

SUMMARY
A carbohydrate-restricted diet is a widely recommended intervention for non-alcoholic fatty liver disease (NAFLD), but a systematic perspective on the multiple benefits of this diet is lacking. Here, we performed a short-term intervention with an isocaloric low-carbohydrate diet with increased protein content in obese subjects with NAFLD and characterized the resulting alterations in metabolism and the gut microbiota using a multi-omics approach. We observed rapid and dramatic reductions of liver fat and other cardiometabolic risk factors paralleled by (1) marked decreases in hepatic de novo lipogenesis; (2) large increases in serum β-hydroxybutyrate concentrations, reflecting increased mitochondrial β-oxidation; and (3) rapid increases in folate-producing Streptococcus and serum folate concentrations. Liver transcriptomic analysis on biopsy samples from a second cohort revealed downregulation of the fatty acid synthesis pathway and upregulation of folate-mediated one-carbon metabolism and fatty acid oxidation pathways. Our results highlight the potential of exploring diet-microbiota interactions for treating NAFLD.
Graphical Abstract
In Brief
Mardinoglu et al. use multi-omics to investigate the effects of a carbohydrate-restricted diet in obese NAFLD patients. They show that the diet improves liver fat metabolism, promotes rapid shifts in the gut microbiota, increases circulating folate, and upregulates expression of genes involved in folate-dependent one-carbon metabolism in the liver.
INTRODUCTION
In the past 30 years, we have seen a marked increase in non-alcoholic fatty liver disease (NAFLD), and it is now the most common cause of chronic liver disease in western countries (Chalasani et al., 2012; Rinella and Sanyal, 2016). NAFLD can progress from simple steatosis to non-alcoholic steatohepatitis (NASH), which is characterized by the additional presence of an inflammatory infiltrate and hepatocellular injury with or without fibrosis (Chalasani et al., 2012; Rinella and Sanyal, 2016), and may further progress to cirrhosis, liver failure, and hepatocellular carcinoma (Marengo et al., 2016). Increasing evidence also indicates that NAFLD is a significant independent risk factor for cardiovascular disease and type 2 diabetes (Lonardo et al., 2015; Targher et al., 2016), and the dyslipidemia that is present in many individuals with NAFLD potentially contributes to the link between these diseases (Gaggini et al., 2013).
The pathophysiology of NAFLD has not been resolved, but it develops when the influx of lipids into the liver exceeds hepatic lipid disposal (by fatty acid oxidation and triglyceride secretion as lipoprotein particles) (Stefan et al., 2008). Potential sources of lipids contributing to fatty liver include fatty acids released into the circulation from peripheral adipose tissue, dietary fatty acids from intestinal chylomicrons, and lipids synthesized (mostly from carbohydrates) in the liver by de novo lipogenesis (DNL) (Donnelly et al., 2005). In hyperinsulinemic subjects with NAFLD, hepatic DNL accounts for approximately 25% of liver triglyceride content (Diraison et al., 2003; Donnelly et al., 2005); thus, carbohydrate restriction, combined with exercise and regular follow-up, has emerged as an effective dietary intervention for obesity (Astrup et al., 2004; Foster et al., 2003) and NAFLD (Rinella and Sanyal, 2016). In addition to their effects on liver fat, carbohydrate-restricted diets have been shown to promote marked shifts in the composition of the gut microbiota (David et al., 2014; Duncan et al., 2007). Furthermore, accumulating evidence suggests that microbial changes are implicated in the development and progression of NAFLD (Le Roy et al., 2013; Leung et al., 2016; Loomba et al., 2017), and fecal microbiota transplantation has been shown to be able to alleviate high-fat-induced steatohepatitis in mice (Zhou et al., 2017).
A systematic perspective integrating dietary intervention, microbial profiling, and in-depth metabolic characterizations in humans with NAFLD is lacking. Given the complexity of NAFLD pathogenesis, in-depth multi-omics profiling—an approach that has recently been used in studies of both human wellness and disease (Chen et al., 2012; Price et al., 2017; Wu et al., 2015)—would likely offer valuable insights into understanding how a carbohydrate-restricted diet promotes reduced hepatic steatosis. Here we performed a 2-week intervention with an isocaloric carbohydrate-restricted diet in obese subjects with NAFLD and used multi-omics profiling to investigate how the diet and associated changes in the gut microbiota contribute to improved liver fat metabolism. In addition, we combined plasma metabolomics and liver transcriptomics in a genomescale metabolic model to further investigate the metabolic responses to this diet intervention.
RESULTS AND DISCUSSION
Reduced Carbohydrate Consumption Has Rapid Effects on Liver Fat
Earlier studies designed to reduce hepatic steatosis in subjects with NAFLD have usually combined carbohydrate restriction with calorie reduction and did not separate out the effect of amplified weight loss (Browning et al., 2006, 2011). To investigate how liver fat metabolism is affected by reduced carbohydrate consumption without a concomitant reduction in calorie intake, we served a pre-prepared isocaloric low-carbohydrate diet with increased protein content (<30 g of carbohydrates and an average of 3,115 kcal per day; Figure 1A; Table S1) for 14 days to ten subjects with obesity and high liver fat (mean ± SEM 16.0% ± 2.3%). The study design is shown in Figure 1B. To minimize the weight loss that is known to occur on a short-term isocaloric carbohydrate-restricted diet (Kekwick and Pawan, 1956), the study subjects were in daily contact with a dietician and were instructed to increase their daily energy intake whenever their weight decreased between two study days by more than 0.2 kg.
Figure 1. Reduced Carbohydrate Consumption Improves Liver Lipid Metabolism and Reduces Inflammation in Obese Subjects with NAFLD.
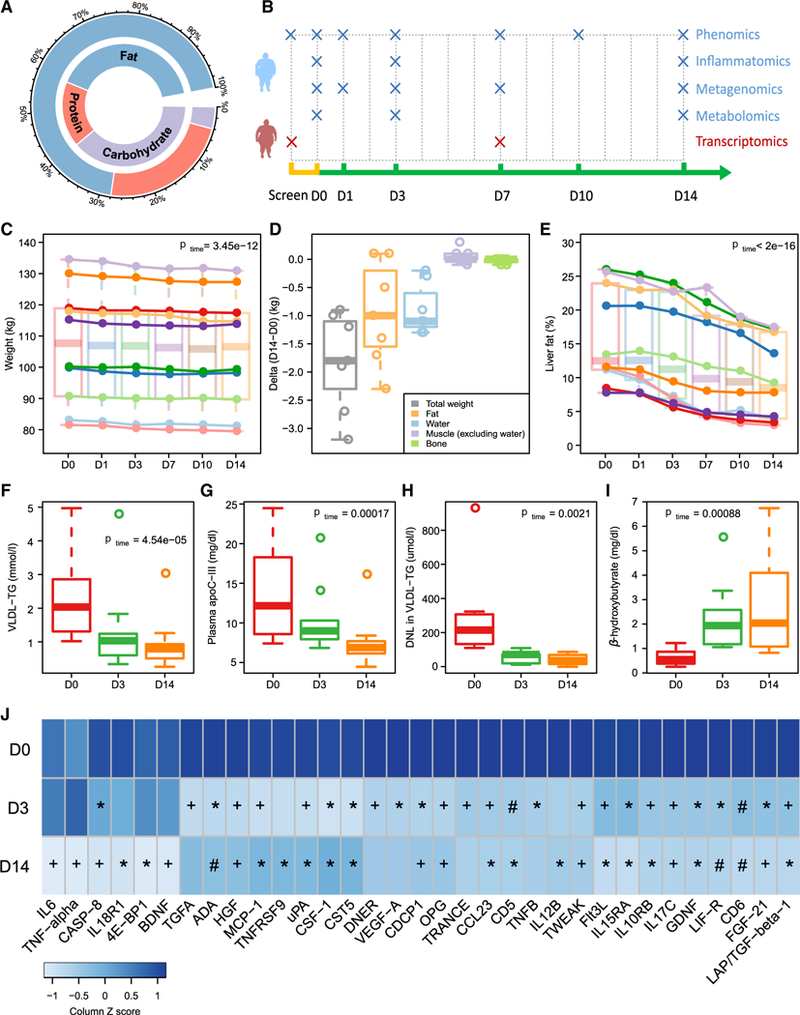
(A) Percentage of energy from carbohydrate, protein, and fat at baseline (inner circle) and during the 14-day dietary intervention (outer circle).
(B) Study design indicating time points used in the multi-omics analysis. D, day.
(C–E) Boxplots (with median) and individual data showing (C) weight changes across the study period, (D) changes in body composition between day 0 and day 14, and (E) liver fat changes across the study period.
(F–I) Boxplots (with median) showing (F) plasma concentrations of VLDL-triglycerides (TG) (n = 10), (G) plasma concentrations of apoC-III (n = 10), (H) DNL(n = 9), and (I) plasma concentrations of β-hydroxybutyrate (n = 10) at days 0, 3, and 14.
(J) Heatmap showing statistically significant reductions in inflammatory markers and FGF-21 over the study period (FDR < 0.05; n = 10). Post hoc group comparisons (D0 versus D3 and D0 versus D14) were performed by paired t test with Bonferroni correction. *p < 0.05; +p < 0.01; #p < 0.001. p or FDR values across time were obtained by one-way ANOVA with repeated measurements.
See also Figures S1 and S2 and Table S1.
There were no changes in waist circumference during the study (Table S1). Despite good compliance, we observed a slight weight loss (1.8% ± 0.2% of their body weight; Figure 1C). Body composition analysis at baseline and after 14 days on the diet revealed that decreases in fat mass and water were the major contributors to this minor weight loss (Figure 1D). In contrast to the small reduction in weight loss, we observed dramatic reductions in liver fat, as measured by magnetic resonance spectroscopy (MRS), in all the individuals over the 14-day study period (mean reduction 43.8%; Figure 1E). Of note, the reduction was significant just 1 day after the start of the diet intervention (p = 0.027) and was paralleled by a significant decrease in total liver volume (Figure S1).
We observed marked reductions in very-low-density lipoprotein (VLDL)-triglycerides (mean reduction 56.7%; Figure 1F) and in fasting plasma triglyceride concentrations (mean reduction 48.4%; Table S1) at the end of the study. In addition, we found a significant reduction of plasma apolipoprotein C-III (apoC-III) (Figure 1G), an inhibitor of VLDL clearance (Ginsberg et al., 1986) and a strong correlation between decreases in the concentration of apoC-III and VLDL-triglycerides over the study period (r = 0.91, p = 0.0015; Figure S2). We also analyzed the composition of VLDL-triglycerides and observed a decreased proportion of saturated fatty acids including myristic acid (14:0) and palmitic acid (16:0) and an increased proportion of unsaturated fatty acids such as oleic acid (18:1) (Table S1). These data are consistent with a significant increase in the number of double bonds per fatty acid chain observed by MRS of the liver over the study period (Table S1).
To test whether the marked reductions in liver fat were linked to the diet intervention, we performed a follow-up MRS in seven of the ten participants 1–3 months after completing the intervention study and returning to their normal diet. We observed that their liver fat content returned to a level similar to that measured before the diet intervention (11.3% ± 1.6% at follow-up versus 13.8% ± 2.5% at baseline, p = 0.08).
Reduced Carbohydrate Consumption Improves Liver Fat Metabolism
To investigate the effects of the diet on processes linked to fat accumulation in the liver, we measured DNL using stable isotope technology and β-hydroxybutyrate (a proxy for β-oxidation). We observed a rapid and dramatic reduction in absolute DNL (Figure 1H), which was paralleled by a rapid increase in hepatic β-oxidation (Figure 1I). At the end of the 14-day diet intervention, DNL was 79.8% lower and β-hydroxybutyrate was 4.9-fold higher than at baseline (Figures 1H and 1I). However, there was no significant change in plasma non-esterified fatty acid (NEFA) concentrations over the study period (Table S1). Furthermore, there was a strong correlation between the reductions in DNL and liver fat (r = 0.96, p = 0.0001) and a trend toward a significant correlation between the reductions in DNL and VLDL-triglycerides (r = 0.67, p = 0.07) (Figure S2). Consistent with improvements in liver function and metabolism, we observed significant reductions in fasting plasma concentrations of alkaline phosphatase and aspartate transaminase, markers of liver damage, as well as significant decreases in fasting insulin and HOMA-IR (homeostasis model assessment of insulin resistance) values over the study period (Table S1).
Studies characterizing the pathogenesis of NAFLD have focused overwhelmingly on the dysregulation of hepatic DNL (Chakravarthy et al., 2005; Fabbrini et al., 2010; Petersen et al., 2007; Wang et al., 2015) and increased delivery of NEFAs from adipose tissue to the liver especially in insulin-resistant subjects (Lewis et al., 2002); according to one estimation (Donnelly et al., 2005), these pathways account for 25% and 60% of liver fat, respectively. In contrast, the roles of the pathways involved in hepatic lipid disposal (i.e., mitochondrial β-oxidation and VLDL-triglyceride export) in NAFLD pathogenesis are less characterized. By restricting carbohydrate intake and thus blunting DNL, we observed a dramatic reduction of liver fat paralleled by a rapid increase of mitochondrial β-oxidation and a marked reduction of VLDL-triglycerides without any change in plasma NEFA concentrations. Thus, the reduced hepatic lipid accumulation could be explained by both blunted DNL and enhanced mitochondrial β-oxidation; however, as their regulation is tightly connected (McGarry et al., 1977) it is difficult to estimate the relative contribution of these two sources in our study.
Reduced Carbohydrate Consumption Decreases Inflammatory Markers and FGF21
NAFLD has been linked to a low-grade systemic inflammation (Asrih and Jornayvaz, 2013; Braunersreuther et al., 2012), and high carbohydrate intake has been shown to associate with higher inflammation (Solga et al., 2004). We therefore investigated the effect of the dietary intervention on a range of inflammation-related plasma proteins. We observed significant reductions in many of the inflammatory markers over the study period (Figure 1J). In particular, we showed that plasma concentrations of interleukin-6 (IL-6) and tumor necrosis factor alpha (TNF-α) were significantly reduced after 14 days of the carbohydrate-restricted diet (Figure 1J). IL-6 and TNF-α have attracted particular attention because the severity of NAFLD and progression to NASH are associated with higher systemic levels of these cytokines (Paredes-Turrubiarte et al., 2016; Wieckowska et al., 2008).However, we did not observe significant reductions in these cytokines after 3 days on the diet, indicating that they are not the main drivers of liver fat.
Several studies have shown that concentrations of the peptide hormone fibroblast growth factor 21 (FGF21) are elevated in subjects with NAFLD and correlate with hepatic fat content (Dushay et al., 2010; Li et al., 2010), and plasma FGF21 has been suggested as a potential diagnostic marker of NAFLD (Li et al., 2013). Consistent with these findings, we showed that plasma concentrations of FGF21 decreased rapidly in response to the carbohydrate-restricted diet, with significant reductions after both 3 and 14 days on the diet (Figure 1J). Paradoxically, plasma FGF21 concentrations in rodents increase in response to low-carbohydrate ketogenic diets (Badman et al., 2007, 2009; Laegeretal., 2014). One potential explanation for this difference is that FGF21 expression in rodents has been shown to be sensitive to the protein content of the diet (Laeger et al., 2014). Ketogenic diets given to rodents typically contain reduced protein levels compared with control diet, in contrast to our study diet with increased protein content. An earlier study also reported diet-induced reductions in FGF21 concentrations in humans upon increased protein intake (Christodoulides et al., 2009).Moreover, plasma concentrations of FGF21 are increased in individuals who have undergone bariatric surgery (Lips et al., 2014), a treatment that is commonly associated with protein malnutrition (Bal et al., 2012). We therefore speculate that the protein content in the diet plays important roles in regulating human FGF21 expression.
Reduced Carbohydrate Consumption Induces Rapid Shifts in the Gut Microbiota Composition
Because of the increasing evidence supporting a link between diet, the gut microbiota, and NAFLD (Leung et al., 2016), we investigated how the gut microbiome was affected by the dietary intervention. We performed whole-genome shotgun sequencing of fecal samples obtained from the participants at five time points across the study (0, 1, 3, 7, and 14 days). On average, we obtained 17.5 million paired-end reads for each sample (ranging from 14.2 million to 21.3 million; Table S2). Ninety-four bacterial strains were significantly altered upon dietary intervention (Figure 2A; Table S2), and major shifts occurred after only 1 day of the dietary intervention (Figure 2A). Principal coordinate analysis (PCoA) of the relative abundance of all these significantly altered strains confirmed the dramatic compositional shifts of the gut microbiome after 1 day (Adonis test; p = 0.049; Figure 2B). These observations are consistent with earlier studies that reported rapid alterations in both microbial composition (David et al., 2014; Wu et al., 2011) and bacterial growth rate (Korem et al., 2015) in response to extreme dietary interventions. We observed further compositional shifts after 7 days, but not between 7 and 14 days (Figure 2B), suggesting that the compositional shifts stabilized after 1 week on the low-carbohydrate diet with increased protein content. Among the top ten most abundant of 25 significantly altered genera, only Streptococcus, Lactococcus, and Eggerthella were increased over the study period (Figure 2C; Table S2). The carbohydrate-degrading bacteria Ruminoccocus, Eubacterium, Clostridium, and Bifidobacterium were all decreased (Figure 2C; Table S2). In parallel, we observed decreased carbohydrate fermentation as shown by reductions in fecal concentrations of short-chain fatty acids (SCFAs) (Figure 2D), in agreement with previous studies (David et al., 2014; Duncan et al., 2007).
Figure 2. Reduced Carbohydrate Consumption Rapidly Alters Gut Microbial Composition.
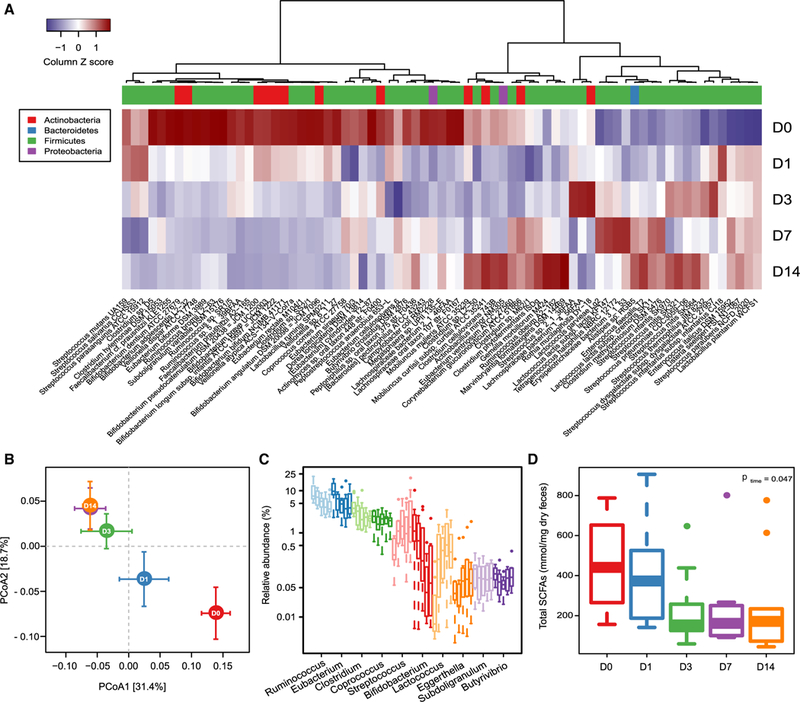
(A) Heatmap showing significant changes in the abundance of bacterial strains over the 14-day study period. D, day. FDR < 0.05; likelihood ratio test.
(B) PCoA plot based on the relative abundance of all bacterial strains that are significantly altered over the study period at the indicated time points (mean ± SEM; p D1 versus D0 = 0.049; Adonis test based on 5,000 permutations).
(C) Boxplots (with median) showing the relative abundance of the ten most abundant genera that were significantly altered by the diet over the study period (n = 10). FDR < 0.05; likelihood ratio test.
(D) Boxplots (with median) showing fecal concentrations of SCFAs at days 0,1,3,7, and 14(n = 10).p values across time were obtained by one-way ANOVA with repeated measurements.
See also Table S2.
Diet-Induced Functional Shifts in the Gut Microbiota Increase Folate Production
To further investigate functional changes in the gut microbiome in response to the dietary intervention, we annotated genes to KEGG orthology (KO) (Kanehisa and Goto, 2000). In total, 598 and 580 KOs were significantly increased and decreased, respectively, after 14 days on the diet (Figure S3; Table S2). Pathway enrichment analysis demonstrated that these 1,178 KOs were involved in 18 KEGG pathways (Figure 3A). Of these, the most upregulated pathways were those involved in folate biosynthesis and the two-component system (which controls gene expression in response to changes in environmental conditions in many bacterial species), whereas the pathways involved in starch and sucrose metabolism and biosynthesis of amino acids including branched-chain amino acids (BCAAs; valine, leucine, and isoleucine) were significantly decreased (Figure 3A). Notably, the folate biosynthesis and starch and sucrose metabolism pathways were significantly altered after only 1 day of dietary intervention (Figure 3A). In-depth analysis of bacterial folate biosynthesis pathways showed that seven of 12 related KOs were significantly increased over the study period (Figure 3B). Of note, folate is a common food fermentation product during the growth of Streptococcus and Lactococcus (LeBlanc et al., 2011), and both of these lactic acid bacteria increased in abundance over the study period (Figure 2C).
Figure 3. Reduced Carbohydrate Consumption Promotes Microbial Shifts toward Folate Production.
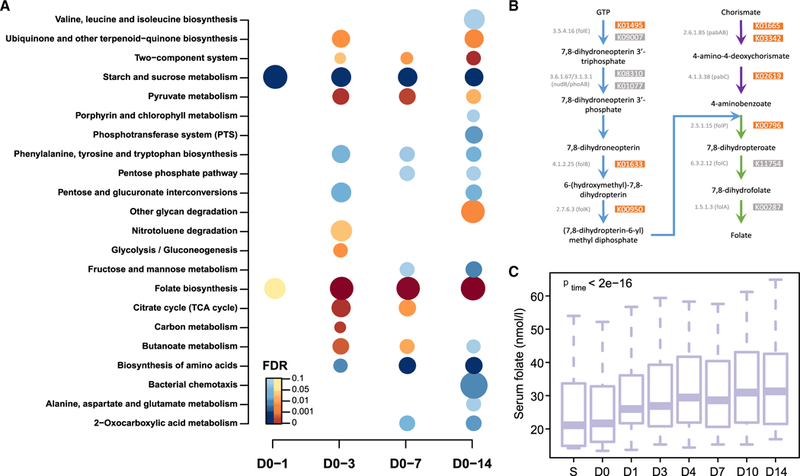
(A) KEGG pathway analysis showing pathways that were significantly altered at the indicated times over the 14-day study period (n = 10). D, day. Pathways that are reduced or increased are shown in blue and brown, respectively. FDR < 0.1; hypergeometric test. The size of the bubble is proportional to the enrichment score for each KEGG pathway term.
(B) Bacterial folate biosynthesis pathway showing KOs that significantly increased over the study period (brown boxes).
(C) Boxplots (with median) showing changes in serum concentrations of folate changes over the study period (n = 10). p values across time were obtained by one-way ANOVA with repeated measurements. S, screen.
See also Figures S3 and S4 and Table S2.
Both the gut microbiota and the diet are sources of serum folate. Although folate is known to be present at higher levels in diets rich in carbohydrates (e.g., from vegetables and cereals) (Witthöft et al., 1999), we did not observe any significant changes in dietary folate when the participants changed to the carbohydrate-restricted diet (Table S1). Furthermore, in contrast to previous studies that reported reduced or no significant changes in serum folate levels in response to carbohydrate-restricted diets (Keogh et al., 2008; Noakes et al., 2005; Nuttall and Gannon, 2006; Wycherley et al., 2010), we observed a significant increase in serum folate concentrations after just 1 day of the diet (Figure 3C; p = 0.0039), in parallel with the rapid effect of the diet on liver fat (Figure 1E). This discrepancy between our study and earlier studies may be explained by differences in the macronutrient composition. Our diet had both increased protein content compared with the baseline diet and very low carbohydrate content (<10% energy from carbohydrates). It is thus likely that the high dietary protein content combined with the reduction in carbohydrate-dependent bacteria would drive the growth of the folate-producing bacteria Streptococcus and Lactococcus, resulting in increased bacterial folate production. We also observed a trend toward an increase in fecal folate when the participants changed to the low-carbohydrate diet with increased protein content (Figure S4), suggesting that bacterially produced folate could contribute to the diet-induced increase in serum folate concentrations.
Our findings thus indicate that the increased circulating folate could be a consequence of the altered microbial composition. In support of this, previous studies have shown that the gut microbiota can produce large quantities of folate (Kim et al., 2004; Rossi et al., 2011; Strozzi and Mogna, 2008; Sugahara et al., 2015) and that folate is absorbable across the colon in humans (Aufreiter et al., 2009). We thus provide the first evidence indicating that a low-carbohydrate diet with increased protein content can shift the gut microbiota toward increased production of folate that is potentially utilized by humans. However, we cannot exclude the possibility that the carbohydrate-restricted diet promoted enhanced dietary folate absorption in the small intestine, resulting in increased circulating concentrations of folate and favoring the growth of bacteria that produce folate.
Folate and Associated Metabolites Are Linked to Improved Liver Fat Metabolism
Animal studies have revealed that folate deficiency increases expression of pro-inflammatory cytokines such as IL-6 and TNF-α (Kolb and Petrie, 2013) and has detrimental effects on hepatic lipid metabolism (Akesson et al., 1982), leading to accumulation of liver fat (Kelley et al., 1950) and steatosis (Christensen et al., 2010; Halsted et al., 2002). Growing evidence also indicates a role for folate deficiency in the pathogenesis of NAFLD in humans (Hirsch et al., 2005; Xia et al., 2017). We therefore investigated whether serum folate correlated with liver fat in our study. We first performed a correlation analysis between these two variables for each individual in the first cohort using data collected at seven time points across the study (0, 1, 3, 4, 7, 10, and 14 days). Despite the limited number of sampling points, we observed significant correlations in five of ten of these individuals and a trend toward significance in a further two (Figure 4A). No correlation between liver fat and serum folate was observed in three individuals (Figure 4A), two of whom had the lowest liver fat in the cohort at baseline (<10%); a lack of correlation in these two is likely explained by the fact that their liver fat had reduced to normal levels (<5%) by day 7 and thus plateaued (Figure 1E). However, by performing a partial regression analysis between liver fat and serum folate based on a linear mixed-effect model, we observed an overall significant association between liver fat and serum folate (p = 0.0001); according to this model, serum folate could explain 35.4% and 19.5% of the variation in liver fat before and after adjusting for BMI, respectively (Figure 4B).
Figure 4. Serum Folate Is Associated with Improved Liver Lipid Metabolism.
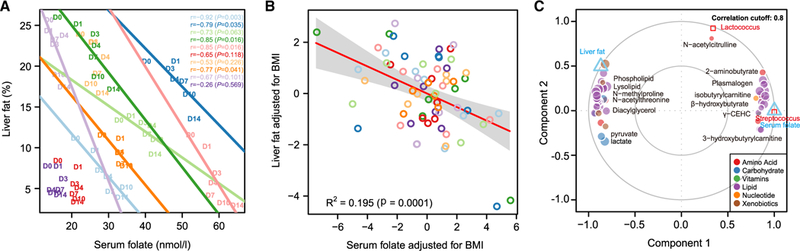
(A) Pearson correlations between liver fat and serum folate in each individual in the first cohort. D, day.
(B) Linear mixed-effect model (with 95% confidence bands highlighted) between liver fat and serum folate after adjusting for contributions from BMI to both variables; samples from the same individual are colored as in (A) (n = 10).
(C) Integration of liver fat, folate-producing bacteria, serum folate, and metabolomics data from the first cohort (n = 10) using a multivariate method (mixDIABLO with repeated measurements) with a correlation cutoff of 0.8. Components are linear combinations of variables from each omics dataset that are maximally correlated using a full-design matrix.
See also Figures S5 and S6 and Table S3.
To investigate whether there are other metabolites that could potentially play a role in improved liver fat metabolism in our study, we also performed untargeted metabolomics on plasma samples from the participants at baseline and after 3 and 14 days on the dietary intervention. We observed significant changes in the relative signal intensity of 202 of 648 detected metabolites over the study period, most of which are involved in lipid or amino acid metabolism (Figure S5; Table S3). In particular, we observed significant decreases in metabolites involved in fatty acid synthesis (including those involved in phospholipid, lysolipid, and diacylglycerol metabolism). Circulating diacylglycerol concentrations have been repeatedly linked with insulin resistance (Petersen et al., 2017); thus, reductions in diacylglycerol are consistent with the improved insulin sensitivity observed in this study (Table S1). We also observed significant increases in metabolites involved in β-oxidation pathways and plasmalogens, which are known antioxidants (Wallner and Schmitz, 2011) (Figure S5; Table S3). Increased oxidative stress is considered to contribute to the pathogenesis of NAFLD (Haas et al., 2016), and antioxidants help to maintain the cellular redox balance by neutralizing the oxidative stress produced by fatty acid oxidation (Gambino et al., 2011), thereby alleviating NAFLD. Nicotinamide and its methylation product 1-methylnicotinamide, which has been shown to suppress liver triglyceride levels in mice (Hong et al., 2015), were also increased during the dietary intervention (Table S3).
By using multivariate dimension reduction and discriminant analysis, we revealed that 48 of the 202 significantly altered metabolites strongly correlated with serum folate (and thus liver fat) as well as with the folate-producing bacteria Streptococcus and Lactococcus (correlation coefficient cutoff 0.8; Figure 4C; Table S3). Of note, β-hydroxybutyrate and most of the identified plasmalogens associated with folate (Figure 4C; Table S3), suggesting that folate may improve liver fat metabolism by promoting an increase in mitochondrial β-oxidation and a reduction of oxidative stress. In agreement, previous studies have demonstrated that folate can confer protective effects against oxidative stress (Sid et al., 2017). However, further efforts are needed to explore the molecular and cellular mechanisms behind the multiple potential benefits provided by folate.
Liver Transcriptional Changes Reflect Improved Lipid Metabolism
We also served the identical isocaloric low-carbohydrate diet with increased protein content to a second cohort of seven subjects for 7 days. In this second cohort of similar age (p = 0.379) and BMI (p = 0.230), we confirmed that the diet reduced DNL, fasting plasma triglycerides, plasma concentrations of alkaline phosphatase and insulin, and HOMA-IR values (Table S4). Analysis of liver biopsies taken from these subjects 7 days before the study start and after 7 days of dietary intervention showed that the diet significantly reduced the hepatic triglycerides by a mean of 43.9% (Table S4). Notably, we also observed a significant increase in serum folate concentrations in this cohort (Table S4).
To investigate whether alterations in hepatic gene expression reflected the diet-induced improvements in lipid metabolism, we also performed a global transcriptomic analysis on the liver samples collected from this second cohort. We observed significant decreases in 9.1% of genes, whereas only 3.8% of genes increased after 7 days on the diet (false discovery rate [FDR] adjusted p value < 0.1; Table S5). KEGG pathway enrichment analysis revealed that 34 pathways were upregulated, including fatty acid degradation, amino acid metabolism, and peroxisome proliferator-activated receptor (PPAR) signaling pathways, whereas only seven pathways were downregulated, such as ribosome and oxidative phosphorylation (Table S5).
Consistent with the diet-induced reductions in plasma apoC-III concentrations and DNL noted earlier, we observed decreased hepatic expression of APOC3 and FASN (although no significant changes in SREBP-1c) in the second cohort after 7 days on the diet (Table S5). We also observed diet-induced increases in the hepatic expression of PPAR-α and its downstream genes (Figure 5), most of which are known to encode proteins involved in the fatty acid oxidation including mitochondrial and peroxisomal β-oxidation as well as microsomal ω-oxidation (Figure 5) (Kersten, 2014; Pawlak et al., 2015). Intriguingly, both the gut microbiota (Crawford et al., 2009) and folate (Tryndyak et al., 2012) have been shown to be able to regulate hepatic lipid metabolism via a PPAR-α-regulated lipid catabolic pathway.
Figure 5. Liver Transcriptome Changes Reflect Improved Hepatic Lipid Metabolism.
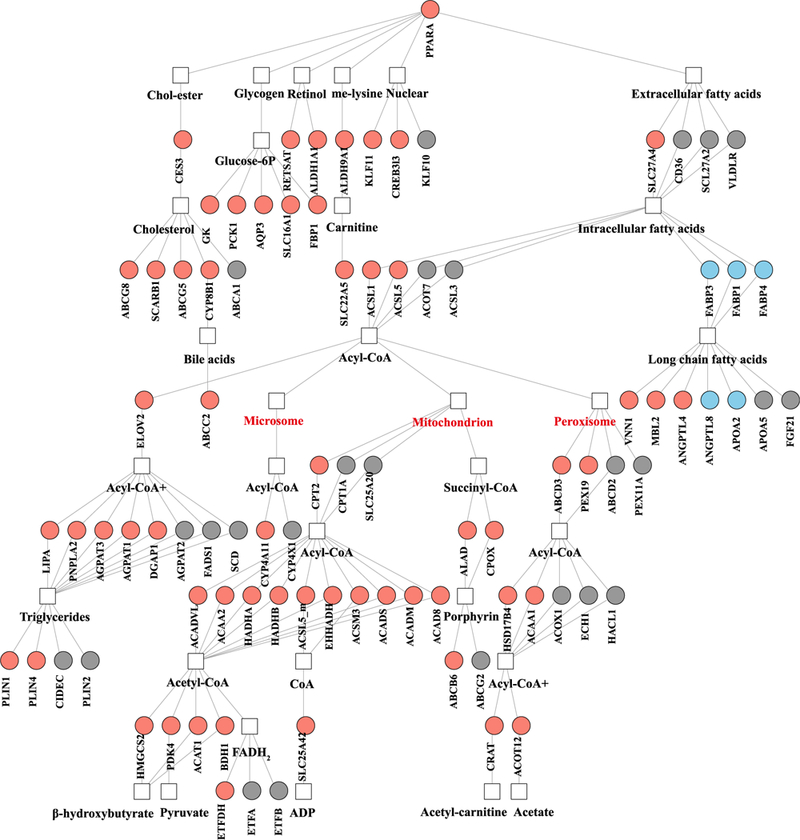
Gene regulatory network for PPARα showing significant increases (salmon) and decreases (light blue) in genes in liver from individuals in the second cohort after 7 days on a low-carbohydrate diet with increased protein content (n = 7). FDR < 0.1; Wald test with paired design.
See also Table S5.
Expression of CAT, the gene encoding catalase, was also up-regulated in response to the diet (Table S5). Hepatic expression of the fatty acid transporter gene CD36 was not significantly changed (Figure 5), in line with the lack of effect of the diet on plasma NEFA concentrations in both cohorts (Tables S1 and S4). Notably, consistent with the diet-induced increase in serum folate concentrations in both cohorts (Figure 3C; Table S4), we found diet-induced increases in the hepatic expression of genes involved in folate-mediated one-carbon metabolism, including MTRR, SHMT1, SHMT2, MTHFD1, and ALDH1L1 (Figure 6A), and the proton-coupled folate transporter gene SLC46A1 (Table S5). Serine hydroxymethyltransferase (SHMT) catalyzes the interconversion of glycine and serine, and serine is required for the generation of glutathione (GSH), an antioxidant that also plays a role in maintaining β-oxidation (Mardinoglu et al., 2017; Nguyen et al., 2013). We have previously shown that individuals with NAFLD have serine deficiency (Mardinoglu et al., 2014, 2017) and that de novo GSH synthesis is altered in humans with hepatic steatosis (Mardinoglu et al., 2017).
Figure 6. Integration of Liver Transcriptomics and Plasma Metabolomics Data.
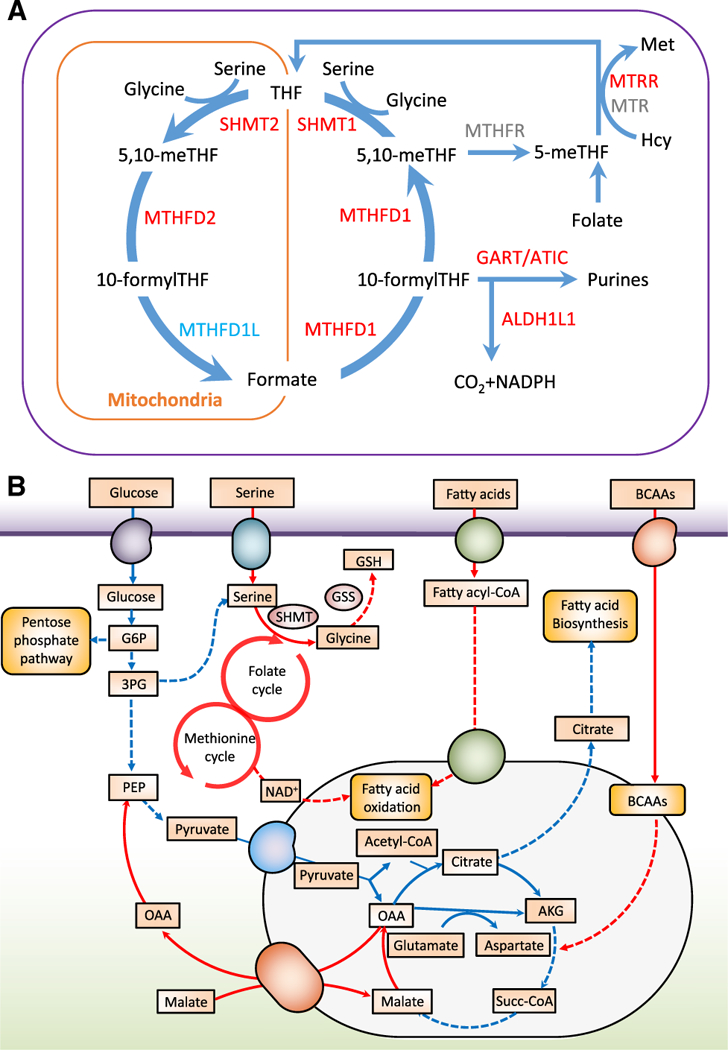
(A) Reactions involved in folate-mediated one-carbon metabolism showing significant increases (red) and decreases (light blue) in genes in liver from individuals after 7 days on a low-carbohydrate diet with increased protein content (n = 7). FDR < 0.1; Wald test with paired design.
(B) A genome-wide metabolic model for the liver integrating both serum metabolomics data and liver transcriptomics data. Red lines indicate increased fluxes, and blue lines indicate decreased fluxes.
Finally, we integrated the liver transcriptomics and plasma metabolomics data using iHepatocytes2322, a functional genome-scale metabolic model of hepatocytes (Mardinoglu et al., 2014). As expected, given the diet-induced reduction in DNL, the model predicted a significant reduction of fluxes carried by the reactions associated with glycolysis, the pentose phosphate pathway, and the tricarboxylic acid (TCA) cycle (Figure 6B; Table S6). The decreased fluxes in the reactions involved in glycolysis and the TCA cycle were partly compensated by increased fluxes in β-oxidation and BCAAs, required to provide sufficient energy as well as substrates for the biosynthesis of essential metabolites. Of note, we observed that the fluxes carried by the reactions involved in folate and methionine metabolism, serine uptake, as well as in the de novo synthesis of GSH were significantly increased in response to the diet. Our data are thus consistent with a central role of folate in mediating one-carbon metabolism and its involvement in a number of biosynthetic processes including synthesis of glycine, serine, and methionine (Ducker and Rabinowitz, 2017; Sid et al., 2017).
Study Limitations
There are several strengths with this study, but there are also limitations. For example, the study lacks a control group, in which a normal carbohydrate diet was provided to subjects. In addition, the types of carbohydrate, protein, and fat provided during the diet intervention were not matched with the diet the subjects consumed before the study. Therefore, it is possible that changes in the types of macronutrients (e.g., refined sugars versus complex carbohydrates, saturated versus unsaturated fats, plant versus animal proteins) could have affected the outcome measures.
Conclusions
In summary, by using a multi-omics approach, we showed that short-term intervention with an isocaloric low-carbohydrate diet with increased protein content promotes multiple metabolic benefits in obese humans with NAFLD. In particular, we observed a dramatic reduction of liver fat resulting from a marked decrease in DNL and increase in β-oxidation. Of note, our data indicate that rapid microbial shifts toward folate production paralleled by increased circulating folate concentrations may be partially behind the improved lipid metabolism, balanced oxidative stress, and reduced inflammation. Taken together, these findings are of important clinical interest in understanding the pathogenesis and prevention of NAFLD. However, although studies in mice have shown that folate supplementation protects against high-fat-diet-induced liver steatosis (Sid et al., 2015, 2017), care should be taken before implementing folate supplementation into clinical practice since long-term effects remain to be elucidated.
STAR★METHODS
Detailed methods are provided in the online version of this paper and include the following:
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Jan Borén (jan.boren@wlab.gu.se).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Clinical Study Design
Ten middle-aged (54±4 years) subjects (2 women, 8 men) with high liver fat were recruited to the first cohort in this study. All subjects had stable (±5 kg) weight in the three months preceding enrollment.
The inclusion criteria were high liver fat (>5.5% measured by MRS), body mass index 27–40 kg/m2, negative viral hepatitis serology, transferrin saturation <40%, normal α1-antitrypsin and ceruloplasmin concentrations, negative autoimmune liver disease markers, negative pregnancy test for pre-menopausal women on enrollment, and no history of medication use associated with hepatic steatosis. The exclusion criteria were liver cirrhosis, portal hypertension, chronic liver disease other than NAFLD, including alcoholic liver disease as defined by daily alcohol intake of >40 g (men) and >20 g (women), respectively, significant endocrine disease including diabetes, any medication acting on nuclear hormone receptors or inducing CYP P450, self-administration of supplements other than calcium, any significant cardiovascular co-morbidity, history of non-compliance, and genetic variations in the two main genetic determinants of liver fat accumulation: PNPLA3 (homozygous for I148M) or TM6SF2 (homozygous or heterozygous for E167K).
Participants in the first cohort followed an isocaloric low-carbohydrate diet with increased protein content for 14 days and came to the study center on days 0, 1, 3, 4, 7, 10 and 14 of the diet after an overnight fast. At each of these visits, subjects were weighed, blood samples were taken and liver fat was measured (by MRS). Fecal samples were collected on days 0, 1, 3, 7 and 14. The study design with time-points for the different analyses performed is shown in Figure 1B.
To obtain liver biopsies from obese individuals with high liver fat before and after the low-carbohydrate diet with increased protein content, we recruited a second cohort (5 men, 2 women; mean age 47.3±14.6 years). These subjects were mildly obese (BMI 32.1±3.8 kg/m2) with ultrasound-verified fatty liver (but without diabetes or any medication) who underwent a Menghini liver biopsy for the diagnosis of NASH. Those individuals from whom we obtained biopsies >4 cm (of which 2 cm was used for histological confirmation of NAFLD/NASH with no advanced fibrosis; see Table S4) were asked to agree to a second biopsy within two weeks and to follow the identical low-carbohydrate diet with increased protein content as the first cohort for 7 days until the morning before the second biopsy. Blood samples were taken from the participants on the same day as their biopsies.
The study was performed in accordance with the Declaration of Helsinki and it was approved by the Helsinki University Central Hospital Ethics Committee (for the first cohort) and the Ethics Committee at the University of Gothenburg (liver biopsy cohort). Each subject gave written informed consent before participation in the study. A CONSORT flow diagram for the study is shown in Figure S7.
Isocaloric Carbohydrate-Restricted Diet
Subjects in the first cohort were instructed to follow an isocaloric low-carbohydrate diet with increased protein content (providing a mean of 3114.6 kcal energy per day) for 14 days. Each subject’s energy requirement was estimated (using the Harris Benedict equation) from the basal metabolic rate (Roza and Shizgal, 1984), which was multiplied by 1.5 corresponding to the subject’s activity level. The study diet provided approximately 4% energy from carbohydrate, 72% energy from fat and 24% energy from protein whereas the corresponding macronutrient composition in the baseline diet was 40%, 42% and 18%, respectively (Figure 1A). The carbohydrate content of the diet was restricted to 23–30 g per day.
To ensure compliance with the diet, subjects underwent a teaching session and had daily contact with an experienced dietitian. Study subjects were asked to keep dietary records for 3 days beforehand to record the macronutrient content of the baseline diet.
All meals and snacks were pre-prepared in a metabolic kitchen and weighed and delivered to the subjects twice a week during the 14-day study period. The meals were frozen and the snacks and vegetables were fresh. The subjects were permitted to consume calorie-free beverages and sweeteners. The dietician also instructed the participants to increase their daily energy intake by 200 kcal from macadamia nuts whenever their weight decreased between two study days by more than 0.2 kg. During the study, all study subjects filled in a checklist of the meal consumed to keep track of meals eaten. The actual macronutrient intake during the low-carbohydrate diet with increased protein content was calculated based on the checklists recorded by the subjects. The dietitian contacted the participants daily (by phone or email) to verify adherence to the meal plan and check weight stability (measured by home scales).
Influence of Gender
The first cohort consisted often obese (BMI 34.1±1.2 kg/m2) middle-aged (54±4 years) subjects (2 women, 8 men) with high liver fat. The second cohort consisted of seven obese (BMI 32.1±3.8 kg/m2) middle-aged (47±15 years) subjects (2 women, 5 men) with high liver fat. All subjects were analyzed as their own control, during and after the diet intervention. Subgroup analysis did not reveal differences between women and men in their responses of liver fat content during the diet intervention.
METHOD DETAILS
Genotyping of PNPLA3 and TM6SF2 Variants
DNA was extracted from whole blood using DNeasy Blood and Tissue kit according to the manufacturer’s instructions. PNPLA3 rs738409 and TM6SF2 rs58542926 variants were genotyped by 5’-nuclease Taqman Assays. Primers and probes (FAM and VIC labelled) were directly supplied by ThermoFisher Scientific (TaqMan Applied Biosystems, Assay ID: C_7241_10 and C_89463510_10 for PNPLA3 rs738409 and TM6SF2 rs58542926, respectively). Post-PCR allelic discrimination was performed on a CFX384 Real-Time System (Bio-Rad) by measuring allele-specific fluorescence. For both variants, genotyping was performed in triplicate with concordance between replicates and success rate of 100%.
Body Composition, Liver Fat and Liver Volume
Body composition of the first cohort was determined using a Tanita MC-980 MA body composition analyzer at baseline and on day 14. Liver fat and liver volume were assessed by MRS and magnetic resonance imaging (Bian et al., 2014) using a clinical 1.5 imager (Avantofit, Siemens, Erlangen, Germany). For determination of liver fat content, PRESS localization technique was used to collect 25×25×25 mm3 spectra with five different echo times (TE): 30, 50, 80, 135 and 200 ms with 4, 4, 4, 8 and 64 number of averages, respectively. Acquisition was triggered to end-expiration (TR > 3000 ms). Intensities of water and methylene resonances were assessed using LCModel v6.3g software. The apparent T2 relaxation times of methylene and water resonances did not change during the study (Table S1). For each individual experiment, the apparent T2 of water and methylene was determined by monoexponential fit to five TEs. T2 relaxation effects on intensities were corrected, and liver fat content was calculated as the ratio methylene: (methylene+water) and further converted to mass fractions as described (Kotronen et al., 2009). Unsaturation indices were determined from 200 ms spectra using jMRUI v3.0 software (Lundbom et al., 2011).
Measurement of Hepatic DNL
DNL was analyzed as described previously (Diraison et al., 1996). Absolute DNL [i.e., the contribution of DNL to triglycerides within plasma very low-density lipoproteins (VLDL-TG)] was calculated by multiplying the relative contribution of newly synthesized palmitate from DNL to VLDL-TG by the concentration of triglycerides in VLDL (Pramfalk et al., 2015). One subject was excluded from the analysis of DNL for technical reasons.
Lipoproteins and Biochemical Analyses
In the first cohort, lipoprotein fractions were separated as described previously (Taskinen et al., 1988). Serum VLDL from the second cohort was isolated by ultracentrifugation using deuterium oxide as described previously (Ståhlman et al., 2008). Plasma concentrations of triglyceride, cholesterol, NEFA, β-hydroxybutyrate, glucose and serum insulin were analyzed using a Konelab 60i analyzer (Thermo Fisher Scientific). Plasma levels of FGF21, IL-6, TNF-α and apoC-III were measured by ELISA (R&D Systems, Minneapolis, USA). Other inflammatory protein biomarkers were analyzed using the Proseek Multiplex Inflammation I96×96 array (Olink Bioscience, Uppsala, Sweden). Serum folate was measured using a chemiluminescent immunoassay (Roche Diagnostics USA).
Fecal Genomic DNA and Genome Sequencing
Fecal genomic DNA was extracted from 100 mg of frozen stools from the first cohort using the QIAamp DNA mini stool kit (Qiagen, Courtaboeuf, France) following repeated bead-beating (6500 r.p.m., 3 × 30 s). The DNA was extracted from 48 fecal samples, obtained from the 10 participants at 5 different time points during the study (2 fecal samples from day 1 were not obtained). DNA fragments of approximately 300 bp were sequenced on an Illumina NextSeq 500 instrument (150 bp; paired-end) at the Genomics Core Facility, Sahlgrenska Academy, University of Gothenburg.
Metagenomics Analyses
We obtained a total of 151 Gb of raw paired-end reads. The taxonomic and KO composition was obtained by using an updated version of the MEDUSA pipeline (Karlsson et al., 2014), as described before (Wu et al., 2017). The mean mapping rates for the genome and gene catalogues were 43.1% and 72.6%, respectively (Table S2). The obtained taxonomic composition and KO profile matrix were further analyzed by DESeq2 package (Love et al., 2014). Pathway enrichment analyses are based on KEGG annotation (Kanehisa and Goto, 2000) and hypergeometric test using goseq (Young et al., 2010). The beta diversity and PCoA analysis were calculated based on the relative abundance of all significant strains (further transformed by square root to reduce the influence of dominant KOs as suggested previously (Kindt and Coe, 2005) using phyloseq (version 1.19.1) (McMurdie and Holmes, 2013).
Measurement of Fecal SCFAs and Folate
Fecal SCFAs were measured using gas chromatography coupled to mass spectrometry detection (GC-MS) as described previously (Wichmann et al., 2013). In brief, approximately 50–250 mg of feces was mixed with internal standards, added to glass vials and freeze dried. All samples were then acidified with HCl, and SCFAs were extracted with two rounds of diethyl ether extraction. The organic supernatant was collected, the derivatization agent N-tert-butyldimethylsilyl- N-methyltrifluoroacetamide (Sigma-Aldrich, Stockholm, Sweden) was added, and samples were incubated overnight. SCFAs were quantified with a 7090A gas chromatograph coupled to a 5975C mass spectrometer (Agilent Technologies 5975C, Santa Clara, CA). SCFA standards were obtained from Sigma-Aldrich (Stockholm, Sweden).
For folate measurements, approximately 100 mg of fecal sample was transferred to pre-weighed Sarstedt tubes along with six ceramic beads. 500 μL methanol:water [80:20] containing 20 mM ammonium acetate, 0.1% ascorbic acid and 100 nM of the internal standard 5-methyl-THF-13C1,d3 (Toronto Research Chemicals, Downsview, ON, Canada) was added and the samples were homogenized for 10 min at 25 Hz using a TissueLyser instrument (Quiagen Nordic, Copenhagen, Denmark). The samples were then centrifuged at 20,000g for 10 min and 300 μL of supernatant was transferred to glass vials and evaporated under a stream of nitrogen. The dried samples were reconstituted in 150 μL of injection solvent (methanol:water [10:90] + 0.1% HCl) and vortexed for 5 min at 1500 rpm. The sample were added to an OSTRO 96-well plate (Waters, Milford, MA) and eluted under positive pressure into the 96-well collection plate.
The samples were then injected into a Waters Acquity UPLC system equipped with a Waters BEH Phenyl column (2.1 × 100 mm; 1.7 μm particle size). Column temperature was maintained at 30°C and the flow rate was 400 μL/min. The temperature of the auto sampler was set at 12°C. The mobile phases consisted of water with 0.1% formic acid (A) and acetonitrile with 0.1% formic acid (B). Gradient elution was performed using a linear gradient from 1–15% B over 3 min. The gradient was then increased linearly to 99% B over 4 min. After 0.5 min of isocratic elution the gradient returned to 1% B and held for 2.5 min for a total runtime of 10 min.
The folates were detected using a Xevo TQ-XS (Waters, Milford, MA) using positive electrospray. Fine tune of the mass spectrometer for each metabolite was performed. After optimization the ion source parameters were: Capillary: 1.00 kV, Desolvation temperature 500°C, Desolvation gas flow 1000 L/h, Cone gas flow 150 L/h, Nebulizer gas flow 7.0 L/h, Cone voltage 20 V, Collision energy 20 eV. For each analyte the most intense precursor/product ion transition was selected. Consequently, the MRM transitions used were: Folic acid m/z 442.2 → 295.2, 5-methyl-THF m/z 460.2 → 313.2, 5-methyl-THF-13C1,d3 m/z 464.2 → 317.2. Dwell time was 44 ms for all analytes.
Untargeted Metabolomics Analyses
Untargeted metabolomic profiling of plasma was performed by Metabolon (Durham, NC), as previously described (Lee et al., 2016). Samples were prepared using the automated MicroLab STAR system from Hamilton Company. A recovery standard was added before the first step in the extraction process for quality control purposes. Sample preparation was conducted using aqueous methanol extraction process to remove the protein fraction while allowing maximum recovery of small molecules. The resulting extract was divided into four fractions: one for analysis by UPLC/MS/MS (positive mode), one for UPLC/MS/MS (negative mode), one for GC/MS, and one for backup. Samples were placed briefly on a TurboVap (Zymark) to remove the organic solvent. Each sample was then frozen and dried under vacuum. Samples were then prepared for the appropriate instrument (either UPLC/MS/MS or GC/MS).
Liver Lipid Analysis
Liver biopsies from the second cohort were collected in tubes with RNAlater (Qiagen, Hilden, Germany) and stored at −80°C. Lipids from liver biopsies were extracted as previously described (Lofgren et al., 2016). Lipid extracts were diluted in chloroform:methanol [1:2] containing 5 mM ammonium acetate and infused into a QTRAP 5500 mass spectrometer (Sciex, Concord, Canada) using a NanoMate Triversa (Advion Biosciences, Ithaca, NY). Phosphatidylcholines and triglycerides were detected in positive mode using precursor ion scanning and neutral loss scanning as previously described (Brugger et al., 1997; Murphy et al., 2007). Quantification was made against internal standards added during the extraction.
Transcript Profiling of Liver Biopsies
Total RNA was extracted from the liver biopsies using the RNeasy Mini Kit (Qiagen, Hilden, Germany) according to the manufacturer’s instructions, and used in the subsequent mRNA sample preparation for sequencing using Illumina HiSeq 2500 systems with the standard Illumina RNA-seq protocol. The RNAseq was performed at GATC Biotech AG [project number NG-10969] (Konstanz, Germany). Gene abundance (in both raw counts and transcripts per million) was quantified using the kallisto pipeline (Bray et al., 2016) based on human genome assembly version GRCh38. The DESeq2 package (Anders and Huber, 2010) was used to identify differentially expressed genes.
Model Simulation
A previously established genome-scale metabolic model for liver, iHepatocyte2322 (Mardinoglu et al., 2014), was used to investigate the metabolic shifts in response to the diet. The substrate uptake of the model is constrained partly based on a previous study (Mardinoglu et al., 2017) and the fluxes are provided in Table S6. To investigate the flux change in response to the diet, both transcriptomic and metabolomic changes were integrated as constraints into the model using a previously developed method, RMetD (Mardinoglu et al., 2015). The flux distribution was calculated based on the average of 10,000 random sampled fluxes as previously described (Mardinoglu et al., 2013).
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical Analysis
All statistical analyses were performed in the R environment (R Core Team, 2015). The Wald test or the Likelihood Ratio Test was used to analyze differential abundances for all count data (for metagenomics and liver transcriptomics) depending on whether the dataset contains samples collected at two time points or at more than two time points from each individual. One-way ANOVA with repeated measurements was used for all other longitudinal datasets (for phenomics and metabolomics). Otherwise, two-tailed Wilcoxon rank–sum tests or Wilcoxon signed–rank tests were used throughout the study, depending on whether the samples were paired. The linear mixed effect model was built using lme4 package (Bates et al., 2015) in which liver fat adjusted for BMI was entered as a response variable whereas serum folate adjusted for BMI was entered for fixed effect and individual IDs as random effect. The model was visualized by visreg (Breheny and Burchett, 2016) and conditional and marginal coefficients of the model were determined using MuMIn (Bartoń, 2016). The residual plot and quantile-quantile plot are shown in Figure S6. The multivariate analysis and integration of phenomics, metagenomics, and metabolomics are based on mixDIABLO (Singh et al., 2016). The Spearman’s rank–order correlation was used to determine the strength and direction of the monotonic relationships between two variables unless strong collinearity was observed, in which case the Pearson product-moment correlation was calculated. Raw P values were adjusted by the Benjamini–Hochberg method (Benjamini and Hochberg, 1995) with a false discovery rate of 5%, unless indicated otherwise. Data are shown as mean±s.e.m. unless otherwise indicated.
DATA AND SOFTWARE AVAILABILITY
Datasets Generated and Deposited
Metagenomics data from the first cohort were deposited at Sequence Read Archive with accession number SRA: PRJNA420817. RNA-seq raw and processed files from double liver needle biopsies taken before and after one-week diet intervention from the second cohort were deposited at NCBI Gene Expression Omnibus with accession number GEO: GSE107650.
ADDITIONAL RESOURCES
The clinical trial was registered at ClinicalTrials.gov with identifier: NCT02558530; https://clinicaltrials.gov/ct2/show/NCT02558530
Supplementary Material
4.1.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Proseek Multiplex Inflammation I96×96 array | Olink Bioscience | article 94300 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| DNeasy Blood and Tissue kit | QIAGEN | Cat# 69506 |
| Critical Commercial Assays | ||
| FGF-21 | R&D Systems | Cat# DF2100 |
| IL-6 | R&D Systems | Cat# D6050 |
| TNF-α | R&D Systems | Cat# DTA00C |
| Triglycerides | Thermo Fisher Scientific | TR22421 |
| Cholesterol | Thermo Fisher Scientific | TR13421 |
| Insulin | Thermo Fisher Scientific | KAQ1251 |
| NEFA-HR(2) Assay | Wako | 434–91795 |
| β-hydroxybutyrate | DiaSys Diagnostic Systems | 1 3701 99 10 930 |
| Folate | Roche Diagnostics USA | A98032 |
| Genotyping PNPLA3 rs738409 | TaqMan Applied Biosystems | ID: C_7241_10 |
| Genotyping TM6SF2 rs58542926 | TaqMan Applied Biosystems | ID: C_89463510_10 |
| Deposited Data | ||
| RNAseq liver data | NCBI Gene Expression Omnibus | GEO: GSE107650 |
| Metagenomics data | Sequence Read Archive | SRA: PRJNA420817 |
| Software and Algorithms | ||
| jMRUI v3.0 software | jMRUI | http://www.jmrui.eu/license-and-download/jmrui-license/ |
| LCModel v6.3g software | LCModel | http://www.lcmodel.com/lcmodel.shtml |
| DESeq2 | bioconductor | http://bioconductor.org/packages/release/bioc/html/DESeq2.html |
| Phyloseq (1.19.1) | bioconductor | https://bioconductor.org/packages/release/bioc/html/phyloseq.html |
| Goseq | bioconductor | https://bioconductor.org/packages/release/bioc/html/goseq.html |
| visreg | R | https://cran.r-project.org/web/packages/visreg/index.html |
| lme4 package | R | https://cran.r-project.org/web/packages/lme4/index.html |
| GRCh38 | Genome Reference Consortium | https://www.ncbi.nlm.nih.gov/grc/human |
| mixDIABLO | mixOmics | http://mixomics.org/mixDIABLO/ |
Highlights.
A low-carbohydrate diet (LCD) improves liver fat metabolism in NAFLD patients
The LCD promotes rapid shifts in the gut microbiota composition of NAFLD patients
The LCD-induced microbial changes are associated with increased circulating folate
The LCD increases folate-dependent one-carbon metabolism gene expression in liver
ACKNOWLEDGMENTS
The authors thank Ara Koh for helpful discussions, Anna Hallén for preparation of the graphical abstract, all staff for excellent laboratory work and patient care, and the clinical biomarker facility at SciLifeLab, Sweden. Whole-genome shotgun sequencing was performed at the Genomics Core Facility at the Sahlgrenska Academy, University of Gothenburg. The computations for metagenomics analyses were performed on resources provided by the Swedish National Infrastructure for Computing (SNIC) through Uppsala Multidisciplinary Center for Advanced Computational Science (UPPMAX). This work was supported by grants from the Swedish Research Council, Swedish Heart-Lung Foundation, Swedish Diabetes Foundation, Sahlgrenska University Hospital ALF funds, Sigrid Jusélius Foundation, Academy of Finland, Finnish Diabetes Research Foundation, and EU Innovative Medicines Initiative EMIF, and by the FP7-HEALTH-2012-INNOVATION-1 program RESOLVE no. 305707. F.B. is a recipient of a European Research Council consolidator grant 615362 - METABASE and Torsten Söderberg Professor in Medicine.
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
SUPPLEMENTAL INFORMATION
Supplemental Information includes seven figures and six tables and can be found with this article online at https://doi.org/10.1016/j-cmet.2018.01.005.
REFERENCES
- Akesson B, Fehling C, Jagerstad M, and Stenram U (1982). Effect of experimental folate deficiency on lipid metabolism in liver and brain. Br. J. Nutr. 47, 505–520. [DOI] [PubMed] [Google Scholar]
- Anders S, and Huber W (2010). Differential expression analysis for sequence count data. Genome Biol. 11, R106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asrih M, and Jornayvaz FR (2013). Inflammation as a potential link between nonalcoholic fatty liver disease and insulin resistance. J. Endocrinol. 218, R25–R36. [DOI] [PubMed] [Google Scholar]
- Astrup A, Meinert Larsen T, and Harper A (2004). Atkins and other low-carbohydrate diets: hoax or an effective tool for weight loss? Lancet 364, 897–899. [DOI] [PubMed] [Google Scholar]
- Aufreiter S, Gregory JF 3rd, Pfeiffer CM, Fazili Z, Kim YI, Marcon N, Kamalaporn P, Pencharz PB, and O’Connor DL (2009). Folate is absorbed across the colon of adults: evidence from cecal infusion of (13)C-labeled [6S]-5-formyltetrahydrofolic acid. Am. J. Clin. Nutr. 90, 116–123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Badman MK, Kennedy AR, Adams AC, Pissios P, and Maratos-Flier E (2009). A very low carbohydrate ketogenic diet improves glucose tolerance in ob/ob mice independently of weight loss. Am. J. Physiol. Endocrinol. Metab. 297, E1197–E1204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Badman MK, Pissios P, Kennedy AR, Koukos G, Flier JS, and Maratos-Flier E (2007). Hepatic fibroblast growth factor 21 is regulated by PPARalpha and is a key mediator of hepatic lipid metabolism in ketotic states. Cell Metab. 5, 426–437. [DOI] [PubMed] [Google Scholar]
- Bal BS, Finelli FC, Shope TR, and Koch TR (2012). Nutritional deficiencies after bariatric surgery. Nat. Rev. Endocrinol. 8, 544–556. [DOI] [PubMed] [Google Scholar]
- Bartoń K (2016). MuMIn: multi-model inference. R package, version 1.15.6. https://CRAN.R-project.org/package=MuMIn.
- Bates D, Mächler M, Bolker B, and Walker S (2015). Fitting linear mixed-effects models using lme4. J. Stat. Software 67, 10.18637/jss.v067.i01. [DOI] [Google Scholar]
- Benjamini Y, and Hochberg Y (1995). Controlling the false discovery rate: a practical and powerful approach to multiple testing. J. Roy. Stat. Soc. B 57, 289–300. [Google Scholar]
- Bian H, Hakkarainen A, Lundbom N, and Yki-Jarvinen H (2014). Effects of dietary interventions on liver volume in humans. Obesity 22, 989–995. [DOI] [PubMed] [Google Scholar]
- Braunersreuther V, Viviani GL, Mach F, and Montecucco F (2012). Role of cytokines and chemokines in non-alcoholic fatty liver disease. World J. Gastroenterol. 18, 727–735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bray NL, Pimentel H, Melsted P, and Pachter L (2016). Near-optimal probabilistic RNA-seq quantification. Nat. Biotechnol. 34, 525–527. [DOI] [PubMed] [Google Scholar]
- Breheny P, and Burchett W (2016). Visualization of regression models using visreg. Version 2.3.0. https://CRAN.R-project.org/package=visreg.
- Browning JD, Baker JA, Rogers T, Davis J, Satapati S, and Burgess SC (2011). Short-term weight loss and hepatic triglyceride reduction: evidence of a metabolic advantage with dietary carbohydrate restriction. Am. J. Clin. Nutr. 93, 1048–1052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Browning JD, Davis J, Saboorian MH, and Burgess SC (2006). A low-carbohydrate diet rapidly and dramatically reduces intrahepatic triglyceride content. Hepatology 44, 487–488. [DOI] [PubMed] [Google Scholar]
- Brugger B, Erben G, Sandhoff R, Wieland FT, and Lehmann WD (1997). Quantitative analysis of biological membrane lipids at the low picomole level by nano-electrospray ionization tandem mass spectrometry. Proc. Natl. Acad. Sci. USA 94, 2339–2344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakravarthy MV, Pan Z, Zhu Y, Tordjman K, Schneider JG, Coleman T, Turk J, and Semenkovich CF (2005). “New” hepatic fat activates PPARalpha to maintain glucose, lipid, and cholesterol homeostasis. Cell Metab. 1, 309–322. [DOI] [PubMed] [Google Scholar]
- Chalasani N, Younossi Z, Lavine JE, Diehl AM, Brunt EM, Cusi K, Charlton M, and Sanyal AJ (2012). The diagnosis and management of non-alcoholic fatty liver disease: practice guideline by the American association for the study of liver diseases, American college of gastroenterology, and the American gastroenterological association. Am. J. Gastroenterol. 107, 811–826. [DOI] [PubMed] [Google Scholar]
- Chen R, Mias GI, Li-Pook-Than J, Jiang L, Lam HY, Chen R, Miriami E, Karczewski KJ, Hariharan M, Dewey FE, et al. (2012). Personal omics profiling reveals dynamic molecular and medical phenotypes. Cell 148, 1293–1307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christensen KE, Wu Q, Wang X, Deng L, Caudill MA, and Rozen R (2010). Steatosis in mice is associated with gender, folate intake, and expression of genes of one-carbon metabolism. J. Nutr. 140, 1736–1741. [DOI] [PubMed] [Google Scholar]
- Christodoulides C, Dyson P, Sprecher D, Tsintzas K, and Karpe F (2009). Circulating fibroblast growth factor 21 is induced by peroxisome proliferator-activated receptor agonists but not ketosis in man. J. Clin. Endocrinol. Metab. 94, 3594–3601. [DOI] [PubMed] [Google Scholar]
- Crawford PA, Crowley JR, Sambandam N, Muegge BD, Costello EK, Hamady M, Knight R, and Gordon JI (2009). Regulation of myocardial ketone body metabolism by the gut microbiota during nutrient deprivation. Proc. Natl. Acad. Sci. USA 106, 11276–11281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- David LA, Maurice CF, Carmody RN, Gootenberg DB, Button JE, Wolfe BE, Ling AV, Devlin AS, Varma Y, Fischbach MA, et al. (2014). Diet rapidly and reproducibly alters the human gut microbiome. Nature 505, 559–563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diraison F, Moulin P, and Beylot M (2003). Contribution of hepatic de novo lipogenesis and reesterification of plasma non esterified fatty acids to plasma triglyceride synthesis during non-alcoholic fatty liver disease. Diabetes Metab. 29, 478–485. [DOI] [PubMed] [Google Scholar]
- Diraison F, Pachiaudi C, and Beylot M (1996). In vivo measurement of plasma cholesterol and fatty acid synthesis with deuterated water: determination of the average number of deuterium atoms incorporated. Metabolism 45, 817–821. [DOI] [PubMed] [Google Scholar]
- Donnelly KL, Smith CI, Schwarzenberg SJ, Jessurun J, Boldt MD, and Parks EJ (2005). Sources of fatty acids stored in liver and secreted via lipoproteins in patients with nonalcoholic fatty liver disease. J. Clin. Invest. 115, 1343–1351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ducker GS, and Rabinowitz JD (2017). One-carbon metabolism in health and disease. Cell Metab. 25, 27–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duncan SH, Belenguer A, Holtrop G, Johnstone AM, Flint HJ, and Lobley GE (2007). Reduced dietary intake of carbohydrates by obese subjects results in decreased concentrations of butyrate and butyrate-producing bacteria in feces. Appl. Environ. Microbiol. 73, 1073–1078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dushay J, Chui PC, Gopalakrishnan GS, Varela-Rey M, Crawley M, Fisher FM, Badman MK, Martinez-Chantar ML, and Maratos-Flier E (2010). Increased fibroblast growth factor 21 in obesity and nonalcoholic fatty liver disease. Gastroenterology 139, 456–463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fabbrini E, Sullivan S, and Klein S (2010). Obesity and nonalcoholic fatty liver disease: biochemical, metabolic, and clinical implications. Hepatology 51, 679–689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foster GD, Wyatt HR, Hill JO, McGuckin BG, Brill C, Mohammed BS, Szapary PO, Rader DJ, Edman JS, and Klein S (2003). A randomized trial of a low-carbohydrate diet for obesity. N. Engl. J. Med. 348, 2082–2090. [DOI] [PubMed] [Google Scholar]
- Gaggini M, Morelli M, Buzzigoli E, DeFronzo RA, Bugianesi E, and Gastaldelli A (2013). Non-alcoholic fatty liver disease (NAFLD) and its connection with insulin resistance, dyslipidemia, atherosclerosis and coronary heart disease. Nutrients 5, 1544–1560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gambino R, Musso G, and Cassader M (2011). Redox balance in the pathogenesis of nonalcoholic fatty liver disease: mechanisms and therapeutic opportunities. Antioxid. Redox Signal. 15, 1325–1365. [DOI] [PubMed] [Google Scholar]
- Ginsberg HN, Le NA, Goldberg IJ, Gibson JC, Rubinstein A, Wang-Iverson P, Norum R, and Brown WV (1986). Apolipoprotein B metabolism in subjects with deficiency of apolipoproteins CIII and AI. Evidence that apolipoprotein CIII inhibits catabolism of triglyceride-rich lipoproteins by lipoprotein lipase in vivo. J. Clin. Invest. 78, 1287–1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haas JT, Francque S, and Staels B (2016). Pathophysiology and mechanisms of nonalcoholic fatty liver disease. Annu. Rev. Physiol. 78, 181–205. [DOI] [PubMed] [Google Scholar]
- Halsted CH, Villanueva JA, Devlin AM, Niemela O, Parkkila S, Garrow TA, Wallock LM, Shigenaga MK, Melnyk S, and James SJ (2002). Folate deficiency disturbs hepatic methionine metabolism and promotes liver injury in the ethanol-fed micropig. Proc. Natl. Acad. Sci. USA 99, 10072–10077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirsch S, Poniachick J, Avendano M, Csendes A, Burdiles P, Smok G, Diaz JC, and de la Maza MP (2005). Serum folate and homocysteine levels in obese females with non-alcoholic fatty liver. Nutrition 21, 137–141. [DOI] [PubMed] [Google Scholar]
- Hong S, Moreno-Navarrete JM, Wei X, Kikukawa Y, Tzameli I, Prasad D, Lee Y, Asara JM, Fernandez-Real JM, Maratos-Flier E, et al. (2015). Nicotinamide N-methyltransferase regulates hepatic nutrient metabolism through Sirt1 protein stabilization. Nat. Med. 21, 887–894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanehisa M, and Goto S (2000). KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 28, 27–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karlsson FH, Nookaew I, and Nielsen J (2014). Metagenomic data utilization and analysis (MEDUSA) and construction of a global gut microbial gene catalogue. PLoS Comput. Biol. 10, e1003706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kekwick A, and Pawan GL (1956). Calorie intake in relation to body-weight changes in the obese. Lancet 271, 155–161. [DOI] [PubMed] [Google Scholar]
- Kelley B, Totter JR, and Day PL (1950). The lipotropic effect of folic acid on rats receiving various purified diets. J. Biol. Chem. 187, 529–535. [PubMed] [Google Scholar]
- Keogh JB, Brinkworth GD, Noakes M, Belobrajdic DP, Buckley JD, and Clifton PM (2008). Effects of weight loss from a very-low-carbohydrate diet on endothelial function and markers of cardiovascular disease risk in subjects with abdominal obesity. Am. J. Clin. Nutr. 87, 567–576. [DOI] [PubMed] [Google Scholar]
- Kersten S (2014). Integrated physiology and systems biology of PPARalpha. Mol. Metab. 3, 354–371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim TH, Yang J, Darling PB, and O’Connor DL (2004). A large pool of available folate exists in the large intestine of human infants and piglets. J. Nutr. 134, 1389–1394. [DOI] [PubMed] [Google Scholar]
- Kindt R, and Coe R (2005). Tree Diversity Analysis. A Manual and Software for Common Statistical Methods for Ecological and Biodiversity Studies (World Agroforestry Centre; ). [Google Scholar]
- Kolb AF, and Petrie L (2013). Folate deficiency enhances the inflammatory response of macrophages. Mol. Immunol. 54, 164–172. [DOI] [PubMed] [Google Scholar]
- Korem T, Zeevi D, Suez J, Weinberger A, Avnit-Sagi T, Pompan-Lotan M, Matot E, Jona G, Harmelin A, Cohen N, et al. (2015). Growth dynamics of gut microbiota in health and disease inferred from single metagenomic samples. Science 349, 1101–1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kotronen A, Peltonen M, Hakkarainen A, Sevastianova K, Bergholm R, Johansson LM, Lundbom N, Rissanen A, Ridderstrale M, Groop L, et al. (2009). Prediction of non-alcoholic fatty liver disease and liver fat using metabolic and genetic factors. Gastroenterology 137, 865–872. [DOI] [PubMed] [Google Scholar]
- Laeger T, Henagan TM, Albarado DC, Redman LM, Bray GA, Noland RC, Munzberg H, Hutson SM, Gettys TW, Schwartz MW, et al. (2014). FGF21 is an endocrine signal of protein restriction. J. Clin. Invest. 124, 3913–3922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Roy T, Llopis M, Lepage P, Bruneau A, Rabot S, Bevilacqua C, Martin P, Philippe C, Walker F, Bado A, et al. (2013). Intestinal microbiota determines development of non-alcoholic fatty liver disease in mice. Gut 62, 1787–1794. [DOI] [PubMed] [Google Scholar]
- LeBlanc JG, Laino JE, del Valle MJ, Vannini V, van Sinderen D, Taranto MP, de Valdez GF, de Giori GS, and Sesma F (2011). B-group vitamin production by lactic acid bacteria-current knowledge and potential applications. J. Appl. Microbiol. 111, 1297–1309. [DOI] [PubMed] [Google Scholar]
- Lee S, Zhang C, Kilicarslan M, Piening BD, Bjornson E, Hallstrom BM, Groen AK, Ferrannini E, Laakso M, Snyder M, et al. (2016). Integrated network analysis reveals an association between plasma mannose levels and insulin resistance. Cell Metab. 24, 172–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leung C, Rivera L, Furness JB, and Angus PW (2016). The role of the gut microbiota in NAFLD. Nat. Rev. Gastroenterol. Hepatol. 13, 412–425. [DOI] [PubMed] [Google Scholar]
- Lewis GF, Carpentier A, Adeli K, and Giacca A (2002). Disordered fat storage and mobilization in the pathogenesis of insulin resistance and type 2 diabetes. Endocr. Rev. 23, 201–229. [DOI] [PubMed] [Google Scholar]
- Li H, Fang Q, Gao F, Fan J, Zhou J, Wang X, Zhang H, Pan X, Bao Y, Xiang K, et al. (2010). Fibroblast growth factor 21 levels are increased in nonalcoholic fatty liver disease patients and are correlated with hepatic triglyceride. J. Hepatol. 53, 934–940. [DOI] [PubMed] [Google Scholar]
- Li H, Zhang J, and Jia W (2013). Fibroblast growth factor 21: a novel metabolic regulator from pharmacology to physiology. Front. Med. 7, 25–30. [DOI] [PubMed] [Google Scholar]
- Lips MA, de Groot GH, Berends FJ, Wiezer R, van Wagensveld BA, Swank DJ, Luijten A, van Dijk KW, Pijl H, Jansen PL, et al. (2014). Calorie restriction and Roux-en-Y gastric bypass have opposing effects on circulating FGF21 in morbidly obese subjects. Clin. Endocrinol. (Oxf.) 81, 862–870. [DOI] [PubMed] [Google Scholar]
- Lofgren L, Forsberg GB, and Stahlman M (2016). The BUME method: a new rapid and simple chloroform-free method for total lipid extraction of animal tissue. Sci. Rep. 6, 27688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lonardo A, Ballestri S, Marchesini G, Angulo P, and Loria P (2015). Nonalcoholic fatty liver disease: a precursor of the metabolic syndrome. Dig. Liver Dis. 47, 181–190. [DOI] [PubMed] [Google Scholar]
- Loomba R, Seguritan V, Li W, Long T, Klitgord N, Bhatt A, Dulai PS, Caussy C, Bettencourt R, Highlander SK, et al. (2017). Gut microbiome-based metagenomic signature for non-invasive detection of advanced fibrosis in human nonalcoholic fatty liver disease. Cell Metab. 25, 1054–1062.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love MI, Huber W, and Anders S (2014). Moderated estimation of fold change and dispersion for RNA-seq datawith DESeq2. Genome Biol. 15, 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lundbom J, Hakkarainen A, Soderlund S, Westerbacka J, Lundbom N, and Taskinen MR (2011). Long-TE 1H MRS suggests that liver fat is more saturated than subcutaneous and visceral fat. NMR Biomed. 24, 238–245. [DOI] [PubMed] [Google Scholar]
- Mardinoglu A, Agren R, Kampf C, Asplund A, Nookaew I, Jacobson P, Walley AJ, Froguel P, Carlsson LM, Uhlen M, et al. (2013). Integration of clinical data with a genome-scale metabolic model of the human adipocyte. Mol. Syst. Biol. 9, 649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mardinoglu A, Agren R, Kampf C, Asplund A, Uhlen M, and Nielsen J (2014). Genome-scale metabolic modelling of hepatocytes reveals serine deficiency in patients with non-alcoholic fatty liver disease. Nat. Commun. 5, 3083. [DOI] [PubMed] [Google Scholar]
- Mardinoglu A, Bjornson E, Zhang C, Klevstig M, Soderlund S, Stahlman M, Adiels M, Hakkarainen A, Lundbom N, Kilicarslan M, et al. (2017). Personal model-assisted identification of NAD+ and glutathione metabolism as intervention target in NAFLD. Mol. Syst. Biol. 13, 916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mardinoglu A, Shoaie S, Bergentall M, Ghaffari P, Zhang C, Larsson E, Backhed F, and Nielsen J (2015). The gut microbiota modulates host amino acid and glutathione metabolism in mice. Mol. Syst. Biol. 11, 834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marengo A, Rosso C, and Bugianesi E (2016). Liver cancer: connections with obesity, fatty liver, and cirrhosis. Annu. Rev. Med. 67, 103–117. [DOI] [PubMed] [Google Scholar]
- McGarry JD, Mannaerts GP, and Foster DW (1977). A possible role for malonyl-CoA in the regulation of hepatic fatty acid oxidation and ketogenesis. J. Clin. Invest. 60, 265–270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McMurdie PJ, and Holmes S (2013). phyloseq: an R package for reproducible interactive analysis and graphics of microbiome census data. PLoS One 8, e61217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy RC, James PF, McAnoy AM, Krank J, Duchoslav E, and Barkley RM (2007). Detection of the abundance of diacylglycerol and triacylglycerol molecular species in cells using neutral loss mass spectrometry. Anal. Biochem. 366, 59–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen D, Samson SL, Reddy VT, Gonzalez EV, and Sekhar RV (2013). Impaired mitochondrial fatty acid oxidation and insulin resistance in aging: novel protective role of glutathione. Aging Cell 12, 415–425. [DOI] [PubMed] [Google Scholar]
- Noakes M, Keogh JB, Foster PR, and Clifton PM (2005). Effect of an energy-restricted, high-protein, low-fat diet relative to a conventional highcarbohydrate, low-fat diet on weight loss, body composition, nutritional status, and markers of cardiovascular health in obese women. Am. J. Clin. Nutr. 81, 1298–1306. [DOI] [PubMed] [Google Scholar]
- Nuttall FQ, and Gannon MC (2006). The metabolic response to a highprotein, low-carbohydrate diet in men with type 2 diabetes mellitus. Metabolism 55, 243–251. [DOI] [PubMed] [Google Scholar]
- Paredes-Turrubiarte G, Gonzalez-Chavez A, Perez-Tamayo R, Salazar-Vazquez BY, Hernandez VS, Garibay-Nieto N, Fragoso JM, and Escobedo G (2016). Severity of non-alcoholic fatty liver disease is associated with high systemic levels of tumor necrosis factor alpha and low serum interleukin 10 in morbidly obese patients. Clin. Exp. Med. 16, 193–202. [DOI] [PubMed] [Google Scholar]
- Pawlak M, Lefebvre P, and Staels B (2015). Molecular mechanism of PPARalpha action and its impact on lipid metabolism, inflammation and fibrosis in non-alcoholic fatty liver disease. J. Hepatol. 62, 720–733. [DOI] [PubMed] [Google Scholar]
- Petersen KF, Dufour S, Savage DB, Bilz S, Solomon G, Yonemitsu S, Cline GW, Befroy D, Zemany L, Kahn BB, et al. (2007). The role of skeletal muscle insulin resistance in the pathogenesis of the metabolic syndrome. Proc. Natl. Acad. Sci. USA 104, 12587–12594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petersen MC, Vatner DF, and Shulman GI (2017). Regulation of hepatic glucose metabolism in health and disease. Nat. Rev. Endocrinol. 13, 572–587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pramfalk C, Pavlides M, Banerjee R, McNeil CA, Neubauer S, Karpe F, and Hodson L (2015). Sex-specific differences in hepatic fat oxidation and synthesis may explain the higher propensity for NAFLD in men. J. Clin. Endocrinol. Metab. 100, 4425–4433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price ND, Magis AT, Earls JC, Glusman G, Levy R, Lausted C, McDonald DT, Kusebauch U, Moss CL, Zhou Y, et al. (2017).A wellness study of 108 individuals using personal, dense, dynamic data clouds. Nat. Biotechnol. 35, 747–756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rinella ME, and Sanyal AJ (2016). Management of NAFLD: a stage-based approach. Nat. Rev. Gastroenterol. Hepatol. 13, 196–205. [DOI] [PubMed] [Google Scholar]
- Rossi M, Amaretti A, and Raimondi S (2011). Folate production by probiotic bacteria. Nutrients 3, 118–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roza AM, and Shizgal HM (1984). The Harris Benedict equation reevaluated: resting energy requirements and the body cell mass. Am. J. Clin. Nutr. 40, 168–182. [DOI] [PubMed] [Google Scholar]
- Sid V, Siow YL, and O K (2017). Role of folate in nonalcoholic fatty liver disease. Can. J. Physiol. Pharmacol. 95, 1141–1148. [DOI] [PubMed] [Google Scholar]
- Sid V, Wu N, Sarna LK, Siow YL, House JD, and O K (2015). Folic acid supplementation during high-fat diet feeding restores AMPK activation via an AMP-LKB1-dependent mechanism. Am. J. Physiol. Regul. Integr. Comp. Physiol. 309, R1215–R1225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh A, Gautier B, Shannon CP, Vacher M, Rohart F, Tebutt SJ, and Le Cao K-A (2016). DIABLO - an integrative, multi-omics, multivariate method for multi-group classification. bioRxiv. 10.1101/067611. [DOI] [Google Scholar]
- Solga S, Alkhuraishe AR, Clark JM, Torbenson M, Greenwald A, Diehl AM, and Magnuson T (2004). Dietary composition and nonalcoholic fatty liver disease. Dig. Dis. Sci. 49, 1578–1583. [DOI] [PubMed] [Google Scholar]
- Ståhlman M, Davidsson P, Kanmert I, Rosengren B, Borén J, Fagerberg B, and Camejo G (2008). Proteomics and lipids of lipoproteins isolated at low salt concentrations in D2O/sucrose or in KBr. J. Lipid Res. 49, 481–490. [DOI] [PubMed] [Google Scholar]
- Stefan N, Kantartzis K, and Haring HU (2008). Causes and metabolic consequences of fatty liver. Endocr. Rev. 29, 939–960. [DOI] [PubMed] [Google Scholar]
- Strozzi GP, and Mogna L (2008). Quantification of folic acid in human feces after administration of Bifidobacterium probiotic strains. J. Clin. Gastroenterol. 42 (Suppl. 3 Pt 2), S179–S184. [DOI] [PubMed] [Google Scholar]
- Sugahara H, Odamaki T, Hashikura N, Abe F, and Xiao JZ (2015). Differences in folate production by bifidobacteria of different origins. Biosci. Microbiota Food Health 34, 87–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Targher G, Byrne CD, Lonardo A, Zoppini G, and Barbui C (2016). Non-alcoholic fatty liver disease and risk of incident cardiovascular disease: a meta-analysis. J. Hepatol. 65, 589–600. [DOI] [PubMed] [Google Scholar]
- Taskinen MR, Kuusi T, Helve E, Nikkila EA, and Yki-Jarvinen H (1988). Insulin therapy induces antiatherogenic changes of serum lipoproteins in noninsulin-dependent diabetes. Arteriosclerosis 8, 168–177. [DOI] [PubMed] [Google Scholar]
- R Core Team. (2015). R: A Language and Environment for Statistical Computing (R Foundation for Statistical Computing; ). [Google Scholar]
- Tryndyak V, de Conti A, Kobets T, Kutanzi K, Koturbash I, Han T, Fuscoe JC, Latendresse JR, Melnyk S, Shymonyak S, et al. (2012). Interstrain differences in the severity of liver injury induced by a choline- and folate-deficient diet in mice are associated with dysregulation of genes involved in lipid metabolism. FASEB J. 26, 4592–4602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallner S, and Schmitz G (2011). Plasmalogens the neglected regulatory and scavenging lipid species. Chem. Phys. Lipids 164, 573–589. [DOI] [PubMed] [Google Scholar]
- Wang Y, Viscarra J, Kim SJ, and Sul HS (2015). Transcriptional regulation of hepatic lipogenesis. Nat. Rev. Mol. Cell Biol. 16, 678–689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wichmann A, Allahyar A, Greiner TU, Plovier H, Lundén GO, Larsson T, Drucker DJ, Delzenne NM, Cani PD, and Backhed F (2013). Microbial modulation of energy availability in the colon regulates intestinal transit. Cell Host Microbe 14, 582–590. [DOI] [PubMed] [Google Scholar]
- Wieckowska A, Papouchado BG, Li Z, Lopez R, Zein NN, and Feldstein AE (2008). Increased hepatic and circulating interleukin-6 levels in human nonalcoholic steatohepatitis. Am. J. Gastroenterol. 103, 1372–1379. [DOI] [PubMed] [Google Scholar]
- Witthöft CM, Forssén K, Johannesson L, and Jägerstad M (1999). Folates - food sources, analyses, retention and bioavailability. Scand. J. Nutr. 43, 138–146. [Google Scholar]
- Wu GD, Chen J, Hoffmann C, Bittinger K, Chen YY, Keilbaugh SA, Bewtra M, Knights D, Walters WA, Knight R, et al. (2011). Linking long-term dietary patterns with gut microbial enterotypes. Science 334, 105–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu H, Esteve E, Tremaroli V, Khan MT, Caesar R, Manneras-Holm L, Stahlman M, Olsson LM, Serino M, Planas-Felix M, et al. (2017). Metformin alters the gut microbiome of individuals with treatment-naive type 2 diabetes, contributing to the therapeutic effects of the drug. Nat. Med. 23, 850–858. [DOI] [PubMed] [Google Scholar]
- Wu H, Tremaroli V, and Backhed F (2015). Linking microbiota to human diseases: a systems biology perspective. Trends Endocrinol. Metab. 26, 758–770. [DOI] [PubMed] [Google Scholar]
- Wycherley TP, Brinkworth GD, Keogh JB, Noakes M, Buckley JD, and Clifton PM (2010). Long-term effects of weight loss with a very low carbohydrate and low fat diet on vascular function in overweight and obese patients. J. Intern. Med. 267, 452–461. [DOI] [PubMed] [Google Scholar]
- Xia MF, Bian H, Zhu XP, Yan HM, Chang XX, Zhang LS, Lin HD, Hu XQ, and Gao X (2017). Serum folic acid levels are associated with the presence and severity of liver steatosis in Chinese adults. Clin. Nutr. 10.1016/j.clnu.2017.06.021. [DOI] [PubMed] [Google Scholar]
- Young MD, Wakefield MJ, Smyth GK, and Oshlack A (2010). Gene ontology analysis for RNA-seq: accounting for selection bias. Genome Biol. 11, R14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou D, Pan Q, Shen F, Cao HX, Ding WJ, Chen YW, and Fan JG (2017). Total fecal microbiota transplantation alleviates high-fat diet-induced steatohepatitis in mice via beneficial regulation of gut microbiota. Sci. Rep. 7, 1529. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Datasets Generated and Deposited
Metagenomics data from the first cohort were deposited at Sequence Read Archive with accession number SRA: PRJNA420817. RNA-seq raw and processed files from double liver needle biopsies taken before and after one-week diet intervention from the second cohort were deposited at NCBI Gene Expression Omnibus with accession number GEO: GSE107650.