

Abstract
Backgound and aims
The the low-density lipoprotein receptor-deficient (Ldlr−/−) mouse has been utilized by cardiovascular researchers for more than two decades to study atherosclerosis. However, there has not yet been a systematic effort to document the ultrastructural changes that accompany the progression of atherosclerotic plaque in this model.
Methods
Employing several different staining and microscopic techniques including immunohistochemistry, as well as electron and polarized microscopy, we analyzed atherosclerotic lesion development in Ldlr−/− mice fed an atherogenic diet over time.
Results
Lipid-like deposits occurred in the subendothelial space after only one week of atherogenic diet. At two weeks, cholesterol crystals (CC) formed and increased thereafter. Lipid, CC, vascular smooth muscles cells and collagen progressively increased over time, while after 4 weeks, relative macrophage content decreased. Accelerated accumulation of plate- and needle-shaped CC accompanied plaque core necrosis. Lastly, CC were surrounded by cholesterol microdomains, which co-localized with CC through all stages of atherosclerosis, indicating that the cholesterol microdomains may be a source of CC.
Conclusions
Here, we have documented for the first time in a comprehensive way, atherosclerotic plaque morphology and composition from early to advanced stages in the Ldlr−/− mouse, one of the most commonly used animal models utilized in atherosclerosis research.
Keywords: cholesterol crystal, atherosclerosis, inflammation, endothelial cells, macrophages, necrotic core, atherogenesis
Graphical Abstract
Introduction
Atherosclerosis, the leading cause of cardiovascular diseases (CVD), was responsible for approximately 32% of the 54 million deaths worldwide in 2013 [1]. Atherosclerosis is widely recognized as a chronic inflammatory disease that is accelerated by hyperlipidemia and involves many aspects of the immune system [2, 3]. Over time, complex plaques develop within the intima of medium and large arteries, which can rupture, leading to thrombosis and possible heart attack or stroke [4].
It is generally believed that one of the first steps in atherogenesis is dysfunction of the endothelial monolayer [5, 6], resulting in a cascade of events that promote white blood cell infiltration into the vessel wall, and accumulation in the subendothelial space. Macrophages contribute to atherosclerosis progression in mice and humans [7, 8], but can also be involved in plaque regression depending on their microenvironment and activation state [9]. Lipid metabolism, cytokine and chemokine secretion, and production of ECM-degrading matrix metalloproteases (MMP) are the preeminent functions of macrophages within the atherosclerotic lesion. These processes have been widely researched to find treatments aimed at preventing or reducing atherosclerosis development [10]. Recently, we have reported that under hyperlipidemic conditions in the initial stages of atherosclerosis formation, endothelial cells (EC) produce and secrete cholesterol crystals (CC) into the subendothelial space. EC exposed to hyperlipidemia accompanied by CC formation leads to increased E-selectin expression, accelerating monocyte adherence and transmigration [11]. Once in the subendothelial space, macrophages encounter endothelial-derived CC, as well as subendothelial lipids including lipid vesicles, lipid droplets and plasma-derived lipoproteins [12, 13], which results in lipid uptake and macrophage foam cell formation.
The effects of CC on macrophages were recently highlighted by publications reporting that CC present within the atherosclerotic lesion activate the macrophage NLRP3 inflammasome via Nrf-2, thus accelerating the progression of atherosclerosis [14–16]. CC have been reported in human atherosclerosis as early as 1959 [17], and in 1963 and 1976 it was demonstrated using X-ray diffraction that cholesterol crystalizes in the form of cholesterol monohydrate [18, 19]. Recent studies using techniques such as Raman spectroscopy and second harmonic generation microscopy have validated these findings [20, 21].
The biological origin of CC within cells of the plaque has been relatively unexplored, with existing data mainly focused on late stages of the disease. The identification of CC being formed by EC [11] within 1–2 weeks of initiating a HFD in atherosclerosis-prone mouse models [11, 15] has been reported. Until very recently, CC were thought to be produced solely by macrophages within the atherosclerotic plaque under the paradigm that macrophage foam cell death within the necrotic core causes the release of CC into the surroundings [22]. Physical factors in the necrotic core, including temperature, pH, hydration, and cholesterol solubilization, may play an important role in CC formation [23]. Active production of CC in macrophages has been shown utilizing primary human and mouse macrophages [24, 25], as well as a macrophage cell line [26, 27]. In addition, another important cell type in plaque formation, vascular smooth muscle cells, were also recently shown to be capable of CC production and secretion in vitro when loaded with cholesterol [28]. Advances have been made in understanding the origin and pathologic consequences of CC within the atherosclerotic lesion, including the identification of several cell types with the capacity to produce CC. However, very little information exists regarding in vivo CC production and its accumulation within plaques in early through late stage atherosclerosis development.
We believe that targeting CC formation and deposition could be a feasible approach for innovative therapies designed to ameliorate atherosclerosis. Therefore, we aimed to further understand the dynamics of CC formation in atherosclerotic lesions. For this purpose, we employed a variety of microscopic and histological techniques to analyze atherosclerosis development over time in high fat diet (HFD)-fed Ldlr−/− mice, one of the most consistent and widely used mouse models of atherosclerosis. These techniques provide a unique insight into atherogenesis and the formation of CC in vivo. Furthermore, we report the close association of cholesterol microdomains with CC in advanced plaque, and speculate as to the importance of these cholesterol microdomains for CC formation and disease progression.
Materials and methods
Mouse atherosclerosis study
All animals were handled in accordance with good animal practice as defined by the Institutional Animal Care and Use Committee (IACUC) and all other relevant animal welfare bodies. Equal numbers of male and female Ldlr−/− mice on C57BL/6 background were put on a high-fat diet (HFD) containing 15.8% (wt/wt) fat and 1.25% cholesterol (diet 94059; Harlan Teklad) for 0, 1, 2, 4, 8, 12, 16, and 36 weeks. 3–5 mice per time point. At sacrifice, aortas and hearts were used for quantification and immunohistological as well as electron microscopy analysis of atherosclerosis. Hearts of these mice were prepared for frozen serial sectioning of the aortic root. The extent of atherosclerosis plaque formation was detected in aortic root sections by staining with Oil Red-O. Plaque areas were measured using ImageJ software as described previously [29].
Scanning and transmission electron microscopy
Aortas were thoroughly cleaned from perivascular fat tissue. For scanning electron microscopy (SEM) the ascending aorta up to the carotid artery was excised and cut open longitudinally. For transmission electron microscopy (TEM) the aortic arch between carotid artery and left subclavian artery was used. Samples were immediately fixed with 4% glutaraldehyde and 2 mM calcium chloride in 0.1 M sodium cacodylate buffer, pH 7.2 for 24h at room temperature (RT). Sample preparation was performed as described previously [11, 29]. For TEM images were acquired on a Hitachi HT7700 transmission electron microscope operating at 100 kV with an AMT XR41 4-megapixel camera (Advanced Microscopy Techniques, Corp.). For SEM, images were acquired with a Hitachi S-4800 field emission scanning electron microscope operating at 5 kV. At least 4 aortas were examined per time point for both SEM and TEM imaging, with at least 25 pictures taken across each aorta, allowing a comprehensive analysis of the excised aortas.
Preparation of aortic root sections
Hearts were fixed using 4% PFA in PBS (pH7.4) for 24h, embedded in Tissue Tek and prepared for frozen sectioning. 5 mm serial sections of the aortic root were collected and subsequently used for further analysis. Analysis and preparation of aortic root sections and all quantifications were performed according to the scientific statement from the American Heart Association [30].
Immunohistological detection of macrophage and smooth muscle cell content
Aortic root sections were stained for macrophages (MOMA-2, ab33451, AbCAM) and smooth muscle cells (SM22a, 10493–1-AP, ProteinTech). Briefly, aortic root sections were rinsed for 5 min in PBS and trypsin antigen retrieval was performed for 15 min at 37°C [31]. Afterwards, sections were blocked for 30 min in 2% BSA/5% normal goat serum in PBS at room temperature. Primary antibodies (MOMA-2: 5 mg/ml, SM22α: 3 μg/ml) were added simultaneously in 2% BSA/PBS over night at 4°C, rinsed 3 times in PBS the following day, and subsequently labeled using cy2/cy3 goat-α-rat/rabbit 1:500 in 2% BSA/PBS for 1h at room temperature in the dark. Nuclei were labeled using DAPI (4’,6-Diamidine-2’-phenylindole dihydrochloride). Sections were rinsed thoroughly at the end of the incubation using PBS and mounted using DAKO Fluorescent mounting media (DAKO/Agilent S3023, USA). Images were taken with a Zeiss Axiovert Fluorescence microscope and quantified with ImageJ analysis.
Detection of cholesterol crystals
The presence of cholesterol crystals (CC) was determined in the 5 mm frozen aortic root sections using a polarized light microscope (Olympus IX81F-2 with IPlab imaging software) as described previously [11].
Quantification of necrotic core area
Routine H&E staining of aortic root frozen sections was performed. Staining was imaged using the NanoZoomer slide scanner (Hamamatsu, USA). Necrotic core size (nuclei-free area) was measured using ImageJ analysis.
Determination of changes in extracellular matrix components
Picro Sirius Red staining was performed on aortic root sections. Bright field images were obtained by scanning the slides using the NanoZoomer (Hamamatsu, USA). Polarized light images were taken using the Olympus IX81F-2 microscope with a Canon 600D attached to an eyepiece adapter.
Immunolabeling of cholesterol microdomains
Frozen paraformaldehyde-fixed aortic tissue sections were placed at room temperature overnight so that the tissue could dry. Drying before immunostaining was helpful for maintaining attachment of the tissue sections during the immunostaining procedure. After drying, sections were rinsed 3 times for 1 minute each rinse with 500 μl DPBS without calcium or magnesium. This rinsing helped remove OCT from the slide. All subsequent procedures were carried out at room temperature. Next, tissue sections were incubated 1 hour with 5 μg/ml purified IgM fraction of mouse anti-cholesterol microdomain MAb 58B1 [32] diluted in DPBS (with Ca2+ and Mg2+ for this and subsequent steps) containing 0.1% BSA. Control immunolabeling was performed with 5 pg/ml of an irrelevant purified mouse IgM fraction of anti-Clavibacter michiganense MAb (clone 9A1) (Agdia, Elkhart, IN) diluted in DPBS and containing 0.1% BSA. Mab IgM fractions were purified as previously described [33]. Cultures were then rinsed 3 times in DPBS, followed by a 30-minute incubation in 5 pg/ml biotinylated goat anti-mouse IgM diluted in DPBS containing 0.1% BSA. After 3 rinses in DPBS, cultures were incubated 10 minutes with 10 pg/ml streptavidin Alexa Fluor 488 diluted in DPBS. After a final 3 rinses with DPBS, slides were mounted in ProLongTM Gold Antifade mounting medium (P36930, Thermo Fisher Scientific, Grand Island, NY) containing DAPI nuclear stain. Sections were imaged with fluorescence and polarization microscopy using an Olympus IX81 microscope (Waltham, MA). Fluorescence of DAPI blue nuclear staining, Alexa Fluor 488 green cholesterol microdomain staining, and red autofluorescence staining were obtained with the following excitation and emission filters, respectively: exciter 360/40 and emitter 460/50 for DAPI; exciter 480/40 and emitter 535/50 for Alexa Fluor 488; and exciter 560/55 and emitter 645/75 for autofluorescence.
Statistical analysis
Statistical comparison between groups was performed using PRISM 7.0 (GraphPad) software. Data are represented as mean ± the standard error of the mean (SEM). Statistical significance was evaluated using appropriate statistical tests. All data sets were tested for Gaussian distribution. Because data were nonparametric and unpaired, comparison of 2 groups was performed using Mann-Whitney test. In cases of multiple parameter comparison, a one-way ANOVA statistical test was done. Significance was established at a p value of p<0.05. (*p<0.05, **p<0.005, ***p<0.0005 indicates significance to 0 weeks HFD; ^p<0.05, ^^p<0.005, ^^^p<0.0005 significance between indicated groups). All experiments were performed with at least an independent experiment number of n=3. Exact n-numbers are given in the Fig. legends. Univariable or multivariable regression analysis were performed using STATA 12 (StataCorp., College Station, TX, USA).
Results
Quantitative histologic assessment of plaque progression
Aortic root sections of Ldlr−/− mice fed a HFD from 0–36 weeks were analyzed to identify various cell types and structural characteristics of atherogenesis. Oil red-O staining of aortic root sections (Fig. 1A) was used to quantify the size of atherosclerotic plaques (Supplementary Fig. 1A). Early atherosclerotic lesions were detected after 2 weeks of HFD (data not shown), increasing in size and complexity with prolonged HFD administration.
Fig. 1:

Histological analysis of the pathophysiology in the atherosclerotic plaque.
Ldlr−/− mice on HFD for several time points were euthanized and frozen aortic root sections prepared. (A) Oil Red-O staining of aortic root sections was used to determine atherosclerotic plaque size. (B) Serial sections of the aortic root were stained with the macrophage marker, Moma-2 (green), to determine macrophage content per plaque size. SM22α was used to stain smooth muscle cells (red). Nuclei shown by DAPI staining in blue. (C) H&E staining of aortic root sections allowed for the quantification of the necrotic core. (n=10 for each given time point). (D) Picro Sirius Red staining of aortic root sections allows for visualization and quantification of total collagen using bright field microscopy, and of collagen type and maturation using polarized light microscopy (E). Under polarized light, collagen III appears green while collagen I shows in yellow, orange and red in order of increasing thickness. n=3; sections from both upper and lower aortic root were analyzed; BF, bright field; PL, polarized light.
Immunofluorescent staining of aortic root sections revealed that macrophages are present within 2 weeks of HFD (data not shown). The macrophage content of the aortic root plaque increased dramatically by 4 weeks of HFD (Fig. 1B, green), accounting for more than 60% of the atherosclerotic plaque area (Supplementary Fig. 1B). Over the following weeks of HFD the MOMA-2-positive macrophage content per plaque size steadily declined. Linear regression analysis of macrophage content compared with plaque size did not reveal any significant correlation.
As the plaque advances with increasing length of HFD, macrophages produce growth factors that induce the proliferation and migration of vascular smooth muscle cells (SMC). An important function of SMC within the atherosclerotic plaque is the production of extracellular matrix components, especially collagen, which serve to form the fibrous cap that largely determines plaque stability. Histological staining for smooth muscle cells (SMC) within aortic root plaques allows visualization of quiescent SMC found within the aortic wall (arrowheads), as well as proliferative SMC comprising the fibrous cap (arrows, Fig. 1B). A thin layer of SMC covering the plaque after 4 weeks of HFD is the first detectable sign of fibrous cap formation. The SMC content at this point constitutes less than 1% of the total plaque area and increases steadily at subsequent time points reaching a maximum of 4% of total plaque area by 16 weeks of HFD (Supplementary Fig. 1C). We found a strong correlation between SMC and plaque size (β=0.525, p<0.0001).
The area comprising the necrotic core was quantified from nuclei-free areas using H&E staining (Fig. 1C, Supplementary Fig. 1D). Prominent necrotic core areas within the atherosclerotic plaque began to appear after 12 weeks of HFD and grew steadily through 36 weeks of HFD. Increase in necrotic core area was positively correlated with plaque size as determined by regression analysis (β=0.720, p<0.0001).
Notable collagen deposition was observed by Sirius Red staining in Ldlr−/− mice on HFD for 4 weeks, increasing over time, and reaching a maximum at 16 weeks (no further increase detectable up to 36 weeks of HFD) (Fig. 1D and E, Supplementary Fig. 1E and F). This staining used for collagen visualization under bright light, has the advantage of being birefringent under polarized light. The emitted color in polarized light reflects the thickness and type of the collagen fibers along a spectrum from green to red [34]. Collagen I is shown by yellow, orange and red, while Collagen III appears in green under polarized light. In Ldlr−/− mice fed HFD, we observed a reduction the contribution of collagen III (green) with increasing collagen I content (yellow, orange, and red) over time. Additionally, we observed thicker collagen I fibers in advanced compared to early plaques, as determined by the shift from yellow to red fibers (Fig. 1E and Supplementary Fig. 1F), revealing advancing collagen deposition and growth at various stages of atherogenesis.
HFD-induced changes in aorta assessed by SEM
Scanning electron microscopy (SEM) was used to visualize the luminal surface of mouse aortas from Ldlr−/− mice on a HFD for various lengths of time (Fig. 2 and Supplementary Fig. 2). EC and the basal lamina form the inner layer of the blood vessel, which separates blood and its contents from the surrounding tissue. Control animals fed a normal chow diet displayed a uniformly contiguous monolayer of endothelial cells, indicated by the presence of interendothelial junctions (yellow arrowheads), covering the underlying tunica media and adventitia, with no monocytes or macrophages detected on the surface of the aorta or beneath the endothelium (Fig. 2A). Control animals also showed normal aorta anatomy with tunica adventitia as the outermost layer, followed by SMC- and collagen-rich tunica media which are separated from the tunica intima by the internal elastic membrane (Fig. 2A). Within 1 week of HFD the aortic surface is no longer smooth, and deposits can be seen collecting beneath the endothelium (Fig. 2B, red arrows). Higher magnifications of these subendothelial areas display extracellular liposomes, and smooth, round particles resembling lipid droplets (LD) of various sizes, similar to those previously described in hyperlipidemic rabbit aortae [12]. These lipid-based structures can be identified by their smooth surface, as opposed to cells of the aorta with visible membranous pores (Supplementary Fig. 3). After 2 weeks of HFD, subendothelial deposits increase in size, and openings on the aortic surface show the first observed infiltrated immune cells (yellow arrows). Increasing the duration of HFD to 4 and 8 weeks resulted in increased disturbance of the aortic surface and increased deposition of lipids, cellular debris, and infiltration of immune cells in a time-dependent manner (Fig. 2D and E). After 12 weeks of HFD, discrimination between intact macrophages and cellular debris in the central areas of the plaque becomes increasingly difficult as the necrotic core continuous to grow (Fig. 2F).
Fig. 2:

Scanning electron microscopy (SEM) to observe atherogenesis.
Ldlr−/− mice were put on HFD for several time points. At the end of each time point comparable areas of the aortic surface were imaged with SEM. (A) An even surface is observed in control animals not on HFD. In cracks through the aorta, typical anatomy of alternating extracellular matrix layers and cells can be seen. Endothelium forms the uppermost layer (roughly 0.5 −1 μm thick) towards the lumen with intact interendothelial junctions (arrowheads). (B-F) Extending the duration of HFD results in increased subendothelial deposition (red arrows) of lipid-based particles. (C-F) Increased complexity of subendothelial deposits and atherosclerotic plaque characteristics is evidenced by presence of infiltrated cells like macrophages or foam cells, and a greater degree of lipid-based particles. Subendothelial depositions grow in number and size with increasing HFD until from 8 weeks HFD on mainly large depositions can be seen. C, collagen; E, internal elastic membrane; EC, endothelial cells; LD, Lipid droplet; M, monocyte; NC, necrotic core; t. media,tunica media. Yellow boxes indicate the location of the higher magnification picture to the right; n=4 individual aortas for each timepoint, with at least 25 pictures taken per aorta.
Monocyte adherence and transmigration
As seen by immunofluorescent staining in Fig. 1, the first subendothelial macrophage foam cells were observed after 2 weeks of HFD. We examined aortas of Ldlr−/− mice at this time point by SEM and TEM to visually investigate adhesive and chemotactic properties of monocytes and macrophages. Various images show monocytes adherent to the aortic wall at different stages of transmigration across the endothelial monolayer (Fig. 3A). Large areas within the subendothelial space are formed within 2 weeks of HFD treatment, at locations where we also observed adherent monocytes by TEM (Fig. 3A, I/II). Further, by using SEM, we detected monocytes adherent on aortic surface fissures (Fig. 3A, III/IV), sometimes observed with tethers (Fig. 3A,VN-VNI). TEM and SEM images show monocytes actively transmigrating through the endothelium (Fig. 3B). We did not observe transmigration further into the aortic wall past the elastin layer, and transmigrated monocytes and macrophages were always observed just beneath the endothelial monolayer (Fig. 3C), where they encounter the accumulated subendothelial lipid deposits shown in Fig. 2.
Fig. 3:

Electron microscopy of monocyte adhesion and transmigration in Ldlr−/− mouse aortas after 2 weeks HFD.
Ldlr−/− mice were fed a HFD for 2 weeks and aortas were studied utilizing SEM and TEM. (A) Monocytes adherent to the luminal side of the endothelial lining can be seen by TEM (A, I-II) especially at sites of underlying subendothelial space. Monocytes adherent to the surface of the endothelium can be seen at various places by SEM (A, III-VIII). Signs of monocyte tethering (A, VII-VIII, magnified area of VII, arrow) are evident among adherent cells. (B) Monocytes transmigrating through the endothelial lining were observed using SEM. (C) After presumably transmigrating, undifferentiated monocytes and/or differentiated macrophages can be found beneath the endothelium. Frames I and II are TEM and frames III and IV are SEM. EC, endothelial cells; E, elastin/internal elastic membrane; SMC, smooth muscle cells; C, collagen; M, monocyte; M/Mac, monocyte/macrophage; T, tethers; ECM, extracellular matrix. Black scale bar=2 μm, n=4 individual aortas for each timepoint with at least 25 pictures taken per aorta
Cholesterol crystals are present in various compartments of the plaque
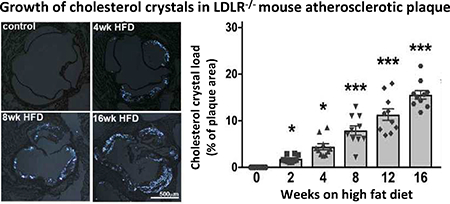
The presence of cholesterol crystals (CC) has been described in both human and mouse atherosclerotic plaques. Large CC found in the necrotic core of advanced plaques are thought to contribute to inflammation and potentially make the plaque less stable. Recently it was shown that endothelial cells can produce CC in the context of hyperlipidemia, and those CC found in early plaques have been shown to drive endothelial dysfunction and inflammation. We used polarized light microscopy to analyze aortic root plaques of Ldlr−/− mice on HFD and identify cholesterol crystals, which grew more abundant with increasing length of HFD treatment (Fig. 4A and B). Regression analysis revealed a highly significant positive correlation between plaque size and CC content (β=0.815, p<0.0001). An overview of an advanced plaque is shown in Fig. 4C, with areas including endothelial cells, macrophages/foam cells, and necrotic core further analyzed by TEM (Fig. 4D–F).
Fig. 4:

CC in atherosclerotic plaques of Ldlr−/− mice at various time points.
(A) Imaging of aortic root sections using polarized light microscopy allows (with subsequent ImageJ quantification) (B) the determination of cholesterol crystal content per plaque area. (n=10 for each time point; ANOVA statistical test, error bar indicates SEM, significance at p<0.05, *significance to 0-week HFD, ^significance between indicated groups; BF - bright field, PL - polarized light) (C) Display of representative areas of atherosclerotic plaques (red squares) visualized by SEM or TEM at various stages of HFD being fed to Ldlr−/− mice. (D) Presence of CC in the endothelial layer can be seen as early as 1 week of HFD. CC within EC can be detected at every stage of atherogenesis. (E) Macrophages with varying degrees of lipid content and CC deposition can be seen in plaques after 2 weeks of HFD. (F) A necrotic core is formed at 8 weeks of HFD and continues to expand with longer duration of HFD. CC are present within the necrotic core. Lipid droplets are located in close proximity to CC and stacking of CC can be seen as well. CC within the necrotic core can reach sizes of 25 μm. CC, cholesterol crystal, E, internal elastic membrane, ECM, extracellular matrix; Mac, macrophage/foam cell; Mi, mitochondria; pCC, possible CC. Orange inserts indicate area of magnification to their right in orange box. Scale bars: black=2.5 μm, blue=10 μm, green=0.5 μm
In the presence of excess lipid, EC have the ability to metabolize lipoproteins and produce CC, which can be observed by TEM within aortic EC after just 1 week of HFD. Early accumulation of lipids and CC beneath the endothelium are thought to drive plaque development by inducing an endothelial inflammatory response, attracting monocytes to the affected areas. These monocytes transmigrate through the endothelial monolayer and differentiate into macrophages, which then begin taking up lipids and CC by phagocytosis. TEM analysis of mouse plaques after 2 weeks of HFD reveals many lipid-laden macrophages, known as foam cells, with prominent intracellular CC observed. After 8 weeks of HFD, a necrotic core is apparent, containing abundant lipid droplets, cellular debris, and very large CC, as shown by TEM and SEM (Fig. 4). The necrotic core area was directly correlated with increases in plaque size, CC content, and smooth muscle cell content, as determined by regression analysis (β=0.720, p<0.0001 and β=0.769, p<0.0001, and β=0.331, p<0.005, respectively).
Ultrastructural analysis of atherosclerotic plaque progression by TEM
Through TEM we were able to observe the size, composition, and location of atherosclerotic plaque formation in Ldlr−/− mice on an unprecedented ultrastructural level. In control aortas from mice that were not fed a HFD, EC were continuously in direct contact with the internal elastic membrane and the subendothelial space was not visible (Fig. 5A). This is in contrast to animals fed a HFD where the subendothelial space was greatly widened with increasing time on HFD (Fig. 5B–E). Intra-endothelial lipid droplets were first observed within 1 week of HFD (Fig. 5B,II). At this stage, CC were also present within the EC, which increased by 2 weeks of HFD, suggesting either endothelium-derived CC production or formation in the extracellular space and subsequent uptake by EC (Fig. 5C). EC appeared misshapen, particularly the curved shape of the nuclei as if the cell layer was being lifted from beneath (Fig. 5B,IV and C,IV). Electron dense material was present in the widened subendothelial space. After an additional week of HFD the content of CC within the EC monolayer increased, and lipid droplets were found fused with crystalline structures (Fig. 5C,II). Furthermore, the area of expanded subendothelial space appeared to grow more complicated between 1 week and 2 weeks of HFD, with macrophages found beneath the endothelium (Fig. 5C,I,II), and an increasing number of subendothelial CC (Fig. 5C,III). Following 4 weeks of HFD, the first substantial atherosclerotic plaques could be identified (Fig. 5D). Macrophages with varying degrees of lipid loading could be seen between the EC and the internal elastic membrane (Fig. 5D,I,IV). At this time point, CC could be found localized not only within EC (Fig. 5D,III), but also within macrophages (Fig. 5D,II). After 8 weeks of HFD (Fig. 5E), plaques became larger in size and morphologically more complex, with the first acellular areas visible, representative of necrotic core formation. From 8 to 12 weeks of HFD, the necrotic core, filled with CC as well as lipid droplets and other cellular remnants, became increasingly prominant (Fig. 5E,I and F,I). CC within the necrotic core were physically associated with lipid droplets, in some cases appearing to fuse with massive lipid droplets up to 10 μm in size. In addition, the elastin layers (including the internal elastic membrane) were no longer organized, beginning to show signs of degradation (arrow head in Fig. 5E,I), allowing SMC to infiltrate into the neointima. At this stage, CC were observed within EC as well as the subendothelial space, and were increasingly abundant within lipid-loaded macrophages. By 12 weeks of HFD (Fig. 5F), the plaques were extremely prominent and accompanied by establishment of mature fibrous caps, formed by SMC and ECM. At this advanced stage, the plaque is comprised of various cell types, a necrotic core, lipid deposits, and abundant CC.
Fig. 5:

Visualization of atherogenesis at the ultrastructural level with transmission electron microscopy (TEM).
(A) Aortas of Ldlr−/− mice with no HFD show an EC layer tightly connected to the underlying internal elastic layer. (B) 1 week of HFD induces the emergence of a subendothelial space. Lipid droplets and CC clefts (spaces where cholesterol crystals have been dissolved during sample preparation) within the EC are also present. (C and D) With longer durations of HFD (2–4 weeks), increasing size of the subendothelial space can be observed. Monocytes infiltrate the subendothelial space, and CC within EC are in close contact with LD. (E and F) 8 and 12 weeks of HFD result in complex atherosclerotic plaque formation, infiltrating SMC, ECM deposition, degradation of the internal elastin layer, and necrotic core formation. C, collagen; CC, cholesterol crystal; E, internal elastic membrane; EC, endothelial cells; ECM, extracellular matrix; L, lipid; LD, lipid droplet; M, monocyte; Mac, macrophage/foam cell; N, nucleus; NC, necrotic core; pCC, possible CC; SMC, smooth muscle cells; subEC, subendothelial space. Arrowheads indicate breaks in elastin layer; black scale bar=2 μm, blue scale bar=5 μm; n=4 individual aortas for each timepoint with at least 25 pictures taken per aorta to allow for comprehensive future analysis; orange inserts indicate area of magnification to their right in orange box
CC are closely associated with cholesterol microdomains
In order to further characterize the CC we observed within various stages of atherosclerotic plaques, we utilized a unique monoclonal antibody (58B1) that labels crystalline cholesterol deposited in the extracellular matrix or that form on the plasma membrane when cells such as macrophages and fibroblasts are enriched with cholesterol [33, 35–38]. We were able to identify cholesterol microdomains in the extracellular matrix surrounding and closely associated with CC (Supplementary Fig. 4). However, because the antibody does not label individual cholesterol molecules [32, 39–42], the antibody’s recognition is limited to the structural motif presented by an ordered array of cholesterol molecules including irregular shaped structures shed from the macrophage plasma membrane [43] and the narrow side of cholesterol monohydrate plates [44]. Whereas in aortic roots from Ldlr−/− mice that were not fed HFD there were no cholesterol microdomains or CC present (Supplementary Fig. 4A), within 2–4 weeks of HFD, early plaque formation could be seen with CC deposits that were surrounded by cholesterol microdomains. After 4 and 8 weeks of HFD, the overlay of CC polarized light signals and the cholesterol microdomain staining exhibited close proximity of CC and microdomains most notably in areas of plaque where macrophages had accumulated (Supplementary Fig. 4B–D). At 12 and 16 weeks of HFD, cholesterol microdomains were increasingly visible in the tunica media and surrounding cells, most likely smooth muscle cells (Supplementary Fig. E and F).
Discussion
To the best of our knowledge we are the first to chronicle in a comprehensive way the timeline of atherosclerotic plaque formation in the Ldlr−/− mouse, one of the most commonly utilized animal models of atherosclerosis. We implemented various microscopic techniques to characterize plaque phenotype as a function of structural morphology. These efforts allowed us to further understand the timeframe of the appearance of various cell types, changes in plaque morphology, and CC formation in atherogenesis. The ubiquitous presence of CC in an advanced plaque is shown in ultrastructural detail, using EM, in the endothelium, macrophage/foam cells, and the necrotic core (Fig. 4). Furthermore, we show that CC formation is present throughout all stages of atherogenesis and directly correlates with various features of plaque development (Table 1). Lastly, we provide insight into the possible role of cholesterol microdomains in CC formation and deposition (Supplementary Fig. 4).
Table 1:
Univariable regression analysis of mouse atherosclerotic plaque morphology (unadjusted)
| Outcome | Exposure | Beta (p-value) |
|---|---|---|
| Plaque size (mm2) | CC content of plaque | 0.82 (p<0.001) |
| Plaque size (mm2) | SMC content of plaque | 0.53 (p<0.001) |
| Plaque size (mm2) | Necrotic core of plaque | 0.72 (p<0.001) |
| CC content of plaque | Necrotic core of plaque | 0.77 (p<0.001) |
| SMC content of plaque | Necrotic core of plaque | 0.33 (p=0.005) |
| SMC content of plaque | CC content of plaque | 0.58 (p<0.001) |
Data represented as standardized beta co-efficient (p-value)
The occurrence of CC in vivo is associated with several diseases and disease states including atherosclerosis [11, 14, 15, 45] and myocardial infarction [46], embolism [47], psoriasis-related cardiovascular diseases [25], gall bladder disease [48], skin xanthomas [49, 50], and age-related macular degeneration [51]. Recently, it was demonstrated in liver cells that CC can be derived from lipid droplets involved in murine NASH development [52]. Despite its involvement in such wide-ranging diseases, how and why cholesterol crystallization occurs within cells or extracellularly is a mystery. Although all 3 major cell types involved in atherosclerosis, endothelial cells [11], macrophages [24–26], and smooth muscle cells [28], are capable of CC production and secretion, macrophages are generally considered to be the main source of CC within the atherosclerotic lesion. In macrophages, the involvement of lysosomes in CC formation has been postulated [24]. The presence of CC in the atherosclerotic plaque is thought to influence inflammasome-dependent cell death [14–16].
Until recently it was widely accepted that EC mostly transcytose LDL from the lumen to the subendothelial space, during which some of the LDL becomes modified and thereby initiates the macrophage-driven inflammatory event that fuels plaque development [13, 53]. We have shown however, that EC themselves are also capable of actively metabolizing the LDL and generating CC when the cells are overburdened with cholesterol. It is likely that both transcytosis and lipid metabolism, including CC formation, occur simultaneously, and contribute to the recruitment of macrophages into the subendothelial space. Infiltrated cells subsequently encounter free lipids and early CC, eliciting a cascade of pro-inflammatory events which ultimately result in atheroma formation and increased CC-induced inflammation [54]. In support of this notion, our data presented here show that monocytes increasingly adhere to the endothelial layer especially at areas with increased subendothelial spaces. This is possibly due to increased endothelial surface expression of E-selectin [11] following perturbation of the EC by the subendotheial deposits, which is known to facilitate endothelial adhesion of circulating monocytes and eventually leads to their transmigration at these specific sites. We believe that hyperlipidemia and subsequent CC deposition in the endothelium could activate additional signaling pathways that amplify atherogenesis.
The necrotic core is a highly inflammatory, lipid-rich region within the atherosclerotic plaque [55]. TEM images depict large lipid droplets (up to 10μm), lipid vesicles and multilamellar liposomes, CC, and large lipid-filled areas that can be over 10 mm in diameter. Regions formed by cholesterol monohydrate crystals can be seen in large quantities. These areas visualized with TEM are “clefts”, or essentially empty spaces, where crystalline structures have been extracted during the preparation process. Sizes and characteristics of the clefts observed here are comparable to what has been reported by Guyton et al. in 1989 [22]. CC within the necrotic core not only impact cellular function but also provide a reactive site for both C-reactive protein (CRP) and the terminal complement complex [56]. These interactions stimulate inflammatory responses and may contribute to accelerating the progression of atherosclerosis. The C-type lectin receptor Mincle has also been shown to bind CC, triggering innate immune responses that enhance inflammation [57]. CC content in advanced lesions appears indicative of plaque vulnerability in human subjects [58], and may have potential as a diagnostic marker for evaluating plaque stability and cardiovascular risk.
The earliest pathological effects of CC within atherosclerotic plaques involve NFR2-dependent activation of the NLRP3 inflammasome, thereby exacerbating atherogenic conditions[14–16]. Within recent years, reports have been published describing several other pathways associated with CC. For example, it was shown that Syk- and PI3-kinases in human macrophages and dendritic cells are activated by CC [59]. Furthermore, exposure to CC induces inflammatory cytokine expression with subsequent NF-kB activation in human retinal epithelium [60]. Inhibiting cholesterol crystallization could be a useful therapeutic intervention that attenuates CC-induced inflammation and its effect on the progression of atherosclerosis [61]. Interestingly, a study in 1995 demonstrated that CC formation could be inhibited by biliary proteins [62], suggesting a potential therapeutic approach to inhibit CC formation. Recently, cyclodextrin has been shown to diminish CC-induced inflammation by altering complement activation [63]. Our recent findings also suggest that treating EC with cAMP-inducing drugs can inhibit LDL-induced CC formation by the EC in vitro [64]. Other literature has emerged discussing how CC released from atheromatous plaques in the aorta affects embolic events [65] and endothelial function [66, 67]. Although the existence of free CC within the serum is still somewhat controversial, a more comprehensive understanding of what impact CC may have on circulating cells is necessary.
It is evident that CC and cholesterol microdomains are closely associated, and the question of whether the formation of one is required for the other remains unanswered [41, 44]. The fact that all the crystals are surrounded by cholesterol microdomains but not all of the cholesterol microdomains are associated with CC indicates that perhaps the cholesterol enrichment in the microdomains leads to formation of large CC once a critical cholesterol level has been reached. Because it is nearly impossible to study the crystallization process in vivo, it is difficult to determine the levels of cholesterol and other requisite conditions that would lead to CC formation. Although it is known that certain factors, such as degree of hydration and solubilization of cholesterol, as well as pH and temperature, can affect cholesterol crystallization [68], exactly how this process occurs in vivo will require innovative new studies to elucidate.
We recognize that ethanol-based SEM preparation techniques and the use of critical point drying might alter appearance of CC and subendothelial lipids. Thus, to preserve the integrity of our findings we developed two critical axioms: First, only particles with lipid-based surface characteristics (no cellular membrane pores visible) and layered in appearance were classified as CC (see Supplementary Fig. 3) especially in very early stage plaques. Furthermore, the correlation of various techniques (PLM, TEM and SEM) further strengthens our interpretation of the obtained data.
In this study we found that macrophages are present within the subendothelial space after just 2 weeks of HFD. We also show with SEM and TEM that monocytes are adherent to the endothelium and transmigrate into the subendothelial space, a process which likely accounts for the majority of macrophages within the plaque. However, it has been discussed in recent literature that other sources for plaque macrophages are possible, e.g. tissue resident macrophages [69–71] and macrophages derived from SMC trans-differentiation [72, 73]. We found that macrophage content per plaque area decreases after 4 weeks of HFD as cells begin dying and the the initial steps of necrotic core formation occur. Plauqe SMC content, however, continues to increase with increasing during of HFD.
Plaque SMC secrete important matrix components that determine plaque stability, especially different types of collagens. In this publication we have used an established staining technique (Sirius Red) which allows discrimination between collagen I and III when viewed in polarized light [34]. Collagen I has been described to be with the major collagen contributor in the atherosclerotic lesion [74], which we have also confirmed with our analysis. Although Sirius Red staining and PL microscopy will not allow for identification of all collagens contributing to the atherosclerotic lesion (collagen I, III, IV, and V) [75], it does allow a detailed look at both collagen I and III, including the relative thickness of collagen I fibers.
In our study, we have opted to use Ldlr−/− mice as these atherosclerotic prone mice do not show spontaneous plaque formation at a young age while on chow diet, whereas ApoE−/− mice do. Interestingly, a study published by Lim et al in 2011 [20] used hyperspectral CARS imaging to determine that the number of cholesterol crystal structures is increased in ApoE−/− mice as compared to Ldlr−/− mice. However, both mouse strains were fed a HFD for 16 weeks which likely resulted in significant differences since ApoE−/− mice develop atherosclerosis more rapidly than Ldlr−/− mice. Differences between these two atherosclerosis-prone mouse strains have been reported by other groups as well, making a direct comparison of findings between the two strains difficult [76].
Given the significant presence of CC within atherosclerotic plaques of humans, as well as mouse and rabbit animal models of the disease, it will be of great importance to determine the exact mechanism of CC formation. Further investigation of the concequences of CC presence within the plaque on various cell types will also be neccesary. The role of cholesterol converting enzymes such as Sterol O-acyltransferase (ACAT-1) and neutral cholesterol ester hydrolase (NCEH1) will also likely be important to this objective. It has been speculated that CC phagocytosed by macrophages can be a source of intracellular free cholesterol through a mechanism involving ACAT-1 activity. It is possible that overexpression of ACAT-1 might be a pathway of CC detoxification, especially as the inhibition of ACAT-1 has been demonstrated to increase atheroma formation accompanied by an increase of extracellular crystalline cholesterol [50, 77].
Data presented here and in previous studies [11, 26, 27, 52] collectively illustrate the formation of CC within cholesterol-enriched cells. Additionally, CC accumulate in the extracellular space and are surrounded by cholesterol microdomains suggesting the possibility that these crystals derive from the cholesterol microdomains. Our findings indicate that CC formation in various cell types and at all stages of atherosclerosis is a useful target for future therapies. Continued investigation of CC formation is imperative in determining the most effective means to limit buildup of inflammatory CC in tissues such as the vascular wall.
Supplementary Material
Highlights.
Ultrastructural detail of atherosclerotic plaque composition throughout all stages of its development in the LDL receptor null mice.
Documentation of the formation and appearance of cholesterol crystals from the very genesis to advanced atherosclerosis.
The use of a monoclonal antibody against crystallized cholesterol and cholesterol microdomain to locate cholesterol crystals in the plaque.
Acknowledgments
We are grateful to Dr. J. Hammer and Dr. E. Hellebrand for the use of the polarized light microscope. We also thank Drs. Lia Addadi and Neta Varsano for providing the anti-cholesterol microdomain MAb and for critical review of the manuscript. Part of this work was performed within the Russian Government Program of Competitive Growth of Kazan Federal University.
Financial support
The Hitachi HT7700 TEM was acquired with NSF grant DBI-1040548. This project was funded by NIH grants R01HL075677 and R01HL081863, by Innovative Research Grant 16IRG27540027 and Grant-in-Aid 16GRNT30810007 from the American Heart Association, and by the Ingeborg v.F. McKee Fund of the Hawaii Community Foundation 17ADVC-86287 to WAB. Additional support for this project was from the Intramural Research Program of the National Institutes of Health and the Binational Science Foundation (Grant 2013045).
Abbreviations
- EC
endothelial cells
- CC
cholesterol crystals
- Ldlr
low-density lipoprotein receptor
- HFD
high fat diet
- SEM
scanning electron microscopy
- TEM
transmission electron microscopy
- PLM
polarized light microscopy
- FC
foam cell
- ECM
extracellular matrix
- SMC
smooth muscle cell
- Mac
macrophage
- M
monocyte
- LD
lipid droplet
Footnotes
Conflict of Interest
The authors declared they do not have anything to disclose regarding conflict of interest with respect to this manuscript.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- [1].Benjamin EJ, Blaha MJ, Chiuve SE, et al. , Heart Disease and Stroke Statistics-2017 Update: A Report From the American Heart Association, Circulation, 2017;135:e146–e603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Libby P, Inflammation in atherosclerosis, Arterioscler Thromb Vasc Biol, 2012;32:2045–2051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Gistera A and Hansson GK, The immunology of atherosclerosis, Nat Rev Nephrol, 2017. [DOI] [PubMed] [Google Scholar]
- [4].Lawler PR, Akinkuolie AO, Chu AY, et al. , Atherogenic Lipoprotein Determinants of Cardiovascular Disease and Residual Risk Among Individuals With Low Low-Density Lipoprotein Cholesterol, J Am Heart Assoc, 2017;6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Gimbrone MA Jr. and Garcia-Cardena G, Endothelial Cell Dysfunction and the Pathobiology of Atherosclerosis, Circ Res, 2016;118:620–636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Cahill PA and Redmond EM, Vascular endothelium - Gatekeeper of vessel health, Atherosclerosis, 2016;248:97–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Cochain C and Zernecke A, Macrophages in vascular inflammation and atherosclerosis, Pflugers Arch, 2017;469:485–499. [DOI] [PubMed] [Google Scholar]
- [8].Biessen EAL and Wouters K, Macrophage complexity in human atherosclerosis: opportunities for treatment?, Curr Opin Lipidol, 2017;28:419–426. [DOI] [PubMed] [Google Scholar]
- [9].Rahman K, Vengrenyuk Y, Ramsey SA, et al. , Inflammatory Ly6Chi monocytes and their conversion to M2 macrophages drive atherosclerosis regression, J Clin Invest, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Geeraerts X, Bolli E, Fendt SM, et al. , Macrophage Metabolism As Therapeutic Target for Cancer, Atherosclerosis, and Obesity, Front Immunol, 2017;8:289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Baumer Y, McCurdy S, Weatherby TM, et al. , Hyperlipidemia-induced cholesterol crystal production by endothelial cells promotes atherogenesis, Nature Communications, 2017;8:1129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Mora R, Simionescu M and Simionescu N, Purification and partial characterization of extracellular liposomes isolated from the hyperlipidemic rabbit aorta, J Lipid Res, 1990;31:1793–1807. [PubMed] [Google Scholar]
- [13].Simionescu M, Implications of early structural-functional changes in the endothelium for vascular disease, Arterioscler Thromb Vasc Biol, 2007;27:266–274. [DOI] [PubMed] [Google Scholar]
- [14].Rajamaki K, Lappalainen J, Oorni K, et al. , Cholesterol crystals activate the NLRP3 inflammasome in human macrophages: a novel link between cholesterol metabolism and inflammation, PLoS One, 2010;5:e11765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Duewell P, Kono H, Rayner KJ, et al. , NLRP3 inflammasomes are required for atherogenesis and activated by cholesterol crystals, Nature, 2011;464:1357–1361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Freigang S, Ampenberger F, Spohn G, et al. , Nrf2 is essential for cholesterol crystal-induced inflammasome activation and exacerbation of atherosclerosis, Eur J Immunol, 2011;41:2040–2051. [DOI] [PubMed] [Google Scholar]
- [17].Stewart GT, Liquid crystals of lipid in normal and atheromatous tissue, Nature, 1959;183:873–875. [DOI] [PubMed] [Google Scholar]
- [18].Katz SS, Shipley GG and Small DM, Physical chemistry of the lipids of human atherosclerotic lesions. Demonstration of a lesion intermediate between fatty streaks and advanced plaques, J Clin Invest, 1976;58:200–211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Bogren H and Larsson K, An X-Ray-Diffraction Study of Crystalline Cholesterol in Some Pathological Deposits in Man, Biochim Biophys Acta, 1963;75:65–69. [DOI] [PubMed] [Google Scholar]
- [20].Lim RS, Suhalim JL, Miyazaki-Anzai S, et al. , Identification of cholesterol crystals in plaques of atherosclerotic mice using hyperspectral CARS imaging, J Lipid Res, 2011;52:2177–2186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Suhalim JL, Chung CY, Lilledahl MB, et al. , Characterization of cholesterol crystals in atherosclerotic plaques using stimulated Raman scattering and second-harmonic generation microscopy, Biophys J, 2012;102:1988–1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Guyton JR and Klemp KF, The lipid-rich core region of human atherosclerotic fibrous plaques. Prevalence of small lipid droplets and vesicles by electron microscopy, Am J Pathol, 1989;134:705–717. [PMC free article] [PubMed] [Google Scholar]
- [23].Vedre A, Pathak DR, Crimp M, et al. , Physical factors that trigger cholesterol crystallization leading to plaque rupture, Atherosclerosis, 2009;203:89–96. [DOI] [PubMed] [Google Scholar]
- [24].Sheedy FJ, Grebe A, Rayner KJ, et al. , CD36 coordinates NLRP3 inflammasome activation by facilitating intracellular nucleation of soluble ligands into particulate ligands in sterile inflammation, Nat Immunol, 2013;14:812–820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Baumer Y, Ng Q, Sanda GE, et al. , Chronic skin inflammation accelerates macrophage cholesterol crystal formation and atherosclerosis, JCI Insight, 2018;3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Tangirala RK, Jerome WG, Jones NL, et al. , Formation of cholesterol monohydrate crystals in macrophage-derived foam cells, J Lipid Res, 1994;35:93–104. [PubMed] [Google Scholar]
- [27].Varsano N, Beghi F, Elad N, et al. , Two polymorphic cholesterol monohydrate crystal structures form in macrophage culture models of atherosclerosis, Proc Natl Acad Sci U S A, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Ho-Tin-Noe B, Vo S, Bayles R, et al. , Cholesterol crystallization in human atherosclerosis is triggered in smooth muscle cells during the transition from fatty streak to fibroatheroma, J Pathol, 2017;241:671–682. [DOI] [PubMed] [Google Scholar]
- [29].Baumer Y, McCurdy S, Alcala M, et al. , CD98 regulates vascular smooth muscle cell proliferation in atherosclerosis, Atherosclerosis, 2017;256:105–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Daugherty A, Tall AR, Daemen M, et al. , Recommendation on Design, Execution, and Reporting of Animal Atherosclerosis Studies: A Scientific Statement From the American Heart Association, Arterioscler Thromb Vasc Biol, 2017;37:e131–e157. [DOI] [PubMed] [Google Scholar]
- [31].McCurdy S, Baumer Y, Toulmin E, et al. , Macrophage-Specific Expression of IL-37 in Hyperlipidemic Mice Attenuates Atherosclerosis, J Immunol, 2017. [DOI] [PubMed] [Google Scholar]
- [32].Perl-Treves D, Kessler N, Izhaky D, et al. , Monoclonal antibody recognition of cholesterol monohydrate crystal faces, Chem Biol, 1996;3:567–577. [DOI] [PubMed] [Google Scholar]
- [33].Kruth HS, Ifrim I, Chang J, et al. , Monoclonal antibody detection of plasma membrane cholesterol microdomains responsive to cholesterol trafficking, J Lipid Res, 2001;42:1492–1500. [PubMed] [Google Scholar]
- [34].Junqueira LC, Cossermelli W and Brentani R, Differential staining of collagens type I, II and III by Sirius Red and polarization microscopy, Arch Histol Jpn, 1978;41:267–274. [DOI] [PubMed] [Google Scholar]
- [35].Ong DS, Anzinger JJ, Leyva FJ, et al. , Extracellular cholesterol-rich microdomains generated by human macrophages and their potential function in reverse cholesterol transport, J Lipid Res, 2010;51:2303–2313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Freeman SR, Jin X, Anzinger JJ, et al. , ABCG1-mediated generation of extracellular cholesterol microdomains, J Lipid Res, 2014;55:115–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Jin X, Dimitriadis EK, Liu Y, et al. , Macrophages Shed Excess Cholesterol in Unique Extracellular Structures Containing Cholesterol Microdomains, Arterioscler Thromb Vasc Biol, 2018;38:1504–1518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Varsano N, Dadosh T, Kapishnikov S, et al. , Development of Correlative Cryo-soft X-ray Tomography and Stochastic Reconstruction Microscopy. A Study of Cholesterol Crystal Early Formation in Cells, J Am Chem Soc, 2016;138:14931–14940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Kessler N, Perl-Treves D and Addadi L, Monoclonal antibodies that specifically recognize crystals of dinitrobenzene, FASEB J, 1996;10:1435–1442. [DOI] [PubMed] [Google Scholar]
- [40].Scheffer L, Futerman AH and Addadi L, Antibody labeling of cholesterol/ceramide ordered domains in cell membranes, Chembiochem, 2007;8:2286–2294. [DOI] [PubMed] [Google Scholar]
- [41].Ziblat R, Fargion I, Leiserowitz L, et al. , Spontaneous formation of two-dimensional and three-dimensional cholesterol crystals in single hydrated lipid bilayers, Biophys. J, 2012;103:255–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Addadi L, Geva M and Kruth HS, Structural information about organized cholesterol domains from specific antibody recognition, Biochim. Biophys. Acta, 2003;1610:208–216. [DOI] [PubMed] [Google Scholar]
- [43].Jin X, Dimitriadis EK, Liu Y, et al. , Macrophages Shed Excess Cholesterol in Unique Extracellular Structures Containing Cholesterol Microdomains, Arterioscler Thromb Vasc Biol, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Ziblat R, Leiserowitz L and Addadi L, Crystalline domain structure and cholesterol crystal nucleation in single hydrated DPPC:cholesterol:POPC bilayers, J Am Chem Soc, 2010;132:9920–9927. [DOI] [PubMed] [Google Scholar]
- [45].Abela GS, Cholesterol crystals piercing the arterial plaque and intima trigger local and systemic inflammation, J Clin Lipidol, 2010;4:156–164. [DOI] [PubMed] [Google Scholar]
- [46].Abela GS, Kalavakunta JK, Janoudi A, et al. , Frequency of Cholesterol Crystals in Culprit Coronary Artery Aspirate During Acute Myocardial Infarction and Their Relation to Inflammation and Myocardial Injury, Am J Cardiol, 2017;120:1699–1707. [DOI] [PubMed] [Google Scholar]
- [47].Ghanem F, Vodnala D, J, KK, et al. , Cholesterol crystal embolization following plaque rupture: a systemic disease with unusual features, J Biomed Res, 2017;31:82–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Jungst D, del Pozo R, Dolu MH, et al. , Rapid formation of cholesterol crystals in gallbladder bile is associated with stone recurrence after laparoscopic cholecystotomy, Hepatology, 1997;25:509–513. [DOI] [PubMed] [Google Scholar]
- [49].Arnaboldi F, Busnelli M, Cornaghi L, et al. , High-density lipoprotein deficiency in genetically modified mice deeply affects skin morphology: A structural and ultrastructural study, Exp Cell Res, 2015;338:105–112. [DOI] [PubMed] [Google Scholar]
- [50].Accad M, Smith SJ, Newland DL, et al. , Massive xanthomatosis and altered composition of atherosclerotic lesions in hyperlipidemic mice lacking acyl CoA:cholesterol acyltransferase 1, J Clin Invest, 2000;105:711–719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Pang CE, Messinger JD, Zanzottera EC, et al. , The Onion Sign in Neovascular Age-Related Macular Degeneration Represents Cholesterol Crystals, Ophthalmology, 2015;122:2316–2326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Ioannou GN, Subramanian S, Chait A, et al. , Cholesterol crystallization within hepatocyte lipid droplets and its role in murine NASH, J Lipid Res, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Simionescu M, Gafencu A and Antohe F, Transcytosis of plasma macromolecules in endothelial cells: a cell biological survey, Microsc Res Tech, 2002;57:269–288. [DOI] [PubMed] [Google Scholar]
- [54].Grebe A and Latz E, Cholesterol crystals and inflammation, Curr Rheumatol Rep, 2013;15:313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Guyton JR and Klemp KF, Development of the atherosclerotic core region. Chemical and ultrastructural analysis of microdissected atherosclerotic lesions from human aorta, Arterioscler Thromb, 1994;14:1305–1314. [DOI] [PubMed] [Google Scholar]
- [56].Pilely K, Fumagalli S, Rosbjerg A, et al. , C-Reactive Protein Binds to Cholesterol Crystals and Co-Localizes with the Terminal Complement Complex in Human Atherosclerotic Plaques, Front Immunol, 2017;8:1040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Kiyotake R, Oh-Hora M, Ishikawa E, et al. , Human Mincle Binds to Cholesterol Crystals and Triggers Innate Immune Responses, J Biol Chem, 2015;290:25322–25332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Koide M, Matsuo A, Shimoo S, et al. , Cholesterol crystal depth in coronary atherosclerotic plaques: A novel index of plaque vulnerability using optical frequency domain imaging, PLoS One, 2017;12:e0180303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Corr EM, Cunningham CC and Dunne A, Cholesterol crystals activate Syk and PI3 kinase in human macrophages and dendritic cells, Atherosclerosis, 2016;251:197–205. [DOI] [PubMed] [Google Scholar]
- [60].Hu Y, Lin H, Dib B, et al. , Cholesterol crystals induce inflammatory cytokines expression in a human retinal pigment epithelium cell line by activating the NF-kappaB pathway, Discov Med, 2014;18:7–14. [PMC free article] [PubMed] [Google Scholar]
- [61].Nidorf SM, Eikelboom JW and Thompson PL, Targeting cholesterol crystal-induced inflammation for the secondary prevention of cardiovascular disease, J Cardiovasc Pharmacol Ther, 2014;19:45–52. [DOI] [PubMed] [Google Scholar]
- [62].Busch N, Lammert F, Marschall HU, et al. , A new subgroup of lectin-bound biliary proteins binds to cholesterol crystals, modifies crystal morphology, and inhibits cholesterol crystallization, J Clin Invest, 1995;96:3009–3015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Bakke SS, Aune MH, Niyonzima N, et al. , Cyclodextrin Reduces Cholesterol Crystal-Induced Inflammation by Modulating Complement Activation, J Immunol, 2017;199:2910–2920. [DOI] [PubMed] [Google Scholar]
- [64].Baumer Y, McCurdy S, Weatherby TM, et al. , Hyperlipidemia-induced cholesterol crystal production by endothelial cells promotes atherogenesis, Nat Commun, 2017;8:1129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Oka H, Kamimura T, Hiramatsu Y, et al. , Cholesterol Crystal Embolism Induced by Direct Factor Xa inhibitor: A First Case Report, Intern Med, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Koka S, Xia M, Chen Y, et al. , Endothelial NLRP3 inflammasome activation and arterial neointima formation associated with acid sphingomyelinase during hypercholesterolemia, Redox Biol, 2017;13:336–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Mani AM, Chattopadhyay R, Singh NK, et al. , Cholesterol crystals increase vascular permeability by inactivating SHP2 and disrupting adherens junctions, Free Radic Biol Med, 2018;123:72–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Vedre A, Pathak DR, Crimp M, et al. , Physical factors that trigger cholesterol crystallization leading to plaque rupture, Atherosclerosis, 2008;203:89–96. [DOI] [PubMed] [Google Scholar]
- [69].Stremmel C, Schuchert R, Wagner F, et al. , Yolk sac macrophage progenitors traffic to the embryo during defined stages of development, Nat Commun, 2018;9:75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Mass E, Ballesteros I, Farlik M, et al. , Specification of tissue-resident macrophages during organogenesis, Science, 2016;353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Chinetti-Gbaguidi G, Colin S and Staels B, Macrophage subsets in atherosclerosis, Nat Rev Cardiol, 2015;12:10–17. [DOI] [PubMed] [Google Scholar]
- [72].Feil S, Fehrenbacher B, Lukowski R, et al. , Transdifferentiation of vascular smooth muscle cells to macrophage-like cells during atherogenesis, Circ Res, 2014;115:662–667. [DOI] [PubMed] [Google Scholar]
- [73].Albarran-Juarez J, Kaur H, Grimm M, et al. , Lineage tracing of cells involved in atherosclerosis, Atherosclerosis, 2016;251:445–453. [DOI] [PubMed] [Google Scholar]
- [74].Murata K, Motayama T and Kotake C, Collagen types in various layers of the human aorta and their changes with the atherosclerotic process, Atherosclerosis, 1986;60:251–262. [DOI] [PubMed] [Google Scholar]
- [75].Rekhter MD, Collagen synthesis in atherosclerosis: too much and not enough, Cardiovasc Res, 1999;41:376–384. [DOI] [PubMed] [Google Scholar]
- [76].Getz GS and Reardon CA, Do the Apoe−/− and Ldlr−/− Mice Yield the Same Insight on Atherogenesis?, Arterioscler Thromb Vasc Biol, 2016;36:1734–1741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Nissen SE, Tuzcu EM, Brewer HB, et al. , Effect of ACAT inhibition on the progression of coronary atherosclerosis, N Engl J Med, 2006;354:1253–1263. [DOI] [PubMed] [Google Scholar]
- [78].Chao FF, Blanchette-Mackie EJ, Chen YJ, et al. , Characterization of two unique cholesterol-rich lipid particles isolated from human atherosclerotic lesions, Am J Pathol, 1990;136:169–179. [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.