

SUMMARY
YAP/TEAD are nuclear effectors of the Hippo pathway regulating organ size and tumorigenesis majorly through promoter-associated function. However, their function as enhancer regulators remains poorly understood. Through an in vivo proximity-dependent labeling (BioID) technique, we identified YAP1 and TEAD4 protein as co-regulators of ERα on enhancers. The binding of YAP1/TEAD4 to ERα-bound enhancers is augmented upon E2 stimulation, and is required for the induction of E2/ERα target genes and E2-induced oncogenic cell growth. Furthermore, their enhancer binding is a prerequisite for enhancer activation marked by eRNA transcription and for the recruitment of the enhancer activation machinery component MED1. Remarkably, the binding of TEAD4 on active ERE-containing enhancers is independent of its DNA-binding behavior, and instead, is through protein-tethering trans-binding. Our data reveals a non-canonical function of YAP1 and TEAD4 as ERα cofactors in regulating cancer growth, highlighting the potential of YAP/TEAD as possible actionable drug targets for ERα-positive breast cancer.
Keywords: enhancer, transcriptional regulation, breast cancer, estrogen signaling, Hippo signaling, YAP/TEAD, ERα
Graphical Abstract
eTOC Blurb
ERα is a key transcription factor that binds to distal enhancers and regulates breast cancer-related gene expression events. Zhu et al. identify the Hippo pathway nuclear effectors YAP/TEAD as ERα cofactors and demonstrate their non-canonical role at enhancer level to regulate enhancer activation, gene transcription, and breast cancer growth.
INTRODUCTION
Enhancers are cis-regulatory elements with well-established roles in regulating cell type-specific gene expression (Plank and Dean, 2014). Despite the wide variation of transcription factors (TFs) bound on enhancers, enhancers share several common genomic features/signatures: open chromatin architecture, a common set of histone modifications such as H3K4me1/2 and H3K27Ac, as well as co-activators including p300/CBP and mediator (Heinz et al., 2015). In addition, many enhancers are actively transcribed, generating noncoding enhancer RNAs (eRNA), which are short transcripts of approximately 50 to 2000 nucleotides in size and often transcribed bi-directionally (De Santa et al., 2010; Hah et al., 2011; Kim et al., 2010). eRNA transcription serves as a robust marker of active enhancers and is widely used to indicate enhancer activity and target gene induction (Hah et al., 2013; Kim and Shiekhattar, 2015; Wang et al., 2011). There are ~100,000 enhancers in each mature cell type, but only a small percent of them have function to activate gene expression. Yet the mechanisms that determine the functionally active enhancers are not fully understood.
ERα is a ligand-regulated transcription factor that is expressed in approximately 70% of breast cancers. ERα is activated by binding to estrogenic ligands, such as 17β-estradiol (E2), to regulate a variety of genes that govern cell proliferation and survival in development and tumors (Moggs and Orphanides, 2001). Upon estrogen binding, ERα changes its conformation and translocates from the cytosol to the nucleus and binds predominantly to distal enhancer regions containing estrogen response elements (EREs) as a regulator of gene transcription (Carroll et al., 2006; Li et al., 2013). ERα requires cis-binding pioneer TFs (such as FOXA1) that bind to their own recognized DNA motifs close to ERE and open chromatin for ERα to localize onto ERE, followed by the recruitment of epigenetic cofactors including histone acetyltransferase P300 and mediator MED1 (Carroll et al., 2005; Hurtado et al., 2011; Jozwik and Carroll, 2012; Kraus and Kadonaga, 1998; Zhang et al., 2005). Notably, we have previously identified a category of ERα TF ‘co-activators’, termed as MegaTrans TFs. Besides binding to their conventional DNA-binding motifs (cis-binding), these MegaTrans TFs are also recruited by ERα through protein-protein interactions (trans-binding) to active ERE enhancers as ‘co-activators’ (Liu et al., 2014). Functionally, the ERα-interacting pioneer TFs facilitate to open the chromatin region and allow ERα to bind ERE motif, and MegaTrans TFs appear to stabilize the whole enhancer activation machinery. Despite the recent advances in understanding the mechanisms of enhancer activation and the subsequent gene regulation, the molecular machinery controlling this process remains elusive.
YAP/TAZ/TEAD have been recently shown to predominantly bind at distal enhancers, instead of promoters, to regulate enhancer function, extending the previous knowledge on transcription regulation by these proteins (Zanconato et al., 2015). YAP and its close paralog TAZ are the major downstream effectors of the Hippo pathway that regulate cell proliferation, cell contact inhibition and organ size (Zhao et al., 2008). YAP/TAZ binds to its target DNA loci by interacting with TEAD proteins (Li et al., 2010), and the transcriptional output of the Hippo pathway is mediated by TEAD to promote expression of Hippo target genes and cell proliferation (Hansen et al., 2015; Marti et al., 2015). In addition to TEAD, a plethora of other TF binding partners have been reported for YAP/TAZ, such as RUNX, p73, KLF4, TBX5, SMAD2/3 and OCT4 (Beyer et al., 2013; Piccolo et al., 2014). YAP/TAZ can also associate with chromatin-remodeling complexes to regulate transcription (Oh et al., 2013; Skibinski et al., 2014). More strikingly, YAP/TAZ/TEAD have recently been demonstrated to interact with AP1 to form a complex at distal enhancers that loop with promoters (Zanconato et al., 2015). However, the roles of YAP/TAZ/TEAD on enhancers beyond their promoter-associated functions have just begun to be recognized.
In this study we identified YAP1 and TEAD4 as cofactors of ERα on estrogen-regulated enhancers by applying the in vivo proximity-dependent labeling (BioID) technique (Roux et al., 2012). Besides binding to the canonical DNA regulatory elements associated with the Hippo pathway targets, we found that YAP1 and TEAD4 also bind to ERα active enhancers. Their non-canonical binding on ERα enhancers is elevated in the presence of E2, and is involved in the regulation of E2-induced transcription and breast cancer cell growth. Our mechanistic studies revealed that YAP1 and TEAD4 facilitate the recruitment of enhancer activation machinery component MED1 and regulate enhancer activation measured by eRNA production. Furthermore, unlike its cis-binding to the canonical sites, TEAD4 is recruited to active ERE-containing enhancers through protein tethering trans-binding in an ERα-dependent manner. Thus, our work provides evidence that Hippo and ER signaling pathways crosstalk at the chromatin level and converge on enhancers to control gene expression, highlighting a non-canonical function of YAP1 and TEAD4 at enhancers with potential therapeutic implications.
RESULTS
Identification of YAP/TEAD as previously unknown ERα-interacting cofactors by proximity BioID proteomics
We have previously identified a number of DNA-binding TFs associated with ERα, including FOXA1, GATA3, AP2γ, AP1 and STAT1, through mass spectrometry analyses on ERα nuclear complex, which was pulled down from MCF7 cells expressing a bacterial biotin ligase (BirA) that could biotinylate the biotin ligase recognition peptide (BLRP)-tagged ERα (Liu et al., 2014). To capture additional ERα-associated proteins, including the weak and transient cofactors, we used the live-cell proximity-dependent biotin identification (BioID) technique (Figure 1A). We engineered MCF7 to stably express ERα protein tagged with both HA and BirA* (a mutant version of BirA) that can biotinylate proximal proteins and ERα itself, followed by standard biotin-affinity pulldown of biotinylated proteins, which include candidate interactors of ERα and ERα itself, and subsequent protein identification by mass spectrometry. To avoid any artifacts due to protein overexpression, we expressed ERα under the control of a Tet-on promoter. We separated the cytoplasmic and nuclear fractions of total proteins and examined protein biotinylation. In the absence of doxycycline, the Tet-on promoter was inactive and we did not detect exogenous ERα expression or biotinylated proteins from either cytoplasmic or nuclear fraction (Figure 1B). When doxycycline was used to induce exogenous ERα expression, we detected the expression of exogenous ERα using either anti-HA or anti-ERα antibody, with an expression level close to endogenous ERα (Figure 1B). The nuclear level of both endogenous and exogenous ERα proteins increased in response to E2 treatment. We detected massive protein biotinylation in both cytoplasmic and nuclear fractions and found that the biotinylation level was higher in the nuclear fraction regardless of E2 treatment (Figure 1B). We then used streptavidin beads to pull down biotin-labeled proteins from nuclear fractions, which were subsequently analyzed with mass spectrometry. Besides the bait protein ERα, we identified many previously known ERα cofactors including TF and epigenetic cofactors, such as FOXA1 and GATA3, as well as previously unknown interactors including YAP1 and TEAD4 (Figure 1C). The peptide numbers of many cofactors from mass spectrometry analyses were significantly higher in the E2-treated group compared to the vehicle condition (Figure 1C), possibly due to E2-induced nuclear localization of ERα. The increased peptide numbers of YAP1/TEAD4 are unlikely due to E2-induced cytoplasm-to-nucleus trans-localization of these two proteins, as we detected no significant changes in the nuclear/cytoplasmic ratio in the presence of E2 (Figure S1A).
Figure 1. Identification of YAP/TEAD as previously unknown ERα-interacting cofactors by proximity BioID proteomics.

(A) Schematic diagram demonstrating BioID (proximity-dependent biotin identification) approach for identification of ERα-interacting nuclear proteins.
(B) Western blots confirming the inducible expression and in vivo biotinylation in the established ERα-BioID tet-on stable cell line. The fractionation of cytoplasmic (Cyto) and nuclear (Nuc) fractions of MCF7 cells was confirmed with Western blots for GAPDH (cytoplasm-specific marker) and Histone H3 (nucleus-specific marker). The doxycycline-inducible ERα-BirA*-HA fusion protein expression was detected by antibodies recognizing HA and ERα (the endogenous ERα was labeled with * and the tagged exogenous ERα was labeled with #). Biotinylated proteins by ERα-BirA* were detected by streptavidin-HRP blot.
(C) Identification of ERα-interacting cofactors by mass spectrometry analyses on the protein complex pulled down from the nuclear fractions of ERα-BioID tet-on stable cell line under the indicated treatments. Biotinylated proteins in nuclear fraction were enriched and purified using streptavidin beads before subjected to mass spectrometry. Besides many listed known cofactors, YAP1 and TEAD4 are two previously unknown ERα-interacting cofactors identified from our studies. Peptide numbers detected from mass spectrometry analyses are listed in the table for each protein.
(D) Co-IP assays in MCF7 cells with the indicated treatments confirming protein-protein interactions of endogenous ERα, YAP1 and TEAD4. Nuclear fractions treated with vehicle or E2 treatment were used for immunoprecipitation with antibodies against ERα, TEAD4 and YAP1 respectively.
(E) Western blots confirming the inducible expression and in vivo biotinylation in the established FOXA1-BioID tet-on stable cell line. The fractionation of cytoplasmic (Cyto) and nuclear (Nuc) fractions of MCF7 cells was confirmed with Western blots for GAPDH and Histone H3 respectively. The doxycycline-inducible Myc-BirA*-FOXA1 fusion protein expression was detected by antibodies recognizing Myc and FOXA1 (the endogenous FOXA1 was labeled with * and the tagged exogenous FOXA1 was labeled with #). Biotinylated proteins by BirA*-FOXA1 were detected by streptavidin-HRP blot.
(F) Identification of FOXA1-interacting factors by mass spectrometry analyses on streptavidin bead pulldowns from the nuclear fractions of FOXA1-BioID tet-on stable cell line with the indicated treatments. Peptide numbers detected from mass spectrometry analyses are listed in the table.
See also Figure S1.
To further confirm the interactions between YAP/TEAD and ERα, we performed a series of coimmunoprecipitation experiments. We detected TEAD4 and YAP1 in the protein complex pulled down from the nuclear lysates by streptavidin beads in the established ERα-BioID stable line (Figure S1B). This confirms that YAP1 and TEAD4 are in close proximity to ERα and biotinylated by ERα-BirA* fusion protein in the nuclei. In the same stable line, we were also able to detect YAP1 and TEAD4 in the ERα complex pulled down by anti-HA antibody (Figure S1C). Additionally, in regular MCF7 cells, we pulled down the protein complexes interacting with ERα, TEAD4 or YAP1 using antibodies recognizing each protein respectively, and we were able to detect the other two proteins in each individual complex as expected (Figure 1D), indicating that these proteins co-exist in the same complex in the nuclei.
FOXA1, a pioneer TF, has been shown to facilitate the binding of ERα to enhancers (Carroll et al., 2005) and is essential for almost all ERα chromatin binding events in breast cancer (Hurtado et al., 2011). Consistent with previous studies, we identified FOXA1 as an ERα-interactor from the BioID mass spectrometry (Figure 1C). We then performed FOXA1 BioID mass spectrometry to identify FOXA1 cofactors and also identified YAP1 and TEAD4 from FOXA1 complex (Figures 1E and 1F), consistent with the notion that FOXA1, ERα, YAP1 and TEAD4 function in the same protein complex for chromatin-associated function. TAZ and YAP are two highly related transcription regulators and often play redundant roles. However, we did not detect TAZ from either ERα or FOXA1 BioID proteomic analyses. Thus, we tested whether TAZ was a low abundance protein in MCF7 cells. Indeed, we found extremely low TAZ expression in MCF7 compared to 293T cells (Figure S1D).
YAP/TEAD bind to both canonical Hippo pathway target genes and non-canonical ERα enhancer regions
To understand the role of YAP1 and TEAD4 as ERα interactors in the nucleus, we first examined their binding on chromatin using ChIP-seq in MCF7 cells and compared their binding sites with those of ERα. Under E2 treatment condition, we identified 7,415 common binding sites of YAP1 and TEAD4, most of which were also decorated with H3K27Ac histone marker and FOXA1 (Figures 2A and 2C). 2,863 out of these 7,415 sites were also bound by ERα, and the binding of YAP1 and TEAD4 to the 2,863 YAP1+/TEAD4+/ERα+ sites was significantly enhanced in response to E2 stimulation (Figures 2A, 2B, and 2C). Notably, we found 2,284 out of these 2,863 YAP1+/TEAD4+/ERα+ sites were enhancers based on enhancer histone marker analyses (see STAR METHODS), and we identified ERE as the #1 enriched motif for these 2863 common binding sites, suggesting that YAP1/TEAD4 predominantly interact with ERα on enhancers containing ERE motif (Figure 2C). In contrast, the binding of YAP1 and TEAD4 to the 4,552 YAP1+/TEAD4+/ERα− sites remained unchanged upon E2 treatment (Figures 2B and 2C). These 4,552 ERα− sites are likely the canonical YAP/TEAD binding sites, as TEAD motif is the #1 enriched motif for these sites (Figure 2C). The differential binding of ERα, YAP1, TEAD4, FOXA1 and H3K27Ac on representative ERα enhancers and Hippo target gene promoters was exemplified by the sites in close proximity to the classic E2-induced gene GREB1 and a well-known Hippo pathway target gene CTGF (Figures 2D and 2E). YAP1 and TEAD4 co-bound with FOXA1 and ERα on GREB1 enhancers marked with H3K27Ac, and their binding was enhanced in response to E2 (Figure 2D). In contrast, on CTGF gene locus, YAP1 and TEAD4 showed strong binding to its promoter independent of E2, with no binding of ERα on this site (Figure 2E). When we compared our ERα ChIP-seq data with published ChIP-seq data of YAP1 and TEAD1 in MCF7 cells (Elster et al., 2018), we also identified 1,523 out of 7,665 YAP1+/TEAD1+ sites as ERα+ sites (Figures S2A and S2B). These results suggest that YAP and TEAD can interact with ERα on ERα enhancers to play a non-canonical role as ERα cofactors.
Figure 2. Identification of non-canonical binding of YAP/TEAD on ERα enhancers.

(A) Venn diagram showing the overlap of ERα binding peaks with those of YAP1 and TEAD4. Two different YAP/TEAD binding groups were identified: the 2,863 YAP1+/TEAD4+/ERα+ group and the 4,552 YAP1+/TEAD4+/ERα− group. The binding peaks in MCF7 cells (+E2) were mapped based on ChIP-seq with antibodies recognizing these three proteins.
(B) Aggregate plots showing the normalized tag density of ERα, YAP1 and TEAD4 ChIP-seq data (±E2) for the 2,863 YAP1+/TEAD4+/ERα+ sites or the 4,552 YAP1+/TEAD4+/ERα− sites. The binding of YAP1, TEAD4 and ERα on the 2,863 YAP1+/TEAD4+/ERα+ sites, but not on the 4,552 YAP1+/TEAD4+/ERα− sites, was significantly enhanced upon E2 stimulation (see fold changes (FC) and P-values (p) between E2 and Veh control).
(C) Heatmaps of ERα, FOXA1, YAP1, TEAD4, and H3K27Ac ChIP-seq data (±E2), supporting the distinct binding patterns for YAP1/TEAD4 in the two groups shown in (B). The top 10 enriched motifs for each group (± 300 bp from the center of ChIP-seq binding sites) are listed.
(D) Genome browser view of ChIP-seq peaks on a well-known ERα target gene GREB1 locus showing the E2-enhanced binding of ERα, FOXA1, YAP1, and TEAD4 at several ERα enhancers.
(E) Genome browser view of ChIP-seq peaks on a well-known Hippo pathway target gene CTGF locus showing that the canonical binding of YAP1 and TEAD4 at CTGF promoter is independent of ERα or E2 stimulation.
See also Figures S2 and S3.
eRNA transcription is known to be associated with E2-induced ERα enhancer activation (Li et al., 2013). Based on the strength of eRNA induction by E2, we sorted the ERα active enhancers into 3 groups (High Activity, Median Activity, and Low Activity) and analyzed the binding of YAP1 and TEAD4 on them. In the High Activity group with the highest eRNA induction (i.e. the most active ERα enhancers), the binding of YAP1 and TEAD4 also showed the strongest induction by E2 (Figures S2C and S2D). Thus, YAP1 and TEAD4 co-bind to ERα enhancers and their binding correlates with E2-induced activation of ERα enhancers, suggesting their possible role in regulating enhancer activity.
YAP1/TEAD4 also interact with ERα on enhancers in T47D cells
We next tested whether the interaction and co-occupancy of YAP1/TEAD4 and ERα is a widespread phenomenon. We first examined coimmunoprecipitation between YAP1/TEAD4 and ERα in another breast cancer cell line T47D. We found that endogenous YAP1 and TEAD4 were detected in the protein complex pulled down by anti-ERα antibody, but not in the IgG control (Figure S3A), indicating that YAP1 and TEAD4 physically interact with ERα in the nuclei. We then mapped the genome-wide binding sites of YAP1, TEAD4 and ERα in T47D cells with ChIP-seq. Like in MCF7 cells, we identified common binding sites of YAP1, TEAD4 and ERα (Figure S3B). The binding of YAP1 and TEAD4 on these 2,015 common sites were significantly enhanced in response to E2 treatment (Figure S3C). In contrast, their binding on non-ERα sites was not affected by the presence of E2 (Figure S3C). We identified ERE and TEAD motif as the #1 motif for these two groups of binding sites respectively (Figure S3C). The differential binding of YAP1, TEAD4 and ERα on a representative E2-induced gene GREB1 and a Hippo pathway target gene CTGF in cells with or without E2 treatment was investigated. Like in MCF7 cells, the E2-enhanced co-occupancy between YAP1/TEAD4 and ERα was only detected at the ERα target gene, but not the Hippo pathway target gene (Figures S3D and S3E).
We further compared MCF7 and T47D cells for the target sites co-occupied by ERα, YAP1 and TEAD4. As shown in the Venn diagram (Fig. S3F), 878 out of the 2,015 YAP1+/TEAD4+/ERα+ sites in T47D cells overlap with those identified in MCF7 cells. The Fisher’s exact test shows that the overlap is statistically significant (P-value: < 2.2e-16; Odds ratio = 1.80). When we examined the YAP1/TEAD4/ERα occupancy in T47D cells on the 2,863 and 4,552 sites defined in MCF7 cells, we observed a similar pattern to that in MCF7 (Fig. S3G). The binding of ERα was much stronger on the 2,863 sites than on the 4,552 sites, while YAP1 and TEAD4 strongly bound on both groups of sites. The binding of all three proteins on the 2,863 sites, but not on the 4,552 sites, was augmented in the presence of E2, although the level of augmentation was not as dramatic as in MCF7 cells, possibly due to the weaker E2 response in T47D cells in general. Together, our data suggest that the interaction of YAP1/TEAD4 and ERα on enhancers might be a common mechanism to regulate the expression of non-Hippo pathway target genes.
The non-canonical binding of YAP/TEAD to ERα enhancers is required for E2-induced transcription and breast cancer cell growth
As YAP1/TEAD4 physically interact and co-bind to a subset of ERα enhancers with ERα, we wondered what are the biological themes associated with the target genes of this subset of ERα enhancers. GREAT analysis showed that the 2,863 YAP1+/TEAD4+/ERα+ sites are enriched for ERα and FOXA1 transcription factor networks, whereas the 4,552 YAP1+/TEAD4+/ERα− sites are enriched for YAP/TAZ-stimulated gene expression pathway (Figure 3A). We then sought to determine whether YAP1 and TEAD4 are required for transcriptional activation of these ERα target genes. We performed RNA-seq in MCF7 cells treated with shRNAs for YAP1 or TEAD4. The shRNAs used were able to greatly abolish YAP1/TEAD4 protein expression without affecting ERα expression (Figure S4A). Of the large cohort of genes that were activated by E2, we found a dramatic inhibition of E2-induced gene expression by YAP1/TEAD4 shRNA knockdown (Figures S4B and S4C), exemplified by a well-known ERα target gene KCNK5 (Figure S4D). We next examined expression changes of those genes associated with the 2,863 YAP1+/TEAD4+/ERα+ target sites. We retrieved the nearest genes within 50 kb from the binding sites, and divided them into different groups based on expression changes in response to E2 in shControl condition. We found that shYAP1 or shTEAD4 significantly attenuated both of the up- and down-regulation effects by E2 (Figure 3B), suggesting the role of YAP1 and TEAD4 in the transcriptional regulation of ERα target genes.
Figure 3. The non-canonical binding of YAP/TEAD to ERα enhancers is required for transcriptional activation of ERα target genes and E2-induced breast cancer growth.

(A) GREAT analysis against MSigDB Pathway identified the potential pathways enriched in genes associated with 2,863 sites and 4,553 sites.
(B) Box plots of RNA-seq showing the effects of YAP1 or TEAD4 knockdown on E2-induced changes in gene expression. Genes associated with the 2,863 sites were divided into 3 groups based on expression changes in response to E2 treatment.
(C) Box plots of RNA-seq showing the E2 effects on expression of the coding genes targeted by 3 groups of ERα enhancers with high, median or low activity (see STAR METHODS for enhancer group definition) in cells transduced with shControl, shYAP1 or shTEAD4. YAP1 or TEAD4 knockdown significantly attenuated the E2-induction in the group of genes associated with “High Activity” enhancers, but not the groups with “Median” or “Low Activity” enhancers.
(D) Cell proliferation assay using CCK-8 showing that YAP1 and TEAD4 are required for E2-induced breast cancer growth. MCF7 cells were knocked down with control shRNA or shYAP1 or shTEAD4 and cultured in medium with or without E2 after stripping.
(E) Colony formation assay confirming that YAP1 and TEAD4 are required for E2-induced breast cancer cell growth. MCF7 cells were treated with control shRNA or shYAP1 or shTEAD4 and cultured in stripping medium with or without E2 followed by fixation and staining with crystal violet. Numbers of colonies were measured using ImageJ.
(F) YAP1 or TEAD4 knockdown inhibited cancer xenograft growth. MCF7 cells treated with shControl, shYAP1 or shTEAD4 were injected into the mammary fat pads of female nude mice with 17β-Estradiol pellets implanted on back underneath skin. Tumors were measured 5 weeks post cell implantation. Statistics for (D-F): ***P < 0.001, **P < 0.01, *P < 0.05, One-way ANOVA.
See also Figure S4.
As the binding of YAP1/TEAD4 was the strongest and the most responsive to E2 on the “High Activity” group of ERα enhancers (Figures S2C and S2D), we hypothesized that YAP1/TEAD4 might predominantly regulate transcription of the most E2-responsive genes controlled by these highly active enhancers. When we classified E2-activated genes into three groups that were regulated by these three groups of ERα enhancers (High Activity, Median Activity and Low Activity), we observed the strongest inhibitory effect on the E2-induced gene expression in the High Activity enhancer group after YAP1 or TEAD4 knockdown (Figure 3C). These results indicate that YAP1 and TEAD4 are particularly important for transcriptional activation of target genes associated with highly active ERα enhancers. We further examined mRNA expression level of representative E2-induced coding genes, including GREB1, KCNK5 and SIAH2, with RT-qPCR. E2-induced transcriptional activation of these representative genes was significantly attenuated following YAP1/TEAD4 knockdown in both MCF7 and T47D cells (Figures S4E–H), confirming the important role of YAP1 and TEAD4 in transcriptional activation of E2/ERα target genes.
Having shown that YAP1 and TEAD4 are required for E2-dependent gene induction, we next tested whether YAP1 and TEAD4 regulate E2-induced breast cancer cell growth by knocking down YAP1 or TEAD4 in MCF7 cells with shRNAs. We found that YAP1 or TEAD4 knockdown in MCF7 cells led to decreased proliferation rate of cells cultured in stripping media containing vehicle control. MCF7 cells that were cultured in stripping medium containing E2 displayed rapid estrogen-induced growth. However, the E2-induced cell growth was greatly dampened by YAP1 or TEAD4 knockdown (Figure 3D). The effect of YAP1 and TEAD4 knockdown on cell growth was further demonstrated by colony formation assay. MCF7 cells infected with shControl, shYAP1 or shTEAD4 were cultured in stripping medium with vehicle or E2 for 10 days before fixation and crystal violet staining. We observed that E2-induced colony formation was diminished by YAP1 or TEAD4 knockdown (Figure 3E). Furthermore, we conducted xenograft experiments by implanting MCF7 cells treated with shControl, shYAP1 or shTEAD4 to nude mice. Consistent with the data from in vitro cell growth assays, we found that knocking down YAP1 or TEAD4 greatly inhibited tumor growth in xenograft models treated with E2 (Figure 3F). These results indicate the roles of YAP1 and TEAD4 in promoting ERα-mediated MCF7 breast cancer cell growth.
YAP1/TEAD4 enhancer binding is a prerequisite for enhancer activation marked by eRNA transcription
Our observation that YAP1 and TEAD4 bind to ERα enhancers and regulate ERα-mediated gene activation and cell growth prompted us to test whether YAP1 and TEAD4 are required for enhancer activation directly. Enhancer activity highly correlates with eRNA synthesis, although it’s still under debate whether eRNAs function as transcriptional activators or they are just the consequence of spurious transcriptional activities (Lam et al., 2014). eRNA transcription can be used to predict active enhancers de novo and has been shown to be a reliable marker for enhancer activity (Hah et al., 2013). To test the role of YAP1 and TEAD4 on ERα enhancer activation, we performed GRO-seq (global run-on coupled with deep sequencing) to detect eRNA production in MCF7 cells transduced with control shRNA or shRNAs against YAP1 or TEAD4 and treated with vehicle or E2. We observed robust E2-induced bi-directionally transcribed eRNAs from the 2,863 YAP1/TEAD4/ERα co-occupied sites (i.e. ERα enhancers) (Figure 4A). Remarkably, knocking down either YAP1 or TEAD4 greatly abolished the E2-induced eRNA transcription from YAP1/TEAD4/ERα co-bound sites (Figures 4A and S5A). The GRO-seq data also revealed a positive correlation between eRNA transcription and the adjacent coding gene transcription. For example, on the representative GREB1 and SMAD7 gene loci, E2 stimulation or YAP1/TEAD4 knockdown affected transcriptional activities on enhancers and target gene bodies in the same direction (Figures 4B and 4C). Therefore, YAP1 and TEAD4 are required for both E2-induced eRNA transcription from ERα enhancers and the expression of the adjacent target coding genes.
Figure 4. YAP1 and TEAD4 are required for ERα enhancer activation.

(A) Aggregate plots for GRO-seq tag density showing both sense (plus) and anti-sense (minus) eRNA expression levels for the 2,863 YAP1+/TEAD4+/ERα+ sites in MCF7 cells transduced with control shRNA, shYAP1 or shTEAD4 (±E2). E2 treatment in control cells significantly increased eRNA transcription, and this E2-induced eRNA production was greatly abolished in cells with YAP1 or TEAD4 knockdown.
(B-C) Genome browser views of GRO-seq signals at representative ERα active enhancers and their target genes GREB1 and SMAD7. Transcriptional activities at enhancers and at target gene bodies were positively correlated (see changes in eRNA level and changes in gene body expression upon E2 stimulation and YAP1/TEAD4 knockdown), suggesting that YAP1 and TEAD4 are required for E2-induced transcription from both ERα enhancers (eRNA) and the adjacent target coding genes (mRNA).
See also Figure S5.
In complement to GRO-seq, we examined eRNA transcription from the enhancers for GREB1, PGR, KCNK5 and SIAH2 genes with RT-qPCR. Consistent with our GRO-seq data, E2 treatment significantly increased eRNA transcription from these active enhancers, and knockdown of YAP1 or TEAD4 was sufficient to attenuate E2-dependent eRNA production (Figures S5B and S5C). Together with the data showing that the binding of YAP1 and TEAD4 on ERα enhancers was positively correlated with E2-induced eRNA production (Figure S2), these results suggest that the co-occupancy of YAP1 and TEAD4 on ERα-bound enhancers is critical for E2-liganded enhancer activation.
YAP1 facilitates the binding of MED1 to ERα-bound enhancers
We next sought to determine the mechanisms of YAP1 and TEAD4 in promoting ERα enhancer activation. We first tested whether knocking down YAP1/TEAD4 could affect the binding of ERα on enhancers. We transduced MCF7 cells with control shRNA or shRNA targeting YAP1 or TEAD4. The cells were then treated with the vehicle control or E2 for one hour, before we performed ERα ChIP-seq. We did not observe any changes in global ERα binding on its enhancers under E2 condition (Figures 5A, S6A, and S6B). Therefore, YAP1 and TEAD4 are dispensable for ERα binding to chromatin.
Figure 5. YAP1 and TEAD4 regulate MED1 binding on active ERα enhancers.

(A) Heatmap of ERα ChIP-seq data (+E2) on the 2,863 YAP1+/TEAD4+/ERα+ sites showing that ERα binding is not dependent on YAP1 or TEAD4. MCF7 cells were treated with control shRNA, shYAP1 or shTEAD4.
(B) E2-induced MED1 binding on enhancers requires YAP1. Aggregate plots showing the normalized tag density of MED1 ChIP-seq data (±E2) on the 2,863 YAP1+/TEAD4+/ERα+ sites. MCF7 cells treated with control shRNA or shYAP1 and maintained in stripping medium with or without E2 were used for ChIP-seq.
(C-D) Genome browser views of ChIP-seq signals for ERα/YAP1/TEAD4 (+E2) and MED1 (±E2) on enhancers at GREB1 and SMAD7 loci. On these YAP1/TEAD4/ERα co-bound enhancers, E2-induced MED1 binding was significantly diminished by shRNA knockdown of YAP1.
See also Figures S6A–B.
The Mediator complex is a central transcriptional coactivator complex that bridges transcriptional activators and RNA polymerase II (Taatjes, 2010). MED1, a subunit in the middle module of the Mediator complex, is known to directly interact with ERα and play an essential role in ERα-mediated enhancer activation. MED1 is required for E2-induced gene expression and breast cancer cell growth (Acevedo et al., 2004; Warnmark et al., 2001; Zhang et al., 2005). Thus, we tested whether YAP1 may play a role in facilitating MED1 binding on ERα enhancers. We performed ChIP-seq of MED1 in cells transfected with control shRNA or shRNA targeting YAP1. As predicted, MED1 binding was detected at the 2,863 YAP1+/TEAD4+/ERα+ sites. In the presence of E2, MED1 binding at the YAP1+/TEAD4+/ERα+ sites was significantly enhanced (Figure 5B). We found that upon YAP1 knockdown, E2-induced MED1 binding at ERα enhancers was greatly dampened (Figure 5B). Genome browser views of the ChIP-seq data of ERα, YAP1, TEAD4 and MED1 on representative gene loci GREB1 and SMAD7 showed E2-induced MED1 binding was significantly reduced upon YAP1 knockdown (Figures 5C and 5D). Together, these data suggest that YAP1 might function to recruit the components of the Mediator complex or stabilize their binding on ERα enhancers.
The binding of YAP1 and TEAD4 on ERα enhancers is dependent on ERα
To further explore the molecular mechanism of the YAP1/TEAD4-dependent enhancer activation, we examined whether binding of YAP1 and TEAD4 on ERα enhancers requires the presence of ERα. ICI 182,780 is an antagonist for ERα, and its binding to ERα leads to a conformational or functional change of ERα. Additionally, the ICI-ERα complex is unstable and undergoes accelerated degradation, resulting in ERα protein level down-regulation (Nicholson et al., 1995). We treated MCF7 cells that were cultured in stripping medium containing DMSO or ICI 182,780 for 24 hours with vehicle or 100 nM E2 for one hour and collected them for ERα degradation test and ChIP-seq of ERα, YAP1 and TEAD4. As expected, ICI 182,780 treatment greatly reduced ERα protein level. In contrast, the expression of YAP1 and TEAD4 was not affected by ICI 182,780 (Figure 6A). The binding of ERα on the 2,863 YAP1+/TEAD4+/ERα+ sites was greatly diminished by ICI 182,780 treatment under E2 treatment condition (Figures 6B and S6C). Interestingly, following the reduction of ERα binding, the binding of YAP1 and TEAD4 on the 2,863 YAP1+/TEAD4+/ERα+ sites were also significantly reduced (Figures 6C, 6D, and S6C), while their binding on the 4,552 YAP1+/TEAD4+/ERα− sites remained unchanged (Figures 6B–6D and S6C). The differential dependence of YAP1, TEAD4 on ERα presence were further examined on two representative E2-induced gene loci GREB1 and SMAD7 and on two Hippo pathway target genes CTGF and AJUBA. Again we found that the ERα-dependent recruitment of YAP1/TEAD4 on chromatin was only detected for the enhancers in the close proximity to GREB1 and SMAD7 genes (Figures 6E–6F and S6D–6E). These experiments demonstrate that the binding of YAP1 and TEAD4 at ERα enhancers requires ERα and thus their enhancer-related function is dependent on the presence of ERα.
Figure 6. Binding of YAP1 and TEAD4 on ERα enhancers requires Erα.

(A) Western blots showing the downregulation effects of ICI 182,780 (ICI) on ERα protein level in the nucleus, especially under E2 stimulation condition. ICI treatment significantly decreased ERα protein level but did not affect YAP1 or TEAD4 protein level in cells in the presence or absence of E2.
(B-D) Aggregate plots showing the normalized tag density of ERα, YAP1 and TEAD4 ChIP-seq data for the two groups of YAP1/TEAD4 binding sites. On the 2,863 YAP1+/TEAD4+/ERα+ non-canonical sites, ICI treatment abolished ERα binding and reduced YAP1/TEAD4 binding at the same time. However, on the 4,552 YAP1+/TEAD4+/ERα− canonical sites, their binding was not affected by ICI treatment (see fold changes (FC) and P-values (p) between DMSO and ICI).
(E-F) Genome browser views of ChIP-seq signals on two well-known E2/ERα target genes loci, GREB1 and SMAD7, confirming that binding of ERα and YAP1/TEAD4 on non-canonical binding sites was greatly reduced in response to ICI-induced ERα degradation.
See also Figures S6C–E.
TEAD4 is recruited to ERα enhancers through protein tethering trans-binding unlike its cis-binding to the canonical sites
We have previously demonstrated that the most strongly activated E2-responsive enhancers are characterized by trans-recruitment of a large protein complex of diverse TFs by ERα (Liu et al., 2014). It’s known that due to the lack of DNA-binding activity, YAP1 must interact with DNA-binding transcription factors, such as TEAD4, to regulate target gene expression. Therefore, the binding of YAP1 to ERα enhancers must be in trans through protein-protein interaction. The dependence on ERα for TEAD4 to bind on ERα enhancers prompted us to test whether TEAD4 binding is in cis or in trans. To this end, we engineered MCF7 cells to stably express BirA, a bacterial biotin ligase that could specifically biotinylate the biotin ligase recognition peptide (BLRP), and BLRP-tagged wildtype (WT) or DNA-binding domain mutant (Mut) version of TEAD4 protein that was controlled by the inducible Tet-on promoter. In this experimental design, the mutation abolishes TEAD4’s cis-binding to the sites containing its cognate TEAD motif as previously reported (Shi et al., 2017). We induced exogenous TEAD4 expression with doxycycline and treated the cells with vehicle or E2. Our coimmunoprecipitation data showed that the interaction between ERα and TEAD4 was not affected by DNA-binding domain mutation (Figure S7A). We then performed ChIP-seq using streptavidin beads to compare the global binding pattern between WT and Mut TEAD4. As expected, the DNA-binding mutation greatly abolished TEAD4 binding, with a fold change (FC) of 2.51, on the 4,552 YAP1+/TEAD4+/ERα− sites (Figure 7A), in which TEAD motif is the #1 ranked motif (Figure 2C). Genome browser views of classic Hippo pathway target genes including CTGF and AJUBA, demonstrated a complete loss of TEAD4 binding when the DNA-binding domain was mutated (Figures 7B and S7B). When we examined TEAD4 binding on the 2,863 sites, we also observed a reduction in binding caused by the mutation, but the reduction was less dramatic (FC = 1.93). The fact that TEAD4 DNA-binding mutation caused a milder reduction in the binding on the 2,863 YAP1+/TEAD4+/ERα+ sites than the 4,552 YAP1+/TEAD4+/ERα− sites suggests that TEAD4 binding on the 2,863 sites might be only partially dependent on its DNA-binding activity. Given that ERE and TEAD are the top two motifs enriched in the 2,863 sites (Figure 2C), it’s likely that the binding of TEAD4 to the 2,863 sites is a mixture of cis-binding to TEAD motif and trans-binding to ERE motif.
Figure 7. TEAD4 is recruited to ERα enhancers through protein tethering trans-binding.

(A) Heatmaps and aggregate plots of the ChIP-seq data for wildtype TEAD4 (WT) and DNA-binding domain mutant TEAD4 (Mut) to compare the effects of DNA-binding domain mutation on TEAD4 binding at three different groups of binding sites: the 4,552 YAP1+/TEAD4+/ERα− sites, the 2,863 YAP1+/TEAD4+/ERα+ sites and the 419 the most active ERα enhancers. The fold changes (FC) and P-values (p) comparing WT and Mut binding (+Dox, +E2) are shown.
(B-C) Genome browser images of WT and Mut TEAD4 ChIP-seq signals on representative gene loci. The DNA-binding mutation completely abolished the binding of TEAD4 on a classic Hippo pathway target gene CTGF, but did not affect its binding on the enhancers associated with the ERα target gene GREB1.
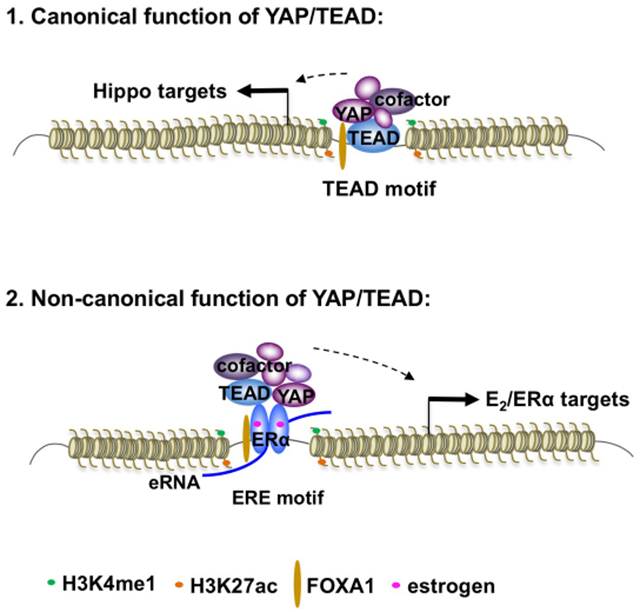
(D) A proposed model for the canonical and non-canonical function of YAP1 and TEAD4. In addition to their canonical role as downstream nuclear effectors of Hippo pathway, YAP1 and TEAD4 can also function as ERα co-regulators at active ERE-containing enhancers to regulate enhancer activity and target gene transcription upon E2 stimulation. The binding of YAP1 and TEAD4 is critical for the assembly of enhancer activation machinery components, such as MED1. Different to its cis-binding to the canonical sites, the binding of TEAD4 on the most active ERα enhancers is in trans through protein-protein tethering interaction, which is independent of the DNA-binding activity of TEAD4.
See also Figure S7.
As the binding strength of YAP1 and TEAD4 on ERα enhancers positively correlated with enhancer activities (Figures S2C and S2D), we then tested if the trans-recruitment of TEAD4 was predominantly associated with those highly active ERα enhancers, most of which contain ERE DNA motif. We analyzed TEAD4 binding on the 419 highly active enhancers defined with E2-induced eRNA FC induction >=2. We found that the binding reduction caused by the DNA-binding mutation of TEAD4 was much less noticeable for the 491 highly active enhancers when compared to the 2,863 sites or the 4,552 sites (FC = 1.34) (Figure 7A). Indeed, when we checked ChIP-seq signals on the classic ERα target genes including GREB1 and SMAD7, we found that Mut TEAD4 had similar binding strength as WT TEAD4 (Figures 7C and S7C). Additionally, we conducted biotin ChIP-qPCR to compare the binding of wildtype and DNA-binding domain mutated TEAD4 proteins on ERα target genes (GREB1 and SIAH2) and Hippo pathway target genes (CTGF and AJUBA). DNA-binding mutation completely diminished the binding of TEAD4 on CTGF and AJUBA, but did not affect that on GREB1 or SIAH2 (Figure S7D). Therefore, at least a significant portion of the binding events of TEAD4 to ERα enhancers, especially those most active ERE-containing enhancers, is independent of the DNA binding activity of TEAD4, and instead might be mediated by protein-protein interaction (trans-recruitment) with ERα and possibly other enhancer components.
DISCUSSION
Here for the first time, our work revealed that besides functioning as the nuclear effectors for the Hippo pathway, YAP/TEAD also work as co-regulators of ERα for estrogen-regulated enhancer activation (see YAP/TEAD working model in Figure 7D). Thus, our studies uncovered a non-canonical role of YAP/TEAD on enhancers, as well as provided a mechanistic explanation for the interplay between the Hippo pathway and ERα signaling, highlighting the potential role of YAP1 and TEAD4 as targets for ERα-positive breast cancer.
First discovered in Drosophila, the Hippo pathway is an evolutionarily conserved governor of cell growth and organ size. Center to this pathway is the MST-LATS kinase cascade, which regulates the nuclear localization of two transcriptional co-activators YAP and TAZ, and subsequently the transcription of downstream targets. Due to the lack of a DNA-binding domain, YAP/TAZ requires transcription factor partners to fulfill their roles in transcriptional regulation. Various partners have been identified, and motif analyses revealed that TEADs are the main platform tethering YAP/TAZ to chromatin (Piccolo et al., 2014). Previous studies have been focused on the promoter-associated transcription regulation by YAP/TAZ/TEAD. More recently, it’s recognized that YAP/TAZ/TEAD bind on distal enhancers and form a complex with AP1 to control gene expression and oncogenic cell growth (Zanconato et al., 2015). However, their function as enhancer regulators remains poorly understood.
In this study, we identified YAP1 and TEAD4 as co-activators of ERα by carrying out a powerful BioID screen for proteins interacting with ERα. Compared to the traditional protein-protein interaction study tools, this in vivo proximity biotin labeling BioID technology can capture in situ interactions including direct and indirect proximity interactions in live cells, therefore identifying additional cofactors. Our ChIP-seq analyses revealed that 2,284 out of the 2,863 YAP1+/TEAD4+/ERα+ overlapping sites were enhancers, supporting a notion that YAP1/TEAD4 may play a key role in enhancer regulation. A recent study has reported that in the absence of LATS, YAP/TAZ and ERα are stabilized and function together to control breast cancer cell fate (Britschgi et al., 2017). But it remains unclear how exactly YAP/TAZ interact with ERα and work together in the nuclei to regulate gene expression. Our observation that YAP/TEAD co-occupied with ERα on enhancers provides a regulatory mechanism at chromatin level that underlies the crosstalk between Hippo pathway and E2/ERα signaling. The co-occupancy of YAP1, TEAD4 and ERα on enhancers was detected in two independent breast cancer cell lines, suggesting that this might be a widespread phenomenon.
Similar to other enhancers, ERα enhancers are associated with various co-regulators. One of the most studied co-regulators is the Mediator multi-protein complex, which directly binds to ERα enhancers through the MED1 subunit, and interacts with Pol II and possibly eRNAs, to promote enhancer-promoter looping (Chen and Roeder, 2011; Lai et al., 2013; Malik and Roeder, 2010). Here we report that YAP1 and TEAD4 are required for E2-induced transcription of ERα target genes, as well as the production of eRNA from ERα enhancers. The effect of YAP1 or TEAD4 knockdown on target gene expression was the strongest for the highly E2-responsive genes, consistent with their binding preference to these highly active enhancers. Therefore, YAP1 and TEAD4 might be more critical for the most active ERα enhancers. In agreement with the important role of Mediator in enhancer activation, we found that MED1 binding on ERα enhancers was significantly reduced when YAP1 was depleted, indicating that Mediator-enhancer interaction requires YAP1. However, we still lack a full understanding how the binding of YAP1/TEAD4 to ERα enhancers influences the recruitment or the maintenance of the components of the enhancer complex.
It’s now well accepted that DNA-binding TFs and components of the enhancer complex can be recruited in trans to either activate or repress specific target gene transcription (Langlais et al., 2012; Pascual et al., 2005; Reichardt et al., 1998). Our previous study revealed that the most active ERα enhancers are characterized by the trans-recruitment and assembly of a large complex of DNA-binding TFs through protein-protein interaction (Liu et al., 2014). This complex, termed MegaTrans complex, is associated with the most potent functional enhancers and is critical for eRNA transcription and recruitment of co-regulators. As YAP1 and TEAD4 are associated with active ERα enhancers and required for eRNA production and MED1 recruitment, we hypothesize that YAP1 and TEAD4 are components of the MegaTrans complex. Further supporting the notion, we found that the binding of TEAD4 to ERα enhancers is mediated by ERα and is in trans, independent of its DNA-binding activity. The fact that the DNA-binding activity of TEAD4 is required for its binding to the Hippo pathway targets but indispensable for binding to ERα targets separates the two groups of YAP/TEAD-regulating target genes: one group with TEAD4 binding directly to DNA through its recognized DNA motif and the other group with TEAD4 binding via interaction with other components on enhancers. The second group represents a non-canonical set of YAP/TEAD target genes that are critical for breast cancer cell growth. Besides ERα, YAP/TEAD may also interact with other TF partners including a recently identified AP1 for enhancer regulation (Zanconato et al., 2015).
It remains unclear how YAP/TEAD is selectively recruited in trans to the highly functional ERα enhancers. It has been previously speculated that the dual roles of the pioneer factor FOXA1, which is selectively recruited to the active enhancers and is required for the binding of ERα. In addition, binding of FOXA1 may cause a conformational change in enhancer DNA architecture, which facilitates the recruitment of the MegaTrans complex. Future investigation will be required to test these possibilities. Phase-separation provides a means to compartmentalize and concentrate biochemical reactions within cells by forming membraneless organelles (Banani et al., 2017). Recent studies have shown that activation domains of various TFs, including ERα, form phase-separated condensates with the mediator coactivator to activate gene expression. And it has been suggested that these condensates facilitate the compartmentalization and concentration of transcriptional components at specific sites (Boija et al., 2018; Sabari et al., 2018). In the future, it will be interesting to test whether YAP/TEAD are recruited to ERα enhancers through the phase-separating properties of their intrinsically disordered regions (IDRs).
It is known that enhancers are bound by a mixture of common and lineage-specific transcription factors to integrate cell signaling to achieve cell-specific transcription regulation. But many questions remain unanswered. How different key signaling pathways crosstalk at the chromatin level and converge on enhancers to control downstream gene expression? How the combinatorial interactions of different TFs determine enhancer activity at individual sites and in response to various signals? Our identification of the non-canonical function of YAP/TEAD as components of enhancer complex extends our knowledge on the transcriptional regulation by YAP/TEAD, and also sheds lights on understanding enhancer regulation. Future studies on the interaction between YAP/TEAD and ERα to control enhancer activity in vivo during the mammary gland normal development and cancer invasive progression will provide further insights on these questions.
STAR METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Zhijie Liu (LiuZ7@uthscsa.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell culture
MCF7 and T47D cell lines were obtained from ATCC. MCF7 cells were cultured in Dulbecco’s Modified Eagle’s Medium (DMEM) supplemented with 10% FBS and penicillin/streptomycin. T47D cells were cultured in Roswell Park Memorial Institute (RPMI) 1640 Medium supplemented with 10% FBS and penicillin/streptomycin. Both cell lines were grown in a humidified incubator with 5% CO2. For E2 stimulation, cells were hormone stripped for 3 days in phenol-free media with 5% charcoal-stripped FBS before receiving 100 nM 17β-estradiol (E2) (Sigma) or ethanol vehicle control treatment for 1 hour or 6 hours for estrogen signaling induction. To knockdown ERα protein level, we used ICI 182,780 (Sigma), a well-known knockdown drug for ERα protein, as previously reported (Ross-Innes et al., 2010; Wakeling et al., 1991), at 100 nM for 24 hours before estrogen stimulation.
Animal in vivo xenograft studies
6-week-old athymic nude female mice (The Jackson Laboratory #002019) were housed in a 12 hours light/dark cycle in the animal facility at the UTHSCSA. All animal xenograft experiments were performed in accordance with a protocol approved by the Institutional Animal Care and Use Committee (IACUC) of the UTHSCSA. The study is compliant with all relevant ethical regulations regarding animal research. 17β-Estradiol pellets (Innovative Research of America, 0.72mg, 60-day release) were implanted underneath the skin on their back three days before cell injection. To study knockdown effects of YAP1 or TEAD4, MCF7 cells with shControl, shYAP1 #1 and shTEAD4 #1 stable knockdown were prepared with puromycin selection in full medium before orthotopic xenograft. For orthotopic xenograft studies, 5×106 MCF7 cells resuspended in 100 μl of 1:1 mix of growth media and Matrigel were injected into the mammary fat pads of the mice. All mice were euthanized 5 weeks after subcutaneous injection. Tumors were then excised, photographed and weighted.
METHOD DETAILS
shRNA Lentivirus Package and infection
Mission shRNA lentiviral plasmids targeting YAP1 (shRNA #1: TRCN0000300282 and shRNA #2: TRCN0000107267) and TEAD4 (shRNA #1: TRCN0000015877 and shRNA #2: TRCN0000274223) and non-target control (SHC002 or SHC202) were purchased from Sigma. Knockdown experiments with shRNA lentiviruses were conducted according to the standard lentivirus package and transduction protocols from Addgene. These pLKO-based lentiviral shRNA plasmids were co-transfected with packaging plasmids (psPAX2 and pMD2.G from Addgene) into 293T cells. Lentiviruses were harvested and used for MCF7 cell infection. Stable knockdown MCF7 cells were selected with 1 μg/ml puromycin and collected for experiments within 5-7 days. If the final cell samples needed to be estrogen treated, these stable knockdown MCF7 cells would be grown in stripping media with 0.5 μg/ml puromycin selection for at least 3 days before hormone treatment.
Western blotting assay
Whole cell lysates or cytoplasmic/nuclear fractions were extracted for Western blotting assays. Proteins were separated by SDS-PAGE gel and identified by antibodies. The antibodies used in this assay were: anti-ERα (sc-543, Santa Cruz), anti-YAP1 (14074S, Cell Signaling Technology), anti-TEAD4 (sc-101184, Santa Cruz), anti-Histone H3 (A01502, GenScript), antiHA (ab9110, Abcam), anti-Myc (2276S, Cell Signaling Technology), anti-FOXA1 (ab5089, Abcam), anti-TAZ (sc-293183, Santa Cruz), anti-Flag (F1804, Sigma) and anti-GAPDH (sc-25778, Santa Cruz). For BioID system, the in vivo proximity biotinylation events mediated by BioID-tagged ERα or FOXA1 can be detected by Western blot using streptavidin-HRP (016-030-084, Jackson ImmunoResearch).
Coimmunoprecipitation
Nuclear pellets were collected and lysed with NP-40 lysis buffer (50 mM Tris-HCl pH 7.4, 150 mM NaCl, 1% NP-40, 0.1% Triton X-100. 1 mM PMSF and 1x protease inhibitor were freshly added.). Sonication was performed for 3 minutes with Q800R sonicator (QSonica) at 4°C to improve lysis before centrifuging to collect nuclear lysate. The nuclear lysate was precleared using 10 μl Dynabeads Protein G (Life Technologies) and antibodies were added to lysate and incubated at 4°C overnight. The protein complex was collected using 20 μl Dynabeads Protein G and washed 6 times with NP-40 lysis buffer before elution with 2x SDS loading buffer. The antibodies used in Co-IP were: anti-ERα (sc-8005, Santa Cruz), anti-TEAD4 (sc-101184, Santa Cruz), anti-YAP1 (14074S, Cell Signaling Technology), anti-HA (ab9110, Abcam) and IgG (ab171870, Abcam).
To study the interaction between ERα and wildtype or DNA-binding domain mutant TEAD4, we used two established doxycycline-inducible stable cell lines expressing BLRP and Flag dual-tagged wildtype or mutant TEAD4 proteins (see the section of “Establishing stable cell lines expressing BLRP-Tagged TEAD4 and biotin ChIP”). The nuclear lysate was prepared and ERα protein complex was pulled down using ERα (sc-8005, Santa Cruz) antibody. The wildtype or mutant TEAD4 proteins were detected with anti-Flag antibody (F1804, Sigma).
BioID system setup and pulldown experiment
pcDNA3.1 mycBioID (Addgene plasmid #35700) and pcDNA3.1 MCS-BirA(R118G)-HA (Addgene plasmid #36047) were gifts from Kyle Roux (Roux et al., 2012). To generate the tet-on inducible BioID constructs, mycBioID from pcDNA3.1 mycBioID plasmid was cloned into pRetroX-Tight-Pur (Clontech) at BamH1 site by PCR to get pRetroX-mycBioID-MCS vector, and BirA(R118G)-HA from pcDNA3.1 MCS-BirA(R118G)-HA plasmid was cloned into pRetroX-Tight-Pur (Clontech) at EcoR I site by PCR to get pRetroX-MCS-BioID-HA vector. The full-length human ERα cDNA was cloned into pRetroX-MCS-BioID-HA at the BamH I and Mlu I sites to get pRetroX-ERα-BioID-HA. The full-length human FOXA1 cDNA was cloned into pRetroX-mycBioID-MCS at the Not I and Mlu I sites by Gibson reaction (NEB E2611L) to get pRetroX-mycBioID-FOXA1. Both pRetroX-ERα-BioID-HA and pRetroX-mycBioID-FOXA1 were co-transfected with pCL-Ampho packaging plasmid into 293T cell line to produce retroviruses. Retroviruses were used to transduce a parental MCF7 stable line that was engineered to stably express a Tet Repressor using a retroviral vector. Hygromycin (200 μg/ml) and puromycin (0.3 μg/ml) were used for stable selection. Multiple stable cell lines for each BioID transduction were isolated, treated with doxycycline, and screened for BioID-tagged protein expression that was similar to the levels of the respective endogenous genes as revealed by immunoblotting with specific antibodies for either ERα or FOXA1. To induce ERα-BioID-HA or mycBioID-FOXA1 protein expression, 2 μg/ml doxycycline was added into culture media approximately 24 hours before estrogen treatment for 1 hour, followed by incubation with 50mM Biotin for another 24 hours to label all proteins in the proximity of ERα or FOXA1 before collection.
To identify ERα- or FOXA1-associated cofactors on chromatin, we purified nuclear fraction for analyses of biotin-labeled proteins. Cytoplasmic and nuclear lysates were isolated as previously described with slight modification (Gagnon et al., 2014). Briefly, cells were scraped from 15 cm dish plates and washed with cold PBS. For every 70 mg of cells, 1 mL of cold Hypotonic Lysis Buffer (HLB) (10 mM Tris-HCl pH 7.5, 3 mM MgCl2, 10 mM NaCl, 0.3% NP-40, 10% glycerol and 1x protease inhibitor) was used to resuspend the cells followed by the incubation on ice for 10 minutes. Cells were then centrifuged at 800 g for 8 minutes at 4°C. The supernatant was collected as cytoplasmic lysate fraction. The precipitated nuclei were washed 4 times with HLB by pipetting and centrifuging at 200 g for 2 minutes at 4 °C. After HLB washes, the nuclei were resuspended in 0.5 mL of ice-cold Modified RIPA buffer (MRB) (50 mM Tris-HCl pH 7.4, 0.1% SDS, 0.5% sodium deoxycholate, 1% NP-40, 6 mM MgCl2, 150 mM NaCl, 1 mM EDTA, 1 mM DTT, and 1x protease inhibitor). The nuclei pellet in MRB was sonicated till the solution turned clear. 25 U/ml Benzonase and 4 U/ml DNase I were added to the solution and incubated at RT for 30 minutes to digest the chromatin. The solution was then centrifuged at 18,000 g for 15 minutes at 4°C to pellet insoluble protein and other components. The supernatant was collected as nuclear lysate fraction for the pulldown experiments. For each sample, a small aliquot for each fraction was kept for fractioning result testing using GAPDH and Histone H3 western blots before proceeding with BioID pulldown experiments below.
For BioID pulldown, each nuclear lysate fraction was incubated with 200 μl PBS-washed MyOne Streptavidin T1 Dynabeads (Life Technologies) with overnight rotation at 4°C to pull down all biotinylated proteins in nuclear lysate. On Day two, the beads were washed 5 times with RIPA buffer (50 mM Tris PH 7.4, 0.4% SDS, 0.1% Triton X-100, 1% NP-40, 300 mM NaCl, 1 mM EDTA, 0.5% sodium deoxycholate, 1 mM DTT) with 10 minutes rotation for each wash (Note: only the first two washes need to use the buffer containing protease inhibitor and be incubated at 4°C. The last three washes can be done at RT without protease inhibitor). After 5X washes with RIPA, the beads were then washed twice more with stringent RIPA buffer containing 5% SDS and then 4X washes with PBS to get rid of SDS residue that might affect trypsin digestion during mass spectrometry. For the 4th wash, only 200 μl PBS was used. 10% (20 μl) PBS with beads was transferred to another tube for western blot tests. 90% (180 μl) PBS with beads was kept in the original tube for mass spectrometry. Both tubes were placed on a magnetic stand for 3 minutes, and then all PBS was removed. For western blot tests, the beads were boiled 10 minutes in 2x protein loading buffer with 5% β-Me to release proteins. For mass spectrometry, beads were stored at −80°C before shipping to the Proteomics Core at Sanford-Burnham-Prebys Medical Discovery Institute.
Mass spectrometry for BioID pulldown beads
Following immunoprecipitation and washes, proteins were digested directly on-beads. Proteins bound to the beads were resuspended with 8 M urea, 50 mM ammonium bicarbonate, and cysteine disulfide bonds were reduced with 5 mM tris(2-carboxyethyl)phosphine (TCEP) at 30°C for 60 minutes followed by cysteine alkylation with 15 mM iodoacetamide (IAA) in the dark at RT for 30 minutes. Following alkylation, urea was diluted to 1 M using 50 mM ammonium bicarbonate, and proteins were finally subjected to overnight digestion with mass spec grade Trypsin/Lys-C mix (Promega). Finally, beads were pulled down and the solution with peptides collected into a fresh tube. The beads were then washed once with 50 mM ammonium bicarbonate to increase peptide recovery. Following overnight digestion, samples were acidified with formic acid (FA) and subsequently desalted using AssayMap C18 cartridges mounted on an Agilent AssayMap BRAVO liquid handling system. C18 cartridges were first conditioned with 100% acetonitrile (ACN), followed by 0.1% FA. Samples were then loaded onto the conditioned C18 cartridge, washed with 0.1% FA, and eluted with 60% ACN, 0.1% FA. Finally, the organic solvent was removed in a SpeedVac concentrator prior to LC-MS/MS analysis. Before injecting in the LC-MS, total sample peptide amount was determined by Pierce Quantitative Colorimetric Peptide Assay (ThermoFisher).
Dried samples were reconstituted with 2% acetonitrile, 0.1% formic acid and analyzed by LC-MS/MS using a Proxeon EASY nanoLC system (ThermoFisher) coupled to Elite mass spectrometer (ThermoFisher). Peptides were separated using an analytical C18 Acclaim PepMap column 75 μm x 500 mm, 2 μm particles (ThermoFisher) in 121 minutes at a flow rate of 300 nL/minute: 1% to 6% B in 1 minute, 6% to 23% B in 56 minutes, 23% to 34% B in 37 minutes, 34% to 48% B in 26 minutes, and 48% to 98% B in 1 minute (A = Formic acid 0.1%; B = 80% ACN: 0.1% Formic acid).
The mass spectrometer was operated in positive data-dependent acquisition mode. MS1 spectra were measured in the Orbitrap with a resolution of 60,000 (AGC target of 1e6, and a mass range from 350 to 1450 m/z). Up to 10 MS2 spectra per duty cycle were triggered, CID-fragmented and acquired in the linear ion trap (AGC target of 1e4, isolation window of 2 m/z, and a normalized collision energy of 35). Dynamic exclusion was enabled with a duration of 30 seconds.
All mass spectra were analyzed with MaxQuant software v1.5.5.1. MS/MS spectra were searched against the Homo sapiens Uniprot protein sequence database (downloaded in July 2018) and GPM cRAP sequences (commonly known protein contaminants). Precursor mass tolerance was set to 20 ppm and 4.5 ppm for the first search where initial mass recalibration was completed and for the main search, respectively. Product ions were searched with a mass tolerance 0.5 Da. The maximum precursor ion charge state used for searching was 7. Carbamidomethylation of cysteine was searched as a fixed modification, while oxidation of methionine and acetylation of protein N-terminal were searched as variable modifications. Enzyme was set to trypsin in a specific mode and a maximum of two missed cleavages was allowed for searching. The target-decoy-based false discovery rate (FDR) filter for spectrum and protein identification was set to 1%.
ChIP-seq
ChIP was performed as previously described (Liu et al., 2014). Briefly, cells were cultured in stripping media for 3 days and treated with ethanol vehicle control or 100 nM E2 for 1 hour. About 1x107 cells were cross-linked with 1% formaldehyde for 10 minutes only or 2 mM DSG crosslinker (CovaChem) at room temperature for 1 hour followed by secondary fixation with 1% formaldehyde for 10 minutes. After neutralization with 0.125 M glycine and nuclear extraction, average length of genomic DNA was sonicated in lysis buffer with Q800R sonicator (QSonica) to get to ~200-500 bp and soluble chromatin was incubated with 3-6 μg of antibody at 4°C overnight. The antibodies used in ChIP were: anti-ERα (sc-543 or sc-7207, Santa Cruz), anti-TEAD4 (sc-101184, Santa Cruz), anti-YAP1 (14074S, Cell Signaling Technology) and anti-MED1 (A300-793A, Bethyl). Immunoprecipitated complexes were pulled down using 50 μl Dynabeads Protein G (Life Technologies) for each ChIP. Beads were washed with washing buffer 6 times to minimize the background. Protein-DNA complexes were then eluted and DNA fragments were purified. The purified DNA was subjected to qPCR directly to confirm target region enrichment before moving on for deep sequencing library preparation. For sequencing, the extracted DNA was used to construct the ChIP-seq library using KAPA Hyper Prep kit (KK8504), followed by deep sequencing with the Illumina’s HiSeq 3000 system according to the manufacturer’s instructions.
Reverse-transcription PCR and quantitative PCR (qPCR)
Total RNA was isolated with RNeasy Mini Kit (Qiagen) according to the manufacture’s protocol and 1 μg RNA was used to convert to cDNA using iScript Select cDNA Synthesis Kit (Bio-Rad) in the presence of both oligo (dT) and random primers. qPCR was conducted with SsoAdvanced Universal SYBR Green Supermix (Bio-Rad) using CFX384 Real-Time PCR Detection System (Bio-Rad) according to the manufacturer’s instructions. We performed qPCR to test E2-induced gene activation from gene bodies and also for enhancer activation by testing the levels of eRNAs that are transcribed from enhancer regions. Primer sets used in qPCR are as follows:
GAPDH: F: ACATCATCCCTGCCTCTACTGG, R: GTTTTTCTAGACGGCAGGTCAGG; GREB1: F: GTGGTAGCCGAGTGGACAAT, R: CGTTGGAAATGGAGACAAGG; KCNK5: F: GATCCCACCCTTCGTATTCA, R: CCAGAGCTCCACGAAGTAGC; SIAH2: F: AGCATCAGGAACCTGGCTAT, R: GTTTCTCCGTATGGTGCAGG; YAP1: F: TGTCTTCTCCCGGGATGTCT, YAP1: R: TCTCGAGAGTGATAGGTGCC; TEAD4: F: GATCTCTTCGAACGGGGACC, TEAD4: R: TGGCTGGAGACCCCATAGAA; GREB1 enhancer: F: TGTCCCGAAGCTGAACACTC, R: AGGGACACAACAGTACCTAAAGT; PGR enhancer: F: CTGACTAAGCCTGGAGAAGGC, R: ACTCCCAAGGGACCATTTCAA; KCNK5 enhancer: F: AACCCAGGGTCTTCCAAGGT, R: GAAACTGAAGTGGAAATGGACCC; SIAH2 enhancer: F: AAATAGGGTGCACCGCAAGA, R: TCTTCCTGCCTGCTCCAATG.
RNA-seq
MCF7 cells with shControl, shYAP1 #1 and shTEAD4 #1 stable knockdown were cultured in stripping media for 3 days and then treated with ethanol vehicle control or 100 nM E2 for 6 hours before collection. Total RNA was extracted using RNeasy Mini Kit (Qiagen). For each sample, 1 μg RNA was used for library construction using KAPA RNA HyperPrep Kit with RiboErase (KK8560). For sequencing, samples with specific adaptors were sequenced with the Illumina’s HiSeq 3000 system according to the manufacturer’s instructions.
Global run-on sequencing (GRO-seq)
To investigate the effect of YAP1 or TEAD4 knockdown on eRNA expression, we performed GRO-seq in MCF7. MCF7 cells with shControl, shYAP1 #2 and shTEAD4 #2 stable knockdown were cultured in stripping media for 3 days and then treated with ethanol vehicle control or 100 nM E2 for 1 hour before collection for GRO-seq. GRO-seq experiments were performed as previously reported (Liu et al., 2014). Briefly, ~5-10 millions of MCF7 cells treated with vehicle or E2 for 1 hour were washed 3 times with cold PBS and then sequentially swelled in swelling buffer (10 mM Tris-HCl pH7.5, 2 mM MgCl2, 3 mM CaCl2) for 10 minutes on ice, harvested, and lysed in lysis buffer (swelling buffer plus 0.5% NP-40, 20 units of SUPERase-In, and 10% glycerol). The resultant nuclei were washed two more times with 10 ml lysis buffer and finally re-suspended in 100 μl of freezing buffer (50 mM Tris-HCl pH8.3, 40% glycerol, 5 mM MgCl2, 0.1 mM EDTA). For the run-on assay, re-suspended nuclei were mixed with an equal volume of reaction buffer (10 mM Tris-HCl pH 8.0, 5 mM MgCl2, 1 mM DTT, 300 mM KCl, 20 units of SUPERase-In, 1% sarkosyl, 500 μM ATP, GTP, and Br-UTP, 2 μM CTP) and incubated for 5 minutes at 30°C. The resultant nuclear-run-on RNA (NRO-RNA) was then extracted with TRIzol LS reagent (Life Technologies) following manufacturer’s instructions. NRO-RNA was fragmented to ~300-500 nt by alkaline base hydrolysis on ice for 30 minutes followed by treatment with DNase I and antarctic phosphatase. At this step, only a small portion of all the RNA species are Br-UTP-labeled. To purify the Br-UTP labeled nascent RNA, the fragmented NRO-RNA was immunoprecipitated with anti-BrdU agarose beads (Santa Cruz Biotechnology) in binding buffer (0.5xSSPE, 1 mM EDTA, 0.05% tween) for 1-3 hours at 4°C with rotation. Subsequently, T4 PNK was used to repair the ends of the immunoprecipitated Br-UTP labeled nascent RNA at 37°C for 1hour. The RNA was extracted and precipitated using acidic phenol-chloroform.
The RNA fragments were subjected to poly-A tailing reaction by poly-A polymerase (NEB) for 30 minutes at 37°C. Subsequently, reverse transcription was performed using oNTI223 primer and SuperScript III RT kit (Life Technologies). The cDNA products were separated on a 10% polyacrylamide TBE-urea gel and only those fragments migrating between 100-500 bp were excised and recovered by gel extraction. Next, the first-strand cDNA was circularized by CircLigase (Epicentre) and re-linearized by APE1 (NEB). Re-linearized single strand cDNA (sscDNA) was separated on a 10% polyacrylamide TBE gel and the appropriately sized product (~120-320 bp) was excised and gel-extracted. Finally, sscDNA template was amplified by PCR using the Phusion High-Fidelity enzyme (NEB) according to the manufacturer’s instructions. The oligonucleotide primers oNTI200 and oNTI201 were used to generate DNA libraries for deep sequencing. The sequences for primers oNTI223, oNTI200 and oNTI201 are as follows:
oNTI223-TruSeqLT:
/5Phos/AGATCGGAAGAGCGTCGTGTA/idSp/CAGACGTGTGCTCTTCCGATC TTTT TTT TTT TTT TTT TTT TVN
oNTI201-TruseqLT:
5’AATGATACGGCGACCACCGAGATCTACACTCTTTCCCTACACGACGCTCTTCCGATCT 3’
oNTI200-TruSeqID1:
5′CAAGCAGAAGACGGCATACGAGAT CGTGAT GTGACTGGAGTT CAGACGTGTGCT CTTCCGATC 3′
Establishing stable cell lines expressing BLRP-Tagged TEAD4 and biotin ChIP
To study binding patterns for wildtype and DNA-binding domain mutant of TEAD4, we used a previously established parental MCF7 stable cell line that expressed BirA enzyme and Tet-Repressor (Liu et al., 2014). We then used this parental cell line to establish doxycycline-inducible stable cell lines expressing BLRP-tagged wildtype or mutant TEAD4 proteins at close to endogenous levels. A DNA-binding domain mutation (R64A/K65A/I66A/L68A) of TEAD4 that has been proven to lose its binding ability to TEAD DNA motif in a previous study (Shi et al., 2017) was constructed using the mutagenesis method described before (Liu et al., 2014). Biotin ChIP experiments for BLRP-tagged wildtype or mutant TEAD4 were performed with our previous published protocol (Liu et al., 2014). Briefly, cross-linked protein-DNA complexes were pulled down by Nanolink Streptavidin Magnetic beads (Solulink) and the washing was performed under much more stringent conditions that included 4 washes with 1% SDS in TE (20 minutes each) and two washes with 1% Triton X-100 in TE. The washed streptavidin beads were then subjected to AcTEV protease (Life Technologies) digestion twice for tagged protein-DNA complex elution before de-crosslinking at 65°C overnight. After de-crosslinking overnight at 65°C, final ChIP DNA was extracted and purified using QIAquick spin columns (Qiagen). BLRP-tagged TEAD4 inducible cell lines without doxycycline induction were used as ChIP-seq control for background detection. The ChIP-seq libraries were constructed using KAPA’s HyperPrep kit (KK8504), followed by deep sequencing with the Illumina’s HiSeq 3000 system according to the manufacturer’s instructions. The purified DNA was also subjected to qPCR directly to confirm target region enrichment. Primer sets used in qPCR are as follows:
GREB1 enhancer: F: TGTCCCGAAGCTGAACACTC, R: AGGGACACAACAGTACCTAAAGT; SIAH2 enhancer: F: AAATAGGGTGCACCGCAAGA, R: TCTTCCTGCCTGCTCCAATG; CTGF promoter: F: ATATGAATCAGGAGTGGTGCGA, R: CAACTCACACCGGATTGATCC; AJUBA 1st intron enhancer: F: GCATTCTCCGAATTCCTGCG, R: TCGTAGGGAAGGGACCAAGT.
Cell growth assay
MCF7 cells that were knocked down with shControl, shTEAD4 #1, and shYAPl #1 were cultured in full medium to 90% confluence and washed with PBS three times before being seeded into 96-well plates in stripping medium. After 24 hours, cells were treated with ethanol vehicle control or 100 nM E2 and cell growth was measured at 0, 24 and 48 hours after E2 treatment. For cell growth measurement, cells were incubated with Cell counting kit-8 reagent (Sigma) for 2 hours at 37°C and absorbance at 450 nm was measured according to the manufacturer’s instructions.
Colony formation assay
MCF7 cells that were knocked down with shControl, shTEAD4 #1, and shYAP1 #1 were cultured in full medium to 90% confluence and seeded in 6-well plates. After cells attached to the bottom of the plate, the media was changed to stripping media and were cultured for 2 days. The cells were treated with ethanol vehicle control or 100 nM E2 for 10 days (change media every 3 days). Cells were stained by Crystal violet for counting. Colony number was measured by ImageJ.
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical analysis
For all RT-qPCR, cell growth assay, colony formation assay, and xenograft experiments were performed with at least three independent biological replicates and three technical replicates for each reaction. Results are reported as mean ± SEM or mean ± SD of three independent experiments. Data were analyzed and statistics were performed using unpaired two-tailed Student’s t-tests or one-way ANOVA (Prism 5 GraphPad). Significant differences between two groups were noted by asterisks (* P<0.05, ** P<0.01, *** P<0.001).
RNA-seq data analysis
The sequencing reads were aligned to human genome (hg19) with STAR v2.5.2b, a spliced-read aligner, and after removing reads mapped to rRNA sequences, read counts for each gene were conducted by featureCounts package with default parameter (Dobin et al., 2013; Liao et al., 2014). Genes with less than 1 read in at least 2 samples were discarded. DESeq2 v1.14.1 was used to call differentially expressed genes with fold expression change >= 1.5 and FDR <= 0.05 as the cutoff (Love et al., 2014).
ChIP-seq data analysis
Reads were aligned to human genome (hg19) using bowtie v1.1.2 with “--best --strata –m 1” parameters (Langmead et al., 2009). Only uniquely mapped and without-duplication reads were selected for subsequent analysis. MACS2 was employed to call peaks by comparing immunoprecipitated chromatin with input chromatin using a standard parameters and a q-value cutoff of 1e-5 (Zhang et al., 2008). For H3K27ac, the broad mode of MACS2 was switched on. The peaks overlapped with the blacklist regions downloaded from UCSC were removed. ChIPseeker was used to annotate the peaks (Yu et al., 2015). GREAT (McLean et al., 2010) was used to annotate the potential functions of the peaks with default parameters by searching against the Broad Molecular Signatures Database (MsigDB) Pathway category (Liberzon et al., 2011; Subramanian et al., 2005).
For motif discovery, findMotifs.pl program in HOMER was used for the regions with 300 bp upstream and downstream from the peak summit against vertebrates motif collector set with parameters “-mset vertebrates –mis 2 –len 6,8,10,12” (Heinz et al., 2010).
For all the aggregation plots, the signal fold change (FC) was calculated as the ratio of average tag density. The library-size normalized tag density within a 1 kb region around the peak center (+/− 500 bp) from each condition was used for calculation. P-value was determined by Wilcoxon signed-rank test.
GRO-seq data analysis
GRO-seq reads were aligned to human genome (hg19) using bowtie v1.1.2 with “--best --strata –m 1 –v 2” parameters. Duplicated reads were eliminated for subsequent analysis. To balance the clonal amplification bias and total useful reads, only three reads at most was allowed for each unique genomic position. When measuring the expression level of genes, mapped reads from the first 30 kb of gene body were counted, excluding promoter-proximal region (transcription start site (TSS) to 1000 bp downstream of TSS; if the length of a gene is shorter than 10 kb, then the reads mapped to the first 10%*length regions were excluded). If the length of a gene is shorter than 30 kb, then mapped reads from the whole gene were counted, excluding promoter-proximal region and gene end (500 bp upstream of transcription termination site (TTS) to TTS). Differential expression analysis was then performed by DESeq2 with a fold change threshold of 2 and FDR<=0.01.
To better understand the activation mechanisms of ERα-bound enhancers, we divided the ERα active enhancers into three groups based on the strength of E2–induced eRNA expression. We defined the 2000 bp centered on ERα-bound enhancers as the eRNA regions. After getting the read counts within eRNA regions for each sample, edgeR 3.16.5 was used to call the differential eRNAs by comparing E2 treatment condition with vehicle condition (Robinson et al., 2010). These ERα enhancers having eRNA regions with fold change >= 2 and FDR <= 0.01 were defined as High Activity group (total 419 ERα enhancers); These ERα enhancers having eRNA regions with fold change within 1.2 to 1.5 were defined as Median Activity group (total 422 enhancers); These ERα enhancers having eRNA regions with fold change within 1 to 1.1 were defined as Low Activity group (total 449 enhancers). These definitions were used in Figures 3, 7, and S2.
Data visualization
All ChIP-seq, RNA-seq, GRO-seq data was visualized in Integrative Genomics Viewer (IGV) (Robinson et al., 2011). For GRO-seq, the reads were separated by strand and extended to a length of 100 nt in the 5’- to −3’ direction; for ChIP-seq, the reads were extended to 200 nt in the 5’- to −3’ direction. ChIP-seq and GRO-seq samples were normalized to 10 million mapped reads per experiment, while RNA-seq samples were normalized to 1 million reads.
DATA AND SOFTWARE AVAILABILITY
The GEO accession number for all deep sequencing data reported in this paper is GSE125609, which includes ChIP-seq data set (GSE125594), RNA-seq data set (GSE125606) and GRO-seq data set (GSE125607). To review GEO accession GSE125609: use https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE125609. (Note: Access token will be provided for review purpose.)
We also used some published ChIP-seq data from the Gene Expression Omnibus database for FoxA1 and ERα under accession number GSE59530 (Franco et al., 2015), H3K27Ac under accession number GSE45822 (Li et al., 2013), YAP1 and TEAD1 under accession number GSE107013 (Elster et al., 2018).
The raw data used in this paper have been deposited to Mendeley Data and are available at: http://dx.doi.org/10.17632/hwz4rdwfr6.1 or https://data.mendeley.com/datasets/hwz4rdwfr6/draft?a=ab20213e-4a26-46e5-b17e-4e3299d3121b.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit polyclonal anti-ERα | Santa Cruz Biotechnology | Cat# sc-543; RRID: AB_631471 |
| Rabbit polyclonal anti-ERα | Santa Cruz Biotechnology | Cat# sc-7207; RRID: AB_640249 |
| Mouse monoclonal anti-ERα | Santa Cruz Biotechnology | Cat# sc-8005; RRID: AB_627556 |
| Rabbit monoclonal anti-YAP1 | Cell Signaling Technology | Cat# 14074S; RRID: AB_2650491 |
| Mouse monoclonal anti-TEAD4 | Santa Cruz Biotechnology | Cat# sc-101184; RRID: AB_2203086 |
| Rabbit polyclonal anti-Histone H3 | GenScript | Cat# A01502; RRID: AB_1968828 |
| Rabbit polyclonal anti-HA | Abcam | Cat# ab9110; RRID: AB_307019 |
| Mouse monoclonal anti-Myc | Cell Signaling Technology | Cat# 2276S; RRID: AB_331783 |
| Mouse monoclonal anti-TAZ | Santa Cruz Biotechnology | Cat# sc-293183; RRID: N/A |
| Goat polyclonal anti-FOXA1 | Abcam | Cat# ab5089; RRID: AB_304744 |
| Rabbit polyclonal IgG, isotype control | Abcam | Cat# ab171870; RRID: AB_2687657 |
| Mouse monoclonal anti-FLAG M2 | Sigma-Aldrich | Cat# F1804; RRID: AB_262044 |
| Rabbit polyclonal anti-GAPDH | Santa Cruz Biotechnology | Cat# sc-25778; RRID: AB_10167668 |
| Streptavidin-HRP | Jackson ImmunoResearch Laboratories | Cat# 016-030-084; RRID: AB_2337238 |
| Rabbit polyclonal anti-MED1 | Bethyl Laboratories | Cat# A300-793A; RRID: AB_577241 |
| Bacterial and Virus Strains | ||
| DH5α Chemically Competent E. coli. | New England Biolabs | Cat# C2987I |
| STBL3 Chemically Competent E. coli. | Life Technologies | Cat# C737303 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| 17β-estradiol | Sigma-Aldrich | Cat# E8875 |
| ICI 182,780 | Sigma-Aldrich | Cat# I4409 |
| 17β-estradiol pellets (0.72mg, 60-day release) | Innovative Research of America | Cat# SE-121 |
| Puromycin | Sigma-Aldrich | Cat# P9620 |
| Hygromycin B | Life Technologies | Cat# 10687010 |
| G418 | Life Technologies | Cat# 10131027 |
| Biotin | Sigma-Aldrich | Cat# B4501 |
| Polybrene | Sigma-Aldrich | Cat# H9268 |
| cOmplete™ Protease Inhibitor Cocktail | Roche | Cat# 11836145001 |
| Lipofectamine™ 2000 | Life Technologies | Cat# 11668019 |
| Formaldehyde | Sigma-Aldrich | Cat# F8775 |
| DSG Crosslinker (Disuccinimidyl glutarate) | CovaChem | Cat# 13301 |
| Dynabeads™ Protein G | Life Technologies | Cat# 10004D |
| Doxycycline | Clontech | Cat# 631311 |
| Trypsin/Lys-C Mix, Mass Spec Grade | Promega | Cat# V5071 |
| SUPERase-In™ RNase Inhibitor | Life Technologies | Cat# AM2696 |
| N-Lauroylsarcosine sodium salt solution (Sarkosyl) | Sigma-Aldrich | Cat# L7414 |
| 5-Bromouridine 5′-triphosphate sodium salt (BrUTP) | Sigma-Aldrich | Cat# B7166 |
| Antarctic phosphatase | New England BioLabs | Cat# M0289S |
| anti-BrdU agarose beads | Santa Cruz Biotechnology | Cat# sc-32323 AC |
| poly-A polymerase | New England BioLabs | Cat# M0276S |
| SuperScript™ III RT kit | Life Technologies | Cat# 18080-051 |
| CircLigase™ ssDNA Ligase | Epicentre | Cat# CL4111K |
| Ape1 | New England BioLabs | Cat# M0282S |
| Phusion® High-Fidelity DNA Polymerase | New England BioLabs | Cat# M0530L |
| Gibson Assembly® Master Mix | New England BioLabs | Cat# E2611L |
| Proteinase K, recombinant, PCR Grade | Roche | Cat# 3115879001 |
| RNase | Sigma-Aldrich | Cat# R6513 |
| TRIzol™ LS Reagent | Life Technologies | Cat# 10296028 |
| T4 PNK | New England BioLabs | Cat# M0201S |
| DNase I | Life Technologies | Cat# AM2222 |
| Benzonase® Nuclease | EMD Millipore | Cat# 70746 |
| MyOne™ Streptavidin T1 Dynabeads™ | Life Technologies | Cat# 65602 |
| Nanolink® Streptavidin Magnetic beads | Solulink | Cat# M-1002-010 |
| AcTEV™ Protease | Life Technologies | Cat# 12575015 |
| Phenol:Chloroform:Isoamyl Alcohol | Sigma-Aldrich | Cat# P2069 |
| Crystal Violet | Sigma-Aldrich | Cat# C0775 |
| Critical Commercial Assays | ||
| Cell Counting Kit-8 (CCK-8) | Sigma | Cat# 96992 |
| RNeasy Mini Kit | Qiagen | Cat# 74106 |
| iScript™ Select cDNA Synthesis Kit | Bio-RAD | Cat# 1708897 |
| SsoAdvanced™ Universal SYBR® Green Supermix | Bio-RAD | Cat# 1725275 |
| QIAquick spin columns | Qiagen | Cat# 28115 |
| KAPA Hyper Prep kit | Kapa Biosystems | Cat# KK8504 |
| KAPA RNA HyperPrep Kit with RiboErase | Kapa Biosystems | Cat# KK8560 |
| Deposited Data | ||
| Raw and processed sequencing data for ChIP-seq | This paper | GEO: GSE125594 |
| Raw and processed sequencing data for RNA-seq | This paper | GEO: GSE125606 |
| Raw and processed sequencing data for GRO-seq | This paper | GEO: GSE125607 |
| FOXA1 and ERα ChIP-seq | Franco et al., 2015 | GEO: GSE59530 |
| H3K27ac ChIP-seq | Li et al., 2013 | GEO: GSE45822 |
| YAP1 and TEAD1 ChIP-seq | Elster et al., 2018 | GEO: GSE107013 |
| Raw data | This paper, Mendeley data | http://dx.doi.org/10.17632/hwz4rdwfr6.1 |
| Experimental Models: Cell Lines | ||
| Human: MCF7 | ATCC | Cat# HTB-22 |
| Human: T47D | ATCC | Cat# HTB133 |
| Human: HEK293T | ATCC | Cat# CRL-3216 |
| Human: MCF7-TetR stable line | This paper | N/A |
| Human: MCF7-BirA-TetR stable line | This paper | N/A |
| Human: MCF7-BirA-TetR-BLRP-TEAD4(wildtype) stable line | This paper | N/A |
| Human: MCF7-BirA-TetR-BLRP-TEAD4(mutant) stable line | This paper | N/A |
| Experimental Models: Organisms/Strains | ||
| Athymic nude female mice | The Jackson Laboratory | Cat# 002019 |
| Oligonucleotides | ||
| Primers or oligos for qPCR, ChIP-qPCR, and GRO-seq see METHOD DETAILS | This paper | N/A |
| Recombinant DNA | ||
| shYAP1 #1 | Sigma-Aldrich | TRCN0000300282 |
| shYAP1 #2 | Sigma-Aldrich | TRCN0000107267 |
| shTEAD4 #1 | Sigma-Aldrich | TRCN0000015877 |
| shTEAD4 #2 | Sigma-Aldrich | TRCN0000274223 |
| shControl | Sigma-Aldrich | SHC002 |
| shControl | Sigma-Aldrich | SHC202 |
| pcDNA3.1 mycBioID | Addgene | Plasmid ID: #35700 |
| pcDNA3.1 MCS-BirA(R118G)-HA | Addgene | Plasmid ID: #36047 |
| pRetroX-Tight-Pur | Clontech | Cat# 632104 |
| pRetroX-mycBioID-MCS | This paper | N/A |
| pRetroX-mycBioID-FOXA1 | This paper | N/A |
| pRetroX-MCS-BioID-HA | This paper | N/A |
| pRetroX-ERα-BioID-HA | This paper | N/A |
| pRetroX-BLRP-MCS | This paper | N/A |
| pRetroX-BLRP-TEAD4 wildtype | This paper | N/A |
| pRetroX-BLRP-TEAD4 mutant | This paper | N/A |
| pCL-Ampho | Novus Biologicals | Cat# NBP2-29541 |
| pMD2.G | Addgene | Plasmid ID: #12259 |
| psPAX2 | Addgene | Plasmid ID: #12260 |
| Software and Algorithms | ||
| STAR v2.5.2b | Dobin et al., 2013 | https://github.com/alexdobin/STAR |
| featureCounts v1.5.1 | Liao et al., 2014 | http://subread.sourceforge.net/ |
| DESeq2 v1.14.1 | Love et al., 2014 | https://bioconductor.org/packages/release/bioc/html/DESeq2.html |
| Broad Molecular Signatures Database (MsigDB) | Subramanian et al., 2005; Liberzon et al., 2011 | http://software.broadinstitute.org/gsea/index.jsp |
| Bowtie v1.1.2 | Langmead et al., 2009 | http://bowtie-bio.sourceforge.net/index.shtml |
| MACS2 v2.1.1.20160309 | Zhang et al., 2008 | https://github.com/taoliu/MACS |
| ChIPseeker v1.10.3 | Yu et al., 2015 | https://bioconductor.org/packages/release/bioc/html/ChIPseeker.html |
| GREAT v3.0.0 | McLean et al., 2010 | http://great.stanford.edu/public/html/index.php |
| HOMER v4.8 | Heinz et al., 2010 | http://homer.ucsd.edu/homer/index.html |
| edgeR v3.16.5 | Robinson et al., 2010 | https://bioconductor.org/packages/release/bioc/html/edgeR.html |
| Integrative Genomics Viewer (IGV) | Robinson et al., 2011 | https://software.broadinstitute.org/software/igv/ |
| MaxQuant software v1.5.5.1 | Max-Planck-Institute of Biochemistry | https://www.biochem.mpg.de/227318/MaxQuant |
| Prism 5 | GraphPad | https://graphpad-prism.software.informer.com/5.0/ |
| ImageJ | NIH | https://imagej.nih.gov/ij/ |
Highlights.
YAP/TEAD non-canonically bind to a group of ERα-bound enhancers
YAP/TEAD are required for estrogen-induced transcription and breast cancer growth
YAP/TEAD regulate enhancer activation via controlling the recruitment of MED1
TEAD is recruited to ERα active enhancers through protein tethering trans-binding
ACKNOWLEDGEMENTS
We would like to thank Dr. Kun-Liang Guan from UC San Deigo for reagents and scientific discussion and all the members of Liu and Chen labs for technical assistance and helpful discussion. The authors are grateful to the Proteomics Core at Sanford-Burnham-Prebys Medical Discovery Institute for assistance in mass spectrometry. All Deep Sequencing Data was generated in the Genome Sequencing Facility at Greehey Children’s Cancer Research Institute in the UT Health San Antonio, which is supported by NIH-NCI P30 CA054174 (NIH Cancer Center at UT Health San Antonio), NIH Shared Instrument grant 1S10OD021805-01 (S10 grant), and CPRIT Core Facility Award (RP160732).
This work was supported by funds from CPRIT RR160017 to Z.L., V Foundation V2016-017 to Z.L., V Foundation DVP2019-018 to Z.L., Voelcker Fund Young Investigator Award to Z.L., UT Rising STARs Award to Z.L., Susan G. Komen CCR Award CCR17483391 to Z.L., NCI U54 CA217297/PRJ001 to Z.L., and the San Antonio Nathan Shock Center (NIA grant 3P30 AG013319-23S2) to L.C..
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
SUPPLEMENTAL INFORMATION
Supplemental information includes seven figures (S1 to S7) and their figure legends.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Acevedo ML, Lee KC, Stender JD, Katzenellenbogen BS, and Kraus WL (2004). Selective recognition of distinct classes of coactivators by a ligand-inducible activation domain. Molecular cell 13, 725–738. [DOI] [PubMed] [Google Scholar]
- Banani SF, Lee HO, Hyman AA, and Rosen MK (2017). Biomolecular condensates: organizers of cellular biochemistry. Nature reviews. Molecular cell biology 18, 285–298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beyer TA, Weiss A, Khomchuk Y, Huang K, Ogunjimi AA, Varelas X, and Wrana JL (2013). Switch enhancers interpret TGF-beta and Hippo signaling to control cell fate in human embryonic stem cells. Cell reports 5, 1611–1624. [DOI] [PubMed] [Google Scholar]
- Boija A, Klein IA, Sabari BR, Dall’Agnese A, Coffey EL, Zamudio AV, Li CH, Shrinivas K, Manteiga JC, Hannett NM, et al. (2018). Transcription Factors Activate Genes through the Phase-Separation Capacity of Their Activation Domains. Cell 175, 1842–1855 e1816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Britschgi A, Duss S, Kim S, Couto JP, Brinkhaus H, Koren S, De Silva D, Mertz KD, Kaup D, Varga Z, et al. (2017). The Hippo kinases LATS1 and 2 control human breast cell fate via crosstalk with ERalpha. Nature 541, 541–545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carroll JS, Liu XS, Brodsky AS, Li W, Meyer CA, Szary AJ, Eeckhoute J, Shao W, Hestermann EV, Geistlinger TR, et al. (2005). Chromosome-wide mapping of estrogen receptor binding reveals long-range regulation requiring the forkhead protein FoxA1. Cell 122, 33–43. [DOI] [PubMed] [Google Scholar]
- Carroll JS, Meyer CA, Song J, Li W, Geistlinger TR, Eeckhoute J, Brodsky AS, Keeton EK, Fertuck KC, Hall GF, et al. (2006). Genome-wide analysis of estrogen receptor binding sites. Nature genetics 38, 1289–1297. [DOI] [PubMed] [Google Scholar]
- Chen W, and Roeder RG (2011). Mediator-dependent nuclear receptor function. Seminars in cell & developmental biology 22, 749–758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Santa F, Barozzi I, Mietton F, Ghisletti S, Polletti S, Tusi BK, Muller H, Ragoussis J, Wei CL, and Natoli G (2010). A large fraction of extragenic RNA pol II transcription sites overlap enhancers. PLoS biology 8, e1000384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, and Gingeras TR (2013). STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elster D, Tollot M, Schlegelmilch K, Ori A, Rosenwald A, Sahai E, and von Eyss B (2018). TRPS1 shapes YAP/TEAD-dependent transcription in breast cancer cells. Nat Commun 9, 3115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franco HL, Nagari A, and Kraus WL (2015). TNFalpha signaling exposes latent estrogen receptor binding sites to alter the breast cancer cell transcriptome. Molecular cell 58, 21–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gagnon KT, Li L, Janowski BA, and Corey DR (2014). Analysis of nuclear RNA interference in human cells by subcellular fractionation and Argonaute loading. Nat Protoc 9, 2045–2060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hah N, Danko CG, Core L, Waterfall JJ, Siepel A, Lis JT, and Kraus WL (2011). A rapid, extensive, and transient transcriptional response to estrogen signaling in breast cancer cells. Cell 145, 622–634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hah N, Murakami S, Nagari A, Danko CG, and Kraus WL (2013). Enhancer transcripts mark active estrogen receptor binding sites. Genome research 23, 1210–1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen CG, Ng YL, Lam WL, Plouffe SW, and Guan KL (2015). The Hippo pathway effectors YAP and TAZ promote cell growth by modulating amino acid signaling to mTORC1. Cell research 25, 1299–1313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinz S, Benner C, Spann N, Bertolino E, Lin YC, Laslo P, Cheng JX, Murre C, Singh H, and Glass CK (2010). Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Molecular cell 38, 576–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinz S, Romanoski CE, Benner C, and Glass CK (2015). The selection and function of cell type-specific enhancers. Nature reviews. Molecular cell biology 16, 144–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hurtado A, Holmes KA, Ross-Innes CS, Schmidt D, and Carroll JS (2011). FOXA1 is a key determinant of estrogen receptor function and endocrine response. Nature genetics 43, 27–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jozwik KM, and Carroll JS (2012). Pioneer factors in hormone-dependent cancers.. Nature reviews Cancer 12, 381–385. [DOI] [PubMed] [Google Scholar]
- Kim TK, Hemberg M, Gray JM, Costa AM, Bear DM, Wu J, Harmin DA, Laptewicz M, Barbara-Haley K, Kuersten S, et al. (2010). Widespread transcription at neuronal activity-regulated enhancers. Nature 465, 182–187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim TK, and Shiekhattar R (2015). Architectural and Functional Commonalities between Enhancers and Promoters. Cell 162, 948–959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kraus WL, and Kadonaga JT (1998). p300 and estrogen receptor cooperatively activate transcription via differential enhancement of initiation and reinitiation. Genes & development 12, 331–342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai F, Orom UA, Cesaroni M, Beringer M, Taatjes DJ, Blobel GA, and Shiekhattar R (2013). Activating RNAs associate with Mediator to enhance chromatin architecture and transcription. Nature 494, 497–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lam MT, Li W, Rosenfeld MG, and Glass CK (2014). Enhancer RNAs and regulated transcriptional programs. Trends in biochemical sciences 39, 170–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langlais D, Couture C, Balsalobre A, and Drouin J (2012). The Stat3/GR interaction code: predictive value of direct/indirect DNA recruitment for transcription outcome. Molecular cell 47, 38–49. [DOI] [PubMed] [Google Scholar]
- Langmead B, Trapnell C, Pop M, and Salzberg SL (2009). Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol 10, R25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W, Notani D, Ma Q, Tanasa B, Nunez E, Chen AY, Merkurjev D, Zhang J, Ohgi K, Song X, et al. (2013). Functional roles of enhancer RNAs for oestrogen-dependent transcriptional activation. Nature 498, 516–520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z, Zhao B, Wang P, Chen F, Dong Z, Yang H, Guan KL, and Xu Y (2010). Structural insights into the YAP and TEAD complex. Genes & development 24, 235–240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao Y, Smyth GK, and Shi W (2014). featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30, 923–930. [DOI] [PubMed] [Google Scholar]
- Liberzon A, Subramanian A, Pinchback R, Thorvaldsdottir H, Tamayo P, and Mesirov JP (2011). Molecular signatures database (MSigDB) 3.0. Bioinformatics 27, 1739–1740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Z, Merkurjev D, Yang F, Li W, Oh S, Friedman MJ, Song X, Zhang F, Ma Q, Ohgi KA, et al. (2014). Enhancer activation requires trans-recruitment of a mega transcription factor complex. Cell 159, 358–373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love MI, Huber W, and Anders S (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15, 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malik S, and Roeder RG (2010). The metazoan Mediator co-activator complex as an integrative hub for transcriptional regulation. Nature reviews. Genetics 11, 761–772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marti P, Stein C, Blumer T, Abraham Y, Dill MT, Pikiolek M, Orsini V, Jurisic G, Megel P, Makowska Z, et al. (2015). YAP promotes proliferation, chemoresistance, and angiogenesis in human cholangiocarcinoma through TEAD transcription factors. Hepatology 62, 1497–1510. [DOI] [PubMed] [Google Scholar]
- McLean CY, Bristor D, Hiller M, Clarke SL, Schaar BT, Lowe CB, Wenger AM, and Bejerano G (2010). GREAT improves functional interpretation of cis-regulatory regions. Nat Biotechnol 28, 495–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moggs JG, and Orphanides G (2001). Estrogen receptors: orchestrators of pleiotropic cellular responses. EMBO reports 2, 775–781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicholson RI, Gee JM, Manning DL, Wakeling AE, Montano MM, and Katzenellenbogen BS (1995). Responses to pure antiestrogens (ICI 164384, ICI 182780) in estrogen-sensitive and -resistant experimental and clinical breast cancer. Annals of the New York Academy of Sciences 761, 148–163. [DOI] [PubMed] [Google Scholar]
- Oh H, Slattery M, Ma L, Crofts A, White KP, Mann RS, and Irvine KD (2013). Genome-wide association of Yorkie with chromatin and chromatin-remodeling complexes. Cell reports 3, 309–318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pascual G, Fong AL, Ogawa S, Gamliel A, Li AC, Perissi V, Rose DW, Willson TM, Rosenfeld MG, and Glass CK (2005). A SUMOylation-dependent pathway mediates transrepression of inflammatory response genes by PPAR-gamma. Nature 437, 759–763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piccolo S, Dupont S, and Cordenonsi M (2014). The biology of YAP/TAZ: hippo signaling and beyond. Physiological reviews 94, 1287–1312. [DOI] [PubMed] [Google Scholar]
- Plank JL, and Dean A (2014). Enhancer function: mechanistic and genome-wide insights come together. Molecular cell 55, 5–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reichardt HM, Kaestner KH, Tuckermann J, Kretz O, Wessely O, Bock R, Gass P, Schmid W, Herrlich P, Angel P, et al. (1998). DNA binding of the glucocorticoid receptor is not essential for survival. Cell 93, 531–541. [DOI] [PubMed] [Google Scholar]
- Robinson JT, Thorvaldsdottir H, Winckler W, Guttman M, Lander ES, Getz G, and Mesirov JP (2011). Integrative genomics viewer. Nat Biotechnol 29, 24–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson MD, McCarthy DJ, and Smyth GK (2010). edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26, 139–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross-Innes CS, Stark R, Holmes KA, Schmidt D, Spyrou C, Russell R, Massie CE, Vowler SL, Eldridge M, and Carroll JS (2010). Cooperative interaction between retinoic acid receptor-alpha and estrogen receptor in breast cancer. Genes & development 24, 171–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roux KJ, Kim DI, Raida M, and Burke B (2012). A promiscuous biotin ligase fusion protein identifies proximal and interacting proteins in mammalian cells. The Journal of cell biology 196, 801–810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabari BR, Dall’Agnese A, Boija A, Klein IA, Coffey EL, Shrinivas K, Abraham BJ, Hannett NM, Zamudio AV, Manteiga JC, et al. (2018). Coactivator condensation at super-enhancers links phase separation and gene control. Science 361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Z, He F, Chen M, Hua L, Wang W, Jiao S, and Zhou Z (2017). DNA-binding mechanism of the Hippo pathway transcription factor TEAD4. Oncogene 36, 4362–4369. [DOI] [PubMed] [Google Scholar]
- Skibinski A, Breindel JL, Prat A, Galvan P, Smith E, Rolfs A, Gupta PB, LaBaer J, and Kuperwasser C (2014). The Hippo transducer TAZ interacts with the SWI/SNF complex to regulate breast epithelial lineage commitment. Cell reports 6, 1059–1072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, et al. (2005). Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A 102, 15545–15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taatjes DJ (2010). The human Mediator complex: a versatile, genome-wide regulator of transcription. Trends in biochemical sciences 35, 315–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wakeling AE, Dukes M, and Bowler J (1991). A potent specific pure antiestrogen with clinical potential. Cancer Res 51, 3867–3873. [PubMed] [Google Scholar]
- Wang D, Garcia-Bassets I, Benner C, Li W, Su X, Zhou Y, Qiu J, Liu W, Kaikkonen MU, Ohgi KA, et al. (2011). Reprogramming transcription by distinct classes of enhancers functionally defined by eRNA. Nature 474, 390–394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warnmark A, Almlof T, Leers J, Gustafsson JA, and Treuter E (2001). Differential recruitment of the mammalian mediator subunit TRAP220 by estrogen receptors ERalpha and ERbeta. The Journal of biological chemistry 276, 23397–23404. [DOI] [PubMed] [Google Scholar]
- Yu G, Wang LG, and He QY (2015). ChIPseeker: an R/Bioconductor package for ChIP peak annotation, comparison and visualization. Bioinformatics 31, 2382–2383. [DOI] [PubMed] [Google Scholar]
- Zanconato F, Forcato M, Battilana G, Azzolin L, Quaranta E, Bodega B, Rosato A, Bicciato S, Cordenonsi M, and Piccolo S (2015). Genome-wide association between YAP/TAZ/TEAD and AP-1 at enhancers drives oncogenic growth. Nature cell biology 17, 1218–1227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Krutchinsky A, Fukuda A, Chen W, Yamamura S, Chait BT, and Roeder RG (2005). MED1/TRAP220 exists predominantly in a TRAP/Mediator subpopulation enriched in RNA polymerase II and is required for ER-mediated transcription. Molecular cell 19, 89–100. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, Nusbaum C, Myers RM, Brown M, Li W, et al. (2008). Model-based analysis of ChIP-Seq (MACS). Genome Biol 9, R137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao B, Lei QY, and Guan KL (2008). The Hippo-YAP pathway: new connections between regulation of organ size and cancer. Current opinion in cell biology 20, 638–646. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The GEO accession number for all deep sequencing data reported in this paper is GSE125609, which includes ChIP-seq data set (GSE125594), RNA-seq data set (GSE125606) and GRO-seq data set (GSE125607). To review GEO accession GSE125609: use https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE125609. (Note: Access token will be provided for review purpose.)
We also used some published ChIP-seq data from the Gene Expression Omnibus database for FoxA1 and ERα under accession number GSE59530 (Franco et al., 2015), H3K27Ac under accession number GSE45822 (Li et al., 2013), YAP1 and TEAD1 under accession number GSE107013 (Elster et al., 2018).
The raw data used in this paper have been deposited to Mendeley Data and are available at: http://dx.doi.org/10.17632/hwz4rdwfr6.1 or https://data.mendeley.com/datasets/hwz4rdwfr6/draft?a=ab20213e-4a26-46e5-b17e-4e3299d3121b.