

Abstract
As a contributor to multidrug resistance, the family of multidrug and toxin extrusion (MATE) transporters couples the efflux of chemically dissimilar compounds to electrochemical ion gradients. Although divergent transport mechanisms have been proposed for these transporters, previous structural and functional analyses of members of the MATE subfamily DinF suggest that the N-terminal domain (NTD) supports substrate and ion binding. In this report, we investigated the relationship of ligand binding within the NTD to the drug resistance mechanism of the H+-dependent MATE from the hyperthermophilic archaeon Pyrococcus furiosus (PfMATE). To facilitate this study, we developed a cell growth assay in Escherichia coli to characterize the resistance conferred by PfMATE to toxic concentrations of the antimicrobial compound rhodamine 6G. Expression of WT PfMATE promoted cell growth in the presence of drug, but amino acid substitutions of conserved NTD residues compromised drug resistance. Steady-state binding analysis with purified PfMATE indicated that substrate affinity was unperturbed in these NTD variants. However, exploiting Trp fluorescence as an intrinsic reporter of conformational changes, we found that these variants impaired formation of a unique H+-stabilized structural intermediate. These results imply that disruption of H+ coupling is the origin of compromised toxin resistance in PfMATE variants. These findings support a model mechanism wherein the NTD mediates allosteric coupling to ion gradients through conformational changes to drive substrate transport in PfMATE. Furthermore, the results provide evidence for diverging transport mechanisms within a prokaryotic MATE subfamily.
Keywords: drug resistance, transporter, cell growth, protein expression, fluorescence, allostery, ion-coupled substrate transport, multidrug and toxin extrusion (MATE)
Introduction
Multidrug resistance has been linked in part to the expression of integral membrane transporters that actively remove cytotoxic molecules from the cell (1). Five multidrug transporter families contribute to this mode of resistance, collectively displaying a remarkable diversity of efflux mechanisms but possessing overlapping substrate specificities that include antibiotics and anticancer agents (2–4). As a consequence, the widespread distribution of these transporters in prokaryotes and eukaryotes presents a serious clinical challenge for successful pharmacological treatment of bacterial infections and cancers (1, 5–9).
Found in all three domains of life (Archaea, Bacteria, and Eukarya), the family of multidrug and toxin extrusion (MATE)2 transporters mediates export of several classes of antimicrobials, chemotherapeutic compounds, and organic cations by coupling uphill transport to the Gibbs energy stored in the Na+ and/or H+ electrochemical gradients (7, 10–16). Further classified as the NorM, DinF, and eukaryotic subfamilies based on the protein sequence, crystal structures of MATEs from these subfamilies reveal a 12-transmembrane (TM) helix architecture organized into separate N- and C-terminal domains (NTDs and CTDs, respectively) related by 2-fold pseudosymmetry in the membrane plane (17–25). In the context of a presumed alternating access mechanism (26, 27), the orientation of the NTD and CTD relative to a bilayer suggests that these structures adopt outward-facing conformations (28).
Although inward-facing states have proved recalcitrant so far, crystal structures with various bound substrates have identified unique interaction motifs implying divergent transport mechanisms between MATE subfamilies (19, 20, 23, 24, 28). Tetraphenylphosphonium, ethidium, and rhodamine 6G (R6G) were found to bind a negatively charged central cavity formed by acidic and polar residues from the NTD and CTD near the membrane–water interface in the Na+-coupled transporter NorM from Neisseria gonorrhoeae (20). Ligand-dependent conformational changes were predicted to facilitate an allosteric coupling transport mechanism (20, 29). In contrast, R6G was found buried within the hydrophobic TM core of H+-coupled DinF from Bacillus halodurans (DinF-BH), in which the majority of contacts are made with NTD residues, including a conserved Asp in TM1 that mediates the only charge–charge interaction with the substrate (19). Disruption of this interaction by substitution (D40N) or protonation of the Asp impaired R6G binding, suggesting an overlapping H+/R6G binding site. Consequently, H+ binding was implicated in drug release by directly competing with the substrate binding site (19).
Despite these fundamental differences between subfamilies, mechanistic diversity within the DinF subfamily has been the subject of controversy. Crystallographic analyses of DinF from the archaeon Pyrococcus furiosus (PfMATE) not only identified a distinct drug (norfloxacin)–binding site deep within the NTD but also a “bent” conformation of TM1 associated with rearrangement of a hydrogen bond network putatively induced by protonation of the conserved Asp-41 (24). A similar conformation of TM1 has yet to be observed in DinF-BH. Thus, in combination with complementary functional analysis, the structures of PfMATE supported a transport model distinct from DinF-BH, invoking a critical role of the NTD in facilitating H+-dependent conformational changes in TM1 that collapse the norfloxacin binding cavity. Similar structural changes have been described recently for another bacterial MATE, arguing that the bent conformation of TM1 is a conserved obligatory intermediate in the transport cycle (18).
Here we explore the function of conserved residues previously implicated in H+ and drug binding within the DinF subfamily to define mechanistic features of the NTD that support drug resistance in PfMATE. Our multifaceted approach incorporates a cell growth assay tailored to uncover PfMATE-mediated resistance to toxic concentrations of R6G in combination with steady-state drug binding and structural analysis. We show that, unlike DinF-BH, high-affinity binding of R6G to PfMATE does not require the charged side chain contributed by a strictly conserved TM1 Asp. However, R6G resistance is compromised by substitution of NTD residues that disrupt formation of an H+-stabilized structural intermediate. The results highlight a role of the NTD in mediating ion coupling to transport through allostery but support the notion of unique drug-binding sites and divergent transport mechanisms within the same subfamily of MATE transporters.
Results
R6G resistance assay: approach and design
Expression of PfMATE promotes cell growth in the presence of the fluoroquinolone antibiotic norfloxacin (24), but resistance to R6G toxicity has not been demonstrated previously. To characterize PfMATE-mediated R6G resistance in vivo, we adapted a cell growth assay used previously to probe the survival of Escherichia coli expressing the Na+/H+-dependent homolog NorM from Vibrio cholerae (NorM-Vc) when subjected to toxic concentrations of doxorubicin (30, 31). This assay was shown to discriminate between variants of NorM-Vc that support or compromise cell growth, facilitating the correlation of apparent drug resistance to ion/drug-dependent conformational dynamics associated with the transport mechanism (30). Similarly, expression of functional PfMATE is expected to increase cell survival in the presence of toxic substrates.
The experimental paradigm is illustrated as a flow diagram in Fig. 1, and additional details are provided under “Experimental procedures.” Briefly, a small seed culture grown overnight from freshly transformed BL21 (DE3) cells was used to inoculate a larger volume of growth medium with a target absorbance at 600 nm (A600 nm) of 0.0375. The cells were allowed to grow for up to 2 h at 37 °C and were then diluted to A600 nm ∼0.25 and induced with IPTG. Following a 2-h incubation period for protein production, the cells were seeded into a 96-well plate containing growth medium and the substrate R6G. Cell growth was monitored by A650 nm, which was measured by a plate reader over a defined period of time. This wavelength (650 nm) was chosen to minimize the contribution of R6G to the overall absorbance at high (>30 μg/ml) R6G concentrations (Fig. S1). The plate format allowed screening of numerous conditions as a function of time, such as IPTG and drug concentrations. Importantly, cell growth in the absence of drug was incorporated as a control for cell health and viability over the course of the assay, as described below. Therefore, A650 nm in the absence of drug was used as a normalization factor to obtain relative A650 nm.
Figure 1.

Flow diagram of the R6G resistance assay. A, comparison of growth for cells harboring the vector (gray line) and WT PfMATE (black line) after 2-h induction with different IPTG concentrations. B, expression of WT PfMATE increases cell survival at elevated R6G concentrations relative to the vector control. Each data point represents the average of two independent experiments. For each experiment, the data were measured in triplicate from separate wells on the plate after 10 h at 37 °C. A650 nm in the presence of R6G was normalized to the 0 μg/ml R6G well. The standard deviation is shown for each data point. The p values were determined by an unpaired t test: **, p = 0.004–0.009; ***, p < 0.0001. C, cells expressing WT PfMATE demonstrate better growth than the vector control at all time points. A650 nm in the presence of R6G was normalized to the 0 μg/ml R6G well. The R6G concentration was 75 μg/ml. The data points are shown with the standard deviation, and each data point was generated from three to six independent experiments as described for B. D, the relative expression of WT PfMATE as a function of IPTG concentration was visualized by SDS-PAGE and staining with InVision His tag stain. Purified PfMATE WT used as a standard is shown for comparison. Vec, vector.
R6G resistance assay: identification of PfMATE expression parameters
Standardization of this assay for screening activity of PfMATE required careful consideration of key protein expression parameters. Cell growth after induction is strongly dependent on the IPTG concentration. In general, cells expressing PfMATE WT grew less than cells harboring vector alone (pET-19b) at all IPTG concentrations tested (Fig. 1A). This pattern of cell growth was inversely correlated with WT expression levels, as visualized by SDS-PAGE analysis, especially in the range of 10–50 μm IPTG, which induced robust expression (Fig. 1D). Furthermore, cells expressing the WT displayed reduced growth relative to the vector in the absence of drug over the course of the assay (Fig. S2A). These observations suggested that WT expression is metabolically challenging and/or toxic to the cell. We thus reasoned that high-level WT expression may reduce the cell capacity to proliferate under additional toxic stress and obfuscate PfMATE-mediated drug resistance. In support of this conclusion, we observed an increase in the growth of cells expressing the WT at elevated R6G concentrations (≥60 μg/ml) relative to vector alone upon induction with 1 μm IPTG (Fig. 1B and Fig. S2B). Enhanced cell survival was seen over the entire time course of the assay at 75 μg/ml R6G after normalizing to growth in the absence of drug (Fig. 1C). The ratio of A650 nm indicated that expression of the WT increased cell growth by 53% ± 9% on average (n = 6) relative to vector at the 10-h time point, where the normalized growth curve began to plateau. In contrast, cell growth was greatly impaired at the identical R6G concentration following induction with 10 μm IPTG (Fig. S3).
In addition to IPTG concentration, potentiation of cell survival was contingent on a full complement of multidrug transporters. Previous applications of this assay with NorM-Vc utilized an E. coli strain devoid of seven endogenous multidrug transporters (BL21 (DE3)-Δ7: macAB, yojHI, acrAB, acrEF, emrAB, emrKY, and mdtEF) (30, 32). However, PfMATE-mediated resistance to R6G toxicity was not observed in this background or in the BL21 (DE3)-Δ3 strain (deletion of macAB, yojHI, and acrAB) relative to vector alone under similar expression conditions (Fig. S4). This result suggested that the elevated R6G resistance conferred by expression of WT PfMATE involved concerted activity of intrinsic transporters.
Amino acid substitutions in the NTD of PfMATE compromise resistance to R6G
Previous crystallographic, computational, and functional studies of PfMATE suggested a critical role of the NTD in binding of target substrates and coupling to the H+ gradient to drive transport (24, 33). In conjunction with a highly conserved Pro (Pro-26 in PfMATE) in TM1, a hydrogen bond network composed of residues that line a cavity in the NTD was proposed to mediate a H+-dependent conformational change in TM1 that facilitates substrate extrusion. Sequence alignment and phylogenetic analysis indicated that these residues are highly conserved (34, 35) (Fig. S5). Consistent with this result, substitution of these residues impaired resistance to norfloxacin and transport of ethidium (24). We tested a subset of these variants to ascertain the contribution of these residues to R6G resistance (Fig. 2A).
Figure 2.

R6G resistance profiles for NTD residue substitutions. A, structure of PfMATE (PDB code 3VVN), highlighting the location of residues chosen for site-directed mutagenesis in the NTD (gray). The CTD is shown in cyan. B, time course of cell growth for the variants relative to both the vector and WT at 37 °C in the presence of 75 μg/ml R6G. The curves were generated from at least three independent experiments as described under “Experimental procedures” and in the legend for Fig 1. A650 nm in the presence of R6G was normalized to the 0 μg/ml R6G well. The standard deviation is shown for each data point. C, the data in B at the 10-h time point were transformed into a cell growth profile relative to WT PfMATE after subtracting the contribution of the vector control. The bar plot highlights the average ± S.D. for the indicated number of measurements. One-way ANOVA indicated that the population means were significantly different at the 0.05 level: F (6, 27) = 28.82, p = 1.57 × 10−10. D, SDS-PAGE followed by InVision His tag staining confirmed similar levels of expression for each construct. The last three lanes on each gel image show purified PfMATE WT used as a standard. Vec, vector.
Site-directed mutagenesis of NTD residues (P26A, Y37A, D41A/D41N, and D184A) significantly impaired cell growth relative to the WT in the presence of 75 μg/ml R6G and instead clustered with the vector control (Fig. 2B and Fig. S2, C and D). In contrast, substitution of a nearby nonconserved surface Glu in TM2 (E51A), designed as a control, demonstrated a similar pattern of growth as the WT. Capturing the pattern of PfMATE-mediated R6G resistance, a growth profile of the variants relative to the WT (Fig. 2C) indicated that substitution of functionally required residues reduced cell growth by ∼80% or more. Importantly, the apparent loss of functional activity could not be attributed to reduced expression levels of the variants (Fig. 2D and Fig. S6), and size exclusion chromatography analysis of the purified variants was consistent with folded transporters (Fig. S7).
NTD variants retain high-affinity R6G binding
We explored the mechanistic basis of compromised R6G resistance by two approaches. First, we investigated the role of targeted residues in R6G binding. Formation of a complex between R6G and PfMATE purified in β-dodecyl maltoside (β-DDM) micelles was monitored by R6G fluorescence anisotropy. Binding of drug to PfMATE was associated with an increase in anisotropy, similar to the change in fluorescence polarization that was observed upon binding to DinF-BH (19). Titration of R6G with increasing concentrations of PfMATE generated a binding isotherm that was fit with a single-site binding model to determine the KD (Fig. 3). The binding curve collected at pH 7.5 indicated that R6G was bound to WT PfMATE with high affinity. A subtle right shift in the curve was observed at pH 4, corresponding to a less than 2-fold increase in KD (Table 1). Notably, introduction of the NTD variants, including D41A/D41N, only marginally perturbed R6G affinity at pH 7.5 relative to the WT. Binding analysis of the E51A variant reported the largest deviation from the WT, but it retained WT-like resistance activity (Fig. 2C). These results therefore suggested that compromised resistance is not likely to be the consequence of impaired substrate binding.
Figure 3.
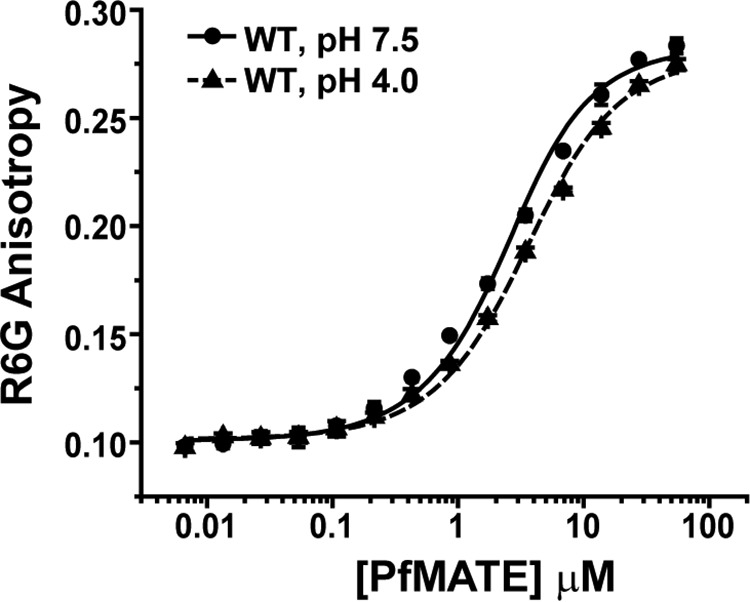
Binding curves of R6G to WT PfMATE. Changes in the fluorescence anisotropy of R6G were plotted as a function of protein concentration in β-DDM buffer at the indicated pH and 23 °C. The data points represent the average ± S.D. of three binding experiments, and the solid and dashed lines are nonlinear least square fits of the curves to obtain the KD (Table 1).
Table 1.
R6G Binding Affinities
| Construct | KD (± S.D.) | n |
|---|---|---|
| μm | ||
| Wildtype, pH 7.5 | 1.59 (0.09) | 6 |
| Wildtype, pH 4 | 2.67 (0.17) | 3 |
| P26A, pH 7.5 | 1.74 (0.07) | 3 |
| Y37A, pH 7.5 | 1.29 (0.26) | 3 |
| D41A, pH 7.5 | 2.06 (0.22) | 3 |
| D41N, pH 7.5 | 1.98 (0.02) | 3 |
| E51A, pH 7.5 | 2.24 (0.10) | 3 |
| D184A, pH 7.5 | 1.98 (0.06) | 3 |
NTD variants disrupt H+-driven conformational changes
Employed as an environmentally sensitive probe, the pattern of Trp fluorescence in transporters has been shown previously to be an effective spectroscopic tool to monitor conformational changes (36–38). PfMATE contains five endogenous Trp residues, two of which are found in the NTD. One of these, Trp-216, is located more than 20 Å away from the NTD cavity and near the intracellular side of TM6. The other Trp residue, Trp-44, is located near the C terminus of TM1, in close proximity to the network of hydrogen-bonded side chains that has been proposed to facilitate TM1 bending (Fig. 4A). Thus, we exploited intrinsic Trp fluorescence as a surrogate reporter of conformational changes induced by H+.
Figure 4.
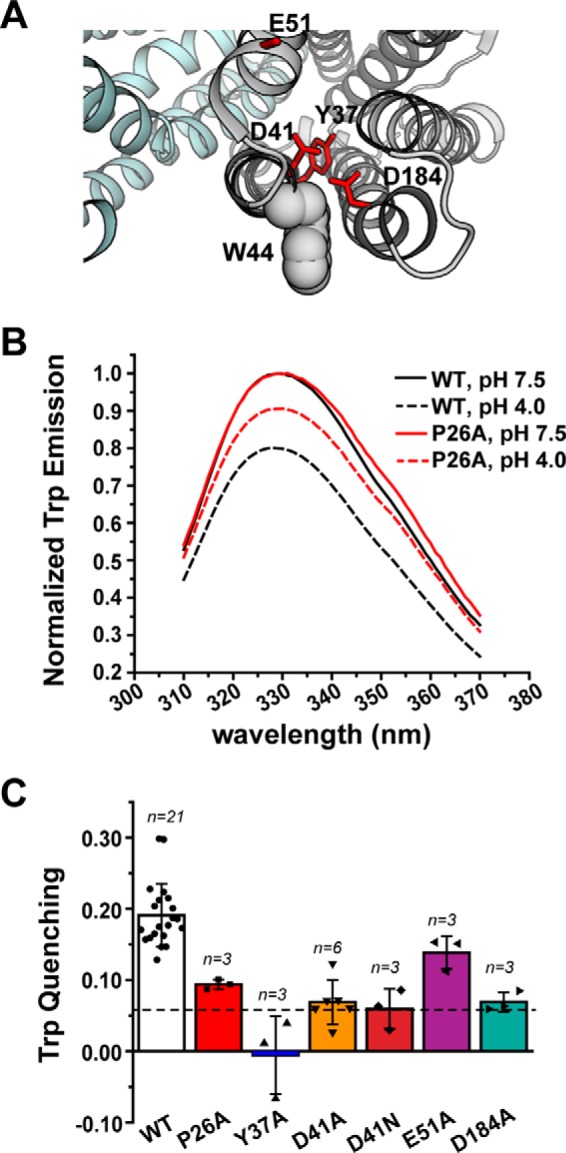
NTD amino acid substitutions disrupt the pattern of H+-dependent Trp quenching. A, structure of PfMATE (PDB code 3VVN), illustrating the location of Trp-44 (shown in space-filling representation) relative to the NTD variants. B, Trp fluorescence is quenched for WT PfMATE at pH 4 (black dashed trace), which is reduced by introduction of P26A (red dashed trace). The P26A spectra are shown as a representative dataset. Spectra were acquired at 23 °C in β-DDM buffer. C, profile of Trp quenching for the NTD variants. The bar plot highlights the mean ± S.D. for the indicated number of measurements. One-way ANOVA indicated that the population means were significantly different at the 0.05 level: F (6, 35) = 20.29, p = 4.53 × 10−10. The horizontal dashed line indicates the quenching observed for the W44C variant. The color code is the same as in Fig. 2.
Fluorescence spectra of WT PfMATE in β-DDM micelles reported an ∼20% reduction in Trp emission intensity at pH 4 relative to pH 7.5 (Fig. 4B), which we attributed to increased solvent exposure of buried Trp side chain(s) at low pH. Mutation of Trp-44 reduced fluorescence quenching to ∼6%, although R6G binding and resistance activity remained relatively unaffected (data not shown), suggesting that Trp-44 contributed to the majority of the signal change in WT PfMATE.
With the exception of E51A, pH-dependent Trp quenching was attenuated substantially in the NTD variants (Fig. 4, B and C). The quenching observed for D41A, D41N, and D184A was reduced to similar levels as the W44C variant. Although quenching was less attenuated for P26A and mostly limited for Y37A, the observed reduction in Trp quenching was consistent with impaired formation of a distinct conformation. Remarkably, the overall quenching profile shown in Fig. 4C echoed the R6G resistance profile shown in Fig. 2C. Thus, the pattern of quenching strongly implied that substitution of relevant residues in the NTD disrupted H+-dependent structural transitions likely associated with the transport cycle.
Discussion
A fundamental property of MATE transporters is the ability to bind and export a broad range of chemically diverse substrates. In addition to H+-dependent ethidium efflux, PfMATE has been described previously to promote bacterial growth in the presence of the fluoroquinolone antibiotic norfloxacin (24). In support of a role in multidrug transport, the results presented here establish that PfMATE also confers enhanced resistance against toxic concentrations of the antimicrobial R6G. Furthermore, R6G resistance is compromised by mutation of residues predicted to be required for transport.
Importantly, PfMATE-mediated resistance to R6G toxicity was observed in E. coli under specific expression conditions. Manipulation of the [IPTG] used for induction was required to balance the metabolic challenge of transporter expression in the host, with the conferred advantage of exporting a toxic substrate present at high concentrations in the growth medium. Moreover, the lack of R6G resistance in hypersensitive E. coli strains supports the notion of coordinated drug efflux mediated by multiple transporters sharing overlapping substrate specificities (39). In this context, PfMATE likely bolsters R6G resistance by integrating with other R6G transport systems, such as the tripartite AcrAB–TolC assembly (40, 41), which was deleted in the BL21 (DE3)-Δ3 and -Δ7 strains (32). According to this model, complete removal of the hydrophobic substrate engenders PfMATE-mediated extrusion into the periplasm, followed by export across the outer membrane by the AcrAB–TolC complex. Thus, the data suggest that heterologous expression of PfMATE in E. coli cannot fully compensate for the loss of major multidrug transporters under the conditions described here.
Combining the cell growth assay with site-directed mutagenesis indicated that R6G resistance depends on the integrity of a conserved network of polar side chains lining a cavity in the NTD. Among these residues, a strictly conserved TM1 Asp has been proposed to form part of the R6G binding site in the homolog DinF-BH (19). Accordingly, R6G binding affinity was reduced by more than 30-fold in a DinF-BH variant predicted to mimic protonation of the carboxylate moiety (D40N). This result was interpreted as evidence for a mutually exclusive H+/drug-binding site, the hallmark of a direct competition mechanism. However, substantial perturbation of R6G binding affinity was not observed with homologous variants in PfMATE (D41A/D41N). Furthermore, the marginal reduction in binding affinity at low pH implies that PfMATE-R6G interactions are not mediated directly by charged side chains that undergo protonation/deprotonation events. These results strongly suggest that R6G is stabilized by distinct binding motifs in PfMATE and DinF-BH. That is, the H+ and R6G binding sites appear to be nonoverlapping in PfMATE.
In contrast to the binding analysis, substitution of functionally required NTD residues altered the pattern of H+-dependent Trp quenching relative to the WT, which we interpret as impaired formation of a unique structural intermediate. Previous crystallographic and computational studies have demonstrated that the conserved residues of the NTD cavity investigated here contribute to an ion-binding site (24, 33, 42). As a reflection of the predicted involvement of these residues in the structural dynamics of TM1, the observed pattern of Trp quenching in the NTD variants highlights the role of conserved side chains in shaping H+-driven conformational changes in PfMATE. Y37A, D41A, D41N, and D184A likely disrupt rearrangement of the hydrogen bond network induced by H+ binding, effectively inhibiting conformational changes. Although not directly involved in H+ binding, Pro-26 in conjunction with Gly-24 was hypothesized to form a hinge in TM1 (24). By extension, P26A may impair TM1 bending in response to H+ binding within the NTD cavity.
Based on the apparent correlation between Trp quenching of NTD variants (Fig. 4C) and the cell growth profile (Fig. 2C), we propose that this residue network is critical to mediate coupling of ion gradients through conformational changes that allosterically modulate substrate binding affinity to trigger substrate release. This conclusion is supported by spectroscopic analysis of NorM-Vc, which revealed the significance of a similar network of residues within the NTD cavity in determining ion-driven conformational dynamics central to drug resistance (30). Experiments that further define the nature and magnitude of ligand-dependent conformational changes and the relationship to R6G binding and resistance in PfMATE are ongoing.
Together, the functional and biochemical analysis of PfMATE emphasizes both common and diverging mechanistic principles of ion-coupled substrate transport in the MATE family. The evidence presented here supports an emerging transport model that envisions ion coupling mediated by specific residues in the NTD to induce conformational changes that culminate in substrate extrusion. However, differences in binding sites for shared substrates of MATE homologs may reflect evolution of transport mechanisms. Subsequent studies with other substrates and transporters are warranted to further uncover shared and unique determinants of MATE transport mechanisms.
Experimental procedures
Site-directed mutagenesis
WT PfMATE was cloned into the pET-19b vector encoding an N-terminal His10 tag under control of an inducible T7 promoter. Mutations were generated using a single-step PCR in which the entire plasmid was replicated from a single mutagenic primer. The template plasmid was subsequently digested by DpnI. Plasmids were propagated using XL-1 Blue or DH5α cells and sequenced using both T7 forward and reverse primers to confirm mutagenesis and the absence of aberrant changes in the protein-coding region.
Expression and purification of PfMATE
C43 (DE3) cells were freshly transformed with pET-19b encoding WT or mutant PfMATE. A single colony was used to inoculate 40 ml of Luria-Bertani (Miller formulation) medium, which was grown overnight (∼15 h) at 34 °C and subsequently used to inoculate 2 liters of minimal medium A. Cultures were incubated at 37 °C with shaking until reaching an A600 nm of ∼0.8, and then the expression of PfMATE was induced by addition of 1 mm IPTG. The cultures were incubated overnight (∼15 h) at 20 °C and then harvested. Cell pellets were resuspended in 20 ml of lysis buffer (20 mm Tris-HCl (pH 8), 20 mm NaCl, 30 mm imidazole, and 10% (v/v) glycerol), including 10 mm DTT, and lysed by five passes through an Avestin C3 homogenizer. Cell debris was removed by centrifugation at 9,000 × g for 10 min. Membranes were isolated from the supernatant by centrifugation at 200,000 × g for 1.5 h. Membrane pellets were resuspended in lysis buffer containing 1.5% (w/v) β-DDM and 0.5 mm DTT and incubated on ice while being stirred for 1 h. Insoluble material was cleared by centrifugation at 200,000 × g for 30 min. The cleared extract was bound to 1.0 ml (bed volume) nickel-nitrilotriacetic acid Superflow resin (Qiagen) at 4 °C for 2 h. After washing with 10 bed volumes of buffer containing 30 mm imidazole, PfMATE was eluted with buffer containing 300 mm imidazole. Elution fractions (4 ml total) were combined and then concentrated to 2.0 ml in a 100,000 molecular weight cut off filter concentrator (Millipore) and injected in 1-ml samples onto a Superdex 200 Increase 10/300 GL column (GE Healthcare) equilibrated with 50 mm Tris/MES (pH 7.5) and 0.05% β-DDM. Peak fractions of purified PfMATE were combined and concentrated using a 100,000 MWCO filter concentrator, and the final concentration was determined by A280 measurement (ϵ = 46870 m−1·cm−1).
R6G binding assay
Stock solutions of rhodamine 6G (Acros) were made in ultrapure water. Drug concentrations for assays were determined by spectrophotometer measurement at 524 nm (ϵ = 116,000 M−1·cm−1) of samples diluted in ethanol. R6G (2.1 μm) was mixed with increasing concentrations of PfMATE WT or variants in 50 mm Tris/MES (pH 7.5) and 0.05% β-DDM buffer in a total volume of 25 μl in a 384-well black fluorescence microplate (Greiner Bio-One) and incubated at room temperature for more than 5 min. R6G fluorescence anisotropy was measured using a BioTek Synergy H4 microplate reader with a 480-nm excitation filter (20-nm band pass) and a 570-nm emission filter (10-nm band pass) (2). Binding isotherms were measured in triplicate, and R6G binding affinity was determined by nonlinear least square analysis in the program Origin (OriginLab). The average KD and standard deviation for each mutant are reported in Table 1.
Tryptophan fluorescence
Purified PfMATE in 50 mm Tris/MES (pH 7.5) and 0.05% β-DDM buffer was adjusted to pH 4 using an empirically determined volume of 1 m citric acid. Samples at pH 7.5 were adjusted with an equivalent volume of buffer to maintain an equal concentration of protein between pH conditions. Samples were placed in a 1-cm quartz fluorometer cell (Starna Cells, Inc.), and tryptophan fluorescence was measured using a T-format fluorometer (Photon Technology International) with excitation and emission slit widths of 4 nm and 1 nm, respectively. The fluorescence spectrum was acquired from 310 to 370 nm following excitation at 295 nm. Spectra were normalized to the peak intensity (329 nm) of the pH 7.5 sample to determine the extent of H+-dependent quenching. Experiments were repeated at least in triplicate, and the mean ± S.D. of fluorescence quenching was determined. To statistically assess the impact of substitutions on Trp quenching, a one-way ANOVA conducted in Origin determined that the population means were significantly different at the 0.05 level.
R6G resistance assay
Resistance to R6G toxicity was carried out as described previously with several modifications. E. coli BL21 (DE3) cells were transformed with empty pET-19b vector, pET19b encoding PfMATE WT, or variants. A dense overnight culture from a single colony was used to inoculate 12 ml of LB broth (Fisher Scientific) containing 0.1 mg/ml ampicillin (Gold Biotechnology) to a starting A600 nm of 0.0375. Cultures were grown for an additional 1.5–2 h at 37 °C and then diluted to A600 nm of 0.25. Expression of the encoded construct was induced with 1–50 μm IPTG (Gold Biotechnology). Expression was allowed to continue at 37 °C for 2 h, and then the A600 nm of the cultures was adjusted to 0.5. The cells were then used to inoculate (1:20 dilution, starting A600 nm = 0.025) a sterile 96-well microplate (Greiner Bio-One) containing 50% LB broth, 0.1 mg/ml ampicillin, and R6G. IPTG was not included on the microplates. Microplates were incubated at 37 °C with shaking at ∼250 rpm for at least 10 h, and cell growth was monitored by A650 nm on a BioTek Synergy H4 microplate reader. Data points were collected every 2 h. The A650 nm in the presence of R6G was normalized to the 0 μg/ml R6G well to obtain a relative A650 nm, which accounts for growth behavior of the vector, WT, and variants in the absence of drug (Fig. S2). Screening of [IPTG] indicated that 1 μm IPTG resulted in optimal resistance, which was used for all subsequent experiments. Because of the narrow dynamic range to observe enhanced R6G resistance (Fig. 1B), each experiment included a vector and WT control. The p values in Fig. 1B were determined by an unpaired t test (GraphPad), indicating that the population means of cell growth for the WT and vector were significantly different at the indicated R6G concentrations. The time course of cell growth for the WT and variants relative to vector was determined at 75 μg/ml R6G. The relative activity of each variant was determined by subtracting the A650 nm of the vector, followed by normalization to the corrected A650 nm of the WT at the 10-h time point. Each data point was acquired from triplicate measurements in three distinct wells on the plate, and the experiment was repeated at least three times to obtain the mean ± S.D. To statistically assess the impact of the substitutions on relative cell growth, a one-way ANOVA conducted in Origin determined that the population means were significantly different at the 0.05 level.
To confirm PfMATE expression, 50 ml of cells harboring the vector, WT, and PfMATE variants was induced with 1 μm IPTG for 2 h at 37 °C and harvested by centrifugation. Cells were resuspended in 1 ml of lysis buffer (20 mm Tris-HCl, 20 mm NaCl, and 10% (v/v) glycerol (pH 8)) with 2.0 mm PMSF and lysed by sonication (60 1-s pulses with 10-s resting intervals). Cell lysates were centrifuged at 3,400 × g for 8 min to remove cell debris. The supernatant was subsequently ultracentrifuged for 30 min at 70,000 × g to obtain the membrane fraction. PfMATE was extracted from a normalized amount of membrane mass in 200 μl of lysis buffer supplemented with 2% (w/v) β-DDM for 1 h at 4 °C, and then the samples were ultracentrifuged at 90,000 × g to clear insoluble material. The supernatant was then mixed with 40 μl (bed volume) of nickel-nitrilotriacetic acid Superflow resin (Qiagen) for 1 h at 4 °C in the presence of 20 mm imidazole. The resin was then applied to a microspin column (Bio-Rad, Bio-spin), and the resin was washed with eight bed volumes of buffer containing 30 mm imidazole. The protein was eluted with 50 μl of buffer containing 300 mm imidazole, and equal-volume samples for SDS-PAGE were prepared from the eluates. Following electrophoresis on a 12.5% acrylamide gel, the protein was visualized by InVision His tag gel stain (Novex).
Author contributions
K. L. J. and D. P. C. data curation; K. L. J. and D. P. C. formal analysis; K. L. J. and D. P. C. investigation; K. L. J., H. S. M., and D. P. C. methodology; K. L. J., H. S. M., and D. P. C. writing-review and editing; H. S. M. and D. P. C. conceptualization; H. S. M. resources; H. S. M. supervision; H. S. M. funding acquisition; D. P. C. writing-original draft.
Supplementary Material
Acknowledgments
We thank Dr. Richard Stein, Dr. Smriti Mishra, Dr. Micheal Leser, and Dr. Brinda Selvaraj for a critical reading of the manuscript and helpful discussions.
This work was supported by NIGMS, National Institutes of Health Grant R01 GM077659 (to H. S. M.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article contains Figs. S1–S7.
- MATE
- multidrug and toxin extrusion
- TM
- transmembrane
- NTD
- N-terminal domain
- CTD
- C-terminal domain
- R6G
- rhodamine 6G
- DinF-BH
- MATE transporter from B. halodurans
- PfMATE
- MATE transporter from P. furiosus
- β-DDM
- β-dodecyl maltoside
- IPTG
- isopropyl 1-thio-β-d-galactopyranoside
- ANOVA
- analysis of variance.
References
- 1. Higgins C. F. (2007) Multiple molecular mechanisms for multidrug resistance transporters. Nature 446, 749–757 10.1038/nature05630 [DOI] [PubMed] [Google Scholar]
- 2. Blair J. M., Webber M. A., Baylay A. J., Ogbolu D. O., and Piddock L. J. (2015) Molecular mechanisms of antibiotic resistance. Nat. Rev. Microbiol. 13, 42–51 [DOI] [PubMed] [Google Scholar]
- 3. Du D., van Veen H. W., Murakami S., Pos K. M., and Luisi B. F. (2015) Structure, mechanism and cooperation of bacterial multidrug transporters. Curr. Opin. Struct. Biol. 33, 76–91 10.1016/j.sbi.2015.07.015 [DOI] [PubMed] [Google Scholar]
- 4. Saier M. H. Jr., and Paulsen I. T. (2001) Phylogeny of multidrug transporters. Semin. Cell Dev. Biol. 12, 205–213 10.1006/scdb.2000.0246 [DOI] [PubMed] [Google Scholar]
- 5. Alibert S., N'Gompaza Diarra J., Hernandez J., Stutzmann A., Fouad M., Boyer G., and Pagès J. M. (2017) Multidrug efflux pumps and their role in antibiotic and antiseptic resistance: a pharmacodynamic perspective. Expert Opin. Drug Metab. Toxicol. 13, 301–309 10.1080/17425255.2017.1251581 [DOI] [PubMed] [Google Scholar]
- 6. Chen Z., Shi T., Zhang L., Zhu P., Deng M., Huang C., Hu T., Jiang L., and Li J. (2016) Mammalian drug efflux transporters of the ATP binding cassette (ABC) family in multidrug resistance: a review of the past decade. Cancer Lett. 370, 153–164 10.1016/j.canlet.2015.10.010 [DOI] [PubMed] [Google Scholar]
- 7. Kaatz G. W., McAleese F., and Seo S. M. (2005) Multidrug resistance in Staphylococcus aureus due to overexpression of a novel multidrug and toxin extrusion (MATE) transport protein. Antimicrob. Agents Chemother. 49, 1857–1864 10.1128/AAC.49.5.1857-1864.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. McAleese F., Petersen P., Ruzin A., Dunman P. M., Murphy E., Projan S. J., and Bradford P. A. (2005) A novel MATE family efflux pump contributes to the reduced susceptibility of laboratory-derived Staphylococcus aureus mutants to tigecycline. Antimicrob. Agents Chemother. 49, 1865–1871 10.1128/AAC.49.5.1865-1871.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Yılmaz Ç., and Özcengiz G. (2017) Antibiotics: Pharmacokinetics, toxicity, resistance and multidrug efflux pumps. Biochem. Pharmacol. 133, 43–62 10.1016/j.bcp.2016.10.005 [DOI] [PubMed] [Google Scholar]
- 10. Chen J., Morita Y., Huda M. N., Kuroda T., Mizushima T., and Tsuchiya T. (2002) VmrA, a member of a novel class of Na+-coupled multidrug efflux pumps from Vibrio parahaemolyticus. J. Bacteriol. 184, 572–576 10.1128/JB.184.2.572-576.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Huda M. N., Chen J., Morita Y., Kuroda T., Mizushima T., and Tsuchiya T. (2003) Gene cloning and characterization of VcrM, a Na+-coupled multidrug efflux pump, from Vibrio cholerae non-O1. Microbiol. Immunol. 47, 419–427 10.1111/j.1348-0421.2003.tb03379.x [DOI] [PubMed] [Google Scholar]
- 12. Kuroda T., and Tsuchiya T. (2009) Multidrug efflux transporters in the MATE family. Biochim. Biophys. Acta 1794, 763–768 10.1016/j.bbapap.2008.11.012 [DOI] [PubMed] [Google Scholar]
- 13. Long F., Rouquette-Loughlin C., Shafer W. M., and Yu E. W. (2008) Functional cloning and characterization of the multidrug efflux pumps NorM from Neisseria gonorrhoeae and YdhE from Escherichia coli. Antimicrob. Agents Chemother. 52, 3052–3060 10.1128/AAC.00475-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Omote H., Hiasa M., Matsumoto T., Otsuka M., and Moriyama Y. (2006) The MATE proteins as fundamental transporters of metabolic and xenobiotic organic cations. Trends Pharmacol. Sci. 27, 587–593 10.1016/j.tips.2006.09.001 [DOI] [PubMed] [Google Scholar]
- 15. Su X. Z., Chen J., Mizushima T., Kuroda T., and Tsuchiya T. (2005) AbeM, an H+-coupled Acinetobacter baumannii multidrug efflux pump belonging to the MATE family of transporters. Antimicrob. Agents Chemother. 49, 4362–4364 10.1128/AAC.49.10.4362-4364.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Yonezawa A., and Inui K. (2011) Importance of the multidrug and toxin extrusion MATE/SLC47A family to pharmacokinetics, pharmacodynamics/toxicodynamics and pharmacogenomics. Br. J. Pharmacol. 164, 1817–1825 10.1111/j.1476-5381.2011.01394.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. He X., Szewczyk P., Karyakin A., Evin M., Hong W. X., Zhang Q., and Chang G. (2010) Structure of a cation-bound multidrug and toxic compound extrusion transporter. Nature 467, 991–994 10.1038/nature09408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kusakizako T., Claxton D. P., Tanaka Y., Maturana A. D., Kuroda T., Ishitani R., Mchaourab H. S., and Nureki O. (2019) Structural basis of H+-dependent conformational change in a bacterial MATE transporter. Structure 27, 293–301.e3 10.1016/j.str.2018.10.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Lu M., Radchenko M., Symersky J., Nie R., and Guo Y. (2013) Structural insights into H+-coupled multidrug extrusion by a MATE transporter. Nat. Struct. Mol. Biol. 20, 1310–1317 10.1038/nsmb.2687 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Lu M., Symersky J., Radchenko M., Koide A., Guo Y., Nie R., and Koide S. (2013) Structures of a Na+-coupled, substrate-bound MATE multidrug transporter. Proc. Natl. Acad. Sci. U.S.A. 110, 2099–2104 10.1073/pnas.1219901110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Miyauchi H., Moriyama S., Kusakizako T., Kumazaki K., Nakane T., Yamashita K., Hirata K., Dohmae N., Nishizawa T., Ito K., Miyaji T., Moriyama Y., Ishitani R., and Nureki O. (2017) Structural basis for xenobiotic extrusion by eukaryotic MATE transporter. Nat. Commun. 8, 1633 10.1038/s41467-017-01541-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Mousa J. J., Yang Y., Tomkovich S., Shima A., Newsome R. C., Tripathi P., Oswald E., Bruner S. D., and Jobin C. (2016) MATE transport of the E. coli-derived genotoxin colibactin. Nat. Microbiol. 1, 15009 10.1038/nmicrobiol.2015.9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Radchenko M., Symersky J., Nie R., and Lu M. (2015) Structural basis for the blockade of MATE multidrug efflux pumps. Nat. Commun. 6, 7995 10.1038/ncomms8995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Tanaka Y., Hipolito C. J., Maturana A. D., Ito K., Kuroda T., Higuchi T., Katoh T., Kato H. E., Hattori M., Kumazaki K., Tsukazaki T., Ishitani R., Suga H., and Nureki O. (2013) Structural basis for the drug extrusion mechanism by a MATE multidrug transporter. Nature 496, 247–251 10.1038/nature12014 [DOI] [PubMed] [Google Scholar]
- 25. Tanaka Y., Iwaki S., and Tsukazaki T. (2017) Crystal structure of a plant multidrug and toxic compound extrusion family protein. Structure 25, 1455–1460.e2 10.1016/j.str.2017.07.009 [DOI] [PubMed] [Google Scholar]
- 26. Jardetzky O. (1966) Simple allosteric model for membrane pumps. Nature 211, 969–970 10.1038/211969a0 [DOI] [PubMed] [Google Scholar]
- 27. Mitchell P. (1967) Translocations through natural membranes. Adv. Enzymol. Relat. Areas Mol. Biol. 29, 33–87 [DOI] [PubMed] [Google Scholar]
- 28. Lu M. (2016) Structures of multidrug and toxic compound extrusion transporters and their mechanistic implications. Channels 10, 88–100 10.1080/19336950.2015.1106654 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Radchenko M., Nie R., and Lu M. (2016) Disulfide cross-linking of a multidrug and toxic compound extrusion transporter impacts multidrug efflux. J. Biol. Chem. 291, 9818–9826 10.1074/jbc.M116.715227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Claxton D. P., Jagessar K. L., Steed P. R., Stein R. A., and Mchaourab H. S. (2018) Sodium and proton coupling in the conformational cycle of a MATE antiporter from Vibrio cholerae. Proc. Natl. Acad. Sci. U.S.A. 115, E6182–E6190 10.1073/pnas.1802417115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Steed P. R., Stein R. A., Mishra S., Goodman M. C., and McHaourab H. S. (2013) Na+-substrate coupling in the multidrug antiporter norm probed with a spin-labeled substrate. Biochemistry 52, 5790–5799 10.1021/bi4008935 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Yamanaka H., Kobayashi H., Takahashi E., and Okamoto K. (2008) MacAB is involved in the secretion of Escherichia coli heat-stable enterotoxin II. J. Bacteriol. 190, 7693–7698 10.1128/JB.00853-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Nishima W., Mizukami W., Tanaka Y., Ishitani R., Nureki O., and Sugita Y. (2016) Mechanisms for two-step proton transfer reactions in the outward-facing form of MATE transporter. Biophys. J. 110, 1346–1354 10.1016/j.bpj.2016.01.027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Ashkenazy H., Abadi S., Martz E., Chay O., Mayrose I., Pupko T., and Ben-Tal N. (2016) ConSurf 2016: an improved methodology to estimate and visualize evolutionary conservation in macromolecules. Nucleic Acids Res. 44, W344–W350 10.1093/nar/gkw408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Landau M., Mayrose I., Rosenberg Y., Glaser F., Martz E., Pupko T., and Ben-Tal N. (2005) ConSurf 2005: the projection of evolutionary conservation scores of residues on protein structures. Nucleic Acids Res. 33, W299–W302 10.1093/nar/gki370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Kozachkov L., and Padan E. (2011) Site-directed tryptophan fluorescence reveals two essential conformational changes in the Na+/H+ antiporter NhaA. Proc. Natl. Acad. Sci. U.S.A. 108, 15769–15774 10.1073/pnas.1109256108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Smirnova I., Kasho V., Sugihara J., and Kaback H. R. (2009) Probing of the rates of alternating access in LacY with Trp fluorescence. Proc. Natl. Acad. Sci. U.S.A. 106, 21561–21566 10.1073/pnas.0911434106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Parashar S., Zou P., and Mchaourab H. S. (2009) Mapping daunorubicin-binding Sites in the ATP-binding cassette transporter MsbA using site-specific quenching by spin labels. J. Biol. Chem. 284, 13904–13913 10.1074/jbc.M900837200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Tal N., and Schuldiner S. (2009) A coordinated network of transporters with overlapping specificities provides a robust survival strategy. Proc. Natl. Acad. Sci. U.S.A. 106, 9051–9056 10.1073/pnas.0902400106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Yu E. W., Aires J. R., and Nikaido H. (2003) AcrB multidrug efflux pump of Escherichia coli: composite substrate-binding cavity of exceptional flexibility generates its extremely wide substrate specificity. J. Bacteriol. 185, 5657–5664 10.1128/JB.185.19.5657-5664.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Yu E. W., McDermott G., Zgurskaya H. I., Nikaido H., and Koshland D. E. Jr. (2003) Structural basis of multiple drug-binding capacity of the AcrB multidrug efflux pump. Science 300, 976–980 10.1126/science.1083137 [DOI] [PubMed] [Google Scholar]
- 42. Ficici E., Zhou W., Castellano S., and Faraldo-Gómez J. D. (2018) Broadly conserved Na+-binding site in the N-lobe of prokaryotic multidrug MATE transporters. Proc. Natl. Acad. Sci. U.S.A. 115, E6172–E6181 10.1073/pnas.1802080115 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.