

Abstract
Mutations in a family with sequence similarity 83 member H (FAM83H) cause autosomal-dominant hypocalcification amelogenesis imperfecta (ADH CAI). All FAM83H ADHCAI-causing mutations terminate translation or shift the reading frame within the specific exon 5 segment that encodes from Ser287 to Glu694. Mutations near Glu694 cause a milder, more localized phenotype. We identified disease-causing FAM83H mutations in two families with ADHCAI: family 1 (g.3115C>T, c.1993 C>T, p.Q665X) and family 2 (g.3151C>T, c.2029 C>T, p.Q677X). We also tested the hypothesis that truncation mutations alter the intracellular localization of FAM83H. Wild-type FAM83H and p.E694X mutant FAM83H fused to green fluorescent protein (GFP) localized in the cytoplasm of HEK293T cells, but the mutant FAM83H proteins (p.R325X, p.W460X, and p.Q677X) fused to GFP localized mainly in the nucleus with slight expression in the cytoplasm. We conclude that nuclear targeting of the truncated FAM83H protein contributes to the severe, generalized enamel phenotype.
Keywords: amelogenesis imperfect, enamel, hypocalcification, protein localization
Introduction
Inherited diseases manifested as isolated (non-syndromic) enamel malformations are grouped under the designation of amelogenesis imperfecta (AI) (Witkop and Sauk, 1977), which breaks down into sub-categories based upon enamel phenotype and mode of inheritance (Witkop, 1988). Inherited enamel malformations, although varied, tend to fall into three clinical patterns. Hypoplastic enamel is thin but hard, hypomaturation enamel is normal in thickness but soft, and hypocalcification enamel is thin and soft, often abrading soon after tooth eruption (Hu et al., 2007).
Genes encoding enamel extracellular matrix proteins and proteases contribute to the etiology of AI (Kim et al., 2006). X-linked AI is caused by mutations in amelogenin (AMELX; MIM *300391) (Lagerström et al., 1991), autosomal-dominant hypoplastic AI by enamelin (ENAM; MIM *606585) (Rajpar et al., 2001), autosomal-recessive hypomaturation AI by enamelysin (MMP20; MIM *604629) (Kim et al., 2005), and kallikrein 4 (KLK4; MIM *603767) (Hart et al., 2004). Some genes encoding intracellular proteins are also associated with AI. Autosomal-dominant hypocalcified AI (ADHCAI) is caused by mutations in a family with sequence similarity 83 member H (FAM83H; MIM *611927 and #130900) (Kim et al., 2008) and autosomal-recessive hypomaturation AI by WD repeat-containing protein 72 (WDR72; MIM 613214) (El-Sayed et al., 2009a). Both of these proteins appear to function in the secretory pathway. About half of AI cases are of unknown etiology.
Among the genes that cause AI, FAM83H causes the highest percentage of cases and the most severe enamel malformations. FAM83H (chromosome 8q24.3) encodes a protein having 1179 amino acids, most of which (933 amino acids) are encoded by the last exon (exon 5). All of the AI-causing FAM83H mutations reported to date are in the last coding exon and cause premature translation termination: p.S287X, pY297X, p.L308fsX323, p.R325X, p.Q398X, p.E415X, p.Q444X, p.Q456X, p.Y458X p.W460X, p.Q470X, p.L625fsX703, pQ677X, and pE694X (Kim et al., 2008; Lee et al., 2008; Ding et al., 2009; El-Sayed et al., 2009b; Hart et al., 2009; Wright et al., 2009). The putative truncated proteins contain between 286 and 693 FAM83H N-terminal amino acids. The majority of FAM83H mutations cause hypocalcified yellowish-brown enamel over the entire crown, while the most downstream mutations (p.L625fsX703 and p.E694X) cause malformations that localize in the cervical half of the crown (Wright et al., 2009).
The observations that all AI-causing mutations in FAM83H are in the last coding exon and truncate the protein, and that downstream mutations result in a localized phenotype provide clues to the underlying pathology. Premature translation termination codons in the last coding exon typically escape nonsense-mediated decay (Shyu et al., 2008), although a downstream region, such as the 3′ UTR, can sometimes trigger nonsense-mediated decay in response to a premature stop codon in the terminal exon (Tan et al., 2008). Nonsense-mediated degradation of the mutant mRNA would result in haploinsufficiency, or half the amount of normal FAM83H protein produced. The dominant pattern of inheritance, however, and the absence of FAM83H mutations that would predictably cause haploinsufficiency strongly suggest that the mutant mRNAs are stable and translate truncated FAM83H proteins that cause dominant-negative effects. These pathological effects lessen when the size of the truncated FAM83H protein is relatively large, that is, when it reaches the length of the longest truncation (p.E694X), which causes a milder, more localized form of ADHCAI.
Here we report 2 ADHCAI-causing mutations (p.Q665X and p.Q677X) that are near the 3′ limit of the observed FAM83H mutations and near the border that shifts from a generalized to a localized enamel phenotype, and we test the hypothesis that FAM83H truncation mutations affect the intracellular localization of the FAM83H protein.
Materials & Methods
Identification of Kindreds and Enrollment of Human Study Population
Two Korean families with hypocalcified-type AI were recruited. Clinical and radiologic examinations were performed, and blood samples were collected with the understanding and written consent of each participant according to the Declaration of Helsinki. This study was independently reviewed and approved by the Institutional Review Board at the Pusan National University Dental Hospital and the Seoul National University Dental Hospital.
Primer Design, Polymerase Chain-reaction (PCR), and DNA Sequencing
Genomic DNA was extracted from peripheral whole blood by means of the QuickGene DNA whole blood kit S with QuickGene-Mini80 equipment (Fujifilm, Tokyo, Japan) according to the manufacturer’s instructions. Based upon a candidate gene approach, exons and exon-intron boundaries of FAM83H were amplified as previously described (Kim et al., 2008). PCR amplifications were performed with the HiPi DNA polymerase premix (ElpisBio, Taejeon, Korea), and the products were purified with a PCR Purification Kit (ElpisBio). DNA sequencing was performed at the DNA sequencing center (Macrogen, Seoul, Korea).
Cloning of Expression Vector and Mutagenesis
Total RNA was isolated from primary cell culture of human enamel organ epithelium with the Trizol reagent (Invitrogen, Carlsbad, CA, USA). First-strand cDNA was made with oligo(dT)18 primer with a Transcriptor First Strand cDNA synthesis kit (Roche, Mannheim, Germany) according to the instructions of the manufacturer. FAM83H coding sequence was amplified (sense primer, 5′-ggagatctATGGCCCGTCGCTCTCAGAGCT; antisense primer, 5′-ggaatTCACTTCTTGCTTTTGAACGTG). The amplified product was cloned into the pEGFP-C1 vector (Clontech, Mountainview, CA, USA) after digestion with BglII and EcoRI restriction endonucleases. The engineering strategy placed the FAM83H translation initiation codon in-frame with an N-terminal green fluorescent protein (GFP) domain. PCR mutagenesis was performed to introduce nonsense mutations: (p.Q677X) sense, 5′-CTGAACCCCCTGGTCTAGCGCAGCTCCAGG, antisense, 5′-CCTGGAGCTGCGCTAGACCAGGGGGTTCAG; (p.E694X) sense, 5′-AGCACGTCACAGGCCTAGGGCGCGGCCGGG, antisense, 5′-CCCGGCCGCGCCCTAGGCCTGTGACGTGCT; (p.R325X) sense, 5′-TCTCCTTCCCTAAATGAGCGCACCTCCTG, antisense, 5′-CAGGAGGTGCGCTCATTTAGGGAAGGAGA; and (p.W460X) sense, 5′-CAGCAGTACCAGTAGGACCCGCAGCTCAC, antisense, 5′-GTGAGCTGCGGGTCCTACTGGTACTGCTG). Correct clones were selected and confirmed by direct sequencing.
Analysis of Intracellular Localization
Plasmid DNA of the pEGFP-C1 vector that expresses GFP and pEGFP-C1 expressing wild-type FAM8hH and FAM83H mutants fused to GFP was purified with the AccuPrep Nano-Plus Plasmid Midi Extraction Kit (Bioneer, Taejeon, Korea). HEK293T cells were cultured in Dulbecco’s Modified Eagle Medium (DMEM) with 10% FBS (GIBCO-BRL, Carlsbad, CA, USA) and antibiotic-antimycotic liquid (GIBCO-BRL). For transfection, cells were grown on 6-well plates and transfected with Lipofectamine2000 (Invitrogen, Carlsbad, CA, USA). After 24 hrs, transfected cells were trypsinized and seeded on poly-L-lysine-coated coverglass in 6-well plates. The next day, the cells were fixed with 4% paraformaldehyde, and nuclear DNA was stained in 20 µM bisBenzimide H 33342 trihydrochloride (Sigma, St. Louis, MO, USA) for 10 min. The samples were washed in PBS and mounted with Fluorescent Mounting Medium (DAKO, Glostrup, Denmark). Confocal laser scanning was performed with an OLYMPUS-FV300 fluorescence microscope.
Results
Mutation Results
Mutational analysis revealed nonsense mutations in exon 5 of the FAM83H gene. A novel nonsense mutation (g.3115C>T, c.1993C>T, p.Q665X) was identified in family 1 (Figs. 1A-1C). The nonsense mutation (g.3151C>T, c.2029C>T, p.Q677X) in family 2 was previously reported in a Caucasian family (Figs. 2A-2C) (Lee et al., 2008). Both mutations showed a dominant pattern of inheritance. These mutations are within the segment of exon 5 that was previously shown to cause ADHCAI and are near the transition zone where the phenotype diminishes to the less severe, localized form (Fig. 3).
Figure 1.
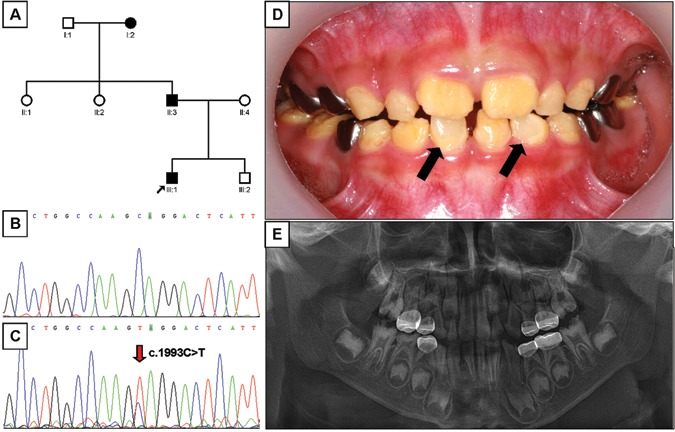
Pedigree, mutational analysis, clinical photo, and panoramic radiograph. (A) Pedigree of family 1. (B) DNA sequencing chromatogram of normal control. (C) DNA sequencing chromatogram of the proband of family 1. Mutated nucleotide (g.3115C>T, c.1993C>T, p.Q665X) is indicated by red arrow. (D) Frontal photograph of the proband of the family 1 taken at age 6.7 yrs. Normal-looking enamel surfaces are indicated by black arrows. (E) Dental panoramic radiograph of the proband of the family 1 taken at age 6.7 yrs.
Figure 2.
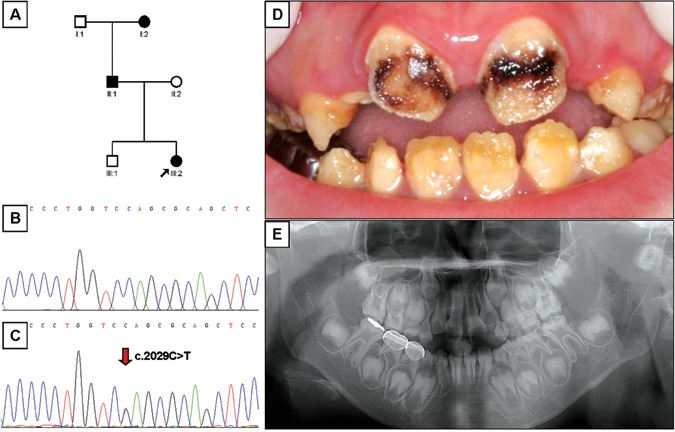
Pedigree, mutational analysis, clinical image, and panoramic radiograph. (A) Pedigree of family 2. (B) DNA sequencing chromatogram of normal control. (C) DNA sequencing chromatogram of the proband of family 2. Mutated nucleotide (g.3151C>T, c.2029C>T, p.Q677X) is indicated by red arrow. (D) Frontal photograph of the proband of the family 2 taken at age 8 yrs. (E) Dental panoramic radiograph of the proband of the family 2 taken at age 8 yrs.
Figure 3.
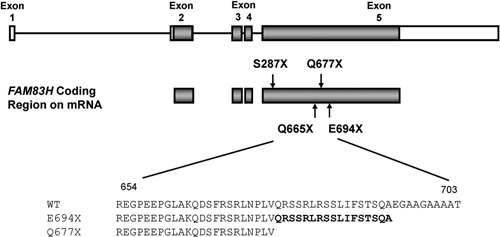
Gene diagram showing the 5 exons (boxes) and 4 introns (bars) of FAM83H. The coding regions in exons are shaded. The p.S287X is the most 5′ nonsense mutation, and the p.E694X is the most 3′ nonsense mutation. The p.Q665X and p.Q677X mutations are identified in this study. Amino acid sequences (WT, wild-type; E694X, Q677X) are aligned, and 17 amino acids (Q677-A693) are marked in bold.
Clinical Findings
The proband 1 of family 1 was a 6.7-year-old boy who presented with rough, yellowish-brown enamel, although normal-looking enamel could be identified in several teeth (Fig. 1D). Radio-graphically, the enamel layer was generally thin and reduced in density (could not readily be distinguished from dentin) (Fig. 1E). The proband of family 2 was an 8-year-old girl with yellowish-brown enamel. Notably, her maxillary incisors had lost most of their enamel and were stained dark brown, presumably due to extrinsic staining (Fig. 2D). As in proband 1, the radiographic density of the enamel was reduced and similar in density to the underlying dentin (Fig. 2E). The permanent first molars of both probands showed signs of taurodontism on radiographs. The diagnosis for both probands was the generalized (severe) form of ADHCAI.
Analysis of Intracellular Localization
The pEGFP-C1 vector expressing GFP showed a diffuse fluorescent signal throughout the transfected HEK293T cells (Fig. 4A). The wild-type FAM83H fused to GFP (Fig. 4B) localized to the cytoplasm of transfected HEK293T cells. But the mutant FAM83H proteins (p.R325X, p.W460X, and p.Q677X) fused to GFP localized mainly in the nucleus, with slight expression in the cytoplasm (Figs. 4C-4E). The p.E694X mutant FAM83H fused to GFP (Fig. 4D) localized to the cytoplasm like the wild-type protein (Fig. 4F).
Figure 4.
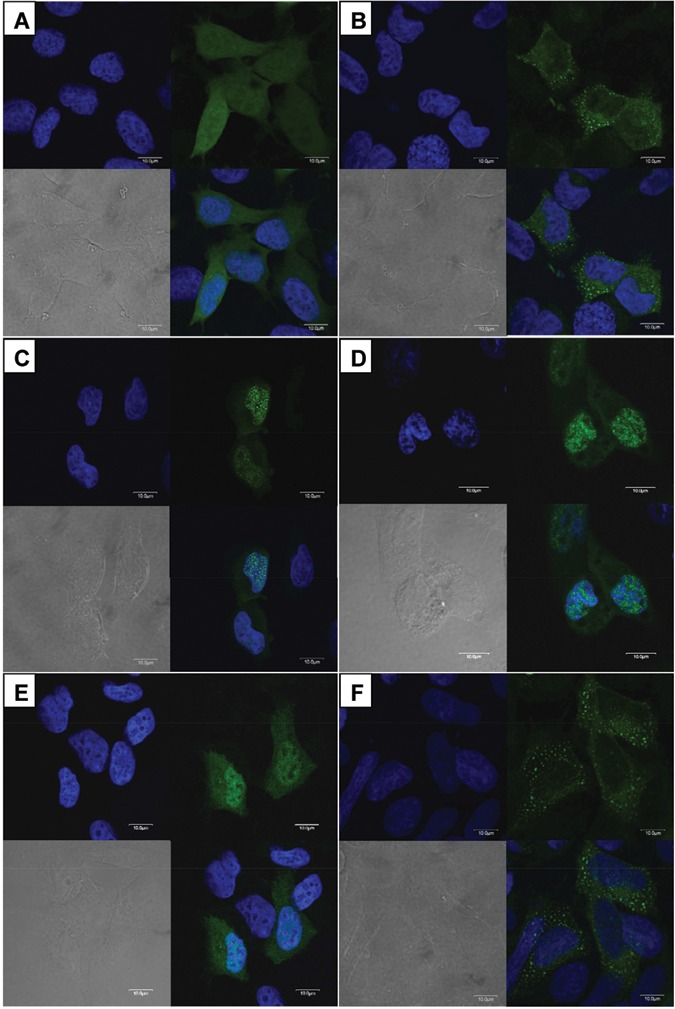
Analysis of intracellular localization. HEK293T cells transfected with pEGFP-C1 vectors expressing wild-type and mutant FAM83H (green). (A) Widespread expression of empty pEGFP-C1 vector. (B) Wild-type FAM83H localized in the cytoplasm. (C) p.Q677X FAM83H localized mainly in the nucleus, with slight expression in the cytoplasm. (D) p.W460X FAM83H localized mainly in the nucleus, with slight expression in the cytoplasm. (E) p.R325X FAM83H showed a localization pattern similar to that of p.Q677X FAM83H. (F) p.E694X FAM83H localized in the cytoplasm like wild-type protein.
Discussion
FAM83H is a gene that was originally discovered during computer analyses of the human genomic sequence and entered the literature in 2008 with the discovery that it is a major contributor to the etiology of AI (Kim et al., 2008). This report brings to 20 the number of families with one of 16 novel FAM83H mutations that cause ADHCAI. Among the 16, 14 introduced nonsense codons and 2 introduced frameshifts. All terminated the protein prematurely between amino acids Ser287 and Glu694. No AI-causing mutations have been identified in the N-terminal region, encoding Met1 to Pro286, which corresponds closely to the part of FAM83H (Arg4 to Leu284) that is included in the conserved domain database (CDD) for the phospholipase D (PLD) superfamily (Marchler-Bauer et al., 2007). This is also the only part of FAM83H that shares amino acid sequence homology with the other 7 members (FAM83A to FAM83G) of the group (Ding et al., 2009). No AI-causing mutations have been found in the downstream part of exon 5 that encodes from Gly695 to Lys1179, which is only homologous with other FAM83H proteins from vertebrates going back to fish (GenBank #CAF99376.1 and #NM_001045090.1). This report adds two more ADHCAI families with defined FAM83H mutations and describes their clinical phenotypes.
Why have FAM83H mutations before p.S287X or after p.E694X not been found to cause ADHCAI? It is possible that mutations affecting a broader segment of FAM83H will be discovered with the characterization of more families. However, 20 families have been characterized, and independent mutations at 3 sites (p.Q398X, p.Q456X, p.Q677X) have already been reported, suggesting that the range of mutations that cause ADHCAI has been mapped out (at least roughly). Alternatively, the repeat mutations could represent mutational ‘hotspots’, and a broader range of disease-causing FAM83H mutations will be discovered with more work. The characterization of FAM83H defects in 20 independent families with ADHCAI has not identified any AI-causing mutations in the 5′ and 3′ regions of FAM83H. Previously, we argued that the premature translation termination codons in the last coding exon of FAM83H probably allows these transcripts to escape nonsense-mediated decay, so that truncated FAM83H protein is translated and the ADHCAI phenotype is not due to a simple 50% reduction in the amount of FAM83H, but is more likely to be caused by the dominant-negative effects of expressing truncated FAM83H that might interact with wild-type FAM83H (expressed from the wild-type allele) or with another protein, forming a non-functional dimer or multimer (Ding et al., 2009). Upstream mutations that cause premature truncation of FAM83H are assumed to cause degradation of the mRNA and a 50% reduction in the amount of wild-type protein, which does not cause the ADHCAI phenotype.
In situ hybridization of developing teeth demonstrated that Fam83h is expressed by cells in the ameloblast (enamel-forming epithelia) lineage (Lee et al., 2009). However, over 100 human FAM83H expressed sequence tags are currently listed in GenBank, and the EST profile shows that, while this protein may not be ubiquitous, it is certainly expressed in many tissues. These observations suggest that FAM83H functions in many cells throughout the body, but the ameloblast lineage is more sensitive than other cell types to FAM83H truncation mutations.
Based upon the apparent dominant-negative mechanism in the pathogenesis of ADHCAI, we hypothesize that the N-terminal phospholipase D homology domain of truncated FAM83H interacts with FAM83H protein expressed from the unmutated allele and interferes with its function. Wild-type human and mouse Fam83h have been shown not to localize in the nucleus (Ding et al., 2009). Our results showed that FAM83H truncations in the region of the protein associated with the most severe, generalized ADHCAI phenotype unmask a nuclear localization signal that greatly reduces the amount of FAM83H protein in the cytoplasm, while increasing its concentration in the nucleus. If the heterodimer of truncated and wild-type FAM83H is transported into the nucleus, it could lower the concentration of wild-type cytosolic FAM83H below what is required for normal function. Other scenarios can also be envisioned. FAM83H protein ectopically located in the nucleus could potentially interact with DNA. Previously, a prediction algorithm (9aa TAD) identified 162DLLSEVLEA as a motif in FAM83H that is common to the transactivation domains of many transcription factors (Kim et al., 2008). Truncated FAM83H might also interact with nuclear proteins and interfere with their function, causing dominant-negative effects.
Mutations in the far 3′ end of FAM83H do not appear to cause enamel defects. Analysis of our data suggests that, as the truncation mutations move past the 3′ end of the region associated with the more severe, generalized ADHCAI phenotype, the truncated FAM83H protein is not transported into the nucleus, but may still cause a less-severe, more localized form of ADHCAI. Perhaps the central domain of FAM83H, extending roughly from Ser287 to Glu694, serves a critical function in ameloblasts, and truncations beyond this central domain do less damage to its function. Further studies are needed to help us understand the complex mechanism of enamel formation and the critical role played by FAM83H in it.
Acknowledgments
We thank all the family members for their cooperation.
Footnotes
This work was supported by a grant from the Korea Science and Engineering Foundation (KOSEF) grant funded by the Korean Government (MEST) [No. R01-2008-000-10174-0 (2010)] and by the Korea Research Foundation Grant funded by the Korean Government (KRF-2008-313-E00597), and a Science Research Center grant to the Bone Metabolism Research Center (20100001741) funded by the Korean Ministry of Education, Science and Technology.
References
- Ding Y, Estrella MR, Hu YY, Chan HL, Zhang HD, Kim JW, et al. (2009). Fam83h is associated with intracellular vesicles and ADHCAI. J Dent Res 88:991-996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El-Sayed W, Parry DA, Shore RC, Ahmed M, Jafri H, Rashid Y, et al. (2009a). Mutations in the beta propeller WDR72 cause autosomal-recessive hypomaturation amelogenesis imperfecta. Am J Hum Genet 85:699-705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El-Sayed W, Shore RC, Parry DA, Inglehearn CF, Mighell AJ. (2009b). Ultrastructural analyses of deciduous teeth affected by hypocalcified amelogenesis imperfecta from a family with a novel Y458X FAM83H nonsense mutation. Cells Tissues Organs 191:235-239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hart PS, Hart TC, Michalec MD, Ryu OH, Simmons D, Hong S, et al. (2004). Mutation in kallikrein 4 causes autosomal recessive hypomaturation amelogenesis imperfecta. J Med Genet 41:545-549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hart PS, Becerik S, Cogulu D, Emingil G, Ozdemir-Ozenen D, Han ST, et al. (2009). Novel FAM83H mutations in Turkish families with autosomal dominant hypocalcified amelogenesis imperfecta. Clin Genet 75:401-404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu JC, Chun YH, Al Hazzazzi T, Simmer JP. (2007). Enamel formation and amelogenesis imperfecta. Cells Tissues Organs 186:78-85. [DOI] [PubMed] [Google Scholar]
- Kim JW, Simmer JP, Hart TC, Hart PS, Ramaswami MD, Bartlett JD, et al. (2005). MMP-20 mutation in autosomal recessive pigmented hypomaturation amelogenesis imperfecta. J Med Genet 42:271-275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JW, Simmer JP, Lin BP, Seymen F, Bartlett JD, Hu JC. (2006). Mutational analysis of candidate genes in 24 amelogenesis imperfecta families. Eur J Oral Sci 114(Suppl 1):3-12. [DOI] [PubMed] [Google Scholar]
- Kim JW, Lee SK, Lee ZH, Park JC, Lee KE, Lee MH, et al. (2008). FAM83H mutations in families with autosomal-dominant hypocalcified amelogenesis imperfecta. Am J Hum Genet 82:489-494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lagerström M, Dahl N, Nakahori Y, Nakagome Y, Backman B, Landegren U, et al. (1991). A deletion in the amelogenin gene (AMG) causes X-linked amelogenesis imperfecta (AIH1). Genomics 10:971-975. [DOI] [PubMed] [Google Scholar]
- Lee MJ, Lee SK, Lee KE, Kang HY, Jung HS, Kim JW. (2009). Expression patterns of the Fam83h gene during murine tooth development. Arch Oral Biol 54:846-850. [DOI] [PubMed] [Google Scholar]
- Lee SK, Hu JC, Bartlett JD, Lee KE, Lin BP, Simmer JP, et al. (2008). Mutational spectrum of FAM83H: the C-terminal portion is required for tooth enamel calcification. Hum Mutat 29:E95-E99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchler-Bauer A, Anderson J, Derbyshire M, DeWeese-Scott C, Gonzales N, Gwadz M, et al. (2007). CDD: a conserved domain database for interactive domain family analysis. Nucleic Acids Res 35:D237-D240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajpar MH, Harley K, Laing C, Davies RM, Dixon MJ. (2001). Mutation of the gene encoding the enamel-specific protein, enamelin, causes autosomal-dominant amelogenesis imperfecta. Hum Mol Genet 10:1673-1677. [DOI] [PubMed] [Google Scholar]
- Shyu AB, Wilkinson MF, van Hoof A. (2008). Messenger RNA regulation: to translate or to degrade. EMBO J 27:471-481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan JT, Kremer F, Freddi S, Bell KM, Baker NL, Lamande SR, et al. (2008). Competency for nonsense-mediated reduction in collagen X mRNA is specified by the 3′ UTR and corresponds to the position of mutations in Schmid metaphyseal chondrodysplasia. Am J Hum Genet 82:786-793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Witkop CJ., Jr (1988). Amelogenesis imperfecta, dentinogenesis imperfecta and dentin dysplasia revisited: problems in classification. J Oral Pathol 17:547-553. [DOI] [PubMed] [Google Scholar]
- Witkop CJ, Jr, Sauk JJ., Jr (1977). Heritable defects of enamel. In: Oral facial genetics. Stewart RE, Prescott GH, editors. St. Louis, MO: CV Mosby Co, pp. 151-226. [Google Scholar]
- Wright JT, Frazier-Bowers S, Simmons D, Alexander K, Crawford P, Han ST, et al. (2009). Phenotypic variation in FAM83H-associated amelogenesis imperfecta. J Dent Res 88:356-360. [DOI] [PMC free article] [PubMed] [Google Scholar]