

Abstract
Recent advances in the understanding of lipid metabolism suggest a critical role of endoplasmic reticulum (ER) stress in obesity‐induced kidney injury. Hepatocyte growth factor (HGF) is a pleiotropic cytokine frequently featured in stem cell therapy with distinct renotropic benefits. This study aims to define the potential link between human induced pluripotent stem cell‐derived mesenchymal stem cells (iPS‐MSCs)/bone marrow‐derived MSCs (BM‐MSCs) and ER stress in lipotoxic kidney injury induced by palmitic acid (PA) in renal tubular cells and by high‐fat diet (HFD) in mice. iPS‐MSCs or BM‐MSCs alleviated ER stress (by preventing induction of Bip, chop, and unfolded protein response), inflammation (Il6, Cxcl1, and Cxcl2), and apoptosis (Bax/Bcl2 and terminal deoxynucleotidyl transferase‐mediated dUTP‐biotin nick end labeling‐positive cells) in renal cortex of animals exposed to HFD thus mitigating histologic damage and albuminuria, via activating HGF/c‐Met paracrine signaling that resulted in enhanced HGF secretion in the glomerular compartment and c‐Met expression in the tubules. Coculture experiments identified glomerular endothelial cells (GECs) to be the exclusive source of glomerular HGF when incubated with either iPS‐MSCs or BM‐MSCs in the presence of PA. Furthermore, both GEC‐derived HGF and exogenous recombinant HGF attenuated PA‐induced ER stress in cultured tubular cells, and this effect was abrogated by a neutralizing anti‐HGF antibody. Taken together, this study is the first to demonstrate that MSCs ameliorate lipotoxic kidney injury via a novel microenvironment‐dependent paracrine HGF/c‐Met signaling mechanism to suppress ER stress and its downstream pro‐inflammatory and pro‐apoptotic consequences. stem cells translational medicine 2019;8:898&910
Keywords: Induced pluripotent stem cells, Mesenchymal stem cells, Hepatocyte growth factor, Endoplasmic reticulum stress, Lipotoxicity
Significance Statement.
Human induced pluripotent stem cell‐derived mesenchymal stem cells (iPS‐MSCs) have potential as an alternative cell‐based therapy to bone marrow‐derived mesenchymal stem cells (BM‐MSCs). However, the therapeutic potential and mechanism of either iPS‐MSCs or BM‐MSCs in treating lipotoxicity‐induced kidney injury remains largely undetermined. This study identified that iPS‐MSCs exhibited equivalent efficacy to BM‐MSCs in ameliorating ER stress, inflammation, and apoptosis in palmitic acid‐treated renal tubular cells and high‐fat diet‐induced obese kidney in mice, via activation of HGF/c‐Met paracrine signaling in the obese kidney microenvironment. The novel findings suggest that iPS‐MSCs and BM‐MSCs alleviate lipotoxicity‐induced chronic kidney injury via a HGF/c‐Met‐dependent mechanism, supporting iPS‐MSCs as a valuable alternative source to BM‐MSCs for therapeutic application in chronic kidney disease.
Introduction
Obesity is a public health problem, of which obesity‐related nephropathy is an important complication 1. A major link between obesity and its complications is the elevated circulating levels of free fatty acids (FFAs), a key hallmark in hyperlipidemia‐related disease. Excessive FFAs not only deposit in adipose tissues, but also accumulate ectopically in other organs to elicit lipotoxicity 2, 3, which is increasingly implicated in the development of obesity‐related kidney injury 2, 4.
Tissues with ectopic FFA deposition are under endoplasmic reticulum (ER) stress 4. ER is an intracellular organelle where proteins are properly folded for maturation, and where calcium homeostasis is modulated and lipids or steroids are synthesized. To cope with stress and re‐establish homeostasis, ER employs an adaptive mechanism called unfolded protein response (UPR) to dynamically expand both ER size and ER capacity to match demand 5. UPR relies on the coordinated action of three canonical pathways defined by the upstream sensors of ER stress: double‐stranded RNA‐activated protein kinase‐like eukaryotic initiation factor 2α kinase (PERK), inositol‐requiring enzyme 1α (IRE1α), and activating transcription factor 6 (ATF6). In physiological conditions, these sensors are bound within an inactive form to ER chaperone binding immunoglobulin protein (BiP), whereas under ER stress, BiP dissociates from these sensors. The activated dissociative sensors then transmit signals to alleviate ER stress by increasing ER folding capacity and suppressing protein translation, as a survival mechanism to reestablish ER homeostasis 5, 6. However, if ER stress fails to be resolved, the signaling switches from pro‐survival to pro‐apoptosis 5. More recently, advances in lipid metabolism suggest ER stress as a common molecular pathway in the pathogenesis of hyperlipidemia‐related diseases 6, 7, 8, 9.
Mesenchymal stem cells (MSCs) derived from induced pluripotent stem cells (known as iPS‐MSCs) is emerging as a potential alternative to bone marrow‐derived MSCs (BM‐MSCs) for cell‐based therapy. Human iPS derives from patient‐specific adult somatic cells, thus iPS‐MSCs are theoretically an inexhaustible source of MSCs for clinical application 10. Moreover, in addition to the preserved differentiation and immunomodulation capacity compared with BM‐MSCs, iPS‐MSCs have been demonstrated to be superior with higher accessibility, less oncogenicity, and greater expandability to overcome the inherent limitations in BM‐MSCs for clinical application 11, 12, 13. Preclinical studies have shown great success in applying iPS‐MSCs for treating renal ischemia/reperfusion injury 14, ischemic cardiac disease 15, chronic obstructive pulmonary disease 16, and osteoarthritis 17. We recently showed that iPS‐MSCs conferred renoprotection in adriamycin nephrosis 18, supporting the potential of iPS‐MSCs in treating kidney diseases. To date, there has been a scarcity of reports on the utility of iPS‐MSCs in obesity‐induced kidney injury.
Hepatocyte growth factor (HGF) is a pleiotropic cytokine derived primarily from cells of mesenchymal origin, and it is the only known ligand of the c‐Met tyrosine kinase transmembrane receptor 19. HGF expression is limited to nonepithelial cells in kidney, such as fibroblasts 20, mesangial cells 21, endothelial cells 22, while c‐Met protein is ubiquitously expressed. HGF/c‐Met signaling has emerged as an endogenous protective factor that maintains renal architecture and reconstructs a microenvironment under various physiologic and pathophysiologic conditions, whereas genetic ablation of c‐Met in tubular or glomerular cells aggravated renal injuries 19, 23, 24. MSCs of different origins are also described to be the sources of HGF 25, 26, and coculture of MSCs with certain stromal cells promoted HGF synthesis in MSCs 19, 27. To date, there has been no report of HGF production in obese kidney treated with iPS‐MSCs infusion, not to mention activation of HGF/c‐Met signaling in the lipotoxic microenvironment. Here, we present the therapeutic value of iPS‐MSCs in obesity‐induced kidney injury, and compare its efficacy with BM‐MSCs to elucidate the underlying regulatory mechanisms of this effect.
Materials and Methods
Reagents and Antibodies
Palmitic acid (PA), oleic acid (OA), fatty acid‐free bovine serum albumin (BSA), recombinant HGF, and other reagents were purchased from Sigma (St. Louis, MO). PA was conjugated with BSA before various experiments. OA was dissolved in phosphate‐buffered saline (PBS) to get a final working concentration of 10 mM. Antibodies to cleaved caspase‐3, p53‐upregulated modulator of apoptosis (PUMA), cleaved poly adenosine diphosphate ribose polymerase (PARP), HGF, c‐Met, phosphorylated IRE1α (p‐IRE1α), and neutralizing antibody to HGF were purchased from Abcam (Carlsbad, CA). Antibodies to activating transcription factor 4 (ATF4), phosphorylated eukaryotic initiation factor 2α (p‐eIF2α), total c‐Jun N‐terminal kinase (JNK), phosphorylated JNK (p‐JNK), total nuclear factor‐κB (NF‐κB), phosphorylated NF‐κB (p‐NF‐κB), total extracellular signal‐regulated kinase (ERK), and phosphorylated ERK (p‐ERK) were purchased from Cell Signaling Technology (Beverly, CA). Horseradish peroxidase (HRP)‐conjugated anti‐mouse and anti‐rabbit secondary antibodies were from Dako (Carpinteria, CA). Anti‐β‐actin antibody was from Lab Vision Co. (Fremont, CA). The antibodies catalogue numbers were provided in Supporting Information Table S1.
Cell Lines
Immortalized human proximal tubule epithelial human kidney‐2 (HK‐2) cells and human mesangial cells (HMCs, American Type Culture Collection, Manassas, VA) were used in vitro as previously described 28. A conditionally immortalized human podocyte cell line obtained from University of Bristol 29 was maintained under permissive conditions (33°C and 5% CO2) in Roswell Park Memorial Institute (RPMI) 1,640 medium (Life Technologies, Inc., Grand Island, NY) supplemented with 10% fetal bovine serum (FBS, Invitrogen, Carlsbad, CA) and 1% insulin–transferrin–selenium (ITS, Life Technologies, Inc.). For differentiation, podocytes were cultured for 14 days under nonpermissive conditions (37°C and 5% CO2) in RPMI 1640 supplemented with 10% FBS and 1% ITS. Primary human glomerular endothelial cells (GECs, Cell Systems, Kirkland, WA) were cultured in Cell Systems Culture Complete Medium containing 10% serum (Cell Systems). All experiments were performed after 12‐hour growth arrest.
Human iPS‐MSCs and BM‐MSCs Preparation
Commercially available BM‐MSCs purchased from Lonza (Verviers, Belgium) were cultured in MesenPro RS Basal Medium (Life Technologies, Inc.) supplemented with 2% MesenPro Growth Supplement (Life Technologies, Inc.) and 1% GlutaMax Supplement (Life Technologies, Inc.) according to the manufacturer's instruction 30. iPS‐MSCs were generated and maintained according to our established protocols as described in our previous studies 31, 32, 33. The iPS‐MSC clone, N1‐iPS‐MSC, was used in our study. This clone was generated from human iPS cells reprogrammed from human fibroblast cells (American Type Culture Collection catalog no. CCL‐186) via viral transduction with human cDNAs of Klf4, Sox2, Oct4, and c‐Myc, as described in our previous studies 18, 31, 32. iPS‐MSCs were derived from iPSCs according to our established clinically compliant protocol 33, 34. iPS‐MSCs within the ninth passages were used throughout this study 18.
Cell Viability Assay and Detection of Intracellular Reactive Oxygen Species
Cell viability was determined by using Mitochondrial Viability Assay Kit (Abcam) and reactive oxygen species (ROS) production was measured by using Oxidative Stress Indicator, 5(‐and‐6)‐chloromethyl‐2′,7′‐dichlorohydrofluorescein diacetate (CM‐H2DCFDA; Invitrogen) according to instructions from manufacturers.
Coculture Experiments
Two coculture systems were established in this study to mimic in vivo situations. (a) Coculture system I was adopted to determine which specific type of glomerular cell produces HGF in the presence of MSCs and lipid stress. Glomerular cells including HMCs, podocytes, and GECs were separately plated into 12‐well plates (Corning, NY) to develop a confluent monolayer, and coculture was initiated by adding either iPS‐MSCs or BM‐MSCs suspension (at a MSCs/glomerular cells ratio of 1:5). After coculture for 48 hours in the absence or presence of PA, supernatant was harvested for detecting HGF protein by human HGF Enzyme‐Linked Immunosorbent Assay (ELISA) Kit (Abcam). Glomerular cells cultured alone without MSCs acted as the control. (b) Coculture system II was used to examine under the interaction among HK‐2, GEC, and MSCs, the synthesis of HGF and HK‐2 response upon PA activation. In this coculture system, GECs were seeded into upper transwell insert (Corning) and HK‐2 cells were plated into lower chamber in 12‐well plates to develop a confluent monolayer, respectively, then MSCs were added to the GEC monolayer in the upper chamber (at a MSCs/GECs ratio of 1:5). PA or BSA was added into both upper insert and lower chamber simultaneously. After coculture for indicated time points, supernatant from both upper inserts and lower chambers was collected for determining HGF concentration by ELISA. Total RNA in HK‐2 cells was isolated to determine genes expression.
Production of MSC‐Conditioned Media for GECs Activation
iPS‐MSCs and BM‐MSCs were maintained and cultured in T75 flasks (Corning) according to previously established protocols 30, 31, 32, 33. At 80%–90% confluence, MSCs were incubated with BSA (0.475%) or PA (500 μM) for 48 hours. The MSC‐conditioned media (CM) was centrifuged for 5 minutes at 3,000g to remove cell debris and was aliquoted and frozen at −80°C until experiment. For activation of GECs by MSC‐CM, confluent GECs on 12‐well plates were cultured with MSC‐CM for 24 hours before extracting total RNA from GECs for determining HGF gene expression.
RNA Extraction and Quantitative Real‐Time PCR
Total RNA was extracted from cells or renal cortex by NucleoSpin RNA II total RNA Isolation Kit or NucleoSpinTriprep Kit (Macherey‐Nagel, Duren, Germany). RNAs were reversely transcribed to cDNAs by High‐Capacity cDNA Reverse Transcription Kit (Applied Biosystems, Foster City, CA) and gene expression was detected by quantitative real‐time PCR (qPCR) using specific primers (Supporting Information Table S2). Relative quantification of genes was normalized to β‐actin expression and all experimental groups were compared with their respective control (CTL) groups using StepOne software v2.3 (Applied Biosystems).
Western Blot Analysis
Total protein lysate was isolated from cells or renal cortex by NucleoSpinTriprep Kit (Macherey‐Nagel), and total protein concentrations were quantified using BCA Protein Assay Kit (Pierce, Rockford, IL). Equal amount of protein lysate was electrophoresed through 4% to 12% gradient polyacrylamide gel (Invitrogen) before transferring to polyvinylidenedifluoride membrane (Millipore, Bedford, MA). Membrane was subjected to overnight primary antibody incubation, thereafter incubated with matched HRP‐conjugated secondary antibody for 2 hours at room temperature (RT). Bands were visualized by ChemiDoc XRS+ system (Bio‐Rad, Hercules, CA) after incubating with Clarity Western ECL Substrate (Bio‐Rad). Densitometries of proteins bands were quantified by Image Lab software (Bio‐Rad).
Immunofluorescence Staining
Immunofluorescence (IF) staining was performed to visualize ER state. Briefly, HK‐2 cells were washed by PBS, fixed by 4% formaldehyde for 15 minutes and permeabilized for 10 minutes with 0.2% Triton X‐100. After blocking nonspecific binding with 5% BSA for 30 minutes, staining with ER stress marker protein disulfide‐isomerase (PDI) was performed by incubating with a mouse monoclonal antibody against PDI (ThermoFisher, CA) for 1.5 hour at RT, thereafter incubated with the fluorescein isothiocyanate‐labeled goat anti‐mouse secondary antibody (Jackson, West Grove, PA) for 2 hours at RT. Fluorescence‐labeled cells were mounted with Vectashield Mounting Medium plus 4′,6‐diamidino‐2‐phenylindole (Vector Laboratories, Burlingame, CA), followed by visualizing under a fluorescence microscope (Olympus, Tokyo, Japan) and analysis by ImageJ software (http://rsb.info.nih.gov/ij) in 20 randomly selected fields for each coverslip at ×400 magnification.
Animal Models
All animal experiments were approved by the Committee on the Use of Live Animal in Teaching and Research of the University of Hong Kong and were conducted in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals. Male 6‐week‐old C57BL6/J mice (Laboratory Animal Unite, The University of Hong Kong, HK) were randomly assigned into normal diet (ND) group (10% of total calorie, n = 24) and high‐fat diet (HFD) group (60% of total calorie, n = 24) for 12‐week feeding. Mice were weighted fortnightly and blood glucose levels were measured by glucometer (Accu‐Check Sensor, Roche Diagnostics, Mannheim, Germany) following overnight fasting period. Urine was collected in metabolic cages over 24‐hour periods, and urine albumin was measured by using a Mouse Albumin ELISA Quantitation Set (Bethyl Laboratories, Montgomery, AL).
MSCs Infusion
At the age of 18 weeks, mice in both ND and HFD groups (n = 24 each group) were, respectively, divided into three subgroups randomly (n = 8 each subgroup): CTL group, iPS‐MSC group, and BM‐MSC group. Mice fed ND were infused either with vehicle control (ND‐CTL), iPS‐MSCs (ND‐iPS‐MSC), or BM‐MSCs (ND‐BM‐MSC). Mice fed HFD were infused either with vehicle control (HFD‐CTL), iPS‐MSCs (HFD‐iPS‐MSC), or BM‐MSCs (HFD‐BM‐MSC). Intravenous infusions of normal saline or MSCs at a dose of 1 × 106 cells per 100 μl normal saline was done in corresponding groups of mice. Mice were sacrificed at age 26 weeks, followed by blood collection through cardiac puncture. Collected kidneys were either fixed in 10% neutral buffered formalin or snap‐frozen in liquid nitrogen and stored at −80°C for further analysis.
Histological Analysis
Paraffin‐embedded kidney tissue sections (4 μm) were stained with Periodic acid‐Schiff (PAS) solution (Sigma) to assess renal pathology alternations. Tubular injury in renal cortex was determined and graded using 10 random nonoverlapping fields from each sample at ×400 magnification and assessment of four categories of damage was made including presence of glycogenated nuclei, tubular cast, vacuolation, and dilatation as previously described 35. Glomerular volume and mesangial expansion were quantified by ImageJ Software in 20 randomly selected cortical glomeruli from each sample at ×400 magnification and analyzed in a blinded fashion.
Immunohistochemistry Staining
Paraffin‐embedded renal sections (4 μm) were deparaffinized, rehydrated, and subjected to microwave‐based antigen retrieval in citrate buffer. Sections were incubated overnight with primary antibodies against HGF and c‐Met, thereafter incubated with HRP‐conjugated secondary antibodies (Dako) and developed by 3,3′‐diaminobenzidine substrate from the Envision Plus system (Dako). Slides were counterstained with hematoxylin before mounting. Quantitative analysis of the immunohistochemistry (IHC) staining in randomly selected 20 fields in the cortex from each sample was measured by ImageJ software and presented as a value of integrated optical density.
In Situ Apoptosis Detection by Terminal Deoxynucleotidyl Transferase‐Mediated dUTP‐biotin Nick End Labeling Assay
Terminal deoxynucleotidyl transferase‐mediated dUTP‐biotin nick end labeling (TUNEL) staining was performed by using an In situ Apoptosis Detection Kit (Abcam) following the manufacturer's instruction. Quantification was performed by calculating the number of TUNEL‐positive cells in five randomly chosen high‐power fields within each slide from each sample and analyzed in a blinded fashion.
Statistical Analysis
Statistical analysis was performed using GraphPad Prism (San Diego, CA). All data were expressed as mean ± SD. Student's two‐tailed t test and one‐way analysis of variance followed by Tukey's procedure for intergroup comparison were conducted where appropriate. p < .05 was considered statistically significant.
Results
PA Induced ER Stress in HK‐2 Cells
PA dose‐dependently reduced HK‐2 cells viability at the time points of 24 and 48 hours of exposure (Fig. 1A). Based on these results, 500 μM PA was selected for subsequent experiments to mimic FFA‐induced lipotoxicity in vitro. PA dose‐dependently increased ROS production in HK‐2 cells at the same time points (Fig. 1C). All these effects were not observed with equivalent doses of BSA, OA, or CTL (Fig. 1B, 1D). PA induced mRNA expression of ER stress mediators (BiP; ER degradation enhancing α‐mannosidase like protein, EDEM; ATF4; CHOP, C/EBP homologous protein; tumor necrosis factor receptor‐associated factor 2, TRAF2; spliced X‐box binding protein 1, s‐XBP1) in HK‐2 cells (Fig. 1E–1J). Protein levels of signaling molecules in UPR including p‐eIF2α, ATF4, and p‐IRE1α were upregulated after PA exposure (Fig. 1K–1N). IF staining revealed that PA exposure significantly increased ER stress in HK‐2 cells, as evidenced by enhanced fluorescent density of PDI (green; Fig. 1O, 1P).
Figure 1.
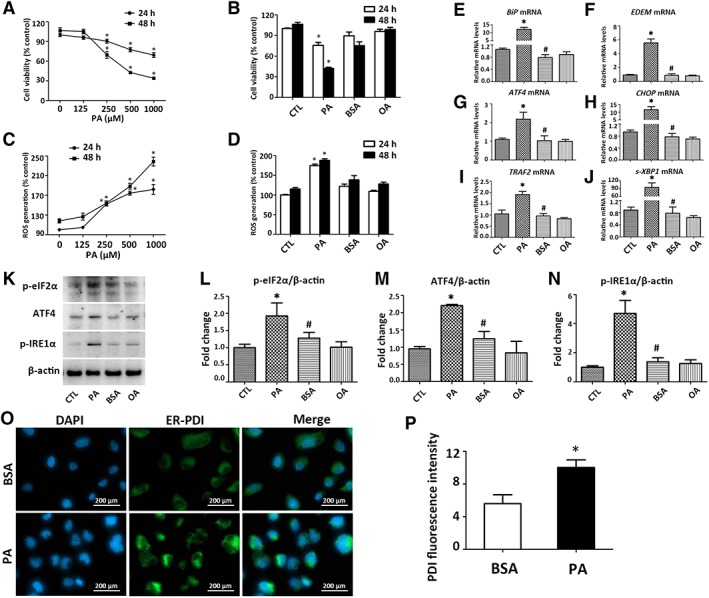
PA induces ER stress in HK‐2 cells. (A, B): Cell viability in HK‐2 cells treated with PA at 0–1,000 μM, or PA (500 μM), BSA (0.475%), and OA (500 μM) for 24 or 48 hours. (C, D): ROS production in HK‐2 cells treated with PA at 0–1,000 μM, or PA (500 μM), BSA (0.475%), and OA (500 μM) for 24 or 48 hours. (E–J): Transcripts of ER stress mediators in HK‐2 cells exposed to PA (500 μM) or OA (500 μM) for 24 hours were quantified by qPCR. (K): Representative blots for p‐eIF2α, ATF4, and p‐IRE1α and β‐actin were shown, with (L–N) results after quantification of each protein normalized to β‐actin. *, p < .05 versus CTL group; #, p < .05 versus PA group. (O): Representative micrographs (×400) from immunofluorescent staining of ER by anti‐PDI (green), (P) with semiquantification of average fluorescence intensity from 20 random fields for each coverslip. *, p < .05 versus BSA group. Results are expressed as mean ± SD. Experiments were performed in triplicate. Abbreviations: ATF4, activating transcription factor 4; BiP, binding immunoglobulin protein; BSA, bovine serum albumin; CHOP, C/EBP homologous protein; CTL, plain culture medium treated control cells; DAPI, 4′,6‐diamidino‐2‐phenylindole; EDEM, endoplasmic reticulum degradation‐enhancing α‐mannosidase‐like protein; ER, endoplasmic reticulum; OA, oleic acid; p‐eIF2α, phosphorylated eukaryotic initiation factor 2α; p‐IRE1α, phosphorylated inositol‐requiring enzyme 1α; PA, palmitic acid; PDI, protein disulfide‐isomerase; qPCR, quantitative real‐time PCR; s‐XBP1, spliced X‐box‐binding protein 1; ROS, reactive oxygen species; TRAF2, tumor necrosis factor receptor‐associated factor 2.
PA‐Induced ER Stress Mediated Inflammation and Apoptosis in HK‐2 Cells
PA upregulated transcripts of pro‐inflammatory cytokines including interleukin‐6 (IL6) and vascular endothelial growth factor (VEGF; Fig. 2A, 2B), pro‐apoptotic mediators including B‐cell lymphoma 2‐associated X protein (BAX), B‐cell lymphoma 2‐antagonist/killer (BAK), and B‐cell lymphoma 2 interacting killer (BIK; Fig. 2D–2F), but downregulated B‐cell lymphoma 2 (BCL2) expression (Fig. 2C) in HK‐2 cells. PA also elevated PUMA, cleaved caspase‐3, and cleaved PARP in HK‐2 cells (Fig. 2G–2J). This was associated with activation of p‐NF‐κB, p‐ERK, and p‐JNK (Fig. 3K–3N).
Figure 2.
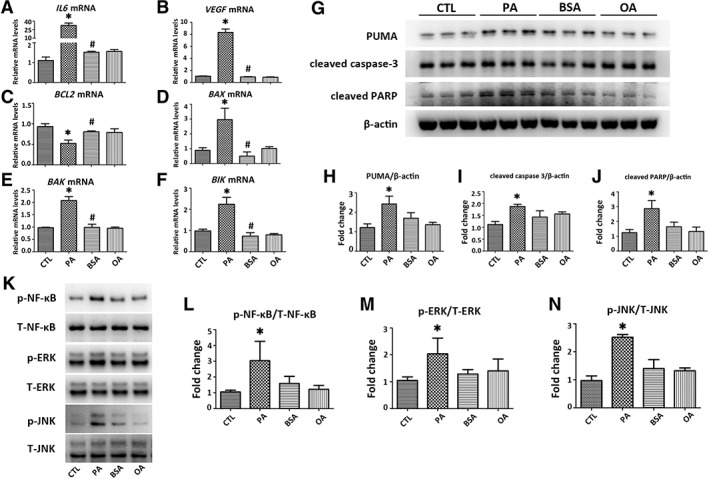
PA induces inflammation and apoptosis in HK‐2 cells. HK‐2 cells were exposed to PA (500 μM) or OA (500 μM) for 24 hours. Transcripts of (A, B) inflammatory and (C–F) apoptotic mediators including IL6, VEGF, BCL2, BAX, BAK, and BIK in HK‐2 cells were determined by qPCR. Results are expressed as fold change relative to CTL. (G): Representative Western blots of apoptotic mediators including PUMA, cleaved caspsae‐3, and cleaved PARP are presented, with (H–J) results after quantification of each protein normalized to β‐actin. (K): Representative Western blots of p‐NF‐κB, T‐NF‐κB, p‐ERK, T‐ERK, p‐JNK, and T‐JNK are presented, with (L–N) results after quantification of each phosphorylated protein normalized to total protein. All the ratios are then normalized with the ratio of CTL. Results are expressed as mean ± SD. Experiments were performed in triplicate. *, p < .05 versus CTL group; #, p < .05 versus PA group. Abbreviations: BAK, Bcl‐2‐antagonist killer; BAX, BCL‐2‐associated X protein; BCL2, B‐cell lymphoma 2; BIK, Bcl‐2‐interacting killer; BSA, bovine serum albumin; CTL, plain culture medium treated control cells; IL6, interleukin 6; OA, oleic acid; p‐ERK, phosphorylated extracellular signal‐regulated kinase; p‐JNK, phosphorylated c‐Jun N‐terminal kinase; p‐NF‐κB, phosphorylated nuclear factor‐κB; PA, palmitic acid; PARP, poly adenosine diphosphate ribose polymerase; PUMA, p53 upregulated modulator of apoptosis; qPCR, quantitative real‐time PCR; T‐ERK, total extracellular signal‐regulated kinase; T‐JNK, total c‐Jun N‐terminal kinase; T‐NF‐κB, total nuclear factor‐κB; VEGF, vascular endothelial growth factor.
Figure 3.
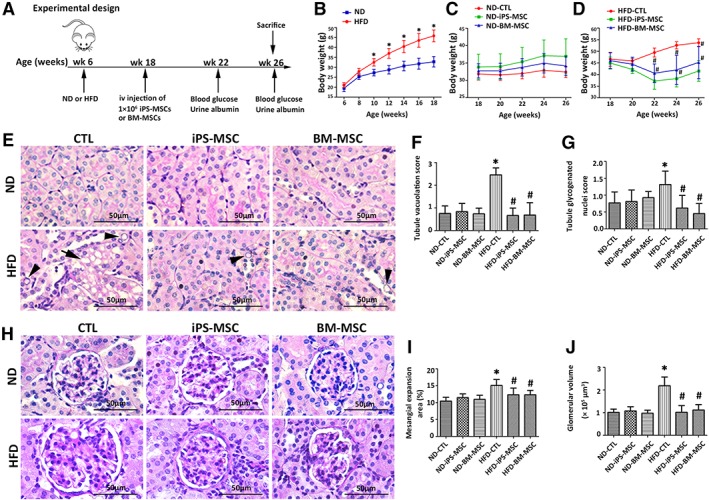
MSCs therapy ameliorates HFD‐induced obesity and kidney injury in mice. (A): Animal experimental design for determining the therapeutic efficiency of MSCs infusion in HFD‐induced obesity mice. Six‐week‐old mice feeding ND or HFD for 12 weeks were randomly divided into CTL, iPS‐MSC, and BM‐MSC subgroups (n = 8 each subgroup), followed by infusion of saline or MSCs via tail vein and fed for further 8 weeks before sacrifice at age 26 weeks. Blood glucose and urine albumin were monitored at age 22 and 26 weeks. (B–D): Body weight was measured biweekly throughout the experiment. Results are expressed as mean ± SD, n = 8 per group.*, p < .05 versus ND group; #, p < .05 versus HFD‐CTL group. (E): Representative micrographs from PAS staining showed tubular injury (arrowheads: glycogenated nuclei; arrows: cytoplasmic vacuolation) with quantitative analysis including (F) tubule vacuolation score and (G) tubule glycogenated nuclei score. (H): Representative micrographs from PAS staining demonstrated glomerular expansion with quantitative analysis including (I) mesangial expansion area and (J) glomerular volume. Results are expressed as mean ± SD, n = 8 per group. *, p < .05 versus ND‐CTL group; #, p < .05 versus HFD‐CTL group. Abbreviations: BM‐MSC, bone marrow‐derived mesenchymal stem cell; CTL, normal saline vehicle control; HFD, high‐fat diet; iPS‐MSC, induced pluripotent stem cell‐derived mesenchymal stem cell; MSCs, mesenchymal stem cells; ND, normal diet; PAS, periodic acid‐Schiff.
MSCs Reduced HFD‐Induced Obesity in Mice
Both ND and HFD mice were randomly assigned treatment with normal saline, iPS‐MSCs, and BM‐MSCs (Fig. 3A). HFD treated animals gained significantly more weight starting from age 10 weeks (Fig. 3B). There was no difference in body weight among the three subgroups of ND (Fig. 3C), whereas MSCs infusion significantly lowered the weight of HFD mice starting from age 22 weeks (Fig. 3D). Similarly, MSCs significantly reduced fasting glucose at age 26 weeks (Supporting Information Table S3). There was no significant difference in body weight and fasting glucose between HFD‐iPS‐MSC and HFD‐BM‐MSC mice.
MSCs Ameliorated HFD‐Induced Kidney Lesions in Obese Mice
Tubular vacuolation and tubular glycogenated nuclei (Fig. 3E) and glomerular expansion (Fig. 3H) were ameliorated by MSC infusion (Fig. 3F, 3G, 3I, 3J). In addition, albuminuria at age 22 weeks (31.72 ± 7.32 μg/day) and 26 weeks (38.51 ± 9.31 μg/day) was markedly diminished in HFD‐iPS‐MSC mice (17.05 ± 6.45 μg/day at 22 weeks; 18.66 ± 3.43 μg/day at 26 weeks) and HFD‐BM‐MSC mice (20.21 ± 5.55 μg/day at 22 weeks; 23.71 ± 8.11 μg/day at 26 weeks; Supporting Information Table S3).
MSCs Alleviated HFD‐Induced ER Stress in Obese Kidney
HFD induced cortical expression of Bip and chop and three other signaling molecules in UPR including p‐eIF2α, ATF4, and p‐IRE1α, which were all downregulated by MSCs treatment (Fig. 4A–4F). iPS‐MSCs and BM‐MSCs had comparable effects.
Figure 4.
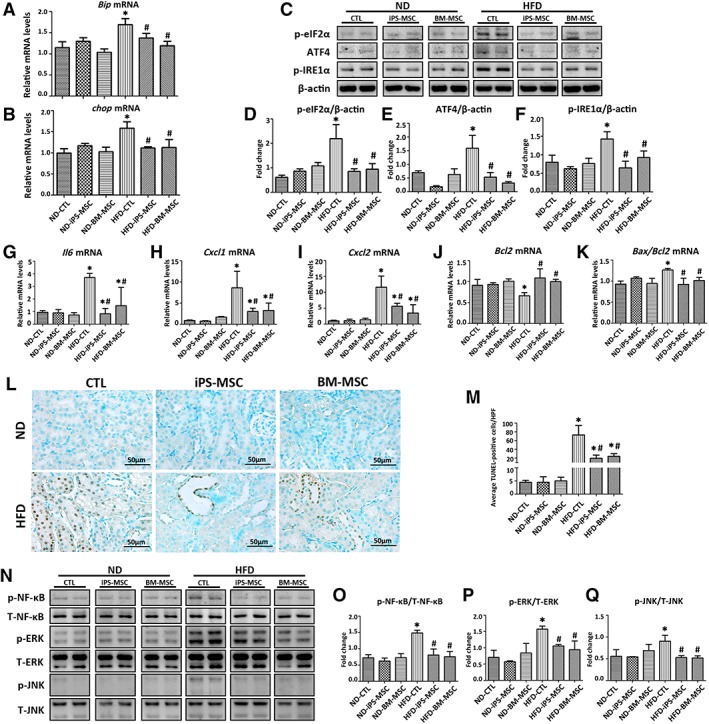
MSCs therapy alleviates HFD‐induced ER stress, inflammation, and apoptosis in obese kidney. (A, B): Bip and chop mRNA expression in kidneys from mice of different subgroups were quantified by qPCR. (C–F): Images from Western blots detecting p‐eIF2α, ATF4, and p‐IRE1α from total kidney tissue protein lysates. (G–K): Transcripts of inflammatory and apoptotic mediators in mice kidneys were quantified by qPCR. (L): Micrographs showed apoptotic cells in kidney cortex as determined by TUNEL assay with (M) quantification of average TUNEL‐positive cells/HPF from five random fields per mouse. (N–Q): Representative Western blots for p‐NF‐κB, T‐NF‐κB, p‐ERK, T‐ERK, p‐JNK, and T‐JNK are presented. Results are expressed as mean ± SD, n = 8 per group. *, p < .05 versus ND‐CTL group; #, p < .05 versus HFD‐CTL group. Abbreviations: ATF4, activating transcription factor 4; Bax, Bcl‐2‐associated X protein; Bcl2, B‐cell lymphoma 2; Bip, binding immunoglobulin protein; BM‐MSC, bone marrow‐derived mesenchymal stem cell; chop, C/EBP homologous protein; CTL, normal saline vehicle control; Cxcl1, C‐X‐C motif chemokine ligand 1; Cxcl2, C‐X‐C motif chemokine ligand 2; HFD, high‐fat diet; HPF, high power field; Il6, interleukin 6; iPS‐MSC, induced pluripotent stem cell‐derived mesenchymal stem cell; MSCs, mesenchymal stem cells; ND, normal diet; p‐eIF2α, phosphorylated eukaryotic initiation factor 2α; p‐IRE1α, phosphorylated inositol‐requiring enzyme 1α; p‐ERK, phosphorylated extracellular signal‐regulated kinase; p‐NF‐κB, phosphorylated nuclear factor‐κB; p‐JNK, phosphorylated c‐Jun N‐terminal kinase; qPCR, quantitative real‐time PCR; T‐ERK, total extracellular signal‐regulated kinase; T‐JNK, total c‐Jun N‐terminal kinase; T‐NF‐κB, total nuclear factor‐κB; TUNEL, terminal deoxynucleotidyl transferase‐mediated dUTP‐biotin nick end labeling.
MSCs Alleviated HFD‐Induced Inflammation and Apoptosis in Obese Kidney
iPS‐MSCs and BM‐MSCs partially reversed or abrogated HFD‐induced cortical upregulation of pro‐inflammatory cytokines Il6, C‐X‐C motif chemokine ligand 1 (Cxcl1), and C‐X‐C motif chemokine ligand 2 (Cxcl2; Fig. 4G–4I), Bax/Bcl2 ratio (Fig. 4K), reduced the numbers of TUNEL‐positive cells (Fig. 4L, 4M), and prevented phosphorylation of NF‐κB, ERK, and JNK (Fig. 4N–4Q).
MSCs Augmented HGF/c‐Met Paracrine Signaling in Obese Kidney Microenvironment
Both Hgf and c‐Met mRNA expression were significantly higher in renal cortex from HFD‐iPS‐MSC and HFD‐BM‐MSC mice compared with HFD‐CTL mice (Fig. 5A, 5B), which was consistent with the results from ELISA (Fig. 5C) and Western blot (Fig. 5D–5F). IHC localized HGF synthesis exclusively in the glomerular compartment (Fig. 5G, 5H), whereas upregulation of c‐Met occurred in the tubular compartment (Fig. 5I, 5J).
Figure 5.
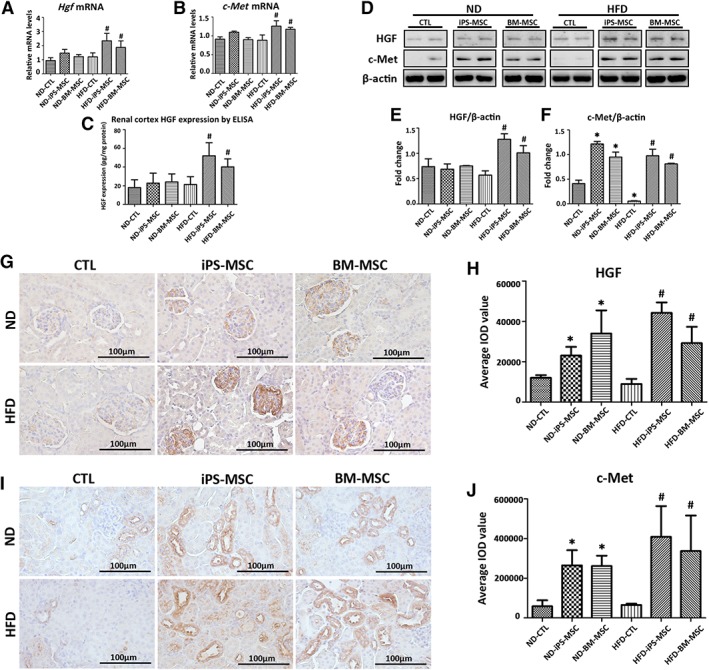
MSCs infusion augments HGF/c‐Met paracrine signaling in obese kidney microenvironment. (A, B): Hgf and c‐Met mRNA expression in mice kidneys were determined by qPCR. Data expressed as fold change relative to CTL. (C): HGF concentration in total renal cortex protein lysatesas measured by ELISA. (D): Representative Western blots for HGF and c‐Met are shown, with (E, F) quantification of each protein normalized to β‐actin. Representative micropraphs from immunohistochemistry staining of (G) HGF and (I) c‐Met in kidney sections of mice, with semiquantification of average IOD value for (H) HGF and (J) c‐Met from 20 random fields per mouse. Results are expressed as mean ± SD, n = 8 per group. *, p < .05 versus ND‐CTL group; #, p < .05 versus HFD‐CTL group. Abbreviations: BM‐MSC, bone marrow‐derived mesenchymal stem cell; CTL, normal saline vehicle control; ELISA, enzyme‐linked immunosorbent assay; HFD, high‐fat diet; HGF, hepatocyte growth factor; iPS‐MSC, induced pluripotent stem cell‐derived mesenchymal stem cell; IOD, integrated optical density; MSCs, mesenchymal stem cells; ND, normal diet.
MSC‐GEC Coculture Protected HK‐2 Cells from Lipotoxicity Through HGF/c‐Met Paracrine Signaling
Cocultures were performed to identify the specific glomerular cell type that contributed to HGF synthesis. HGF was found undetectable in supernatants from either iPS‐MSCs or BM‐MSCs cultured alone (Supporting Information Table S4), confirmed by the unvaried HGF transcripts in MSCs under PA stimulation (Fig. 6A, 6B). In direct‐contact cocultures, HGF was undetectable in either supernatants of HMCs, podocytes, or GECs cultured alone, or supernatants of HMCs or podocytes cocultured with MSCs (Supporting Information Table S4). HGF was detected in supernatants of GECs cocultured with either iPS‐MSCs or BM‐MSCs (Supporting Information Table S4 and Fig. 6C, 6D), which was further increased by PA but not BSA treatment (Fig. 6D). HGF mRNA expression in GECs was upregulated when cocultured with MSC‐CM (Fig. 6E, 6F).
Figure 6.
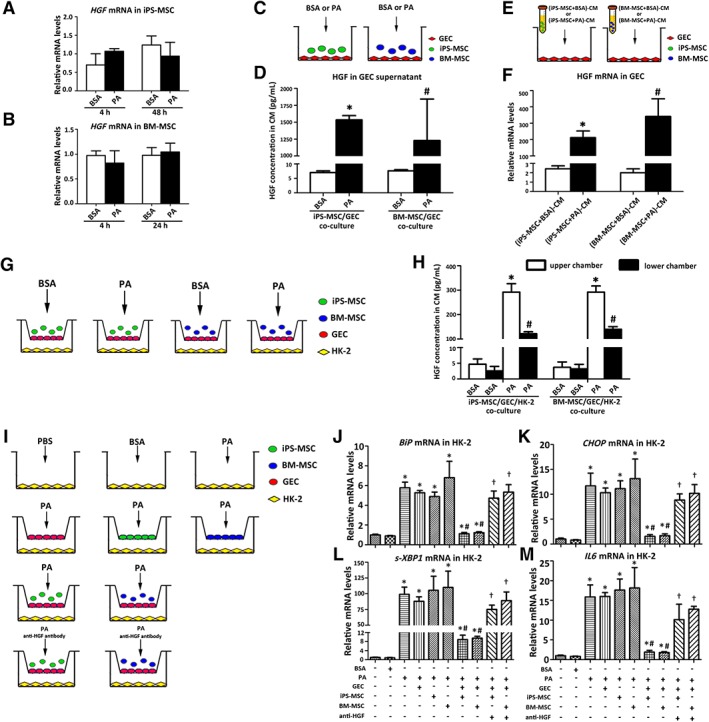
MSC‐GEC coculture protects HK‐2 cells from lipotoxicity through HGF/c‐met paracrine signaling. (A, B): Transcripts of HGF in MSCs exposed to BSA (0.475%) or PA (500 μM). (C): GECs were cocultured with or without MSCs in the presence of PA (500 μM) or BSA (0.475%) and (D) HGF concentration in supernatant was measured by ELISA after 48‐hour incubation. Results are expressed as mean ± SD. * and #, p < .05 versus the corresponding BSA intervention under the same coculture condition. (E): GECs were cultured with CM derived from MSCs in the presence of PA (500 μM) or BSA (0.475%) and (F) HGF gene expression in GECs was determined by qPCR after 24‐hour incubation. Results are expressed as mean ± SD. * and #, p < .05 versus the corresponding BSA intervention under the same coculture condition. (G): Coculture system in which HK‐2 cells were seeded into lower chambers, whereas GECs were cocultured with MSCs in upper chambers, and (H) HGF concentrations in supernatant from both upper and lower chambers were measured by ELISA after 48‐hour incubation. Results are expressed as mean ± SD. * and #, p < .05 versus the corresponding BSA intervention under the same coculture condition. (I): Coculture systems were further used to clarify the effects of MSC‐GEC coculture on HK‐2 cells, and (J–M) gene expression in HK‐2 cells was assessed by qPCR after 48‐hour incubation. Experiments were performed in triplicate. Results are expressed as mean ± SD. *, p < .05 versus HK‐2 cells treated by plain culture medium; #, p < .05 versus HK‐2 cells treated by only PA; †, p < .05 versus corresponding HK‐2 cells cocultured with either iPS‐MSC or BM‐MSC. Abbreviations: BM‐MSC, bone marrow‐derived mesenchymal stem cell; BSA, bovine serum albumin; BiP, binding immunoglobulin protein; CHOP, C/EBP homologous protein; CM, conditioned medium; ELISA, enzyme‐linked immunosorbent assay; GEC, glomerular endothelial cell; HGF, hepatocyte growth factor; IL6, interleukin 6; iPS‐MSC, induced pluripotent stem cell‐derived mesenchymal stem cell; MSC, mesenchymal stem cell; PA, palmitic acid; qPCR, quantitative real‐time PCR; s‐XBP1, spliced X‐box‐binding protein 1.
To explore if GEC‐secreted HGF acts in a paracrine fashion on tubular epithelial cells to exert biological functions, a coculture system with HK‐2 cells in the presence of MSCs was set up (Fig. 6G). PA but not BSA stimulated HGF synthesis by GECs predominantly in the apical direction (Fig. 6H). In addition, PA‐induced ER stress, inflammation and apoptosis in HK‐2 cells were significantly alleviated under the triple coculture setting (Fig. 6I–6M), and this favorable effect was partially abrogated by neutralizing anti‐HGF antibody (Fig. 6I–6M).
Recombinant HGF Alleviated PA‐Induced ER Stress, Inflammation, and Apoptosis in HK‐2 Cells
Pretreatment with exogenous HGF dose‐dependently restored viability (Fig. 7A), inhibited ROS generation (Fig. 7B), reduced mRNA upregulation of BiP, EDEM, CHOP, and s‐XBP1 (Fig. 7C–7F), diminished p‐eIF2α, ATF4, and p‐IRE1α synthesis (Fig. 7G–7J) in HK‐2 cells after PA stimulation. PA‐induced pro‐inflammatory (IL6, VEGF) and pro‐apoptotic transcripts (BAK) and phosphorylation of NF‐κB, ERK, and JNK were downregulated by HGF pretreatment (Fig. 7K–7Q).
Figure 7.
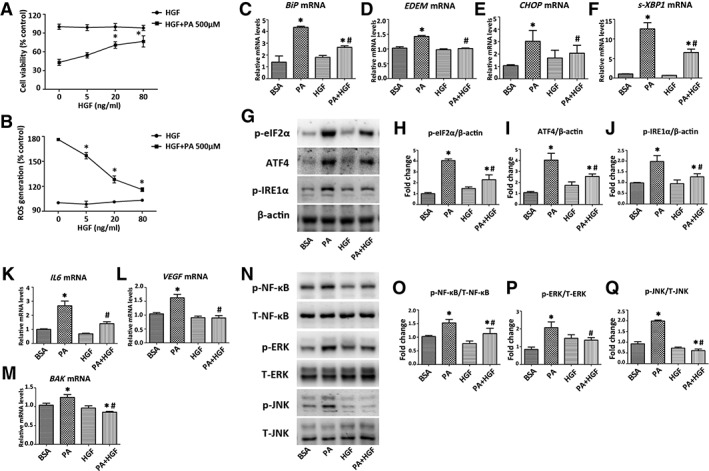
HGF alleviates PA‐induced ER stress, inflammation and apoptosis in HK‐2 cells. HK‐2 cells pretreated with 0–80 ng/ml HGF overnight were stimulated with or without PA (500 μM) for another 24 hours, and (A) cell viabilities and (B) ROS production were determined. HK‐2 cells pretreated with or without HGF (20 ng/ml) overnight were stimulated with PA (500 μM) for 24 or 48 hours for studying mRNA or protein expression. (C–F): Transcripts in HK‐2 cells were determined by qPCR. (G–J): Proteins expression of p‐eIF2α, ATF4, and p‐IRE1α were determined by Western blot. Transcripts of (K, L) inflammatory and (M) apoptotic mediators in HK‐2 cells were determined by qPCR. (N–Q): Western blots for proteins expression of p‐NF‐κB, T‐NF‐κB, p‐ERK, T‐ERK, p‐JNK, and T‐JNK are presented. Results are expressed as mean ± SD. Experiments were performed in triplicate. *, p < .05 versus BSA group; #, p < .05 versus PA group. Abbreviations: ATF4, activating transcription factor 4; BAK, Bcl‐2‐antagonist killer; BiP, binding immunoglobulin protein; CHOP, C/EBP homologous protein; BSA, bovine serum albumin; EDEM, endoplasmic reticulum degradation‐enhancing α‐mannosidase‐like protein; HGF, hepatocyte growth factor; IL6, interleukin 6; p‐eIF2α, phosphorylated eukaryotic initiation factor 2α; p‐ERK, phosphorylated extracellular signal‐regulated kinase; p‐IRE1α, phosphorylated inositol‐requiring enzyme 1α; p‐JNK, phosphorylated c‐Jun N‐terminal kinase; p‐NF‐κB, phosphorylated nuclear factor‐κB; PA, palmitic acid; qPCR, quantitative real‐time PCR; ROS, reactive oxygen species; s‐XBP1, spliced X‐box‐binding protein 1; T‐ERK, total extracellular signal‐regulated kinase; T‐JNK, total c‐Jun N‐terminal kinase; T‐NF‐κB, total nuclear factor‐κB; VEGF, vascular endothelial growth factor.
Discussion
We showed for the first time the therapeutic potential of iPS‐MSCs and BM‐MSCs in obesity‐induced kidney injury. To address the mechanism underlying this benefit, we hypothesized that MSCs modulate the paracrine HGF/c‐Met signaling within the obese kidney microenvironment to reduce lipotoxicity. Coculture experiments revealed a novel renoprotective HGF secreting response in GECs prompted by MSCs, which led to HGF/c‐Met activation in tubular epithelial cells to mitigate ER stress. These data suggest that the therapeutic benefit of MSCs in the obese kidney could be attributed, at least in part, to the induction of HGF/c‐Met signaling.
Growing evidences suggest lipotoxicity‐induced ER stress in the pathogenesis of obesity‐related nephropathy 5, 36. Our in vitro and in vivo data are consistent with this observation in that lipotoxicity caused ER stress and activated UPR in both cultured tubular cells and the renal cortex of HFD fed mice at both gene and protein levels. The PERK and IRE1α associated cascades in UPR are the two dominant arms preferentially activated by lipid stress in the kidney, whereas the ATF6 branch appeared relatively insensitive. In addition, overexpression of BiP, EDEM, and s‐XBP1 and the ER‐resident protein folding catalyst PDI indicated that lipid stress resulted in accumulation of nascent proteins in ER lumen of tubular cells. Failing to restore ER homeostasis by inducing BiP, EDEM, and s‐XBP1 to diminish deposition of nascent proteins induced by lipotoxicity, tubular cells overexpress CHOP driven by ATF4 to trigger apoptosis to avoid catastrophic outcomes. This is reflected by our observation of activated pro‐apoptotic signaling in cultured tubular cells and renal cortex, as well as increased TUNEL‐positive cells in kidney sections from obese mice. The link between ER stress and lipid metabolism is also supported by observations in genetically modified PERK/eIF2α/ATF4/CHOP or IRE1α/XBP1 signal transduction in the context of lipid stress in liver and other organs 37, 38, 39, 40.
Inhibiting ER stress has been suggested to preserve kidney function by mitigating inflammation and apoptosis 41, 42. Our data demonstrated that in addition to aggravating ER stress, lipid stress triggered transition of HK‐2 cells into a pro‐inflammatory and pro‐apoptotic phenotype, and promoted HFD‐induced inflammation and apoptosis in renal cortex of obese mice. Of note, overexpression of IL6 and BCL2 was simultaneously observed in PA‐treated HK‐2 cells and renal cortex of obese mice. IL6 is triggered by aberrant ER stress 43, 44, and mechanistically this is likely the result of NF‐κB activation 42, 43, which was also observed here. Equally important, antiapoptotic BCL2 is suppressed by ER stress 45, which represents an imbalance in the BCL2 family of proteins induced by the PERK branch of CHOP and the IRE1α branch of JNK signaling 42, 46, as also observed in the present study. Intriguingly, we observed activation of ERK that is known to counteract ER stress‐induced death 47, suggesting the dynamics of pro‐survival and pro‐apoptosis signaling in tubular cells under lipid stress.
Pharmacological attempts targeting ER stress are still premature 5. iPS‐MSCs may be a superior alternative to BM‐MSCs for MSC‐based therapy 13. Here, we showed that both iPS‐MSCs and BM‐MSCs partially reversed renal pathology and albuminuria through suppression of renal ER stress induced by lipotoxicity. Mechanistically, this effect is mediated through diminished UPR activation, downregulation of Bip, chop, inflammatory genes Il6, chemokines Cxcl1 and Cxcl2, and pro‐apoptotic signals (Bax/Bcl2 ratio) with reversal of Bcl2 gene expression, decreased activation of NF‐κB, JNK kinase and TUNEL‐positive renal cells. Our finding of equivalent efficacy between iPS‐MSCs and BM‐MSCs in ameliorating HFD‐induced kidney injury is in agreement with that observed in other disease models 12, 13, 17.
Although an expanding body of the literature has shown the renotropic function of HGF 18, 19, 23, 24, its mechanism of action remains largely unknown. Here, HGF and c‐Met was found to be selectively upregulated in the glomerular and tubular compartments, respectively, in obese mice infused with MSCs, implying that infused MSCs could foster a microenvironment in which synthesis and secretion of HGF was enhanced in neighboring glomerular cells to counteract HFD‐induced lipotoxicity in tubular cells via a HGF/c‐Met‐dependent pathway. To identify the glomerular cell type responsible for this, we found that HGF was only detected in the supernatants of GECs, but not HMCs nor podocytes, when cocultured with MSCs and that this effect was exaggerated by PA. This finding is in alignment with previously published literature in the ability of GECs 21, 48 and disability of podocytes in generating HGF 24, 49, but shows disagreement in the ability of mesangial cells in producing HGF 50, 51. This inconsistence may be explained by the bluntness of mesangial cells to lipotoxicity, or the possibility of suppressive HGF secretion in mesangial cells by lipotoxicity‐induced transforming growth factor‐β 21, 52. Given that MSCs of different origins could also synthesize HGF 25, 26, 27, we investigated whether MSCs per se could also produce HGF, and found that exposure to PA did not alter HGF gene expression in either iPS‐MSCs or BM‐MSCs, and HGF protein remained undetectable in their supernatants. The discrepancy from previous studies might be due to the different cell types (like BM, adipose tissue or umbilical cord blood) used for study that may comprise heterogeneous populations of stem cells with variable degrees of plasticity. Collectively, our data suggest GECs to be the origin of HGF in the microenvironment of obese kidney after MSC in fusion, akin to lung‐derived MSCs that promoted HGF secretion in fibroblasts to exert antifibrotic benefits 53.
Finally, we confirmed in vitro that HGF secreted by GECs prevented overexpression of BiP, CHOP, s‐XBP1, and IL6 in cocultured HK‐2 cells exposed to PA, and this phenomenon was partially abolished by adding a neutralizing anti‐HGF antibody. Furthermore, pretreating HK‐2 cells with recombinant HGF suppressed ER stress, inflammation, and apoptosis in HK‐2 cells exposed to PA.
Despite extending our current understanding in the therapeutic effect of iPS‐MSCs, several limitations pose the need for further investigation. Firstly, we did not fully dissect the molecular mechanistic insight how either iPS‐MSCs or BM‐MSCs interplay with GECs to augment HGF/c‐Met paracrine signaling. Of particular interest, a growing body of evidence showed that MSCs derived vesicles exert comparable therapeutic effect as intact MSCs themselves 14, strongly challenging our research hypothesis that only through endocrine crosstalk plus physical contact could MSCs promote HGF/c‐Met paracrine signaling in obese kidney microenvironment. Extracellular vesicles (EVs) are extensively involved in cell–cell communication via transferring mRNA, microRNA, protein or other signaling molecules, thus more investigations are warranted to further characterize the role of EV released from MSCs on HGF/c‐Met signaling activation, with the ultimate goal for developing novel therapeutic strategies that harness these mechanisms to obtain more efficient outcomes. An additional limitation of this work is the lack of in vivo and in vitro experiments examining the effects of epigenetic or pharmacologic inhibition of HGF/c‐Met signaling in tubular cells under lipotoxicity injury. Data from these experiments may identify novel insight to tackle lipotoxicity‐induced tubular injury. Finally, although we did observe a significant decrease in weight gain, blood glucose and glomerular injury after MSCs infusion, obviously we cannot attribute these findings solely to attenuated ER stress in tubular cells. MSCs are previously described to improve insulin resistance through attenuating ER stress 54, and HGF/c‐Met signaling pathway reportedly interacts with insulin receptor to improve insulin resistance and energy metabolism 55. These findings might partially help explain the improved body weight and blood glucose in our current study, but considering the complex mechanism linking obesity and renal damage, a better understanding of how MSCs and HGF systemically improve whole‐body metabolic homeostasis in obesity will hopefully strengthen the possibilities for therapeutic application.
Conclusion
This study is the first to demonstrate the comparable renoprotective potential of iPS‐MSCs to BM‐MSCs in lipotoxicity‐induced kidney injury through suppression of ER stress and the associated downstream events by engaging HGF/c‐Met paracrine signaling in the kidney microenvironment. Furthermore, this observation supports the rationale of further exploiting HGF/c‐Met signaling 19, 56 as an adjunctive therapeutic strategy to MSC‐based therapy in protecting the kidney from lipid insult.
Author Contributions
B.L., J.C.K.L., L.Y.Y.C.: conception and design, collection and/or assembly of data, data analysis and interpretation, manuscript writing; W.H.Y., Y.L., S.W.Y.L., W.H.L., K.W.C.: collection and/or assembly of data, data analysis and interpretation; H.F.T.: provision of study material and technical advice; K.N.L., S.C.W.T.: conception and design, financial support, data analysis and interpretation, manuscript revision, final approval of the manuscript.
Disclosure of Potential Conflicts of Interest
The authors indicated no potential conflicts of interest.
Supporting information
Supplemental Table 1 Antibodies catalogue numbers.
Supplemental Table 2 Primers used for quantitative real‐time PCR.
Supplemental Table 3 Metabolic measures at 22 and 26 weeks of age.
Supplemental Table 4 Determination of HGF in culture supernatant.
Acknowledgments
This study was supported by the Health and Medical Research Fund (ref. #03143726, #05163596), the Research Grants Council (C7018‐16G, 17151716, 17119818), and by philanthropic donations from Rita T. Liu SBS of L&T Charitable Foundation, Ltd., Mr. Winston Leung, Mr. K.K. Chan of Hong Kong Concrete Co. Ltd., Ms. Lau Siu Suet, Ms. Jeny Yeung, and HUI Hoy & CHOW Sin Lan Charity Fund and the Family of Mr HUI Ming, and an Endowment Fund established at the University of Hong Kong for the Yu Professorship in Nephrology awarded to S.C.W.T. Part of the results from this study has been submitted for presentation in abstract form at the American Society of Nephrology Kidney Week, 2018, San Diego, CA.
References
- 1. Ng M, Fleming T, Robinson M et al. Global, regional, and national prevalence of overweight and obesity in children and adults during 1980–2013: A systematic analysis for the Global Burden of Disease Study 2013. Lancet 2014;384:766–781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Tanaka Y, Kume S, Araki S et al. Fenofibrate, a PPAR alpha agonist, has renoprotective effects in mice by enhancing renal lipolysis. Kidney Int 2011;79:871–882. [DOI] [PubMed] [Google Scholar]
- 3. Yamamoto T, Takabatake Y, Takahashi A et al. High‐fat diet‐induced lysosomal dysfunction and impaired autophagic flux contribute to lipotoxicity in the kidney. J Am Soc Nephrol 2017;28:1534–1551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Szeto HH, Liu S, Soong Y et al. Protection of mitochondria prevents high‐fat diet‐induced glomerulopathy and proximal tubular injury. Kidney Int 2016;90:997–1011. [DOI] [PubMed] [Google Scholar]
- 5. Cybulsky AV. Endoplasmic reticulum stress, the unfolded protein response and autophagy in kidney diseases. Nat Rev Nephrol 2017;13:681–696. [DOI] [PubMed] [Google Scholar]
- 6. Cnop M, Foufelle F, Velloso LA. Endoplasmic reticulum stress, obesity and diabetes. Trends Mol Med 2012;18:59–68. [DOI] [PubMed] [Google Scholar]
- 7. Wires ES, Trychta KA, Back S et al. High fat diet disrupts endoplasmic reticulum calcium homeostasis in the rat liver. J Hepatol 2017;67:1009–1017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Martinez‐Sanchez N, Seoane‐Collazo P, Contreras C et al. Hypothalamic AMPK‐ER stress‐JNK1 axis mediates the central actions of thyroid hormones on energy balance. Cell Metab 2017;26:212.e212–229.e212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Palomer X, Pizarro‐Delgado J, Barroso E et al. Palmitic and oleic acid: The Yin and Yang of fatty acids in type 2 diabetes mellitus. Trends Endocrinol Metab 2018;29:178–190. [DOI] [PubMed] [Google Scholar]
- 10. Takahashi K, Yamanaka S. A decade of transcription factor‐mediated reprogramming to pluripotency. Nat Rev Mol Cell Biol 2016;17:183–193. [DOI] [PubMed] [Google Scholar]
- 11. Nagaishi K, Mizue Y, Chikenji T et al. Umbilical cord extracts improve diabetic abnormalities in bone marrow‐derived mesenchymal stem cells and increase their therapeutic effects on diabetic nephropathy. Sci Rep 2017;7:8484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Chow L, Johnson V, Regan D et al. Safety and immune regulatory properties of canine induced pluripotent stem cell‐derived mesenchymal stem cells. Stem Cell Res 2017;25:221–232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Wang LT, Jiang SS, Ting CH et al. Differentiation of mesenchymal stem cells from human induced pluripotent stem cells results in downregulation of c‐Myc and DNA replication pathways with immunomodulation toward CD4 and CD8 cells. Stem Cells 2018;36:903–914. [DOI] [PubMed] [Google Scholar]
- 14. Yuan X, Li D, Chen X et al. Extracellular vesicles from human‐induced pluripotent stem cell‐derived mesenchymal stromal cells (hiPSC‐MSCs) protect against renal ischemia/reperfusion injury via delivering specificity protein (SP1) and transcriptional activating of sphingosine kinase 1 and inhibiting necroptosis. Cell Death Dis 2017;8:3200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Winters AA, Bou‐Ghannam S, Thorp H et al. Evaluation of multiple biological therapies for ischemic cardiac disease. Cell Transplant 2016;25:1591–1607. [DOI] [PubMed] [Google Scholar]
- 16. Li X, Michaeloudes C, Zhang Y et al. Mesenchymal stem cells alleviate oxidative stress‐induced mitochondrial dysfunction in the airways. J Allergy Clin Immunol 2018;141:1634.e1635–1645.e1635. [DOI] [PubMed] [Google Scholar]
- 17. Zhu Y, Wang Y, Zhao B et al. Comparison of exosomes secreted by induced pluripotent stem cell‐derived mesenchymal stem cells and synovial membrane‐derived mesenchymal stem cells for the treatment of osteoarthritis. Stem Cell Res Ther 2017;8:64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Wu HJ, Yiu WH, Wong DWL et al. Human induced pluripotent stem cell‐derived mesenchymal stem cells prevent adriamycin nephropathy in mice. Oncotarget 2017;8:103640–103656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Zhou D, Tan RJ, Lin L et al. Activation of hepatocyte growth factor receptor, c‐met, in renal tubules is required for renoprotection after acute kidney injury. Kidney Int 2013;84:509–520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Li Y, Spataro BC, Yang J et al. 1,25‐Dihydroxyvitamin D inhibits renal interstitial myofibroblast activation by inducing hepatocyte growth factor expression. Kidney Int 2005;68:1500–1510. [DOI] [PubMed] [Google Scholar]
- 21. Yo Y, Morishita R, Yamamoto K et al. Actions of hepatocyte growth factor as a local modulator in the kidney: Potential role in pathogenesis of renal disease. Kidney Int 1998;53:50–58. [DOI] [PubMed] [Google Scholar]
- 22. Morisako T, Takahashi K, Kishi K et al. Production of hepatocyte growth factor from human lung microvascular endothelial cells induced by interleukin‐1beta. Exp Lung Res 2001;27:675–688. [DOI] [PubMed] [Google Scholar]
- 23. Dai CS, Saleem MA, Holzman LB et al. Hepatocyte growth factor signaling ameliorates podocyte injury and proteinuria. Kidney Int 2010;77:962–973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Dai C, Yang J, Bastacky S et al. Intravenous administration of hepatocyte growth factor gene ameliorates diabetic nephropathy in mice. J Am Soc Nephrol 2004;15:2637–2647. [DOI] [PubMed] [Google Scholar]
- 25. Lee EJ, Hwang I, Lee JY et al. Hepatocyte growth factor improves the therapeutic efficacy of human bone marrow mesenchymal stem cells via RAD51. Mol Ther 2018;26:845–859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Bartolucci J, Verdugo FJ, Gonzalez PL et al. Safety and efficacy of the intravenous infusion of umbilical cord mesenchymal stem cells in patients with heart failure: A Phase 1/2 Randomized Controlled Trial (RIMECARD Trial [randomized clinical trial of intravenous infusion umbilical cord mesenchymal stem cells on cardiopathy]). Circ Res 2017;121:1192–1204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Chen QH, Liu AR, Qiu HB et al. Interaction between mesenchymal stem cells and endothelial cells restores endothelial permeability via paracrine hepatocyte growth factor in vitro. Stem Cell Res Ther 2015;6:44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Seleznik G, Seeger H, Bauer J et al. The lymphotoxin beta receptor is a potential therapeutic target in renal inflammation. Kidney Int 2016;89:113–126. [DOI] [PubMed] [Google Scholar]
- 29. Saleem MA, O'Hare MJ, Reiser J et al. A conditionally immortalized human podocyte cell line demonstrating nephrin and podocin expression. J Am Soc Nephrol 2002;13:630–638. [DOI] [PubMed] [Google Scholar]
- 30. Wu HJ, Yiu WH, Li RX et al. Mesenchymal stem cells modulate albumin‐induced renal tubular inflammation and fibrosis. PLoS One 2014;9:e90883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Sun YQ, Deng MX, He J et al. Human pluripotent stem cell‐derived mesenchymal stem cells prevent allergic airway inflammation in mice. Stem Cells 2012;30:2692–2699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Zhang J, Lian Q, Zhu G et al. A human iPSC model of Hutchinson Gilford Progeria reveals vascular smooth muscle and mesenchymal stem cell defects. Cell Stem Cell 2011;8:31–45. [DOI] [PubMed] [Google Scholar]
- 33. Lian Q, Zhang Y, Zhang J et al. Functional mesenchymal stem cells derived from human induced pluripotent stem cells attenuate limb ischemia in mice. Circulation 2010;121:1113–1123. [DOI] [PubMed] [Google Scholar]
- 34. Lian Q, Lye E, Suan Yeo K et al. Derivation of clinically compliant MSCs from CD105+, CD24− differentiated human ESCs. Stem Cells 2007;25:425–436. [DOI] [PubMed] [Google Scholar]
- 35. Glastras SJ, Chen H, Teh R et al. Mouse models of diabetes, obesity and related kidney disease. PLoS One 2016;11:e0162131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Hummasti S, Hotamisligil GS. Endoplasmic reticulum stress and inflammation in obesity and diabetes. Circ Res 2010;107:579–591. [DOI] [PubMed] [Google Scholar]
- 37. Suzuki T, Gao JH, Ishigaki Y et al. ER stress protein CHOP mediates insulin resistance by modulating adipose tissue macrophage polarity. Cell Rep 2017;18:2045–2057. [DOI] [PubMed] [Google Scholar]
- 38. Lee AH, Scapa EF, Cohen DE et al. Regulation of hepatic lipogenesis by the transcription factor XBP1. Science 2008;320:1492–1496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Zhang KZ, Wang SY, Malhotra J et al. The unfolded protein response transducer IRE1 alpha prevents ER stress‐induced hepatic steatosis. EMBO J 2011;30:1357–1375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Xiao GZ, Zhang T, Yu SB et al. ATF4 protein deficiency protects against high fructose‐induced hypertriglyceridemia in mice. J Biol Chem 2013;288:25350–25361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Fan Y, Xiao W, Li Z et al. RTN1 mediates progression of kidney disease by inducing ER stress. Nat Commun 2015;6:7841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Zhang K, Kaufman RJ. From endoplasmic‐reticulum stress to the inflammatory response. Nature 2008;454:455–462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Li Y, Schwabe RF, DeVries‐Seimon T et al. Free cholesterol‐loaded macrophages are an abundant source of tumor necrosis factor‐alpha and interleukin‐6: Model of NF‐kappaB‐ and map kinase‐dependent inflammation in advanced atherosclerosis. J Biol Chem 2005;280:21763–21772. [DOI] [PubMed] [Google Scholar]
- 44. Gargalovic PS, Gharavi NM, Clark MJ et al. The unfolded protein response is an important regulator of inflammatory genes in endothelial cells. Arterioscl Throm Vas 2006;26:2490–2496. [DOI] [PubMed] [Google Scholar]
- 45. Heath‐Engel HM, Chang NC, Shore GC. The endoplasmic reticulum in apoptosis and autophagy: Role of the BCL‐2 protein family. Oncogene 2008;27:6419–6433. [DOI] [PubMed] [Google Scholar]
- 46. Marciniak SJ, Yun CY, Oyadomari S et al. CHOP induces death by promoting protein synthesis and oxidation in the stressed endoplasmic reticulum. Genes Dev 2004;18:3066–3077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Gall JM, Wang Z, Bonegio RG et al. Conditional knockout of proximal tubule mitofusin 2 accelerates recovery and improves survival after renal ischemia. J Am Soc Nephrol 2015;26:1092–1102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Patry C, Betzen C, Fathalizadeh F et al. Endothelial progenitor cells accelerate endothelial regeneration in an in vitro model of Shigatoxin‐2a‐induced injury via soluble growth factors. Am J Physiol Renal Physiol 2018;315:F861–F869. [DOI] [PubMed] [Google Scholar]
- 49. Fu J, Lee K, Chuang PY et al. Glomerular endothelial cell injury and cross talk in diabetic kidney disease. Am J Physiol Renal Physiol 2015;308:F287–F297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Li Y, Wen X, Spataro BC et al. Hepatocyte growth factor is a downstream effector that mediates the antifibrotic action of peroxisome proliferator‐activated receptor‐gamma agonists. J Am Soc Nephrol 2006;17:54–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Yo Y, Morishita R, Nakamura S et al. Potential role of hepatocyte growth factor in the maintenance of renal structure: Anti‐apoptotic action of HGF on epithelial cells. Kidney Int 1998;54:1128–1138. [DOI] [PubMed] [Google Scholar]
- 52. Tsai PJ, Chang ML, Hsin CM et al. Antilipotoxicity activity of Osmanthus fragrans and Chrysanthemum morifolium flower extracts in hepatocytes and renal glomerular mesangial cells. Mediators Inflamm 2017;2017:4856095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Hostettler KE, Gazdhar A, Khan P et al. Lung‐derived mesenchymal stem cells induce the secretion of hepatocyte growth factor in fibroblasts. Eur Respir J 2017;50:PA343. [Google Scholar]
- 54. Sun XY, Hao HJ, Han QW et al. Human umbilical cord‐derived mesenchymal stem cells ameliorate insulin resistance by suppressing NLRP3 inflammasome‐mediated inflammation in type 2 diabetes rats. Stem Cell Res Ther 2017;8:241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Fafalios A, Ma JH, Tan XP et al. A hepatocyte growth factor receptor (Met)‐insulin receptor hybrid governs hepatic glucose metabolism. Nat Med 2011;17:U1577–U1587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Herrero‐Fresneda I, Torras J, Franquesa M et al. HGF gene therapy attenuates renal allograft scarring by preventing the profibrotic inflammatory‐induced mechanisms. Kidney Int 2006;70:265–274. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Table 1 Antibodies catalogue numbers.
Supplemental Table 2 Primers used for quantitative real‐time PCR.
Supplemental Table 3 Metabolic measures at 22 and 26 weeks of age.
Supplemental Table 4 Determination of HGF in culture supernatant.