

Abstract
Reduced activity of vagal efferents has long been implicated in schizophrenia and appears to be responsible for diminished parasympathetic activity and associated peripheral symptoms such as low heart rate variability and cardiovascular complications in affected individuals. In contrast, only little attention has been paid to the possibility that impaired afferent vagal signaling may be relevant for the disorder's pathophysiology as well. The present study explored this hypothesis using a model of subdiaphragmatic vagal deafferentation (SDA) in male rats. SDA represents the most complete and selective vagal deafferentation method existing to date as it leads to complete disconnection of all abdominal vagal afferents while sparing half of the abdominal vagal efferents. Using next-generation mRNA sequencing, we show that SDA leads to brain transcriptional changes in functional networks annotating with schizophrenia. We further demonstrate that SDA induces a hyperdopaminergic state, which manifests itself as increased sensitivity to acute amphetamine treatment and elevated accumbal levels of dopamine and its major metabolite, 3,4-dihydroxyphenylacetic acid. Our study also shows that SDA impairs sensorimotor gating and the attentional control of associative learning, which were assessed using the paradigms of prepulse inhibition and latent inhibition, respectively. These data provide converging evidence suggesting that the brain transcriptome, dopamine neurochemistry, and behavioral functions implicated in schizophrenia are subject to visceral modulation through abdominal vagal afferents. Our findings may encourage the further establishment and use of therapies for schizophrenia that are based on vagal interventions.
SIGNIFICANCE STATEMENT The present work provides a better understanding of how disrupted vagal afferent signaling can contribute to schizophrenia-related brain and behavioral abnormalities. More specifically, it shows that subdiaphragmatic vagal deafferentation (SDA) in rats leads to (1) brain transcriptional changes in functional networks related to schizophrenia, (2) increased sensitivity to dopamine-stimulating drugs and elevated dopamine levels in the nucleus accumbens, and (3) impairments in sensorimotor gating and the attentional control of associative learning. These findings may encourage the further establishment of novel therapies for schizophrenia that are based on vagal interventions.
Keywords: behavior, dopamine, next-generation mRNA sequencing, nucleus accumbens, schizophrenia, vagal afferents
Introduction
Schizophrenia is a heterogeneous and multifactorial psychiatric disorder affecting ∼1% of the population worldwide (Owen et al., 2016). Its clinical manifestation involves impairments in cognitive functions, emotions, and behavior, which together undermine basic processes of perception and judgment (Tandon et al., 2009). It is believed that disturbances in various neural and glial systems, along with a functional disconnection of discrete brain regions, are responsible for the emergence of multiple symptoms characteristic of schizophrenia (Insel, 2010; Landek-Salgado et al., 2016).
In addition to the pathologies that relate to CNS dysfunctions, schizophrenia has often been linked to peripheral anomalies (Kirkpatrick, 2009; Severance et al., 2016), including functional alterations in the autonomic nervous system (ANS; Clamor et al., 2016). Reduced activity of vagal efferents is one of the well known ANS pathologies in schizophrenia and appears to be responsible for diminished parasympathetic activity and associated peripheral symptoms, such as low heart rate variability and cardiovascular complications (Bär et al., 2010; Bär, 2015; Montaquila et al., 2015; Clamor et al., 2016). In addition to its parasympathetic contribution, the vagus nerve consists of 80% afferent sensory fibers and is critical for conveying visceral signals to the CNS via neuronal routes (Berthoud and Neuhuber, 2000). Vagal afferent neurons synapse bilaterally on the nucleus tractus solitarii (NTS), from where visceral signals can be transferred to various brainstem nuclei and forebrain structures (Berthoud and Neuhuber, 2000; Khalsa et al., 2009).
Despite the wide recognition of reduced activity of vagal efferents in schizophrenia, only little attention has been paid to the possibility that impaired afferent vagal signaling may be relevant for the disorder's pathophysiology as well (Hasan et al., 2015). Using a rat model of subdiaphragmatic vagal deafferentation (SDA), the present study sought preclinical evidence for this hypothesis by examining the influence of vagal afferents on gene expression profiles, behaviors, and neurochemistry implicated in schizophrenia. SDA leads to complete disconnection of all abdominal vagal afferents while sparing approximately half of the abdominal vagal efferents (Norgren and Smith, 1994; Arnold et al., 2006). It is the most complete and selective vagal deafferentation method existing to date and critically differs from total subdiaphragmatic vagotomy (TVX), which leads to a disconnection of both the afferent and efferent fibers of the vagus nerve below the diaphragm (Bercik et al., 2011; Bravo et al., 2011). Unlike TVX (Kraly et al., 1986), SDA allows for a discrimination of the relative functional contribution of vagal afferents versus efferents in the absence of severe side effects such as disturbances in gastrointestinal motility and secretion, hypophagia, and subsequent body weight loss (Arnold et al., 2006; Azari et al., 2014; Klarer et al., 2014, 2017).
First, we used unbiased next-generation mRNA sequencing to explore genome-wide transcriptional changes in SDA rats relative to Sham controls. Two brain regions relevant for schizophrenia were selected for this purpose, namely the prefrontal cortex (PFC) and nucleus accumbens (NAc; Selemon and Zecevic, 2015; Chuhma et al., 2017). We then compared SDA and Sham rats in a number of translational, cross-species behavioral paradigms used to assess schizophrenia-relevant phenotypes (Peleg-Raibstein et al., 2012), including sensorimotor gating (Swerdlow et al., 2008), selective attentional learning (Weiner and Arad, 2009), and sensitivity to the psychostimulant drug amphetamine (Laruelle, 2000). Finally, we measured dopamine and its metabolites in the PFC and NAc in relation to the well known role of dopaminergic imbalances in schizophrenia and related psychotic disorders (Howes and Kapur, 2009).
Materials and Methods
Animals.
Adult (280–320 g) male Sprague-Dawley (Crl:CD) rats were obtained from Charles River Laboratories. Animals were housed in acrylic stainless-steel grid-floor cages (60 × 38 × 20 cm) and kept under a reversed light/dark cycle (lights on from 20:00 to 08:00) at 22 ± 2°C and 55–60% humidity. The animals had ad libitum access to water and standard chow (Kliba 3436, Provimi Kliba) unless otherwise specified. Before surgery (see below), the animals were allowed to acclimatize to the new animal holding facility for 3 weeks, during which they were handled on a daily basis to habituate them to the experimenter. Throughout all testing, the animals were housed in groups of two to four rats per cage to avoid confounds arising from isolation stress (Zhang et al., 2012). All experiments and investigations were performed during the dark phase of the reversed light/dark cycle. All procedures were approved by the Cantonal Veterinarian Office of Zurich. All efforts were made to minimize the number of animals used as well as their suffering.
SDA and Sham surgeries.
To prevent excessive weight loss postsurgery, rats were nursed with special diets presurgery and postsurgery as described before (Arnold et al., 2006; Labouesse et al., 2012; Klarer et al., 2017), and were readapted to standard rodent ground chow thereafter. The SDA and Sham surgeries were performed as established and described in detail previously (Arnold et al., 2006; Labouesse et al., 2012; Klarer et al., 2014). In brief, SDA consisted of a left-side intracranial vagal rhizotomy and a transection of the second, dorsally located subdiaphragmatic trunk of the vagus nerve (Fig. 1A). Sham surgery consisted of similarly exposing both the vagal rootlets and dorsal subdiaphragmatic vagus, but without manipulating them. The SDA results in a complete disconnection of abdominal vagal afferents, whereas half of the abdominal vagal efferents are spared (Fig. 1A; Norgren and Smith, 1994; Arnold et al., 2006). Complete and uniquely vagal deafferentation at the cervical level through bilateral rhizotomy causes severe breathing complications after surgery, and as a consequence of this, most rats will die within 48 h postsurgery (M. Arnold, unpublished observations). Given these limitations of bilateral cervical rhizotomy, the SDA procedure is still the most complete and selective vagal deafferentation method existing to date.
Figure 1.
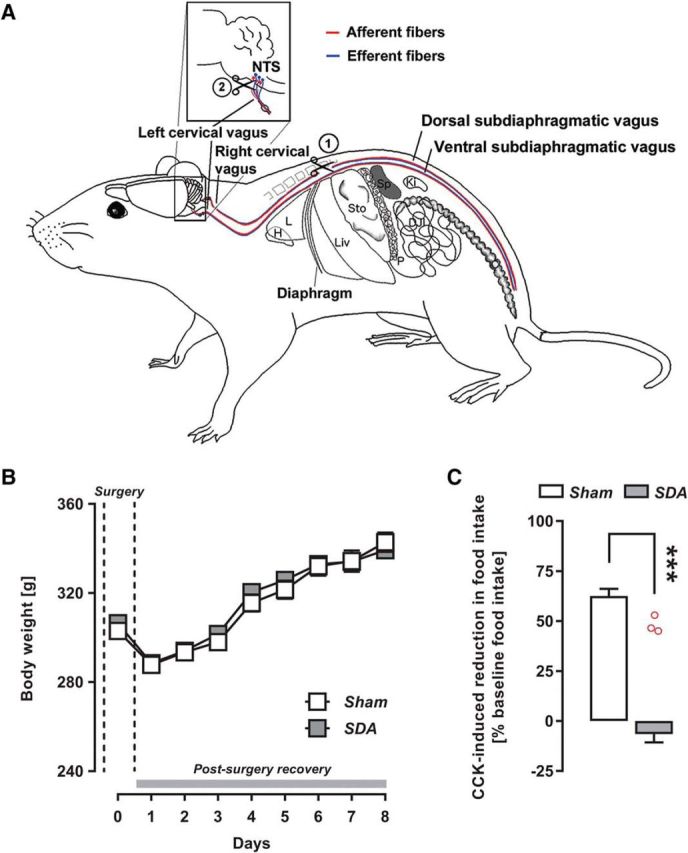
A, Schematic illustration of afferent and efferent vagal fibers targeted by the SDA procedure. Afferent and efferent fibers are represented in red and blue, respectively. In the SDA procedure, the dorsal (right) subdiaphragmatic trunk of the vagus nerve is fully transected (indicated by the first scissor symbol), leading to a disconnection of both afferent and efferent fibers in the dorsal trunk of the vagus nerve. The ventral (left) subdiaphragmatic trunk of the vagus nerve is left intact at this level. In addition, a left-sided intracranial vagal rhizotomy is performed (indicated by the second scissor symbol) to selectively disconnect the remaining vagal afferents. This SDA procedure induces a complete (100%) disconnection of vagal afferents in the abdomen while leaving 50% of the abdominal vagal efferents functionally intact. Vagally innervated organs: C, colon; DJI, duodenum, jejunum, ileum; H, heart; Ki, kidney; L, lung; Liv, liver; P, pancreas; Sp, spleen; Sto, stomach. B, Body weights before surgery (day 0; indicated by the dashed lines) and during the first recovery week (days 1–8). C, Verification of SDA completeness in a CCK satiation test. The bar plot shows the percentage reduction in CCK-induced food intake (percentage baseline food intake) during a 30 min test session. Note that SDA rats displayed no reduction in food intake in response to CCK administration relative to their baseline food intake, confirming the completeness of SDA. In contrast, Sham control rats displayed a 55–65% reduction in food intake in response to CCK relative to their baseline food intake. Against these backgrounds, three SDA animals (with 46.3%, 47.1%, and 52.3% reduction in food intake in response to CCK; red circles) were excluded from the final analysis and presentation of the data. ***p < 0.001. All values are mean ± SEM and based on n(Sham) = 42 and n(SDA) = 46.
The body weight of each animal was monitored before surgery and for a period of 1 week postsurgery. The total number of animals assigned to Sham and SDA surgery was n = 42 and n = 49, respectively, which were then randomly allocated to the different investigations of interest (Table 1).
Table 1.
Summary of the number of Sham and SDA animals included in each series of investigations
| Cohort | Test | n(Sham) | n(SDA) |
|---|---|---|---|
| 1 | mRNA sequencing | 5 | 5 |
| 2 | PPI | 14 | 13 |
| LI CTA | 14 | 13 | |
| NPE | 7 | 6 | |
| PE | 7 | 7 | |
| 3a | Low-dose Amph 1 mg/kg | 7 | 7 |
| 3b | High-dose Amph 2 mg/kg | 6 | 10 |
| 4 | Neurochemistry | 10 | 11 |
| Pooled | BW | 42 | 46 |
| CCK | 42 | 46 |
Animals in Cohort 1 were used for next-generation mRNA sequencing and subsequent gene expression validation by quantitative RT-PCR. Animals in Cohort 2 were first tested in the paradigm of PPI and were then subjected to the test of LI in CTA. For the latter, Sham and SDA animals were equally distributed to NPE and PE subgroups. Animals in Cohort 3 were used to asses the sensitivity to the psychostimulant drug, Amph. A subcohort of animals (3a) was treated with 1 mg/kg Amph, whereas another subcohort (3b) was treated with 2 mg/kg Amph. Animals in Cohort 4 were used for postmortem analyses of dopamine neurochemistry using HPLC. Animals from all cohorts were pooled for the assessment of postsurgery body weight (BW) and for the verification of SDA completeness in a CCK satiation test. All animals were adult male Sprague-Dawley (Crl:CD) rats.
Functional verification of SDA completeness.
SDA completeness was verified using an established functional test ascertaining the lack of cholecystokinin (CCK) satiation that depends on intact abdominal vagal afferent fibers (Smith and Gibbs, 1985). The CCK test was performed as described previously (Klarer et al., 2014, 2017). In short, the animals were food deprived overnight (13 h) and injected intraperitoneally (i.p.) with 4 μg/kg CCK-8 (Bachem) or vehicle (PBS) using a within-subject crossover design. Individual food intake was monitored for 30 min.
CCK-8 treatment in Sham rats typically leads to a robust reduction in food intake within the first 30 min after food presentation (Arnold et al., 2006; Labouesse et al., 2012; Klarer et al., 2014, 2017). Therefore, the inclusion criterion for SDA rats was set at 40% ± SEM of CCK-induced reduction in food intake during the first 30 min. Only data from animals that passed this functional verification were included in the final analysis and presentation of data.
Next-generation mRNA sequencing.
The animals were anesthetized with a lethal overdose of sodium pentobarbital (20 mg/kg) and perfused with ice-cold, oxygenated and glucose-supplemented artificial CSF (Notter et al., 2014) to obtain fresh brain tissue for the subsequent isolation of total RNA and next-generation mRNA sequencing (see below). The brains were rapidly extracted from the skull (within <2 min) and placed on an ice-chilled plate. Coronal sections were prepared using razorblade cuts along the following coordinates with respect to bregma: anterior–posterior, PFC: +3.2 to +2.2 and NAc: +1.7 to +0.7. PFC (including prelimbic and infralimbic cortices) and NAc (including core and shell subregions) samples were then generated via micropunches of 1 mm diameter as previously described (Richetto et al., 2017).
For each brain region of interest, mRNA was extracted and isolated using the Qiagen AllPrep DNA/RNA Mini Kit according to the manufacturer's instructions. The integrity and amount of mRNA was determined using the Agilent 4200 TapeStation system (Agilent Technologies). Only samples with an RNA integrity number >8 were further processed and included in the analysis.
To produce library constructs, we used the Illumina TruSeq stranded mRNA protocol with the TruSeq Stranded mRNA Sample preparation kit as described before (Weber-Stadlbauer et al., 2017). In brief, 500 ng of total RNA per sample was polyA enriched, fragmented, and reverse-transcribed into double-stranded cDNA, and ligated with TruSeq adapters. Using PCR, fragments containing TruSeq adapters on both ends were selectively enriched. Quality and quantity of enriched libraries were analyzed using the Agilent 4200 TapeStation system (Agilent Technologies) with DNA-specific chip. The DNA library contained fragments in the size range of 100–800 bp with a mean fragment size of ∼260 bp. Diluted libraries (10 nm) were pooled and further used for cluster generation according to the manufacturer's recommendations using TruSeq SR Cluster Kit v4-cBot-HS reagents. The libraries were sequenced with TruSeq SBS Kit v-4-HS reagents on Illumina HiSeq 4000 in the high output mode.
Bioinformatic analyses were performed using SUSHI as described in detail by Hatakeyama et al. (2016). All reads were first quality-checked using FastQC, and low-quality ends were clipped (5′: 3 bases; 3′: 10 bases). Trimmed reads (length >20 nt, average quality >Q10) were aligned with the STAR aligner with additional options that require at least 30 bp matching, accept at most 10 mismatches, and at most 5% of mismatches (Dobin et al., 2013). Read alignments were only documented for reads with <50 valid alignments. As reference, we used the rat genome build and annotation from Ensembl (Rnor_6.0). Whereas expression counts were computed using featureCounts within the Bioconductor package “Rsubread” (Liao et al., 2013), differential expression was computed for pairwise comparisons (SDA vs Sham) using the Bioconductor package Deseq2 (Love et al., 2014).
Hierarchical clustering and gene-expression network analyses.
Differentially expressed genes (DEGs) revealed by next-generation mRNA sequencing (see above) were loaded into Partek Genomics Suite (Partek) and were clustered according to the “hierarchical clustering” function of the program. For this purpose, the data were normalized with standardization (each column mean is 0, and the SD is scaled to 1), and then multidimensional scaling with a Euclidian distance metric was performed on the normalized samples to allow visualization of the distance between them.
Qiagen's Ingenuity Pathway Analysis (IPA) was used to predict functional networks (Thomas and Bonchev, 2010). IPA uses the curated Ingenuity Knowledge Base to identify the involvement of differentially expressed genes in specific diseases and cellular pathways, and to establish functional networks of direct and indirect interactions between differentially expressed genes based on a functional analysis algorithm (Thomas and Bonchev, 2010). For IPA, we used the DEGs identified by the Bioconductor package Deseq2 as described above.
Validation of differential gene expression by quantitative RT-PCR.
To further validate the next-generation mRNA sequencing data, the expression of selected candidate genes was verified using quantitative RT-PCR analyses. Total RNA was isolated from PFC and NAc samples, and checked for quantity and quality, as described above. To avoid DNA contamination, an aliquot of each RNA sample was treated with DNase. mRNA was analyzed by TaqMan qRT-PCR instrument (CFX384 real-time system; Bio-Rad Laboratories) after conversion into cDNA using the iScript cDNA synthesis kit (Bio-Rad Laboratories). The samples were run in 384-well formats in triplicates as multiplexed reactions with a normalizing internal control (β-actin) using the iTaq Universal Probes Supermix (Bio-Rad Laboratories). We chose β-actin as internal standard for gene expression analyses because its expression was not affected by the lesion. Thermal cycling was initiated by 95°C for 5 min (TaqMan polymerase activation). After this initial step, 39 cycles of PCR were performed. Each PCR cycle consisted of heating the samples at 95°C for 10 s to enable the melting process and then for 30 s at 60°C for the annealing and extension reaction. Relative target gene expression was calculated according to the 2−ΔCT method (Livak and Schmittgen, 2001). Probe and primer sequences of genes selected for validation were purchased from ThermoFisher Scientific (Gjb1: Rn01641031_s1; Gjc2: Rn01750744_m1; Grik5: Rn00568522_m1; Mag: Rn01457782_m1; Reln: Rn00589609_m1; Sst: Rn00561967_m1). The selection of genes for RT-PCR validation was based on both the IPA and availability of primers. β-actin was used as internal reference gene (forward: 5′CACTTTCTACAATGAGCTGCG-3′; reverse: 5′-CTGGATGGCTACGTACATGG-′3; probe: 5′-TCTGGGTCATCTTTTCACGGTTGGC-3′).
Sensorimotor gating.
To examine schizophrenia-related sensorimotor gating abnormalities (Braff et al., 2001), we used the paradigm of prepulse inhibition (PPI) of the acoustic startle reflex. PPI of the acoustic startle reflex reflects the reduction of startle reaction in response to a startle-provoking pulse stimulus when it is shortly preceded by a weak prepulse stimulus (Swerdlow et al., 2008). The test apparatus consisted of a sound-attenuated acoustic startle chamber (SR-LAB; San Diego Instruments) as described previously (Pothuizen et al., 2006). The test procedures used to assess PPI have been validated before (Meyer et al., 2005; Pothuizen et al., 2006). In brief, rats underwent one 45 min PPI test session, which consisted of 11 distinct stimuli types, including pulse-alone stimuli, prepulse-alone stimuli at all five distinct prepulse intensities, and all possible combinations of a preceding prepulse with the startle-eliciting pulse (prepulse-plus-pulse stimuli). The test session also included no-stimulus trials, in which no discrete stimulus other than the constant background noise (65 dBA) was presented. The pulse trials consisted of 40 ms pulses of white noise (120 dBA), which were either presented alone (pulse-alone trials) or in conjunction with one of five different prepulses (prepulse-plus-pulse). Prepulses were a 20 ms burst of white noise at five different intensities (69, 73, 77, 81, and 85 dBA, which corresponded to +4, +8, +12, +16, and +20 dBA above background). The stimulus onset asynchrony of the prepulse and pulse stimuli in prepulse-plus-pulse trials was 100 ms. Following a 2 min acclimatization period, six consecutive pulse-alone trials were presented to habituate and stabilize the animals' startle response. Subsequent to this habituation phase, each trial stimulus was presented 12 times in a pseudorandom order with an average interval between successive trials of 15 ± 5 s. The session was concluded with six consecutive pulse-alone trials. The chamber was cleansed with warm tap water and dried between each animal.
For each rat and at each of the five possible prepulse intensities, PPI was indexed by percentage inhibition of startle response obtained in the prepulse-plus-pulse-trials compared with pulse-alone trials by following expression: [1 − (mean reactivity on prepulse-plus-pulse trials/mean reactivity on pulse-alone trials) × 1/100]. In addition to percentage PPI, reactivity to pulse-alone trials and prepulse-alone trials were also recorded and analyzed. Habituation to the startle reaction was assessed by a comparison between the reactivity obtained on the first and the last block of 6 consecutive pulse-alone trials.
Attentional control of associative learning.
We used the paradigm of latent inhibition (LI) to examine schizophrenia-relevant deficits in the attentional control of associative learning (Weiner and Arad, 2009). In LI, healthy subjects who are pre-exposed (PE) to a to-be-conditioned stimulus (CS) typically show reduced conditioning between the CS and an unconditioned stimulus (US) compared with subjects who were not pre-exposed (NPE; Lubow, 2005). This LI effect is reduced or absent in patients with schizophrenia, especially in those with marked positive symptoms (Lubow, 2005; Weiner and Arad, 2009), which reflects the patients' difficulties in ignoring irrelevant stimuli and directing their attentional resources to relevant associations.
We assessed the LI effect using a conditioned taste avoidance (CTA) paradigm. The CTA paradigm was selected for two main reasons. First, CTA-LI has previously been validated in various preclinical rodent models relevant to schizophrenia and related disorders involving dopaminergic imbalances (Russig et al., 2003; Meyer et al., 2004, 2006). Second, deficits in CTA-LI are extendable to other LI paradigms and thus allow a certain degree of generalization of the findings from one paradigm to another (Meyer et al., 2004, 2006).
In the CTA-LI test, 5% sucrose solution served as the CS, whereas nausea induced by lithium chloride (LiCl) served as the US (see below). All animals were naive to the CS and US before commencing the LI test. To assess LI in CTA, half of the SDA and Sham rats (i.e., the PE subgroups) were exposed to sucrose solution (= CS) before conditioning for CTA took place, whereas the other half (i.e., the NPE subgroups) were not subjected to sucrose presentation before conditioning (see below).
For this test, the animals were temporarily single caged for a period of 10 d, after which they were rehoused with their previous cage-mates. The apparatus consisted of acrylic cages (48 × 27 × 20 cm) with sawdust bedding and flat grid tops, which allowed the efficient placement or removal of the drinking bottles (Russig et al., 2003). Each cage was provided with two 250 ml drinking bottles (Tecniplast). The placement was such that the animals could easily switch drinking from one bottle to the other (Russig et al., 2003). The rats were progressively familiarized to a restrictive water regimen over a period of 5 d until they were finally maintained on a scheduled total access to water for 2 h per d. Access to water was divided into two drinking sessions: the first one (30 min) corresponded to the time required for the actual test sessions (see below), and the second one (1.5 h) was given 5 h after the test sessions. The test sessions were always conducted between 11:00 and 13:00. The animals' body weights were monitored throughout the entire experiment on a daily basis. All animals maintained their body weight that was at least 85% of their basal body weight. Liquid consumption was measured by calculating the difference in the weight of the drinking bottles before and after each drinking session. Before assigning the animals to the NPE and PE conditions (see below), basal water intake was measured on two preceding days (days 6 and 7) using a bottle that was placed in the middle of the cage. To this aim, water access was exactly scheduled for a 30 min period, analogous to the test sessions.
The test procedure of the LI test then consisted of three phases, namely the pre-exposure phase (day 8), conditioning phase (day 9), and expression phase (day 10) of CTA, which were conducted 24 h apart from each other. In the pre-exposure session, half of the SDA and Sham animals had access to a drinking bottle filled with a 5% sucrose solution (PE subgroups) for a 30 min period, whereas the other half (NPE subgroups) had access to water only for the same amount of time. The drinking bottles were located either on the left- or right-holding of the cage. The bottle's position was switched after 15 min to minimize the possible development of a side preference (Russig et al., 2003). One day after the pre-exposure phase, the conditioning session was conducted, in which all rats first had a 30 min access to 5% sucrose solution. Again, the position of the bottle was switched after 15 min. Immediately after the 30 min drinking period, the animals received an intraperitoneal injection of LiCl (0.25 m, 0.75% of body weight; dissolved in sterile 0.9% saline solution) and were gently returned to their home cages. The CTA-LI effect was then assessed on the next day. For this purpose, all animals had simultaneous 30 min access to both water and 5% sucrose solution presented in two different bottles. The positions of the bottles were again switched after 15 min and counterbalanced between experimental groups. CTA was indexed by the sucrose consumption as a proportion (in percentage) of total liquid (sucrose solution and water) consumed when the animals were confronted with a choice between sucrose and water. In this experimental setup, LI is demonstrated when PE animals show reduced CTA compared with NPE animals, i.e., when PE animals consume a higher proportion of sucrose solution during the expression phase compared with NPE subjects (Russig et al., 2003; Meyer et al., 2004).
Sensitivity to amphetamine.
Based on the findings showing increased sensitivity of schizophrenic patients to the dopamine-releasing drug, amphetamine (Amph; Laruelle, 2000), we compared SDA and Sham rats in terms of Amph-induced behavioral modifications in the open field. The apparatus consisted of two identical square arenas (80 × 80 cm) surrounded by walls (50 cm high). It was made of gray Plexiglas and was located in a testing room under diffused lighting (30 lux as measured in the center of the arenas). A digital camera was mounted directly above the two arenas. Images were captured at a rate of 5 Hz and transmitted to a PC running the EthoVision (Noldus) tracking system.
The amphetamine test was performed using a within-subject design, where all rats were first treated with saline (Sal) solution (Phase 1) and then with Amph (Phase 2). To begin the test, the animals first received an intraperitoneal injection of an isotonic 0.9% saline (NaCl) solution (= Sal) and were placed in the open field to measure basal locomotor activity for 20 min. Subsequently, the animals were briefly removed from the apparatus and injected intraperitoneally with Amph (d-amphetamine sulfate, Sigma-Aldrich; dissolved in isotonic 0.9% NaCl). They were then immediately returned to the same arena again for a total amount of 60 min. Based on previous dose–response studies in rats (Mueller et al., 1989a,b), Amph was either given at a dose of 1 or 2 mg/kg. For both doses, the animals' locomotor responses to the drug were indexed in terms of distance moved and quantified using the EthoVision tracking system (see above) as a function of 10 min bins. Because Amph treatment at doses >1 mg/kg often induce noticeable behavioral stereotypies in rats (Mueller et al., 1989a,b), we also analyzed stereotypic behaviors in animals treated with 2 mg/kg Amph. The latter was achieved by manual scores obtained from video recordings using a scan sampling technique, which encompassed stereotypic scores during the first and sixth minutes in every 10 min bin. Scoring was performed by an experimenter who was blinded to experimental conditions. The dependent variables of primary interest were repetitive head waving and circling movement (Mueller et al., 1989a,b), which were integrated into one single stereotypy score and analyzed as a function of 10 min bins.
All solutions were freshly prepared on the day of testing and injected using an injection volume of 1 ml/kg. Each animal was treated with Amph only once to avoid possible confounds arising from behavioral sensitization (Robinson and Becker, 1986). Hence, two different subgroups of SDA and Sham rats were used to assess the behavioral effects of Amph given at the low (1 mg/kg) or high (2 mg/kg) dose (Table 1).
Dopamine neurochemistry.
Dopamine (DA) and its major metabolite 3,4-dihydroxyphenylacetic acid (DOPAC) were measured in the postmortem PFC and NAc samples of Sham and SDA animals using HPLC. The animals were decapitated using a rat guillotine to obtain fresh brain tissue for the subsequent neurochemical analyses. To avoid possible confounds arising from circadian effects on DA neurochemistry (Sleipness et al., 2008), the order of decapitation was counterbalanced between experimental groups and was performed during the dark phase of the reversed light/dark cycle (not earlier or later than 3 h before or after the light-cycle switch). All animals had ad libitum access to food and water before decapitation. The brains were extracted from the skull within <2 min, immediately snap-frozen in −30°C cold isopentane, and stored at −80°C until further use. Frozen coronal sections (1 mm) were prepared at the following bregma coordinates according to Paxinos and Watson (1998): anterior–posterior, PFC: +3.2 to +2.2; NAC: +1.7 to +0.7.
PFC (including prelimbic and infralimbic cortices) and NAc samples from both hemispheres were generated via micropunches of 1 mm diameter as previously described (Winter et al., 2009; Klarer et al., 2014). Tissue samples from each brain area were homogenized by ultrasonication in 500 μl 0.1 m perchloric acid at 4°C immediately after micropunching. One hundred microliters of each homogenate was stored at −80°C for subsequent protein determination. The remaining homogenates were centrifuged at 17,000 × g and 4°C for 10 min. DA and DOPAC were quantified using HPLC with electrochemical detection as previously described (Winter et al., 2009; Klarer et al., 2014). Briefly, the perchloric acid extracts were separated on a column (Prontosil 120-C18-SH; 5 μm, 200 × 2 mm; Bischoff Analysentechnik und-Geräte) at a flow rate of 0.3 ml/min. The mobile phase consisted of 80 mm sodium dihydrogen phosphate, 0.9 mm octane-1-sulfonic acid sodium salt, 0.5 mm EDTA disodium salt, 0.92 mm phosphoric acid, and 4% 2-propanol (all chemicals: Merck). DA and DOPAC were detected using an electrochemical detector (Decade II; Antec Leyden) at an electrode potential of 0.65 V. For calibration, two standard solutions containing 10 nm DA and DOPAC, respective 100 nm in 0.1 m perchloric acid, were injected into the HPLC system before and after sample analysis. Sample analysis was performed based on peak areas using a computer-based chromatography data system (LCSolution v1.23, Shimadzu) in relation to the mean of the applied calibration solutions. DA and DOPAC levels (nm) were normalized to the protein contents measured in the brain areas of interest, and DA turnover was assessed by analyzing the DOPAC–DA ratio.
Statistical analyses.
Body weights were analyzed by a 2 × 8 (lesion × day) ANOVA. CCK-induced reduction in food intake was expressed relative to basal food intake and analyzed using independent Student's t test (two-tailed). This analysis was supplemented with additional one-way ANCOVA with basal food intake as covariate. Differential gene expression in the next-generation mRNA sequencing datasets was computed for pairwise comparisons (Sham vs SDA) using the Bioconductor package Deseq2 and was analyzed separately for PFC and NAc regions. Follow-up validations of differential gene expression were performed by analyzing the 2−ΔCT levels of selected genes of interest using independent Student's t test (two-tailed). Percentage PPI and prepulse-induced reactivity were analyzed using 2 × 5 (lesion × prepulse level) ANOVAs, whereas the pulse-induced startle reactivity was analyzed using independent Student's t test (two-tailed). Raw-score reactivity levels, which assess whether the presentation of a prepulse preceding the pulse is efficient in reducing pulse reactivity toward the succeeding pulse, were analyzed using a 2 × 6 (lesion × prepulse intensity) ANOVA. Habituation to the pulse-induced startle reaction was analyzed using a 2 × 2 (lesion × block) ANOVA to allow for a comparison between the reactivity obtained on the first and the last block of six consecutive pulse-alone trials. Percentage sucrose intake in the expression phase of the CTA test was analyzed using a 2 × 5 (lesion × PE condition) ANOVA, followed by one-way ANOVAs restricted to lesion and PE conditions. Sucrose or water intake during the PE phase, as well as total fluid intake during the PE, conditioning and expression phases of the CTA test, were separately analyzed using independent Student's t tests (two-tailed). In the Amph sensitivity test, the distance moved in response to the drug challenge was first analyzed using a 2 × 2 × 6 (lesion × dose × 10 min bin) ANOVA, followed by 2 × 6 (lesion × 10 min bin) ANOVAs restricted to each dose of Amph. Behavioral stereotypy scores after administration of the higher Amph dose were also analyzed using a 2 × 6 (lesion × 10 min bin) ANOVA, followed by independent Student's t tests (two-tailed) restricted to individual bins if appropriate. The behavioral stereotypy data were first LN-transformed to better conform to the assumption of data homogeneity and normality by parametric ANOVA. The distance moved and LN-transformed behavioral stereotypy scores during the initial saline administration phase were analyzed using 2 × 2 (lesion × 10 min bin) ANOVAs. DA, DOPAC, and DOPAC/DA were all analyzed using 2 × 2 (lesion × brain region) ANOVAs, followed by Fisher's least significant difference test whenever appropriate. Statistical significance was set at p < 0.05. With the exception of the sequencing data (see above), all statistical analyses were performed using the statistical software SPSS Statistics v22.0 (IBM).
Results
Body weights and functional verification of SDA
Following an initial drop in body weight induced by the surgical procedures, all animals regained their presurgery body weight within 4 d after surgery (Fig. 1B). Importantly, there were no group differences in body weight between rats undergoing SDA or Sham surgery at any presurgical or postsurgical time points (Fig. 1B; F(1,86) = 0.123, p = 0.73).
SDA completeness was functionally verified using a test assessing CCK-induced suppression of food intake. As expected (Arnold et al., 2006; Labouesse et al., 2012; Klarer et al., 2014, 2017), Sham rats showed a 55–65% (t(1,86) = 12.59, p < 0.001) reduction in food intake after intraperitoneal CCK injection relative to their baseline food intake (Fig. 1C). Forty-six of 49 SDA rats displayed no such reduction in food intake in response to CCK administration relative to their baseline food intake, confirming the completeness of SDA in these animals. Three SDA rats, however, showed a 40–50% reduction in food intake following CCK administration, indicating a lack of SDA completeness (Arnold et al., 2006; Labouesse et al., 2012; Klarer et al., 2014, 2017). These three SDA rats were therefore excluded from the final analyses. SDA rats showed a small (Sham: 6.7 ± 0.29 g; SDA: 5.6 ± 0.20 g) but significant (t(86) = 3.07, p < 0.01) reduction in the baseline level of feeding, that is, in basal food intake following initial Sal administration. To test whether the difference in CCK-induced suppression of food intake between SDA and Sham rats is accounted for by the SDA-induced reduction in the baseline level of feeding, we performed additional ANCOVA of relative food intake following CCK administration with basal food intake as covariate. This analysis led to results that were consistent with the conclusion above. Hence, the main effect of lesion remained highly significant (F(1,85) = 132.84, p < 0.001) despite the inclusion of the covariate, the latter of which attained statistical significance as well (F(1,85) = 4.15, p < 0.05). Together, these results confirm the completeness of SDA in 46 of 49 animals even after controlling for differences in basal food intake.
SDA induces changes in the CNS transcriptome
Next-generation mRNA sequencing showed that SDA induced genome-wide transcriptional changes in two brain regions implicated in schizophrenia and related disorders, namely the NAc and PFC (Selemon and Zecevic, 2015; Chuhma et al., 2017). We revealed 908 and 426 DEGs in the NAc and PFC of SDA, respectively (p < 0.05; deposited at the European Nucleotide Archive: PRJEB20027). In both brain regions, these DEGs clustered according to lesion (Fig. 2A,D).
Figure 2.

Brain transcriptomic changes in SDA rats as revealed by next-generation mRNA sequencing. A, Heat map of all DEGs in the NAc of SDA rats relative to Sham controls. The color-coded key denotes upregulation (purple) and downregulation (yellow) in terms of fold-changes. B, Graphical representation of the functional entity most strongly affected by SDA in the NAc, as revealed by IPA under the category “top diseases and biological functions”. C, Graphical illustration of the STAT3 pathway, which is deregulated in the NAc of SDA animals. Colored elements in the pathway denote significant changes in gene expression. D, Heat map of all DEGs in the PFC of SDA rats relative to Sham controls. The color-coded key denotes upregulation (purple) and downregulation (yellow) in terms of fold-changes. E, Graphical representation of the functional entity most strongly affected by SDA in the PFC, as revealed by IPA under the category “top diseases and biological functions”. F, Graphical illustration of the IFN signaling pathway, which is deregulated in the PFC of SDA animals. Colored elements in the pathway denote significant changes in gene expression. All data in the NAc are based on n(Sham) = 5 and n(SDA) = 5; all data in the PFC are based on n(Sham) = 4 and n(SDA) = 5.
We then analyzed the DEGs with IPA to examine which functional entities are most strongly affected by SDA under the category “top diseases and biological functions”. This analysis revealed “neurological disease” as the top-1 subcategory in both brain areas (p value range for NAc: 4.71E-02 to 3.88E-03; p value range for PFC: 4.54E-02 to 3.90E-05). Intriguingly, within the subcategory “neurological disease”, the functional entity most strongly affected by SDA in the NAc annotated with the term “schizophrenia” (p = 3.88E-03). This functional entity encompassed 16 genes, the expression of which is known to be deregulated in schizophrenia (Fig. 2B), including semaphorin 3a (Sema3a), somatostatin (Sst), calbindin 1 (Calb1), reelin (Reln), metabotropic glutamate receptor 3 (Grm3), and glutamate ionotropic receptor kainate type subunit 5 (Grik5; Eastwood et al., 2003; Egan et al., 2004; Shibata et al., 2006; Fung et al., 2010). In the PFC region, the functional entity most strongly affected by SDA under the subcategory “neurological disease” annotated with the term “degeneration of the nervous system” (p = 3.90E-05), which included 16 DEGs (Fig. 2E). This functional entity included several genes involved in neurodegenerative processes, myelination, and formation of gap junctions, including Bcl-2-like 1 (Bcl2l1), myelin-associated glycoprotein (Mag), kallikrein-related peptidase 6 (Klk6), gap junction protein β 1 (Gjb1), and gap junction protein gamma 2 (Gjc2; Llorens et al., 2011; Abrams and Scherer, 2012; Yoon et al., 2013).
Venn analysis showed that only 59 DEGs (4.6% of all DEGs) were common to the PFC and NAc, suggesting that SDA induced mostly distinct gene expression changes in these two brain areas. Interestingly, however, transcriptional changes in cytokine signaling networks were present in both the NAc (Fig. 2C) and the PFC (Fig. 2F). In the NAc, SDA primarily caused differential expression of genes encompassed in the signal transducer and activator of transcription 3 (STAT3) pathway, including janus kinase 2 (Jak2), suppressor of cytokine signaling 2 (Socs2), serine/threonine-protein kinase pim-1 (Pim1), and protein inhibitor of activated STAT3 (Pias3; Fig. 2C). In the PFC, SDA caused differential expression of genes involved in the interferon (IFN) signaling, including upregulation of IFN-gamma receptor 1 (IFNγRa), IFN-induced transmembrane protein 3 (Ifitm3), and IFN-induced protein with tetratricopeptide repeats 3 (Ifit3; Fig. 2F).
To further validate the next-generation mRNA sequencing data, we used quantitative RT-PCR analyses to measure the mRNA levels of several candidate genes. To this aim, we focused on DEGs that were annotated with the functional entity “schizophrenia” (NAc region) and “degeneration of the nervous system” (PFC region) as revealed by IPA (Fig. 2B,E). Consistent with the next-generation mRNA sequencing data (Fig. 2B), we found that the mRNA levels of Grik5 (t(8) = 3.24, p < 0.05) and Sst (t(8) = 2.75, p < 0.05) were significantly increased in the NAc of SDA rats compared with Sham controls, whereas the accumbal mRNA levels of Reln were decreased (t(8) = 2.90, p < 0.05) in the former relative to the latter group (Fig. 3A). Quantitative RT-PCR analyses further confirmed significantly reduced mRNA levels of Gjb1 (t(8) = 3.22, p < 0.05), Gjc2 (t(8) = 2.63, p < 0.05), and Mag (t(8) = 2.58, p < 0.05) in the PFC region of SDA rats compared with Sham controls (Fig. 3B), as previously revealed by next-generation mRNA sequencing (Fig. 2E).
Figure 3.

Validation of differential gene expression by quantitative RT-PCR. Candidate genes were selected based on the outcomes of the preceding next-generation mRNA sequencing and availability of primers. A, The scatter dot plots show individual messenger RNA levels (in terms of 2−ΔCT levels) for Grik5, Reln, and Sst in the nucleus accumbens of Sham and SDA rats. *p < 0.05. B, The scatter dot plots depict individual messenger RNA levels (in terms of 2−ΔCT levels) for Gjb1, Gjc2, and Mag in the PFC of Sham and SDA rats. *p < 0.05. All data are based on n(Sham) = 5 and n(SDA) = 5.
SDA disrupts sensorimotor gating and attentional control of associative learning
To examine whether abdominal vagal afferents modulate behaviors relevant to schizophrenia, we compared SDA and Sham rats in behavioral paradigms used to assess schizophrenia-associated phenotypes in animal models (Peleg-Raibstein et al., 2012). First, we measured sensorimotor gating in the form of PPI of the acoustic startle reflex, which is attenuated in patients with schizophrenia (Braff et al., 2001). As expected (Yee et al., 2005), percentage PPI increased with increasing prepulse stimulus intensities for both experimental groups, as supported by the significant main effect of prepulse intensity (F(4,100) = 16.36, p < 0.001; Fig. 4A). Most importantly, SDA rats displayed a significant overall reduction in percentage PPI compared with Sham controls (main effect of lesion: F(1,25) = 4.86, p < 0.05; Fig. 4A). The interaction between lesion and prepulse intensity did not reach statistical significance (F(4,100) = 0.77, p = 0.55), suggesting that the SDA-induced reduction in percentage PPI emerged independently of prepulse intensity. SDA did not affect the responses to pulse-alone (t(25) = 1.21, p = 0.24; Fig. 4B) or prepulse-alone stimuli (main effect of lesion: F(1,25) = 1.25, p = 0.27; interaction between lesion × prepulse intensity: F(4,100) = 0.77, p = 0.55; Fig. 4B), indicating that the PPI-attenuating effects of SDA represent a genuine sensorimotor gating impairment. To seek additional support for this notion, we analyzed the raw-score reactivity levels, which assess whether the presentation of a prepulse preceding the pulse is efficient in reducing pulse reactivity toward the succeeding pulse (Swerdlow et al., 2000; Yee et al., 2005). This additional analysis revealed a significant interaction between lesion and prepulse intensity (F(5,125) = 4.86, p < 0.05), thus further supporting the notion that the extent to which the presentation of a prepulse is efficient in reducing subsequent pulse reactivity (i.e., PPI) is reduced in SDA relative to Sham controls. On the other hand, SDA did not affect habituation to the pulse-induced startle response, which was analyzed by a comparison between the reactivity obtained on the first and the last block of six consecutive pulse-alone trials. This analysis only revealed a significant main effect of block (F(1,25) = 50.62, p < 0.001), whereas the main effect of lesion (F(1,25) = 1.63, p = 0.21) or its interaction with block (F(1,25) = 1.39, p = 0.25) did not attain statistical significance. The mean ± SEM reactivity (in arbitrary units) during the first and the last block of six consecutive pulse-alone trials was 227.4 ± 22.0 and 103.6 ± 22.3 in Sham rats, and 285.9 ± 34.5 and 120.4 ± 35.4 in SDA rats.
Figure 4.

Deficient sensorimotor gating and attentional control of associative learning in SDA rats. Sensorimotor gating was assessed using the paradigm of PPI of the acoustic startle reflex, whereas attentional control of associative learning was measured using the paradigm of LI in conditioned taste avoidance. A, The line plot shows percentage PPI (mean ± SEM) as a function of prepulse intensity (dBA above background of 65 dBA); the bar plot depicts the mean percentage PPI (mean ± SEM) across all prepulse levels. *p < 0.05, n(Sham) = 14 and n(SDA) = 13. B, The bar plot depicts the reactivity to pulse-alone trials (i.e., startle reaction; mean ± SEM); the line plot shows the prepulse-induced reactivity (mean ± SEM) as a function of prepulse intensity. n(Sham) = 14 and n(SDA) = 13. C, Relative sucrose solution intake (mean ± SEM) during the test session of LI, when NPE and PE animals were given free choice between sucrose solution and water. *p < 0.05, reflecting the difference between PE Sham and PE SDA animals; **p < 0.01, representing the difference between NPE Sham and PE Sham animals (i.e., the presence of LI in Sham animals). n(Sham/NPE) = 7, n(Sham/PE) = 7, n(SDA/NPE) = 6, and n(SDA/PE) = 7. D, Sucrose intake (in ml; mean ± SEM) during the PE phase in SDA and Sham PE animals. n(Sham/PE) = 7, and n(SDA/PE) = 7. E, Total fluid intake (mean ± SEM) during the initial PE day (NPE = water, PE = sucrose solution), conditioning (Cond) day (all animals: sucrose solution), and test day (all animals: water and sucrose solution). n(Sham/NPE) = 7, n(Sham/PE) = 7, n(SDA/NPE) = 6, and n(SDA/PE) = 7.
In a next step, we explored the effects of SDA on attentional control of associative learning using the paradigm of LI in CTA (Russig et al., 2003; Meyer et al., 2004). In this paradigm, LI reflects intact attentional control over associative learning and is manifest when NPE animals show reduced intake of sucrose solution (= CS) after CS-US conditioning compared with animals that were previously exposed to the CS without negative consequences (= PE animals; Weiner and Arad, 2009). This LI effect was clearly present in Sham rats, but was fully abolished by SDA (Fig. 4C). ANOVA of percentage sucrose intake during the test day revealed a significant interaction between lesion and PE condition (F(1,23) = 4.51, p < 0.05), and subsequent analyses restricted to Sham or SDA rats confirmed the presence of a significant PE effect in the former (F(1,12) = 13.15, p < 0.01) but not in the latter (F(1,11) = 0.30, p = 0.60). Restricted analyses to NPE or PE animals further showed that the disruption of LI by SDA was attributable to selective effects in PE animals, demonstrating that SDA modulates the attentional control of associative learning without affecting associative learning per se. Hence, although NPE Sham and SDA rats did not differ (F(1,11) = 0.01, p = 0.95), percentage sucrose intake during the test day was significantly (F(1,12) = 6.37, p < 0.05) reduced in PE SDA rats compared with PE Sham rats (Fig. 4C). As summarized in Fig. 4D, SDA and Sham rats assigned to the PE condition did not differ in sucrose intake (milliliters) during PE (t(12) = 0.99, p = 0.34). There were also no group differences with regards to total fluid (sucrose solution or water) intake during the initial PE and conditioning (sucrose solution) phases, or during the subsequent test (sucrose solution and water) phase (Fig. 4E). These results show that the LI-disrupting effect of SDA is not associated with changes in sucrose or general fluid intake.
SDA increases the sensitivity to amphetamine
In view of the findings of increased Amph sensitivity in schizophrenia (Laruelle, 2000), we compared the drug's behavioral effects in SDA and Sham rats. First, we evaluated the animals' locomotor reactions to Sal administration to control for possible differences in basal activity. The two groups did not differ in terms of the distance traveled after Sal administration, neither when it preceded the administration of the low (F(1,12) = 0.09, p = 0.77; Fig. 5A) or high (F(1,14) = 0.01, p = 0.94; Fig. 5B) dose of Amph. As expected (Mueller et al., 1989a,b), acute Amph administration increased forward locomotor activity (Fig. 5A,B). The locomotor-enhancing effects of Amph were influenced by dose, as indicated by the significant main effect of dose (F(1,26) = 9.16, p < 0.01) and its interaction with bins (F(5,130) = 2.30, p < 0.05). When analyzing the lower (1 mg/kg) dose of Amph, we found that SDA rats displayed a significant increase in drug-induced locomotor activity compared with Sham controls (main effect of lesion: F(1,12) = 5.16, p = 0.04; Fig. 5A). There was, however, no significant group difference when analyzing the distance moved after administration of the higher (2 mg/kg) dose of Amph (main effect of lesion: F(1,14) = 0.05, p = 0.83; Fig. 5B). These data thus show that SDA potentiates Amph-induced hyperactivity when using a low but not high dose of the psychostimulant drug. The lack of group differences when using the higher dose of Amph may be related to ceiling effects in forward locomotion. In support of this notion, we revealed a dose-dependent effect in Amph-induced hyperactivity for Sham controls (main effect of amphetamine dose: F(1,11) = 10.43, p < 0.01) but not in SDA animals (main effect of amphetamine dose: F(1,15) = 1.91, p = 0.19).
Figure 5.

Increased amphetamine sensitivity in SDA rats. A, The line plot depicts the distance moved (in cm; mean ± SEM) in an open field as a function of 10 min bins in response to initial Sal administration and subsequent Amph treatment at 1 mg/kg (i.p.). The bar plot shows the total distance moved (in cm; mean ± SEM) during the entire (60 min) period after Amph (1 mg/kg) treatment. *p < 0.05. n(Sham) = 7 and n(SDA) = 7. B, The line plot depicts the distance moved (in cm; mean ± SEM) in an open field as a function of 10 min bins in response to initial Sal administration and subsequent Amph treatment at 2 mg/kg (i.p.). The bar plot shows the total distance moved (in cm; mean ± SEM) during the entire (60 min) period after Amph (2 mg/kg) treatment. n(Sham) = 6 and n(SDA) = 10. C, The line plot depicts the LN-transformed stereotypy scores (mean ± SEM) in an open field as a function of 10 min bins in response to initial Sal administration and subsequent Amph treatment at 2 mg/kg (i.p.). The stereotypy scores encompass cumulative head waving and circling movements. The bar plot shows the total LN-transformed stereotypy scores (% Sal baseline; mean ± SEM) during the entire (60 min) period after Amph (2 mg/kg) treatment. *p < 0.05, §p = 0.05. n(Sham) = 6 and n(SDA) = 10.
In addition to its effects on forward locomotor activity, Amph treatment at high doses (>1 mg/kg in rats) can induce considerable behavioral stereotypies such as repetitive head waving and circling movements (Mueller et al., 1989a,b). Therefore, we analyzed such repetitive behaviors following treatment with the high dose (2 mg/kg) of Amph. As shown in Fig. 5C, the stereotypy scores in SDA animals increased with time after Amph treatment, whereas those measured for Sham controls decreased as a function of time, leading to a significant interaction between lesion and 10 min bins (F(5,70) = 2.73, p < 0.03). Subsequent post hoc tests at individual bins revealed significantly increased stereotypy scores in SDA animals relative to Sham controls during the last 20 min of the test (bin 7: t(14) = 4.42, p < 0.05; bin 8: t(14) = 4.91, p < 0.05; Fig. 5C).
Together, these findings demonstrate increased sensitivity of SDA animals to the psychostimulant drug Amph. Depending on the dose, the SDA-induced potentiation of Amph sensitivity can manifest itself as greater Amph-induced locomotor hyperactivity or induction of behavioral stereotypies.
SDA changes dopamine neurochemistry
In a last step, we investigated the effects of SDA on basal dopamine neurochemistry by measuring DA and its major metabolite, DOPAC, in postmortem NAc and PFC samples using HPLC. As expected (Winter et al., 2009), DA and DOPAC contents in the NAc were generally higher than those in the PFC (Fig. 6A,B), as supported by the main effect of brain area (DA: F(1,19) = 209.01, p < 0.001; DOPAC: F(1,19) = 190.10, p < 0.001). SDA rats showed increased DA and DOPAC contents specifically in the NAc, as indicated by the presence of a significant interaction between lesion and brain area (DA: F(1,19) = 4.91, p < 0.05; DOPAC: F(1,19) = 12.22, p < 0.01) and by the subsequent post hoc tests revealing a significant elevation of DA (p < 0.05) and DOPAC (p < 0.01) in the NAc of SDA relative to Sham animals (Fig. 6A,B). No significant group differences were detected with regards to DA and DOPAC contents in the PFC (DA: p = 0.11; DOPAC: p = 0.42).
Figure 6.

Altered postmortem dopamine neurochemistry in SDA rats. A, DA contents (nanomoles per milligram of protein; mean ± SEM) in the NAc and PFC of Sham and SDA rats. *p < 0.05, ***p < 0.001. n(Sham) = 10 and n(SDA) = 11. B, DOPAC contents (nanomoles per milligram of protein; mean ± SEM) in the NAc and PFC of Sham and SDA rats. **p < 0.01, ***p < 0.001. n(Sham) = 10 and n(SDA) = 11. C, DOPAC–DA ratio (mean ± SEM) as an index of DA turnover in the NAc and PFC of Sham and SDA rats. ***p < 0.001, §p < 0.05, reflecting the significant main effect of lesion. n(Sham) = 10 and n(SDA) = 11.
The analysis of the DOPAC/DA ratio, which was used to index DA turnover, revealed significant main effects of brain area (F(1,19) = 61.11, p < 0.001) and lesion (F(1,19) = 4.87, p < 0.05). The interaction between the two factors did not attain statistical significance (F(1,19) = 1.81, p = 0.18). Hence, as shown in Fig. 6C, DA turnover was generally higher in the PFC compared with NAc, and in SDA animals relative to Sham controls.
Discussion
The present study demonstrates that abdominal vagal afferents modulate the brain transcriptome, neurochemistry, and behaviors relevant to schizophrenia and related psychotic disorders. Using the most complete and selective vagal deafferentation model, we identified transcriptional changes in functional networks annotating with schizophrenia. Furthermore, disconnection of abdominal vagal afferents was associated with a hyperdopaminergic state, which manifested itself as increased sensitivity to the DA-releasing drug Amph and increased accumbal levels of DA and DOPAC. Our study further shows that the disruption of abdominal vagal afferents by SDA impairs sensorimotor gating and the attentional control of associative learning, which were assessed using the paradigms of PPI and LI, respectively. Increased Amph sensitivity and deficits in PPI and (to a lesser extent) LI are hallmarks of neurobehavioral alterations in schizophrenia and related disorders (Laruelle, 2000, Braff et al., 2001; Lubow, 2005; Swerdlow et al., 2008; Weiner and Arad, 2009) and have been widely described in a number of animal models that are relevant to these disorders (Meyer and Feldon, 2010; Peleg-Raibstein et al., 2012).
Influence of vagal afferents on the brain transcriptome
Our study represents the first unbiased approach toward the identification of genome-wide transcriptional abnormalities in a model of disrupted afferent vagal signaling. We found that SDA led to quantitatively more transcriptional changes in the NAc compared with the PFC, and only a few DEGs were common to both brain areas. Furthermore, whereas the SDA-induced transcriptional changes in the NAc were annotated with the functional entity “schizophrenia”, the entity most strongly affected by SDA in the PFC was associated with the term “degeneration of the nervous system”. It thus appears that the transcriptional alterations emerging in the NAc of SDA animals may be more directly related to schizophrenia than those emerging in the PFC. It should be noted, however, that some of the SDA-induced prefrontal changes are clearly relevant for schizophrenia as well. For example, we found that SDA caused an upregulation of Ifitm3, which is a viral restriction factor that inhibits processes involved in viral entry and replication (Bailey et al., 2014). Consistent with our findings, the expression of Ifitm3 is upregulated in the PFC of patients with schizophrenia (Arion et al., 2007; Saetre et al., 2007; Volk et al., 2015). SDA rats also showed a downregulated expression of the myelination-related gene, Mag, and the receptor tyrosine-protein kinase, Erbb3, which is consistent with the findings of reduced cortical expression of these genes in patients with schizophrenia (Aston et al., 2005; McCullumsmith et al., 2007).
Altered dopamine functions in relation to schizophrenia and SDA-induced behavioral dysfunctions
Increased striatal DA release is thought to be a core pathophysiological mechanism in schizophrenia and related psychotic disorders (Howes and Kapur, 2009). Historically, dopaminergic imbalances in the NAc and allied mesolimbic pathway have been considered a key region for DA dysfunctions in schizophrenia (O'Donnell and Grace, 1998). However, recent neuroimaging studies point to a more prominent role of the dorsal (caudate–putamen) rather than the ventral (NAc) striatum in this context (Kegeles et al., 2010; Egerton et al., 2013). Hence, although focusing on mesolimbic DA abnormalities may be an oversimplification in view of these recent findings, it does not make the NAc irrelevant (Chuhma et al., 2017). On speculative grounds, DA abnormalities in the NAc may contribute to some of the schizophrenia-related behavioral dysfunctions emerging in SDA rats. Indeed, increased DA signaling in the NAc and allied mesolimbic circuitries has been repeatedly associated with the emergence of PPI deficits (Swerdlow et al., 1990, 1992; Wan et al., 1995), loss of LI (Solomon and Staton, 1982; Weiner et al., 1996; Murphy et al., 2000), and increased sensitivity to Amph (Creese and Iversen, 1975; Pijnenburg et al., 1976; Heidbreder and Feldon, 1998). Hence, increased dopaminergic activity within mesolimbic structures seems to be a common neural mechanism underlying the disruption of attentional control of associative learning, sensorimotor gating, and Amph hypersensitivity. It should be noted, however, that dorsal parts of the striatum have been implicated in the modulation of PPI as well (Baldan Ramsey et al., 2011), suggesting that (DA-related) neuronal mechanisms beyond those existing in the NAc could contribute to sensorimotor gating deficits in SDA rats (De Koning et al., 2014). Additional studies are thus warranted to ascertain causal relationships between the changes in DA functions and schizophrenia-relevant behaviors in the SDA model.
Additional research will also be required to delineate the neuronal routes by which SDA can induce dopaminergic changes in the NAc. The primary target region of vagal afferents is the NTS, from where visceral signals are further conveyed to other brainstem nuclei (e.g., locus ceruleus) and the forebrain (Berthoud and Neuhuber, 2000). Hence, the dopaminergic changes emerging in the NAc of SDA rats likely represent secondary neurochemical responses to primary changes in upstream neuronal signaling pathways that are directly targeted by vagal afferents. A key neuronal route by which SDA increases DA in the NAc may involve the monosynaptic projections from the NTS to the ventral tegmental area (VTA; Zheng et al., 2015), the latter of which contains the majority of DA cells projecting to the NAc (Björklund and Dunnett, 2007). Hypothetically, SDA may thus attenuate signaling between the NTS and VTA, thereby altering the firing rate of VTA DA neurons (Manta et al., 2013) and eventually increasing DA outputs in the NAc.
Possible role of vagal afferents in schizophrenia
Although vagal efferents have long been a focus of interest in schizophrenia (Bär et al., 2010; Bär, 2015; Montaquila et al., 2015; Clamor et al., 2016), the role of vagal afferents have received less attention in this context. Only a few studies have thus far explored the possible involvement of vagal afferents in schizophrenia. Using a neurodevelopmental disruption model that is based on prenatal methylazoxymethanol acetate (MAM) exposure in rats, Perez et al. (2014) recently found that chronic (2 week) vagus nerve stimulation (VNS) normalized MAM-induced increases in DA neuron firing and Amph hypersensitivity. This study thus showed that activation of vagal afferents by VNS could attenuate schizophrenia-related DA hyperactivity (Perez et al., 2014), which is congruent with our findings showing associations between disrupted vagal afferent signaling by SDA and signs of hyperdopaminergia in the ventral striatum. Collectively, the preclinical data suggest that visceral signals mediated via vagal afferents can influence mesolimbic DA signaling and DA-driven behaviors associated with schizophrenia and related psychotic disorders. Thus far, however, only one clinical study examined the effects of VNS in patients with chronic schizophrenia (Hasan et al., 2015). In this study, application of VNS was well tolerated, but did not improve schizophrenia symptoms in a 26 week trial (Hasan et al., 2015). It should be noted, however, that patients receiving VNS displayed relatively mild psychopathology at baseline, which might have masked potential beneficial effects of the neurostimulatory procedure (Hasan et al., 2015). Additional studies examining the possible benefits of VNS in schizophrenia are thus clearly warranted, especially in view of the promising preclinical findings reported by Perez et al. (2014) using the MAM model of schizophrenia.
Limitations
A notable limitation of our study is that we examined the effects of SDA in male rats only. Hence, we do not know at this stage whether the disruption of vagal afferents induces similar transcriptional, neurochemical and behavioral effects in males and females. An extension of our investigations to the female sex appears crucial in view of the noticeable sex differences in schizophrenia (Flor-Henry, 1990; Castle et al., 1998; Roy et al., 2001; Aleman et al., 2003). Another limitation relates to the difficulties in translating the preclinical LI data to human subjects in general, and to cases with schizophrenia in particular. Not only are reports of disrupted LI equivocal, but also are there various (and sometimes controversial) theoretical accounts of LI in both animals and in humans (Swerdlow et al., 1996, 2005; Escobar et al., 2002; Weiner, 2003; Gray and Snowden, 2005). Hence, an extension to other behavioral and cognitive tests measuring attentional functions (Young et al., 2009) is warranted to further corroborate our findings.
Conclusion
Our findings suggest the disruption of afferent visceral signaling may contribute to schizophrenia-related abnormalities by modifying the brain transcriptome, neurochemistry, sensorimotor gating, and attention-related neurocognitive processes. An emerging question that awaits exploration is whether the behavioral, neurochemical, and transcriptional consequences of SDA may reflect a failure of the organism to convey gut-derived signals form the viscera to the CNS (Cryan and Dinan, 2012). This hypothesis is particularly intriguing in view of the recent findings demonstrating that germ-free mice lacking commensal gut-associated bacteria show several transcriptional alterations in the PFC that are similar to those revealed in SDA animals, including altered expression of Ifit3, Mag, and NR4A1 (Hoban et al., 2016). Future investigations of the possible links between SDA and gut-derived signals may thus help to gain more in-depth insights into the intricate mechanisms whereby visceral signals can influence complex brain and behavioral functions relevant to schizophrenia and beyond.
Footnotes
This work was supported by ETH Zurich, Switzerland (ETH Research Grant ETH-25_13-2) awarded to U.M. and W.L., with additional financial support from the Swiss National Science Foundation (Grant 310030_169544) awarded to U.M. We thank Weihong Qi and Jelena Kühn-Georgijevic from the Functional Genomics Center Zurich, Switzerland, for their technical assistance in RNA sequencing.
The authors declare no competing financial interests.
References
- Abrams CK, Scherer SS (2012) Gap junctions in inherited human disorders of the central nervous system. Biochim Biophys Acta 1818:2030–2047. 10.1016/j.bbamem.2011.08.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aleman A, Kahn RS, Selten JP (2003) Sex differences in the risk of schizophrenia: evidence from meta-analysis. Arch Gen Psychiatry 60:565–571. 10.1001/archpsyc.60.6.565 [DOI] [PubMed] [Google Scholar]
- Arion D, Unger T, Lewis DA, Levitt P, Mirnics K (2007) Molecular evidence for increased expression of genes related to immune and chaperone function in the prefrontal cortex in schizophrenia. Biol Psychiatry 62:711–721. 10.1016/j.biopsych.2006.12.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnold M, Mura A, Langhans W, Geary N (2006) Gut vagal afferents are not necessary for the eating-stimulatory effect of intraperitoneally injected ghrelin in the rat. J Neurosci 26:11052–11060. 10.1523/JNEUROSCI.2606-06.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aston C, Jiang L, Sokolov BP (2005) Transcriptional profiling reveals evidence for signaling and oligodendroglial abnormalities in the temporal cortex from patients with major depressive disorder. Mol Psychiatry 10:309–322. 10.1038/sj.mp.4001565 [DOI] [PubMed] [Google Scholar]
- Azari EK, Ramachandran D, Weibel S, Arnold M, Romano A, Gaetani S, Langhans W, Mansouri A (2014) Vagal afferents are not necessary for the satiety effect of the gut lipid messenger oleoylethanolamide. Am J Physiol Regul Integr Comp Physiol 307:R167–R178. 10.1152/ajpregu.00067.2014 [DOI] [PubMed] [Google Scholar]
- Bailey CC, Zhong G, Huang IC, Farzan M (2014) IFITM-family proteins: the cell's first line of antiviral defense. Annu Rev Virol 1:261–283. 10.1146/annurev-virology-031413-085537 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baldan Ramsey LC, Xu M, Wood N, Pittenger C (2011) Lesions of the dorsomedial striatum disrupt prepulse inhibition. Neuroscience 180:222–228. 10.1016/j.neuroscience.2011.01.041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bär KJ. (2015) Cardiac autonomic dysfunction in patients with schizophrenia and their healthy relatives: a small review. Front Neurol 6:139. 10.3389/fneur.2015.00139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bär KJ, Berger S, Metzner M, Boettger MK, Schulz S, Ramachandraiah CT, Terhaar J, Voss A, Yeragani VK, Sauer H (2010) Autonomic dysfunction in unaffected first-degree relatives of patients suffering from schizophrenia. Schizophr Bull 36:1050–1058. 10.1093/schbul/sbp024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bercik P, Park AJ, Sinclair D, Khoshdel A, Lu J, Huang X, Deng Y, Blennerhassett PA, Fahnestock M, Moine D, Berger B, Huizinga JD, Kunze W, McLean PG, Bergonzelli GE, Collins SM, Verdu EF (2011) The anxiolytic effect of bifidobacterium longum NCC3001 involves vagal pathways for gut-brain communication. Neurogastroenterol Motil 23:1132–1139. 10.1111/j.1365-2982.2011.01796.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berthoud HR, Neuhuber WL (2000) Functional and chemical anatomy of the afferent vagal system. Auton Neurosci 85:1–17. 10.1016/S1566-0702(00)00215-0 [DOI] [PubMed] [Google Scholar]
- Björklund A, Dunnett SB (2007) Dopamine neuron systems in the brain: an update. Trends Neurosci 30:194–202. 10.1016/j.tins.2007.03.006 [DOI] [PubMed] [Google Scholar]
- Braff DL, Geyer MA, Swerdlow NR (2001) Human studies of prepulse inhibition of startle: normal subjects, patient groups, and pharmacological studies. Psychopharmacology (Berl) 156:234–258. 10.1007/s002130100810 [DOI] [PubMed] [Google Scholar]
- Bravo JA, Forsythe P, Chew MV, Escaravage E, Savignac HM, Dinan TG, Bienenstock J, Cryan JF (2011) Ingestion of lactobacillus strain regulates emotional behavior and central GABA receptor expression in a mouse via the vagus nerve. Proc Natl Acad Sci U S A 108:16050–16055. 10.1073/pnas.1102999108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castle D, Sham P, Murray R (1998) Differences in distribution of ages of onset in males and females with schizophrenia. Schizophr Res 33:179–183. 10.1016/S0920-9964(98)00070-X [DOI] [PubMed] [Google Scholar]
- Chuhma N, Mingote S, Kalmbach A, Yetnikoff L, Rayport S (2017) Heterogeneity in dopamine neuron synaptic actions across the striatum and its relevance for schizophrenia. Biol Psychiatry 81:43–51. 10.1016/j.biopsych.2016.07.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clamor A, Lincoln TM, Thayer JF, Koenig J (2016) Resting vagal activity in schizophrenia: meta-analysis of heart rate variability as a potential endophenotype. Br J Psychiatry 208:9–16. 10.1192/bjp.bp.114.160762 [DOI] [PubMed] [Google Scholar]
- Creese I, Iversen SD (1975) The pharmacological and anatomical substrates of the amphetamine response in the rat. Brain Res 83:419–436. 10.1016/0006-8993(75)90834-3 [DOI] [PubMed] [Google Scholar]
- Cryan JF, Dinan TG (2012) Mind-altering microorganisms: the impact of the gut microbiota on brain and behaviour. Nat Rev Neurosci 13:701–712. 10.1038/nrn3346 [DOI] [PubMed] [Google Scholar]
- De Koning MB, Bloemen OJ, Van Duin ED, Booij J, Abel KM, De Haan L, Linszen DH, Van Amelsvoort TA (2014) Pre-pulse inhibition and striatal dopamine in subjects at an ultra-high risk for psychosis. J Psychopharmacol 28:553–560. 10.1177/0269881113519507 [DOI] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, Gingeras TR (2013) STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29:15–21. 10.1093/bioinformatics/bts635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eastwood SL, Law AJ, Everall IP, Harrison PJ (2003) The axonal chemorepellant semaphorin 3A is increased in the cerebellum in schizophrenia and may contribute to its synaptic pathology. Mol Psychiatry 8:148–155. 10.1038/sj.mp.4001233 [DOI] [PubMed] [Google Scholar]
- Egan MF, Straub RE, Goldberg TE, Yakub I, Callicott JH, Hariri AR, Mattay VS, Bertolino A, Hyde TM, Shannon-Weickert C, Akil M, Crook J, Vakkalanka RK, Balkissoon R, Gibbs RA, Kleinman JE, Weinberger DR (2004) Variation in GRM3 affects cognition, prefrontal glutamate, and risk for schizophrenia. Proc Natl Acad Sci U S A 101:12604–12609. 10.1073/pnas.0405077101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egerton A, Chaddock CA, Winton-Brown TT, Bloomfield MA, Bhattacharyya S, Allen P, McGuire PK, Howes OD (2013) Presynaptic striatal dopamine dysfunction in people at ultra-high risk for psychosis: findings in a second cohort. Biol Psychiatry 74:106–112. 10.1016/j.biopsych.2012.11.017 [DOI] [PubMed] [Google Scholar]
- Escobar M, Oberling P, Miller RR (2002) Associative deficit accounts of disrupted latent inhibition and blocking in schizophrenia. Neurosci Biobehav Rev 26:203–216. 10.1016/S0149-7634(01)00067-7 [DOI] [PubMed] [Google Scholar]
- Flor-Henry P. (1990) Influence of gender in schizophrenia as related to other psychopathological syndromes. Schizophr Bull 16:211–227. 10.1093/schbul/16.2.211 [DOI] [PubMed] [Google Scholar]
- Fung SJ, Webster MJ, Sivagnanasundaram S, Duncan C, Elashoff M, Weickert CS (2010) Expression of interneuron markers in the dorsolateral prefrontal cortex of the developing human and in schizophrenia. Am J Psychiatry 167:1479–1488. 10.1176/appi.ajp.2010.09060784 [DOI] [PubMed] [Google Scholar]
- Gray NS, Snowden RJ (2005) The relevance of irrelevance to schizophrenia. Neurosci Biobehav Rev 29:989–999. 10.1016/j.neubiorev.2005.01.006 [DOI] [PubMed] [Google Scholar]
- Hasan A, Wolff-Menzler C, Pfeiffer S, Falkai P, Weidinger E, Jobst A, Hoell I, Malchow B, Yeganeh-Doost P, Strube W, Quast S, Müller N, Wobrock T (2015) Transcutaneous noninvasive vagus nerve stimulation (tVNS) in the treatment of schizophrenia: a bicentric randomized controlled pilot study. Eur Arch Psychiatry Clin Neurosci 265:589–600. 10.1007/s00406-015-0618-9 [DOI] [PubMed] [Google Scholar]
- Hatakeyama M, Opitz L, Russo G, Qi W, Schlapbach R, Rehrauer H (2016) SUSHI: an exquisite recipe for fully documented, reproducible and reusable NGS data analysis. BMC Bioinformatics 17:228. 10.1186/s12859-016-1104-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heidbreder C, Feldon J (1998) Amphetamine-induced neurochemical and locomotor responses are expressed differentially across the anteroposterior axis of the core and shell subterritories of the nucleus accumbens. Synapse 29:310–322. [DOI] [PubMed] [Google Scholar]
- Hoban AE, Stilling RM, Ryan FJ, Shanahan F, Dinan TG, Claesson MJ, Clarke G, Cryan JF (2016) Regulation of prefrontal cortex myelination by the microbiota. Transl Psychiatry 6:e774. 10.1038/tp.2016.42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howes OD, Kapur S (2009) The dopamine hypothesis of schizophrenia: version III: the final common pathway. Schizophr Bull 35:549–562. 10.1093/schbul/sbp006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Insel TR. (2010) Rethinking schizophrenia. Nature 468:187–193. 10.1038/nature09552 [DOI] [PubMed] [Google Scholar]
- Kegeles LS, Slifstein M, Xu X, Urban N, Thompson JL, Moadel T, Harkavy-Friedman JM, Gil R, Laruelle M, Abi-Dargham A (2010) Striatal and extrastriatal dopamine D2/D3 receptors in schizophrenia evaluated with [18F]fallypride positron emission tomography. Biol Psychiatry 68:634–641. 10.1016/j.biopsych.2010.05.027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khalsa SS, Rudrauf D, Feinstein JS, Tranel D (2009) The pathways of interoceptive awareness. Nat Neurosci 12:1494–1496. 10.1038/nn.2411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirkpatrick B. (2009) Schizophrenia as a systemic disease. Schizophr Bull 35:381–382. 10.1093/schbul/sbn183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klarer M, Arnold M, Günther L, Winter C, Langhans W, Meyer U (2014) Gut vagal afferents differentially modulate innate anxiety and learned fear. J Neurosci 34:7067–7076. 10.1523/JNEUROSCI.0252-14.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klarer M, Weber-Stadlbauer U, Arnold M, Langhans W, Meyer U (2017) Cognitive effects of subdiaphragmatic vagal deafferentation in rats. Neurobiol Learn Mem 142:190–199. 10.1016/j.nlm.2017.05.006 [DOI] [PubMed] [Google Scholar]
- Kraly FS, Jerome C, Smith GP (1986) Specific postoperative syndromes after total and selective vagotomies in the rat. Appetite 7:1–17. 10.1016/S0195-6663(86)80038-1 [DOI] [PubMed] [Google Scholar]
- Labouesse MA, Stadlbauer U, Weber E, Arnold M, Langhans W, Pacheco-López G (2012) Vagal afferents mediate early satiation and prevent flavour avoidance learning in response to intraperitoneally infused exendin-4. J Neuroendocrinol 24:1505–1516. 10.1111/j.1365-2826.2012.02364.x [DOI] [PubMed] [Google Scholar]
- Landek-Salgado MA, Faust TE, Sawa A (2016) Molecular substrates of schizophrenia: homeostatic signaling to connectivity. Mol Psychiatry 21:10–28. 10.1038/mp.2015.141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laruelle M. (2000) The role of endogenous sensitization in the pathophysiology of schizophrenia: implications from recent brain imaging studies. Brain Res Rev 31:371–384. 10.1016/S0165-0173(99)00054-5 [DOI] [PubMed] [Google Scholar]
- Liao Y, Smyth GK, Shi W (2013) The subread aligner: fast, accurate and scalable read mapping by seed-and-vote. Nucleic Acids Res 41:e108. 10.1093/nar/gkt214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Livak KJ, Schmittgen TD (2001) Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods 25:402–408. 10.1006/meth.2001.1262 [DOI] [PubMed] [Google Scholar]
- Llorens F, Gil V, del Río JA (2011) Emerging functions of myelin-associated proteins during development, neuronal plasticity, and neurodegeneration. FASEB J 25:463–475. 10.1096/fj.10-162792 [DOI] [PubMed] [Google Scholar]
- Love MI, Huber W, Anders S (2014) Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15:550. 10.1186/s13059-014-0550-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lubow RE. (2005) Construct validity of the animal latent inhibition model of selective attention deficits in schizophrenia. Schizophr Bull 31:139–153. 10.1093/schbul/sbi005 [DOI] [PubMed] [Google Scholar]
- Manta S, El Mansari M, Debonnel G, Blier P (2013) Electrophysiological and neurochemical effects of long-term vagus nerve stimulation on the rat monoaminergic systems. Int J Neuropsychopharmacol 16:459–470. 10.1017/S1461145712000387 [DOI] [PubMed] [Google Scholar]
- McCullumsmith RE, Gupta D, Beneyto M, Kreger E, Haroutunian V, Davis KL, Meador-Woodruff JH (2007) Expression of transcripts for myelination-related genes in the anterior cingulate cortex in schizophrenia. Schizophr Res 90:15–27. 10.1016/j.schres.2006.11.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer U, Feldon J (2010) Epidemiology-driven neurodevelopmental animal models of schizophrenia. Prog Neurobiol 90:285–326. 10.1016/j.pneurobio.2009.10.018 [DOI] [PubMed] [Google Scholar]
- Meyer U, Chang DL, Feldon J, Yee BK (2004) Expression of the CS- and US-pre-exposure effects in the conditioned taste aversion paradigm and their abolition following systemic amphetamine treatment in C57BL/6J mice. Neuropsychopharmacology 29:2140–2148. 10.1038/sj.npp.1300522 [DOI] [PubMed] [Google Scholar]
- Meyer U, Feldon J, Schedlowski M, Yee BK (2005) Towards an immuno-precipitated neurodevelopmental animal model of schizophrenia. Neurosci Biobehav Rev 29:913–947. 10.1016/j.neubiorev.2004.10.012 [DOI] [PubMed] [Google Scholar]
- Meyer U, Schwendener S, Feldon J, Yee BK (2006) Prenatal and postnatal maternal contributions in the infection model of schizophrenia. Exp Brain Res 173:243–257. 10.1007/s00221-006-0419-5 [DOI] [PubMed] [Google Scholar]
- Montaquila JM, Trachik BJ, Bedwell JS (2015) Heart rate variability and vagal tone in schizophrenia: a review. J Psychiatr Res 69:57–66. 10.1016/j.jpsychires.2015.07.025 [DOI] [PubMed] [Google Scholar]
- Mueller K, Hollingsworth EM, Cross DR (1989a) Another look at amphetamine-induced stereotyped locomotor activity in rats using a new statistic to measure locomotor stereotypy. Psychopharmacology (Berl) 97:74–79. 10.1007/BF00443416 [DOI] [PubMed] [Google Scholar]
- Mueller K, Kunko PM, Whiteside D, Haskett C (1989b) Time course of amphetamine-induced locomotor stereotypy in an open field. Psychopharmacology (Berl) 99:501–507. 10.1007/BF00589899 [DOI] [PubMed] [Google Scholar]
- Murphy CA, Pezze M, Feldon J, Heidbreder C (2000) Differential involvement of dopamine in the shell and core of the nucleus accumbens in the expression of latent inhibition to an aversively conditioned stimulus. Neuroscience 97:469–477. 10.1016/S0306-4522(00)00043-9 [DOI] [PubMed] [Google Scholar]
- Norgren R, Smith GP (1994) A method for selective section of vagal afferent or efferent axons in the rat. Am J Physiol 267:R1136–R1141. 10.1152/ajpregu.1994.267.4.R1136 [DOI] [PubMed] [Google Scholar]
- Notter T, Panzanelli P, Pfister S, Mircsof D, Fritschy JM (2014) A protocol for concurrent high-quality immunohistochemical and biochemical analyses in adult mouse central nervous system. Eur J Neurosci 39:165–175. 10.1111/ejn.12447 [DOI] [PubMed] [Google Scholar]
- O'Donnell P, Grace AA (1998) Dysfunctions in multiple interrelated systems as the neurobiological bases of schizophrenic symptom clusters. Schizophr Bull 24:267–283. 10.1093/oxfordjournals.schbul.a033325 [DOI] [PubMed] [Google Scholar]
- Owen MJ, Sawa A, Mortensen PB (2016) Schizophrenia. Lancet 388:86–97. 10.1016/S0140-6736(15)01121-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paxinos G, Watson C (1998) The rat brain in stereotaxic coordinates, Ed 4 San Diego, CA: Academic. [Google Scholar]
- Peleg-Raibstein D, Feldon J, Meyer U (2012) Behavioral animal models of antipsychotic drug actions. Handb Exp Pharmacol 212:361–406. 10.1007/978-3-642-25761-2_14 [DOI] [PubMed] [Google Scholar]
- Perez SM, Carreno FR, Frazer A, Lodge DJ (2014) Vagal nerve stimulation reverses aberrant dopamine system function in the methylazoxymethanol acetate rodent model of schizophrenia. J Neurosci 34:9261–9267. 10.1523/JNEUROSCI.0588-14.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pijnenburg AJ, Honig WM, Van der Heyden JA, Van Rossum JM (1976) Effects of chemical stimulation of the mesolimbic dopamine system upon locomotor activity. Eur J Pharmacol 35:45–58. 10.1016/0014-2999(76)90299-5 [DOI] [PubMed] [Google Scholar]
- Pothuizen HH, Jongen-Rêlo AL, Feldon J, Yee BK (2006) Latent inhibition of conditioned taste aversion is not disrupted, but can be enhanced, by selective nucleus accumbens shell lesions in rats. Neuroscience 137:1119–1130. 10.1016/j.neuroscience.2005.10.032 [DOI] [PubMed] [Google Scholar]
- Richetto J, Massart R, Weber-Stadlbauer U, Szyf M, Riva MA, Meyer U (2017) Genome-wide DNA methylation changes in a mouse model of infection-mediated neurodevelopmental disorders. Biol Psychiatry 81:265–276. 10.1016/j.biopsych.2016.08.010 [DOI] [PubMed] [Google Scholar]
- Robinson TE, Becker JB (1986) Enduring changes in brain and behavior produced by chronic amphetamine administration: a review and evaluation of animal models of amphetamine psychosis. Brain Res 396:157–198. 10.1016/0165-0173(86)90002-0 [DOI] [PubMed] [Google Scholar]
- Roy MA, Maziade M, Labbé A, Mérette C (2001) Male gender is associated with deficit schizophrenia: a meta-analysis. Schizophr Res 47:141–147. 10.1016/S0920-9964(99)00231-5 [DOI] [PubMed] [Google Scholar]
- Russig H, Kovacevic A, Murphy CA, Feldon J (2003) Haloperidol and clozapine antagonise amphetamine-induced disruption of latent inhibition of conditioned taste aversion. Psychopharmacology (Berl) 170:263–270. 10.1007/s00213-003-1544-5 [DOI] [PubMed] [Google Scholar]
- Saetre P, Emilsson L, Axelsson E, Kreuger J, Lindholm E, Jazin E (2007) Inflammation-related genes up-regulated in schizophrenia brains. BMC Psychiatry 7:46. 10.1186/1471-244X-7-46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selemon LD, Zecevic N (2015) Schizophrenia: a tale of two critical periods for prefrontal cortical development. Transl Psychiatry 5:e623. 10.1038/tp.2015.115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Severance EG, Yolken RH, Eaton WW (2016) Autoimmune diseases, gastrointestinal disorders and the microbiome in schizophrenia: more than a gut feeling. Schizophr Res 176:23–35. 10.1016/j.schres.2014.06.027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shibata H, Aramaki T, Sakai M, Ninomiya H, Tashiro N, Iwata N, Ozaki N, Fukumaki Y (2006) Association study of polymorphisms in the GluR7, KA1 and KA2 kainate receptor genes (GRIK3, GRIK4, GRIK5) with schizophrenia. Psychiatry Res 141:39–51. 10.1016/j.psychres.2005.07.015 [DOI] [PubMed] [Google Scholar]
- Sleipness EP, Jansen HT, Schenk JO, Sorg BA (2008) Time of day differences in dopamine clearance in the rat medial prefrontal cortex and nucleus accumbens. Synapse 62:877–885. 10.1002/syn.20552 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith GP, Gibbs J (1985) The satiety effect of cholecystokinin: recent progress and current problems. Ann N Y Acad Sci 448:417–423. 10.1111/j.1749-6632.1985.tb29936.x [DOI] [PubMed] [Google Scholar]
- Solomon PR, Staton DM (1982) Differential effects of microinjections of d-amphetamine into the nucleus accumbens or the caudate putamen on the rat's ability to ignore an irrelevant stimulus. Biol Psychiatry 17:743–756. [PubMed] [Google Scholar]
- Swerdlow NR, Braff DL, Geyer MA (1990) GABAergic projection from nucleus accumbens to ventral pallidum mediates dopamine-induced sensorimotor gating deficits of acoustic startle in rats. Brain Res 532:146–150. 10.1016/0006-8993(90)91754-5 [DOI] [PubMed] [Google Scholar]
- Swerdlow NR, Caine SB, Geyer MA (1992) Regionally selective effects of intracerebral dopamine infusion on sensorimotor gating of the startle reflex in rats. Psychopharmacology (Berl) 108:189–195. 10.1007/BF02245306 [DOI] [PubMed] [Google Scholar]
- Swerdlow NR, Braff DL, Hartston H, Perry W, Geyer MA (1996) Latent inhibition in schizophrenia. Schizophr Res 20:91–103. 10.1016/0920-9964(95)00097-6 [DOI] [PubMed] [Google Scholar]
- Swerdlow NR, Braff DL, Geyer MA (2000) Animal models of deficient sensorimotor gating: what we know, what we think we know, and what we hope to know soon. Behav Pharmacol 11:185–204. 10.1097/00008877-200006000-00002 [DOI] [PubMed] [Google Scholar]
- Swerdlow NR, Stephany N, Wasserman LC, Talledo J, Sharp R, Minassian A, Auerbach PP (2005) Intact visual latent inhibition in schizophrenia patients in a within-subject paradigm. Schizophr Res 72:169–183. 10.1016/j.schres.2004.03.022 [DOI] [PubMed] [Google Scholar]
- Swerdlow NR, Weber M, Qu Y, Light GA, Braff DL (2008) Realistic expectations of prepulse inhibition in translational models for schizophrenia research. Psychopharmacology (Berl) 199:331–388. 10.1007/s00213-008-1072-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tandon R, Nasrallah HA, Keshavan MS (2009) Schizophrenia, “just the facts”: 4. Clinical features and conceptualization. Schizophr Res 110:1–23. 10.1016/j.schres.2009.03.005 [DOI] [PubMed] [Google Scholar]
- Thomas S, Bonchev D (2010) A survey of current software for network analysis in molecular biology. Hum Genomics 4:353–360. 10.1186/1479-7364-4-5-353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volk DW, Chitrapu A, Edelson JR, Roman KM, Moroco AE, Lewis DA (2015) Molecular mechanisms and timing of cortical immune activation in schizophrenia. Am J Psychiatry 172:1112–1121. 10.1176/appi.ajp.2015.15010019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wan FJ, Geyer MA, Swerdlow NR (1995) Presynaptic dopamine-glutamate interactions in the nucleus accumbens regulate sensorimotor gating. Psychopharmacology (Berl) 120:433–441. 10.1007/BF02245815 [DOI] [PubMed] [Google Scholar]
- Weber-Stadlbauer U, Richetto J, Labouesse MA, Bohacek J, Mansuy IM, Meyer U (2017) Transgenerational transmission and modification of pathological traits induced by prenatal immune activation. Mol Psychiatry 22:102–112. 10.1038/mp.2016.41 [DOI] [PubMed] [Google Scholar]
- Weiner I. (2003) The “two-headed” latent inhibition model of schizophrenia: modeling positive and negative symptoms and their treatment. Psychopharmacology (Berl) 169:257–297. 10.1007/s00213-002-1313-x [DOI] [PubMed] [Google Scholar]
- Weiner I, Arad M (2009) Using the pharmacology of latent inhibition to model domains of pathology in schizophrenia and their treatment. Behav Brain Res 204:369–386. 10.1016/j.bbr.2009.05.004 [DOI] [PubMed] [Google Scholar]
- Weiner I, Gal G, Rawlins JN, Feldon J (1996) Differential involvement of the shell and core subterritories of the nucleus accumbens in latent inhibition and amphetamine-induced activity. Behav Brain Res 81:123–133. 10.1016/S0166-4328(96)00051-4 [DOI] [PubMed] [Google Scholar]
- Winter C, Djodari-Irani A, Sohr R, Morgenstern R, Feldon J, Juckel G, Meyer U (2009) Prenatal immune activation leads to multiple changes in basal neurotransmitter levels in the adult brain: implications for brain disorders of neurodevelopmental origin such as schizophrenia. Int J Neuropsychopharmacol 12:513–524. 10.1017/S1461145708009206 [DOI] [PubMed] [Google Scholar]
- Yee BK, Chang T, Pietropaolo S, Feldon J (2005) The expression of prepulse inhibition of the acoustic startle reflex as a function of three pulse stimulus intensities, three prepulse stimulus intensities, and three levels of startle responsiveness in C57BL/6J mice. Behav Brain Res 163:265–276. 10.1016/j.bbr.2005.05.013 [DOI] [PubMed] [Google Scholar]
- Yoon H, Radulovic M, Wu J, Blaber SI, Blaber M, Fehlings MG, Scarisbrick IA (2013) Kallikrein 6 signals through PAR1 and PAR2 to promote neuron injury and exacerbate glutamate neurotoxicity. J Neurochem 127:283–298. 10.1111/jnc.12293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young JW, Powell SB, Risbrough V, Marston HM, Geyer MA (2009) Using the MATRICS to guide development of a preclinical cognitive test battery for research in schizophrenia. Pharmacol Ther 122:150–202. 10.1016/j.pharmthera.2009.02.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang LF, Shi L, Liu H, Meng FT, Liu YJ, Wu HM, Du X, Zhou JN (2012) Increased hippocampal tau phosphorylation and axonal mitochondrial transport in a mouse model of chronic stress. Int J Neuropsychopharmacol 15:337–348. 10.1017/S1461145711000411 [DOI] [PubMed] [Google Scholar]
- Zheng H, Stornetta RL, Agassandian K, Rinaman L (2015) Glutamatergic phenotype of glucagon-like peptide 1 neurons in the caudal nucleus of the solitary tract in rats. Brain Struct Funct 220:3011–3022. 10.1007/s00429-014-0841-6 [DOI] [PMC free article] [PubMed] [Google Scholar]