

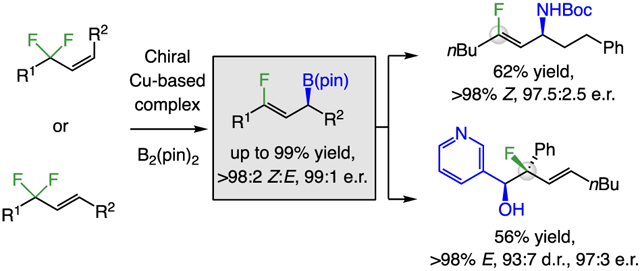
Abstract
The first catalytic method for diastereo- and enantioselective synthesis of allylic boronates bearing a Z-trisubstituted alkenyl fluoride is disclosed. Boryl substitution is performed with a Z- or an E-allyldifluoride and is catalyzed by bisphosphine–Cu complexes, affording products in up to 99% yield, >98:2 Z:E selectivity, and 99:1 enantiomeric ratio. A variety of subsequent modifications are feasible; notable examples are diastereoselective additions to aldehydes/aldimines to access homoallylic alcohols/amines containing a fluoro-substituted stereogenic quaternary center.
Keywords: Boron, Copper, Enantioselective catalysis, Fluorine, Synthetic methods
Graphical Abstract
Stereoselective strategies for synthesis of organofluorine compounds are in high demand, as such entities are capable of exhibiting entirely distinct modes of, and/or improved, activity.[1] Catalytic enantioselective methods that may be used to prepare fluorine-containing organic molecules are therefore key, and compounds that contain a single fluorine atom are particularly valuable.[2] For instance, trisubstituted alkenyl fluorides are peptide bond mimics (Scheme 1a),[3] and those bearing a fluoro-substituted stereogenic quaternary carbon center are isosteric with a more common tertiary stereogenic carbon center (Scheme 1a).[4] Still, effective methods that generate such organofluorine compounds with high diastereo- and enantioselectivity remain uncommon.[5,6]
Scheme 1.
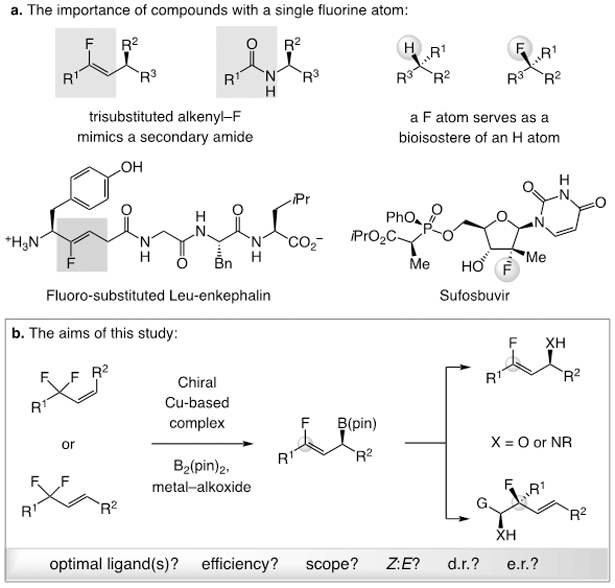
The significance and aims of this study. Abbreviations: R1-R3 = C-based moieties; pin = pinacolato.
Catalytic approaches to cleaving one of several C–F bonds in a fluoro-organic moiety has recently received significant attention.[1c,7-8] Directly relevant to the present work are two reports on catalytic γ-boryl substitution of trifluoromethyl-substituted alkenes to generate enantiomerically enriched gem-difluoroallylic boronates.[8c-d] We reasoned that a practical catalytic method for enantioselective synthesis of allyl–B(pin) products that contain a trisubstituted alkenyl fluoride[9] unit would represent a notable advance. The resulting products might then be used to prepare an assortment of otherwise difficult-to-access unsaturated fluoro-organic entities efficiently and in high stereochemical purity (Scheme 1b). Herein, we disclose the first catalytic strategy for enantioselective synthesis of (Z)-γ-monofluoroallylic boronates by boryl substitution reactions[10] involving a Z- or an E-allyldifluoride substrate; reactions are catalyzed by easily accessible bisphosphine–Cu complexes (Scheme 1b), and the products can be converted to numerous desirable derivatives, including those with a fluoro-substituted stereogenic quaternary carbon center.
We began by searching for an optimal ligand for the transformation of Z-alkene 1a, which was accessed in three steps and 51% overall yield by using previously reported protocols[11] (Scheme 2a). We began by evaluating (R,S)-josiphos as the ligand, largely because it had proved to be optimal in our previous studies on γ-boryl substitution reactions with trifluoromethyl-substituted alkenes. While appreciable efficiency and stereoselectivity was observed [93:7 Z:E, 83.5:16.5 enantiomeric ratio (e.r.)], further screening revealed that benzP* is a superior option. Thus at 5.0 mol % catalyst loading, 2a was generated in 93% yield, 95:5 Z:E selectivity, and 97:3 e.r. Although the reaction with the closely related (R,R)-quinoxP* was similarly effective (93% yield, 95:5 Z:E, 96:4 e.r.), catalysts derived from other ligands were much less so (e.g., josiphos, binap, or segphos).[12] With E-alkene 4a as the substrate (Scheme 2b), the reaction with phenyl-bpe and LiOtBu was optimal, affording 2b with the opposite sense of enantioselectivity in 51% yield[13], >98:2 Z:E, and 95:5 e.r. The reaction with Z-alkene 1a under these latter conditions, afforded preferentially the same (S)-2a enantiomer that was generated with benzP*, albeit in lower yield (70% yield, 94:6 Z:E, 96:4 e.r.), indicating that a stereoisomerically pure Z- or E-alkene starting material is needed for high enantioselectivity.
Scheme 2.
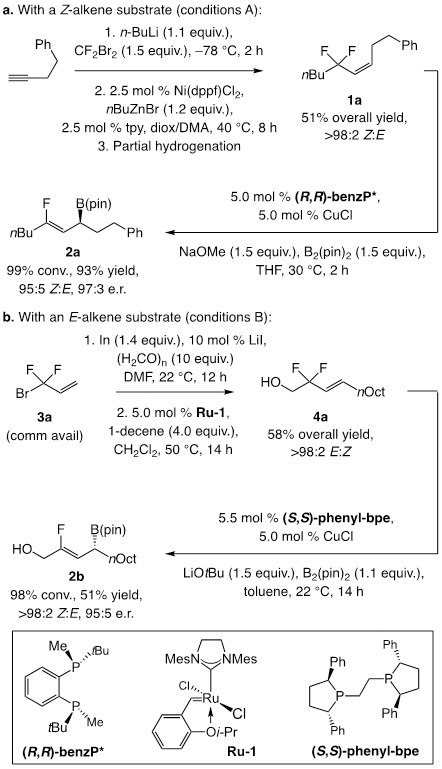
The initial studies involving representative E and Z Substrates. Reactions performed under N2 atm. Conversion was determined by 19F NMR analysis of unpurified mixtures. Z:E by GC or 1H NMR spectra of unpurified mixtures (±2%). Enantioselectivity was determined by HPLC analysis of derived alcohols (±1%). Yield of purified products (±5%). See the Supporting Information for details. tpy = 2,6-bis(2-pyridyl)pyridine.
Importantly, the above routes are complementary. Under conditions A, the reaction time is two hours (vs 14 h, conditions B), and the yield is higher. On the other hand, the E-alkenes for conditions B can be prepared in two steps, and because cross-metathesis with Ru-1 is utilized, allylic alcohol products can be directly and more easily prepared.[14]
The method has considerable scope (Scheme 3). Benzyl ether 2c, silyl ether 2d, and acetate 2e were isolated in 90–99% yield, 90:10–95:5 Z:E, and 96:4 e.r. (Scheme 3a). Phthalimide 2f was obtained in somewhat lower yield (56%) owing to partial decomposition during purification.[13] Other products included those bearing two n-alkyl moieties (2g). Reaction of a substrate with a more hindered cyclohexyl alkenyl substituent was efficient and highly stereoselective (2h); crystallographic analysis of 2h allowed us to confirm the identity of the major enantiomer formed. Me-substituted 2i was synthesized with similar efficiency and er but the Z:E ratio was lower (83:17). A plausible rationale (Figure 1) is that, with a smaller substituent (G), there is less energy gap between I and II; similar arguments apply if Cu–F elimination proceeded with syn stereochemistry. Thus, in cases where the alkene bears a relatively diminutive moiety, such as 2i, the Z:E ratio will be lower.
Scheme 3.

Scope of chiral allylic boronates. The same conditions were used as in Scheme 2. Conversion was determined by analysis of the 19F NMR spectra of the unpurified mixtures. Z:E ratios were determined by GC or 19F NMR analysis (±2%). Yields of purified products (±5%). Enantioselectivity was established by HPLC analysis of the saturated alcohols or corresponding ester derived from boronates (±1%). ds = diastereospecificity. See the Supporting Information for details.
Figure 1.
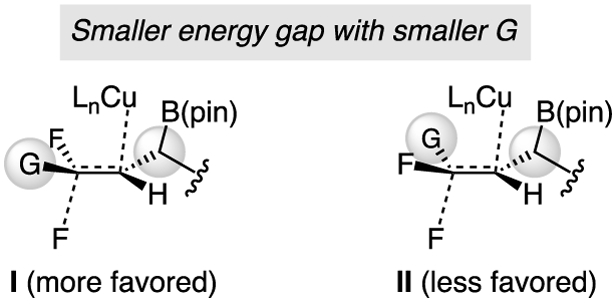
Origin of variations in Z:E ratios.
Products 2j-l were generated under conditions B in 52–63% yield[13], 98:2 Z:E, and 93.5:5–96:4 e.r.; these allylic alcohols provide the opportunity for investigation of various hydroxy-directed transformations. We synthesized 2m under conditions A with excellent Z:E ratio, albeit in lower yield (30%) and diminished e.r. (88.5:11.5), presumably due to the sizeable silyl group. The reaction with a difluoro-substituted terminal alkene delivered 2n in 95% yield, and 94:6 e.r. but, as expected (see Figure 1), as a mixture of olefin isomers. Finally, we prepared aryl-substituted 2o and 2p; the formerly disclosed boryl substitution reactions with F3C-substituted alkene substrates are confined to aliphatic variants.[8c-d] The ability to prepare allylic difluorides efficiently by cross-metathesis is key, rendering reactions with more functionalized substrates feasible; the examples presented in Scheme 3b (4→5a or 5b) are illustrative.
For a rationale regarding the high enantioselectivities, DFT studies were performed (M06L/def2-TZVPP//M06L/def2-SVP level) on model substrates (Me-substituted; Figure 2).[12] These investigations point to significant steric pressure in the transition states leading to the minor enantiomers (IV and VI vs III and V).
Figure 2.
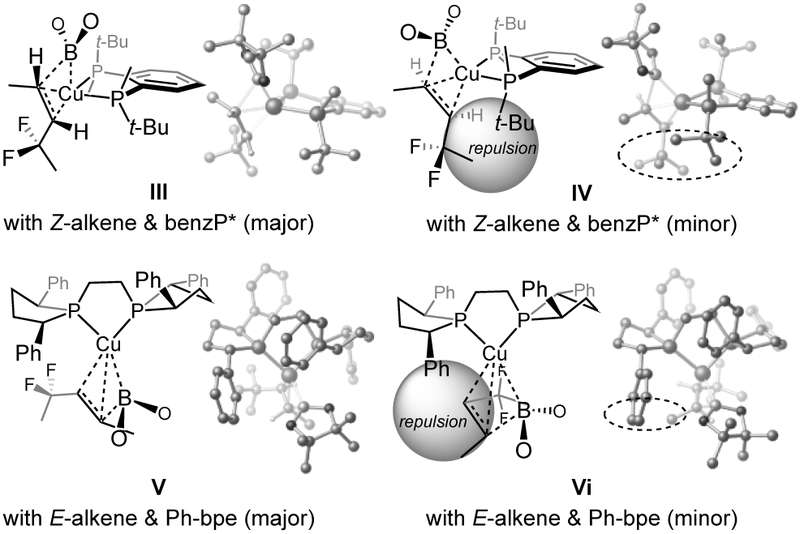
Stereochemical models based on DFT studies.
The enantiomerically enriched products may be modified stereospecifically (other than C–B oxidation). For instance, homoallylic boronate 6 and allylic amide 7 (Scheme 4a) were obtained in 98% and 62% yield, respectively,[15,16] without detectable loss in stereoisomeric purity. Such organofluorides cannot be readily accessed by an alternative method (catalytic or otherwise).
Scheme 4.

Representative functionalization of chiral allylic boronates.E:Z by analysis of 1H NMR spectra of unpurified mixtures. Yields of purified products (±5%). Enantioselectivity was determined by HPLC analysis (±1%). For compounds 8f and 10, isolated by pentane wash of solid product; e.r. might be higher than allylic boronate. See the Supporting Information for details.
The method may be used for synthesis of homoallylic alcohols with a fluoro-substituted allylic stereogenic quaternary carbon center (Scheme 4b). Under the conditions introduced by Aggarwal,[17] 2o was transformed to fluorohydrin[18] 8a, likely via A, in 54% yield, >98:2 E:Z ratio, and 99:1 e.r. The absolute stereochemistries of 2o and 8a were strongly suggested by crystallography with Cu X-ray source.[12] We synthesized other fluorohydrins with similar efficiency and complete stereospecificity. Reaction of 2o with sterically demanding 2-methylbenzaldehyde and 2-bromobenzaldehyde afforded 8b and 8c. Heterocyclic aldehydes (8d-e) and an enal (8f) were suitable substrates. Because compounds with F-substituted quaternary allylic carbon centers that contain only aliphatic substituents are unstable to chromatography, 8g was obtained after catalytic hydrogenation. Perhaps even more valuable are β-fluoroamines, as a β-fluorosubstituent can lower the pKa of a neighboring improving pharmacological properties.[19] We used the reaction of 2o with N-trimethylsilyl benzaldimine 9 (Scheme 4c) to generate homoallylic amide 10 in 59% overall yield, >98:2 E:Z ratio, >98:2 d.r., and without any loss in e.r. (98:2).
In closing, we put forth a bisphosphine–copper-catalyzed method for γ-boryl substitution of difluoro-substituted Z- or E-alkenes. The approach offers facile access to an assortment of (Z)- γ-monofluoroallylic boronates in high selectivity. The applicability of the approach is illustrated by stereospecific synthesis of several key types of organo-fluorine compounds. In view of the general importance of F-containing organic molecules, and the paucity of broadly applicable catalytic strategies for their diastereo- and enantioselective synthesis, this advance represents a key step forward.
Supplementary Material
Acknowledgements
This work was supported by the Japan Society for the Promotion of Science (JSPS) through KAKENHI grant 18H03907, 17H06370 and 19K15547 with the Institute for Chemical Reaction Design and Discovery (ICReDD), and the NIH (GM-130395). S. A., Q. L. thank the JSPS (grant 18J20858) and Shanghai Institute of Organic Chemistry for a scholarship and a postdoctoral fellowship, respectively. Y. Z. is a LaMattina Graduate Fellow. We are grateful to Dr. T. Seki and T. Sumitani for their assistance in analyzing the X-ray data, and to Dr. S. Torker for advice on the DFT studies.
Footnotes
Supporting information for this article is given via a link at the end of the document.
References
- [1] a).Yang X, Wu T, Phipps RJ, Toste FD, Chem. Rev 2015, 115, 826; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Ma JA, Cahard D, Chem. Rev 2008, 108, PR1; [DOI] [PubMed] [Google Scholar]; c) Ahrens T, Kohlmann J, Ahrens M, Braun T, Chem. Rev 2015, 115, 931. [DOI] [PubMed] [Google Scholar]
- [2] a).Fustero S, Sedgwick DM, Román R, Barrio P, Chem. Commun 2018, 54, 9706; [DOI] [PubMed] [Google Scholar]; b) Champagne PA, Desroches J, Hamel JD, Vandamme M, Paquin JF, Chem. Rev 2015, 115, 9073. [DOI] [PubMed] [Google Scholar]
- [3] a).Drouin M, Paquin JF, Beilstein J. Org. Chem 2017, 13, 2637; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Altman RA, Sharma KK, Rajewski LG, Toren PC, Baltezor MJ, Pal M, Karad SN, ACS Chem. Neurosci 2018, 9, 1735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].For reviews regarding preparation of compounds bearing a fluorine-substituted stereogenic quaternary carbon center, see:Zhu Y, Han J, Wang J, Shibata N, Sodeoka M, Soloshonok VA, Coelho JAS, Toste FD, Chem. Rev 2018, 118, 3887;Cahard D, Xu X, Couve-Bonnaire S, Pannecoucke X, Chem. Soc. Rev 2010, 39, 558. For selected papers about clinical candidates bearing a fluorine-substituted stereogenic quaternary carbon center, see:Synthetic routes to sofosbuvir in Synthesis of Heterocycles in Contemporary Medicinal Chemistry. Topics in Heterocyclic Chemistry, Vol. 44 (Eds.: Časar Z), Springer, 2015, pp. 51–88;Fried J, Sabo EF, J. Am. Chem. Soc 1954, 76, 1455.
- [5].For examples of synthesis monofluoro-substituted alkenes, see:Dutheuil G, Couve-Bonnaire S, Pannecoucke X, Angew. Chem. Int. Ed 2007, 46, 1290; Angew. Chem. 2007, 119, 1312;Nakamura Y, Okada M, Sato A, Horikawa H, Koura M, Saito A, Taguchi T, Tetrahedron 2005, 61, 5741;Zygalski L, Middel C, Harms K, Koert U, Org. Lett 2018, 20, 5071;Kondoh A, Koda K, Terada M, Org. Lett 2019, 21, 2277.
- [6].For examples of synthesis of compounds bearing a fluoro-substituted stereogenic quaternary carbon center, see:Yang X, Phipps RJ, Toste FD, J. Am. Chem. Soc 2014, 136, 5225;Phipps RJ, Toste FD, J. Am. Chem. Soc 2013, 135, 1268;Butcher TW, Hartwig JF, Angew. Chem. Int. Ed 2018, 57, 13125; Angew. Chem. 2018, 130, 13309;Topczewski JJ, Tewson TJ, Nguyen HM, J. Am. Chem. Soc 2011, 133, 19318;Liang Y, Fu GC, J. Am. Chem. Soc 2014, 136, 5520;Shibatomi K, Kitahara K, Okimi T, Abe Y, Iwasa S, Chem. Sci 2016, 7, 1388.
- [7].For reviews on C–F bond activation, see:Unzner TA, Magauer T, Tetrahedron Lett. 2015, 56, 877;Shen Q, Huang YG, Liu C, Xiao JC, Chen QY, Guo Y, J. Fluor. Chem 2015, 179, 14;Hamel JD, Paquin JF, Chem. Commun 2018, 54, 10224;Fujita T, Fuchibe K, Ichikawa J, Angew. Chem. Int. Ed 2019, 58, 390; Angew. Chem. 2019, 131, 396.
- [8].For examples of the transition metal catalyzed reactions of various B-based compounds with CF3-substituted alkenes, see:Corberán R, Mszar NW, Hoveyda AH, Angew. Chem. Int. Ed 2011, 50, 7079; Angew. Chem. 2011, 123, 7217;Liu Y, Zhou Y, Zhao Y, Qu J, Org. Lett 2017, 19, 946;Kojima R, Akiyama S, Ito H, Angew. Chem. Int. Ed 2018, 57, 7196; Angew. Chem. 2018, 130, 7314;Gao P, Yuan C, Zhao Y, Shi Z, Chem 2018, 4, 2201;Zhao X, Li C, Wang B, Cao S, Tetrahedron Lett. 2019, 60, 129.
- [9].For selected examples of the synthesis of fluorine-containing organoboron compounds, see:Ito H, Seo T, Kojima R, Kubota K, Chem. Lett 2018, 47, 1330;Sakaguchi H, Uetake Y, Ohashi M, Niwa T, Ogoshi S, Hosoya T, J. Am. Chem. Soc 2017, 139, 12855;Zhang J, Dai W, Liu Q, Cao S, Org. Lett 2017, 19, 3283;Hu J, Han X, Yuan Y, Shi Z, Angew. Chem. Int. Ed 2017, 56, 13342; Angew. Chem. 2017, 129, 13527;Tan DH, Lin E, Ji WW, Zeng YF, Fan WX, Li Q, Gao H, Wang H, Adv. Synth. Catal 2018, 360, 1032;Argintaru OA, Ryu D, Aron I, Molander GA, Angew. Chem. Int. Ed 2013, 52, 13656; Angew. Chem. 2013, 125, 13901;Guo W-H, Zhao H-Y, Luo Z-J, Zhang S, Zhang X, ACS Catal. 2019, 9, 38.
- [10].For representative initial studies regarding catalytic enantioselective boryl substitution reactions with Cu-based complexes, see: [Google Scholar]; a) Ito H, Ito S, Sasaki Y, Matsuura K, Sawamura M, J. Am. Chem. Soc 2007, 129, 14856; [DOI] [PubMed] [Google Scholar]; b) Guzman-Martinez A, Hoveyda AH, J. Am. Chem. Soc 2010, 132, 10634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11] a).Xu B, Mae M, Hong JA, Li Y, Hammond GB, Synthesis 2006, 803; [Google Scholar]; b) An L, Xu C, Zhang X, Nat. Commun 2017, 8, 1460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].See the supporting information for details.
- [13].The products with polar functionalities tend to show lower yield after isolation because stacking and partial decomposition occurs in silica-gel chromatography.
- [14].Moreover, whereas NaOMe is optimal for conditions A (significant diminution in efficiency with LiOMe), there was 30–40% conversion to the desired product when NaOtBu was used under conditions B. The mechanistic origin of these differences is under study.
- [15].Sadhu KM, Matteson DS, Organometallics 1985, 4, 1687. [Google Scholar]
- [16].Mlynarski SN, Karns AS, Morken JP, J. Am. Chem. Soc 2012, 134, 16449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17] a).Chen JLY, Scott HK, Hesse MJ, Willis CL, Aggarwal VK, J. Am. Chem. Soc 2013, 135, 5316; [DOI] [PubMed] [Google Scholar]; b) Chen JLY, Aggarwal VK, Angew. Chem. Int. Ed 2014, 53, 10992; Angew. Chem. 2014, 126, 11172. [Google Scholar]
- [18].For selected reviews on fluorohydrines, see:Haufe G, J. Fluor. Chem 2004, 125, 875;Sedgwick DM, Kobayashi N, Fustero S, López I, Xu B, Román R, Hammond GB, Okoromoba OE, Barrio P, Org. Lett 2018, 20, 2338;Cresswell AJ, Davies SG, Lee JA, Roberts PM, Russell AJ, Thomson JE, Tyte MJ, Org. Lett 2010, 12, 2936.
- [19].For examples of β-fluoroamine synthesis, see:Pupo G, Vicini AC, Ascough DMH, Ibba F, Christensen KE, Thompson AL, Brown JM, Paton RS, Gouverneur V, J. Am. Chem. Soc 2019, 141, 2878;Trost BM, Saget T, Lerchen A, Hung CI, Angew. Chem. Int. Ed 2016, 55, 781; Angew. Chem. 2016, 128, 791;Cresswell AJ, Davies SG, Lee JA, Roberts PM, Russell AJ, Thomson JE, Tyte MJ, Org. Lett 2010, 12, 2936;Kalow JA, Schmitt DE, Doyle AG, J. Org. Chem 2012, 77, 4177.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.