

Abstract
After traumatic brain injury (TBI), neurons surviving the initial insult can undergo chronic (secondary) degeneration via poorly understood mechanisms, resulting in long-term cognitive impairment. Although a neuroinflammatory response is promptly activated after TBI, it is unknown whether it has a significant role in chronic phases of TBI (>1 year after injury). Using a closed-head injury model of TBI in mice, we showed by MRI scans that TBI caused substantial degeneration at the lesion site within a few weeks and these did not expand significantly thereafter. However, chronic alterations in neurons were observed, with reduced dendritic spine density lasting >1 year after injury. In parallel, we found a long-lasting inflammatory response throughout the entire brain. Deletion of one allele of CX3CR1, a chemokine receptor, limited infiltration of peripheral immune cells and largely prevented the chronic degeneration of the injured brain and provided a better functional recovery in female, but not male, mice. Therefore, targeting persistent neuroinflammation presents a new therapeutic option to reduce chronic neurodegeneration.
SIGNIFICANCE STATEMENT Traumatic brain injury (TBI) often causes chronic neurological problems including epilepsy, neuropsychiatric disorders, and dementia through unknown mechanisms. Our study demonstrates that inflammatory cells invading the brain lead to secondary brain damage. Sex-specific amelioration of chronic neuroinflammation rescues the brain degeneration and results in improved motor functions. Therefore, this study pinpoints an effective therapeutic approach to preventing secondary complications after TBI.
Keywords: 3D imaging, axon, degeneration, inflammation, spine, trauma
Introduction
Traumatic brain injury (TBI) accounts for one-third of all injury-related deaths and has become a leading cause of disability in children and young adults (Langlois et al., 2005; Thompson et al., 2006). Individuals surviving severe brain trauma or repetitive mild traumas such as concussions often show delayed chronic brain degeneration affecting the entire brain. Despite the high incidence and cost burden on society, there is little understanding of the underlying mechanisms of chronic neurodegeneration after TBI and a lack of effective treatments (Olesen et al., 2012).
Neurodegeneration after TBI occurs both acutely and chronically. During acute stages (a few days after injury), severely damaged neurons die via necrosis and apoptosis (Rink et al., 1995; Raghupathi, 2004). Excessive calcium influxes, glutamate excitotoxicity, disruption of the blood–brain barrier (BBB), vasogenic edema, and complex changes in local cerebral blood flow also contribute to acute neuronal death (Cortez et al., 1989; Bramlett and Dietrich, 2007). Neurodegeneration can continue long after TBI, perhaps throughout a person's remaining lifetime, albeit at a smaller scale compared with the acute cell loss (Pierce et al., 1998; Bramlett and Dietrich, 2002). Recent studies identifying TBI as a risk factor for the development of dementia and neuropsychiatric diseases suggest the occurrence of long-term maladaptive changes in the injured brain (Lye and Shores, 2000; Saulle and Greenwald, 2012; Kamper et al., 2013). How neurons that survive the acute trauma are affected long after injury is not well understood.
TBI also triggers a rapid inflammatory response involving activated astrocytes and microglia (Kreutzberg, 1996; Chen and Swanson, 2003; Ekmark-Lewén et al., 2013). In addition, peripheral immune cells in the blood such as macrophages and lymphocytes, which are normally excluded from the brain, infiltrate into the injured brain tissue and potentially further amplify the immune response. Neuroinflammation of the injured brain can be both beneficial (reparative) and destructive (contributing to disease) (Jeong et al., 2013). Human genetic studies have demonstrated that inflammation-related microglial genes such as TREM2 (Jonsson et al., 2013) and CD33 (Naj et al., 2011) are risk factors for Alzheimer's disease (AD), which suggests a possible causative relationship between inflammation and chronic neurodegeneration.
Microglia, the resident immune cells of the brain, have long been thought to be main contributors to inflammation in chronic brain diseases (Aguzzi et al., 2013). Recent studies have highlighted differences between microglia and macrophages: microglia are derived from primitive erythromyeloid progenitors in the yolk sac, whereas macrophages are derived from hematopoiesis in the bone marrow (Salter and Beggs, 2014). In addition, it has been shown that microglia and macrophages have different gene expression signatures and functions (Butovsky et al., 2014; Prinz and Priller, 2014). Therefore, it is possible that resident microglia react differently in the injured brain compared with invading macrophages and other blood-derived immune cells. In the healthy brain, microglia play critical functions, such as monitoring and controlling neuronal activity, spine pruning, regulating apoptosis, and engulfing remnants of dead cells during CNS development (Salter and Beggs, 2014). Microglia are also involved in vital roles in the diseased brain, including uptake and clearance of amyloid plaques in AD, neurotrophin secretion, and clearance of debris (Aguzzi et al., 2013; Parkhurst et al., 2013).
In this study using a closed-head mouse model of TBI, we report that dendritic spine loss and neuroinflammation occur, not only shortly after brain injury, but continue into chronic stages of TBI and take place in widespread regions beyond the site of acute trauma. Targeting the inflammatory response via deletion of one allele of CX3CR1 reduced the infiltration of blood-derived peripheral immune cells into the injured brain, protected the brain from chronic degeneration, and improved the recovery of motor functions in female mice. Therefore, long-term inflammation is a critical component of chronic neurodegeneration and thus targeting chronic neuroinflammation has potential for treating the long-term effects of TBI.
Materials and Methods
Animals
In this study, we performed TBI on ∼3-month-old C57BL/6-GFP-M (Feng et al., 2000) and C57BL/6-CX3CR1 (Jung et al., 2000) mice compared with littermate wild-type controls. Males weighed 29.8 ± 3.0 g (n = 21 mice) and females 22.8 ± 2.6 g (n = 17 mice). We used mixed-sex mice (∼1:1 ratio) in the experiments shown in Figures 1A–G, 2, 3, 4, 5, 6, 7, 8, and 9 and only females for the behavior tasks (Fig. 1H–J) to reduce the number of mice used in long-term behavior experiments. This was done because the controlled cortical impact (CCI) model induced more consistent lesions in female mice. All experimental animals were handled in accordance with Institutional Animal Care and Use Committee regulations and were approved by the Genentech Institutional Animal Care and Use Committee. The mice were housed in ventilated microisolator cages in a 12/12 h dark/light cycle. To reduce the usage of animals, we used the minimum number of mice required to obtain biologically meaningful data with the statistical tests that were used.
Figure 1.

CCI injury model and motor assessment of mice after CCI TBI. Shown are examples of mild (A) and moderate (B) brain injury induced by CCI impact (see Materials and Methods) as assessed by T2-MRI. Arrowheads indicate edematous tissue, as defined by T2 signal. C, D, Brains of the same mice imaged in A and B fixed at ∼4 months after injury to confirm location and extent of macroscopic tissue loss. E, Quantification of tissue edema volume as assessed by MRI acutely (4 h after injury) for mice with and without a skull fracture during the impact. There is a strong correlation between the presence of skull fracture and tissue loss. Each data point represents one animal. n = 5 and 6 mice for the mild and moderate groups, respectively. The mean values are shown by the horizontal lines; ***p < 0.001 calculated by Student's t test. F, G, IgG antibody labeled demonstrating the extent of the BBB leakage after the mild and moderate injuries, respectively. H–J, Motor performance of mice with mild and moderate injury. H, Quantification of average latency to fall in the rotarod test for mild injury (blue dots) and moderate injury animals (red dots), before the lesion (preinjury) and at the indicated times postinjury. n = 4 and 5 mice for the mild and moderate groups, respectively. I, Quantification of average foot slips in the balance beam test for mild (blue dots) and moderate (red dots) injury. n = 5 and 4 mice for the mild and moderate groups, respectively. J, Quantification of total beam breaks in open field, a measure of horizontal activity, for mild (blue dots) and moderate (red dots) injury. n = 5 and 4 mice for the mild and moderate groups, respectively. Each data point represents one animal. The mean values are shown by the horizontal lines. *p < 0.05, **p < 0.01, ***p < 0.001 calculated by one-way ANOVA test with Tukey–Kramer post hoc test at the indicated time point compared with their preinjury performance.
Figure 2.

Long-term tissue degeneration on moderately injured mice by MRI. A, Serial T2 maps obtained with MRI from a representative mouse with moderate CCI TBI taken over 18 weeks. Blue arrow indicates approximate site of CCI impact. The elevated T2 at 4 h and 1 d after injury (arrowheads) represents tissue edema. By 1 week, T2 hyperintensity has dissipated and the edematous region is replaced by degenerating tissue (black signal, arrowhead at 1 week) starting from the lesioned area of the cortex and spreading toward inner brain structures including corpus callosum, hippocampus, and thalamus. When the injury cavity reached the lateral ventricle, it was filled with CSF, seen as a homogenous white region of elevated T2 in MRI (arrowheads at 4, 8, and 18 weeks). B, Top row, 3D rendering (top view) of the MRI scans from A is used to show the location and calculate the volume of tissue edema (yellow region) and tissue degeneration (red region) over time. The normal appearing tissue in MRI is shown in green (spared tissue). Bottom row shows edematous (yellow) and lost tissue (red) only at higher magnification in coronal view. Note that much of the edematous tissue (yellow) degenerated (red) over time. C, Volume quantification of edematous (yellow) and lost tissue (red). n = 5 mice. ***p < 0.001 calculated by one-way ANOVA test with Tukey–Kramer post hoc test at the indicated time point for moderate injury compared with the 4 h time point after injury.
Figure 3.

Dendritic spine loss at acute and chronic stages after moderate CCI TBI. A–F, Representative images of dendritic spines from apical secondary branches of hippocampal CA1 pyramidal neurons: unlesioned young mice (3-month-old age-matched controls for acute CCI TBI) (A), mice at 1 week after CCI TBI, ipsilateral (B), or contralateral (C), unlesioned old mice (1.5-year-old age-matched controls for chronic CCI TBI) (D), and at 1.5 years after CCI TBI (E, F). G, Quantification of dendritic spine density from the hippocampus of mice that were unlesioned or at indicated time-points after CCI injury. Each data point represents average from one animal; n = 5, 6, 7, 4, 6, and 5 mice, respectively. les, Lesioned; unles, unlesioned; n.s., not significant. *p < 0.05 and ***p < 0.001 calculated by one-way ANOVA test with Tukey–Kramer post hoc test between indicated groups.
Figure 4.
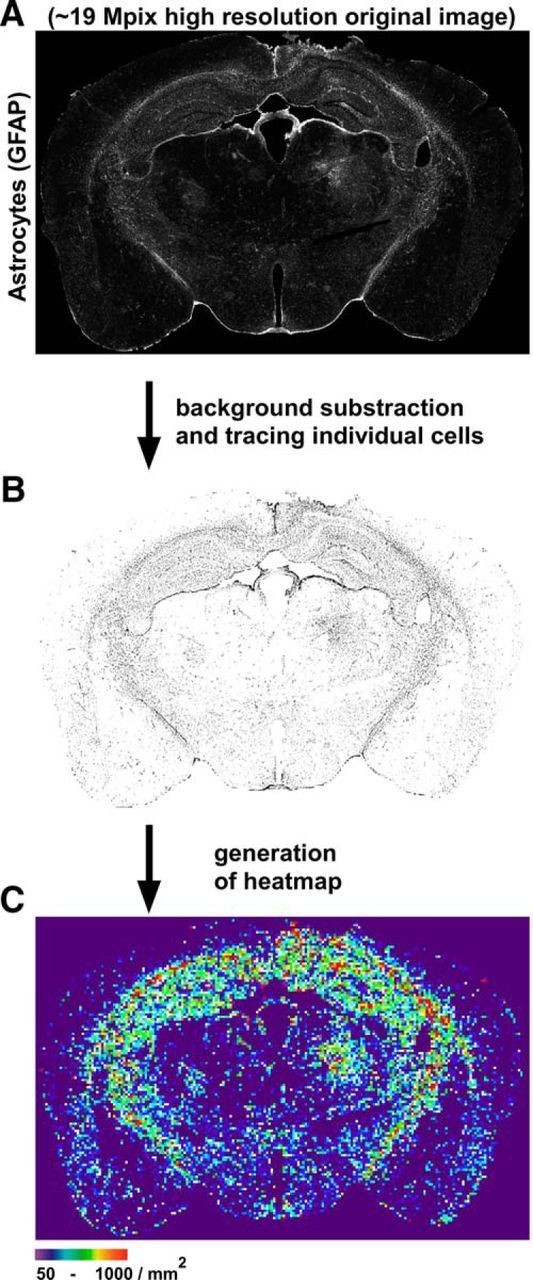
Method of quantification of astrocyte and microglia density in brain sections. ImageJ macros were written to measure cell density on immunohistochemistry sections of brain in an automated and blinded fashion. Coronal brain sections were immunostained for GFAP or Iba1, imaged by epifluorescence microscopy using the same acquisition parameters, and stitched together into a composite whole section (A). The workflow of the macro is as follows: a background subtraction to eliminate noise signal is applied to the original image and all individual cells above the defined uniform threshold are identified and segmented (B). The number of cells is then determined and a heat map is generated based on the density of the cells per 25 × 25 pixel squares (equivalent to 2123 μm2) (C).
Figure 5.
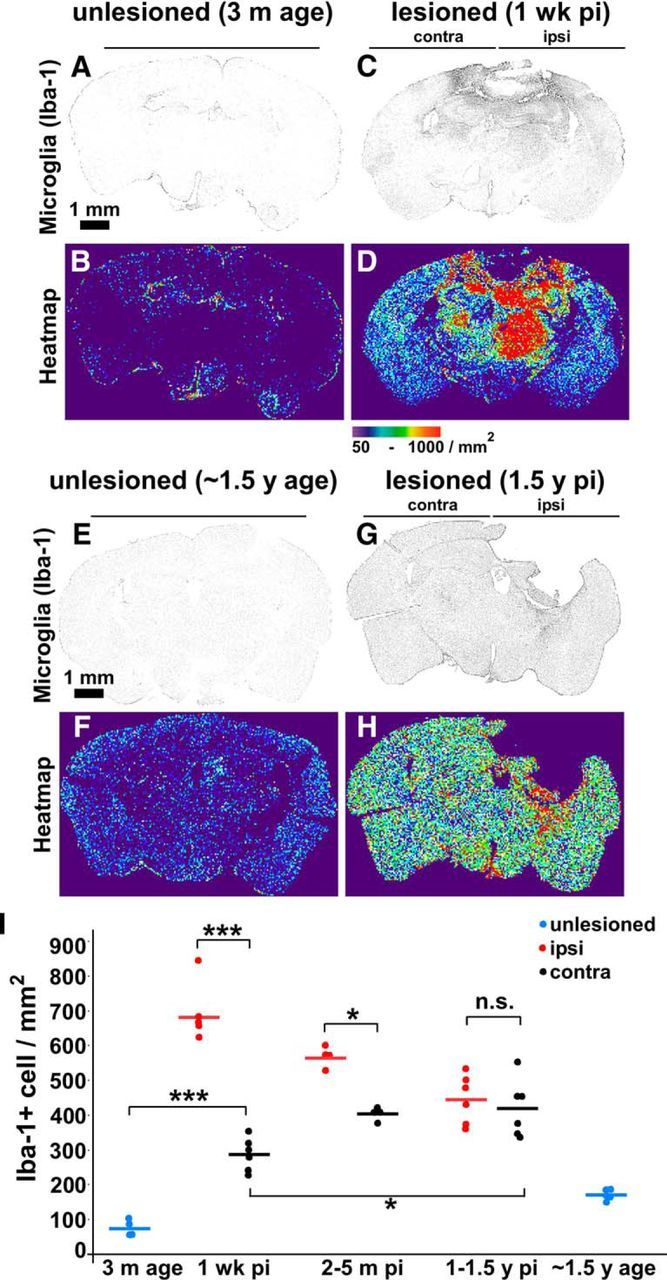
Microglia/macrophage density increases at distal brain regions from acute to chronic stages of TBI. Iba-1+ cell (microglia/macrophage) density and distribution were assessed in coronal brain sections by Iba-1 immunostaining. A–H, Representative images in grayscale and heat map: unlesioned young mice (A, B), mice at 1 week after CCI TBI (C, D), unlesioned old mice (E, F), and at 1.5 years after CCI TBI (G, H). I, Quantification of Iba-1+ cell density (average of entire hemibrain) from mice that were unlesioned or at indicated time points after CCI injury. Each dot represents one animal, n = 4, 6, 6, 3, 3, 6, 6, and 5 mice, respectively; ***p < 0.001, *p < 0.05 calculated by one-way ANOVA test with Tukey–Kramer post hoc test between indicated groups.
Figure 6.

Chronic activation of microglia in the injured brain. Microglia cell count and activity and cerebral lymphocyte subpopulations were analyzed by flow cytometry in the acute (7 d after injury) and chronic (4 months after injury) phase after TBI. Both hemispheres, ipsilateral (ipsi) and contralateral (contra), were investigated at the indicated time points after TBI and compared with age-matched naive animals (acute and chronic control). A, Absolute cell counts of microglia (CD11b+CD45low/int) were quantified per one hemisphere of the indicated group after gating for single cells and excluding dead cells. B, Microglial activation was analyzed by expression of MHC class II molecules on microglia cells (MHCII). Data are presented as the percentage of MHCIIhigh-expessing cells of total CD11b+CD45low/int microglia. Note that microglia activation persisted several months after injury. C, Absolute cell counts of total lymphocytes (CD45highCD11b−) were quantified per hemisphere. n = 4 and 5 mice for the acute and chronic groups, respectively. ***p < 0.001, **p < 0.01 and *p < 0.05 calculated by one-way ANOVA test with Tukey–Kramer post hoc testing for multiple comparisons. C–H, High-magnification confocal images of the microglia: unlesioned old mice (∼1.5 years age) (C), mice at 1.5 years after CCI TBI, contralateral (D) or ipsilateral (E), unlesioned young mice (3 months age) (F), and mice at 1 week after CCI TBI, contralateral (G) or ipsilateral (H). Iba-1+ cells in unlesioned young mice have many long processes (F). One week after injury, microglia retract most of their processes and show enlargement of cell bodies (G). In chronic stages, most of the microglia/macrophages still show increased cell body size and fewer processes (D, E) compared with unlesioned age-matched mice (C).
Figure 7.

Increased astrocyte density in widespread brain regions in acute and chronic stages of TBI. GFAP+ astrocyte density and distribution were assessed in coronal brain sections. A–H, Representative images in grayscale and heat map: unlesioned young mice (A, B), mice at 1 week after CCI TBI (C, D), unlesioned old mice (E, F) and at 1.5 years after CCI TBI (G, H). I, Quantification of GFAP+ astrocyte density (average of entire hemibrain) from mice that were unlesioned or at indicated time points after CCI injury. Each dot represents one animal; n = 4, 5, 5, 5, 5, 5, 5, and 4 mice, respectively; mean values are indicated by horizontal lines. ***p < 0.001, *p < 0.05 calculated by one-way ANOVA test with Tukey–Kramer post hoc test between indicated groups. J–O, High-magnification confocal images of the astrocytes: unlesioned young mice (3 months age) (J), mice at 1 week after CCI TBI, contralateral (K) or ipsilateral (L), unlesioned old mice (∼1.5 years age) (M), and mice at 1.5 years after CCI TBI, contralateral (N) or ipsilateral (O). Note that astrocytes in unlesioned young mice have a star-shaped morphology with long processes (J). Acutely after injury, cell bodies of astrocytes become enlarged (L). In chronic stages (>1 year after injury), most of the astrocytes preserve their increased volume (N, O) compared with unlesioned age-matched mice (M).
Figure 8.

Lymphocytes are persistently increased in the chronically injured brain. A, Absolute cell counts of total lymphocytes (CD45highCD11b−) were quantified per hemisphere. Note that significantly increased lymphocyte counts still persist at 4 months after TBI. B, Lymphocytes were further characterized to their main subpopulations of NK cells (NK1.1+CD3−), B cells (B220+), Thelper cells (CD3+CD4+), and Tcytotoxic cells (CD3+CD8+) after gating for single, live lymphocytes (CD45+CD11b−). Results are presented as percentage of total lymphocytes of the respective treatment group. We observed a nonsignificant expansion in the fraction of NK cells and Thelper cells acutely, which did not last into the chronic stage. n = 4 and 5 mice for acute and chronic groups, respectively. **p < 0.01 and *p < 0.05 calculated by one-way ANOVA test with Tukey–Kramer post hoc testing for multiple comparisons.
Figure 9.

Female CX3CR1+/− mice are protected from chronic degeneration after TBI. A, CX3CR1 immunostaining on unlesioned and lesioned brain tissues at acute and chronic stages. Note that increased CX3CR1 levels acutely (yellow arrows) were kept in the chronic stage (yellow arrowheads). The lesion size quantifications on fixed brains from male CX3CR1+/− (B) and female CX3CR1+/− (C) mice compared with wild-type controls at the indicated after injury times. D–I, Examples of fixed brains from female CX3CR1+/− and wild-type control mice at ∼1 year after injury demonstrating the lesion size. J–O, Fixed brains from lesioned wild-type and lesioned and unlesioned CX3CR1+/− female mice were cleared and autofluorescence signal was imaged with a light-sheet microscope to visualize gross brain anatomy. 3D reconstruction of a lesioned wild-type brain is shown in a surface-rendering view (J) and a close-up view (K) showing large lesion cavity and degeneration of inner brain structures at the lesion site. 3D reconstruction of a lesioned CX3CR1+/− brain at 1 year after injury is shown in surface-rendering view (L) and with a corner-cut view (M) versus an unlesioned CX3CR1+/− brain is shown in surface rendering view (N) and with a corner-cut (O) demonstrating inner brain structures. White arrowheads in M and O mark the hippocampi that are preserved long after injury in lesioned CX3CR1+/− compared with unlesioned CX3CR1+/− mice. n = 7, 5, 4, and 5 mice for lesioned CX3CR1+/− at 4 months after injury, aged-matched controls, lesioned CX3CR1+/− at 1 year after injury, and unlesioned aged-matched controls, respectively. ***p < 0.001 and *p < 0.05 Student's t test between the groups at each time line.
TBI
We performed TBI using a CCI device (TBI-0310 Impactor; Precision Systems) under 3% isoflurane anesthesia. Briefly, the mouse was placed under general anesthesia and then placed in a stereotaxic apparatus with a temperature-controlled heating blanket to keep the body temperature at ∼37°C. The incision area was shaved and aseptically prepped with an appropriate disinfectant (e.g., betadine followed by alcohol wipe). Then, a longitudinal incision of the skin was made between the occiput and forehead and the periosteum underneath was scraped off to the temporal crests. The right somatosensory cortex target area (interaural 2.5 mm and bregma 0.5 mm) was identified using a stereotaxic apparatus and sagittal and coronal sutures on the exposed skull were identified visually. The injury was then applied using the apparatus with the following parameters: impact speed: 3.5 m/s; impact duration: 350 ms; and impact depth: 2 mm. After the impact, the skin was closed with either silk suture (for MRI animals) or metallic wound closure clips (for non-MRI animals) and the animals were placed on a heating pad overnight for recovery. Animals with and without skull fracture had moderate and mild lesions, respectively, which was confirmed with MRI. Approximately one-fifth of the mice died shortly after the impact (within hours); these were considered as having severe injury and were excluded from the study. To reduce stress and pain, analgesics (buprenorphine: 0.05–0.1 mg/kg, SC) were injected every 12 h up to 3 d. The animals were monitored for 5 d (at least once per day) initially and twice a week afterward. We did not observe any important adverse events.
Magnetic resonance imaging
Animal preparation.
The animals were anesthetized using 3% isoflurane and placed in a custom-built head holder with ear bars and a tooth bar. During imaging, anesthesia was maintained using 1.5–2% isoflurane to keep the respiration rate at ∼80 breaths/min. The rectal temperature was monitored and maintained at 36.5–37.5°C using a feedback system with warm air (SA Instruments).
MRI protocol.
Experiments were performed on a 9.4 T/210 mm horizontal imaging system (Agilent Technologies) using a 40-mm-inner-diameter quadrature volume coil. The relaxation rate T2 was measured using a 2D multiecho, multislice experiment with a field of view of 20 × 20 mm2, 16 coronal slices of 0.8 mm thickness, matrix size of 64 × 64 (zero-filled to 128 × 128), repetition time (TR) of 3000 ms, echo time (TE) of 10, 20, 30, 40, 50, 60, 70, and 80 ms, and 4 averages. For each image pixel, T2 was calculated from the following equation:
 |
Rotarod motor task
The rotarod task was performed as described previously (Hamm et al., 1994) using an EZ-ROD instrument (Accuscan Instruments). Briefly, the pretraining was 2–3 sessions of 4 trials each (5–10 min intervals) over 2–3 d. For the actual test, the mice underwent a single, fixed-speed warm-up and three trials of the accelerating rotarod, with a maximum length of 360 s. Each mouse also had a 1 h rest between each accelerating trial. Assessments were limited to once/week.
Balance beam motor task
The balance beam motor task was performed as described previously (Luong et al., 2011). In brief, animals were placed on a beam to traverse an 80 cm beam (11 mm diameter) on an upward incline to reach a dark, enclosed escape box. After pretraining, they were given two trials with a maximum latency of 180 s and the observer counted occurring foot slips.
Open field motor task
To assess general locomotor activity, the mice were placed individually in a PAS open-field box (16 × 16 inches) from San Diego Instruments for 15 minutes. The horizontal distance covered by the mouse, the time it spent in the periphery versus the center (a measure of anxiety), the number of rearings, and its speed of locomotion were detected automatically as interruptions of infrared photobeams.
3D imaging of the entire brain
3D imaging of entire unsectioned brains of mice of either sex expressing GFP in microglia was performed as described previously (Ertürk et al., 2012a; Ertürk et al., 2012b). In brief, after perfusion, the mouse brains were dissected and postfixed in 4% PFA overnight. Subsequently, brains were cleared using the following protocol: 50% tetrahydrofuran (THF; Sigma-Aldrich, 186562) for 2 h, 70% THF for 2 h, 80% for 2 h, 100% THF for 2 h, 100% THF overnight, 100% THF for 2 h, and dibenzyl ether for 2 × 3 h or overnight. Tissue clearing rendered mouse brains transparent within 1–2 d, but preserved the GFP signal of microglia. We also noted that clearing caused tissue shrinkage, which was ∼20% for each dimension. After clearing, we used a light-sheet ultramicroscope (LaVision Biotec) with two-side illumination to scan the entire cleared brains at once (Dodt et al., 2007; Ertürk et al., 2014a) with a resolution of 4 × 4 × 3 μm for the x, y, z dimensions, respectively. A scan of the entire brain with 2-side illumination took ∼60 min.
Tissue sectioning and immunohistochemistry (IHC)
After overnight postfixation, the brains were transferred into sucrose gradients: 15% for 6 h and 30% for 2–3 d at 4°C. We then used a sliding Leica microtome to section the brains at 50 μm in coronal orientation. The tissue sections were collected into cryro-freezing medium (30% glycerol + 30% ethylene glycol + 40% PBS).
For IHC, brain sections were stained in a free-floating way using 12-well dishes. The tissue sections were washed twice times with PBS before staining and incubated with blocking buffer (10% goat serum in PBS) for 2 h at room temperature. Primary antibody incubation was performed in blocking buffer overnight at 4°C. After washing 3–4 times with PBS, 4 h of secondary antibody incubation was done at room temperature. Finally, after washing three to four times with PBS, tissue sections were mounted onto glass imaging slides using Prolong Gold Antifade Reagent (Invitrogen). We used the following antibodies in this study: anti-rabbit GFAP (Dako) 1:1000, anti-rabbit Iba-1 (Wako) 1:1000, anti-rabbit CX3CR1 1:250 (Abcam, ab8020), and Alexa Fluor-555 or Alexa Fluor-568 anti-rabbit or anti-rat secondary (1:400).
Confocal microscopy and spine analysis
The 50 μm brain sections coming from the same anatomical region of the brains were imaged using a 40× oil objective (numerical aperture 1.25) on a Leica SP5 confocal or a Zeiss LSM880 with Airyscan. We quantified spine density on the initial 50 μm of secondary branches of CA1 neurons. The spine density shown in Figure 2 (defined as protrusions of length between 0.5 and 2 μm) was measured blindly using ImageJ as described previously (Ertürk et al., 2014b).
Flow cytometry
The flow cytometric analysis shown in Figure 8 was performed as described previously (Liesz et al., 2011). In brief, brains were removed after transcardial perfusion with saline and collected in DMEM + 10% FCS. Mononuclear cells from the brain were isolated by incubating the samples in 2 ml of digestion mixture [DMEM + 10% FCS + 0.4% DNase 1 (#11284932001, Roche) + 3% collagenase D (#11088866001, Roche)] for 10 min at 37°C and then dissociated mechanically. Cerebral mononuclear cells were subsequently isolated using a 70% and 40% discontinuous Percoll gradient. To quantify the various cell populations, cells were stained with the following monoclonal antibodies in accordance with the manufacturer's protocols: anti-CD3 FITC (17A2), anti-CD4 PerCP5.5 (RM4–5), anti-CD8 PE (53–6.7), anti-CD19 APC-eFlur 780 (eBisID3), anti-CD45 eF450 (30-F11), anti-NK1.1 PE Cy7 (PK136), anti-F4/80 FITC (BM8), anti-CD11b PerCP5.5 (M1/70), anti-MHCII PE (NIMR-4), anti-Ly6C APC (HK1.4), and anti-Ly6G PE Cy7 (RB6–8C5). Data were acquired on a FACSVerse flow cytometer (BD Biosciences) and analyzed using FACSuite software (BD Biosciences).
For flow cytometric analysis of results demonstrated in Figure 11, the mice were perfused with 20 ml of PBS (at a speed of 3 ml/min) and brains were dissected out immediately. After splitting brains into two hemispheres, they were chopped with a razor blade 10–20 times (on ice) and transferred into safe-lock tubes (Eppendorf, 022363417), which were freshly filled with 1.6 ml of Accutase solution (Millipore, SCR005). Samples were rotated in the cold room for 20 min and centrifuged at 4°C for 2 min at 300 × g. After discarding the supernatant, the pellets were resuspended in 1.3 ml of Hibernate A buffer (Brainbits, HA-Lf) by triturating using a 1 ml pipette (the tip slightly cut) ∼6–8 times, letting the large pieces to settle down on ice and transferring 1 ml of supernatant into a 50 ml of Falcon tube through a cell strainer (70 μm). This step was repeated three to four times (with 800 μl of Hibernate A buffer) until the supernatant was a clear solution. Small cellular debris were reduced by density centrifugation through a three-density step gradient of Percoll (Sigma-Aldrich, P1644). One milliliter of each solution was carefully layered in a 15 ml Falcon tube, with the highest density solution on the bottom (high-density solution: 3.426 ml of Hibernate A + 824.5 μl of Percoll + 97.8 μl of 1 m NaCl; medium-density solution: 3.600 ml of Hibernate A + 650.5 μl of Percoll + 76.5 μl of 1 m NaCl; low-density solution: 3.770 ml of Hibernate A + 480.3 μl of Percoll + 59.5 μl of 1 m NaCl). The filtered cell suspension was applied to the top of this gradient and centrifuged at 430 × g for 3 min. The cloudy top layer (∼2 ml) containing debris was removed and discarded. Cells in the remaining layers were pelleted by centrifugation at 550 × g for 5 min, resuspended in 1 ml of Hibernate A, and incubated with 1:1000 DAPI (Biotium), 1:250 PE rat anti-mouse CD45 (BD Pharma, 553081), 1:250 APC rat anti-mouse CD11b (BD Pharma, 553312) in a cold room for 30 min on a rotator. After a washing step with Hibernate A buffer, the pellet was resuspended in 1 ml of Hibernate A and transferred into FACS Falcon tubes (BD Biosciences, 352235) prefilled with 3 ml of Hibernate A. Cells were scanned with a FACSAria (BD Biosciences) and analyzed with FlowJo software is not listed in the references. Please either add this reference to the list or delete the citation from the text (TreeStar).
Figure 11.

Frequency of peripheral immune cells is reduced to unlesioned levels in the brains of CX3CR1+/− mice. Flow cytometry analysis of immune cells gated on CD45+ from unlesioned old mice (A) versus lesioned mice analyzed chronically at 1 year after injury: shown is the ipsilateral brain hemisphere (B) and the contralateral brain hemisphere (C). D, Histograms indicating the percentage of each cell population among CD45+ cells. n ≥ 3 mice for unlesioned young and lesioned mice analyzed chronically. **p < 0.01 and *p < 0.05 calculated by one-way ANOVA test with Tukey–Kramer post hoc test. Shown is the flow cytometry analysis of immune cells gated on CD45+ from unlesioned old CX3CR1+/− mice (E) versus lesioned CX3CR1+/− mice analyzed chronically at 1 year after injury in the ipsilateral brain hemisphere (F) and the contralateral brain hemisphere (G). Data from one representative experiment are shown. H, Histograms indicating the percentage of each cell population among CD45+ cells. n ≥ 3 mice for lesioned CX3CR1+/− mice analyzed chronically and unlesioned aged-matched controls. One-way ANOVA test with Tukey–Kramer post hoc test did not show any significant difference between the groups.
Image processing and statistical analysis
The individual images were made using ImageJ. Images were cropped, resized (if necessary) with Adobe Photoshop, and assembled to figures using Canvas (ACD Systems). For MRI and 3DISCO, segmentation on consecutive 2D images and their 3D reconstructions were obtained using Amira (Visage Imaging). All statistical analysis was performed with JMP (SAS Institute). The quantifications were measured by investigators blinded to the experimental conditions using ImageJ.
Results
Chronic TBI induced by a CCI instrument
To model TBI in mice, we used a CCI instrument, which allows the control of impact force. We performed the injury on mice with an intact skull, creating a so-called closed head injury. This model is more representative of human TBI than open skull injury models, in which the brain is exposed with a craniotomy (Xiong et al., 2013). However, the severity of injury induced is more variable in the closed head injury model. Therefore, we first optimized the injury severity to study mice with a similar degree of injury. To begin, we consistently used the following parameters to induce CCI injury over the somatosensory cortex of mice: impact depth: 2 mm, impact speed: 3.5 m/s, and impact duration: 350 ms. By examining the mouse brains 4 h after injury with MRI, we could group animals into two different levels of injury severity: (1) mild injury, which showed little or no edematous tissue as measured by enhanced T2 signal (arrowhead in Fig. 1A; MRI quantification of tissue edema is shown in Fig. 1E), or (2) moderate injury, which showed extensive edematous tissue (Fig. 1B,E). We noted a strong correlation between the incidence of a skull fracture (as visually observed immediately after CCI impact) and injury severity: whereas all animals in moderate injury group had an obvious skull fracture (broken and collapsed), none of the animals in mild injury group had any visible skull fracture (Fig. 1E). Assessment of fixed brains 18 weeks after injury confirmed the absence of gross tissue loss in mild lesions (Fig. 1C), whereas moderate lesions resulted in extensive tissue loss around the site of CCI impact (Fig. 1D). Finally, we assessed BBB integrity using IgG immunostaining at 48 h after injury. We found that mildly injured brains had a small amount of BBB leakage confined to lesion site (Fig. 1F, arrowhead) and moderately injured brains had much larger BBB leakage extending to distal cortical and hippocampal regions (Fig. 1G, arrows). We conclude that observation of the skull fracture immediately after injury is sufficient to sort animals into mild or moderate injury severity groups.
As functional readouts, we measured the effect of CCI TBI on various motor behavior tests (rotarod, balance beam, and mobility in open field). To this end, the mice were pretrained (before TBI) in the motor task until they could perform the task consistently. Their performance was tested a few days before the TBI and at 5 additional times during the course of 8 weeks after injury (Fig. 1H–J). In the rotarod test (the ability of mice to stay on a rotating rod; see Materials and Methods), the mice with mild TBI injury showed minor residual deficits (Fig. 1H, blue dots), whereas mice with moderate injury exhibited severe deficits that persisted for 8 weeks after injury (Fig. 1H, red dots). In the balance beam test (measured by number of foot slips during traversal of the balance beam), moderately injured mice were strongly affected but recovered partially over 8 weeks (Fig. 1I, red dots), whereas no significant performance change was observed in the mild injury group (Fig. 1I, blue dots). In the open-field test, the moderate TBI mice showed marked diminution of movement immediately after trauma (4 h after injury), but had largely recovered by 4 d after head injury, as measured by beam breaks (Fig. 1J, red dots). Mildly injured mice did not show any significant deficit in open-field testing (Fig. 1J, blue dots). Even mice with moderate injury were able to move around and feed themselves shortly after CCI TBI.
Because there was observable long-term functional impairment in the moderate injury group, we next aimed to understand brain pathophysiology of moderately injured mice and underlying causes leading to functional deficits over months in this group.
MRI shows no gross degeneration chronically after TBI
To assess the time course of brain tissue damage longitudinally, mice with moderate CCI injury were followed by T2 mapping with MRI over several months (Fig. 2A). The injury was readily detectable by 4 h after the impact in the form of widespread, diffuse elevated T2 signal, most likely marking edema in the brain tissue (arrowheads at the 4 h and 1 d time point images in Fig. 2A and yellow in representative 3D reconstruction in Fig. 2B). Quantifications show that the elevated T2 (presumed acute edema) persisted for >1 d, but had largely resolved by 1 week after injury (Fig. 2C, yellow). In the same time frame (1 d to 1 week), the edematous tissue at the site of injury began to degenerate (Fig. 2A, arrowhead at 1 week time point; black signal in MRI image marks the absence of tissue). At ∼4 weeks after injury, the tissue cavity had typically expanded to include the ipsilateral corpus callosum, hippocampus, and lateral ventricle and was filled with fluid (presumably CSF, which appears as a smaller, homogenous region of elevated T2 in the MRI, indicated by arrowheads in the 4, 8, and 18 week images in Fig. 2A and represented by red volume in the 3D reconstruction in Fig. 2B). There was a consistent boundary between the lesion cavity and spared brain tissue from 4 weeks on, suggesting that the lesion cavity was stabilized by this time and did not increase significantly in volume thereafter (quantified in Fig. 2C, red). Overall, these data show that gross tissue loss in the moderate CCI TBI model occurred during the acute stage (within initial weeks of injury).
Chronic neurodegeneration occurs at the subcellular level
Could there be neurodegeneration at a smaller scale that was not detectable by MRI? To address this question, we quantified dendritic spine density in hippocampal CA1 neurons of GFP-M mice, in which a subset of neurons express GFP under control of the Thy-1 promoter (Feng et al., 2000). We found a >50% reduction in dendritic spine density acutely (1 week after injury) in the ipsilateral hippocampus compared with unlesioned controls (Fig. 3A,B, quantified in G). The ipsilateral loss of spines persisted in animals that were 1–1.5 years after injury (Fig. 3E,G). Surprisingly, although there was a slight but nonsignificant decrease in spine density in the contralateral hippocampus at 1 week after injury, we observed a marked reduction at 1–1.5 years after injury (Fig. 3A,C,D,F,G). These data reveal a loss of spines in the contralateral hippocampus after TBI that is delayed relative to the ipsilateral side and that persists beyond 1 year after TBI.
Widespread chronic neuroinflammation after TBI
Although previous reports have consistently demonstrated a role of the neuroinflammatory response in the pathophysiology of brain injury, including TBI (Perry et al., 2010; Ramlackhansingh et al., 2011), the impact of neuroinflammatory mechanisms in the chronic phase after acute brain injury are still elusive.
To assess temporal and spatial distribution of neuroinflammation and astrogliosis, we immunostained inflammatory cells and astrocytes and imaged coronal brain sections from the same anatomical region at a resolution that allowed us to discriminate individual glial cells. A total of 15–20 microscope images covering the entire coronal section were stitched together (Fig. 4A). The resulting images were analyzed in an automated and blinded fashion, using a custom ImageJ macro that first performed a background subtraction and then identified individual cells (Fig. 4B) and quantified the average cell density above the threshold in the entire ipsilateral or contralateral side of the brain section. Finally, it produced a heat map representing cell density that enabled quantitative analysis of cell counts per section (Fig. 4C).
As reported previously (Kreutzberg, 1996; Jeong et al., 2013), we observed a massive increase in Iba-1 signal intensity (denoting microglia/macrophages) during the acute stage (1 week after injury) ipsilaterally, which then fell to intermediate levels at 1–1.5 years after injury (Fig. 5A–H, quantified in I). Conversely, Iba-1+ cells increased moderately on the contralateral side at 1 week after injury, but continued to rise from acute stage to chronic stages (Fig. 5A–I). At 1–1.5 years after injury, there was no significant difference in the Iba-1+ cells on the ipsilateral versus the contralateral side (Fig. 5I). Therefore, whereas activated microglia/macrophage numbers subsided with time around the lesion site, they increased in brain regions distant from the lesion. We also noted an increase in microglia/macrophage density (which did not reach statistical significance) in old mice (∼1.5 years age) compared with young mice (3 months age) that was independent of TBI (Fig. 5B,F,I). We obtained similar histological results using CD68, another marker of myeloid cells including microglia and macrophages (data not shown). Using fluorescence-activated cell sorting (FACS), we found that the number of microglia (CD11b+CD45low/int) actually did not alter significantly either acutely or chronically after TBI, suggesting that the majority of the Iba-1+ cells were infiltrating macrophages (Fig. 6A). Importantly, the fraction of MHC-IIhigh (proinflammatory) microglia (CD11b+CD45low/int) was increased (Fig. 6B), demonstrating substantial microglial activation both acutely and chronically after TBI despite nearly unaltered absolute cell counts. High-magnification confocal images of microglia/macrophages demonstrated an amoeboid morphology (retracted processes) at chronic stages compared with unlesioned animals (Fig. 6C–E) that was similar to the activated microglia/macrophages observed in the acute stage of TBI (Fig. 6F–H). We also found that astrogliosis, assessed with the GFAP marker, persisted a remarkably long time (>1 year) after brain injury (Fig. 7). Overall, these data suggest the presence of inflammatory cells in the injured brain months/years after injury, which is indicative of chronic neuroinflammation.
Blocking immune cell infiltration rescues degeneration of the injured brain in a sex-specific manner
TBI induces massive infiltration of blood-derived peripheral immune cells into the brain acutely. Whether they are also present in the chronic stage of TBI and contribute to neurodegeneration has not been explored. Using flow cytometry, we found a significant elevation in the total number of lymphocytes (CD45highCD11b−) acutely (7 d after injury) (Fig. 8A), which persisted chronically (4 months after injury). Analysis of lymphocyte subpopulations infiltrating the brain at different time points after TBI revealed an increase in Thelper (CD3+CD4+) and NK cells (NK1.1+CD3−) acutely, which subsided chronically (Fig. 8B). Overall, these data demonstrate that blood-borne peripheral immune cells are a significant part of chronic neuroinflammation after TBI.
To assess whether infiltration of the peripheral immune cells is involved in chronic degeneration, we used CX3CR1-GFP mice, in which CX3CR1 (fractalkine receptor), crucial for immune cell recruitment through its ligand CX3CL1 (Luster et al., 2005), is knocked out with a GFP insertion (Jung et al., 2000). Previous reports demonstrated that heterozygous deletion of this gene (CX3CR1+/−) is sufficient to cause behavioral deficits similar to homozygous KOs (CX3CR1−/−) (Rogers et al., 2011; Sheridan and Murphy, 2013). Using immunolabeling, we found an increase in CX3CR1 after TBI acutely (2 d after injury) compared with unlesioned controls, which persisted chronically (1.5 years after injury) (Fig. 9A), suggesting that CX3CR1 might indeed be involved in chronic neuroinflammation. Analyzing the brain tissue, we found that CX3CR1+/− male mice did not have protection compared with controls (Fig. 9B), whereas female mice had significantly less degeneration of brain tissue compared with control mice at the 4 and 18 months postinjury times (Fig. 9C). Whereas the lesion cavity extended from cortex into deeper brain regions (including hippocampus and thalamic nuclei) in wild-type controls (Fig. 9D–F), it was mostly restricted to the superficial cortex regions in the female CX3CR1+/− mice after TBI (Fig. 9G–I). 3D imaging of entire brains after tissue clearing (Ertürk et al., 2012a; Ertürk et al., 2012b) showed that, whereas the inner brain structures were largely degenerated in mice with TBI (Fig. 9J,K), they were preserved in CX3CR1+/− females after TBI (Fig. 9L,M, white arrows mark preserved hippocampal structures) similar to unlesioned CX3CR1+/− mice (Fig. 9N,O). Next, we investigated whether the CX3CR1+/− mice with decreased tissue degeneration had better functional recovery over months. Our data again demonstrated a sex-dependent difference: female CX3CR1+/− mice recovered better on the rotarod test compared with wild-type females (Fig. 10A), whereas the male CX3CR1+/− mice did not perform better compared with wild-type males (Fig. 10B).
Figure 10.

Female CX3CR1+/− mice show better recovery on rotarod test. Fine motor performance of CX3CR1+/− and wild-type control mice was assessed using rotarod test. Whereas CX3CR1+/− females showed significantly better recovery over time compared with female controls (A), CX3CR1+/− males did not (B). *p < 0.05.
Finally, to assess whether there is indeed a change in the frequency of peripheral immune cells in the brains of wild-type verseus CX3CR1+/− mice after TBI, we performed flow cytometric analysis of cerebral immune cells. At 1 year after injury, we observed first that the frequency of macrophages was significantly higher in both ipsilateral and contralateral sides than unlesioned controls; moreover, we detected a significant increase in the frequency of lymphocytes on both the ipsilateral and contralateral sides (Fig. 11A–D). Conversely, the percentage of peripheral immune cell populations (macrophages and lymphocytes) in the injured CX3CR1+/− brain at 1 year after injury (Fig. 11F–H) was similar to age-matched unlesioned CX3CR1+/− mice (Fig. 11E,H) or wild-type mice (Fig. 11A,D), suggesting that TBI-induced infiltration of peripheral immune cells was significantly blocked in CX3CR1+/−. Altogether, the above findings demonstrate the presence of a previously underrated chronic neuroinflammation that is mediated by infiltration of peripheral immune cells, as well as microglia and astrocyte activation, and that is causally linked to chronic brain degeneration after TBI.
Discussion
In the current study, we aimed to assess the degree of chronic neurodegeneration and neuroinflammation after TBI and to determine whether neuroinflammation contributes significantly to chronic neurodegeneration after TBI. In a closed head CCI injury model in mice, we found that the major loss of brain tissue (as measured by MRI) took place within a few weeks after moderate TBI and was largely localized to the brain region around the impact site. In the subsequent months after injury, there appeared to be no further major gross anatomical changes as detected by MRI. Confocal microscopy, however, showed a reduction in dendritic spine density in the contralateral hippocampus in the chronic phase (>1 year after injury), located far from the initial impact. Consistent with a widespread, long-term detrimental impact on the brain, we observed an increase in gliosis induced by microglia, macrophages, and astrocytes that occurred mainly around the lesion site during the acute stage, but that had spread to distant brain regions at >1 year after injury. These morphological findings suggest that trauma induces chronic neuroinflammation in the brain, which not only does not fully recover near the lesion, but also seems to spread over time to brain regions distant from the impact site. Heterozygous deletion of CX3CR1 (a chemokine receptor involved in immune cell recruitment) reduced the number of infiltrating immune cells, rescued chronic degeneration of the injured brain, and improved functional recovery after TBI in a sex-specific way.
Spine degeneration after TBI
The majority of TBI cases in humans is caused by a single impact to the head, often resulting from falls (∼35%), motor vehicle accidents (∼17%), or being struck by hard objects (∼16%) (Langlois et al., 2005; Rutland-Brown et al., 2006). Unlike most TBI studies, which expose the brain by removal of a piece of skull, we opted to use a closed skull model for CCI TBI. Although a closed head injury model may increase the variability of the severity of brain injury, it better mimics the vast majority of human TBI cases. In addition, craniotomy itself can cause additional stress to the brain, such as inflammation, which can further complicate interpretation of the data. Here, we tried to circumvent the variability of closed head CCI TBI by selecting a group that suffered skull fracture and showed relatively consistent damage to brain tissue (substantial acute brain degeneration) and major acute functional deficits after the brain trauma.
Tissue degeneration via necrotic and apoptotic cell death are hallmarks of acute neurodegeneration after TBI (Keane et al., 2001; Werner and Engelhard, 2007; Zhou et al., 2012). In the following days to weeks after the initial trauma, acute complications are largely reduced by elimination of damaged cells and cellular debris as well as repair of the BBB (Shlosberg et al., 2010). In our moderate CCI TBI injury, a tissue cavity formed within days of injury encompassing a region from the injured cortex and hippocampus to the thalamus, consistent with previous studies (Dietrich et al., 1994; Hicks et al., 1996; Hall et al., 2008). As reported previously, TBI also caused a substantial spine loss acutely 1 d after TBI (Winston et al., 2013) and 1 week after TBI (Campbell et al., 2012). In the subsequent months, there was no significant change in the lesion size by MRI, suggesting that large-scale tissue loss was restricted to the acute stage in our injury model. Surprisingly, however, we found a significant spine loss in brain regions far away from the initial injury site a year after TBI. Even mild TBI without noticeable tissue loss can lead to long-term cognitive deficits in humans (Lipton et al., 2008) and in animal models (Rubovitch et al., 2011). For example, neuropsychiatric problems and posttraumatic epilepsy can arise months to years after injury (Malt, 1988; Kharatishvili and Pitkänen, 2010; Masel and DeWitt, 2010; Masel et al., 2012). Although an acute injury naturally causes spine loss in stressed neurons around the lesion, chronic spine reduction more than a year after injury in distant brain regions was unexpected and is likely to be mediated by different mechanisms than direct injury.
Recent studies suggest a role for microglia in the phagocytosis and pruning of synapses and spines via complement system in the developing brain (Stevens et al., 2007; Ricklin et al., 2010; Paolicelli et al., 2011; Schafer et al., 2012; Stephan et al., 2012; Kettenmann et al., 2013) and neurodegeneration in a mouse model of AD (Hong et al., 2016). Therefore, it is possible that the broadly increased microglia and infiltrating macrophage and lymphocyte density in the TBI brain could also be contributing to the reduced spine density that we observed bilaterally in the hippocampus. It will be important to identify the exact subtype of immune cell and the underlying molecular mechanisms that are damaging healthy dendritic spines/synapses. For example, Campbell et al. (2012) demonstrated that calcineurin inhibition by FK506 can prevent the loss of spines occurring 24 h after a lateral fluid percussion TBI. However, whether FK506 has any effect on chronic spine degeneration is unknown. We observed a substantial opening of the BBB in our CCI model. It is also plausible that hemorrhages occurring due to BBB leakage contribute to secondary neurodegeneration in TBI (Kurland et al., 2012).
TBI induces a persistent neuroinflammation
Upon brain injury, glia cells promptly become activated, proliferate, and migrate to the lesioned area (Silver and Miller, 2004; Perry et al., 2010; Smith et al., 2013; Clarke and Barres, 2013). Whereas inflammatory cells, particularly resident microglia, with increased phagocytosis (clearing harmful debris) and neurotrophin secretion are considered to be neuroprotective, inflammatory cells secreting excessive proinflammatory cytokines (including TNF-α and IL-1), nitric oxide, and superoxide free radicals are thought to be neurotoxic (Chao et al., 1992; Banati et al., 1993; Satake et al., 2000; Allan and Rothwell, 2001; Chan, 2001; Lourbopoulos et al., 2015). Interestingly, acute inflammation can both block regeneration by forming a glial scar and promote regeneration by clearing the toxic debris, secreting neurotrophic factors and even enhancing neurogenesis (Ramlackhansingh et al., 2011; Kyritsis et al., 2012). Our data demonstrate a state of pervasive and persistent neuroinflammation after TBI beyond a year after injury. We currently do not know how gliosis occurs in brain regions distant from the lesion site. It is also possible that microglia use axonal tracks including the corpus callosum for long-distance migration or a local proliferation of immune cells, as well as local infiltration of immune cells through compromised BBB, occur in distant brain regions.
Reducing the inflammatory response limits degeneration in a sex-specific way
The receptor for the chemokine fractalkine (CX3CR1), which is expressed in peripheral immune cells including macrophages and lymphocytes (Imai et al., 1997; Mionnet et al., 2010), is important for migration and infiltration of these cells (Sheridan and Murphy, 2013). CX3CR1 is also expressed in microglia and is critical for microglial function (Jung et al., 2000). In CX3CR1 KOs, a significant neuroprotection has been observed in spinal cord injury models acutely within a few days/weeks of injury by restricting recruitment and activation of peripheral macrophages (Donnelly et al., 2011). Various studies also pointed out the neuroprotective effect of the immune cell trafficking modulation, including TNF (Longhi et al., 2013). However, whether a chronic manipulation of CX3CR1 could affect neuroinflammation and associated neurodegeneration long after injury (months to years after injury), when the disease progression is evident in human patients, was not known. In this study, we showed that sex-specific heterozygous deletion of CX3CR1 limited the populations of infiltrating macrophages and lymphocytes, which was associated with a large protection of the injured brain and functional recovery of fine motor skills. It has been shown that mice lacking CX3CR1 have reduced proinflammatory cytokine and iNOS expression in microglia/macrophages after CNS injury (Donnelly et al., 2011; Aguzzi et al., 2013). Therefore, it is possible that CX3CR1 deficiency also contributes to neuroprotection by reducing the secretion of potentially neurotoxic proinflammatory cytokines (Dénes et al., 2008; Donnelly et al., 2011) in CNS-resident microglia in addition to interfering with infiltration of circulating immune cells. Interestingly, homozygous deletion of CX3CR1 dysregulates the microglial response and results in more neurotoxicity in Parkinson's disease and amyotrophic lateral sclerosis mouse models (Cardona et al., 2006), suggesting that a complete lack of CX3CR1 can aggravate neurodegeneration rather than being more neuroprotective.
Our study reveals the anatomic spread and remarkably enduring presence of neuroinflammation and spine loss after brain trauma. We believe that a chronic manipulation of inflammatory response, but not an acute manipulation alone such as via a chemokine inhibitor, is more likely to result in long-lasting therapeutic effects.
Footnotes
The authors declare no competing financial interests.
References
- Centers for Disease Control and Prevention (CDC) CDC grand rounds: reducing severe traumatic brain injury in the united states. MMWR Morb Mortal Wkly Rep. 2013;62:549–552. [PMC free article] [PubMed] [Google Scholar]
- Aguzzi A, Barres BA, Bennett ML. Microglia: scapegoat, saboteur, or something else? Science. 2013;339:156–161. doi: 10.1126/science.1227901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allan SM, Rothwell NJ. Cytokines and acute neurodegeneration. Nat Rev Neurosci. 2001;2:734–744. doi: 10.1038/35094583. [DOI] [PubMed] [Google Scholar]
- Banati RB, Gehrmann J, Schubert P, Kreutzberg GW. Cytotoxicity of microglia. Glia. 1993;7:111–118. doi: 10.1002/glia.440070117. [DOI] [PubMed] [Google Scholar]
- Bramlett HM, Dietrich WD. Quantitative structural changes in white and gray matter 1 year following traumatic brain injury in rats. Acta Neuropathol. 2002;103:607–614. doi: 10.1007/s00401-001-0510-8. [DOI] [PubMed] [Google Scholar]
- Bramlett HM, Dietrich WD. Progressive damage after brain and spinal cord injury: pathomechanisms and treatment strategies. Prog Brain Res. 2007;161:125–141. doi: 10.1016/S0079-6123(06)61009-1. [DOI] [PubMed] [Google Scholar]
- Butovsky O, Jedrychowski MP, Moore CS, Cialic R, Lanser AJ, Gabriely G, Koeglsperger T, Dake B, Wu PM, Doykan CE, Fanek Z, Liu L, Chen Z, Rothstein JD, Ransohoff RM, Gygi SP, Antel JP, Weiner HL. Identification of a unique TGF-beta-dependent molecular and functional signature in microglia. Nat Neurosci. 2014;17:131–143. doi: 10.1038/nn.3599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell JN, Register D, Churn SB. Traumatic brain injury causes an FK506-sensitive loss and an overgrowth of dendritic spines in rat forebrain. J Neurotrauma. 2012;29:201–217. doi: 10.1089/neu.2011.1761. [DOI] [PubMed] [Google Scholar]
- Cardona AE, Pioro EP, Sasse ME, Kostenko V, Cardona SM, Dijkstra IM, Huang D, Kidd G, Dombrowski S, Dutta R, Lee JC, Cook DN, Jung S, Lira SA, Littman DR, Ransohoff RM. Control of microglial neurotoxicity by the fractalkine receptor. Nat Neurosci. 2006;9:917–924. doi: 10.1038/nn1715. [DOI] [PubMed] [Google Scholar]
- Chan PH. Reactive oxygen radicals in signaling and damage in the ischemic brain. J Cereb Blood Flow Metab. 2001;21:2–14. doi: 10.1097/00004647-200101000-00002. [DOI] [PubMed] [Google Scholar]
- Chao CC, Hu S, Molitor TW, Shaskan EG, Peterson PK. Activated microglia mediate neuronal cell injury via a nitric oxide mechanism. J Immunol. 1992;149:2736–2741. [PubMed] [Google Scholar]
- Chen Y, Swanson RA. Astrocytes and brain injury. J Cereb Blood Flow Metab. 2003;23:137–149. doi: 10.1097/01.WCB.0000044631.80210.3C. [DOI] [PubMed] [Google Scholar]
- Clarke LE, Barres BA. Emerging roles of astrocytes in neural circuit development. Nat Rev Neurosci. 2013;14:311–321. doi: 10.1038/nrn3484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cortez SC, McIntosh TK, Noble LJ. Experimental fluid percussion brain injury: vascular disruption and neuronal and glial alterations. Brain Res. 1989;482:271–282. doi: 10.1016/0006-8993(89)91190-6. [DOI] [PubMed] [Google Scholar]
- Dénes A, Ferenczi S, Halász J, Környei Z, Kovács KJ. Role of CX3CR1 (fractalkine receptor) in brain damage and inflammation induced by focal cerebral ischemia in mouse. J Cereb Blood Flow Metab. 2008;28:1707–1721. doi: 10.1038/jcbfm.2008.64. [DOI] [PubMed] [Google Scholar]
- Dietrich WD, Alonso O, Busto R, Globus MY, Ginsberg MD. Post-traumatic brain hypothermia reduces histopathological damage following concussive brain injury in the rat. Acta Neuropathol. 1994;87:250–258. doi: 10.1007/BF00296740. [DOI] [PubMed] [Google Scholar]
- Dodt HU, Leischner U, Schierloh A, Jahrling N, Mauch CP, Deininger K, Deussing JM, Eder M, Zieglgänsberger W, Becker K. Ultramicroscopy: three-dimensional visualization of neuronal networks in the whole mouse brain. Nat Methods. 2007;4:331–336. doi: 10.1038/nmeth1036. [DOI] [PubMed] [Google Scholar]
- Donnelly DJ, Longbrake EE, Shawler TM, Kigerl KA, Lai W, Tovar CA, Ransohoff RM, Popovich PG. Deficient CX3CR1 signaling promotes recovery after mouse spinal cord injury by limiting the recruitment and activation of Ly6Clo/iNOS+ macrophages. J Neurosci. 2011;31:9910–9922. doi: 10.1523/JNEUROSCI.2114-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ekmark-Lewén S, Flygt J, Kiwanuka O, Meyerson BJ, Lewén A, Hillered L, Marklund N. Traumatic axonal injury in the mouse is accompanied by a dynamic inflammatory response, astroglial reactivity and complex behavioral changes. J Neuroinflammation. 2013;10:44. doi: 10.1186/1742-2094-10-44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ertürk A, Becker K, Jährling N, Mauch CP, Hojer CD, Egen JG, Hellal F, Bradke F, Sheng M, Dodt HU. Three-dimensional imaging of solvent-cleared organs using 3DISCO. Nat Protoc. 2012a;7:1983–1995. doi: 10.1038/nprot.2012.119. [DOI] [PubMed] [Google Scholar]
- Ertürk A, Mauch CP, Hellal F, Forstner F, Keck T, Becker K, Jahrling N, Steffens H, Richter M, Hubener M, Kramer E, Kirchhoff F, Dodt HU, Bradke F. Three-dimensional imaging of the unsectioned adult spinal cord to assess axon regeneration and glial responses after injury. Nat Med. 2012b;18:166–171. doi: 10.1038/nm.2600. [DOI] [PubMed] [Google Scholar]
- Ertürk A, Lafkas D, Chalouni C. Imaging cleared intact biological systems at a cellular level by 3DISCO. J Vis Exp. 2014a:89. doi: 10.3791/51382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ertürk A, Wang Y, Sheng M. Local pruning of dendrites and spines by caspase-3-dependent and proteasome-limited mechanisms. J Neurosci. 2014b;34:1672–1688. doi: 10.1523/JNEUROSCI.3121-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng G, Mellor RH, Bernstein M, Keller-Peck C, Nguyen QT, Wallace M, Nerbonne JM, Lichtman JW, Sanes JR. Imaging neuronal subsets in transgenic mice expressing multiple spectral variants of GFP. Neuron. 2000;28:41–51. doi: 10.1016/S0896-6273(00)00084-2. [DOI] [PubMed] [Google Scholar]
- Hall ED, Bryant YD, Cho W, Sullivan PG. Evolution of post-traumatic neurodegeneration after controlled cortical impact traumatic brain injury in mice and rats as assessed by the de Olmos silver and fluorojade staining methods. J Neurotrauma. 2008;25:235–247. doi: 10.1089/neu.2007.0383. [DOI] [PubMed] [Google Scholar]
- Hamm RJ, Pike BR, O'Dell DM, Lyeth BG, Jenkins LW. The rotarod test: an evaluation of its effectiveness in assessing motor deficits following traumatic brain injury. J Neurotrauma. 1994;11:187–196. doi: 10.1089/neu.1994.11.187. [DOI] [PubMed] [Google Scholar]
- Hicks R, Soares H, Smith D, McIntosh T. Temporal and spatial characterization of neuronal injury following lateral fluid-percussion brain injury in the rat. Acta Neuropathol. 1996;91:236–246. doi: 10.1007/s004010050421. [DOI] [PubMed] [Google Scholar]
- Hong S, Beja-Glasser VF, Nfonoyim BM, Frouin A, Li S, Ramakrishnan S, Merry KM, Shi Q, Rosenthal A, Barres BA, Lemere CA, Selkoe DJ, Stevens B. Complement and microglia mediate early synapse loss in Alzheimer mouse models. Science. 2016;352:712–716. doi: 10.1126/science.aad8373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imai T, Hieshima K, Haskell C, Baba M, Nagira M, Nishimura M, Kakizaki M, Takagi S, Nomiyama H, Schall TJ, Yoshie O. Identification and molecular characterization of fractalkine receptor CX3CR1, which mediates both leukocyte migration and adhesion. Cell. 1997;91:521–530. doi: 10.1016/S0092-8674(00)80438-9. [DOI] [PubMed] [Google Scholar]
- Jeong HK, Ji K, Min K, Joe EH. Brain inflammation and microglia: facts and misconceptions. Exp Neurobiol. 2013;22:59–67. doi: 10.5607/en.2013.22.2.59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jonsson T, et al. Variant of TREM2 associated with the risk of Alzheimer's disease. N Engl J Med. 2013;368:107–116. doi: 10.1056/NEJMoa1211103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung S, Aliberti J, Graemmel P, Sunshine MJ, Kreutzberg GW, Sher A, Littman DR. Analysis of fractalkine receptor CX(3)CR1 function by targeted deletion and green fluorescent protein reporter gene insertion. Mol Cell Biol. 2000;20:4106–4114. doi: 10.1128/MCB.20.11.4106-4114.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamper JE, Pop V, Fukuda AM, Ajao DO, Hartman RE, Badaut J. Juvenile traumatic brain injury evolves into a chronic brain disorder: behavioral and histological changes over 6months. Exp Neurol. 2013;250:8–19. doi: 10.1016/j.expneurol.2013.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keane RW, Kraydieh S, Lotocki G, Alonso OF, Aldana P, Dietrich WD. Apoptotic and antiapoptotic mechanisms after traumatic brain injury. J Cereb Blood Flow Metab. 2001;21:1189–1198. doi: 10.1097/00004647-200110000-00007. [DOI] [PubMed] [Google Scholar]
- Kettenmann H, Kirchhoff F, Verkhratsky A. Microglia: new roles for the synaptic stripper. Neuron. 2013;77:10–18. doi: 10.1016/j.neuron.2012.12.023. [DOI] [PubMed] [Google Scholar]
- Kharatishvili I, Pitkänen A. Posttraumatic epilepsy. Curr Opin Neurol. 2010;23:183–188. doi: 10.1097/WCO.0b013e32833749e4. [DOI] [PubMed] [Google Scholar]
- Kreutzberg GW. Microglia: a sensor for pathological events in the CNS. Trends Neurosci. 1996;19:312–318. doi: 10.1016/0166-2236(96)10049-7. [DOI] [PubMed] [Google Scholar]
- Kurland D, Hong C, Aarabi B, Gerzanich V, Simard JM. Hemorrhagic progression of a contusion after traumatic brain injury: a review. J Neurotrauma. 2012;29:19–31. doi: 10.1089/neu.2011.2122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kyritsis N, Kizil C, Zocher S, Kroehne V, Kaslin J, Freudenreich D, Iltzsche A, Brand M. Acute inflammation initiates the regenerative response in the adult zebrafish brain. Science. 2012;338:1353–1356. doi: 10.1126/science.1228773. [DOI] [PubMed] [Google Scholar]
- Langlois JA, Rutland-Brown W, Thomas KE. The incidence of traumatic brain injury among children in the United States: differences by race. J Head Trauma Rehabil. 2005;20:229–238. doi: 10.1097/00001199-200505000-00006. [DOI] [PubMed] [Google Scholar]
- Liesz A, Zhou W, Mracskó É, Karcher S, Bauer H, Schwarting S, Sun L, Bruder D, Stegemann S, Cerwenka A, Sommer C, Dalpke AH, Veltkamp R. Inhibition of lymphocyte trafficking shields the brain against deleterious neuroinflammation after stroke. Brain. 2011;134:704–720. doi: 10.1093/brain/awr008. [DOI] [PubMed] [Google Scholar]
- Lipton ML, Gellella E, Lo C, Gold T, Ardekani BA, Shifteh K, Bello JA, Branch CA. Multifocal white matter ultrastructural abnormalities in mild traumatic brain injury with cognitive disability: a voxel-wise analysis of diffusion tensor imaging. J Neurotrauma. 2008;25:1335–1342. doi: 10.1089/neu.2008.0547. [DOI] [PubMed] [Google Scholar]
- Longhi L, Perego C, Ortolano F, Aresi S, Fumagalli S, Zanier ER, Stocchetti N, De Simoni MG. Tumor necrosis factor in traumatic brain injury: effects of genetic deletion of p55 or p75 receptor. J Cereb Blood Flow Metab. 2013;33:1182–1189. doi: 10.1038/jcbfm.2013.65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lourbopoulos A, Ertürk A, Hellal F. Microglia in action: how aging and injury can change the brain's guardians. Front Cell Neurosci. 2015;9:54. doi: 10.3389/fncel.2015.00054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luong TN, Carlisle HJ, Southwell A, Patterson PH. Assessment of motor balance and coordination in mice using the balance beam. J Vis Exp. 2011;49:2376. doi: 10.3791/2376. pii. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luster AD, Alon R, von Andrian UH. Immune cell migration in inflammation: present and future therapeutic targets. Nat Immunol. 2005;6:1182–1190. doi: 10.1038/ni1275. [DOI] [PubMed] [Google Scholar]
- Lye TC, Shores EA. Traumatic brain injury as a risk factor for Alzheimer's disease: a review. Neuropsychol Rev. 2000;10:115–129. doi: 10.1023/A:1009068804787. [DOI] [PubMed] [Google Scholar]
- Malt U. The long-term psychiatric consequences of accidental injury: a longitudinal study of 107 adults. Br J Psychiatry. 1988;153:810–818. doi: 10.1192/bjp.153.6.810. [DOI] [PubMed] [Google Scholar]
- Masel BE, DeWitt DS. Traumatic brain injury: a disease process, not an event. J Neurotrauma. 2010;27:1529–1540. doi: 10.1089/neu.2010.1358. [DOI] [PubMed] [Google Scholar]
- Masel BE, Bell RS, Brossart S, Grill RJ, Hayes RL, Levin HS, Rasband MN, Ritzel DV, Wade CE, DeWitt DS. Galveston Brain Injury Conference 2010: clinical and experimental aspects of blast injury. J Neurotrauma. 2012;29:2143–2171. doi: 10.1089/neu.2011.2258. [DOI] [PubMed] [Google Scholar]
- Mionnet C, Buatois V, Kanda A, Milcent V, Fleury S, Lair D, Langelot M, Lacoeuille Y, Hessel E, Coffman R, Magnan A, Dombrowicz D, Glaichenhaus N, Julia V. CX3CR1 is required for airway inflammation by promoting T helper cell survival and maintenance in inflamed lung. Nat Med. 2010;16:1305–1312. doi: 10.1038/nm.2253. [DOI] [PubMed] [Google Scholar]
- Naj AC, et al. Common variants at MS4A4/MS4A6E, CD2AP, CD33 and EPHA1 are associated with late-onset Alzheimer's disease. Nat Genet. 2011;43:436–441. doi: 10.1038/ng.801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olesen J, Gustavsson A, Svensson M, Wittchen HU, Jönsson B, CDBE2010 study group; European Brain Council The economic cost of brain disorders in Europe. Eur J Neurol. 2012;19:155–162. doi: 10.1111/j.1468-1331.2011.03590.x. [DOI] [PubMed] [Google Scholar]
- Paolicelli RC, Bolasco G, Pagani F, Maggi L, Scianni M, Panzanelli P, Giustetto M, Ferreira TA, Guiducci E, Dumas L, Ragozzino D, Gross CT. Synaptic pruning by microglia is necessary for normal brain development. Science. 2011;333:1456–1458. doi: 10.1126/science.1202529. [DOI] [PubMed] [Google Scholar]
- Parkhurst CN, Yang G, Ninan I, Savas JN, Yates JR, 3rd, Lafaille JJ, Hempstead BL, Littman DR, Gan WB. Microglia promote learning-dependent synapse formation through brain-derived neurotrophic factor. Cell. 2013;155:1596–1609. doi: 10.1016/j.cell.2013.11.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perry VH, Nicoll JA, Holmes C. Microglia in neurodegenerative disease. Nat Rev Neurol. 2010;6:193–201. doi: 10.1038/nrneurol.2010.17. [DOI] [PubMed] [Google Scholar]
- Pierce JE, Smith DH, Trojanowski JQ, McIntosh TK. Enduring cognitive, neurobehavioral and histopathological changes persist for up to one year following severe experimental brain injury in rats. Neuroscience. 1998;87:359–369. doi: 10.1016/S0306-4522(98)00142-0. [DOI] [PubMed] [Google Scholar]
- Prinz M, Priller J. Microglia and brain macrophages in the molecular age: from origin to neuropsychiatric disease. Nat Rev Neurosci. 2014;15:300–312. doi: 10.1038/nrn3722. [DOI] [PubMed] [Google Scholar]
- Raghupathi R. Cell death mechanisms following traumatic brain injury. Brain Pathol. 2004;14:215–222. doi: 10.1111/j.1750-3639.2004.tb00056.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramlackhansingh AF, Brooks DJ, Greenwood RJ, Bose SK, Turkheimer FE, Kinnunen KM, Gentleman S, Heckemann RA, Gunanayagam K, Gelosa G, Sharp DJ. Inflammation after trauma: microglial activation and traumatic brain injury. Ann Neurol. 2011;70:374–383. doi: 10.1002/ana.22455. [DOI] [PubMed] [Google Scholar]
- Ricklin D, Hajishengallis G, Yang K, Lambris JD. Complement: a key system for immune surveillance and homeostasis. Nat Immunol. 2010;11:785–797. doi: 10.1038/ni.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rink A, Fung KM, Trojanowski JQ, Lee VM, Neugebauer E, McIntosh TK. Evidence of apoptotic cell death after experimental traumatic brain injury in the rat. Am J Pathol. 1995;147:1575–1583. [PMC free article] [PubMed] [Google Scholar]
- Rogers JT, Morganti JM, Bachstetter AD, Hudson CE, Peters MM, Grimmig BA, Weeber EJ, Bickford PC, Gemma C. CX3CR1 deficiency leads to impairment of hippocampal cognitive function and synaptic plasticity. J Neurosci. 2011;31:16241–16250. doi: 10.1523/JNEUROSCI.3667-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubovitch V, Ten-Bosch M, Zohar O, Harrison CR, Tempel-Brami C, Stein E, Hoffer BJ, Balaban CD, Schreiber S, Chiu WT, Pick CG. A mouse model of blast-induced mild traumatic brain injury. Exp Neurol. 2011;232:280–289. doi: 10.1016/j.expneurol.2011.09.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rutland-Brown W, Langlois JA, Thomas KE, Xi YL. Incidence of traumatic brain injury in the United States, 2003. J Head Trauma Rehabil. 2006;21:544–548. doi: 10.1097/00001199-200611000-00009. [DOI] [PubMed] [Google Scholar]
- Salter MW, Beggs S. Sublime microglia: expanding roles for the guardians of the CNS. Cell. 2014;158:15–24. doi: 10.1016/j.cell.2014.06.008. [DOI] [PubMed] [Google Scholar]
- Satake K, Matsuyama Y, Kamiya M, Kawakami H, Iwata H, Adachi K, Kiuchi K. Nitric oxide via macrophage iNOS induces apoptosis following traumatic spinal cord injury. Brain Res Mol Brain Res. 2000;85:114–122. doi: 10.1016/S0169-328X(00)00253-9. [DOI] [PubMed] [Google Scholar]
- Saulle M, Greenwald BD. Chronic traumatic encephalopathy: a review. Rehabil Res Pract. 2012;2012:816069. doi: 10.1155/2012/816069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schafer DP, Lehrman EK, Kautzman AG, Koyama R, Mardinly AR, Yamasaki R, Ransohoff RM, Greenberg ME, Barres BA, Stevens B. Microglia sculpt postnatal neural circuits in an activity and complement-dependent manner. Neuron. 2012;74:691–705. doi: 10.1016/j.neuron.2012.03.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheridan GK, Murphy KJ. Neuron-glia crosstalk in health and disease: fractalkine and CX3CR1 take centre stage. Open Biol. 2013;3:130181. doi: 10.1098/rsob.130181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shlosberg D, Benifla M, Kaufer D, Friedman A. Blood-brain barrier breakdown as a therapeutic target in traumatic brain injury. Nat Rev Neurol. 2010;6:393–403. doi: 10.1038/nrneurol.2010.74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silver J, Miller JH. Regeneration beyond the glial scar. Nat Rev Neurosci. 2004;5:146–156. doi: 10.1038/nrn1326. [DOI] [PubMed] [Google Scholar]
- Smith C, Gentleman SM, Leclercq PD, Murray LS, Griffin WS, Graham DI, Nicoll JA. The neuroinflammatory response in humans after traumatic brain injury. Neuropathol Appl Neurobiol. 2013;39:654–666. doi: 10.1111/nan.12008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stephan AH, Barres BA, Stevens B. The complement system: an unexpected role in synaptic pruning during development and disease. Annu Rev Neurosci. 2012;35:369–389. doi: 10.1146/annurev-neuro-061010-113810. [DOI] [PubMed] [Google Scholar]
- Stevens B, Allen NJ, Vazquez LE, Howell GR, Christopherson KS, Nouri N, Micheva KD, Mehalow AK, Huberman AD, Stafford B, Sher A, Litke AM, Lambris JD, Smith SJ, John SW, Barres BA. The classical complement cascade mediates CNS synapse elimination. Cell. 2007;131:1164–1178. doi: 10.1016/j.cell.2007.10.036. [DOI] [PubMed] [Google Scholar]
- Thompson HJ, McCormick WC, Kagan SH. Traumatic brain injury in older adults: epidemiology, outcomes, and future implications. J Am Geriatr Soc. 2006;54:1590–1595. doi: 10.1111/j.1532-5415.2006.00894.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Werner C, Engelhard K. Pathophysiology of traumatic brain injury. Br J Anaesth. 2007;99:4–9. doi: 10.1093/bja/aem131. [DOI] [PubMed] [Google Scholar]
- Winston CN, Chellappa D, Wilkins T, Barton DJ, Washington PM, Loane DJ, Zapple DN, Burns MP. Controlled cortical impact results in an extensive loss of dendritic spines that is not mediated by injury-induced amyloid-beta accumulation. J Neurotrauma. 2013;30:1966–1972. doi: 10.1089/neu.2013.2960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiong Y, Mahmood A, Chopp M. Animal models of traumatic brain injury. Nat Rev Neurosci. 2013;14:128–142. doi: 10.1038/nrn3407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou H, Chen L, Gao X, Luo B, Chen J. Moderate traumatic brain injury triggers rapid necrotic death of immature neurons in the hippocampus. J Neuropathol Exp Neurol. 2012;71:348–359. doi: 10.1097/NEN.0b013e31824ea078. [DOI] [PMC free article] [PubMed] [Google Scholar]