biphasic action potential spikes from noise. Active electrodes were defined as >1 spike over a 200 s analysis period. Firing frequencies were averaged among all active electrodes from wells expressing either construct. MEA data were analyzed using Axion Integrated Studio AxIS2.1 (Axion Biosystems) and NeuroExplorer (Nex Technologies).
To assess the effects of temperature on DRG neurons expressing either Nav1.7 WT or Nav1.7-A1632G mutant channels, we used the precise temperature control within the MEA system, which enables continuous recording of action potentials during a temperature ramp. DRG neurons expressing either WT or Nav1.7-A1632G mutant channels were plated on the same MEA plate for temperature manipulation and assessed by an investigator blinded to the identities of the wells. The MEA plate was ramped and maintained at 33°C, 37°C, 40°C, and 45°C for 7–10 min at each temperature to allow analysis of firing of these neurons at temperature steady state.
Data analysis.
Unless otherwise noted, statistical significance was determined using an independent t test. Two-proportion z test was used for comparing populations of spontaneously firing neurons. Mann–Whitney test was used for comparison of firing frequencies between neurons expressing WT and Ala1632Gly hNav1.7 channels in response to stimuli ranging from 25 pA to 500 pA. Repeated-measures two-way ANOVA with Bonferroni corrections was used for comparison of temperature sensitivity of firing frequencies between neurons expressing WT and Ala1632Gly hNav1.7 channels. Pairwise Mann–Whitney test with FDR corrections and two-way ANOVA were used for data collected during MEA experiments. Results are presented as mean ± SEM.
Results
Clinical description and molecular genetics
The female proband was first evaluated in rheumatology clinic at age 14 for bilateral leg pain. She manifested activity- and heat-associated neuropathic pain and color change of the distal extremities consistent with erythromelalgia. For ∼2 years, the patient had experienced episodic burning, aching, activity-related pain starting in the feet and radiating up both legs to the knees, exacerbated by warm temperature. The pain was accompanied by light red dorsal and plantar foot discoloration and a sensation of warmth in the feet. There was also occasional aching in the palms. Her symptoms resolved within 10–15 min of stopping activity and removal of socks and footwear. There was no extremity numbness, tingling, or swelling, and no symptoms elsewhere. The symptoms worsened in hot weather. Physical examination was notable for a normal general, neurologic, and musculoskeletal examination, with the exception of mild acral acrocyanosis in the toes. Gabapentin was not helpful. A trial of amitriptyline was stopped due to side effects and failure to improve. Bilateral topical lidocaine patches yielded partial pain relief when applied to the feet.
Family history was notable for appearance of similar symptoms in multiple members of this family across three generations, with the common feature of warmth-triggered pain attacks. The pedigree of this family contains 22 members: 11 subjects (one deceased) with IEM phenotype and 11 subjects without IEM phenotype (Fig. 1A). Family members with IEM phenotype have a similar age of onset (∼5–12 years old) compared with the proband (∼7 years old). We have previously reported a comparable range of age of onset in affected members of the family with IEM due to the Nav1.7-F1449V IEM mutation (McDonnell et al., 2016). Genetic analysis of SCN9A, the gene that encodes Nav1.7, from the proband showed a heterozygous (c. 4895C>G) mutation. This mutation substitutes alanine (Ala) by glycine (Gly) at position 1632 of the polypeptide (p. Ala1632Gly). Ala1632 is conserved in all human sodium channels (Nav1.1-Nav1.8), except for Nav1.9 where serine is present; Nav1.9 shows the lowest sequence conservation compared with other family members (Dib-Hajj et al., 1998). This mutation was confirmed by Sanger sequencing in every affected member of the family who consented to genetic analysis, but was absent from the asymptomatic father of the proband, suggesting high penetration of this mutation (Fig. 1A).
Figure 1.
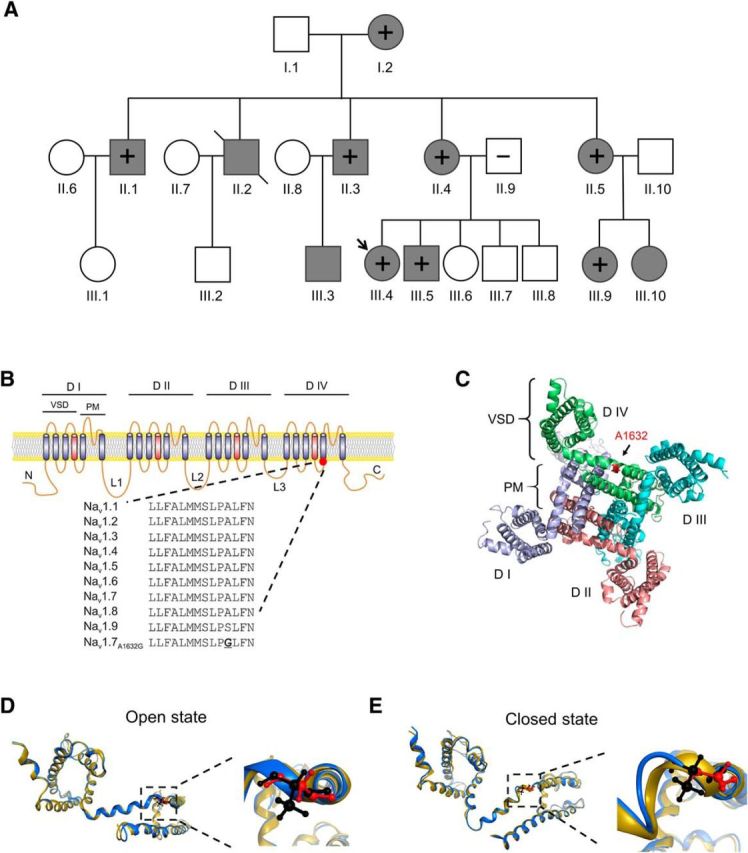
Ala1632Gly family pedigree and structural modeling of Ala1632Gly mutation in the Nav1.7 channel. A, Circles represent females. Squares represent males. Arrow indicates the proband. Filled symbols represent subjects with IEM. +, subjects tested positive for the Ala1632Gly mutation; −, subjects tested negative for the Ala1632Gly mutation. Slash indicates one deceased subject. Age of onset (years) of consenting subjects: I.2: preteen; II.1: ∼10; II.3: ∼5; II.4: ∼10; II.5: ∼7; III.4: ∼7; III.5: not sure; III.9: ∼8. B, Schematic of Nav1.7 channel topology showing the Ala1632Gly mutation. Nav1.7 consists of four homologous domains (DI–DIV), joined by three loops (L1–L3). Sequence alignment shows the alignment of Nav1.1-Nav1.9-containing Ala1632. The equivalent residue is conserved in all human voltage-gated sodium channels, except for Nav1.9, in which the analogous residue is a serine (Ser1496). C, Cytosolic view of the structural model of Nav1.7 channel transmembrane domains. Ala1632 is located in domain IV, at the hinge between S4–S5 linker and the S5 helix. Red represents Ala1632. D, E, Nav1.7 domain III multi-state structural models. Blue represents Nav1.7 WT model. Black represents Ala1632 residue. Yellow represents Nav1.7-A1632G model. Red represents Gly1632 residues. The models are superimposed. D, Nav1.7 WT and Ala1632Gly models in open state. The region containing Ala1632 or Gly1632 is boxed and enlarged on the right side. E, Nav1.7 WT and Ala1632Gly models in closed state. The region containing Ala1632 or Gly1632 is boxed and enlarged on the right side. The position and orientation of Gly1632 have a radial tuning compared with Ala1632.
Effect of Ala1632Gly on the local structure of the S4–5 linker in Nav1.7
Nav1.7 channel shares the hallmarks of the voltage-gated sodium channel family, including the four homologous domains (I-IV) linked by three intracellular loops (L1-L3), with each domain comprised of six transmembrane helices (S1-S6) (Catterall et al., 2005). The S1-S4 helices form the voltage-sensing domain, whereas the S5 and S6 helices, together with a small pore loop between S5 and S6, form the pore module (Fig. 1B). To visualize Ala1632Gly in the folded channel, we constructed a Nav1.7 structural model (Yang et al., 2012, 2013a) (Fig. 1C) and observed that Ala1632 is positioned at the turn between the S4-S5 linker and the S5 helix of domain IV (Fig. 1C). Sequence alignment and our structural modeling suggest that, for DIV, Ala1632 is likely the “hinge” residue between S4-S5 linker and S5 helix (Fig. 1C). It has been suggested that, during channel activation, the voltage-sensing domain and S4-S5 linker move together as a modular unit, transducing energy via a molecular hinge (e.g., Ala1632) to mediate pore transition from the closed state to open state (Payandeh et al., 2011). As glycine is a more flexible residue than alanine due to its smaller side chain, the Ala1632Gly substitution could thus impact the hinge movement.
To further explore how the Ala1632Gly mutation may affect channel structure and gating, we constructed a multistate model of Nav1.7 WT and Nav1.7-A1632G channels in their open and closed states (Amaral et al., 2012; Yang et al., 2013b; Huang et al., 2014a). The structural models of Nav1.7 WT and Nav1.7-A1632G in the open state largely superimpose in this part of the linker (Fig. 1D), whereas in the closed state, Gly1632 induces a notable twist of the helix compared with Ala1632 (Fig. 1E). These data suggest that the Ala1632Gly mutation causes a structural change in a critical hinge region of the channel that participates in gating.
Ala1632Gly confers gain-of-function attributes on the Nav1.7 channel
To understand the biophysical changes that the Ala1632Gly mutation confers on the Nav1.7 channel, we studied the voltage dependence of activation, steady-state fast-inactivation, and slow-inactivation in HEK cells expressing WT or Nav1.7-A1632G channels. Representative traces of recorded current families from cells expressing WT or Ala1632Gly are shown in Figure 2A, B. Notably, Nav1.7-A1632G caused a significant hyperpolarizing shift (∼7 mV) in the V1/2 of activation compared with WT channel (Ala1632Gly: −27.17 ± 1.41 mV, n = 8; WT: −20.68 ± 1.71 mV, n = 8; p < 0.05, Student's t test; Fig. 2C). Steady-state fast-inactivation was assessed in response to a 500 ms depolarizing potential. Ala1632Gly markedly shifted channel fast-inactivation V1/2 by ∼9 mV in a depolarizing direction (Ala1632Gly: −76.91 ± 2.42, n = 5; WT: −86.32 ± 3.04, n = 5, p < 0.05, Student's t test; Fig. 2D). The Ala1632Gly mutation did not significantly affect the channel's slow-inactivation V1/2 (Ala1632Gly: −72.01 ± 3.81, n = 4; WT: −70.03 ± 5.89, n = 5; p = 0.79, Student's t test). The hyperpolarizing shift of activation and the depolarizing shift of fast-inactivation are both gain-of-function attributes at the channel level, likely making the mutant channel easier to open and harder to inactivate.
Figure 2.

Biophysical properties of Nav1.7-A1632G mutant channels. A, B, Representative current traces recorded from HEK293 cells expressing WT (A) and Ala1632Gly mutant (B) channels. C, Voltage dependence of activation of WT and Ala1632Gly was plotted and fitted with a single Boltzmann equation. D, Voltage dependence of steady-state fast inactivation of WT and Ala1632Gly was plotted and fitted with a single Boltzmann equation.
Current-clamp characterization of DRG neurons expressing WT or Ala1632Gly
As one approach to assessing the potential impact on neuronal excitability resulting from altered biophysical properties in the hNav1.7-A1632G mutant channel, we performed current-clamp experiments on DRG neurons expressing either WT or Ala1632Gly at room temperature. After the formation of whole-cell configuration, neurons were allowed to stay at their resting membrane potential. We observed that 25% of DRG neurons carrying hNav1.7-A1632G mutant channels (12 of 48) fired action potentials spontaneously, whereas 7.9% of DRG neurons expressing WT hNav1.7 channels (3 of 38) fired spontaneously (p < 0.05, two-proportion z test), showing that the mutant channel tripled the population of small DRG neurons with spontaneous electrical activity. A representative recording trace (Fig. 3A) illustrates 30 s of continuous spontaneous firing in a DRG neuron expressing hNav1.7-A1632G channel with no external stimulus. For neurons that required external stimuli to fire an all-or-none action potential, we analyzed the following parameters and compared them between neurons expressing WT and Ala1632Gly channels: input resistance, current threshold, voltage threshold, action potential amplitude, and action potential width at 0 mV. No significant differences were found between WT (n = 35) and Ala1632Gly (n = 36) for these parameters (Table 1). Notably, there was a 35% statistically significant reduction in action potential current threshold (the intensity of external stimulus, which was required to initiate an all-or-none action potential) in neurons expressing Ala1632Gly (WT, 158 ± 20 pA; Ala1632Gly, 102 ± 16 pA; p < 0.05) (Fig. 3B). Representative action potentials recorded from small DRG neurons demonstrating the different current threshold for neurons expressing WT and Ala1632Gly hNav1.7 channels are shown in Figure 3C and Figure 3D, respectively. In Figure 3C, the neurons expressing WT channels did not fire an action potential when the injected current was <195 pA, which was defined as the threshold. In contrast, the neuron expressing Ala1632Gly mutant channels fired an action potential upon reaching its threshold of 105 pA (Fig. 3D). Action potential waveforms for WT and Ala1632Gly mutant channel-expressing neurons do not appear to be different, as we found that two primary parameters for the action potential waveforms (action potential amplitude and action potential width) were similar (Table 1). In addition, when the action potential waveforms for WT and mutant channel-expressing neurons were superimposed, we found them to largely overlap (Fig. 3D, inset).
Figure 3.

Ala1632Gly increases the number of spontaneously firing small DRG neurons and reduces current threshold for evoked firing. A, Representative recording of a spontaneously firing small DRG neuron expressing Ala1632Gly. The trace is a record of activity over 30 s without current injection. Bar graph represents a 3.2-fold increase in the proportion of spontaneously firing DRG neurons expressing Ala1632Gly (orange) compared with WT hNav1.7 channels (blue). B, Current threshold was significantly reduced in neurons expressing Ala1632Gly channels. C, Responses of a current-clamped small DRG neuron transfected with WT hNav1.7 channels to a series of subthreshold (180–190 pA) and suprathreshold depolarizing current steps (195 and 200 pA). Starting at a subthreshold stimulus, the current amplitude was increased in 5 pA increments to stimulus strength above threshold. The current threshold was 195 pA for this neuron. D, The same threshold protocol was applied to a small DRG neuron transfected with hNav1.7-A1632G channel. The current threshold was 105 pA for this neuron. Arrows indicate the current amplitude used to elicit the labeled response. Inset, Superimposed action potential waveforms of WT (blue) and Ala1632Gly expressing neurons (orange). Error bars indicate SEM. *p < 0.05.
Table 1.
Action potential characterization for WT and A1632G Nav1.7 channels in small DRG neurons
| Nav1.7 | Spontaneously firing neurons | Resting membrane potential (mV) | Input resistance (mΩ) | Voltage threshold (mV) | Current threshold (pA) | Action potential amplitude (mV) | Action potential width at 0 mV (ms) |
|---|---|---|---|---|---|---|---|
| WT | 3 of 38, 7.9% | −52.0 ± 0.82 | 641 ± 71 | −28.6 ± 1.2 | 158 ± 20 | 122 ± 1.9 | 3.33 ± 0.30 |
| A1632G | 12 of 48, 25%* | −50.2 ± 1.0 | 618 ± 55 | −31.6 ± 1.3 | 102 ± 16* | 126 ± 1.6 | 3.44 ± 0.27 |
| p value | 0.0380 | 0.189 | 0.792 | 0.0953 | 0.0309 | 0.0941 | 0.782 |
*p < 0.05 A1632G versus WT channels.
We further investigated whether there might be differences in the number of action potentials evoked by external stimuli between WT and Ala1632Gly by injecting a series of graded currents ranging from 25 to 500 pA in 25 pA increments into small DRG neurons and counting the total number of action potentials fired during the 500 ms stimulation period. As shown in Figure 4A–C, neurons expressing WT channels were more likely to generate only one action potential when injected with currents of amplitudes twofold or threefold of the current threshold. In contrast, neurons expressing Ala1632Gly tended to fire multiple action potentials in response to similar stimuli (Figs. 4D–F). Overall, neurons expressing hNav1.7-A1632G mutant channel fired substantially more action potentials than those expressing WT, and there was a statistically significant difference at each stimulus intensity (Fig. 4G). Additionally, as RMP is known to affect DRG neuron excitability (Harty et al., 2006), we also assessed RMP of DRG neurons expressing WT or Ala1632Gly mutant channels. The average RMP for neurons expressing Nav1.7-A1632G was depolarized by 1.8 mV compared with WT; the difference, however, did not reach statistical significance (WT, −52.0 ± 0.82 mV; Ala1632Gly, −50.2 ± 1.0 mV; p = 0.189, Student's t test) (Fig. 4H).
Figure 4.

The Ala1632Gly mutation increases evoked firing frequency in small DRG neurons. Responses of a small DRG neuron expressing WT (A–C) or Ala1632Gly (D–F) hNav1.7 channels to 500 ms depolarizing current steps that are onefold (A, D), twofold (B, E), and threefold (C, F) of its threshold, respectively. G, Summary of firing frequency in response to graded inputs. Error bars indicate SEM. *p < 0.05. H, Resting membrane potential of neurons expressing Ala1632Gly displays a mild depolarization of 1.8 mV compared with neurons expressing WT, but this depolarization does not reach statistical significance (p = 0.189).
Thermal stimuli increase the spontaneous firing of DRG neurons expressing hNav1.7-A1632G mutant channels
Warmth evokes intense burning pain in patients with IEM (Drenth and Waxman, 2007). Although it has been suggested that thermally triggered pain in IEM may be related to the altered firing of DRG neurons expressing mutant Nav1.7 channels (Dib-Hajj et al., 2013), the temperature sensitivity of firing properties of DRG neurons expressing IEM mutations is not well studied. To understand how warmth affects DRG neurons, we studied the firing properties of DRG neurons at 33°C and 40°C in current-clamp configuration. It has been suggested that 33°C represents skin temperature and 40°C represents warmth (Vriens et al., 2014). After the formation of whole-cell configuration, neurons were allowed to remain at their RMP. When we increased the temperature from room temperature to 33°C or 40°C, the percentage of spontaneously firing neurons expressing Nav1.7-A1632G increased from 25% (room temperature) to 42% (33°C) and 80% (40°C) (Fig. 5A,B). However, the percentage of spontaneously firing neurons expressing WT channel only increases minimally (room temperature: 7.5%; 33°C: 9%; 40°C: 10%; Fig. 5A,B). The percentage of spontaneously firing neurons expressing Nav1.7-A1632G mutant channels was significantly higher than those expressing Nav1.7 WT channels at both 33°C (p = 0.04) and 40°C (p = 0.0008) temperature (two-proportion z test).
Figure 5.

Increased temperature enhances firing of DRG neurons expressing hNav1.7-A1632G mutant channels. Current-clamp recordings of DRG neurons at different temperatures. A, B, Percentages of spontaneously firing DRG neurons expressing Ala1632Gly (A, left panel) or WT (A, right panel) at 33°C, and Ala1632Gly (B, left panel) or WT (B, right panel) at 40°C. C, Responses of DRG neurons to depolarizing current steps at a set holding membrane potential (Vm) = −60 mV at 33°C (C, left panel), and 40°C (C, right panel). *p < 0.05 (repeated-measures two-way ANOVA with Bonferroni corrections; n = 8–10).
To further understand the effect of increased temperature on the firing of DRG neurons, we studied evoked firing of DRG neurons expressing WT Nav1.7 or Nav1.7-A1632G mutant channels at a set holding membrane potential (−60 mV). This configuration eliminates the impact of variable RMP on firing properties of these neurons. Our results show that DRG neurons expressing Nav1.7-A1632G channels are significantly more excitable than those expressing WT Nav1.7 channels at 33°C and 40°C (Fig. 5C; p < 0.0005, two-way repeated-measures ANOVA).
Spontaneous firing is increased in intact DRG neurons expressing hNav1.7-A1632G
Whole-cell current-clamp study requires rupturing the cell membrane and dialyzing the cells' intracellular contents with pipette solution. To determine whether the Ala1632Gly mutation can change the excitability of intact nondialyzed DRG neurons, we assessed the firing of DRG neurons using MEA, a noninvasive, high-throughput, extracellular recording approach (Spira and Hai, 2013), which allows assessment of the firing behavior of the same neuron at multiple temperatures. For each experiment, neurons expressing either WT Nav1.7 or Nav1.7-A1632G mutant channels, all derived from the same preparation of neurons, were plated in three wells (with ∼192 available electrodes for recording) on the same MEA recording plate, which was assessed by a blinded investigator. The number of active electrodes (defined as electrode displaying at least 1 spike per 200 s) and mean firing frequency of active electrodes were analyzed at 37°C.
We have reported that DRG neurons transfected with WT or mutant Nav1.7 channels remain viable for several weeks after transfection (Persson et al., 2013; Estacion et al., 2015). Nonetheless, to rule out a possibility that Ala1632Gly mutation may have an effect on cell viability, we performed a cell viability assay for the neurons expressing WT or mutant channel after 3 d in culture, the time point when we performed MEA recordings. While transfection by electroporation causes cell death due to the electroporation procedure itself, we observed that the majority of neurons transfected with WT (82.9 ± 1.2%, n = 9 randomly selected fields from four independent experiments) were viable, and a comparable percentage of neurons transfected with the Ala1632Gly mutant were viable (84.1 ± 2.1%, n = 9 randomly selected fields from four independent experiments; p = 0.41 compared with WT, Student's t test). We also applied KCl to elicit firing of a population of DRG neurons (Shields et al., 2012; Estacion et al., 2015); In experiments where we studied the response of the DRG neurons to KCl (20 mm) with MEA, we found that the numbers of KCl-responsive DRG neurons/electrode were comparable between WT (36.5 ± 6.7, n = 8 wells) and Ala1632Gly (38.5 ± 4.4, n = 6 wells, p = 0.79, Student's t test), suggesting that recordable neurons on electrodes are comparable between the two conditions.
As shown in Figure 6, DRG neurons expressing WT Nav1.7 (Fig. 6A) and Nav1.7-A1632G mutant channels (Fig. 6B) both produced clearly identifiable spikes, which exhibited a reproducible waveform in the MEA recordings in the absence of stimulation. Representative raw traces of a 10 s epoch show that DRG neurons expressing Nav1.7-A1632G mutant channels fired at a higher level than those expressing WT Nav1.7 channel (Fig. 6C,D). Importantly, at the population level, well-wide analysis demonstrated more active electrodes in wells containing DRG neurons expressing Nav1.7-A1632G mutant channels compared with those expressing WT Nav1.7 channel. Among the active electrodes, higher firing frequencies were recorded from DRG neurons expressing Nav1.7-A1632G mutant channels. A representative well-wide raster plot of DRG neurons expressing WT Nav1.7 had 6 active electrodes over a 200 s period with the three highest firing frequencies at 0.65, 0.21, and 0.03 Hz (Fig. 6E). In contrast, a representative well-wide raster plot of DRG neurons expressing Nav1.7-A1632G mutant channels had 10 active electrodes over a 200 s period with the three highest firing frequencies at 1.67, 1.62, and 0.98 Hz (Fig. 6F). On average, wells containing DRG neurons expressing WT Nav1.7 channel had 8 ± 1 active electrodes with a mean firing frequency of 0.04 ± 0.02 Hz (4 independent experiments from a total of 8 rats), and wells with DRG neurons expressing Nav1.7-A1632G mutant channels had 24 ± 2 active electrodes with mean firing frequency of 0.18 ± 0.04 Hz (4 independent experiments with a total of 8 rats; p < 0.01 for numbers of active electrodes, two-way ANOVA with Bonferroni corrections; and p < 0.05 for mean firing frequency compared with WT, pairwise Mann–Whitney test with FDR corrections).
Figure 6.

MEA recording of spontaneous firing of DRG neurons expressing WT or Ala1632Gly mutant channels. A, B, Representative spike waveforms recorded using MEA from DRG neurons expressing WT Nav1.7 channels (A) and Nav1.7-A1632G mutant channels (B). Black line indicates the averaged waveform from 8 individual spikes. Dark cyan lines indicate each individual spike. C, D, Raw traces from a single electrode recording DRG neurons expressing WT Nav1.7 channels (C) and Nav1.7-A1632G mutant channels (D). DRG neurons expressing Nav1.7-A1632G mutant channels produce more spikes compared with those expressing WT Nav1.7 in the same recording period. E, F, Well-wide (64 electrodes) raster plot of MEA recordings of DRG neurons expressing WT (E) and hNav1.7-A1632G (F) at 37°C. Each horizontal plot represents recording from one electrode. More electrodes recorded spikes, and at higher frequencies from DRG neurons expressing Nav1.7-A1632G mutant channels. G–N, Heat maps of representative MEA recordings from DRG neurons expressing WT or hNav1.7-A1632G mutant channel. The firing frequency of each active electrode is color-coded: White/red represents high firing frequency. Blue/black represents low firing frequency. Each circle represents an active electrode within an 8 × 8 electrode array. For DRG neurons expressing Nav1.7-A1732G mutant channels (G–J), three active electrodes were evident at 33°C (G), four at 37°C (H), five at 40°C (I), and six at 45°C (J). Neurons expressing hNav1.7-A1632G mutant channel fired at a relatively high firing frequency. For DRG neurons expressing WT channels (K–N), three active electrodes were evident at 33°C (K), 37°C (L), and 40°C (M). Four active electrodes were seen at 45°C (N). These neurons fired at a relatively low frequency. O, Average firing frequencies of neurons expressing Nav1.7-A1632G and WT channel at the four temperatures (33°C, 37°C, 40°C, and 45°C). Forty active electrodes recorded firing from DRG neurons expressing WT channels (pooled from a total of 8 rats), whereas 162 active electrodes recorded firing from DRG neurons expressing Ala1632Gly mutant channels (pooled from a total of 8 rats). *p < 0.05 (pairwise Mann–Whitney test with FDR corrections). **p < 0.01 (pairwise Mann–Whitney test with FDR corrections). P, Average numbers of active electrodes from 3 wells containing DRG neurons expressing Nav1.7-A1632G and WT channels at the four temperatures. *p < 0.05 (repeated-measures two-way ANOVA with Bonferroni corrections). **p < 0.01 (repeated-measures two-way ANOVA with Bonferroni corrections). ***p < 0.01 (repeated-measures two-way ANOVA with Bonferroni corrections).
Thermal stimuli evoke firing in DRG neurons expressing hNav1.7-A1632G mutant channels
MEA recording is capable of accurately monitoring neuronal firing under a wide temperature range. To assess the effect of thermal stimuli on the firing of DRG neurons expressing Nav1.7-A1632G mutant channels, neuronal firing from the cultures described above was studied at four different temperatures (33°C, 37°C, 40°C, and 45°C). It is suggested that 33°C represents skin temperature, 37°C represents core body temperature, and 40°C/45°C represent different levels of warmth/hot (Vriens et al., 2014). DRG neurons expressing WT Nav1.7 channels or Nav1.7-A1632G mutant channels were plated in separate wells on the same MEA plate and assessed by a blinded investigator.
We found that the mean firing frequencies and the number of active electrodes within MEA wells containing DRG neurons expressing Nav1.7-A1632G mutant channels displayed a clear temperature-dependent response. In the representative heat-map plot, the number of colored circles, each representing an active electrode, clearly increased when the temperature was raised from 33°C to 45°C (Fig. 6G–J). Firing frequencies, which were represented by colors as a heatmap, also displayed a remarkable increase in response to elevated temperature (Fig. 6G–J). In contrast, the number of active electrodes and mean firing frequencies only slightly increased with elevated temperature for DRG neurons expressing WT Nav1.7 channels (Fig. 6K–N), which is consistent with our current-clamp data. Figure 6O, P summarizes data from 4 independent experiments with neurons from a total of 8 rats. On average, for DRG neurons expressing WT Nav1.7 channels, the numbers of active electrodes per 3 wells were 9 ± 1 (33°C), 8 ± 1 (37°C), 8 ± 2 (40°C), and 10 ± 2 (45°C), whereas the mean firing rates were 0.05 ± 0.04, 0.04 ± 0.02, 0.06 ± 0.03, and 0.07 ± 0.03 Hz, respectively. For DRG neurons expressing Nav1.7-A1632G mutant channels at these temperatures, the numbers of active electrodes were 20 ± 3 (33°C; p < 0.01 compared with WT, two-way ANOVA with Bonferroni corrections), 24 ± 2 (37°C; p < 0.01), 33 ± 3 (40°C; p < 0.001), and 40 ± 6 (45°C; p < 0.001), whereas the mean firing frequencies were 0.14 ± 0.02 Hz (33°C; p < 0.05 compared with WT, pairwise Mann–Whitney test with FDR corrections), 0.18 ± 0.04 Hz (37°C; p < 0.05), 0.35 ± 0.06 Hz (40°C; p < 0.05), and 0.65 ± 0.09 Hz (45°C; p < 0.01) (Fig. 6O,P). Together, our data demonstrate a clear increase in the number of neurons firing action potentials and in mean firing frequencies in response to an increase in temperature from 33°C to 45°C in DRG neurons expressing mutant but not WT Nav1.7 channel, suggesting a cellular basis of warmth-triggered pain in IEM.
Discussion
Using MEA as well as current- and voltage-clamp recordings, we have studied a newly identified Nav1.7 mutation, Ala1632Gly, from a multigenerational family with IEM. We found by voltage clamp that the Ala1632Gly mutation enhances channel activation and impairs fast-inactivation, and observed by current-clamp that it renders DRG neuron hyperexcitable, as manifested by a higher number of spontaneously firing neurons, lower current threshold, and enhanced evoked firing. Using noninvasive, extracellular MEA recordings, we studied the spontaneous firing of intact nonperfused DRG neurons and found that MEA distinguishes DRG neurons expressing Nav1.7-A1632G mutant channels from those expressing WT Nav1.7 channels. Our results also reveal a previously undocumented effect of Nav1.7 mutant channels on excitability of DRG neurons and demonstrate a higher number of active DRG neurons expressing Nav1.7-A1632G mutant channels, which displayed higher average firing frequencies, in response to increased temperature.
The Ala1632 residue appears to be a hot spot for disease-causing Nav1.7 mutations. We have previously reported the Ala1632Glu mutation from a patient with a mixed clinical phenotype that includes characteristics of both IEM and another distinct pain syndrome, paroxysmal extreme pain disorder (PEPD) (Estacion et al., 2008). More recently, the Ala1632Thr mutation was reported from two patients with IEM (Eberhardt et al., 2014). Here, we describe the Ala1632Gly, from a much larger multigeneration family. As our structural model suggests that the Ala1632 residue is likely to be a critical “hinge” between the S4-S5 linker and S5 helix of DIV, it is reasonable to suggest that mutations of this residue have a strong impact on channel function. We found that the Ala1632Glu mutation hyperpolarizes channel activation (suggested as a molecular basis for IEM disease phenotype) and depolarizes fast-inactivation (suggested as a molecular basis for PEPD disease phenotype) (Estacion et al., 2008). Interestingly, the Ala1632Thr mutation of IEM does not change channel activation but depolarizes channel fast-inactivation, suggesting that depolarized fast-inactivation alone may not necessarily correlate with PEPD phenotype, and that it can also be linked to IEM (Eberhardt et al., 2014). In the current study, we found that the Ala1632Gly mutation hyperpolarizes channel activation and depolarizes fast-inactivation in a family in which affected members display an IEM disease phenotype. Together, these data indicate that multiple substitutions of residue 1632 can cause pain syndromes. How a particular change in channel biophysical properties may correlate with different pain phenotypes needs further investigation.
We have studied the biophysical properties of the naturally occurring variants of the Ala1632 residues (Ala1632Glu, A1632Ser, and Ala1632Gly), and Eberhardt et al. (2014) have studied those of Ala1632Thr (naturally occurring) and Ala1632Asp, Ala1632Lys, and Ala1632Val variants. Interestingly, activation and fast-inactivation of Nav1.7-Ala1632Ser, Nav1.7-Ala1632Lys, and Nav1.7-Ala1632Val are similar to those of WT channel, whereas Nav1.7-Ala1632Asp displays a significant shift of activation and fast-inactivation comparable with the changes documented for the Nav1.7-Ala1632Glu. Together with data from Eberhardt et al. (2014), our current results argue that the effects of Ala1632Glu, Ala1632Asp, and Ala1632Gly on channel activation and fast-inactivation are probably achieved by different molecular pathways, which destabilize both the closed state and the inactivated state of the channel. By contrast, the sole effect of Ala1632Thr on fast-inactivation argues for another molecular pathway that destabilizes the inactivated state of the channel, sparing the closed state.
The size of the side-chain at the 1632 position does not appear to be the critical determinant for the mutant phenotype because Gly1632 produces robust effects on both activation and fast-inactivation, whereas Val1632 and Lys1632 do not alter these properties despite bulkier side-chains and the introduction a positive charge in the case of Lys1632. Additionally, a Nav1.7-Ser1632 channel, containing the residue present in the Nav1.9 channels, did not alter gating properties of Nav1.7 (Estacion et al., unpublished data), whereas Ala1632Thr shifted the inactivation but not activation curve. In contrast, Ala1632Gly, Ala1632Glu, and Ala1632Asp alter both activation and inactivation. A glutamic or aspartic acid residue at the hinge between the S4-S5 linker and S5 may engage in novel interactions or cause electrostatic repulsion due to the introduction of a negative charge, causing the mutant phenotype. By contrast, a glycine residue at position 1632, with its small side-chain, is more likely to cause a similar mutant phenotype by introducing flexibility to this hinge. Thus, the effect on channel activation and inactivation produced by Ala1632Gly and Ala1632Glu might be achieved by distinct mechanisms.
We previously demonstrated that Nav1.7-F1449V mutant channels cause hyperexcitability of DRG neurons without significantly altering RMP (Dib-Hajj et al., 2005), whereas some other Nav1.7 mutations depolarize RMP (Harty et al., 2006; Dib-Hajj et al., 2013). We have also shown that depolarizing resting potential of neurons by 5–10 mV contributes 25%–50% of the reduction in current threshold associated with Nav channel mutations (Harty et al., 2006; Huang et al., 2014b). In the current study, we found that the Ala1632Gly mutation depolarizes the membrane potential by ∼2 mV. As an ∼2 mV depolarization might play a role in DRG neuron hyperexcitability, to further understand the contribution of other properties of Nav1.7-A1632G mutant channels, we studied the evoked firing of DRG neurons at elevated temperatures in current-clamp experiments where we held the neurons at a set membrane potential (−60 mV), which eliminates the impact of variable RMP. In this setting, we found that DRG neurons expressing Nav1.7-A1632Gly mutant channels are still significantly more excitable than those expressing WT Nav1.7.
Multiwell MEA recording provides the opportunity to assess the firing of multiple intact neurons simultaneously within their normal culture condition without rupturing their membrane or dialyzing the recorded neurons (Wainger et al., 2014; Cesca et al., 2015). This approach, compared with manual whole-cell current-clamp, substantially increases throughput. The assay uses extracellular recording, and temperature ramps can be readily applied during the recording, so that the effects of thermal stimuli on the firing of intact neurons can be studied. On the other hand, current-clamp, while dialyzing the cells, can measure RMP, current threshold, and evoked firing of individual neurons (Yang et al., 2012; Huang et al., 2013; Han et al., 2015). The two approaches thus complement each other. In this study, the two techniques have provided information about different aspects of the effect of mutant Nav1.7 channels on DRG neurons.
Temperature effects on wild-type neurons have been studied previously. Although increased temperature may trigger activation of temperature-sensing transient receptor potential (TRP) channels (Vriens et al., 2014), which produce generator potentials that are amplified by Nav1.7 to evoke action potential firing, temperature also has marked effects on many potassium channels, leading to a reduction of input resistance of DRG neurons, adding a factor that inhibits action potential firing. For example, currents generated by TREK2 and TRAAK potassium channels, which are expressed in DRG neurons, increase ∼20-fold upon an increase in temperature from 24°C to 42°C (Kang et al., 2005). Studies on mouse superficial dorsal horn neurons found that raising temperature from 22°C to 32°C not only reduces input resistance but also increases the percentage of neurons that do not fire action potentials from ∼2% to ∼13% (Graham et al., 2008). Therefore, temperature effect on wild-type DRG neurons is likely to be the consequence of the combined impact of temperature on TRP channels, potassium channels, and Nav channels among others. A subgroup of temperature-sensing neurons could display significant increase in firing frequency upon temperature raise. However, within the overall DRG neuron population, neurons that do not respond to temperature may mask the responses of temperature-sensing neurons.
We found that, upon thermal stimulation, both the number of active DRG neurons expressing Nav1.7-A1632G mutant channel and their mean firing frequencies increased considerably. It is generally regarded that temperature-sensing in DRG neurons is achieved via a variety of TRP channels (Clapham, 2003; Julius, 2013). Different DRG neurons may have different subsets and expression levels of TRP channels, thereby transducing thermal stimuli of different strengths to produce generator potentials (Vriens et al., 2014). Nav1.7, on the other hand, is suggested to set the gain on DRG neurons by boosting subthreshold stimuli (e.g., generator potentials produced by TRP channels) and assisting in action potentials initiation (Cummins et al., 1998; Waxman, 2006; Rush et al., 2007). We suggest that, in the presence of gain-of-function IEM mutations that reduce action potential threshold and increase firing frequency of DRG neurons, warmth may activate a larger-than-normal set of DRG neurons (including pain-sensing neurons) and cause them to fire at higher-than-normal frequencies, consistent with a role in producing thermal hyperalgesia. This notion is supported by our observation that, even at the physiological temperature of 37°C and non-noxious 40°C temperature, there are significantly more active DRG neurons and higher mean firing frequency in neurons expressing Nav1.7-A1632G mutant channels. Although future work will be needed to delineate the precise contributions of potassium, sodium, and TRP conductances to the temperature responses of DRG neurons, the possible engagement of pain-sensing neurons, and high-frequency firing of these neurons, by subnoxious (mildly warm) thermal stimuli in the presence of Nav1.7 mutant channels provides evidence for a cellular mechanism within peripheral sensory neurons for warmth-triggered pain episodes in IEM.
Footnotes
This work was supported by the Medical Research Service and Rehabilitation Research Service, Department of Veterans Affairs, and the Kenneth Rainin Foundation. Y.Y. was supported by Connecticut Stem Cell Research Grants Program (CT Innovations). M.E. was supported by Department of Veterans Affairs SPiRE Grant. The Center for Neuroscience and Regeneration Research is a Collaboration of the Paralyzed Veterans of America with Yale University. We thank Dr. Chongyang Han and Dr. Andrew Tan for valuable comments; and Fadia Dib-Hajj and Peng Zhao for technical support.
The authors declare no competing financial interests.
References
- Amaral C, Carnevale V, Klein ML, Treptow W. Exploring conformational states of the bacterial voltage-gated sodium channel NavAb via molecular dynamics simulations. Proc Natl Acad Sci U S A. 2012;109:21336–21341. doi: 10.1073/pnas.1218087109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atkins JF, Wills NM, Loughran G, Wu CY, Parsawar K, Ryan MD, Wang CH, Nelson CC. A case for “StopGo”: reprogramming translation to augment codon meaning of GGN by promoting unconventional termination (Stop) after addition of glycine and then allowing continued translation (Go) RNA. 2007;13:803–810. doi: 10.1261/rna.487907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Catterall WA, Goldin AL, Waxman SG. International Union of Pharmacology: XLVII. Nomenclature and structure-function relationships of voltage-gated sodium channels. Pharmacol Rev. 2005;57:397–409. doi: 10.1124/pr.57.4.4. [DOI] [PubMed] [Google Scholar]
- Cesca F, Satapathy A, Ferrea E, Nieus T, Benfenati F, Scholz-Starke J. Functional interaction between the scaffold protein Kidins220/ARMS and neuronal voltage-gated Na+ channels. J Biol Chem. 2015;290:18045–18055. doi: 10.1074/jbc.M115.654699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clapham DE. TRP channels as cellular sensors. Nature. 2003;426:517–524. doi: 10.1038/nature02196. [DOI] [PubMed] [Google Scholar]
- Cox JJ, Reimann F, Nicholas AK, Thornton G, Roberts E, Springell K, Karbani G, Jafri H, Mannan J, Raashid Y, Al-Gazali L, Hamamy H, Valente EM, Gorman S, Williams R, McHale DP, Wood JN, Gribble FM, Woods CG. An SCN9A channelopathy causes congenital inability to experience pain. Nature. 2006;444:894–898. doi: 10.1038/nature05413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cummins TR, Waxman SG. Downregulation of tetrodotoxin-resistant sodium currents and upregulation of a rapidly repriming tetrodotoxin-sensitive sodium current in small spinal sensory neurons after nerve injury. J Neurosci. 1997;17:3503–3514. doi: 10.1523/JNEUROSCI.17-10-03503.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cummins TR, Howe JR, Waxman SG. Slow closed-state inactivation: a novel mechanism underlying ramp currents in cells expressing the hNE/PN1 sodium channel. J Neurosci. 1998;18:9607–9619. doi: 10.1523/JNEUROSCI.18-23-09607.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cummins TR, Dib-Hajj SD, Waxman SG. Electrophysiological properties of mutant Nav1.7 sodium channels in a painful inherited neuropathy. J Neurosci. 2004;24:8232–8236. doi: 10.1523/JNEUROSCI.2695-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dib-Hajj SD, Tyrrell L, Black JA, Waxman SG. NaN, a novel voltage-gated Na channel, is expressed preferentially in peripheral sensory neurons and down-regulated after axotomy. Proc Natl Acad Sci U S A. 1998;95:8963–8968. doi: 10.1073/pnas.95.15.8963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dib-Hajj SD, Rush AM, Cummins TR, Hisama FM, Novella S, Tyrrell L, Marshall L, Waxman SG. Gain-of-function mutation in Nav1.7 in familial erythromelalgia induces bursting of sensory neurons. Brain. 2005;128:1847–1854. doi: 10.1093/brain/awh514. [DOI] [PubMed] [Google Scholar]
- Dib-Hajj SD, Choi JS, Macala LJ, Tyrrell L, Black JA, Cummins TR, Waxman SG. Transfection of rat or mouse neurons by biolistics or electroporation. Nat Protoc. 2009;4:1118–1126. doi: 10.1038/nprot.2009.90. [DOI] [PubMed] [Google Scholar]
- Dib-Hajj SD, Yang Y, Black JA, Waxman SG. The Na(V)1.7 sodium channel: from molecule to man. Nat Rev Neurosci. 2013;14:49–62. doi: 10.1038/nrc3640. [DOI] [PubMed] [Google Scholar]
- Drenth JP, Waxman SG. Mutations in sodium-channel gene SCN9A cause a spectrum of human genetic pain disorders. J Clin Invest. 2007;117:3603–3609. doi: 10.1172/JCI33297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dudley SC, Jr, Chang N, Hall J, Lipkind G, Fozzard HA, French RJ. mu-conotoxin GIIIA interactions with the voltage-gated Na(+) channel predict a clockwise arrangement of the domains. J Gen Physiol. 2000;116:679–690. doi: 10.1085/jgp.116.5.679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eberhardt M, Nakajima J, Klinger AB, Neacsu C, Hühne K, O'Reilly AO, Kist AM, Lampe AK, Fischer K, Gibson J, Nau C, Winterpacht A, Lampert A. Inherited pain: sodium channel Nav1.7 A1632T mutation causes erythromelalgia due to a shift of fast inactivation. J Biol Chem. 2014;289:1971–1980. doi: 10.1074/jbc.M113.502211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Estacion M, Dib-Hajj SD, Benke PJ, Te Morsche RH, Eastman EM, Macala LJ, Drenth JP, Waxman SG. NaV1.7 gain-of-function mutations as a continuum: A1632E displays physiological changes associated with erythromelalgia and paroxysmal extreme pain disorder mutations and produces symptoms of both disorders. J Neurosci. 2008;28:11079–11088. doi: 10.1523/JNEUROSCI.3443-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Estacion M, Yang Y, Dib-Hajj SD, Tyrrell L, Lin Z, Yang Y, Waxman SG. A new Nav1.7 mutation in an erythromelalgia patient. Biochem Biophys Res Commun. 2013;432:99–104. doi: 10.1016/j.bbrc.2013.01.079. [DOI] [PubMed] [Google Scholar]
- Estacion M, Vohra BP, Liu S, Hoeijmakers J, Faber CG, Merkies IS, Lauria G, Black JA, Waxman SG. Ca2+ toxicity due to reverse Na+/Ca2+ exchange contributes to degeneration of neurites of DRG neurons induced by a neuropathy-associated Nav1.7 mutation. J Neurophysiol. 2015;114:1554–1564. doi: 10.1152/jn.00195.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faber CG, Hoeijmakers JG, Ahn HS, Cheng X, Han C, Choi JS, Estacion M, Lauria G, Vanhoutte EK, Gerrits MM, Dib-Hajj S, Drenth JP, Waxman SG, Merkies IS. Gain of function Nanu1.7 mutations in idiopathic small fiber neuropathy. Ann Neurol. 2012;71:26–39. doi: 10.1002/ana.22485. [DOI] [PubMed] [Google Scholar]
- Fertleman CR, Baker MD, Parker KA, Moffatt S, Elmslie FV, Abrahamsen B, Ostman J, Klugbauer N, Wood JN, Gardiner RM, Rees M. SCN9A mutations in paroxysmal extreme pain disorder: allelic variants underlie distinct channel defects and phenotypes. Neuron. 2006;52:767–774. doi: 10.1016/j.neuron.2006.10.006. [DOI] [PubMed] [Google Scholar]
- Graham BA, Brichta AM, Callister RJ. Recording temperature affects the excitability of mouse superficial dorsal horn neurons, in vitro. J Neurophysiol. 2008;99:2048–2059. doi: 10.1152/jn.01176.2007. [DOI] [PubMed] [Google Scholar]
- Han C, Hoeijmakers JG, Liu S, Gerrits MM, te Morsche RH, Lauria G, Dib-Hajj SD, Drenth JP, Faber CG, Merkies IS, Waxman SG. Functional profiles of SCN9A variants in dorsal root ganglion neurons and superior cervical ganglion neurons correlate with autonomic symptoms in small fibre neuropathy. Brain. 2012;135:2613–2628. doi: 10.1093/brain/aws187. [DOI] [PubMed] [Google Scholar]
- Han C, Yang Y, de Greef BT, Hoeijmakers JG, Gerrits MM, Verhamme C, Qu J, Lauria G, Merkies IS, Faber CG, Dib-Hajj SD, Waxman SG. The domain II S4–S5 linker in Nav1.9: a missense mutation enhances activation, impairs fast inactivation, and produces human painful neuropathy. Neuromolecular Med. 2015;17:158–169. doi: 10.1007/s12017-015-8347-9. [DOI] [PubMed] [Google Scholar]
- Harty TP, Dib-Hajj SD, Tyrrell L, Blackman R, Hisama FM, Rose JB, Waxman SG. Na(V)1.7 mutant A863P in erythromelalgia: effects of altered activation and steady-state inactivation on excitability of nociceptive dorsal root ganglion neurons. J Neurosci. 2006;26:12566–12575. doi: 10.1523/JNEUROSCI.3424-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang J, Yang Y, Zhao P, Gerrits MM, Hoeijmakers JG, Bekelaar K, Merkies IS, Faber CG, Dib-Hajj SD, Waxman SG. Small-fiber neuropathy Nav1.8 mutation shifts activation to hyperpolarized potentials and increases excitability of dorsal root ganglion neurons. J Neurosci. 2013;33:14087–14097. doi: 10.1523/JNEUROSCI.2710-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang J, Yang Y, Dib-Hajj SD, van Es M, Zhao P, Salomon J, Drenth JP, Waxman SG. Depolarized inactivation overcomes impaired activation to produce DRG neuron hyperexcitability in a Nav1.7 mutation in a patient with distal limb pain. J Neurosci. 2014a;34:12328–12340. doi: 10.1523/JNEUROSCI.2773-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang J, Han C, Estacion M, Vasylyev D, Hoeijmakers JG, Gerrits MM, Tyrrell L, Lauria G, Faber CG, Dib-Hajj SD, Merkies IS, Waxman SG. Gain-of-function mutations in sodium channel Na(v)1.9 in painful neuropathy. Brain. 2014b;137:1627–1642. doi: 10.1093/brain/awu079. [DOI] [PubMed] [Google Scholar]
- Julius D. TRP channels and pain. Annu Rev Cell Dev Biol. 2013;29:355–384. doi: 10.1146/annurev-cellbio-101011-155833. [DOI] [PubMed] [Google Scholar]
- Kang D, Choe C, Kim D. Thermosensitivity of the two-pore domain K+ channels TREK-2 and TRAAK. J Physiol. 2005;564:103–116. doi: 10.1113/jphysiol.2004.081059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klugbauer N, Lacinova L, Flockerzi V, Hofmann F. Structure and functional expression of a new member of the tetrodotoxin-sensitive voltage-activated sodium channel family from human neuroendocrine cells. EMBO J. 1995;14:1084–1090. doi: 10.1002/j.1460-2075.1995.tb07091.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li RA, Ennis IL, French RJ, Dudley SC, Jr, Tomaselli GF, Marbán E. Clockwise domain arrangement of the sodium channel revealed by (mu)-conotoxin (GIIIA) docking orientation. J Biol Chem. 2001;276:11072–11077. doi: 10.1074/jbc.M010862200. [DOI] [PubMed] [Google Scholar]
- Lossin C, Wang DW, Rhodes TH, Vanoye CG, George AL., Jr Molecular basis of an inherited epilepsy. Neuron. 2002;34:877–884. doi: 10.1016/S0896-6273(02)00714-6. [DOI] [PubMed] [Google Scholar]
- Luke GA, de Felipe P, Lukashev A, Kallioinen SE, Bruno EA, Ryan MD. Occurrence, function and evolutionary origins of ‘2A-like’ sequences in virus genomes. J Gen Virol. 2008;89:1036–1042. doi: 10.1099/vir.0.83428-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDonnell A, Schulman B, Ali Z, Dib-Hajj SD, Brock F, Cobain S, Mainka T, Vollert J, Tarabar S, Waxman SG. Inherited erythromelalgia due to mutations in SCN9A: natural history, clinical phenotype and somatosensory profile. Brain. 2016;139:1052–1065. doi: 10.1093/brain/aww007. [DOI] [PubMed] [Google Scholar]
- Payandeh J, Scheuer T, Zheng N, Catterall WA. The crystal structure of a voltage-gated sodium channel. Nature. 2011;475:353–358. doi: 10.1038/nature10238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Persson AK, Liu S, Faber CG, Merkies IS, Black JA, Waxman SG. Neuropathy-associated Nav1.7 variant I228M impairs integrity of dorsal root ganglion neuron axons. Ann Neurol. 2013;73:140–145. doi: 10.1002/ana.23725. [DOI] [PubMed] [Google Scholar]
- Roy A, Xu D, Poisson J, Zhang Y. A protocol for computer-based protein structure and function prediction. J Vis Exp. 2011;57:e3259. doi: 10.3791/3259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rush AM, Dib-Hajj SD, Liu S, Cummins TR, Black JA, Waxman SG. A single sodium channel mutation produces hyper- or hypoexcitability in different types of neurons. Proc Natl Acad Sci U S A. 2006;103:8245–8250. doi: 10.1073/pnas.0602813103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rush AM, Cummins TR, Waxman SG. Multiple sodium channels and their roles in electrogenesis within dorsal root ganglion neurons. J Physiol. 2007;579:1–14. doi: 10.1113/jphysiol.2006.121483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryan MD, Drew J. Foot-and-mouth disease virus 2A oligopeptide mediated cleavage of an artificial polyprotein. EMBO J. 1994;13:928–933. doi: 10.1002/j.1460-2075.1994.tb06337.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shields SD, Ahn HS, Yang Y, Han C, Seal RP, Wood JN, Waxman SG, Dib-Hajj SD. Nav1.8 expression is not restricted to nociceptors in mouse peripheral nervous system. Pain. 2012;153:2017–2030. doi: 10.1016/j.pain.2012.04.022. [DOI] [PubMed] [Google Scholar]
- Spira ME, Hai A. Multi-electrode array technologies for neuroscience and cardiology. Nat Nanotechnol. 2013;8:83–94. doi: 10.1038/nnano.2012.265. [DOI] [PubMed] [Google Scholar]
- Vasylyev DV, Han C, Zhao P, Dib-Hajj S, Waxman SG. Dynamic-clamp analysis of wild-type human Nav1.7 and erythromelalgia mutant channel L858H. J Neurophysiol. 2014;111:1429–1443. doi: 10.1152/jn.00763.2013. [DOI] [PubMed] [Google Scholar]
- Vriens J, Nilius B, Voets T. Peripheral thermosensation in mammals. Nat Rev Neurosci. 2014;15:573–589. doi: 10.1038/nrn3784. [DOI] [PubMed] [Google Scholar]
- Wainger BJ, Kiskinis E, Mellin C, Wiskow O, Han SS, Sandoe J, Perez NP, Williams LA, Lee S, Boulting G, Berry JD, Brown RH, Jr, Cudkowicz ME, Bean BP, Eggan K, Woolf CJ. Intrinsic membrane hyperexcitability of amyotrophic lateral sclerosis patient-derived motor neurons. Cell Rep. 2014;7:1–11. doi: 10.1016/j.celrep.2014.03.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waxman SG. Neurobiology: a channel sets the gain on pain. Nature. 2006;444:831–832. doi: 10.1038/444831a. [DOI] [PubMed] [Google Scholar]
- Yang Y, Wang Y, Li S, Xu Z, Li H, Ma L, Fan J, Bu D, Liu B, Fan Z, Wu G, Jin J, Ding B, Zhu X, Shen Y. Mutations in SCN9A, encoding a sodium channel alpha subunit, in patients with primary erythermalgia. J Med Genet. 2004;41:171–174. doi: 10.1136/jmg.2003.012153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y, Dib-Hajj SD, Zhang J, Zhang Y, Tyrrell L, Estacion M, Waxman SG. Structural modelling and mutant cycle analysis predict pharmacoresponsiveness of a Na(V)1.7 mutant channel. Nat Commun. 2012;3:1186. doi: 10.1038/ncomms2184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y, Estacion M, Dib-Hajj SD, Waxman SG. Molecular architecture of a sodium channel S6 helix: radial tuning of the voltage-gated sodium channel 1.7 activation gate. J Biol Chem. 2013a;288:13741–13747. doi: 10.1074/jbc.M113.462366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y, Vasylyev DV, Dib-Hajj F, Veeramah KR, Hammer MF, Dib-Hajj SD, Waxman SG. Multistate structural modeling and voltage-clamp analysis of epilepsy/autism mutation Kv10.2-R327H demonstrate the role of this residue in stabilizing the channel closed state. J Neurosci. 2013b;33:16586–16593. doi: 10.1523/JNEUROSCI.2307-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yarov-Yarovoy V, DeCaen PG, Westenbroek RE, Pan CY, Scheuer T, Baker D, Catterall WA. Structural basis for gating charge movement in the voltage sensor of a sodium channel. Proc Natl Acad Sci U S A. 2012;109:E93–E102. doi: 10.1073/pnas.1118434109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Liang Y, Zhang Y. Atomic-level protein structure refinement using fragment-guided molecular dynamics conformation sampling. Structure. 2011;19:1784–1795. doi: 10.1016/j.str.2011.09.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Yang J, Jang R, Zhang Y. GPCR-I-TASSER: a hybrid approach to G protein-coupled receptor structure modeling and the application to the human genome. Structure. 2015;23:1538–1549. doi: 10.1016/j.str.2015.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]