

Abstract
Historically, efforts to assess ‘zoonotic risk’ have focused mainly on quantifying the potential for cross-species emergence of viruses from animal hosts. However, viruses clearly differ in relative burden, both in terms of morbidity and mortality (virulence) incurred and the capacity for sustained human-to-human transmission. Extending previously published databases, we delineated host and viral traits predictive of human mortality associated with viral spillover, viral capacity to transmit between humans following spillover and the probability of a given virus being zoonotic. We demonstrate that increasing host phylogenetic distance from humans positively correlates with human mortality but negatively correlates with human transmissibility, suggesting that the virulence induced by viruses emerging from hosts at high phylogenetic distance may limit capacity for human transmission. Our key result is that hosts most closely related to humans harbour zoonoses of lower impact in terms of morbidity and mortality, while the most distantly related hosts—in particular, order Chiroptera (bats)—harbour highly virulent zoonoses with a lower capacity for endemic establishment in human hosts. As a whole, our results emphasize the importance of understanding how zoonoses manifest in the human population and also highlight potential risks associated with multi-host transmission chains in spillover.
This article is part of the theme issue ‘Dynamic and integrative approaches to understanding pathogen spillover’.
Keywords: zoonoses, emerging infectious diseases, virulence, bridge hosts, host breadth, Chiroptera
1. Introduction
Emerging infectious diseases (EIDs) pose a significant threat to public health with the frequency of emergence events increasing in recent decades [1]. The vast majority of EIDs are zoonotic, meaning they transmit from animal to human hosts [2]. Several studies have conducted meta-analyses to characterize trait profiles associated with zoonotic hosts and viruses [2–6]. The majority of this work has focused on quantifying ‘zoonotic potential’—the probability that a pathogen, or a pathogen originating from an animal species or region of interest, could emerge into the human population. Such a binary categorization effectively treats all zoonoses as risks of equal magnitude, but it is clear that not all zoonoses are created equal. Emerging zoonoses vary both in the severity of disease they engender in the human population (virulence) and in their capacity for sustained human-to-human transmission post-emergence. A more nuanced understanding of how zoonoses establish in their human hosts will be critical to any public health effort to combat EIDs—as diseases with different severities and transmissibilities will require uniquely targeted intervention and control strategies.
Recent meta-analyses have begun to explore the impact of zoonoses post-spillover, though, to date, only two studies have assessed variation in zoonotic severity. Geoghegan et al. [7] and Brierley et al. [8] demonstrated that zoonoses engendering higher case fatality rates (CFRs) are associated with limited ability for human-to-human transmission. Brierley et al. [8] identified viral traits predictive of virulence but categorized zoonotic severity only in binary terms—‘severe’ or ‘non-severe’. A slightly larger body of work has identified viral traits predictive of a zoonotic pathogen's capacity for between-human transmission post-spillover [7–10]. However, the majority of these studies use a binary classification scheme (i.e. can transmit between humans or cannot) in their analyses. In nature, there is considerable variation in a pathogen's capacity for human-to-human transmission, ranging from none (humans are dead-end hosts) to stuttering chains to sustained transmission [11]. Woolhouse et al. [12] and Lloyd-Smith et al. [13] classify this variation according to a zoonotic pathogen's basic reproduction number (R0) among human hosts, delineating the number of cases engendered by a single primary case in the human population. In this classification scheme, both dead-end and stuttering chain zoonoses are described by subcritical (less than 1) R0 values, while zoonoses that sustain human-to-human transmission have R0 values greater than 1. For example, human-to-human transmission has been reported for Andes virus, but transmission chains are rare and typically short-lived (R0 < 1) [14], while other viruses, such as Ebola, have the capacity for effective between-human transmission, resulting in long transmission chains and large epidemics (R0 > 1) [15]. Additionally, some zoonotic pathogens, such as hepatitis E, can establish endemic transmission in human populations [16]. To our knowledge, Brierley et al. [8] is the only meta-analysis to date that begins to capture this nuance, using a three-point ranking system to distinguish between zoonotic pathogens without human transmission, and those with limited and sustained transmission.
All previous meta-analyses investigating zoonotic severity and transmissibility have focused almost exclusively on identifying viral predictors of zoonotic impact post-spillover. Currently, no comparative studies of zoonotic virulence and transmissibility consider the role of the animal host, though both virulence and transmission are shaped by viruses' interactions with their hosts [17–19]. A virus must replicate to overcome host defences and transmit to other individuals in the host population, but replication can also produce maladaptive virulence that damages the host, thus shortening the infectious period and jeopardizing opportunities for future transmission [20,21]. According to this virulence–transmission trade-off, viruses optimize virulence incurred in their hosts so as to effectively transmit to new hosts. This optimal balance depends on how hosts respond to the virus (host selective pressure) [22]. If increased viral replication is needed to overcome more robust host defences, host immunity will select for higher virulence [23–25]. Host population structure can also facilitate or hinder transmission, further influencing the evolution of virulence [26,27].
In this study, we addressed the need for a more nuanced understanding of how emerging zoonoses manifest in the human population. Focusing on directly transmitted viruses with a recent history of spillover to humans, we extended databases published by Olival et al. [28], detailing the mortality (virulence) induced by a given virus upon spillover to humans and quantifying each virus's capacity for human-to-human transmission. We used generalized additive models (GAMs) to identify mammalian host and viral traits predictive of: (i) the human CFRs induced by viral zoonosis, (ii) the extent of human-to-human transmission resulting from zoonotic spillover, and (iii) the probability of a given virus being zoonotic.
Our analysis of zoonotic risk extends beyond the simple probability of emergence and improves on previous analogous studies to investigate both host and viral trait predictors for non-binary metrics of virulence and transmissibility. This is, to our knowledge, the first meta-analysis of its type to distinguish between viruses' reservoir host species (i.e. primary selective environment), and secondary host species that have been infected, but do not maintain zoonotic transmission. We hypothesized an inverse relationship between CFR and human transmissibility and anticipated that both reservoir host and viral traits would significantly predict zoonotic risk. In particular, we investigated the hypothesis that host species distantly related to humans would host more virulent viruses with lower capacities for between-human transmission.
2. Methods
Detailed methods and datasets are given in the electronic supplementary material, Methods and Data and Results.
(a). Compiling the databases
We compiled a list of 420 associations between 67 directly transmitted zoonotic viruses and 278 mammalian hosts, drawing primarily from an extensive database of virus–mammal associations published by Olival et al. [28] (electronic supplementary material, Data and Results, table S1). For each virus–mammal association, we conducted literature searches to collect two metrics of zoonotic risk: CFR in the human population, following viral spillover (a proxy for virulence), and viral capacity for human-to-human transmission, which we ranked according to a four-point scale, ranging from ‘1’ for viruses never recorded as transmitting between humans to ‘4’ for viruses known to maintain endemic human transmission [8,11,13,29].
In addition to collecting targeted metrics of virulence and transmissibility, we classified each virus–mammal association according to the mammal's role in viral transmission. We used a binary code to distinguish between mammal species that maintain viruses endemically (reservoir hosts) and species that harbour the virus but are not implicated in zoonotic maintenance (secondary hosts, see table 1 for definitions of all terms in this study). We assigned a second binary code to define each host's role in zoonotic spillover (spillover capacity), distinguishing between mammal species that serve as human infection sources and species with no record of transmission to humans. Combining these two codes, we defined a third ‘spillover type’ code to distinguish between ‘primary spillover’ from reservoir host species and ‘secondary spillover’ from secondary host species, or ‘bridge hosts’.
Table 1.
Definition of terms used in this study.
| term | definition |
|---|---|
| spillover | transmission of animal viruses to humans |
| spillback | transmission of human viruses to animals |
| zoonotic potential | probability that a pathogen—or a pathogen originating from an animal species or region of interest—could emerge into the human population |
| spillover host | host species that has been recorded to infect humans and thus has served as a source of human infection |
| reservoir host | primary host species that is responsible for maintaining zoonotic transmission |
| primary spillover | spillover to humans from a reservoir host species |
| secondary host | host species that has been infected, but does not maintain zoonotic transmission |
| bridge host | secondary host species that has been recorded to infect humans |
| secondary spillover | spillover to humans from a secondary host species |
| transmission cost | a trade-off that compromises a virus's capacity to transmit between hosts |
In addition to the virus–mammal association database, we extracted a dataset of 345 directly transmitted mammalian viruses (both zoonotic and non-zoonotic) from Olival et al. [28] (electronic supplementary material, Data and Results, table S2).
Using previously published databases [7,9,28–33], we next collected a series of host and viral traits that we hypothesized might predict the observed variation in zoonotic virus dynamics in humans. For hosts, we focused on four life-history traits, quantifiable across mammal species, that have been linked to host–pathogen coevolution: body mass, litter size, gestation duration and lifespan. Host body mass has been linked with the rate of disease progression [34], reservoir competence [35] and pathogen replication rate [36,37]; host reproductive effort trades off with immune investment and shapes susceptible host population demography [38,39]; and protracted host lifespans are associated with heightened population-level transmission [40,41]. Replicating Olival et al. [28], we additionally considered host phylogenetic order and distance from humans. Evolutionary distance between novel and previously documented host species has been identified as a predictor of disease-induced mortality post-spillover in domesticated animals [42]. For viral traits, we focused on traits that Olival et al. [28] previously linked to zoonotic infectivity, collecting viruses' host phylogenetic breadths as proxies for viral host ranges. We additionally included the position of a virus’s host breadth relative to humans by considering the maximum host phylogenetic distance from humans across a virus's host range. All datasets with metadata and references are available in the electronic supplementary material, Data and Results, tables S1–S4. Table 2 describes predictor and response variables used in our analysis.
Table 2.
Description of predictor and response variables used in the GAM analyses.
| term | type | description |
|---|---|---|
| response variables | ||
| human case fatality rate | numeric | the proportion of human cases for a given virus that are fatal |
| human transmissibility | categorical ranking (1–4) | a virus’s capacity for human-to-human transmission see the electronic supplementary material, Methods for a description of the ranking system |
| zoonotic potential | binary (0,1) | the probability that a virus has the capacity to infect humans (i.e. is or is not zoonotic) |
| host predictors | ||
| length of gestation period | numeric | length of pregnancy (in days) |
| litter size | numeric | average number of offspring produced at one time |
| maximum lifespan | numeric | maximum recorded longevity (in months) |
| body mass | numeric | average body mass (in grams) |
| phylogenetic distance from humans | numeric | distance from humans on a cytochrome b phylogenetic tree |
| taxonomic order | categorical | taxonomic classification |
| number of disease-related citations | numeric | number of PubMed citations relevant to zoonotic diseases; to control for any potential publication bias |
| viral predictors | ||
| whether or not the virus is enveloped | binary | does the virus have an external envelope? |
| whether or not the virus replicates in the cytoplasm | binary | does the virus replicate in the cytoplasm? |
| average genome length | numeric | metric for genome size |
| DNA or RNA | binary | is the virus DNA or RNA? |
| genome composition | categorical | viral genome classification |
| maximum host phylogenetic distance from humans | numeric | phylogenetic distance from humans of the most distantly related known host for a given virus |
| maximum host phylogenetic breadth | numeric | maximum phylogenetic distance between the two most distantly related known hosts for a given virus |
| number of citations | numeric | number of relevant PubMed citations; control for research effort bias |
| categories for virus–mammal associations | ||
| reservoir status | binary (1,2) | host species role in the maintenance of zoonotic transmission. Species that maintain viruses endemically are reservoir hosts (reservoir status = 1). Species that harbour the virus but are not implicated in zoonotic maintenance are secondary hosts (reservoir status = 2) |
| spillover capacity | binary (1,0) | host species' role in the spillover of zoonoses to humans. Spillover hosts are (1) defined as species that are a source of human infection. Non-spillover hosts are (0) defined as species that have no record of transmission to humans |
| spillover type | binary (1,2) | chain of spillover transmission for a given virus–host system. Spillover to humans from a reservoir host is ‘primary spillover’ (reservoir status = 1, spillover capacity = 1, spillover type = 1). Spillover to humans from a secondary host (bridge host) is ‘secondary spillover’ (reservoir status = 2, spillover capacity = 1, spillover type = 2) |
(b). Statistical analysis
Because nonlinear relationships were anticipated, we used GAMs in the mgcv package in R [43] to assess host and viral predictors of zoonotic risk. GAMs are flexible generalized linear models that, rather than manually specifying higher order polynomial functions, use smooth functions to capture nonlinear relationships between response and predictor variables. We fitted two sets of GAMs, assessing host predictors of zoonotic risk in one group of models and viral predictors in the other.
Our global models included all host and viral trait predictors outlined in table 2. We used automated term selection by double penalty smoothing for variable selection. This method constructs an additional penalty for each GAM smooth function, effectively removing terms without predictive power and has been recognized as superior or comparable to alternative approaches [44]. We set an effective degree of freedom cut-off of 0.001 to identify which terms had been penalized and effectively removed from the model [28].
(i). Host models
We restricted our analysis of host predictors of zoonotic risk to known reservoir host species with demonstrated evidence of animal-to-human spillover, thus only considering species implicated as both the primary selective environment and source of human infection for a given virus. Because the specific host species responsible for a spillover event is not always identified, we were frequently unable to collect human CFR and transmissibility data that varied depending on the specific spillover host. Thus, to avoid pseudoreplication, we further restricted our analysis to include only unique entries for each host order per virus in a simplified dataset, summarizing information across hosts encapsulated in each unique entry by taking the maximum value for each host trait metric.
Using this simplified dataset, we first asked what host traits best predict CFRs in human hosts following spillover? Specifically, we used a GAM to query the predictive capacity of the host-specific traits outlined in table 2 on the response variable of the mean CFR in a human host.
We next asked, what host variables best predict the extent of human-to-human transmission of a given virus following spillover? In this case, we used a GAM to query the predictive capacity of the same host traits outlined in table 2 on the response variable of human transmissibility.
While our previous GAMs only included reservoir host species, we next investigated both reservoir and secondary hosts with evidence of spillover to humans (bridge hosts). Fitting a separate GAM for each response variable, we explored the relationship between host phylogenetic distance from humans and both CFR and transmissibility as a function of ‘spillover type’ (‘primary spillover’ from reservoir hosts versus ‘secondary spillover’ from bridge hosts). In each case, we queried the response variable against the predictor variable of host phylogenetic distance, modelled as two distinct smoothers separated by spillover type [44].
(ii). Virus models
For our analysis of viral predictors of zoonotic risk, we first asked what viral traits best predict the probability that a virus is zoonotic? We constructed a binomial GAM, testing the predictive capacity of viral traits outlined in table 2 against the response variable of zoonotic status (0–1: is versus is not).
Our analysis of viral predictors of CFR and human transmissibility largely mirrored that of our host analysis. To avoid pseudoreplication, we again only considered unique entries, grouping trait information by discrete CFR and transmissibility values per virus.
As with our host models, we then applied GAMs to this simplified viral dataset to ask, what viral traits best predict case mortality rates in human hosts following spillover? and what viral traits best predict the extent of human-to-human transmission of a given virus following spillover?
3. Results
(a). Host predictors of human case fatality rates and capacity for human-to-human transmission
The selected model to predict human CFR explained 51.4% of the total deviance and included maximum host body mass, gestation period, lifespan and phylogenetic distance from humans as significant predictors, as well as a term for host phylogenetic order (electronic supplementary material, Data and Results, table S5a). We observed a correlation between increasing CFR and increasing mammalian host phylogenetic distance from humans (figure 1a): hosts most distantly related to humans harbour the most virulent viruses. The model additionally indicated that CFR decreases with increasing host body mass and decreasing host gestation period, though host life-history traits are inherently correlated with each other and with host phylogenetic distance from humans (electronic supplementary material, Data and Results, table S6).
Figure 1.
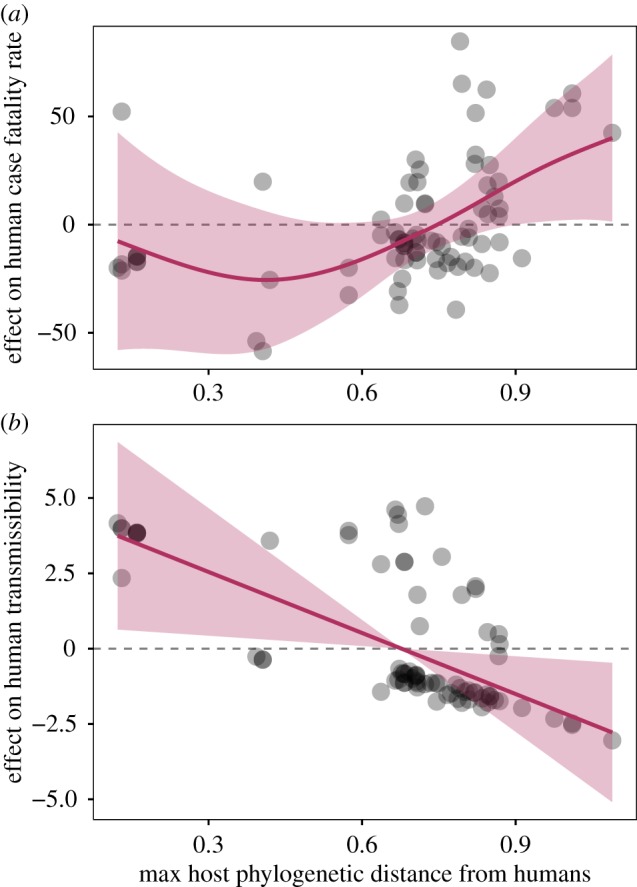
Host phylogenetic distance from humans predicts zoonotic risk. Partial effect plots from our selected GAMs show the relative effect of host phylogenetic distance from humans on (a) human CFR (virulence); and (b) capacity for human-to-human transmission (human transmissibility). Data points represent partial residuals, and shaded regions represent 95% confidence intervals around the mean partial effects. Full model descriptions are provided in the electronic supplementary material, Data and Results, table S5a,b.
The selected host model for human transmissibility, which explained 26.5% of the deviance, included maximum host lifespan, litter size and phylogenetic distance from humans as significant predictors, as well as a term for maximum host body mass (electronic supplementary material, Data and Results, table S5b). In contrast with CFR, capacity for human-to-human transmission decreases as host phylogenetic distance increases (figure 1b).
(b). Effect of phylogenetic distance on case fatality rate and transmissibility as a function of spillover type
While our first analysis focused on primary spillover events, in which viruses spill over to humans directly from reservoir hosts, we added important nuance to our analysis by running additional GAMs that considered all virus–mammal associations, including cases of secondary spillover. We explored whether zoonotic risk changes with the position of the immediate spillover source in the transmission chain from reservoir host to human.
The selected model to predict human CFR with interaction terms for host phylogenetic distance and spillover type explained 66.8% of the deviance and included host maximum lifespan, gestation period and phylogenetic order as significant terms (electronic supplementary material, Data and Results, table S5c). Spillover type affects the relationship between host phylogenetic distance from humans and human CFR (figure 2). For primary spillovers from reservoir hosts, CFR increases with host phylogenetic distance from humans (figure 2a), consistent with results in figure 1a. By contrast, spillovers from secondary (bridge) hosts are associated with elevated CFR across all secondary host species, irrespective of phylogenetic distance from humans. The largest positive effect sizes are recovered from host species both closely and distantly related to humans (figure 2b). Heightened CFR in secondary hosts at high phylogenetic distances is consistent with the trends observed for reservoir hosts—host species most distantly related to humans harbour the most virulent viruses. However, elevated virulence in secondary hosts at low phylogenetic distances conflicts with our prior observation for reservoir hosts that species most closely related to humans harbour zoonoses of lower impact in terms of morbidity and mortality.
Figure 2.

Spillover type modulates host predictors of zoonotic risk. Partial effect plots from our selected GAMs show how primary (orange) versus secondary (blue) spillover changes the relative effect of host phylogenetic distance from humans on (a,b) human CFR (virulence); and (c,d) the capacity for human-to-human transmission (human transmissibility). Data points represent partial residuals, and shaded regions represent 95% confidence intervals by standard error around mean partial effects. Full model descriptions are provided in the electronic supplementary material, Data and Results, table S5c,d.
The selected model for human transmissibility with interaction terms for host phylogenetic distance and spillover type explained 34.5% of the deviance and included host order and maximum body mass as significant terms (electronic supplementary material, Data and Results, table S5d). With primary spillover, transmissibility decreases with increasing host phylogenetic distance from humans, consistent with the virulence–transmission relationship outlined in figure 1. However, variable selection effectively omitted the secondary spillover predictor from this model, suggesting that gaps in data—particularly our limited ability to trace viral outcomes in humans to specific spillover hosts—or a more muddled evolutionary trade-off among viruses that have a less extensive shared evolutionary history with their secondary host species may limit our inference capacity.
(c). Examining bats as ‘special’ viral reservoirs
Broadly, we observed that viruses within a given viral family preferentially infect a discrete subset of host orders (i.e. some viral families are predominantly primate or rodent or bat viruses, etc.) and that distinct mammalian host orders cluster at distinct phylogenetic distances from humans (figure 3). Of the five major animal source orders for zoonotic viruses (figure 3a: Primates, Cetartiodactyla, Carnivora, Rodentia and Chiroptera), order Chiroptera (bats) is, on average, the most phylogenetically distant from humans (figure 3b). As such, bats could underpin the heightened virulence of viruses harboured by hosts at high phylogenetic distance (figure 1a). Here, we examine bats as a possible ‘special’ host order in our database. Bat-derived zoonoses cluster in the upper right-hand corner of the plane of phylogenetic distance and human CFR (figure 4); our GAM for host traits predictive of virulence demonstrated that order Chiroptera has the strongest positive effect on CFR (electronic supplementary material, Data and Results, table S7).
Figure 3.
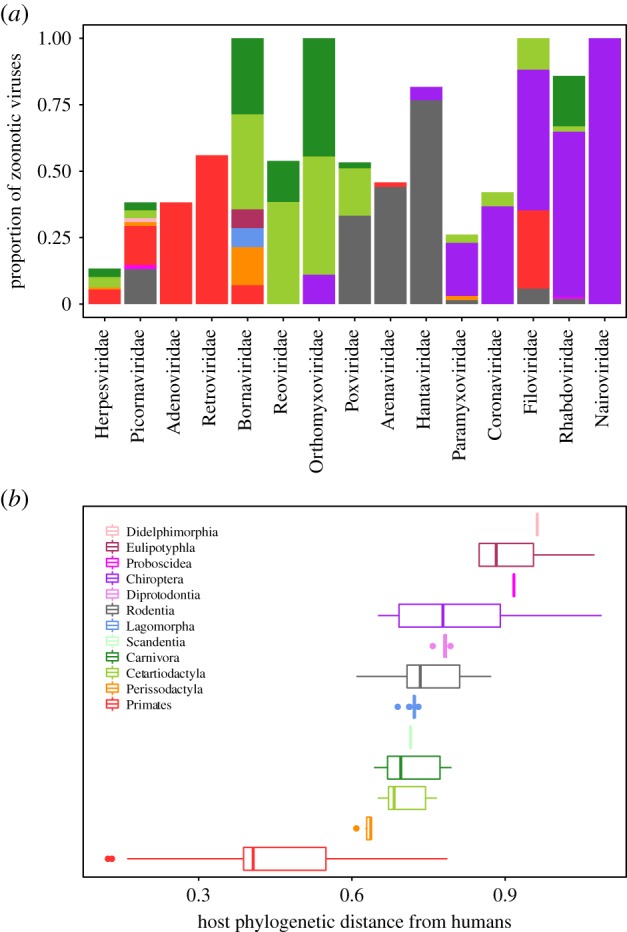
Correlation between virus and host types. Across host phylogenetic distance from humans, the distributions of (a) zoonotic virus species, grouped by family; and (b) mammalian host species, grouped by host order. Colours correspond to host order.
Figure 4.
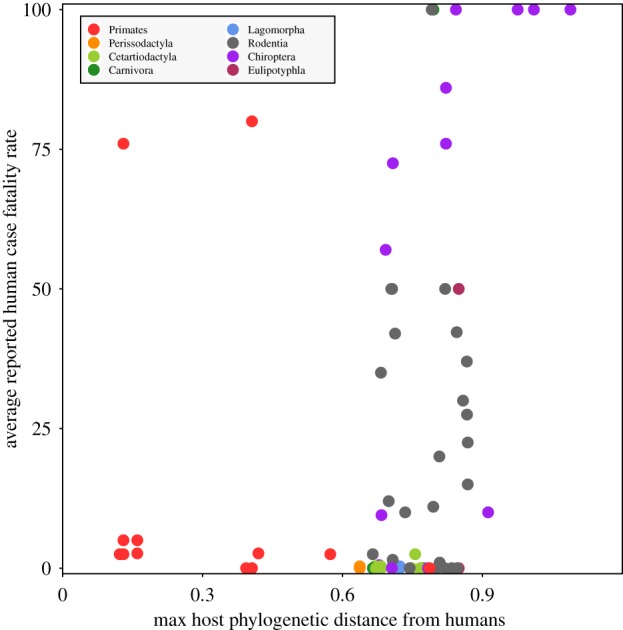
Bat-borne viruses are the most virulent of all mammalian zoonoses. Phylogenetic distance and CFR data for all mammalian zoonoses with colours indicating host order. Each point represents a unique CFR per host order for each virus.
(d). Viral predictors of virus zoonotic potential, human case fatality rates and capacity for human-to-human transmission
Because particular viral families associate non-randomly with particular host orders, variation in zoonoses derived from these orders could be equally attributed both to traits of the hosts themselves or traits of the viruses that infect those hosts. Incorporation of viral predictors into our GAM framework demonstrated significant predictive power for several viral traits on three metrics of zoonotic risk. In our probability analysis of a virus's zoonotic potential, the selected model, which explained 32.5% of variation in the data, included significant predictors for cytoplasmic replication capacity, viral genome composition, maximum host phylogenetic distance from humans and maximum host phylogenetic breadth (electronic supplementary material, Data and Results, table S5e). Our results mirror previous findings which indicate that viruses with cytoplasmic replication and broader host phylogenetic breadths are more likely to be zoonotic (figure 5b) [6,28]. We further observed that viruses derived from hosts at the closest phylogenetic distances to humans had the highest zoonotic potential (figure 5a), consistent with Olival et al.'s [28] finding that hosts closely related to humans harboured the most zoonoses. Both host breadth and the phylogenetic position of that breadth relative to humans significantly predict whether a virus is zoonotic.
Figure 5.

Viral predictors of zoonotic risk. Partial effect plots from our selected GAMs show the relative effect of maximum host phylogenetic distance from humans (red) and host phylogenetic breadth (blue) on (a,b) the probability that a virus is zoonotic; (c,d) human CFR (virulence); and (e,f) capacity for human-to-human transmission (human transmissibility). Data points represent partial residuals, and shaded regions represent 95% confidence intervals by standard error around mean partial effects. Full model descriptions are provided in the electronic supplementary material, Data and Results, table S5e–g.
Using the zoonotic subset of our virus database, we next considered the effect of viral traits on the response variables of human CFR and capacity for sustained human-to-human transmission. Our selected viral trait model for human CFR explained 53.8% of observed variation in the data and included terms for genome composition, disease-related citations, the presence/absence of a viral envelope, and the maximum phylogenetic distance from humans of all hosts observed for a given virus (electronic supplementary material, Data and Results, table S5f). The absence of viral envelope was associated with lower virulence upon spillover to humans. Consistent with trends reported in figure 1a, host phylogenetic distance was positively correlated with higher human CFRs post-spillover (figure 5c; though note that viral host phylogenetic distance was calculated differently than the host distances used in figure 1 analyses, see the electronic supplementary material, Methods). Notably, maximum host phylogenetic breadth per virus was not a significant predictor in our final CFR model (figure 5d).
Finally, the selected viral model for human transmissibility described 55.1% of variation in the data and included terms for genome and DNA/RNA composition, the presence/absence of a viral envelope, the maximum host phylogenetic distance from humans and the maximum host phylogenetic breadth observed for each viral species (electronic supplementary material, Data and Results, table S5g) (figure 5e,f). Consistent with Geoghegan et al. [7], the absence of viral envelope was positively associated with increased capacity for human-to-human transmission. Consistent with our host model in figure 1b, we observed that capacity for human-to-human transmission decreased with increasing host phylogenetic distance from humans. The relationship between host phylogenetic breadth and human transmissibility was more complicated: human-to-human transmissibility increased with heightened host phylogenetic breadth up to a point (0.85), then declined sharply at extremely high breadths. This trend is probably driven by only a few viruses—most notably, rabies, for which zoonotic infection and transmission dynamics are complex [45]. Human-to-human transmission may be limited for rabies owing to effective control strategies and a bite-dependent transmission route rather than lack of innate viral capacity; rabies virus is known to be present in human tissues relevant for transmission, and, indeed, between-human transmission has been suspected but not confirmed for a few infections [46,47].
4. Discussion
Our work introduces a novel multi-dimensional framework for assessing and predicting zoonotic risk. Building off previous analyses (electronic supplementary material, Data and Results, table S8), we extended databases published by Olival et al. [28] to delineate host and viral traits predictive of (i) the mortality burden associated with viral spillover into the human population, (ii) the extent of sustained human-to-human transmission following zoonosis, and (iii) the probability that a virus is zoonotic. For each metric of zoonotic risk, we identified a unique set of host and viral predictor variables, which highlight the critical role of host phylogenetic distance and virus–host evolutionary history in driving zoonotic outcomes.
Our work uncovers a positive correlation between host phylogenetic distance and the mortality incurred by viruses derived from these hosts upon spillover to human populations; zoonoses emerging from more distantly related hosts are more virulent. This result is consistent with empirical work that has demonstrated an association between host phylogeny and disease-induced mortality in a novel host following a pathogen host shift [48,49]. Nonetheless, while distantly related hosts may harbour more virulent viruses, our results suggest that these viruses are less likely to engender zoonoses capable of establishing sustained human-to-human transmission. Virulence appears to constrain transmission, limiting zoonotic spread within the human population. Host phylogenetic distance from humans appears to modulate this relationship, predicting virus-induced mortality in a human host and the transmission ‘cost’ associated with that virulence.
The correlation between increasing phylogenetic distance, elevated virulence, and diminishing capacity for transmission is additionally modulated based on the pathway of zoonotic emergence. This study represents, to our knowledge, the first meta-analysis of its type to distinguish between primary spillover from reservoir hosts—the primary selective environment of viruses—and secondary spillover from bridge host species that have been infected, but do not maintain zoonotic transmission. The primary spillover from reservoir hosts to humans follows the general observed trends, with the highest transmission cost incurred at high phylogenetic distance and high virulence. Intriguingly, secondary spillover hosts, including those closely related to humans, produce elevated CFRs across phylogenetic distances. Because our analysis did not identify secondary spillover as a significant predictor of human transmissibility—potentially owing to a limited sample size or more muddled evolutionary relationships between viruses and their secondary hosts—we cannot infer whether spillover from secondary hosts is constrained by the same virulence–transmission trade-off observed in reservoir hosts. However, increased CFRs in secondary host species closely related to humans suggest that these hosts could function as ‘gateways’ for virulent viruses originally derived from more distant reservoirs. The virulence-inducing viral traits driving CFR upon spillover from secondary hosts may largely reflect viruses' long-term evolutionary history in more phylogenetically distant reservoirs. By contrast, short-term history within more closely related secondary hosts could facilitate pre-adaptation to human transmission. Thus, secondary spillover—by which virulent viruses from distant phylogenetic backgrounds spill over to humans through more closely related, secondary hosts—may pose the greatest zoonotic threat.
Across mammalian host orders, host species most closely related to humans harbour zoonoses of lower impact in terms of morbidity and mortality but with an elevated propensity for human-to-human transmission. More distantly related hosts—in particular, order Chiroptera—harbour highly virulent zoonoses that nonetheless have a lower capacity for between-human transmission. Chiroptera, previously posited to represent a ‘special’ order of mammalian hosts for zoonotic viruses [50], does appear to uniquely shift zoonoses into otherwise uncharted territory on the virulence axis. Though occupying the same general region of the host phylogenetic distance axis as order Rodentia, bat zoonoses yield CFRs more than double those resulting from rodents.
Results from the viral trait models support previous findings that multi-host pathogens are more likely to emerge and transmit in human populations [2,10,28,51,52]. Consistent with findings from Olival et al. [28], we identify maximum phylogenetic host breadth, a marker of viral generalism, as a powerful predictor of zoonotic behaviour for the directly transmitted mammalian viruses queried in our dataset. However, our results indicate that, in addition to the breadth of a virus's host range, the positioning of that range, with respect to phylogenetic distance from humans, has important predictive capacity. Thus, predictions of zoonotic risk will differ for a generalist virus that evolved in a reservoir environment that is phylogenetically close to humans (i.e. primates) versus one that is distant (i.e. bats). This distinction is important, because our work further emphasizes that distinct viral families tend to preferentially infect distinct host orders. Furthermore, in keeping with our host model results, we observed that viral traits predicting zoonotic behaviour following emergence demonstrate opposite impacts on virulence and transmissibility in human hosts—indicating trade-offs between these two traits. Host phylogenetic distance, corresponding to the evolutionary host environment in which a virus evolved, is positively correlated with mortality incurred by an emerging virus in human hosts, but negatively correlated with viral capacity for human-to-human transmission.
The multi-dimensional definition of zoonotic risk considered in this analysis highlights limitations in our understanding of emerging zoonoses to date. Historically, most work in this field has been restricted to descriptions and predictions of viral, host and geographical ‘hotspots’ for zoonosis, with little consideration of variation in the impact of a given spillover event post-emergence. Our work emphasizes the power of a more nuanced approach to assessing zoonotic risk, but also uncovers gross gaps in data and reporting which will present challenges for future research extensions. To facilitate tracking of directionality in transmission chains, we restricted our database to mammal host–virus associations previously confirmed via polymerase chain reaction or viral isolation, excluding unconfirmed or serologically identified mammalian hosts. For example, compelling serological evidence suggests that bats probably function as the natural reservoir host for Middle Eastern respiratory syndrome (MERS-CoV), a coronavirus similar to bat-derived SARs-CoV [53], but evidence is insufficient to support inclusion of this association in our study. Indeed, many animal hosts for emerging zoonoses remain unidentified or unconfirmed, and for the majority of known hosts, we lack data regarding transmission and infection dynamics. These gaps in reporting limit our capacity for inference.
The extent to which human mortality and transmissibility may differ depending on the reservoir or spillover host in question has yet to be documented for the vast majority of zoonoses—indeed, we identified distinct measures of CFR and transmissibility that could be traced to specific spillover hosts only for two viruses: Nipah and Marburg. As both of these examples support our finding that changes in a spillover transmission chain are associated with shifts in virulence and transmissibility, collection of data from additional systems demonstrating both secondary and primary zoonotic sources is thus a major research priority. Ebola virus outbreaks, for example, have varied extensively in both virulence and transmissibility across the past four decades of emergence [15,54]. The extent to which these variations may be attributable to phylogenetic differences in the source host or transmission chain remains, to our knowledge, unexplored. A more nuanced framework for understanding variation in severity and transmissibility across all potential zoonotic hosts will nonetheless be central to any future effort to develop targeted and effective public health strategies for intervention and control.
Supplementary Material
Supplementary Material
Acknowledgements
The authors thank Kevin J. Olival for laying the groundwork for this study and providing feedback on the initial project proposal. The authors also thank Samuel Alizon and two anonymous reviewers for their valuable comments; Whitney Mgbara for guidance, stimulating conversation and thoughtful comments throughout the development of the study design; Anecia Gentles and Kimberly Rivera for feedback on the data compilation methods; and Lawrence Uricchio for statistical recommendations.
Data accessibility
All datasets used in the analyses outlined in this article have been included in the electronic supplementary material.
Authors' contributions
S.G., C.E.B. and M.B. conceived and designed the study. S.G. and C.E.B. collected the data and conducted the analyses. All authors participated in writing the manuscript.
Competing interests
We declare we have no competing interests.
Funding
S.G. and E.V. are supported by National Science Foundation Graduate Research Fellowships; M.B. is supported by the National Institutes of Health (grant no. GM122061) and Bioscience for the Future (grant no. BB/L010879/1); and C.E.B. is supported by the Miller Institute for Basic Research at the University of California, Berkeley. This work is additionally supported by DARPA PREEMPT program Cooperative Agreement no. D18AC00031.
Disclaimer
The content of the information does not necessarily reflect the position or the policy of the US government, and no official endorsement should be inferred.
References
- 1.Woolhouse M, Scott F, Hudson Z, Howey R, Chase-Topping M. 2012. Human viruses: discovery and emergence. Phil. Trans. R. Soc. B 367, 2864–2871. ( 10.1098/rstb.2011.0354) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Woolhouse MEJ, Gowtage-Sequeria S. 2005. Host range and emerging and reemerging pathogens. Emerg. Infect. Dis. 11, 1842–1847. ( 10.3201/eid1112.050997) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gomez JM, Nunn CL, Verdu M. 2013. Centrality in primate-parasite networks reveals the potential for the transmission of emerging infectious diseases to humans. Proc. Natl Acad. Sci. USA 110, 7738–7741. ( 10.1073/pnas.1220716110) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Han BA, Schmidt JP, Bowden SE, Drake JM. 2015. Rodent reservoirs of future zoonotic diseases. Proc. Natl Acad. Sci. USA 112, 7039–7044. ( 10.1073/pnas.1501598112) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Han BA, Schmidt JP, Alexander LW, Bowden SE, Hayman DTS, Drake JM. 2016. Undiscovered bat hosts of filoviruses. PLoS Negl. Trop. Dis. 10, e0004815 ( 10.1371/journal.pntd.0004815) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pulliam JRC, Dushoff J. 2009. Ability to replicate in the cytoplasm predicts zoonotic transmission of livestock viruses. J. Infect. Dis. 199, 565–568. ( 10.1086/596510) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Geoghegan JL, Senior AM, Di Giallonardo F, Holmes EC. 2016. Virological factors that increase the transmissibility of emerging human viruses. Proc. Natl Acad. Sci. USA 113, 4170–4175. ( 10.1073/pnas.1521582113) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Brierley L, Pedersen AB, Woolhouse MEJ.2019. Tissue tropism and transmission ecology predict virulence of human RNA viruses. bioRxiv. (Cited 16 May 2019). See http://biorxiv.org/lookup/doi/10.1101/581512 . [DOI] [PMC free article] [PubMed]
- 9.Walker JW, Han BA, Ott IM, Drake JM. 2018. Transmissibility of emerging viral zoonoses. PLoS ONE 13, e0206926 ( 10.1371/journal.pone.0206926) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kreuder Johnson C, et al. 2015. Spillover and pandemic properties of zoonotic viruses with high host plasticity. Sci. Rep. 5, 14830 ( 10.1038/srep14830) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wolfe ND, Dunavan CP, Diamond J. 2007. Origins of major human infectious diseases. Nature 447, 279–283. ( 10.1038/nature05775) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Woolhouse MEJ, Haydon DT, Antia R. 2005. Emerging pathogens: the epidemiology and evolution of species jumps. Trends Ecol. Evol. 20, 238–244. ( 10.1016/j.tree.2005.02.009) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lloyd-Smith JO, George D, Pepin KM, Pitzer VE, Pulliam JRC, Dobson AP, Hudson PJ, Grenfell BT. 2009. Epidemic dynamics at the human–animal interface. Science 326, 1362–1367. ( 10.1126/science.1177345) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Martinez VP, Bellomo C, San Juan J, Pinna D, Forlenza R, Elder M, Padula PJ. 2005. Person-to-person transmission of Andes virus. Emerg. Infect. Dis. 11, 1848–1853. ( 10.3201/eid1112.050501) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Urbanowicz RA, et al. 2016. Human adaptation of Ebola virus during the West African outbreak. Cell 167, 1079–1087.e5. ( 10.1016/j.cell.2016.10.013) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pavio N, Meng X-J, Renou C. 2010. Zoonotic hepatitis E: animal reservoirs and emerging risks. Vet. Res. 41, 46 ( 10.1051/vetres/2010018) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.May RM, Anderson RM. 1990. Parasite–host coevolution. Parasitology 100, S89–S101. ( 10.1017/S0031182000073042) [DOI] [PubMed] [Google Scholar]
- 18.Woolhouse MEJ, Webster JP, Domingo E, Charlesworth B, Levin BR. 2002. Biological and biomedical implications of the co-evolution of pathogens and their hosts. Nat. Genet. 32, 569–577. ( 10.1038/ng1202-569) [DOI] [PubMed] [Google Scholar]
- 19.Little TJ, Shuker DM, Colegrave N, Day T, Graham AL. 2010. The coevolution of virulence: tolerance in perspective. PLoS Pathog. 6, e1001006 ( 10.1371/journal.ppat.1001006) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ewald PW. 1983. Host–parasite relations, vectors, and the evolution of disease severity. Annu. Rev. Ecol. Syst. 14, 465–485. ( 10.1146/annurev.es.14.110183.002341) [DOI] [Google Scholar]
- 21.Anderson RM, May RM. 1982. Coevolution of hosts and parasites. Parasitology 85, 411 ( 10.1017/S0031182000055360) [DOI] [PubMed] [Google Scholar]
- 22.Izhar R, Ben-Ami F. 2015. Host age modulates parasite infectivity, virulence and reproduction. J. Anim. Ecol. 84, 1018–1028. ( 10.1111/1365-2656.12352) [DOI] [PubMed] [Google Scholar]
- 23.Mackinnon MJ, Read AF. 2004. Immunity promotes virulence evolution in a malaria model. PLoS Biol. 2, e230 ( 10.1371/journal.pbio.0020230) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Andre J-B, Ferdy J-B, Godell B. 2003. Within-host parasite dynamics, emerging trade-off, and evolution of virulence with immune system. Evolution 57, 1489–1497. ( 10.1111/j.0014-3820.2003.tb00357.x) [DOI] [PubMed] [Google Scholar]
- 25.Fleming-Davies AE, Williams PD, Dhondt AA, Dobson AP, Hochachka WM, Leon AE, Ley DH, Osnas EE, Hawley DM. 2018. Incomplete host immunity favors the evolution of virulence in an emergent pathogen. Science 359, 1030–1033. ( 10.1126/science.aao2140) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Boots M, Sasaki A. 1999. ‘Small worlds’ and the evolution of virulence: infection occurs locally and at a distance. Proc. R. Soc. Lond. B 266, 1933–1938. ( 10.1098/rspb.1999.0869) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lipsitch M, Herre EA, Nowak MA. 1995. Host population structure and the evolution of virulence: a ‘law of diminishing returns'. Evolution 49, 743–748. ( 10.1111/j.1558-5646.1995.tb02310.x) [DOI] [PubMed] [Google Scholar]
- 28.Olival KJ, Hosseini PR, Zambrana-Torrelio C, Ross N, Bogich TL, Daszak P. 2017. Host and viral traits predict zoonotic spillover from mammals. Nature 546, 646–650. ( 10.1038/nature22975) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Woolhouse MEJ, Brierley L. 2018. Epidemiological characteristics of human-infective RNA viruses. Sci. Data 5, 180017 ( 10.1038/sdata.2018.17) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Parr CS, et al. 2014. The encyclopedia of life v2: providing global access to knowledge about life on earth. Biodivers. Data J. 2, e1079 ( 10.3897/BDJ.2.e1079) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jones KE, et al. 2009. PanTHERIA: a species-level database of life history, ecology, and geography of extant and recently extinct mammals. Ecology 90, 2648 ( 10.1890/08-1494.1) [DOI] [Google Scholar]
- 32.Tacutu R, et al. 2018. Human ageing genomic resources: new and updated databases. Nucleic Acids Res. 46(Database issue), D1083–D1090. ( 10.1093/nar/gkx1042) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Myers P, Espinosa R, Parr CS, Jones T, Hammond GS, Dewey TA. 2019. The animal diversity web (online). See https://animaldiversity.org.
- 34.Cable JM, Enquist BJ, Moses ME. 2007. The allometry of host–pathogen interactions. PLoS ONE 2, e1130 ( 10.1371/journal.pone.0001130) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Huang ZYX, de Boer WF, van Langevelde F, Olson V, Blackburn TM, Prins HHT. 2013. Species' life-history traits explain interspecific variation in reservoir competence: a possible mechanism underlying the dilution effect. PLoS ONE 8, e54341 ( 10.1371/journal.pone.0054341) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Banerjee S, Perelson AS, Moses M. 2017. Modelling the effects of phylogeny and body size on within-host pathogen replication and immune response. J. R. Soc. Interface. 14, 20170479 ( 10.1098/rsif.2017.0479) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Althaus CL. 2015. Of mice, macaques and men: scaling of virus dynamics and immune responses. Front. Microbiol. 6, 3 ( 10.3389/fmicb.2015.00355) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gandon S, Jansen VAA, Van Baalen M. 2007. Host life history and the evolution of parasite virulence. Evolution 55, 1056–1062. ( 10.1111/j.0014-3820.2001.tb00622.x) [DOI] [PubMed] [Google Scholar]
- 39.Koella JC, Restif O. 2001. Coevolution of parasite virulence and host life history. Ecol. Lett. 4, 207–214. ( 10.1046/j.1461-0248.2001.00213.x) [DOI] [Google Scholar]
- 40.Hily JM, García A, Moreno A, Plaza M, Wilkinson MD, Fereres A, Fraile A, García-Arenal F. 2014. The relationship between host lifespan and pathogen reservoir potential: an analysis in the system Arabidopsis thaliana—cucumber mosaic virus. PLoS Pathog. 10, e1004492 ( 10.1371/journal.ppat.1004492) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lidsky PV, Andino R. 2017. Protection from epidemics is a driving force for evolution of lifespan setpoints. bioRxiv. 6 November 2017 (Cited 22 May 2019). See http://biorxiv.org/lookup/doi/10.1101/215202.
- 42.Farrell MJ, Davies J. 2019. Disease mortality in domesticated animals is predicted by host evolutionary relationships. Proc. Natl Acad. Sci. USA 116, 7911–7915. ( 10.1073/pnas.1817323116) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wood SN. 2001. mgcv: GAMs and generalized ridge regression for R. R News 1, 20–25. [Google Scholar]
- 44.Marra G, Wood SN. 2011. Practical variable selection for generalized additive models. Comput. Stat. Data Anal. 55, 2372–2387. ( 10.1016/j.csda.2011.02.004) [DOI] [Google Scholar]
- 45.Mollentze N, Nel LH, Townsend S, le Roux K, Hampson K, Haydon DT, Soubeyrand S. 2014. A Bayesian approach for inferring the dynamics of partially observed endemic infectious diseases from space-time-genetic data. Proc. R. Soc. B 281, 20133251 ( 10.1098/rspb.2013.3251) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Fekadu M, Endeshaw T, Alemu W, Bogale Y, Teshager T, Olson JG. 1996. Possible human-to-human transmission of rabies in Ethiopia. Ethiop. Med. J. 34, 123–127. [PubMed] [Google Scholar]
- 47.Helmick CG, Tauxe RV, Vernon AA. 1987. Is there a risk to contacts of patients with rabies? Rev. Infect. Dis. 9, 511–518. ( 10.1093/clinids/9.3.511) [DOI] [PubMed] [Google Scholar]
- 48.Longdon B, Hadfield JD, Webster CL, Obbard DJ, Jiggins FM. 2011. Host phylogeny determines viral persistence and replication in novel hosts. PLoS Pathog. 7, e1002260 ( 10.1371/journal.ppat.1002260) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Longdon B, Hadfield JD, Day JP, Smith SCL, McGonigle JE, Cogni R, Cao C, Jiggins FM. 2015. The causes and consequences of changes in virulence following pathogen host shifts. PLoS Pathog. 11, e1004728 ( 10.1371/journal.ppat.1004728) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Brook CE, Dobson AP. 2015. Bats as ‘special’ reservoirs for emerging zoonotic pathogens. Trends Microbiol. 23, 172–180. ( 10.1016/j.tim.2014.12.004) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Cleaveland S, Laurenson MK, Taylor LH. 2001. Diseases of humans and their domestic mammals: pathogen characteristics, host range and the risk of emergence. Phil. Trans. R. Soc. Lond. B 356, 991–999. ( 10.1098/rstb.2001.0889) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Garamszegi LZ. 2006. The evolution of virulence and host specialization in malaria parasites of primates. Ecol. Lett. 9, 933–940. ( 10.1111/j.1461-0248.2006.00936.x) [DOI] [PubMed] [Google Scholar]
- 53.Memish ZA, et al. 2013. Middle East respiratory syndrome coronavirus in bats, Saudi Arabia. Emerg. Infect. Dis. 19, 1819–1823. ( 10.3201/eid1911.131172) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Holmes EC, Dudas G, Rambaut A, Andersen KG. 2016. The evolution of Ebola virus: insights from the 2013–2016 epidemic. Nature 538, 193–200. ( 10.1038/nature19790) [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All datasets used in the analyses outlined in this article have been included in the electronic supplementary material.